


Abstract
Sex is a modulator of health that has been historically overlooked in biomedical research. Recognizing this knowledge gap, funding agencies now mandate the inclusion of sex as a biological variable with the goal of stimulating efforts to illuminate the molecular underpinnings of sex biases in health and disease. DNA methylation (DNAm) is a strong molecular candidate for mediating such sex biases; however, a robust and well characterized annotation of sex differences in DNAm is yet to emerge. Beginning with a large (n = 3795) dataset of DNAm profiles from normative adult whole blood samples, we identified, validated and characterized autosomal sex-associated co-methylated genomic regions (sCMRs). Strikingly, sCMRs showed consistent sex differences in DNAm over the life course and a subset were also consistent across cell, tissue and cancer types. sCMRs included sites with known sex differences in DNAm and links to health conditions with sex biased effects. The robustness of sCMRs enabled the generation of an autosomal DNAm-based predictor of sex with 96% accuracy. Testing this tool on blood DNAm profiles from individuals with sex chromosome aneuploidies (Klinefelter [47,XXY], Turner [45,X] and 47,XXX syndrome) revealed an intimate relationship between sex chromosomes and sex-biased autosomal DNAm.
INTRODUCTION
Sex is a biological variable that shapes human development, health and disease, yet has been historically overlooked in biomedical research (1). This omission has resulted in a limited understanding of the molecular underpinnings of sex differences in disease risk, onset and progression, and manifested in inequalities in the prevention, diagnosis and treatment of a wide range of conditions. Underscoring this knowledge gap, funding agencies now mandate the inclusion of sex as a variable in biological research (2), seeking to stimulate efforts to understand the role of sex in shaping diverse aspects of biology.
Large population-based studies have identified sex differences in several molecular processes thought to underlie sex biases in health and disease (3), including DNA methylation (DNAm) and gene expression (e.g. (4–6)). DNAm, a chemical modification that occurs most often on cytosines in the context of cytosine-guanine (CpG) dinucleotides, is emerging as a strong molecular candidate for mediating the relationship between sex and disease (3). DNAm is altered in a wide range of diseases (7) and plays key roles in several sex-associated molecular processes including genomic imprinting (i.e. DNAm-based monoallelic gene silencing based on the sex of the parent of origin) (8) and X chromosome inactivation (i.e. DNAm-based silencing of most genes on a single X chromosome in XX females to equalize gene expression levels to XY males) (9).
Despite growing evidence of a role for DNAm in mediating the relationship between sex and disease (10,11), epigenome wide association studies (EWAS) are often underpowered to support sex-stratified analyses. As a result, EWAS often have to prioritize the detection of robust DNAm-disease associations and control for possible sex differences using statistical approaches. While powerful, controlling for sex overlooks its contribution to health and disease and yields effect estimates that are averages between males and females.
One way to stimulate the integration of sex in epigenome studies is to improve our understanding of the effects of this biological variable on DNAm. In this regard, several efforts have highlighted sex differences in DNAm in blood (6,10,12–17) and other tissues and cell types (18–23). Collectively, these studies support the existence of sex-biased DNAm signatures across the autosomes, highlight CpG-dense regions (CpG islands) as key sites of sex differences, and emphasize that autosomal loci showing sex-biased DNAm most often show higher DNAm levels in females compared to males. Despite these advances, many questions remain regarding the effect of sex on DNAm and a clear picture of genomic loci showing sex-biased DNAm is yet to emerge.
To improve our understanding of the role of sex on DNAm, we set out to generate a robust annotation of autosomal genomic regions showing sex-biased DNAm patterns. We began by comparing sites previously reported to show male-female differences in DNAm and found inconsistencies across studies, an observation that prompted us to re-examine this question using a stringent and well-powered (n = 3795) approach. In this study, we focused on genomic regions rather than individual sites, as the former have been suggested to yield more reproducible associations. Regions of correlated DNAm levels were defined in normative whole blood samples using the CoMeBack method (24) and tested for sex differences in DNAm using strict criteria. In this way, 179 sex co-methylated regions (denoted sCMRs) were identified and these showed consistent sex differences in DNAm across the lifespan and several cell, tissue and tumor types. Importantly, sCMRs identified using Illumina Infinium Human Methylation 450K BeadChip data validated in a reduced representation bisulfite sequencing (RRBS) dataset, suggesting transferability of findings across genomic platforms. Encouraged by the reproducibility of sCMRs, we built an easy to use and remarkably accurate (96%) autosomal DNAm-based predictor of sex, a tool that can be used to impute or evaluate sex information without the need for sex chromosome DNAm data. Using this tool in a unique dataset of individuals with sex chromosome aneuploidies [47,XXY (Klinefelter Syndrome), 45,X (Turner Syndrome) and 47,XXX syndrome patients] revealed a strong link between sex chromosome complement and sex differences on autosomal DNAm. Altogether, our study provides a reproducible and highly detailed annotation of autosomal genomic regions showing sex-DNAm associations and implicates several pathways in their establishment.
MATERIALS AND METHODS
Software
Preprocessing, quality control, analysis, replication and enrichment analyses were done using R version 3.6.3 (25).
Datasets
All datasets used in this study are listed in Table 1.
Table 1.
Datasets used in this study
| Dataset | Tissue | N | Females | Males | Age | Analysis | Reference |
|---|---|---|---|---|---|---|---|
| GSE55763 | Whole blood | 2669 | 860 | 1809 | 31–75 | CMR construction, sCMR discovery and construction of sex predictor | (26) |
| GSE84727 | Whole blood | 377 | 92 | 285 | 25–66 | (27) | |
| GSE80417 | Whole blood | 224 | 117 | 107 | 26–79 | (95) | |
| GSE111629 | Whole blood | 175 | 68 | 107 | 26–55 | (47) | |
| GSE72680 | Whole blood | 350 | 244 | 106 | 26–77 | (98) | |
| GSE125105 | Whole blood | 697 | 388 | 309 | 17–87 | sCMR Validation and testing of sex predictor | (96) |
| GSE132203 | Whole blood | 794 | 571 | 223 | 18–76 | Testing of sex predictor | (97) |
| ARIES | Cord blood | 905 | 465 | 440 | 0 | sCMR concordance across the life span and testing of sex predictor | |
| Whole blood/white blood cells | 907/63 | 454/36 | 453/27 | 7–9 | (41) | ||
| White blood cells | 970 | 502 | 648 | 14.5–19 | |||
| GSE79100 | Kidney | 31 | 16 | 15 | sCMR concordance across tissues. *Testing of sex predictor | (43) | |
| GSE80261 | Buccal* | 96 | 57 | 39 | (44) | ||
| GSE61258 | Liver | 79 | 34 | 45 | (45) | ||
| GSE64509 | Brain | 25–41 | 17–22 | 8–11 | (47) | ||
| Cerebellum | 32 | 21 | 11 | ||||
| Frontal Cortex | 41 | 22 | 19 | ||||
| Hippocampus | 25 | 17 | 8 | ||||
| Occipital lobe | 33 | 22 | 11 | ||||
| Temporal lobe | 29 | 21 | 8 | ||||
| GSE87640 | Immune cells | 19–20 | 7–8 | 12 | (46) | ||
| Monocytes | 20 | 8 | 12 | ||||
| CD4T | 20 | 8 | 12 | ||||
| CD8T | 19 | 7 | 12 | ||||
| GSE132513 | E2 containing medium | 9 | sCMR regulation by estrogen in a MCF-7 HTB-22 breast cancer cell line | ||||
| E2-deprived medium | 6 | ||||||
| E2-deprived to E2 containing | 3 | ||||||
| TCGA: Thyroid | Thyroid carcinoma (THCA) | 498 | 364 | 137 | sCMR concordance across cancer types and matched control tissues | ||
| normals | 54 | 40 | 14 | ||||
| TCGA: Lung | Lung adenocarcinoma (LUAD) | 455 | 243 | 212 | |||
| normals | 32 | 15 | 17 | ||||
| TCGA: Kidney | Kidney renal clear cell carcinoma | 316 | 112 | 204 | |||
| normals | 160 | 54 | 106 | ||||
| TCGA: Liver | Liver hepatocellular carcinoma (LIHC) | 375 | 121 | 254 | |||
| normals | 51 | 20 | 31 | ||||
| TCGA: Colon | Colon adenocarcinoma (COAD) | 250 | 133 | 157 | |||
| normals | 38 | 17 | 21 | ||||
| TCGA: Bladder | Bladder Urothelial carcinoma | 409 | 107 | 302 | |||
| normals | 21 | 10 | 11 | ||||
| TCGA: Skin | Skin Cutaneous Melanoma (SKCM) | 105 | 43 | 62 | |||
| TCGA: Stomach | Stomach adenocarcinoma (STAD) | 393 | 135 | 257 | |||
| Sex chromosome abnormalities | 46,XX | 33 | Sex predictor and concordance with normative samples | (63); (64); (65) | |||
| 46,XY | 67 | ||||||
| 47,XXY | 67 | ||||||
| 45,X | 33 | ||||||
| 47,XXX | 7 | ||||||
| GSE136849 | Peripheral blood | 158 | 79 | 79 | 27–40 | Reduced representation bisulfite (RRBS) sequencing validation | (37) |
Discovery of sCMRs
Sex differences in DNAm levels were identified in a discovery cohort composed of five publicly available whole blood Illumina Infinium Human Methylation 450K BeadChip datasets (GSE55763, GSE80417, GSE72680, GSE84727, GSE111629) (Table 1) (26–31). Whole blood samples were selected for their clinical relevance and abundance. Samples annotated to a disease state or with potential sex mislabels (see below) were removed from the analysis, resulting in a merged dataset of 3795 (2414 males and 1381 female) normative adult (age 25–80 years old) whole blood samples (Figure 1A). Datasets were processed as described previously including batch correction and normalization (24). X and Y chromosome probes, genotyping probes, probes overlapping polymorphic loci at the CpG or single-base extension site, and probes predicted to cross-hybridize to the X and Y chromosome were removed, leaving 404 779 probes for analysis (32,33).
Figure 1.

sCMRs were reproducible, detected across the autosomes and included sites with known sex differences in DNAm levels. (A) Schematic of the sCMR discovery, validation and characterization steps. An aggregate cohort of 3795 (2414 males and 1381 females) adult whole blood DNAm samples was assembled and used to identify CMRs. Five sex-balanced random subsamples were generated and used to identify sCMRs. sCMRs were defined as CMRs having an FDR < 0.05% and a composite beta difference > 4% when comparing males and females. In addition, for a CMR to be considered all probes had to show the same sex-biased DNAm pattern, either higher DNAm levels in males compared to females or vice versa. (B) Manhattan plot showing the distribution of sCMRs across all the autosomes. (C) Boxplot showing male versus female DNAm levels for the three sites included in the SLC6A4 sCMR (chr17:28521337–28562986). cg05951817, cg22584138 and cg03363743 showed significant methylation (beta) differences of 6%, 8% and 3%, respectively, when comparing males and females (P-values of 0). The SLC6A4 sCMR was found at the 5′ UTR, a region previously shown to be differentially methylated between males and females in brain tissue (76).
Co-methylated regions (CMRs) were constructed using CoMeBack with default parameters and a Spearman correlation cut-off of 30% (24) (See Supplementary Table S1 for all CMRs). For each CMR, a composite beta value was constructed using a weighted sum of all the individual probe betas contained within a CMR. Individual probe weights were proportional to the variability of their DNAm levels and calculated as their scaled (to sum to one) loadings of the first principal component (see code availability). CMR composite betas were used to identify sex-associated CMRs using the following model:
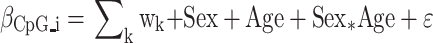 |
where wk (Σk wk = 1) are the blood cell type counts estimated with the Houseman method (34) and sex was coded as 0/1 (see below). Since the discovery cohort had more males than females, we ran the model on five sex-balanced random subsamples (Figure 1A). In all five subsample comparisons, 205 CMRs were differentially methylated between males and females as defined by a strict significance criterion: (i) a false discovery rate (FDR) <0.05, (ii) a composite beta difference > 4% between the sexes and (iii) being fully composed of probes with higher mean DNAm levels in males compared to females or vice versa. A 4% composite beta threshold was selected because it captured 99% of the absolute difference between technical replicates present in the GSE55763 dataset used in the discovery of sCMRs. To ensure the robust performance of our model in terms of the numerical methods used in the R software (25), the analysis was verified twice, flipping the coding of males and females between zero and one.
