


Abstract
Macrolides have been effective clinical antibiotics for over 70 years. They inhibit protein biosynthesis in bacterial pathogens by narrowing the nascent protein exit tunnel in the ribosome. The macrolide class of natural products consist of a macrolactone ring linked to one or more sugar molecules. Most of the macrolides used currently are semi-synthetic erythromycin derivatives, composed of a 14- or 15-membered macrolactone ring. Rapidly emerging resistance in bacterial pathogens is among the most urgent global health challenges, which render many antibiotics ineffective, including next-generation macrolides. To address this threat and advance a longer-term plan for developing new antibiotics, we demonstrate how 16-membered macrolides overcome erythromycin resistance in clinically isolated Staphylococcus aureus strains. By determining the structures of complexes of the large ribosomal subunit of Deinococcus radiodurans (D50S) with these 16-membered selected macrolides, and performing anti-microbial studies, we identified resistance mechanisms they may overcome. This new information provides important insights toward the rational design of therapeutics that are effective against drug resistant human pathogens.
INTRODUCTION
Antibiotic resistant bacterial pathogens are ubiquitous in hospitals as well as community medical facilities. According to the CDC report the number of recalcitrant infections, owing to drug resistant bacteria, increases by 2.8M annually, as reported in (https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf, https://www.cdc.gov/media/releases/2019/p1113-antibiotic-resistant.html https://www.who.int/drugresistance/documents/surveillancereport/en/, https://www.who.int/news/item/17–01–2020-lack-of-new-antibiotics-threatens-global-efforts-to-contain-drug-resistant-infectionshttps://ec.europa.eu/health/sites/health/files/antimicrobial_resistance/docs/amr_2017_action-plan.pdf). Antibacterial agents often target essential processes of the cell, which impairs function and halts cellular metabolism and integrity (1,2). One of the main targets of antibacterial agents is the ribosome, the cellular organelle that translates the genetic code into proteins in all living cells. As it is essential for cellular viability, its inhibition leads to growth arrest or cell death. Structural analyses of bacterial ribosome particles in complex with paralyzing antibiotics shed light on their mode of action and basis of selectivity, thus elucidating clinical efficacy. These studies have revealed that clinical antibiotics target select functional sites in the ribosome, including the decoding region, the peptidyl transferase center (PTC), the nascent chain exit tunnel, the inter-subunit bridges and the tRNA accommodation corridor (3). These efforts have also illuminated diverse resistance mechanisms that have evolved in human pathogens to evade various antibiotic agents (3,4).
Erythromycin, the first macrolide developed as an antibacterial agent is employed clinically to treat Gram-positive and a limited number of Gram-negative bacterial infections (5). The macrolide chemical structure consists of a macrolactone ring of various sizes (typically 12–20 atoms), to which one or more sugar moieties of various types, including amino sugars, deoxy sugars, or both, are attached by an α- or β-glycosidic bond (Figure 1A) (5). Overall, macrolides inhibit protein synthesis by binding to the nascent peptide exit tunnel (NPET) in close proximity to the PTC, thus interfering with protein nascent chain progression (6). The mechanism of action of each specific macrolide is determined by the location of the sugar(s) and specific functional groups on the macrolactone ring, which affect the interactions with ribosomal RNA (rRNA) nucleotides that constitute most of the walls of the protein exit tunnel (7). Notably, whereas macrolides are efficient inhibitors of prokaryotic ribosomes, they do not bind to eukaryotic ribosomes owing to the existence of G2058 instead of the prokaryotic A2058 that forms a vital hydrogen bond between the desosamine sugar moiety C2 hydroxyl and the N1 imine of rRNA nucleotide adenosine 2058 (A2058) (Escherichia coli numbering used throughout) (8). Pikromycin, was the first macrolide discovered as a natural product in 1950 (9,10). Subsequently, additional macrolides were identified from several Streptomyces species and related actinomycetes, including erythromycin A (ery), which is the prototype for several clinically relevant antibiotics. Second generation macrolides (semi-synthetic derivatives) clarithromycin, and azithromycin (azi), are among the most widely prescribed antiinfective agents, and third generation ketolide cethromycin is clinically approved while solithromycin remains under development. Following its FDA approval, safety issues emerged with telithromycin and it was withdrawn from the US market (Figure 1). The ery-derived semisynthetic derivatives bearing a 14- or 15-membered ring core are more commonly used than other macrolide classes (Figure 1B). Tylosin, josamycin, carbomycin A, and spiramycin are 16-membered macrolide antibiotics that include a disaccharide at position C-5, with tylosin also bearing a mycinose substituent at position C-23. They are mainly used in veterinary medicine and are less studied despite their impressive antibacterial profiles and potent activity against Gram-positive and a limited range of Gram-negative organisms, similar to the 14-membered macrolides. From a molecular perspective, the 16-membered ring macrolides extend deeper in the protein exit tunnel, and their clinical advantages include better gastrointestinal tolerance, lack of drug-drug interactions, and activity against certain drug resistant bacterial strains (11). The mycinamicins (mycs) are comprised of a 16-membered macrolactone ring with O-linked sugars desosamine and mycinose located at the C-5 and C-21 positions, respectively. They were first discovered in 1980 from Micromonospora griseorubida (12), and were shown to inhibit the growth of Gram-positive bacteria, including drug-resistant Staphylococcus aureus (SA) strains (12). The wild-type M. griseorubida strain mainly generates mycs I, II, IV and V, whereas the industrial strain has been optimized for production of myc I and II as the two major products. Mycs biosynthesis occurs by the action of seven polyketide synthase (PKS) modules, including MycAI and MycAIII that contain two modules each, whereas MycAII, MycAIV and MycAV constitute monomodules. The PKS system encompasses 37 catalytic domains for assembly of the 16-membered ring protomycinolide-IV aglycone (13). Mycs I, II, IV, and V are produced by late-stage oxidation steps from the initial macrolide myc IV. Thus, myc IV can be oxidized to a terminal product myc I (through C12—C13 olefin epoxidation) or to myc V (C-14 hydroxylation). The myc V intermediate is subsequently converted to the completely oxidized myc II as the terminal pathway product resulting from a final epoxidation step (14).
Figure 1.

(A) Chemical structures of macrolide antibiotics: natural product (first generation) erythromycin A contains a 14-membered macrolactone, second generation azithromycin includes a 15-membered aza-macrolactone ring, third generation tylosin, compared to mycinamicin I, II (also called mirosamycin), and IV. Chemical variations among the antibiotics are indicated in red. (B) Classification of natural and semisynthetic macrolides based on macrolactone ring size and chemical modifications (15) shows that the majority of clinical agents approved or under development comprise 14-membered macrolides and their derivatives.
