

Abstract
Tuberculosis (TB) is one of the leading infectious diseases worldwide even with the ravaging COVID-19 pandemic in recent times. This mandated further search and exploration of more possible anti-TB drug candidates against M. tuberculosis strains. As an extension of our previous work on the homology modeled cytochrome b subunit of the bc1 complex (QcrB) of Mycobacterium tuberculosis, an in-silico design was carried out in order to further explore more newly potential anti-TB compounds. Ligand 26 was selected as the lead template (scaffold A) based on our previous docking results and its less bulky structure. Successively, eight (8) new ligands (A1–A8) were designed with better binding affinities in comparison to the scaffold template (−6.8 kcal/mol) and isoniazid standard drug (−6.00 kcal/mol) respectively. In addition, three (3) designed ligands namely, A6, A2, and A7 with higher binding affinities were validated via ADME and toxicity prediction analysis, and the results showed zero violations of Lipinski rules with similar bioavailability, and high rate in gastrointestinal absorption, while toxicity parameters such as carcinogenicity and cytotoxicity were all predicted as non-toxic (inactiveness). The designed IPA compounds in the present study could serve as a promising gateway that could help the medicinal and synthetic chemist in the exploration of a new set of derivatives as anti-TB agents. Therefore, this research strongly recommends further experimental consideration of the newly designed IPA compounds through synthesis, in-vitro and in-vivo studies to validate the theoretical findings.
Keywords: In-silico design, Tuberculosis, Binding affinity, Pharmacokinetics, Molecular interactions, Hydrogen bond
1. Introduction
Mycobacterium tuberculosis is the organism that causes one of the chronic infectious diseases popularly known as Tuberculosis (TB) responsible for the global high mortality rate [1]. The emergence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as the cursor of the COVID-19 pandemic has continued to dominate the scientific research community and other media outlets in recent times [2], [3]. Scientific evidence based on clinical perspective indicates that COVID-19 materializes regardless of TB manifestation, either after, during, or before an active diagnosis [2]. Therefore, TB should be given utmost attention even with its global declining rate of cases [1]. An imidazo [1, 2-a] pyridine-3-carboxamide (IPA) candidate (Q203) was reported to exhibit robust inhibitory activity against extensively drug-resistant (XDR) and multidrug-resistant (MDR) strains and it is currently in clinical trials [4]. Researchers are currently developing a keen interest in the synthesis of diverse series of compounds as anti-TB agents. Recently, benzo[d]imidazole-2-carboxamides and benzimidazoquinazoline derivatives as new anti-TB agents were designed, synthesized, and tested for biological responses respectively [5], [6]. Hence, the rapid increase in the occurrences of TB drug resistance attracts the need to find new therapeutics as well to discover novel drug targets that could effectively kill M. tuberculosis when exploited. Some of the promiscuous targets inhibited by more than one compound include DprE1, MmpL3, QcrB, etc [7]. The novel derivatives of Q203 (IPAs) as anti-TB agents were also reported to have the ability to block the growth of MDR and XDR strains of M. tuberculosis by targeting the respiratory cytochrome bc1 complex (QcrB) [7]. The QcrB subunit is an important component of the electron transport chain necessary for the synthesis of ATP as it catalyzes the transfer of an electron from the ubiquinol to the cytochrome c [8]. However, the interaction of bonded ligand to the QcrB subunit receptor remains unclear and the crystal structure is not available in the Protein Data Bank (PDB) [9]. The search for more potent compounds is very tedious, costly, and time-consuming [10]. As such, the use of computational chemistry tools based on theoretical insights could come in handy with the aim to modify and design new compounds with better bioactivities. Some of the computational methods employed in computer-aided drug design include homology modeling, molecular docking simulation, pharmacokinetic predictions, and QSAR analysis amongst others. These computational approaches have been employed over the years to improve existing anti-tubercular agents through virtual screening for the identification and modification of potential hits [11], [12]. Structure-based drug design (SBDD) solemnly depends on the knowledge and information of the 3D crystal structure of the targeted protein to design the ligands that can serve as better inhibitors [13]. In the case where the 3D experimental structure of the targeted protein is not reported, the experimental amino acid sequence can be used to build a homology model [14]. The homology modeling technique predicts the 3D structure of the targeted protein sequence based on the alignment of an experimentally known homologous protein as a template [15]. In our previous report, homology modeling and molecular docking studies were carried out on some IPAs anti-TB agents targeting the QcrB subunit. The homology modeling of the receptor built and predicted a new 3D structure of QcrB target in M. tuberculosis using QcrB subunit of M. smegmatis as template [12], [16]. Furthermore, the results of molecular docking in the study further revealed the binding profiling of the 35 IPA ligands docked with the modeled protein. In the current study, the same 3D crystal structure of the QcrB modeled protein in M. tuberculosis was used to analyze the binding profiling and ADMET prediction of some newly designed compounds as potential hits of anti-TB candidates.