The 205 CMRs with significant sex differences in DNAm were validated using an independently processed Illumina Infinium Human Methylation 450K BeadChip dataset of 699 adult whole blood samples (312 males and 387 females) (GSE125105) (Table 1) (Figure 1A). After quality control, 201 CMRs were present in the validation dataset, 44 were truncated and 14 were represented by a single probe. All CMRs represented by at least one probe in the validation dataset were examined, resulting in 179 (89%) validated sex-CMRs (sCMRs). Similar to the discovery cohort, validated CMRs were strictly defined: (i) a q-value FDR < 0.05, (ii) an adjusted composite beta fold change > 4% and (iii) being fully composed of probes that showed higher mean DNAm levels (within one standard error of the mean) in males compared to females or vice versa. To account for the smaller sample size of the validation dataset, the fold change cut-off was adjusted by subtracting the standard errors of the estimated validation cohort betas. The 179 sCMRs were used for all subsequent analyses (Supplementary Table S2).
To determine if inclusion of an effect size cut-off led to higher rates of reproducibility across studies, we repeated the discovery and validation approach with a 3% and without an effect size cut-off. With a 3% cut-off, 471 sCMRs were discovered and 372 (79%) validated in the independent cohort (Supplementary Table S3). Without an effect size cut-off, 1595 sCMRs were identified and 496 (31%) validated in the independent cohort (Supplementary Table S4).
Identification of potential sex mislabels
The discovery and validation datasets were assessed independently for potential sex mislabels. Two previously described checks were performed to test concordance between the metadata sex variable and the expected sex chromosome DNAm patterns (35). The first check clusters samples based on the beta values of all probes mapping to the X and Y chromosomes (11 648 probes). The second check clusters samples based on the beta values of five probes mapping to the XIST promoter (cg03554089, cg12653510, cg05533223, cg11717280, cg20698282). In the first check, X and Y chromosome probes separate males (46,XY) and females (46,XX) by the presence of male-specific Y chromosome DNAm and female-specific X-chromosome mono-allelic methylation resulting from X chromosome inactivation (35). In the second check, XIST promoter probes separate samples by the number of X chromosomes, providing evidence that samples classified as male (46,XY) and female (46,XX) are not affected by X chromosome aneuploidies (35). For all datasets two main clusters emerged for both checks. Nine samples clustered with the opposite sex in the discovery dataset and were removed from the analysis. In the validation dataset, three male samples showed the expected clustering pattern when considering the XY probes but clustered with female samples when considering the XIST probes. These males were suspected to have a XXY sex chromosome complement, a possibility ruled out in all situations via genomic copy number estimates by the R package conumee (36). These samples were deemed to possess a XY sex chromosome complement and retained in the analysis as genetic males.
Reduced representation bisulfite sequencing (RRBS) analysis
Normalized normative adult peripheral blood RRBS profiles were obtained from GSE136849 (Table 1) (37). Because the RRBS dataset coordinates are based on the GRCh38/hg38 human genome assembly, they were converted to the GRCh37/hg19 assembly using liftover from the UCSC. Only sites present in at least half of the samples were considered. Sites in the RRBS dataset contained within sCMRs were deemed validated if they had a q-value FDR < 0.05 and an adjusted composite beta fold change > 4% when comparing males and females and showed the same direction of change observed in the discovery and validation cohort.
Stability of sCMRs across the life course and several cell, tissue and tumor types
The Avon Longitudinal Study of Parents and Children (ALSPAC) is a large, prospective cohort study that recruited 14 541 pregnant women residing in Avon, UK with expected dates of delivery between 1 April 1991 and 31 December 31 1992 (38–40). Of these initial pregnancies, there was a total of 14 676 fetuses, resulting in 14 062 live births and 13 988 children who were alive at 1 year of age. Further details of the study and available data are provided on the study website through a fully searchable data dictionary (http://www.bristol.ac.uk/alspac/). Ethical approval for the study was obtained from the ALSPAC Law and Ethics Committee and the Local Research Ethics Committees. Consent for biological samples was collected in accordance with the Human Tissue Act (2004). All data are available by request from the Avon Longitudinal Study of Parents and Children Executive Committee for researchers who meet the criteria for access to confidential data (http://www.bristol.ac.uk/alspac/researchers/our-data/).
The ALSPAC generated blood-based DNAm profiles at birth, 7 and 15 years of age are part of the Accessible Resource for Integrated Epigenomic Studies (ARIES), a subsample of 1018 mother-child pairs from the ALSPAC cohort (41). DNA samples were extracted from cord blood on delivery, and from peripheral blood samples in childhood (age 7) and adolescence (age 15–17) according to established procedures (41). Background correction and functional normalization were performed using the R-package meffil (42). Samples with > 10% of sites with a detection P-value > 0.01 or a bead count < 3 were removed from further analysis. All samples passing quality control procedures were used in subsequent analyses (Table 1). In the ARIES cohort, the concordance of sCMRs was tested with a linear model accounting for cell types predicted using the Houseman method (34).
The consistency of sex differences in DNAm levels at sCMRs across tissues and immune cell types was examined using a series of datasets (43–47) (Table 1). For these analyses only samples labeled ‘control’ were used. All the datasets were normalized as described previously (42).
To examine if sex differences in DNAm at sCMRs detected in blood were recapitulated in tumor and matched control samples, preprocessed and normalized (TCGA level-3) Illumina Infinium Human Methylation 450K BeadChip data for several cancer datasets was retrieved from the Firebrowse repository (firebrowse.org, version 2016_01_28) (Table 1). Only datasets with at least 100 samples and 25% of samples representing any one sex were considered: Thyroid carcinoma (THCA), skin cutaneous melanoma (SKCM), stomach adenocarcinoma (STAD), lung adenocarcinoma (LUAD), kidney renal clear cell carcinoma (KIRC), liver hepatocellular carcinoma (LIHC), colon adenocarcinoma (COAD) and bladder urothelial carcinoma (BLCA).
For the cell, tissue, and cancer analyses, a sCMR was deemed to show significant sex differences in DNAm if it satisfied a Welch test P-value < 0.05 and showed the same direction of the sex-biased DNAm observed in blood.
Characterization and enrichment of sCMRs for various genomic features
Gene coordinates based on the human GRCh37/hg19 reference genome were derived from ENSEMBL (release 99) (48). A gene was defined as the genomic region from the 5′ to the 3′ UTR and an overlap with a CMR was reported if there was any intersection between the CMR coordinates and that of the gene coordinates plus 5 kb upstream or downstream. GO terms and KEGG pathways were annotated using the missMethyl R-package (49). Homo sapiens imprinted genes were obtained from www.geneimprint.com (accessed July 2019). Imprinting control centers and their genomic ranges were reported previously (50,51). A comprehensive list of lncRNAs was used to map CMRs to lncRNAs (52). Genes with significant gene expression differences between males and females were reported previously (4,5,53). Chromatin states estimated with ChromHMM were obtained from the Roadmap Epigenomics project (54) and CpG feature information from the Illumina Infinium Human Methylation 450K BeadChip manifest. Transcription factor binding site (TFBS) motifs were obtained from the HOCOMOCO v11 database (55). Transcription factors were annotated to CMRs by scanning a region 200 bp upstream or downstream of every site contained within a CMR using the FIMO tool from the MEME software suite (56). An overlap was reported if the region contained a TFBS motif. The effect of DNAm on TF binding affinity was reported previously (57). DNAm quantitative trait loci (mQTLs) were reported previously (58,59). For the ARIES database, we focused on adult mQTLs. Correlated regions of systemic interindividual variation were reported previously (60). Throughout, enrichments were evaluated using Fisher exact tests comparing sCMRs to the CMR background (Supplementary Table S1). A q-value < 0.05 following Benjamini–Hochberg multiple test correction was deemed significant. Trait enrichment was done by comparing sCMR sites to the CMR background using the EWAS atlas (61).
Analysis of sCMRs in relation to sex hormone biology
Sites with significant associations (FDR < 0.05) to changes in reproductive hormones over the puberty transition were reported previously (62). A Fisher exact test q-value < 0.05 following Benjamini–Hochberg multiple test correction was deemed significant when comparing sCMRs to the CMR background (Supplementary Table S1).
The effect of estrogen exposure on sCMR DNAm levels was examined using GSE132513, an Illumina Infinium Human Methylation EPIC BeadChip dataset of MCF-7 HTB-22 breast cancer cell lines grown for 4 or 14 days in media with and without estradiol (Table 1). Composite betas were compared using a Welch test and P-values < 0.05 were considered significant.
To examine the relationship between sCMR DNAm levels and estrogen and progesterone receptor status, female breast invasive carcinoma (BRCA) preprocessed and normalized (TCGA level-3) Illumina Infinium Human Methylation EPIC BeadChip data and the associated estrogen and progesterone receptor status metadata were retrieved from the Firebrowse repository (firebrowse.org, version 2016_01_28). The R package umap (63) was used to obtain the Uniform Manifold Approximation and Projection (UMAP) plot with the following parameters: random_state = 123, n_neighbors = 40, min_dist = 0.2. UMAP clustering patterns were inspected for evidence that sCMR DNAm values were associated with estrogen/progesterone receptor status.
Analysis of sCMRs in relation to sex chromosomes
Klinefelter (47,XXY), Turner (45,X) and 47,XXX syndrome patient DNAm data was described previously (63–65) (Table 1). These datasets were preprocessed and normalized independently of the discovery and validation cohort, using the R package minfi (66). Detection P-values were calculated and used to identify failed probes (P-value cut-off > 0.01). Probes that failed in >20% of samples were removed from the analysis (183 probes were removed from the Klinefelter analysis and 361 from the Turner and 47,XXX analysis). No samples had a proportion of failed probes exceeding 1% or a median methylated or unmethylated probe intensity below 11 (66,67). Raw data were normalized using default parameters in Genome Studio® which includes background and control probe normalization. Subset-quantile-within-array-normalization (SWAN) was used for correcting technical differences between Illumina Infinium type I and II assay design, allowing both within-array and between-sample normalization. Cross-hybridizing probes, probes overlapping polymorphic loci at the C or G residue of the target DNA sequence and probes on the sex chromosomes were excluded from the analysis.
Autosomal DNAm-based predictor of sex
Focusing on sCMRs and their probes, we generated probe and region-based predictors of sex using the elastic net machine learning algorithm, whose parameters were tuned with 7-fold cross-validation using data from the discovery cohort. Predictors were tested on several independent blood-based datasets: normative samples from the GSE132203, GSE125105 and ARIES cohorts, as well as clinical and matching control samples from the Klinefelter (47,XXY), Turner (45,X) and 47,XXX syndrome cohorts (63–65). The region-based predictor was also tested in the blood RRBS dataset (37). All test results are presented in Table 3. To ensure that predictor tests were completely independent from the training dataset, we tested for overlapping samples between the GSE132203 test dataset and the GSE72680 training dataset using the R package ewastools (68). An overlap was suspected because both studies profiled individuals in the Grady Trauma Project. In total, 122 samples were deemed overlapping and removed from the GSE132203 dataset prior to testing the predictor (69).
Table 3.
Sex predictor results
| Accuracy | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Predictor | No. probes | No. of sCMRs | No. of mQTL probes | Validation cohort (GSE125105) | EPIC BeadChip dataset (GSE143303) | ARIES birth | ARIES 7yr | ARIES 15yr | RRBS | Overall accuracy |
| 63-probe | 63 | 51 | 18 | 98.1 (6 females; 7 males misclassified) | 95.0 (38 females; 2 males misclassified) | 97.9 (13 females; 6 males misclassified) | 98.9 (9 females; 1 male misclassified) | 98.3 (14 females; 2 males misclassified) | n/a | 97.64 |
| 45-probe | 45 | 35 | 0 | 97.8 (10 females; 5 males misclassified) | 94.6 (43 females; 0 male misclassified) | 94.7 (45 females; 3 males misclassified) | 98.5 (14 females; 1 male misclassified) | 96.7 (32 females; 0 male misclassified) | n/a | 96.46 |
| 11-probe/sparse | 11 | 0 | 93.3 (27 females; 20 males misclassified) | 91.7 (61 females; 5 males misclassified) | 51.4 (440 females; 0 males misclassified) | 70.5 (286 females; 0 male misclassified) | 77.5 (217 females; 1 male misclassified) | n/a | 76.88 | |
| region-based | n/a | 31 | n/a | 94.1 (2 females; 40 males misclassified) | 94.1 (46 females; 1 male misclassified) | 89.8 (2 females; 90 males misclassified) | 91.9 (0 female; 79 males misclassified) | 92.9 (0 female; 69 males misclassified) | 91.8 (11 females; 1 male misclassified) | 92.45 |
Software availability
Code to implement the DNAm-based sex predictors is available as a free open-source R package, whatsex, at bitbucket.com/flopflip/whatsex. Code for constructing CMRs and computing composite beta measures is available at bititbucket.org/flopflip/comeback/.