A critical challenge reflects the urgent need for new antibiotics to overcome emerging resistance in human bacterial pathogens. Four distinct macrolide resistance mechanisms have been described for SA: rRNA mutational substitutions in position 2058 or 2059, modification of the target site, that is, by rRNA methylation, activation of efflux pumps that reduces cellular concentrations of the drug, and drug inactivation by enzymatic modification or cleavage. Macrolides, lincosamides, and streptogramins B (MLSB) share an overlapping binding site in the large ribosomal subunit (6) in which they interact with A2058. Thus, methylation or demethylation of A2058 affects the ability of these three classes of compounds to form a hydrogen bond with the above mentioned nucleotide and enables resistance against MLSB (16–18). Erythromycin-resistant methylase (erm) genes that trigger mono- and di-methylation of A2058 represent a common resistance mechanism. To date, over 40 known erm genes, constitutive or inducible, were identified in bacteria (19) and it has been shown that inducible MLSB (MLSBi) is associated with exposure to 14- and 15-membered ring macrolide antibiotics (20,21). The lack of a cladinose sugar at the C-3 position of the lactone ring can potentially contribute to the ability of these compounds to remain active towards bacterial pathogens that bear inducible erm genes by preventing their activation (21). Most efflux pumps that enable a macrolide resistant phenotype are compound-specific and hence affect only a small number of drugs. Currently, approximately 25 bacterial genes are known to encode antibiotic efflux pumps, each of which is highly specific to a specific set of compounds. These primarily mediate resistance against 14- and 15-membered ring macrolides rather than 16-membered ring macrolides (http://faculty.washington.edu/marilynr/) (18). Antibiotic target protection mechanisms typically involve a protein that directly interacts and blocks an antibiotic binding site to safeguard its function (22). Such blockers are the ABC-F transporters that are widespread in Gram-positive bacteria and have been shown to act towards 14-membered-ring macrolides (23). Mechanisms that involve antibiotic inactivation (as opposed to target modification or efflux) through structural degradation or modification are less common in bacterial pathogens. They are mediated by esterases that hydrolyze the macrolactone of 14- and 15-membered ring macrolides, or by a phosphotransferase that phosphorylates one of the sugar moieties (http://faculty.washington.edu/marilynr/) (18,24,25).
In the current study, we were motivated to elucidate the structural basis for the myc group of molecules due to their ability to overcome resistance toward 14-membered macrolides. The high-resolution structures of the large ribosomal subunit of the non-pathogenic bacterium Deinococcus radiodurans (D50S), which serves as a model organism, in complex with myc I, II, and IV were determined. Anti-microbial assays were performed using these compounds to assess their in vivo potency to overcome common macrolide resistance mechanisms using antibiotic-resistant bacterial pathogens. Our studies, together with current and developing methods (14) to apply semi-synthetic and chemo-enzymatic synthesis of mycs (26), demonstrate the clinical potential of these underexplored 16-membered ring macrolide antibiotics.
MATERIALS AND METHODS
Mycinamicins biosynthesis
Mycinamicin I, II, and IV, were produced in M. griseorubida, purified and characterized as previously described (27,28).
Broth microdilution assay
Twenty seven strains of SA were tested, including twenty six isolates from human patients obtained from hospitals and community clinics and one reference strain (ATCC 29213). Spa types (29) and Panton-Vaentine Leukocidin (pvl) PCR (30) are reported in Table 1. Spa typing and pvl PCR were performed as previously described (31). The range of the antibiotic concentrations used for biological assays was 16–0.125 μg/ml. The concentration range for macrolides was selected according to the examined range of ery and azi in commercially available SensititreTM susceptibility plates (Gram-positive GPALL1F AST Plate). A stock solution of 3.2 mg/ml in DMSO was prepared for all compounds. In every 96-well plate, variable concentration antibiotic solutions were introduced in a total volume of 50μl/well. A serial dilution in the range of 32–0.25 μg/ml was prepared for every antibiotic (myc I, II, IV, ery and azi). One of the control experiments included bacterial growth in media containing DMSO without antibiotics, therefore one row was prepared accordingly, without the addition of antibiotics (Supplementary Figure S1). Four to eight colonies were suspended in 4 ml sterile milli-Q water to a turbidity of 0.5 McFarland. Then, 10 μl of the suspension was transferred into an 11 ml stock of cation-adjusted Mueller-Hinton (MH) broth (cat. Number T3462) media. The plates were inoculated manually by adding 50 μl of MH media to every well, including positive control wells. As a negative control, we used the MH media from the same stock prior to the addition of bacteria. The plates were incubated for 24 h. Minimum inhibitory concentrations (MIC) were determined according to CLSI guidelines (32) (Supplementary Figure S1). S/R was determined according to The Clinical & Laboratory Standards Institute (CLSI) guidelines.
Table 1.
Summary of the MIC values [μg/ml] of the growth inhibition assay using 27 SA strains
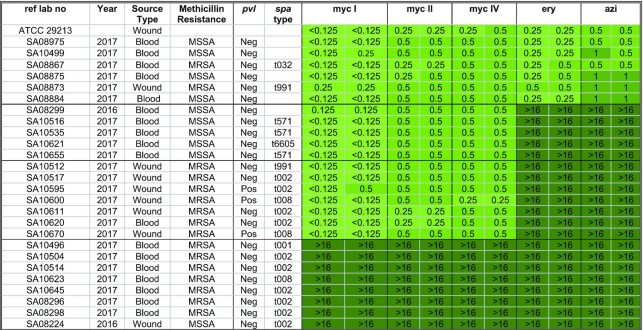
|
Disk diffusion assay
The disk diffusion assay was conducted according to the CLSI guidelines (32) testing the iMLSB resistance mechanism where clindamycin (cli) was employed as a representative lincosamide. For each strain, the inducible effect of ery over cli (ery – cli) and ery over myc I (ery – myc I) was tested, as well as the effect of myc I over cli (myc I – cli). Commercial disks were used for ery and cli (Oxoid), and for myc I, 15 μg lab-made disks was used. As a control experiment, 15 μg lab-made disks of ery were prepared. Whatman (Cat. No. 1001 125) paper disks were cut into 5 mm diameter disks, autoclaved, and soaked with myc I or ery solutions (3.2 mg/ml in DMSO) as well as a DMSO (as control). Two MH agar plates were used for every bacterial strain, for each repeat. The plates were inoculated with bacterial strains as follows: four to eight colonies were suspended in 4 ml of NANOpure Diamond® sterile water in a test tube to a turbidity of 0.5 McFarland, a swab was soaked into the tube and was used to streak the plate with a gentle back-and-forth motion, which was moved across and down the plate. The plate was rotated 60° twice and the streaking action was repeated, respectively. This ensures an even distribution of the inoculum for a confluent lawn of growth. For commercial disks, a dispenser that splits the disks into spaces of 25 mm was employed. The lab-made disks were placed manually in the same space. In the first agar plate, five disks were placed: DMSO, ery, cli, lab-made ery, cli (Supplementary Figure S2). In the second plate, four disks were placed: myc I, cli, ery, myc I (Supplementary Figure S2). All plates were incubated for 16 h at 37°C and the diameter of the clearance zone was measured (Supplementary Figure S2).
Polymerase Chain Reaction (PCR) of ermA and ermC
PCR primers were used to detect ermA and ermC genes according to previously described protocol (33). The following sequence, AAG CGG TAA ACC CCT CTG A, and TTC GCA AAT CCC TTC TCA AC for ermA and AAT CGT CAA TTC CTG CAT GT and TAA TCG TGG AAT ACG GGT TTG for ermC were used. Primers were synthesized by Syntezza (Israel). PCR amplifications were performed using PCRBIO HS Taq DNA Polymerase (PCRBIOSYSTEMS) in a 25 μl reaction mixture containing approximately 10 ng of template DNA and 2.5 pmol of tested gene primers. A sample of primers and reaction reagents, without DNA, was used as a negative control. Initial denaturation at 94°C for 3 min was followed by 30 cycles of amplification with 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s (except for the final cycle, which had an extension step of 4 min). PCR products were analyzed on a 2% agarose gel using GeneDireX 100bp DNA Ladder RTU (Supplementary Figure S3).