2. Methodology
2.1. Template selection and structural modifications
In our previous report, we have successfully carried out virtual screening of thirty-five (35) N-(2-phenoxy) ethyl imidazo[1,2-a] pyridine-3-carboxamides (IPAs) synthesized by Wang et al., (2019) with our homology modeled QcrB protein as the active target in the Mycobacterium tuberculosis [7], [16]. As such, ligand 26 was selected as the template scaffold for further structural modification and rigorous molecular docking simulation. The structure of the newly designed ligands was drawn (Table 1) and optimized accurately at the density functional level of theory (B3LYP/6-31G**) in a vacuum using Spartan 14 [17].
Table 1.
Chemical structures of the designed imidazo[1,2-a] pyridine-3-carboxamides (IPAs).
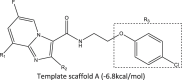 Template scaffold A (−6.8 kcal/mol) | |||
|---|---|---|---|
| Compound code | R1 | R2 | R3 |
| A1 | Cl | Me | 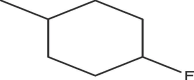 |
| A2 | Cl | Me | 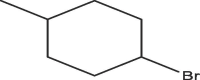 |
| A3 | H | Me | 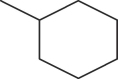 |
| A4 | H | Et | |
| A5 | H | Me |  |
| A6 | H | n-Pr | 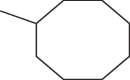 |
| A7 | H | OMe | 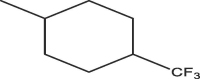 |
| A8 | H | c-Pr | 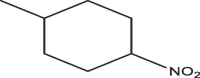 |
2.2. Molecular docking, ADME analysis, and toxicity prediction
Molecular docking is the most preferable technique in structure-based drug design to predict the binding free energy and the binding mode of the protein and ligand compound [18]. Therefore, molecular docking simulation was carried out to determine the binding affinities and the residual interactions when the ligand molecules bind with the active pockets of the protein as macromolecule using AutoDock 4.2 module implemented in PyRx 0.8. Blind docking was performed for all the designed ligand molecules to predict the active binding pockets of the modeled QcrB protein as the targeted macromolecule [19]. To ensure that all ligand molecules are properly docked, the 3D grid box dimensions were adjusted as X: 203.60, Y: 177.43, Z: 211.23 for grid center, and X: 88.26, Y: 86.09, Z: 82.38 for the number of points at the spacing of 1.875 Å on the whole protein structure to predict the best outcome of the docking task. Furthermore, the docking algorithm used was the Lamarckian Genetic Algorithm at default parametrized settings. After docking, protein and the ligands were obtained in PDBQT format, and complexes were formed using UCSF Chimera software while the visualization of residual interactions was done using Discovery Studio 2020 and UCSF Chimera software accordingly. The Swiss ADME online server (http://www.swissadme.ch/) was applied to predict absorption, distribution, metabolism, and excretion properties of the best ligands while ProTox-II online server (https://tox-new.charite.de/protox_II/) was also used to determine their toxicity.
3. Results and discussions
3.1. Molecular docking analysis
The docking results of ligand molecules with the targeted protein showed the binding affinity ranging from (−8.5 kcal/mol to −11 kcal/mol). To compare the best binding affinity of the ligand molecules, we docked the standard drug with the modeled QcrB protein in M. tuberculosis and showed binding affinity as (−6.00 kcal/mol). All binding amino acid residues including non-bond interactions and binding affinities of the stable complexes formed were shown in Table 2.