RESULTS
Reproducibility of previously reported sex differences in DNAm
To define a set of autosomal genomic regions showing robust sex differences in DNAm, we examined previous studies. Because DNAm profiles are dependent on the age of the participant and the cell or tissue-type assayed (70–73), we focused our analysis on adult whole blood DNAm, for which there are several reports. To ensure that comparisons were robust and not affected by outliers or differences in coverage between DNAm technologies, we restricted our analysis to datasets of at least 50 male and 50 female DNAm profiles generated with the Illumina Infinium Human Methylation 450K BeadChip technology. Four studies met these criteria (Singmann et al., (6), McCartney et al., (12), Shah et al., (13) and Zaghlool et al., (14)) and reported 1184, 1687, 69 384 and 274 sites with significant sex differences in autosomal DNAm respectively (Supplementary Table S5). We note that the McCartney et al., study only provided information for the top 1000/69384 sites showing significant sex differences in DNAm, thus comparisons are based on this lower number. Furthermore, the McCartney et al., and Zaghlool et al., studies contained among their hits 42 and 83 probes respectively that cross-hybridize to other genomic regions, including the sex chromosomes (33). Because these probes may not reliably measure DNAm at their annotated site and were removed in the other studies, they were also excluded from our comparisons.
Overall, the four studies represented 3234 unique differentially methylated sites, of which 10 (0.3%) were detected in all the studies, 124 (3%) in three studies, 508 (16%) in two studies and 2592 (80%) were unique. Since none of these studies employed effect size cut-offs in the identification of sex-biased DNAm sites, we wondered if reproducibility was linked to the magnitude of the sex difference. Leveraging the effect sizes provided by Shah et al., we found that although DNAm differences between males and females were generally small (85% of sites showed a delta beta difference <10%) (Supplementary Figure S1A), sites reported as sex-biased in more than one study had significantly higher effect sizes when compared to loci reported in a single study (Supplementary Figure S1B) (Wilcoxon P-value < 0.05). In fact, effect sizes significantly increased as probes were reported in more studies.
Discovery and validation of robust sex-associated co-methylated regions (CMRs) in whole blood
The limited reproducibility of previously reported autosomal sex-differentially methylated sites suggested that more research is needed to identify genomic regions with consistent sex differences in DNAm. To this end, we employed a region-based approach and included a modest effect size cut-off (74). A well-powered discovery cohort of 3795 (2414 males and 1381 females) normative adult (25-80 years) whole blood samples was aggregated from 5 Illumina HumanMethylation 450K array datasets (Table 1, Figure 1A) (see Materials and Methods section). To ensure that our analyses were not confounded by sex mislabels (35,75), we used X and Y chromosome DNAm information to identify samples whose metadata sex label did not correspond to the expected sex chromosome DNA methylation profiles. All samples with potential mislabels were removed from the analysis. Because DNAm-sex associations can be confounded by probes that map to or cross-hybridize to the X and Y chromosomes, these probes were also removed from the dataset prior to analysis, as were probes containing or adjacent to single nucleotide polymorphisms with a minor allele frequency ≥ 5% (32,33).
From this filtered dataset, we defined a total of 26 434 regions of co-variable DNAm, termed co-methylated regions (CMRs). This was done using the CoMeBack method, which groups sites based on DNAm correlation and CpG background density (24) (Figure 1A and Supplementary Table S1). Composite beta values were calculated for each CMR by summing all of the individual probe betas using a weighted method based on the first principal component loadings of each probe’s DNAm levels (see code availability). Composite beta values were tested for sex differences in DNAm, yielding 205 CMRs (0.8% of all CMRs) with significant and consistent sex differences in DNAm levels based on strict criteria: FDR < 0.05; absolute composite beta differences > 4%; and composed entirely of probes with higher mean DNAm levels in one sex compared to the other. A 4% composite beta threshold was selected because it captured 99% of the technical variability in the discovery dataset.
To validate the sex differences at the 205 CMRs, we used an independently processed cohort of 312 males and 387 females aged 17–87 (GSE125105). After quality control, 201 (98%) CMRs were covered in the preprocessed validation dataset and 179 (87%) showed significant sex differences in DNAm as defined by: a q-value FDR < 0.05; adjusted absolute composite beta differences > 4%; and composed entirely of probes showing the same direction of sex-biased DNAm levels seen in the discovery cohort (Figure 1A and Supplementary Table S2). An 87% rate of validation is particularly striking considering that previous studies reported validation rates of 11–52% (6,12–14), and pairwise comparisons between studies yielded rates between 2 and 69% (Supplementary Table S5). To determine if the increased rate of validation seen by our approach was dependent on the inclusion of an effect size cut-off, we repeated our discovery and validation procedure with a 3% effect size cut-off and without an effect size cut-off (for a full list of sites meeting these criteria see Supplementary Table S3 and S4). Not surprisingly, reducing or removing the effect size cut-off resulted in the identification of more CMRs showing sex differences in DNAm, however validation rates dropped to 78% and 31%, respectively (Supplementary Table S6), as may be expected from a sensitivity/specificity trade-off. Thus, inclusion of even a modest effect size threshold increased reproducibility across studies, likely by limiting spurious findings related to technical variability.
Characteristics of sCMRs
Focusing on the 179 sCMRs showing effect sizes > 4%, we found that they were distributed across the autosomes (Figure 1B) and ranged in size from 2 to 15 CpG probes (Supplementary Figure S2A). Most (152 or 85%) showed higher mean DNAm levels in females compared to males (Supplementary Table S2), effects consistent with previous reports (10,20,21). sCMRs also captured sites previously reported to show sex differences in DNAm levels (6,12–14) (Table 2), including the 5′ UTR of the SLC6A4 gene (Figure 1C), a gene implicated in sex-biased depression and bipolar disorder (76). A set of sCMRs also overlapped regions previously reported to show sex-biased DNAm levels. In fact, 136 (76%) sCMRs overlapped regions detected with DMRcate in a dataset of 53 male and 58 female cord blood samples (Table 2 and Supplementary Table S6) (15). This substantial overlap is consistent with higher rates of reproducibility seen with regional based approaches, although we note that our approach was more stringent and only captured 5.5% of the previously reported sex-biased regions.
Table 2.
sCMRs overlapped individual sites and regions previously reported to show sex-biased DNAm
| Overlap with this study | |||
|---|---|---|---|
| Study | No. of reported sex-linked probes | CpGs (out of 654) | sCMRs (out of 179) |
| Shah et al., 2014 | 1687 | 219 | 112 |
| Singmann et al., 2015 | 1184 | 59 | 35 |
| Zaghlool et al., 2015 | 274 | 36 | 21 |
| McCarthy et al., 2020 | 1000 | 153 | 72 |
| all studies combined | 3354 | 299 | 129 |
| Yousefi et al., 2015* | 2471 | 136 | |
*Differentially methylated region (DMR)-based analysis. Any overlap in genomic coordinates is reported.
A link between sCMRs and sex was further corroborated by trait analysis from the EWAS Atlas, one of the largest collections of EWAS (Supplementary Figure S2B) (61). Overall, sCMRs were enriched for DNAm sites previously associated with sex (labeled gender in the EWAS atlas), sex chromosome aneuploidies (Klinefelter syndrome), X-linked intellectual disability syndromes (Claes–Jensen syndrome), mendelian disorders caused by mutations on chromatin remodelers that also show dysregulated DNAm (Nicolaides–Baraitser syndrome and SETD1B-related syndromes) and Alzheimer’s disease which has known sex-biased characteristics (77).
We also examined the overrepresentation of specific genomic features among the sCMRs. Compared to the entire CMRs background (26 434), sCMRs were enriched for sites of systemic interindividual variation (CoRSIVs) (16 sCMRs overlapped CoRSIVs—Fisher’s exact test P-value 0.046) (60), CpG islands and shores; mQTLs (58,59) and repressed chromatin states (54) (Figure 2A–C). In fact, 117 (65%) sCMRs were contained within a single chromatin state (Supplementary Table S2) and another 9 were located at the boundary of largely similar chromatin states (‘Polycomb-repressed’ and ‘Weak polycomb repressed’), findings suggestive of sCMRs functioning as cohesive units. The sCMRs were not enriched for transcription factor binding motifs, although we note that VEZF1, MAZ, KLF12 and KLF15 were overrepresented based on a nominal P-value (Supplementary Figure S2C). Of note, MAZ has been suggested to contribute to sex-biased gene expression (78).
Figure 2.
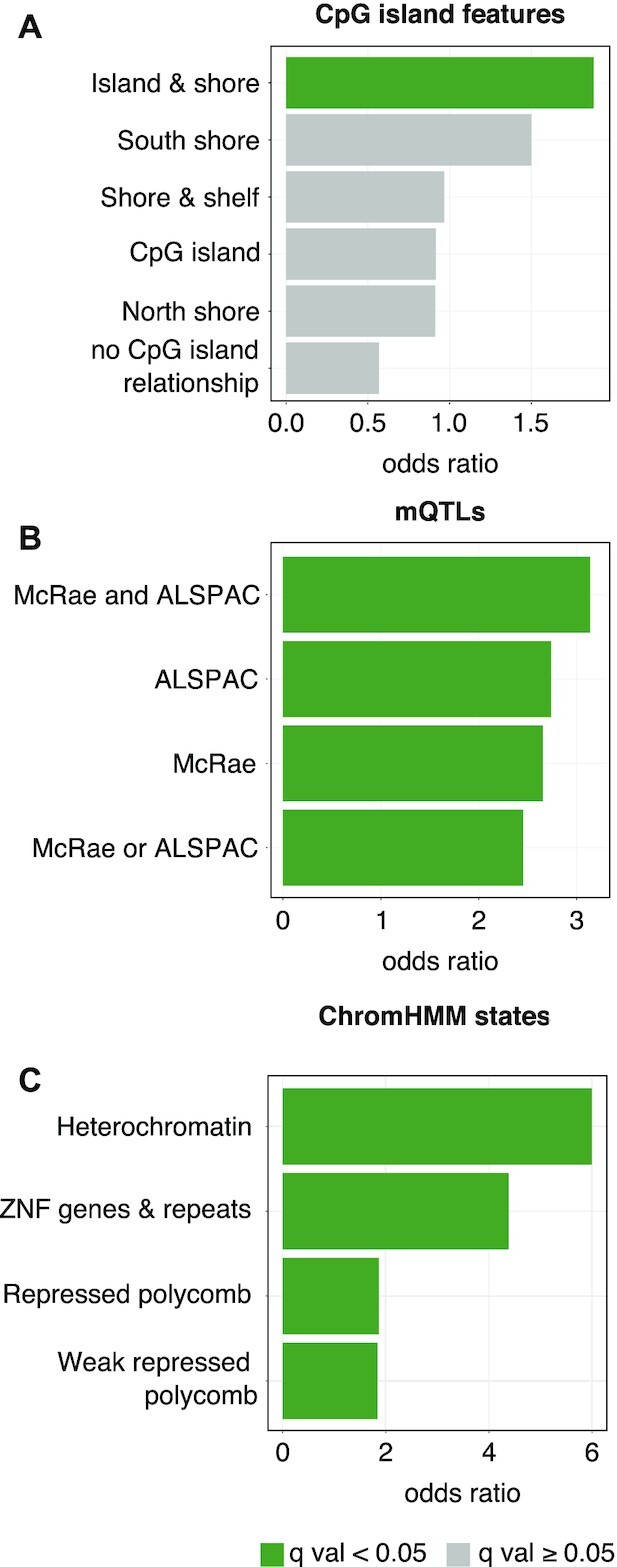
sCMRs were enriched for CpG islands, mQTLs and repressive chromatin states. (A) sCMRs were enriched for CpG island and shores based on the annotation from the Infinium Human Methylation 450K BeadChip manifest. sCMRs were also enriched for sites previously reported to be under genetic influence (mQTLs) in adult whole blood samples (B) (58,59) as well as repressed chromatin states (C) (54). In (A), categories shown in dark green are those with significant enrichments based on a Fisher exact test q value < 0.05 following Benjamini–Hochberg multiple test correction. In (B and C), only categories with significant enrichments are shown.
sCMR genes were not enriched for functional pathways or sex-biased gene expression
Next, we examined if genes mapping to sCMRs (Supplementary Table S2) were enriched for specific gene or functional categories. In total, 94 genes mapped to sCMRs and these were not enriched for imprinted genes, imprinting control centers (50,51), lncRNAs (52), or any particular GO category or KEGG pathway. sCMR genes were also not enriched for genes reported to show sex-biased gene expression in blood, a finding that was perhaps not surprising given that previous studies highlighted blood as a tissue with limited sex differences in mRNA levels (4,5,53).