Deinococcus radiodurans (DR) growth and ribosomes purification
Methodology was based on previously published protocols (34,35). Briefly, DR were grown at 30°C in a lysogeny broth (LB) Oxoid medium (pH 7.3). The culture was initiated from a 1:60 dilution of glycerol stock to LB oxoid, and incubation for 24 h. The bacteria were then diluted 1:10 and incubated overnight. The bacteria were diluted again in a 1:10 dilution, incubated for 6 h and then further diluted in a 1:33 ratio. The final growth stage was performed in a 10 l fermentor after a 1:20 dilution, under constant conditions of pH (7.3) and oxygen (80%). The culture was grown to a final OD600 of 3.0. The bacterial cells were harvested by continuous-flow centrifugation, flash frozen in liquid nitrogen and stored at –80°C.
The purification procedure was based on the protocol developed by Pfizer (35). The bacterial cells were suspended in H10M30N150β6 buffer (10 mM Hepes pH 7.8, 30 mM MgCl2, 150 mM NH4Cl, 6 mM β-mercaptoethanol) and DNase RNase free (Roche Diagnostics) was added (1 μl per 1 ml of buffer). Cells were lysed using one passage through a French press at 10 000 PSI. The cell lysate was centrifuged for 45 min at 20 000 rpm in a Ti-70 rotor. The clear supernatant was collected and directly loaded onto a 10–40% linear sucrose gradient and centrifuged at 25 000 rpm for 16.5 h. The sucrose gradients were prepared using H10M30N150β6 buffer. The gradient was pumped out through an absorbance monitor and the fractions containing the 70S ribosomes were collected and concentrated by ultra-filtration using a Pelicon device (Millipore). The sucrose concentration in the samples was adjusted to 4% and loaded onto a second (10–40%) linear sucrose gradient in H10M1N100β6 (10 mM HEPES pH 7.8, 1 mM MgCl2, 100 mM NH4Cl, 6 mM β-mercaptoethanol) and centrifuged at 27 000 rpm for 17.5 h in Ti-15 zonal rotor. Fractions contained the small and large ribosomal subunits were collected separately, and the ones containing the 50S subunits were used for 50S crystallization. The magnesium concentration of the sample was adjusted to 10 mM and centrifuged for 19 h in a Ti-70 rotor. The pellets were re-suspended in the H10M15N75 buffer (10 mM HEPES pH 7.8, 15 mM MgCl2, 75 mM NH4Cl). The samples were centrifuged again in a TLA100 rotor at 75 000 RPM for 1.5 h. The pellets were re-suspended in the H10M15N75 buffer to a final concentration of 800–1000 A260 ml–1. The samples were flash-frozen in liquid nitrogen for storage at –80°C.
Crystallization of large ribosomal subunit of Deinococcus radiodurans (D50S) ribosomes
Crystals were obtained following the protocol described in Harms et al. (34) at 20°C by the hanging-drop vapor diffusion technique. Each crystallization drop (5 μl) included 10 mM spermidine, 1.25–1.5% 2-ethylhexan-1,3 diol (EHD): ethanol (ratio 1:2), 7.9–9 μM D50S subunits and H10M15N75 buffer. The reservoir solution contained 14–15% 1:2 EHD–ethanol and H10M15N75 buffer. The D50S subunits were heat activated for 30 min at 37°C before crystallization. These conditions usually yield both single crystals and clusters. After the crystals reached their maximal size (2–3 weeks), they were transferred to stabilizing solution (30% EHD–ethanol 1:2 in the crystallization buffer).
Soaking of D50S crystals in stabilization solutions containing macrolide antibiotics
To obtain crystals of D50S in complex with select antibiotics, the 50S crystals were soaked into a stabilization solution (30% EHD-ethanol 1:2 in the crystallization buffer) containing the compound. Prior to data collection, the crystals were transferred to a stabilizing solution (30% EHD–ethanol 1:2 in the crystallization buffer), which includes antibiotic (final concentration of 450 μM = ∼×50 to ribosomes) for 6 h. Afterwards, the soaked crystals were transferred to a cryo-protecting solution (15% EHD–ethanol 1:2, 15% MPD) for 10 min, which contains the same antibiotic concentration. The crystals were then flash frozen in liquid nitrogen for data collection.
Data collection and processing of D50S ribosome complexes
Crystallographic data were collected at the microfocus beamlines ID23-2 and ID30A-3, of the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Diffraction data were measured at 100 K to minimize radiation damage. Crystallographic data were collected for each D50S complex with one of the three mycs antibiotics. Due to the rapid decay of the crystals in the X-ray beam, several crystals were needed to achieve high completeness of the datasets. Data were processed using HKL2000 (36) and CCP4 package suite (37,38).
Map calculation, model building and refinement
The D50S structure (PDB code: 2ZJR) was used as a starting model for calculating difference electron density maps using PHENIX (39). A clear positive density, in both DFo – mFc and 2DFo – mFc maps, could be immediately identified and attributed to the antibiotic molecule (Figure 2D–F). The antibiotic structures were modeled using ChemDraw and their respective crystallographic Information Files (cif) was generated via Grade Web Server (40). Individual antibiotic structures were then docked into their electron density using Coot (41). Each ribosome complex was refined using alternate cycles of refinements with Phenix (42) and Coot (41). For R-free calculations, random 5% of the data were omitted during refinement. Figures were generated using Chimera (43).
Figure 2.

(A, B) D50S in complex with myc I (D50S-myc I) (magenta) superposed by three tRNA (A-tRNA in blue, P- tRNA in green, E- tRNA in yellow, from PDBID 3I8G) and a polypeptide chain (light blue) modeled in the tunnel for indicating the location of the mycinamicin binding site within the context of an active large subunit. (A) and (B) are 180° rotated images. (C) A zoom into the NPET/antibiotic binding site. The yellow star marks the PTC. (D–F) Superimposing of myc I (D), myc II (E) and myc IV (F) on difference electron density map, Fo– Fc, σ = 3.0 (light blue), prior to the refinement with the macrolide. Surrounding NPET nucleotides are shown in grey. (G) Superposition of myc I, II and IV structures from each complex shows high similarity in the overall orientation of the molecules.
RESULTS
Minimum inhibitory concentration assay using clinical isolates of SA resistant strains
In order assess the potency of mycs against SA resistant strains MSSA as well as MRSA, broth microdilution assays were performed. We tested and determined minimum inhibitory concentration (MIC) of 26 clinically isolated SA strains, including Methicillin-sensitive S. aureus (MSSA) and Methicillin-resistant S. aureus (MRSA) with some pvl positive and a single ATCC strain (described in Materials and Methods). The susceptibility range of the strains in response to each antibiotic tested is represented by a green heat map (Table 1). Twenty strains were resistant to ery and azi (ery/azi), among these, 12 were susceptible to myc I, II and IV (mycs). Six clinically isolated strains were ery-azi as well as myc I, II and IV susceptible whereas eight strains were resistant toward all compounds tested. From these broth microdilution assays, we concluded that mycs MIC values are in the same range compared to ery and azi, highlighting their clinical potential. Furthermore, a dozen strains were ery–azi resistant, yet mycs susceptible. These results encouraged us to investigate the mechanism by which myc may overcome the resistance mechanism.