Table 2.
Binding affinity (kcal/mol) and non-bonding interactions of the complexes.
| Compounds | Binding affinity (kcal/mol) | Bonding types | Interacting amino acid residues | Distance (Å) |
|---|---|---|---|---|
| Standard drug | −6.00 | Conventional Hydrogen Bond | LEU58 | 2.09388 |
| Conventional Hydrogen Bond | LEU59 | 2.84072 | ||
| Pi-Anion | GLU159 | 3.32022 | ||
| Pi-Alkyl | LEU58 | 3.97204 | ||
| Pi-Alkyl | PRO221 | 5.18191 | ||
| A1 | −8.5 | Conventional Hydrogen Bond | ALA385 | 2.52924 |
| Halogen (Fluorine) | LEU348 | 2.87618 | ||
| Pi-Sigma | PHE133 | 3.61502 | ||
| Pi-Sigma | ALA385 | 3.67506 | ||
| Pi-Sigma | ALA385 | 3.60692 | ||
| Pi-Pi T-shaped | PHE133 | 4.99664 | ||
| Amide-Pi Stacked | ALA385 | 4.12602 | ||
| Amide-Pi Stacked | ILE386 | 4.12602 | ||
| Alkyl | LEU129 | 5.40777 | ||
| Alkyl | ILE386 | 4.18787 | ||
| Alkyl | VAL345 | 4.30783 | ||
| Alkyl | ALA385 | 4.43333 | ||
| Alkyl | ALA385 | 4.32462 | ||
| Pi-Alkyl | ILE386 | 5.06303 | ||
| Pi-Alkyl | LEU129 | 5.19201 | ||
| Pi-Alkyl | PHE133 | 4.1159 | ||
| Pi-Alkyl | PHE134 | 4.35564 | ||
| Pi-Alkyl | PHE388 | 4.7971 | ||
| Pi-Alkyl | TYR389 | 4.44871 | ||
| A2 | −10.5 | Conventional Hydrogen Bond | GLY62 | 2.08894 |
| Halogen (Fluorine) | GLU159 | 3.59989 | ||
| Pi-Anion | GLU159 | 4.31326 | ||
| Alkyl | LEU59 | 3.92938 | ||
| Alkyl | PRO221 | 4.39931 | ||
| Alkyl | LEU65 | 4.57881 | ||
| Alkyl | ARG111 | 4.54332 | ||
| Alkyl | PRO167 | 4.47863 | ||
| Alkyl | LEU65 | 4.48087 | ||
| Alkyl | LEU166 | 5.41423 | ||
| Alkyl | PRO167 | 5.16489 | ||
| Pi-Alkyl | ILE217 | 4.59328 | ||
| Pi-Alkyl | PRO221 | 4.71614 | ||
| Pi-Alkyl | PHE69 | 5.14437 | ||
| Pi-Alkyl | PHE69 | 4.72374 | ||
| A3 | −10.0 | Halogen (Fluorine) | HIS114 | 3.36308 |
| Pi-Anion | GLU159 | 3.94788 | ||
| Alkyl | LEU58 | 3.81904 | ||
| Alkyl | LEU59 | 4.09035 | ||
| Alkyl | PRO221 | 4.4197 | ||
| Alkyl | LEU65 | 4.40346 | ||
| Alkyl | LEU166 | 4.97691 | ||
| Pi-Alkyl | LEU58 | 5.39169 | ||
| Pi-Alkyl | LEU59 | 5.27014 | ||
| Pi-Alkyl | PRO221 | 4.32695 | ||
| Pi-Alkyl | PHE69 | 4.72942 | ||
| Pi-Alkyl | HIS114 | 5.15802 | ||
| Pi-Alkyl | HIS216 | 5.28912 | ||
| A4 | −9.1 | Carbon Hydrogen Bond | GLY163 | 3.31031 |
| Halogen (Fluorine) | GLY163 | 3.31031 | ||
| Halogen (Fluorine) | HIS114 | 3.68598 | ||
| Halogen (Fluorine) | HIS216 | 3.05615 | ||
| Pi-Sigma | LEU65 | 3.7055 | ||
| Alkyl | ALA97 | 3.69526 | ||
| Alkyl | ILE100 | 4.33314 | ||
| Alkyl | ARG111 | 4.58662 | ||
| Alkyl | PRO167 | 4.85181 | ||
| Alkyl | ILE217 | 4.56014 | ||
| Alkyl | PRO221 | 5.48313 | ||
| Pi-Alkyl | PRO167 | 5.10454 | ||
| Pi-Alkyl | PHE69 | 5.29162 | ||
| Pi-Alkyl | HIS114 | 4.68175 | ||
| Pi-Alkyl | HIS216 | 5.24304 | ||
| A5 | −10.3 | Halogen (Fluorine) | HIS114 | 3.50679 |
| Alkyl | LEU58 | 4.04364 | ||
| Alkyl | PRO221 | 4.89791 | ||