Despite these findings, we note that one sCMR mapped to DDX43, a gene previously reported to show sex-biased DNAm and mRNA levels (79). Importantly, since we removed all probes that cross-hybridize to the sex chromosomes (33), the sex differences in DNAm levels at the DDX43 sCMR are unlikely to be driven by homology to the X chromosome DDX3X gene. The DDX43 sCMR mapped to a region shared with the OOEP gene and contained five sites, two of which were previously reported to show significant sex differences in DNAm (79) (Figure 3). Leveraging DNAm data for several cell and tissue types, we found that sex differences at this site extended beyond blood. In fact, despite very small samples sizes, all of the cell types and tissues examined showed higher DNAm in females compared to males, an effect that was significant in 8 of the 11 cell and tissue types tested (Figure 3). Using gene expression data from the GTEx database, we also examined if there were sex-biases in DDX43 and OOEP mRNA levels across tissues. Limiting our mRNA analysis to tissues for which we had DNAm information revealed significantly lower DDX43 mRNA levels in females compared to males. Sex-biased mRNA levels were also seen for OOEP, although this was only significant for 2 out of the 6 tissues examined. Observing similar patterns of sex-biased DDX43 and OOEP gene expression is consistent with nearby genes showing co-expression patterns perhaps due to shared regulation.
Figure 3.

Sex-biased DNAm levels at the DDX43-OOEP sCMR was observed across several tissues and associated with sex-biased gene expression at the DDX43 and OOEP genes. Sex-biased DNAm levels were detected across several tissues for the sCMR found at the overlapping and divergent OOEP and DDX43 genes (top). Tissues in bold are those that were present in the GTEx database and for which we could examine mRNA levels. Of these, several showed sex-biased mRNA levels (bottom). Asterisks indicate significant comparisons (P-value < 0.05).
Validation of sCMRs using reduced representation bisulfite sequencing profiles
To determine if sex differences at sCMRs were recapitulated in DNAm profiles generated with a different technology, we examined a dataset of 158 normative adult peripheral blood DNAm profiles based on reduced representation bisulfite sequencing (RRBS) (Table 1) (37). In this dataset, 4354 sites, mapping to 106 (59%) sCMRs, had DNAm information in at least half of the samples. In agreement with sCMRs representing regions of correlated DNAm, DNAm values at RRBS sites located within a sCMR were positively correlated (Figure 4A). In fact, the mean correlation within sCMRs ranged from 9% to 76%, centering around 29%, just below the 30% cut-off used in CoMeBack to define CMRs in the first place (Figure 4B). Importantly, shifting the sCMR coordinates by just 2500 bps in either direction significantly decreased this correlation (one-way anova P-value < 2.2e16), indicating that the sCMR coordinates captured most of the region showing correlated DNAm. With regards to sex differences, we found that sites mapping to sCMRs were able to separate male and female RRBS samples when subjected to Uniform Manifold Approximation and Projection (UMAP) (Figure 4C), findings in line with sCMRs representing regions of sex differences in DNAm. Of the 4354 sites, 1784 (41%) mapping to 90 (85%) sCMRs passed a q-value threshold < 0.05, a delta beta cut off > 4% and showed the same direction of change observed in the discovery and validation cohort (Supplementary Figure S3). Furthermore, of the significantly differentially methylated sites, most (1333 or 74%) had higher DNAm in females compared to males, effects consistent with findings based on Illumina HumanMethylation 450K array datasets (10,20,21). Altogether, the independent RRBS analysis supported the classification of sCMRs as regions of correlated and sex-biased DNAm.
Figure 4.
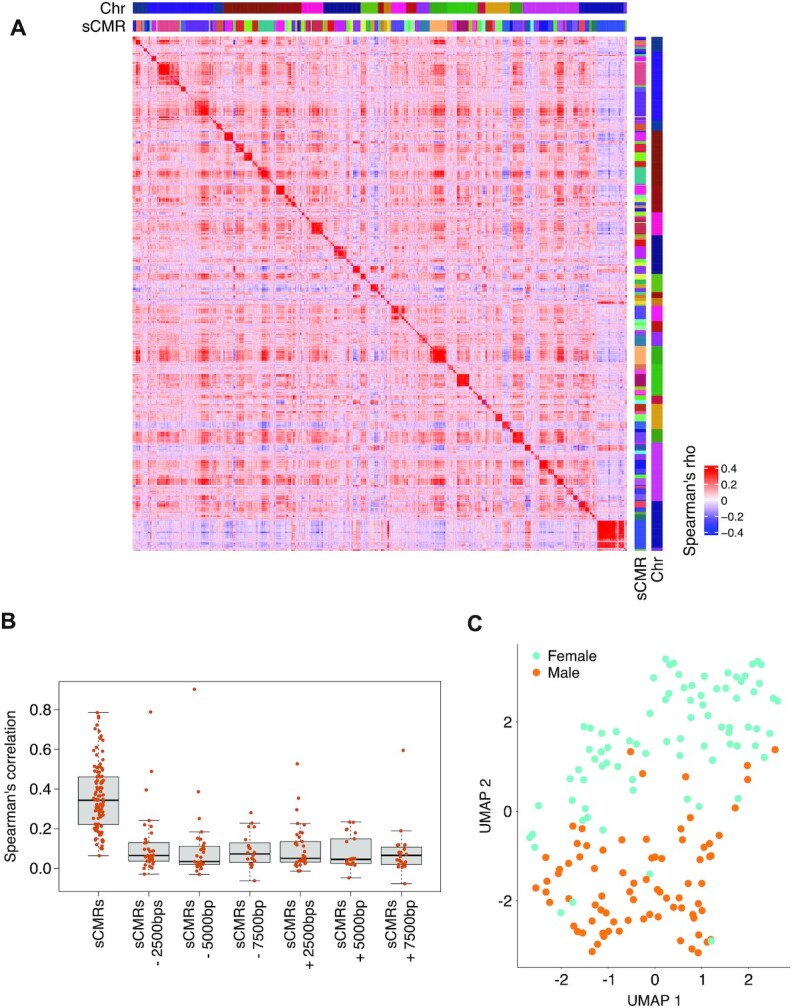
An independent RRBS dataset supports the categorization of sCMRs as correlated units with sex-biased DNAm. (A) Heatmap of spearman correlations for the 4354 sites mapping to sCMRs and present in at least half of the RRBS samples showed clusters of high correlation mapping to individuals sCMRs. (B) Boxplot of average internal correlation values for the sCMRs covered by the RRBS dataset. Internal correlations significantly decreased when sCMR coordinates were shifted by as little as 2500 bps on either direction (one-way anova P-value < 2.2e-16). (C) sCMR sites present in the RRBS dataset could separate male and female samples based on UMAP.
Blood sCMRs were consistent across the life span
Given that DNAm levels vary during development and across the lifespan (70,71), we wondered if sex-biased DNAm levels observed in adults were recapitulated in younger individuals. To test this, we used the ARIES cohort, which contains DNAm profiles for over 900 males and females sampled at birth, 7 and 15 years of age (Table 1) (41). We note that within the AIRES cohort there are slight differences in the type of blood-based sample assayed at each time point (cord blood, whole blood and white blood cells sampled at birth, age 0, 7 and 15, respectively), an important caveat given that DNAm patterns also vary between cell and tissue types (72,73). Examining each time point individually, we tested if sCMRs showed significant sex differences in DNAm using a linear model that accounted for differences in cell type proportions using the Houseman method (34). Overall, we found that at all three timepoints 170 (95%) sCMRs had significant sex differences in DNAm that also matched the direction of change observed in the discovery cohort (Figure 5A) (Supplementary Table S2). Of the remaining 9 sCMRs, none showed significant differences in DNAm levels between males and females at birth, but 8 became significantly differentially methylated by sex at later time points (age 7 and 15, respectively) (Figure 5B). The latter finding indicates that some sex-associated patterns in DNAm emerge later during childhood or adolescence, findings consistent with recent reports (80). Collectively, these results highlight remarkable stability of sCMRs across the life course, indicating that the majority of genomic regions showing consistent sex-biased DNAm patterns are established prior to birth and remain stable throughout the lifespan.
Figure 5.
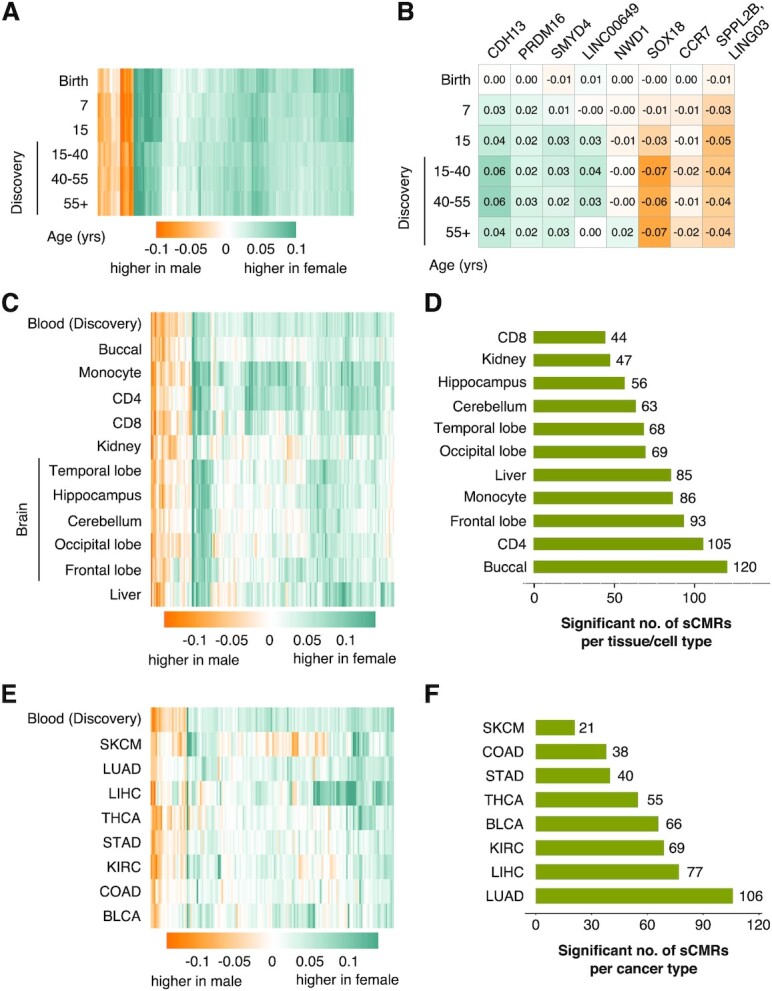
sCMRs were remarkably stable across the life span and a subset were cell, tissue, and cancer status agnostic. (A) Heatmap of the mean composite betas difference (female minus male) for the 179 validated sCMRs split by age categories. (B) Heatmap of the 8 sCMRs that changed during postnatal development. These sCMRs mapped to the genes included in the figure. (C) Heatmap of the mean composite beta difference (female minus male) for the 179 validated sCMRs across a variety of tissues. Blood is included for comparison. (D) Bar graphs showing the number of sCMRs that were significantly different between males and females cross the indicated cells and tissues. (E) Heatmap showing the mean composite beta differences (female minus male) for sCMRs across a variety of cancer types: thyroid carcinoma (THCA), skin cutaneous melanoma (SKCM), stomach adenocarcinoma (STAD), lung adenocarcinoma (LUAD), kidney renal clear cell carcinoma (KIRC), liver hepatocellular carcinoma (LIHC), colon adenocarcinoma (COAD) and bladder urothelial carcinoma (BLCA). The heatmap only shows 148 sCMRs that were present in all of the cancer samples and includes normative blood samples for comparison. (F) Bar graphs showing the number of sCMRs significantly different between males and females in the indicated cancer types.
A subset of sCMRs were cell type, tissue, and cancer status agnostic
Given that DNAm patterns vary widely between tissues and even between cell types within a tissue (72,73), we examined if sCMRs discovered and validated in blood also showed significant sex differences in DNAm in other tissues and cell types. We leveraged multiple publicly available datasets of somatic tissues and regions (buccal, kidney, liver and brain— hippocampus, cerebellum, temporal lobe, occipital lobe and frontal lobe) as well as isolated blood cell types (monocytes, CD4 and CD8 T cells) (43–47). In each dataset, the DNAm levels of each sCMR were compared between males and females and sites having a Welch test P-value < 0.05 and showing the same direction of change observed in blood were deemed significant (Figure 5C and D) (Supplementary Table S2). Overall, we found that each cell type and tissue showed a different degree of sex-biased DNAm at sCMRs. CD8 T cells had the least number of significant sCMRs (44 sCMRs or 25%), while buccal cells had the greatest number (120 sCMRs or 67%). Of the significant sCMRs, 5 met the significance threshold in all of the tissues and cell types examined (Supplementary Table S2). These cell type and tissue agnostic sCMRs were associated with the GLI4, ZFP41, NUP58 and FIGNL1 genes. Despite only a few sites meeting the significance threshold in all of the tissue and cell types tested, we note that sCMR composite beta differences across tissues and cell types were positively correlated with the patterns observed in blood. Of all of the tissues, the cerebellum showed the lowest correlation (Pearson’s correlation of 0.599) while monocytes showed the highest correlation (Pearson’s correlation of 0.832).