Disk diffusion assay for detection of erythromycin-inducible resistance to clindamycin
In order to study the mechanism that allows the mycs to overcome resistance against ery, and their potential for the treatment of clinically isolated resistant SA bacteria, a disk diffusion assay was performed. Cli, a lincosamide antibiotic, which cannot activate the inducible MLSB genes, is a common choice in the treatment of skin and soft-tissue infections caused by staphylococci, particularly for patients allergic to penicillin (44). Although cli and ery target the ribosome, both have unique binding pockets yet share the A2058 binding site (45). Misidentification of MLSBi, caused by the ery-resistance inducible methylase (erm) may lead to failed cli treatment (46). Thus, a D-test was performed to assess whether MLSBi resistance by the erm gene was induced by the presence of ery but not by cli. In this test, a D-shaped inhibition zone indicates an induced enzyme phenotype, and this confers cross-resistance to both drugs (44) (see Table 2 and Supplementary Figure S2).
Table 2.
Summary of disk diffusion assay
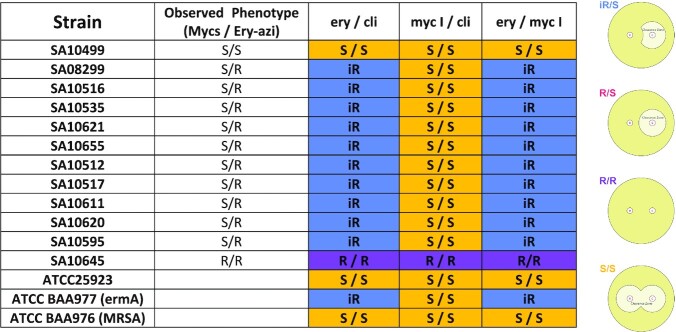
|
The MIC test (Table 1) indicated that 12 strains that are resistant to ery-azi remain susceptible to myc I, II and IV. These strains were then employed in a disk diffusion assay to visualize their activity. For the control experiment, we used a strain (SA10499) that was susceptible towards ery-azi and mycs, a strain (SA10645) that was resistant to ery-azi and mycs, and three ATCC strains: ATCC 25923 (a susceptible strain), ATCC BAA977 (bearing inducible ermA (MLSBi)) and ATCC BAA976 (known MRSA ery susceptible strain).
The pattern of the clearance zone of each tested strain is described according to the experimental results obtained for each plate tested and is explained using the disk diffusion test scheme (Table 2, Right). A MLSBi inducible resistance mechanism affects the shape of the clearance zone, and a D shaped curve is observed when it occurs. The scheme describes all possible expected results due to variability in susceptibility/resistance scenarios where (1) and (2) represent disks soaked with antibiotics (as specified in the header of each row), placed in close proximity on an agar plate. Possible results are iR (inducible resistance) induced by macrolides, most probably by MLSBi mechanism, where the binding site of compound no. 2 is overlapping the binding site of compound no. 1. Therefore, the low concentrations of compound no. 1 triggered resistance towards compound no. 2. R/S—no inducible resistance on compound no. 2 by compound no. 1. R/R—resistance towards compound no. 1 and compound no. 2, S/S—susceptible towards both compounds. The different colors in the table correlate to the colors of the scheme labels. Comparing the disk diffusion assay results of the ery/cli and ery/myc I assays, we observed that ery induced equivalent levels of resistance toward cli and myc I. Thus, we conclude that ery is the inducer antibiotic that activates this specific resistance mechanism. In the myc I/cli plates none of these compounds activate the MLSBi inducible resistance mechanism.
Polymerase chain reaction (PCR) of ermA and ermC
The ability of a macrolide to overcome resistance mechanisms is critical for efficacy, and thus, we sought to identify the specific resistance mechanisms induced by ery, in these ten strains. The ermA and ermC resistance genes are frequently present in SA clinically isolated strains (44), therefore a direct PCR reaction was conducted to determine their presence.
Table 3 summarises the PCR assays performed to identify the presence of ermA and ermC using ten ery-azi resistant/myc susceptible strains as well as a single ery-azi/myc susceptible (strain SA10499), and a single ery-azi/myc resistant (strain SA10645) strains. Two strains were identified as ermC positive, four strains were identified as ermA positive and the remaining four strains were ermA and ermC negative.
Table 3.
Summary of polymerase chain reaction (PCR) assay
| Ref lab no | Observed phenotype (myc/ery-azi) | Methicillin resistance | Cli | ermA PCR* | ermC PCR* | Resistance mechanism |
|---|---|---|---|---|---|---|
| SA10499 | S / S | MSSA | S | N | N | |
| SA08299 | S / R | MSSA | S | N | N | I |
| SA10516 | S / R | MSSA | S | N | N | I |
| SA10535 | S / R | MSSA | S | N | N | I |
| SA10621 | S / R | MSSA | S | N | N | I |
| SA10655 | S / R | MSSA | S | N | N | I |
| SA10512 | S / R | MRSA | S | N | P | II |
| SA10517 | S / R | MRSA | S | P | N | III |
| SA10611 | S / R | MRSA | S | P | N | III |
| SA10620 | S / R | MRSA | S | P | N | III |
| SA10595 | S / R | MRSA | S | N | P | II |
| SA10645 | R / R | MRSA | R | P | N | IV |
Abbreviations: myc – mycinamicin, ery – erythromycin, azi – azithromycin, cli – clindamycin, N – negative, P – positive, S – susceptible, R –resistant.
*PCR reactions were performed in duplicate.
Based on the MIC tests, disk diffusion assays (Tables 1 and 2), and PCR results (Table 3), the strains were sorted into four classes: (I) SA strains where mycs overcome ery resistance by an unknown ery-inducible mechanism (II) SA strains where mycs overcome ery resistance from an inducible ermC mechanism, (III) SA strains where mycs overcome ery resistance from an inducible ermA mechanism, (IV) SA strain with constitutive ermA resistance or combination of ermA with other unknown mechanism that mycs do not overcome. Furthermore, most MSSA strains were negative towards ermA and ermC, yet susceptible to mycs.
Structural studies
Complete crystallographic data sets of D50S in complex with myc I, myc II and myc IV yielded electron density maps at 3.22, 3.42 and 3.42 Å resolution (Table 4), respectively. The crystal structures indicate that the mycs bind at the upper rim of the nascent peptide exit tunnel (NPET) in close proximity to the PTC (Figure 2A–C is shown for orienting the reader in the mycs binding site with respect to the binding sites of modelled tRNA molecules and a nascent polypeptide chain), in agreement with other known macrolides bound to the large ribosomal subunit (6,7,47–50). In each example, the locations and conformations of the bound antibiotic could be unambiguously resolved as observed from the difference electron density map (Figure 2D–F).
Table 4.