| Alkyl | LEU65 | 4.70392 | ||
| Alkyl | ILE217 | 5.46661 | ||
| Alkyl | LEU65 | 4.52788 | ||
| Alkyl | LEU65 | 4.86007 | ||
| Alkyl | LEU166 | 4.60995 | ||
| Alkyl | PRO221 | 5.42632 | ||
| Pi-Alkyl | LEU59 | 5.39063 | ||
| Pi-Alkyl | PRO221 | 4.44757 | ||
| Pi-Alkyl | PHE69 | 4.87213 | ||
| Pi-Alkyl | HIS114 | 5.14963 | ||
| Pi-Alkyl | HIS114 | 5.12793 | ||
| Pi-Alkyl | HIS216 | 5.28053 | ||
| A6 | −11.0 | Conventional Hydrogen Bond | GLY62 | 2.39142 |
| Halogen (Fluorine) | GLU159 | 3.66252 | ||
| Amide-Pi Stacked | LEU58 | 4.97455 | ||
| Amide-Pi Stacked | LEU59 | 4.97455 | ||
| Alkyl | LEU58 | 4.97214 | ||
| Alkyl | VAL63 | 4.49813 | ||
| Alkyl | ILE217 | 4.54423 | ||
| Alkyl | LEU65 | 4.95044 | ||
| Alkyl | LEU166 | 5.47454 | ||
| Alkyl | LEU65 | 4.41666 | ||
| Alkyl | PRO167 | 5.21434 | ||
| Alkyl | PRO221 | 4.89313 | ||
| Pi-Alkyl | ILE217 | 4.84499 | ||
| Pi-Alkyl | PHE69 | 5.17173 | ||
| Pi-Alkyl | PHE69 | 5.12895 | ||
| Pi-Alkyl | TYR213 | 5.39932 | ||
| A7 | −10.5 | Carbon Hydrogen Bond | HIS216 | 3.78978 |
| Halogen (Fluorine) | HIS114 | 3.60387 | ||
| Alkyl | LEU58 | 4.03498 | ||
| Alkyl | LEU59 | 3.97007 | ||
| Alkyl | LEU65 | 5.01232 | ||
| Alkyl | LEU65 | 4.53948 | ||
| Alkyl | PRO167 | 5.11711 | ||
| Alkyl | PRO221 | 5.46251 | ||
| Pi-Alkyl | LEU59 | 5.39657 | ||
| Pi-Alkyl | PRO221 | 4.5088 | ||
| Pi-Alkyl | PHE69 | 5.20022 | ||
| Pi-Alkyl | HIS114 | 5.13828 | ||
| Pi-Alkyl | HIS114 | 4.95511 | ||
| Pi-Alkyl | HIS216 | 5.23027 | ||
| Pi-Alkyl | HIS216 | 5.00678 | ||
| A8 | −9.0 | Conventional Hydrogen Bond | ALA385 | 2.16555 |
| Amide-Pi Stacked | ALA385 | 4.63904 | ||
| Amide-Pi Stacked | ILE386 | 4.63904 | ||
| Alkyl | LEU129 | 4.97501 | ||
| Alkyl | MET126 | 4.10023 | ||
| Alkyl | VAL345 | 4.66888 | ||
| Alkyl | VAL345 | 4.80002 | ||
| Alkyl | LEU348 | 5.44256 | ||
| Alkyl | ALA385 | 4.26799 | ||
| Alkyl | ALA385 | 4.0649 | ||
| Alkyl | ALA385 | 4.78506 | ||
| Pi-Alkyl | LEU129 | 5.14748 | ||
| Pi-Alkyl | ALA385 | 4.62964 | ||
| Pi-Alkyl | ILE386 | 4.76785 | ||
| Pi-Alkyl | PHE133 | 4.51175 | ||
| Pi-Alkyl | PHE388 | 4.91468 | ||
| Pi-Alkyl | TYR389 | 3.85255 |
A6 showed the best binding affinity (−11.0 kcal/mol) as a complex with the respected modeled QcrB protein and formed one conventional hydrogen bond with the amino acid residue of (GLY62 at a distance of 2.39142 Å) and Halogen (Fluorine), Amide-Pi Stacked, Alkyl, Pi-Alkyl bonds with the amino acid residues of (LEU58, LEU59, VAL63, ILE217, LEU65, LEU166, PRO167, PRO221, PHE69, TYR213) showed in Fig. 1. The complex of the A2 ligand molecule with the targeted modeled QcrB protein showed (−10.5 kcal/mol) binding affinity and formed one Conventional Hydrogen Bond with the amino acid residue (GLY62 at a distance of 2.08894 Å). Four different types of bonds such as