To further examine the effect of tissue on sCMR DNAm patterns, we compared sex differences in DNAm levels in normal lung, kidney, liver, thyroid, bladder and colon samples available through The Cancer Genome Atlas (TCGA) Research Network (Table 1). We note that only 148 of the 179 sCMRs were represented in the TCGA dataset due to differences in probe filtering protocols, thus comparisons are based on this smaller sCMR subset. Similar to our findings described above, 39 to 95 out of 148 sCMRs (26–64%) showed significant sex differences in DNAm (Welch test P-value < 0.05) and matched the direction of change seen in the discovery blood cohort (Supplementary Figure S4A).
The samples available through the TCGA also made it possible to examine whether cancer status affected sex differences in DNAm at sCMRs. We focused this analysis on thyroid carcinoma (THCA), skin cutaneous melanoma (SKCM), stomach adenocarcinoma (STAD), lung adenocarcinoma (LUAD), kidney renal clear cell carcinoma (KIRC), liver hepatocellular carcinoma (LIHC), colon adenocarcinoma (COAD) and bladder urothelial carcinoma (BLCA), as these datasets had large sample sizes (n > 100) and a good representation of both male and female samples (Table 1). For these datasets, we found between 21 and 106 (14–71%) sCMRs showing significant sex differences in DNAm (Welch test P-value < 0.05 and the same direction of change seen in the discovery blood cohort) (Figure 5E and F and Supplementary Figure S4A and B). Of these, thyroid carcinoma (THCA) and kidney renal clear cell carcinoma (KIRC) had fewer sCMRs that recapitulated the sex differences observed in blood when compared to their respective matching normal samples, indicating that disruptions in sex-biased DNAm levels at sCMRs may occur in a subset of cell malignancies.
Altogether, our findings indicate that sex differences in DNAm at sCMRs are partially agnostic to the cell or tissue of origin as well as whether these were derived from healthy or tumor samples. Importantly, our analysis does not rule out the possibility that additional sites show sex differences in DNAm in a cell, tissue or cancer-status-specific manner. However, the paucity of available data prevents us from examining this possibility as a minimum of 500 samples are needed to reliably detect correlations as low as 30% and build robust CMRs (24).
Assessing the link between sCMRs and sex hormones
Given that sex hormones may contribute to the establishment of sex-biased DNAm (62,81), we examined if sCMRs were linked to sex hormones or their biology. We began by examining whether sCMRs included sites whose DNAm levels were previously associated with changes in sex hormones over the puberty transition (62). Indeed, compared to the CMR background, sCMRs were significantly enriched for sites whose DNAm levels correlate with changes in inhibin B, luteinizing hormone, testosterone, follicle-stimulating hormone and anti-müllerian hormone (Figure 6A). Using a publicly available dataset, we also explored if DNAm levels at sCMRs changed in response to estradiol treatment. Estradiol is an estrogen hormone known to inhibit the DNA methyltransferase DNMT1 and lower DNAm levels (82). Examining DNAm profiles for HTB-22 breast cancer cells cultured with or without estradiol for 4–14 days (Table 1) revealed 11 (6%) sCMRs that displayed significant changes in response to estradiol exposure (Welch test P-value < 0.05) (Figure 6B). Of these, seven sCMRs showed significant DNAm differences regardless of the length of estradiol exposure. Finally, we assessed if DNAm levels at sCMR were linked to estrogen or progesterone receptor status by leveraging female breast cancer DNAm profiles from the TCGA. Overall, sCMR composite beta values could separate breast cancer samples based on estrogen and progesterone receptor status when a dimensionality reduction algorithm (UMAP) was applied (left side of Figure 6C) although this separation was mostly driven by three sCMRs (chr2:121269292–121269349, chr3:134031551–134031686 and chr8:144371537–144371780) (right side of Figure 6C). While limited in scope, our findings support a relationship between DNAm at sCMRs and sex hormones but suggest that this effect may be limited to a just few sCMRs.
Figure 6.
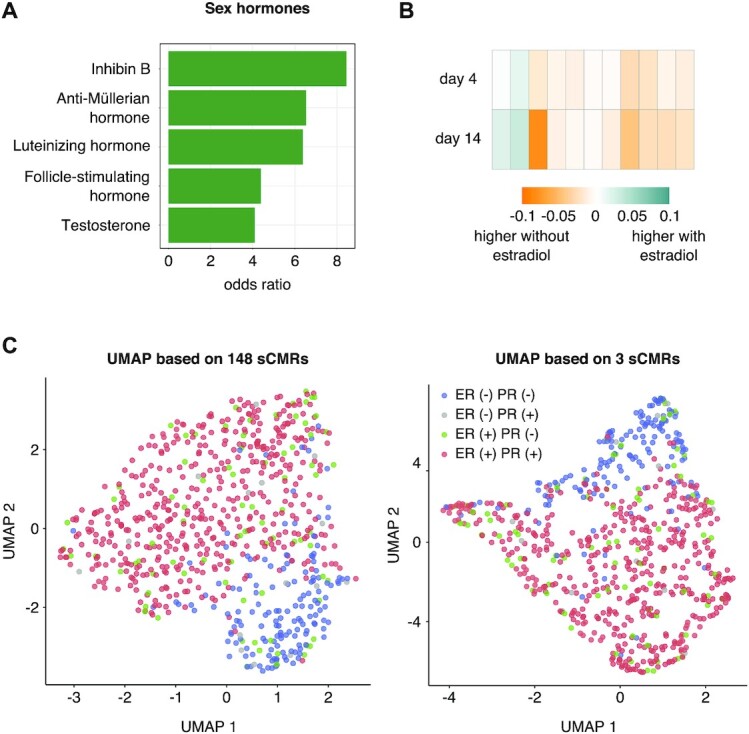
sCMRs were linked to sex hormone biology. (A) sCMR were enriched for sites whose DNAm levels are linked to sex hormone levels. Significance was based on a Fisher exact test q value < 0.05 following Benjamini–Hochberg multiple test correction. (B) Eleven sCMRs showed significant changes in DNAm levels in HTB-22 breast cancer cells based on estradiol treatment. (C) UMAP of breast cancer tissue samples showed a strong separation based on estrogen and progesterone receptor status when the sCMR composite beta values for the 148 sCMRs present in the dataset (left) were used. This effect was largely driven by three sCMRs (right)
Sex chromosome complement rather than physical sexual characteristics are predictive of sCMR DNAm status
Having observed a limited relationship between DNAm levels at sCMRs and sex hormones, we turned our attention to the potential influence of sex chromosomes. Previous work using clinical samples from individuals with sex chromosome aneuploidies [males with Klinefelter syndrome (47,XXY) and females with Turner (45,X) and 47,XXX syndrome] revealed a relationship between sex chromosome complement and DNAm patterns (63–65). Using these samples, we examined the relationship between DNAm at sCMRs and sex chromosome complement. Validating our findings once again, we found that the DNAm profiles of normative females (46,XX) and males (46,XY) in this clinical dataset recapitulated the sex differences observed in the discovery cohort (Figure 7A). We also found that sCMR DNAm levels in females with 47,XXX syndrome patients recapitulated the normative female profile. By contrast, sCMR DNAm levels in males with Klinefelter syndrome (47,XXY) closely mirrored the normative female rather than the male DNAm pattern, whereas DNAm levels in females with Turner syndrome (45,X) mirrored but did not completely match the male rather than the female DNAm pattern (Figure 7A and Supplementary Figure S5A). Taken together, these findings point at an intimate relationship between sex chromosomes and DNAm profiles at sCMRs.
Figure 7.

sCMR DNAm levels reflected sex chromosome complement information. (A) Heatmap of the mean composite betas difference for the indicated genotypes compared to control normative male samples. (B) UMAP of male and female samples and samples from individuals with sex chromosome abnormalities revealed a strong relationship between DNAm levels at sCMRs and sex chromosome complement.
Building upon these findings, we applied a UMAP dimensionality reduction algorithm to sCMRs in the dataset of Klinefelter (47,XXY), Turner (45,X) and 47,XXX syndrome patients. This analysis produced three main clusters: cluster 1 was composed exclusively of males (46,XY); cluster 2 of Turner syndrome patients (45,X); and cluster 3 of karyotypically normal (46,XX) and 47,XXX syndrome females, as well as Klinefelter syndrome (47,XXY) males (Figure 7B). We note that within cluster 3, karyotypically normal females (46,XX) and Klinefelter (47,XXY) syndrome patient samples also separated from one another, while 47,XXX syndrome patient samples were dispersed throughout both normative female and Klinefelter syndrome patient sample clusters. These findings indicate that sCMR DNAm levels are strongly influenced by the number of X chromosomes and, to a lesser extent, by the presence of a Y chromosome.
An accurate sex predictor based on autosomal DNAm
Having identified sCMRs that showed remarkable consistency across the lifespan and recapitulated patterns of sex-biased DNAm in a variety of tissues and cell types, we reasoned that autosomal DNAm levels at sCMRs may be sufficient to predict sample sex. Although methods that predict sex based on sex chromosome DNAm levels have been described (68), an autosomal DNAm-based predictor of sex fills an important gap, it makes it possible to assess sex in DNAm datasets lacking raw IDAT files or other types of sex chromosome information, as is the case for many publicly available datasets (35) and for most datasets at later stages of the pre-processing and normalization pipeline. For ease of application, we focused on developing a probe-based predictor, removing the need to calculate composite betas.
Using elastic net regression (83), we created three predictors and benchmarked them against a previously described method to assess sex based on XY chromosome DNAm levels (see Materials and Methods section) (35). The most accurate predictor relied on 63 probes from 51 sCMRs and had an overall accuracy of 98% when tested in three independent blood datasets (GSE132203, GSE125105, ARIES) that varied in age and DNAm array platform (Illumina Infinium Human Methylation 450K BeadChip versus Illumina Infinium Human Methylation EPIC BeadChip) (Tables 1 and 3 and Figure 8A). Since this predictor contained sites annotated as mQTLs and could be influenced by genetic background, we also generated a ‘no-mQTL’ predictor that relied on 45 probes from 35 sCMRs and showed an overall accuracy of 96% (Figure 8A). Encouraged by these results, we also generated a predictor that relied on a minimal set of probes and may be useful for targeted approaches. The ‘minimal’ predictor relied on just 11 non-mQTL probes and although it had an accuracy of 92.5% in adult blood samples, its accuracy dropped to 66.5% in younger individuals (Table 3). Based on performance and a reduced possibility of genetic background effects, we recommend the use of the 45-probe based tool for testing or inputting sex information in DNAm datasets. Nevertheless, both the 63-probe and ‘no-mQTL’ 45-probe predictors are freely available as an open-source R package whatsex at bitbucket.com/flopflip/whatsex.
Figure 8.
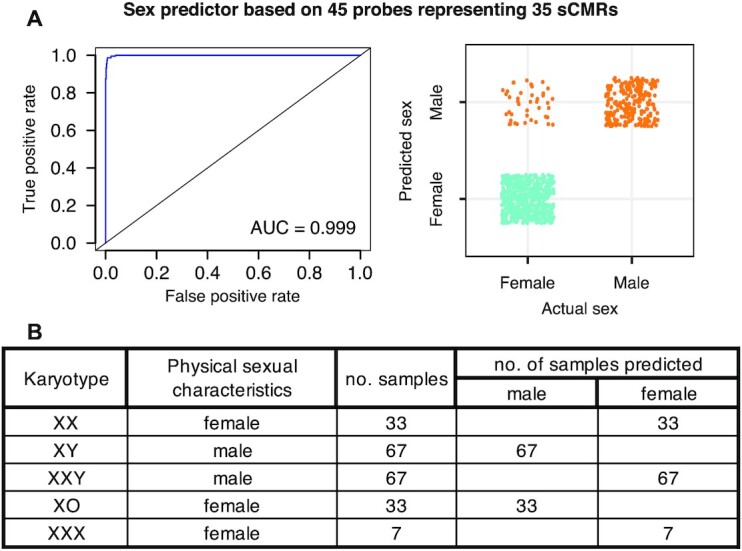
A subset of sCMRs were sufficient to predict biological sex with high accuracy. (A) Receiver operating curve (left) and graphical representation of the distribution of true positive and false negatives (right) for the biological sex predictor based on 45 probes representing 35 sCMRs. Figures are based on test performed on the GSE125105 dataset. (B) Testing the DNAm sex predictor in blood samples from individuals with sex chromosome abnormalities revealed discordance between physical sexual characteristics and predicted sex. Klinefelter (47,XXY) syndrome patients were predicted to be female, while Turner (45,X) syndrome patients were predicted to be male.