Crystallographic data and refinement statistics of the merged data sets for D50S in complex with myc I, myc II and myc IV. Values for the highest-resolution shells are in parentheses
| D50S-myc I | D50S-myc II | D50S-myc IV | |
|---|---|---|---|
| Crystal information | |||
| Space group | I 2 2 2 (no. 23) | I 2 2 2 | I 2 2 2 |
| a (Å) | 170.0 | 170.5 | 169.6 |
| b (Å) | 410.7 | 408.9 | 410.1 |
| c (Å) | 697. 8 | 697.3 | 690.5 |
| α = β = γ (°) | 90 | 90 | 90 |
| Number of molecules per asymmetric unit | 1 | 1 | 1 |
| Z | 8 | 8 | 8 |
| VM (V/Mw) | 3.8 | 3.8 | 3.8 |
| Solvent content (%) | 68.2% | 68.0% | 67.6% |
| Diffraction data statistics | |||
| X-ray source (ESRF) | ID23-2 | ID23-2 | ID30A-3 |
| Wavelength (Å) | 0.873 | 0.873 | 0.968 |
| Number of crystals used | 3 | 2 | 6 |
| Num. of data sets merged | 15 | 12 | 23 |
| Resolution (Å) | 50–3.21 (3.27–3.21) | 50–3.42 (3.48–3.42) | 50–3.40 (3.48–3.42) |
| Observed reflections | 1 143 022 | 1 224 927 | 1 716 395 |
| Unique reflections | 306 799 | 359 719 | 344 576 |
| Redundancy | 4.5 (4.5) | 3.4 (3.3) | 4.1 (3.2) |
| Completeness (%) | 99.6 (99.6) | 98.4 (99.1) | 94.1 (90.6) |
| <I>/<σ(I)> | 13.28 (1.0) | 5.47 (1.1) | 4.17 (1.18) |
| R-merge (%) | 14.2 (96.7) | 18.2 (88.8) | 29.5 (92.0) |
| Refinement statistics | |||
| R-factor (%) | 22.50 | 21.51 | 28.71 |
| R-free (%) | 25.59 | 24.92 | 31.82 |
| RMSD bonds (Å) | 0.008 | 0.008 | 0.008 |
| RMSD angles (°) | 1.351 | 1.378 | 1.394 |
Mycs binding pocket description
All three mycs bind the ribosome at the upper part of the NPET, in a very similar orientation (Figure 2G), and form hydrogen bonds between the desosamine C2 hydroxyl and N1 of A2058 and between the mycinose C4 hydroxyl and O6 of A752 (Figure 3B–D). The macrolactone ring and the desosamine sugar interact through Van der Waals interactions with the rRNA domain V, residues A2058, A2059, A2062, G2505, U2609 and U/C2611 (Figure 3A). The mycinose orientation reveals Van der Waals interactions with rRNA domain II, helix 35, residues U746, G748, A751, A752 (Figure 3A), which are part of the NPET.
Figure 3.

(A) Mycs molecular interactions with binding site nucleotides in the D50S NPET. 2D structure diagram (51) of the 23S rRNA where the nucleotides that directly interact with mycs are marked in orange) (B–D) Stereo-view of the binding pocket of myc I (yellow), myc II (cyan) and myc IV (magenta) at the ribosome NPET (grey). Hydrogen bonds (black dashes) between the desosamine C2 hydroxyl and A2058 N1 (A2041) and the mycinose C4 hydroxyl and A752 N4 (C765) are marked. (E, F) Superposition of the apo D50S structure (PDB code 2ZJR, grey) and the D50S-myc I (yellow), D50S-myc II (cyan) and D50S-myc IV (magenta). Only the nucleotide with variable orientation in the three crystal structures are shown. uL22 R111 shown (light blue) is part of the apo structure. (A) and (B) are 90° apart.
Orientation of nucleotides at the mycs binding site
Superposition of the structures of the three mycs (myc I, myc II and myc IV) complexes reveals that the spatial orientations of the three antibiotics are very similar (Figure 3E, F and Supplementary Figure S5). Structural changes due to antibiotic binding could be identified by superpositioning of the complex structures of D50S with myc I (D50S-myc I), myc II (D50S-myc II) and myc IV (D50S-myc IV) and the apo ribosome structure (PDB: 2ZJR) (Figure 3E and F). Upon mycs binding, the nitrogen base of nucleotide U746 (C759) shifts by ∼3Å relative to the apo D50S subunit. In addition, the side chain of rProtein uL22 R111 (SA numbering, K90 in E. coli) may also be displaced to avoid clashing with the antibiotic molecules. R111 could not be modeled in the mycs complexes due to lack of an electron density map for this bulky side chain, seemingly because the arginine side chain becomes flexible upon antibiotic binding. Nucleotide A2062 (A2045), a flexible moiety within the PTC (34) is flipped 90° and interacts hydrophobically with the mycs desosamine sugar. Furthermore, U790 (C803), a more distant domain II 23S rRNA nucleotide, that is located in the tunnel's upper rim, is a first shell nucleotide, without direct interaction with the antibiotic, which appears to be dynamic in the tunnel of the D50S-mycs complex structures.
DISCUSSION
The structures of the complexes of 16-membered ring macrolides myc I, II and IV with the large ribosomal subunit indicate that the antibiotics bind to the known macrolide binding site. These three mycinamicin derivatives bind the ribosome in a similar orientation and their superposition indicates that the C12–C14 macrolactone substituents are of minor importance for the interactions of the drug with the ribosome (Figure 2G) since this moiety is facing the tunnel void. However, these atoms may be important for the modes of their specific antibiotic action as they may interact with the nascent chain in a differential manner.
Inspecting the view of the tunnel from its interior towards the PTC (Figure 5A–E), as well as the view from the PTC toward the tunnel (Figure 5F–J), show that the mycs narrow the tunnel as opposed to causing a complete block. However, despite this binding mode, the potency of these compounds as antibacterial agents is reflected by their MIC values (Table 1). Furthermore, to confirm the similarity of the binding location and orientation of the mycinamicins to the S. aureus ribosome and for supporting our claims about the potential of these compounds for clinical development against SA infections, we used single particle cryo-EM for studying the complex structure of the SA ribosome with myc I (to be published). Thus, the 3.2 Å resolution structure of the SA50S-myc I complex was superimposed on the D50S-myc I structure and indicates that the antibiotic binds the SA ribosome in a highly similar location and orientation to the DR ribosome (Supplementary Figure S4A).
Figure 5.

Macrolides bound to the ribosomal tunnel. (A–E) A view at the tunnel entrance from the internal side of the tunnel showing the electrostatic surface of the tunnel (A) and its blockage by ery (B) PDB: 4V7U, myc I (C), myc II (D) and myc IV (E), on D50S crystal structure. The green arrows represent the directionality of the nascent protein chain in the tunnel. (F–J) A view at the tunnel entrance from the PTC showing the electrostatic surface of the tunnel (F) and its blockage by ery (G) PDB: 4V7U, myc I (H), myc II (I) and myc IV (J) on D50S crystal structures. The blue arrows represent the directionality of the nascent protein chain in the tunnel.
The MIC assays indicated that among these three antibiotics, myc I, which includes a C12—C13 epoxide moiety (but lacks the C14 hydroxyl group of myc II and myc V) is the most active compound (Table 1). Further studies are required to determine whether other substituents or chemical modifications can improve the pharmacokinetics qualities and in vivo potency of these compounds. Furthermore, the MIC assays of various SA strains and some ery-resistant strains (Table 1), indicate that the compounds are active in vivo and may overcome specific SA resistance mechanisms including ermA and ermC. Thus, 16-membered ring macrolides bearing mycinose and desosamine sugar (as opposed to cladinose and desosamine in ery) can potentially become a prototype for the next generation of this class of antibiotics.
The focus of these structural studies was to decipher the inhibitory mechanism of mycs I, II and IV, and highlight their interactions with 23S rRNA domain II nucleotides (Figure 3A), in addition to the typical macrolide structural interactions in the NPET at the 23S domain V nucleotides (Figure 3A). We hypothesize that although resistance may be inevitable in the long-term, additional interactions of the antibiotic drugs at the NPET might delay emergence and attenuate resistance, compared to methylation of nucleotide A2058 at the NPET.