Although the predictor was not designed to prioritize sites with sex-biased DNAm across cell and tissue types, 34 out of the 35 sCMRs included in the 45-probe predictor were significant in more than one tissue (Supplementary Figure S2). In fact, 24 out of 35 sCMRs were significant in at least 5 tissues, suggesting that the most predictive sites may also be the most consistent across cells and tissues. Despite these findings, the accuracy of the predictor was limited to blood samples, as testing in buccal DNAm profiles revealed poor performance. Thus, more publicly available data are needed to develop a tissue agnostic predictor of sex based on autosomal DNAm information.
Given that sCMRs also showed sex-biased DNAm levels in the RRBS dataset, we reasoned that a region-based predictor developed with Illumina Infinium Human Methylation 450K BeadChip data may generalize to other platforms, like RRBS. A region-based predictor of sex was generated using 31 sCMRs and it showed an overall accuracy of 92% in the Illumina Infinium Human Methylation 450K/EPIC BeadChip testing datasets as well as the RRBS dataset. Although promising, more calibration and testing in other platform-types is needed before a true platform-agnostic predictor of sex based on autosomal DNAm becomes available.
Finally, we examined the performance of the 45-probe predictor in individuals with sex chromosome abnormalities. Applying the predictor to the blood DNAm profiles of patients affected by common sex chromosome aneuploidies (63–65) revealed some interesting patterns. Importantly, the predictor correctly classified all the normative male (46,XY) and female (46,XX) samples (Figure 8B and Supplementary Figure S5B), again highlighting its accuracy independent of differences in preprocessing and normalization pipelines. In line with the strong relationship between sCMR DNAm and sex chromosomes described above, we found that the predictor classified all females with 47,XXX syndrome as female, but it classified all males with Klinefelter (47,XXY) syndrome as female and all females with Turner (45,X) syndrome as male, revealing that this tool may help identify mislabeled samples or samples from individuals with chromosomes aneuploidies.
DISCUSSION
To facilitate the consideration of sex in epigenetic studies, we defined and deeply characterized a set of reproducible autosomal genomic regions showing sex differences in DNAm. Using a well-powered dataset of 3795 normative adult whole blood DNAm profiles and applying a region-based approach grounded in strict criteria, we identified and validated 179 sex-associated co-methylated regions (sCMRs) in adult blood samples, patterns that were also observed at earlier timepoints in development (birth, age 7 and age 15). Sex differences in sCMR DNAm patterns were also observed across a range of normative and oncogenic tissues, although deviations in DNAm patterns were observed at particular sCMRs and should be explored further. Importantly, the categorization of sCMRs as regions of sex-biased co-variable DNAm in blood was supported by several DNAm platforms including the Illumina Infinium Human Methylation 450K and EPIC BeadChips, as well as RRBS. Functionally, sCMRs were linked to repressive chromatin states and a transcription factor proposed to modulate sex-specific gene expression (57,78). They also contained sites that show altered DNAm levels in sex chromosome aneuploidy syndromes, or syndromes emerging from altered X-linked genes or epigenetic modifiers. sCMR DNAm status was strongly associated with sex chromosome complement indicating that sex chromosomes and their associated regulatory mechanisms may influence DNAm at the autosomes. Finally, the robustness of sCMRs allowed us to develop an accurate, easy-to-use and robust predictor of sex that does not rely on raw DNAm data, or other sex chromosome information, i.e. data often missing in public datasets, especially at later stages of data normalization pipelines. Our sex predictor was accurate regardless of sample age or preprocessing and normalization methods, and it can be used to impute sex and assess data quality.
Although sex differences in DNAm levels have been reported previously (6,12–16) a consensus of affected regions has failed to emerge. In fact, initial examination of studies reporting sex-biased DNAm levels revealed limited reproducibility despite profiling similar populations using the same technology. To re-examine the issue of sex-biased DNAm we made two important changes in the definition of sex-associated DNAm: we used a region-based approach and included an effect size cut-off.
At present there is no standard for the inclusion of effect size cut-offs in DNAm studies. In fact, most studies examining sex differences in DNAm to date did not included an effect size threshold when reporting significant results. Despite this, we found that reproducibility across studies was linked to the magnitude of the sex difference in DNAm, whereby larger DNAm changes were more likely to replicate across studies. Although our findings indicate that such thresholds should be more widely applied, selecting an effect size cut-off remains challenging because the magnitude of DNAm change associated with a functional biological consequence is unknown. Nevertheless, determining effect size cut-offs based on technical, rather than biological, variability can help limit associations below the error rate of the assay (Illumina Infinium Human Methylation 450K BeadChIP) and increase the likelihood of observing true biological signal. Importantly, we acknowledge that meaningful small-magnitude sex-differences in DNAm may exist, as has been noted for gene expression (4), however, considering the limitations of the techniques used is important in the context of reproducibility.
We focused on regions of variable DNAm rather than individual CpG sites because growing evidence indicates that region-based approaches are more powerful at detecting small effects and produce more robust associations with phenotypes of interest (74). Consistent with this, we found that 87% of the 201 sCMRs we discovered, validated in an independent cohort, well above the 10–53% internal validation rates reported previously (6,12,13). Furthermore, 136 sCMRs (76%) overlapped with 2471 regions reported in a previous study (Table 2 and Supplementary Table S6) (15). The overlap between this study and ours underscores the superior reproducibility of region-based approaches and is particularly striking considering that different methods were used to define the regions: DMRcate versus CoMeBack (24,84). Nevertheless, our approach identified far fewer regions with sex differences in DNAm than previously reported (15). This likely reflects the stringency of our selection criteria, which required regions to behave as cohesive units and pass both statistical and regional effect size thresholds, criteria that can be easily implemented with CoMeBack. We acknowledge that while additional genomic regions may have DNAm levels influenced by sex, our goal was to produce a high-confidence annotation of human genomic regions exhibiting the most reproducible sex differences. As such, we employed large and diverse discovery and validation cohorts and thoroughly characterized the robustness of our findings across life stages, cell and tissue types and cancer states.
Beyond sex, factors like age, ancestry and tissue or cell type also affect DNAm patterns. Accordingly, findings in adult blood may not generalize to DNAm states in other tissues or in younger individuals (85). With regard to age, sCMRs were consistently differentially methylated by sex across the lifespan in blood and are distinct from recently reported age-associated sites (80). Nevertheless, we recognize that by prioritizing sample size and grouping together 25–80 years olds in the discovery cohort, our approach may have prevented the identification of age-specific sex-biased DNAm and obscured links to sex hormones, which vary over that age range. In relation to ancestry, a lack of detailed ancestry information or the necessary probe data to infer it, precluded us from examining the relationship between sex-biased DNAm at sCMRs and genotype. Nevertheless, because the discovery cohort included individuals of Caucasian, African American and Hispanic ancestry, and the RRBS dataset was likely composed primarily of individuals of Chinese ancestry based on the recruitment hospital, sCMRs likely generalize to these populations. Nevertheless, further research is clearly needed to address this important question. Additionally, we found that some sCMRs maintained sex differences in DNAm across various tissues and cell types. However, these findings were based on small sample sizes and will need to be revisited as larger non-blood datasets become available. Indeed, the identification of cell- or tissue-specific sex-biased DNAm is a question that cannot be robustly addressed with the current publicly available data.
To identify well-defined genomic regions showing robust sex differences in DNAm, we deliberately used discovery and validation cohorts comprised exclusively of normative adult samples. Despite the lack of pathology in our discovery and validation datasets, the sCMRs were enriched for sites showing altered DNAm in syndromes or disorders of sex chromosomes, X-linked loci or chromatin modellers. We also found that a subset of sCMRs had altered DNAm patterns in tumor samples, an effect that may reflect chromosomal abnormalities typical of cancer cells or the microenvironment of tumors (86). Nevertheless, it remains unknown whether sCMRs underscore sex differences in disease or susceptibility to the environment. It is also unclear whether disease states and harmful exposures result in additional genomic regions showing sex differences in DNAm. Although these are important questions, our goal was to identify regions that should be carefully considered in all blood EWAS when sample sizes prevent sex-stratified analysis, or when phenotypes of interest are confounded by sex.
To determine whether physical sexual characteristics and or sex chromosomes were tightly related to DNAm levels at sCMRs, we considered DNAm profiles of individuals with sex chromosome aneuploidies: females with Turner (45,X) and 47,XXX syndrome, and males with Klinefelter syndrome (47,XXY). Our analyses revealed that sCMR DNAm was highly dependent on sex chromosome complement and not on physical sexual characteristics. In other words, females with Turner syndrome showed male DNAm profiles at sCMRs, while males with Klinefelter syndrome (47,XXY) and females with 47,XXX syndrome showed female DNAm profiles. Our findings are consistent with previous reports showing a strong association between widespread alterations in autosomal DNAm patterns and sex chromosome complement, specifically X chromosome aneuploidy (10,17,18,63–65). This work highlights the value of samples from individuals with sex chromosome aneuploidies to disentangle the mechanisms giving rise to sex biases in biology. In this regard, the remarkable relationship between sCMR DNAm signatures and sex chromosome complement implicates several mechanisms in the establishment of autosomal sex-biased DNAm. This includes X chromosome inactivation pathways like XIST expression levels, 3D X/Y chromosome-autosome contacts (87), X- or Y-linked genes or variants of epigenetic regulators (e.g. ASTML) (88) or autosomal genes or variants that modulate X or Y chromosome DNAm (89,90). By providing a reproducible set of autosomal regions showing robust sex-differences in DNAm, our work provides a set of loci that can be used to test the contribution of each of these mechanisms.
Leveraging sCMRs, we constructed a predictor of genetic sex based on autosomal DNAm levels, an easy-to-use, robust tool that can be implemented at any stage of preprocessing pipelines to identify sex mislabels or impute sex information if unavailable (35). Although several efforts to predict sex from DNAm data have been reported (20,66,87,91,92), previous tools rely on X and Y chromosomes information, which is often missing in publicly available datasets, or in data that have undergone normalization. We note that sex can also be predicted from autosomal gene expression data, though a recently reported predictor achieved 84% accuracy, compared the 96% seen for the autosomal DNAm-based predictor of sex (4,93).
Finally, we acknowledge that examining the role of sex in health and disease is difficult because of the complex relationship between sex and gender (gender defined by the Canadian Institutes of Health Research [CIHR] as the ‘socially constructed roles, expectations, relationships, behaviours, relative power, and other traits that societies ascribe to women, men and people of diverse gender identities’ (http://www.cihr-irsc.gc.ca/e/32019.html). While gender was not directly examined in this study, the discordance between physical sexual characteristics and ‘epigenetic sex’ observed in individuals with sex chromosome aneuploidy suggests that sex rather than gender may be driving DNAm differences at these sites. Our findings in this regard underscore the importance of correctly using the terms ‘sex’ and ‘gender’ in biomedical research and argue for a need to more explicitly describe how sex as a biological variable was ascertained in research samples (e.g. patient survey of sex assigned at birth, genetic testing and/or external medical examination) (1,94).
Altogether, our study highlights autosomal regions with DNA methylation levels that consistently associate with sex. Importantly, these regions should be carefully considered in future EWAS to prevent spurious associations driven by sex rather than phenotypes of interest. While it remains to be fully determined how these epigenetic modifications are established and contribute to sex differences in health and disease, we hope to facilitate the future exploration of these questions by providing a high-confidence annotation of regions with sex-biased DNAm and an autosomal DNAm-based predictor of sex.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Wendy Robinson, Dr. Carolyn Brown and Sarah Goodman for helpful discussions and critical comments on the manuscript. The results shown here are in whole or part based upon data generated by the TCGA Research Network (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). We thank all of the researchers who made their dataset publicly available and made this research possible. We are extremely grateful to all the families who took part in the ALSPAC study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. The UK Medical Research Council and Wellcome (grant ref: 217065/Z/19/Z) and the University of Bristol provide core support for ALSPAC. This publication is the work of the authors and will serve as guarantors for the contents of this paper. C.H.G. and A.S. are members of the European Reference Network on Rare Endocrine Conditions (ENDO-ERN), Project ID number 739543.