Furthermore, our current study provides new insights regarding the mode of 16-membered ring macrolide binding to its prokaryotic biological target. The superposition of the D50S-myc I structure with the previously available structures of 16-membered ring macrolides bound to H. marismortui 50S ribosomal subunit (H50S) (PDBIDs 1K9M, 1K8A and 1KD1), indicates that tylosin, carbomycin and spiramycin (Figure 4A–C) bind in a similar orientation despite the fact that the identity of nucleotide 2058 in archea is G, which should preclude their binding. Thus, the available complex structures reveal that H50S-tylosin, H50S-carbomycin and H50S-spiramycin include a steric clash between the macrolide's mycaminose sugar moiety and the G2058 nucleotide residue. Tylosin, carbomycin, and spiramycin share an identical disaccharide, composed of mycaminose and mycarose at C5 (Figure 4), which is pointing toward the PTC and narrows the upper rim of the tunnel. Tylosin shares a mycinose sugar moiety with the mycinamycins and it also interacts with domain II rRNA nucleotides (52). By contrast, 16-membered ring macrolides spiramycin and carbomycin do not interact with domain II of the rRNA nucleotides due to the lack of an additional sugar substituent oriented in this direction. Furthermore, in comparison to these 16-membered ring macrolides where tylosin, carbomycin and spiramycin form a covalent imine bond with their C6 aldehyde moiety and the rRNA A2062 nucleotide, a key hydrogen bond interaction with the myc I and the rRNA A2062 nucleotide is formed.
Figure 4.

(A–C) A superposition of D50S-myc I (yellow) on the 16-membered ring macrolides bound to the H. marismortui large ribosomal subunit, the orange arrow directs the PTC; (A) tylosin (green), (B) carbomycin (pink) and (C) spiramycin (blue). The distances between the bound antibiotics and the G2058 rRNA nucleotide indicate a clash between the bound drugs and the guanine, which does not represent the clinical scenario of a macrolide bound to a prokaryotic ribosome. (D) Superposition of D50S-myc I (yellow) and the E. coli 70S structure with A2058 N6 dimethylation (brown), which shows a clash with the macrolide and preventing its binding to the pocket (the orange arrow indicates direction to the PTC).
Currently no antibiotic belonging to the 16-membered ring macrolide class has been clinically approved for human use, and thus a robust resistance mechanism has not emerged. On the other hand, SA resistance to tylosin, which has been employed for decades as an animal health product (e.g. antibiotic and growth promotion), is primarily ermB (53). Additional known synergistically acting tylosin self-resistance genes from the producing organism Streptomyces fradiae (54), tlrA, tlrB, tlrC and tlrD confer A2058 and G748 methylation (16). The mobilization of these genes could result in cross-over mycinamycin resistance, thus limiting its long-term clinical efficacy if tylosin is persistently used in veterinary medicine (55). Fortunately, various recent studies suggest how the potential cross-over resistance burden may be deliberately managed and reduced (56,57).
The screen of MSSA and MRSA clinical isolates demonstrated the potential of these compounds as antibacterial agents. In these MICs assays, using 20 ery-resistant MSSA and MRSA isolates, 12 strains were mycs susceptible. The t008 strains that are pvl positive belong to the pandemic USA300 lineage which is a major cause of skin infection outbreaks globally (strains SA10600, SA10670). The t002 strain that is pvl positive is a common lineage in the Bedouin population in southern Israel (strain SA10595). It is interesting to note that both of these lineages are ery-resistant azi-resistant and myc-susceptible.
PCR identified a single resistance mechanism that the mycs cannot overcome, namely the structural changes in position A2058 in the ribosomal NPET. This was rationalized by modeling the outcome of various common resistance mechanisms on the mycs-ribosome complex structures. A2058 N6 dimethylation (58) (Figure 4D), as well as a single mutation in position A2058 may cause a steric clash between the desosamine sugar of the drug and the modified nucleotide (similar to the H50S and 16-membered ring macrolides shown in Figure 4). It has been shown that tylosin (a 16-membered ring macrolide) escapes this steric clash due to its slightly different ribosome binding orientation compared to erythromycin (59), where the C6 aldehyde appears to be located ideally to form a covalent imine bond with A2062 (Figure 4A–C). The formation of this covalent bond with A2062 is unique to tylosin, carbomycin and spiramycin 16-membered lactone ring macrolides due to their size and their common C6 aldehyde moiety, which is lacking in the 14-membered macrolide antibiotics. A double mutation A2058G and A2062U resulted in a higher level of drug resistance towards 16-membered macrolides in M. smegmatis (59), which underscores the significance of the macrolide A2062 interaction. In this respect, the mycinamycins are also interacting with A2062, via a hydrogen bond, but do not escape resistance due to A2058 modifications. Nevertheless, the clinical use of 14-membered ring macrolides as a treatment against bacterial pathogens having MLSBi resistance triggers the erm genes and severely limits the use of lincosamides and streptogramins B (60). Using PCR, we identified other myc I, II and IV-susceptible ery-resistance strains with resistance mechanisms that do not involve ermA and ermC, and further investigation is needed to determine their molecular mechanisms.
Our studies on various S. aureus MLSBi bacterial pathogens demonstrates that the major advantage of mycs includes their inability to induce erm expression. Therefore, our results combined with the current ability to generate new 16-membered macrolides by chemoenzymatic approaches (26) provides a promising path for future discovery and clinical development of this underexplored class of therapeutics for fighting MLSBi resistance in human bacterial pathogens.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank (PDB) under accession codes 7A0R, 7A0S and 7A18.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff members at Beamlines ID23 and ID30A of the European Synchrotron Radiation Facility/European Molecular Biology Laboratory for their assistance during data collection. We thank Dr Moshe Peretz, Andre Rivalta, Shoshana Tel-Or, Miriam Lachever and Dr Maggie Kessler for their interest and experimental support. We also wish to express our gratitude to Ghil Jona and Amnon Naziri from the Weizmann Bacteriology Unit for performing the large-scale fermentation of DR.
Author contributions: E.B.-G. performed ribosome purification and crystallization. A.N.-L., Y.A. and J.J.-S. produced the compounds under supervision of D.H.S. E.B.-G., Z.E., D.M., Y.H., G.C. H.R., E.Z. and A.B. collected X-ray data. E.B.-G. determined the structures with the help of D.M. and Z.E. under supervision of H.R., A.B. and A.Y., E.B.-G., M.B. and A.V.E. preformed in vivo studies under supervision of A.R. and L.V. A.Y. and D.H.S. initiated the study. E.B.-G., A.B. and A.Y. prepared the manuscript with input from D.H.S., L.V. and A.R.
Notes
Present address: Zohar Eyal, Department of Planet and Environmental Science, The Weizmann Institute of Science, Rehovot 760001, Israel.
Contributor Information
Elinor Breiner-Goldstein, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
Zohar Eyal, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
Donna Matzov, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
Yehuda Halfon, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
Giuseppe Cimicata, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
Moti Baum, Government Central Laboratories, Ministry of Health, Jerusalem 91342, Israel.
Assaf Rokney, Government Central Laboratories, Ministry of Health, Jerusalem 91342, Israel.
Analia V Ezernitchi, Government Central Laboratories, Ministry of Health, Jerusalem 91342, Israel.
Andrew N Lowell, Life Sciences Institute and Departments of Medicinal Chemistry, Chemistry, Microbiology & Immunology, University of Michigan, Ann Arbor, MI 48109-2216, USA.
Jennifer J Schmidt, Life Sciences Institute and Departments of Medicinal Chemistry, Chemistry, Microbiology & Immunology, University of Michigan, Ann Arbor, MI 48109-2216, USA.
Haim Rozenberg, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
Ella Zimmerman, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
Anat Bashan, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
Lea Valinsky, Government Central Laboratories, Ministry of Health, Jerusalem 91342, Israel.
Yojiro Anzai, Department of Microbiology, Faculty of Pharmaceutical Sciences, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-0072, Japan.
David H Sherman, Life Sciences Institute and Departments of Medicinal Chemistry, Chemistry, Microbiology & Immunology, University of Michigan, Ann Arbor, MI 48109-2216, USA.
Ada Yonath, Department of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot 760001, Israel.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
University of Michigan-Israel Partnership for Research, the European Research Council [322581 (NOVRIB)]; Kimmelman Center for Macromolecular Assemblies, and the National Institutes of Health [R35 GM118101]. Funding for open access charge: European Research Council Grants [322581].
Conflict of interest statement. None declared.
REFERENCES
- 1.Lewis K.Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013; 12:371. [DOI] [PubMed] [Google Scholar]
- 2.Coates A., Hu Y., Bax R., Page C.. The future challenges facing the development of new antimicrobial drugs. Nat. Rev. Drug Discov. 2002; 1:895. [DOI] [PubMed] [Google Scholar]
- 3.Auerbach-Nevo T., Baram D., Bashan A., Belousoff M., Breiner E., Davidovich C., Cimicata G., Eyal Z., Halfon Y., Krupkin M.et al.. Ribosomal antibiotics: contemporary challenges. Antibiotics. 2016; 5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crowe-McAuliffe C., Graf M., Huter P., Takada H., Abdelshahid M., Nováček J., Murina V., Atkinson G.C., Hauryliuk V., Wilson D.N.. Structural basis for antibiotic resistance mediated by the Bacillus subtilis ABCF ATPase VmlR. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:8978–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiomi K., Ōmura S., Ōmura S.Macrolide Antibiotics: Chemistry, Biology, and Practice. 2003; 2nd ednSan Diego: Academic Press; 1–56. [Google Scholar]
- 6.Schlünzen F., Zarivach R., Harms J., Bashan A., Tocilj A., Albrecht R., Yonath A., Franceschi F.. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001; 413:814. [DOI] [PubMed] [Google Scholar]
- 7.Dunkle J.A., Xiong L., Mankin A.S., Cate J.H.. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:17152–17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Böttger E.C., Springer B., Prammananan T., Kidan Y., Sander P.. Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO Rep. 2001; 2:318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brockmann H., Henkel W.. Pikromycin, ein neues antibiotikum aus actinomyceten. Naturwissenschaften. 1950; 37:138–139. [Google Scholar]
- 10.Brockmann H., Henkel W.. Pikromycin, ein bitter schmeckendes Antibioticum aus Actinomyceten (Antibiotica aus Actinomyceten, VI. Mitteil. Chem. Ber. 1951; 84:284–288. [Google Scholar]
- 11.Arsic B., Barber J., Čikoš A., Mladenovic M., Stankovic N., Novak P.J.I.j.o.a.a.. 16-membered macrolide antibiotics: a review. Int. J. Antimicrob. Agents. 2018; 51:283–298. [DOI] [PubMed] [Google Scholar]
- 12.Satoi S., Muto N., Hayashi M., Fujii T., Otani M.. Mycinamicins, new macrolide antibiotics. I. J. Antibiot. (Tokyo). 1980; 33:364–376. [DOI] [PubMed] [Google Scholar]
- 13.Anzai Y., Saito N., Tanaka M., Kinoshita K., Koyama Y., Kato F.J.F.m.l.. Organization of the biosynthetic gene cluster for the polyketide macrolide mycinamicin in Micromonospora griseorubida. FEMS Microbiol. Lett. 2003; 218:135–141. [DOI] [PubMed] [Google Scholar]
- 14.Li S., Tietz D.R., Rutaganira F.U., Kells P.M., Anzai Y., Kato F., Pochapsky T.C., Sherman D.H., Podust L.M.. Substrate recognition by the multifunctional cytochrome P450 MycG in mycinamicin hydroxylation and epoxidation reactions. J. Biol. Chem. 2012; 287:37880–37890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon J.H.Cohen J., Powderly W.G., Opal S.M.. Infectious Diseases. 2017; 4th ednElsevier; 1217–1229. [Google Scholar]
- 16.Liu M., Douthwaite S.. Resistance to the macrolide antibiotic tylosin is conferred by single methylations at 23S rRNA nucleotides G748 and A2058 acting in synergy. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:14658–14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vester B., Douthwaite S.. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob. Agents Chemother. 2001; 45:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leclercq R.Mechanisms of resistance to macrolides and in lincosamides: Nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 2002; 34:482–492. [DOI] [PubMed] [Google Scholar]
- 19.Roberts M.C., Schwarz S., Aarts H.J.. Acquired antibiotic resistance genes: an overview. Front. Microbiol. 2012; 3:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pechère J.-C.J.I.j.o.a.a.Macrolide resistance mechanisms in Gram-positive cocci. Int. J. Antimicrob. Agents. 2001; 18:25–28. [DOI] [PubMed] [Google Scholar]
- 21.Bailey M., Chettiath T., Mankin A.S.J.A.a.chemotherapy. . Induction of erm (C) expression by noninducing antibiotics. Antimicrob. Agents Chemother. 2008; 52:866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharkey L.K., O’Neill A.J.. Antibiotic resistance ABC-F proteins: bringing target protection into the limelight. ACS Infect. Dis. 2018; 4:239–246. [DOI] [PubMed] [Google Scholar]
- 23.Reynolds E., Ross J.I., Cove J.H.. Msr (A) and related macrolide/streptogramin resistance determinants: incomplete transporters?. Int. J. Antimicrob. Agents. 2003; 22:228–236. [DOI] [PubMed] [Google Scholar]
- 24.Barthelemy P., Autissier D., Gerbaud G., Courvalin P.. Enzymic hydrolysis of erythromycin by a strain of Escherichia coli. J. Antibiot. (Tokyo). 1984; 37:1692–1696. [DOI] [PubMed] [Google Scholar]
- 25.Ounissi H., Courvalin P.. Nucleotide sequence of the gene ereA encoding the erythromycin esterase in Escherichia coli. Gene. 1985; 35:271–278. [DOI] [PubMed] [Google Scholar]
- 26.Lowell A.N., DeMars M.D., Slocum S.T., Yu F., Anand K., Chemler J.A., Korakavi N., Priessnitz J.K., Park S.R., Koch A.A.. Chemoenzymatic total synthesis and structural diversification of tylactone-based macrolide antibiotics through late-stage polyketide assembly, tailoring, and C-H functionalization. J. Am. Chem. Soc. 2017; 139:7913–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anzai Y., Tsukada S., Sakai A., Masuda R., Harada C., Domeki A., Li S., Kinoshita K., Sherman D.H., Kato F.. Function of cytochrome P450 enzymes MycCI and MycG in Micromonospora griseorubida, a producer of the macrolide antibiotic mycinamicin. Antimicrob. Agents Chemother. 2012; 56:3648–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anzai Y., Li S., Chaulagain M.R., Kinoshita K., Kato F., Montgomery J., Sherman D.H.. Functional analysis of MycCI and MycG, cytochrome P450 enzymes involved in biosynthesis of mycinamicin macrolide antibiotics. Chem. Biol. 2008; 15:950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koreen L., Ramaswamy S.V., Graviss E.A., Naidich S., Musser J.M., Kreiswirth B.N.. spa typing method for discriminating among Staphylococcus aureus isolates: implications for use of a single marker to detect genetic micro-and macrovariation. J. Clin. Microbiol. 2004; 42:792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szmigielski S., Prevost G., Monteil H., Colin D., Jeljaszewicz J.. Leukocidal toxins of staphylococci. Zentral. Bakteriol. 1999; 289:185–201. [DOI] [PubMed] [Google Scholar]