Contributor Information
Evan Gatev, Institute of Molecular Biology “Roumen Tsanev”, Bulgarian Academy of Sciences, Sofia 1113, Bulgaria; Graduate program in Bioinformatics, University of British Columbia, Vancouver, British Columbia V5T 4S6, Canada; Beedie School of Business, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada; BC Children's Hospital Research Institute Vancouver, British Columbia V5Z 4H4, Canada; Department of Medical Genetics, University of British Columbia, Vancouver, British Columbia V6H 3N1, Canada; Centre for Molecular Medicine and Therapeutics, Vancouver, British Columbia V6H 0B3, Canada.
Amy M Inkster, BC Children's Hospital Research Institute Vancouver, British Columbia V5Z 4H4, Canada; Department of Medical Genetics, University of British Columbia, Vancouver, British Columbia V6H 3N1, Canada.
Gian Luca Negri, Canada's Michael Smith Genome Sciences Centre, BC Cancer, Vancouver, British Columbia V5Z 1L3, Canada.
Chaini Konwar, BC Children's Hospital Research Institute Vancouver, British Columbia V5Z 4H4, Canada; Department of Medical Genetics, University of British Columbia, Vancouver, British Columbia V6H 3N1, Canada; Centre for Molecular Medicine and Therapeutics, Vancouver, British Columbia V6H 0B3, Canada.
Alexandre A Lussier, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Psychiatry, Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
Anne Skakkebaek, Department of Clinical Genetics, Aarhus University Hospital, Aarhus 8200, Denmark; Department of Molecular Medicine, Aarhus University Hospital, Aarhus 8200, Denmark.
Marla B Sokolowski, Department of Ecology and Evolutionary Biology, University of Toronto, Toronto, ON M5S 3B2, Canada; Program in Child and Brain Development, CIFAR, MaRS Centre, West Tower, 661 University Ave, Suite 505, Toronto, ON M5G 1M1, Canada.
Claus H Gravholt, Department of Molecular Medicine, Aarhus University Hospital, Aarhus 8200, Denmark; Department of Endocrinology, Aarhus University Hospital, Aarhus 8200, Denmark.
Erin C Dunn, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Psychiatry, Harvard Medical School, Boston, MA 02114, USA; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
Michael S Kobor, BC Children's Hospital Research Institute Vancouver, British Columbia V5Z 4H4, Canada; Department of Medical Genetics, University of British Columbia, Vancouver, British Columbia V6H 3N1, Canada; Centre for Molecular Medicine and Therapeutics, Vancouver, British Columbia V6H 0B3, Canada; Program in Child and Brain Development, CIFAR, MaRS Centre, West Tower, 661 University Ave, Suite 505, Toronto, ON M5G 1M1, Canada.
Maria J Aristizabal, BC Children's Hospital Research Institute Vancouver, British Columbia V5Z 4H4, Canada; Department of Medical Genetics, University of British Columbia, Vancouver, British Columbia V6H 3N1, Canada; Centre for Molecular Medicine and Therapeutics, Vancouver, British Columbia V6H 0B3, Canada; Department of Ecology and Evolutionary Biology, University of Toronto, Toronto, ON M5S 3B2, Canada; Program in Child and Brain Development, CIFAR, MaRS Centre, West Tower, 661 University Ave, Suite 505, Toronto, ON M5G 1M1, Canada; Department of Biology, Queen’s University, Kingston ON K7L 3N6, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Aase og Einar Danielsen Foundation; Familien Hede Nielsens Fond; Fonden til Lægevidenskabens Fremme; Natural Sciences and Engineering Research Council of Canada; One Mind; Aarhus University; Central Denmark Region; Fonden af 17-12-1981; National Institute of Mental Health, National Institutes of Health [R01MH113930]; Independent Research Fund Denmark [0134-00406A, 0134-00130B]; Jacobs Foundation; Canadian Institute for Advanced Research; Augustinus Fonden; Lundbeckfonden; Novo Nordisk Foundation Center for Basic Metabolic Research [NNF13OC0003234, NNF15OC0016474, NNF20OC0060610]. Funding for open access charge: Canadian Institute for Advanced Research / Child and Brain Development [FS22-148].
Conflict of interest statement. None declared.
REFERENCES
- 1.Mauvais-Jarvis F., Merz N.B., Barnes P.J., Brinton R.D., Carrero J.-J., DeMeo D.L., Vries G.J.D., Epperson C.N., Govindan R., Klein S.L.et al.. Sex and gender: modifiers of health, disease, and medicine. Lancet North Am. Ed. 2020; 396:565–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee S.K.Sex as an important biological variable in biomedical research. BMB Rep. 2018; 51:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mooney M.A., Ryabinin P., Wilmot B., Bhatt P., Mill J., Nigg J.T.. Large epigenome-wide association study of childhood ADHD identifies peripheral DNA methylation associated with disease and polygenic risk burden. Transl. Psych. 2020; 10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oliva M., Muñoz-Aguirre M., Kim-Hellmuth S., Wucher V., Gewirtz A.D.H., Cotter D.J., Parsana P., Kasela S., Balliu B., Viñuela A.et al.. The impact of sex on gene expression across human tissues. Science. 2020; 369:eaba3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gershoni M., Pietrokovski S.. The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol. 2017; 15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singmann P., Shem-Tov D., Wahl S., Grallert H., Fiorito G., Shin S.-Y., Schramm K., Wolf P., Kunze S., Baran Y.et al.. Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenetics Chromatin. 2015; 8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiong Y., Wei Y., Gu Y., Zhang S., Lyu J., Zhang B., Chen C., Zhu J., Wang Y., Liu H.et al.. DiseaseMeth version 2.0: a major expansion and update of the human disease methylation database. Nucleic Acids Res. 2017; 45:D888–D895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monk D., Mackay D.J.G., Eggermann T., Maher E.R., Riccio A.. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019; 20:235–248. [DOI] [PubMed] [Google Scholar]
- 9.Lyon M.F.Gene Action in the X-chromosome of the Mouse (Mus musculus L.). Nature. 1961; 190:372–373. [DOI] [PubMed] [Google Scholar]
- 10.Maschietto M., Bastos L.C., Tahira A.C., Bastos E.P., Euclydes V.L.V., Brentani A., Fink G., de Baumont A., Felipe-Silva A., Francisco R.P.V.et al.. Sex differences in DNA methylation of the cord blood are related to sex-bias psychiatric diseases. Sci. Rep. 2017; 7:44547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtis S.W., Gerkowicz S.A., Cobb D.O., Kilaru V., Terrell M.L., Marder M.E., Barr D.B., Marsit C.J., Marcus M., Conneely K.N.et al.. Sex-specific DNA methylation differences in people exposed to polybrominated biphenyl. Epigenomics. 2020; 12:757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCartney D.L., Zhang F., Hillary R.F., Zhang Q., Stevenson A.J., Walker R.M., Bermingham M.L., Boutin T., Morris S.W., Campbell A.et al.. An epigenome-wide association study of sex-specific chronological ageing. Genome Medicine. 2020; 12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah S., McRae A.F., Marioni R.E., Harris S.E., Gibson J., Henders A.K., Redmond P., Cox S.R., Pattie A., Corley J.et al.. Genetic and environmental exposures constrain epigenetic drift over the human life course. Genome Res. 2014; 24:1725–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaghlool S.B., Al-Shafai M., Al Muftah W.A., Kumar P., Falchi M., Suhre K.. Association of DNA methylation with age, gender, and smoking in an Arab population. Clin. Epigenet. 2015; 7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yousefi P., Huen K., Davé V., Barcellos L., Eskenazi B., Holland N.. Sex differences in DNA methylation assessed by 450 K BeadChip in newborns. BMC Genomics. 2015; 16:911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoshita M., Numata S., Tajima A., Kinoshita M., Umehara H., Yamamori H., Hashimoto R., Imoto I., Ohmori T.. Sex differences of leukocytes DNA methylation adjusted for estimated cellular proportions. Biol. Sex Differ. 2015; 6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen A., Heredia N., Senut M.-C., Hess M., Land S., Qu W., Hollacher K., Dereski M.O., Ruden D.M.. Early life lead exposure causes gender-specific changes in the DNA methylation profile of DNA extracted from dried blood spots. Epigenomics. 2015; 7:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davegårdh C., Hall Wedin E., Broholm C., Henriksen T.I., Pedersen M., Pedersen B.K., Scheele C., Ling C.. Sex influences DNA methylation and gene expression in human skeletal muscle myoblasts and myotubes. Stem Cell Res. Ther. 2019; 10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaz A.M., Wong C.-J., Varadan V., Willis J.E., Chak A., Grady W.M.. Global DNA methylation patterns in Barrett's esophagus, dysplastic Barrett's, and esophageal adenocarcinoma are associated with BMI, gender, and tobacco use. Clin. Epigenet. 2016; 8:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy N.S., Melton P.E., Cadby G., Yazar S., Franchina M., Moses E.K., Mackey D.A., Hewitt A.W.. Meta-analysis of human methylation data for evidence of sex-specific autosomal patterns. BMC Genomics. 2014; 15:981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall E., Volkov P., Dayeh T., Esguerra J.L.S., Salö S., Eliasson L., Rönn T., Bacos K., Ling C.. Sex differences in the genome-wide DNA methylation pattern and impact on gene expression, microRNA levels and insulin secretion in human pancreatic islets. Genome Biol. 2014; 15:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J., Morgan M., Hutchison K., Calhoun V.D.. A study of the influence of sex on genome wide methylation. PLoS One. 2010; 5:e10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu H., Wang F., Liu Y., Yu Y., Gelernter J., Zhang H.. Sex-biased methylome and transcriptome in human prefrontal cortex. Hum. Mol. Genet. 2014; 23:1260–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gatev E., Gladish N., Mostafavi S., Kobor M.S.. CoMeBack: DNA methylation array data analysis for co-methylated regions. Bioinformatics. 2020; 36:2675–2683. [DOI] [PubMed] [Google Scholar]
- 25.R Core Team and R Foundation for Statistical Computing R: A language and environment for statistical computing. 2019; Vienna, Austria. [Google Scholar]
- 26.Lehne B., Drong A.W., Loh M., Zhang W., Scott W.R., Tan S.-T., Afzal U., Scott J., Jarvelin M.-R., Elliott P.et al.. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015; 16:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannon E., Lunnon K., Schalkwyk L., Mill J.. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015; 10:1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horvath S., Ritz B.R.. Increased epigenetic age and granulocyte counts in the blood of Parkinson's disease patients. Aging. 2015; 7:1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wahl S., Drong A., Lehne B., Loh M., Scott W.R., Kunze S., Tsai P.C., Ried J.S., Zhang W., Yang Y.et al.. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017; 541:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chuang Y.-H., Paul K.C., Bronstein J.M., Bordelon Y., Horvath S., Ritz B.. Parkinson's disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017; 9:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chuang Y.-H., Lu A.T., Paul K.C., Folle A.D., Bronstein J.M., Bordelon Y., Horvath S., Ritz B.. Longitudinal epigenome-wide methylation study of cognitive decline and motor progression in Parkinson’s disease. J. Parkinsons Dis. 2019; 9:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pidsley R., Wong C.C.Y., Volta M., Lunnon K., Mill J., Schalkwyk L.C.. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013; 14:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price M.E., Cotton A.M., Lam L.L., Farré P., Emberly E., Brown C.J., Robinson W.P., Kobor M.S.. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013; 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Houseman E.A., Accomando W.P., Koestler D.C., Christensen B.C., Marsit C.J., Nelson H.H., Wiencke J.K., Kelsey K.T.. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012; 13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cotton A.M., Price E.M., Jones M.J., Balaton B.P., Kobor M.S., Brown C.J.. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet. 2014; 24:1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hovestadt V., Remke M., Kool M., Pietsch T., Northcott P.A., Fischer R., Cavalli F.M.G., Ramaswamy V., Zapatka M., Reifenberger G.et al.. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. (Berl). 2013; 125:913–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen W., Peng Y., Ma X., Kong S., Tan S., Wei Y., Zhao Y., Zhang W., Wang Y., Yan L.et al.. Integrated multi-omics reveal epigenomic disturbance of assisted reproductive technologies in human offspring. EBioMed. 2020; 61:103076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boyd A., Golding J., Macleod J., Lawlor D.A., Fraser A., Henderson J., Molloy L., Ness A., Ring S., Davey Smith G.. Cohort profile: The ‘children of the 90′s’- the index offspring of the avon longitudinal study of parents and children. Int. J. Epidemiol. 2012; 42:111–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraser A., Macdonald-Wallis C., Tilling K., Boyd A., Golding J., Davey Smith G., Henderson J., Macleod J., Molloy L., Ness A.et al.. Cohort profile: the avon longitudinal study of parents and children: ALSPAC mothers cohort. Int. J. Epidemiol. 2013; 42:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Golding J., Pembrey M., Jones R.the ALSPAC Study Team . ALSPAC: The avon longitudinal study of parents and children I. Study methodology. Paediatr. Perinat. Epidemiol. 2001; 15:74–87. [DOI] [PubMed] [Google Scholar]