- 31.Rokney A., Baum M., Ben-Shimol S., Sagi O., Anuka E., Agmon V., Greenberg D., Valinsky L., Danino D.. Dissemination of the methicillin-resistant staphylococcus aureus pediatric clone (ST5-T002-IV-PVL+) as a major cause of community-associated staphylococcal infections in bedouin children, Southern Israel. Pediatr. Infect. Dis. J. 2019; 38:230–235. [DOI] [PubMed] [Google Scholar]
- 32.CLSI Performance Standards for AntimicrobialSusceptibility Testing. 2018; 28 ednWayne, Pennsylvania, USA: Clinical and Laboratory Standards Institute; 296. [Google Scholar]
- 33.Strommenger B., Kettlitz C., Werner G., Witte W.. Multiplex PCR assay for simultaneous detection of nine clinically relevant antibiotic resistance genes in Staphylococcus aureus. J. Clin. Microbiol. 2003; 41:4089–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harms J., Schluenzen F., Zarivach R., Bashan A., Gat S., Agmon I., Bartels H., Franceschi F., Yonath A.. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001; 107:679–688. [DOI] [PubMed] [Google Scholar]
- 35.McLellan T.J., Marr E.S., Wondrack L.M., Subashi T.A., Aeed P.A., Han S., Xu Z., Wang I.-K., Maguire B.A.. A systematic study of 50S ribosomal subunit purification enabling robust crystallization. Acta Crystallogr. Sect. D. 2009; 65:1270–1282. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 37.Collaborative C.P.The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, Biol. Crystallogr. 1994; 50:760. [DOI] [PubMed] [Google Scholar]
- 38.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A.. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smart O., Womack T., Sharff A., Flensburg C., Keller P., Paciorek W., Vonrhein C., Bricogne G.. Grade,version 1.2.20. 2011; Cambridge, UK: Global Phasing Ltd; https://www.globalphasing.com. [Google Scholar]
- 41.Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. Sect. D. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liebschner D., Afonine P.V., Baker M.L., Bunkóczi G., Chen V.B., Croll T.I., Hintze B., Hung L.-W., Jain S., McCoy A.J.. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D: Struct. Biol. 2019; 75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 44.Fiebelkorn K., Crawford S., McElmeel M., Jorgensen J.. Practical disk diffusion method for detection of inducible clindamycin resistance in Staphylococcus aureus and coagulase-negative staphylococci. J. Clin. Microbiol. 2003; 41:4740–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matzov D., Eyal Z., Benhamou R.I., Shalev-Benami M., Halfon Y., Krupkin M., Zimmerman E., Rozenberg H., Bashan A., Fridman M.J.N.a.r.. Structural insights of lincosamides targeting the ribosome of Staphylococcus aureus. Nucleic Acids Res. 2017; 45:10284–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drinkovic D., Fuller E.R., Shore K.P., Holland D.J., Ellis-Pegler R.. Clindamycin treatment of Staphylococcus aureus expressing inducible clindamycin resistance. J. Antimicrob. Chemother. 2001; 48:315–316. [DOI] [PubMed] [Google Scholar]
- 47.Belousoff M.J., Shapira T., Bashan A., Zimmerman E., Rozenberg H., Arakawa K., Kinashi H., Yonath A.. Crystal structure of the synergistic antibiotic pair, lankamycin and lankacidin, in complex with the large ribosomal subunit. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen J.L., Ippolito J.A., Ban N., Nissen P., Moore P.B., Steitz T.A.. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell. 2002; 10:117–128. [DOI] [PubMed] [Google Scholar]
- 49.Almutairi M.M., Svetlov M.S., Hansen D.A., Khabibullina N.F., Klepacki D., Kang H.-Y., Sherman D.H., Vázquez-Laslop N., Polikanov Y.S., Mankin A.S.. Co-produced natural ketolides methymycin and pikromycin inhibit bacterial growth by preventing synthesis of a limited number of proteins. Nucleic Acids Res. 2017; 45:9573–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hansen J.L., Ippolito J.A., Ban N., Nissen P., Moore P.B., Steitz T.A.. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell. 2002; 10:117–128. [DOI] [PubMed] [Google Scholar]
- 51.Cannone J.J., Subramanian S., Schnare M.N., Collett J.R., D'Souza L.M., Du Y., Feng B., Lin N., Madabusi L.V., Müller K.M.. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002; 3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poulsen S.M., Kofoed C., Vester B.J.J.o.m.b.. Inhibition of the ribosomal peptidyl transferase reaction by the mycarose moiety of the antibiotics carbomycin, spiramycin and tylosin. J. Mol. Biol. 2000; 304:471–481. [DOI] [PubMed] [Google Scholar]
- 53.Bahraminia F., Emadi S.R., Emaneini M., Farzaneh N., Rad M., Khoramian B.. Veterinary Research Forum. Faculty of Veterinary Medicine. 2017; 8:Urmia, Iran: Urmia University; 121. [PMC free article] [PubMed] [Google Scholar]
- 54.Zalacain M., Cundliffe E.. Methylation of 23S rRNA caused by tlrA (ermSF), a tylosin resistance determinant from Streptomyces fradiae. J. Bacteriol. 1989; 171:4254–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cazer C., Eldermire E., Lhermie G., Murray S.A., Scott H.M., Grohn Y.T.. The effect of tylosin on antimicrobial resistance in beef cattle enteric bacteria: a systematic review and meta-analysis. Prev. Vet. Med. 2020; 176:104934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weinroth M.D., Martin J.N., Doster E., Geornaras I., Parker J.K., Carlson C.R., Metcalf J.L., Morley P.S., Belk K.E.. Investigation of tylosin in feed of feedlot cattle and effects on liver abscess prevalence, and fecal and soil microbiomes and resistomes. J. Anim. Sci. 2019; 97:4567–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao H., Zhao Q., Cui P., Chen Z., Yu Z., Geisen S., Friman V.-P., Zhou S.. Efficient reduction of antibiotic residues and associated resistance genes in tylosin antibiotic fermentation waste using hyperthermophilic composting. Environ. Int. 2019; 133:105203. [DOI] [PubMed] [Google Scholar]
- 58.Svetlov M.S., Syroegin E.A., Aleksandrova E.V., Atkinson G.C., Gregory S.T., Mankin A.S., Polikanov Y.S.J.N.C.B.. Structure of Erm-modified 70S ribosome reveals the mechanism of macrolide resistance. Nat. Chem. Biol. 2021; 17:412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pfister P., Jenni S., Poehlsgaard J., Thomas A., Douthwaite S., Ban N., Böttger E.C.J.J.o.m.b.. The structural basis of macrolide–ribosome binding assessed using mutagenesis of 23 S rRNA positions 2058 and 2059. J. Mol. Biol. 2004; 342:1569–1581. [DOI] [PubMed] [Google Scholar]
- 60.Lewis J.S. II, Jorgensen J.H.. Inducible clindamycin resistance in staphylococci: should clinicians and microbiologists be concerned?. Clin. Infect. Dis. 2005; 40:280–285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank (PDB) under accession codes 7A0R, 7A0S and 7A18.