- 41.Relton C.L., Gaunt T., McArdle W., Ho K., Duggirala A., Shihab H., Woodward G., Lyttleton O., Evans D.M., Reik W.et al.. Data resource profile: accessible resource for integrated epigenomic studies (ARIES). Int. J. Epidemiol. 2015; 44:1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min J.L., Hemani G., Davey Smith G., Relton C., Suderman M.. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018; 34:3983–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joseph S., George N., Green-Knox B., Nicolson T.J., Hammons G., Word B., Huang S., Lyn-Cook B.. Epigenome-Wide association (DNA Methylation) study of sex differences in normal human kidney. J. Drug Metab. Toxicol. 2017; 8:1–14. [Google Scholar]
- 44.Portales-Casamar E., Lussier A.A., Jones M.J., MacIsaac J.L., Edgar R.D., Mah S.M., Barhdadi A., Provost S., Lemieux-Perreault L.-P., Cynader M.S.et al.. DNA methylation signature of human fetal alcohol spectrum disorder. Epigenetics Chromatin. 2016; 9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horvath S., Erhart W., Brosch M., Ammerpohl O., von Schönfels W., Ahrens M., Heits N., Bell J.T., Tsai P.-C., Spector T.D.et al.. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:15538–15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ventham N.T., Kennedy N.A., Adams A.T., Kalla R., Heath S., O’Leary K.R., Drummond H., Wilson D.C., Gut I.G., Nimmo E.R.et al.. Integrative epigenome-wide analysis demonstrates that DNA methylation may mediate genetic risk in inflammatory bowel disease. Nat. Commun. 2016; 7:13507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horvath S., Mah V., Lu A.T., Woo J.S., Choi O.-W., Jasinska A.J., Riancho J.A., Tung S., Coles N.S., Braun J.et al.. The cerebellum ages slowly according to the epigenetic clock. Aging (Albany NY). 2015; 7:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S.et al.. Ensembl 2019. Nucleic Acids Res. 2019; 47:D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phipson B., Maksimovic J., Oshlack A.. missMethyl: an R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics. 2015; 32:286–288. [DOI] [PubMed] [Google Scholar]
- 50.Court F., Tayama C., Romanelli V., Martin-Trujillo A., Iglesias-Platas I., Okamura K., Sugahara N., Simon C., Moore H., Harness J.V.et al.. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014; 24:554–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pervjakova N., Kasela S., Morris A.P., Kals M., Metspalu A., Lindgren C.M., Salumets A., Mägi R.. Imprinted genes and imprinting control regions show predominant intermediate methylation in adult somatic tissues. Epigenomics. 2016; 8:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z., Yang B., Zhang M., Guo W., Wu Z., Wang Y., Jia L., Li S.Cancer Genome Atlas Research Network Cancer Genome Atlas Research Network Xie W.et al.. lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell. 2018; 33:706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bongen E., Lucian H., Khatri A., Fragiadakis G.K., Bjornson Z.B., Nolan G.P., Utz P.J., Khatri P.. Sex differences in the blood transcriptome identify robust changes in immune cell proportions with aging and influenza infection. Cell Rep. 2019; 29:1961–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J.et al.. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A.et al.. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2017; 46:D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yin Y., Morgunova E., Jolma A., Kaasinen E., Sahu B., Khund-Sayeed S., Das P.K., Kivioja T., Dave K., Zhong F.et al.. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017; 356:eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gaunt T.R., Shihab H.A., Hemani G., Min J.L., Woodward G., Lyttleton O., Zheng J., Duggirala A., McArdle W.L., Ho K.et al.. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016; 17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McRae A.F., Marioni R.E., Shah S., Yang J., Powell J.E., Harris S.E., Gibson J., Henders A.K., Bowdler L., Painter J.N.et al.. Identification of 55, 000 replicated DNA methylation QTL. Sci. Rep. 2018; 8:17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gunasekara C.J., Scott C.A., Laritsky E., Baker M.S., MacKay H., Duryea J.D., Kessler N.J., Hellenthal G., Wood A.C., Hodges K.R.et al.. A genomic atlas of systemic interindividual epigenetic variation in humans. Genome Biol. 2019; 20:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li M., Zou D., Li Z., Gao R., Sang J., Zhang Y., Li R., Xia L., Zhang T., Niu G.et al.. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019; 47:D983–D988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Almstrup K., Johansen M.L., Busch A.S., Hagen C.P., Nielsen J.E., Petersen J.H., Juul A.. Pubertal development in healthy children is mirrored by DNA methylation patterns in peripheral blood. Sci. Rep. 2016; 6:28657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trolle C., Nielsen M.M., Skakkebæk A., Lamy P., Vang S., Hedegaard J., Nordentoft I., Ørntoft T.F., Pedersen J.S., Gravholt C.H.. Widespread DNA hypomethylation and differential gene expression in Turner syndrome. Sci. Rep. 2016; 6:34220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skakkebæk A., Nielsen M.M., Trolle C., Vang S., Hornshøj H., Hedegaard J., Wallentin M., Bojesen A., Hertz J.M., Fedder J.et al.. DNA hypermethylation and differential gene expression associated with Klinefelter syndrome. Sci. Rep. 2018; 8:13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nielsen M.M., Trolle C., Vang S., Hornshøj H., Skakkebaek A., Hedegaard J., Nordentoft I., Pedersen J.S., Gravholt C.H.. Epigenetic and transcriptomic consequences of excess X-chromosome material in 47, XXX syndrome-A comparison with Turner syndrome and 46, XX females. Am. J. Med. Genet. C Semin. Med. Genet. 2020; 184:279–293. [DOI] [PubMed] [Google Scholar]
- 66.Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A.. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014; 30:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fortin J.-P., Labbe A., Lemire M., Zanke B.W., Hudson T.J., Fertig E.J., Greenwood C.M., Hansen K.D.. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014; 15:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murat K., Grüning B., Poterlowicz P.W., Westgate G., Tobin D.J., Poterlowicz K.. Ewastools: Infinium Human Methylation BeadChip pipeline for population epigenetics integrated into Galaxy. GigaScience. 2020; 9:giaa049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heiss J.A., Just A.C.. Identifying mislabeled and contaminated DNA methylation microarray data: an extended quality control toolset with examples from GEO. Clin. Epigenet. 2018; 10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McEwen L.M., O’Donnell K.J., McGill M.G., Edgar R.D., Jones M.J., MacIsaac J.L., Lin D.T.S., Ramadori K., Morin A., Gladish N.et al.. The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells. Proc. Natl Acad. Sci. 2020; 117:23329–23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jones M.J., Goodman S.J., Kobor M.S.. DNA methylation and healthy human aging. Aging Cell. 2015; 14:924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Christensen B.C., Houseman E.A., Marsit C.J., Zheng S., Wrensch M.R., Wiemels J.L., Nelson H.H., Karagas M.R., Padbury J.F., Bueno R.et al.. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009; 5:e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blake L.E., Roux J., Hernando-Herraez I., Banovich N., Perez R.G., Hsiao C.J., Eres I., Cuevas C., Marques-Bonet T., Gilad Y.. A comparison of gene expression and DNA methylation patterns across tissues and species. Genome Res. 2020; 30:250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robinson M.D., Kahraman A., Law C.W., Lindsay H., Nowicka M., Weber L.M., Zhou X.. Statistical methods for detecting differentially methylated loci and regions. Front. Genet. 2014; 5:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim J.H., Park J.-L., Kim S.-Y.. Non-negligible occurrence of errors in gender description in public data sets. Genomics Inform. 2016; 14:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dukal H., Frank J., Lang M., Treutlein J., Gilles M., Wolf I.A., Krumm B., Massart R., Szyf M., Laucht M.et al.. New-born females show higher stress- and genotype-independent methylation of SLC6A4 than males. Borderline Person. Disorder Emotion Dysregul. 2015; 2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Podcasy J.L., Epperson C.N.. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin. Neurosci. 2016; 18:437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lopes-Ramos C.M., Chen C.-Y., Kuijjer M.L., Paulson J.N., Sonawane A.R., Fagny M., Platig J., Glass K., Quackenbush J., DeMeo D.L.. Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Rep. 2020; 31:107795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lam L.L., Emberly E., Fraser H.B., Neumann S.M., Chen E., Miller G.E., Kobor M.S.. Factors underlying variable DNA methylation in a human community cohort. PNAS. 2012; 109:17253–17260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moore S.R., Humphreys K.L., Colich N.L., Davis E.G., Lin D.T.S., MacIsaac J.L., Kobor M.S., Gotlib I.H.. Distinctions between sex and time in patterns of DNA methylation across puberty. BMC Genomics. 2020; 21:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arathimos R., Sharp G.C., Granell R., Tilling K., Relton C.L.. Associations of sex hormone-binding globulin and testosterone with genome-wide DNA methylation. BMC Genet. 2018; 19:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nugent B.M., Wright C.L., Shetty A.C., Hodes G.E., Lenz K.M., Mahurkar A., Russo S.J., Devine S.E., McCarthy M.M.. Brain feminization requires active repression of masculinization via DNA methylation. Nat. Neurosci. 2015; 18:690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zou H., Hastie T.. Regularization and variable selection via the elastic net. J. R. Stat. Soc. 2005; 67:301–320. [Google Scholar]
- 84.Peters T.J., Buckley M.J., Statham A.L., Pidsley R., Samaras K., Lord R.V., Clark S.J., Molloy P.L.. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015; 8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aristizabal M.J., Anreiter I., Halldorsdottir T., Odgers C.L., McDade T.W., Goldenberg A., Mostafavi S., Kobor M.S., Binder E.B., Sokolowski M.B.et al.. Biological embedding of experience: A primer on epigenetics. Proc. Natl. Acad. Sci. 2019; 117:23261–23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun B., Hyun H., Li L., Wang A.Z.. Harnessing nanomedicine to overcome the immunosuppressive tumor microenvironment. Acta Pharmacol. Sin. 2020; 41:970–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou W., Triche T.J. Jr, Laird P.W., Shen H.. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018; 46:e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pandya-Jones A., Plath K.. The “lnc” between 3D chromatin structure and X chromosome inactivation. Semin. Cell Dev. Biol. 2016; 56:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Massah S., Hollebakken R., Labrecque M.P., Kolybaba A.M., Beischlag T.V., Prefontaine G.G.. Epigenetic characterization of the growth hormone gene identifies SmcHD1 as a regulator of autosomal gene clusters. PLoS One. 2014; 9:e97535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luijk R., Wu H., Ward-Caviness C.K., Hannon E., Carnero-Montoro E., Min J.L., Mandaviya P., Müller-Nurasyid M., Mei H., van der Maarel S.M.et al.. Autosomal genetic variation is associated with DNA methylation in regions variably escaping X-chromosome inactivation. Nat. Commun. 2018; 9:3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jung C.-H., Park D.J., Georgeson P., Mahmood K., Milne R.L., Southey M.C., Pope B.J.. sEst: Accurate sex-estimation and abnormality detection in methylation microarray data. Int. J. Mol. Sci. 2018; 19:3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Assenov Y., Müller F., Lutsik P., Walter J., Lengauer T., Bock C.. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods. 2014; 11:1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ellis S.E., Collado-Torres L., Jaffe A., Leek J.T.. Improving the value of public RNA-seq expression data by phenotype prediction. Nucleic Acids Res. 2018; 46:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Clayton J.A., Tannenbaum C.. Reporting sex, gender, or both in clinical research. JAMA. 2016; 316:1863–1864. [DOI] [PubMed] [Google Scholar]
- 95.Hannon E., Dempster E., Viana J.. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome. Biol. 2016; 17:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arloth J., Eraslan G., Andlauer T.F.M., Martins J.. DeepWAS: Multivariate genotype-phenotype associations by directly integrating regulatory information using deep learning. PLoS Comput. Biol. 2020; 16:e1007616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kilaru V., Knight A.K., Katrinli S., Cobb D.. Critical evaluation of copy number variant calling methods using DNA methylation. Genet. Epidemiol. 2020; 44:148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zannas A.S., Jia M., Hafner K., Baumert J., Wiechmann T., Pape J.C., Arloth J., Ködel M., Martinelli S., Roitman M.et al.. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB-driven inflammation and cardiovascular risk. Proc. Natl. Acad. Sci. 2019; 116:11370–11379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.