Halogen (Fluorine), Pi-Anion, Alkyl, Pi-Alkyl were visualized in the complex with the amino acid residues of (GLY62, GLU159, LEU59, PRO221, LEU65, ARG111, PRO167, LEU65, LEU166, ILE217, PHE69) showed in Fig. 2. A7 as a ligand compound expressed (−10.5 kcal/mol) binding affinity with the targeted modeled QcrB protein. Complex showed one Carbon Hydrogen Bond with the amino acid residue of (HIS216 at a distance of 3.78978 Å) and three different types of bonds such as Halogen (Fluorine), Alkyl, Pi-Alkyl with the amino acid residues of (HIS114, LEU58, LEU59, LEU65, PRO167, PRO221, LEU59, PHE69, HIS114, HIS216) showed in Fig. 3. Furthermore, A3, A4, A5, A8 ligand molecules as complexes with the targeted modeled QcrB protein also revealed higher binding affinity than the template molecule and standard drug respectively. Based on the highest molecular docking scores as binding affinity, non-bond interactions and in comparison with the binding affinity of the standard drug, three ligand compounds (A6, A2, and A7) were considered for further analysis.
Fig. 1.

(a) Schematic representation of predicted A6 ligand with protein complex interactions in the 2D diagram. Interactions are colored depending on their type. (b) The three-dimensional representation of the binding pose, interactions, H bond donor, and acceptor surface of predicted A6 ligand with the protein complex. (c) Targeted protein is depicted in surface view and A6 ligand compound as the stick in the binding pocket.
Fig. 2.

(a) Schematic representation of predicted A2 ligand with protein complex interactions in the 2D diagram. Interactions are colored depending on their type. (b) The three-dimensional representation of the binding pose, interactions, H bond donor, and acceptor surface of predicted A2 ligand with protein complex. (c) Targeted protein is depicted in surface view and A2 ligand compound as a stick in the binding pocket.
Fig. 3.

(a) Schematic representation of predicted A7 ligand with protein complex interactions in the 2D diagram. Interactions are colored depending on their type. (b) The three-dimensional representation of the binding pose, interactions, H bond donor, and acceptor surface of predicted A7 ligand with protein complex. (c) Targeted protein is depicted in surface view and A7 ligand compound as the stick in the binding pocket.
3.2. ADME and toxicity prediction
Molecular weight (acceptable range: ≤500), number of hydrogen bond acceptors (acceptable range: ≤10), lipophilicity (Log P) ≤ 5, and molar refractivity (40–130) indicates the five rules of Lipinski, are crucial parameters for a successful drug candidate [20]. All the ADME parameters including drug-likeness, pharmacokinetic profile, and water solubility were analyzed for the selected ligand molecules showed in Table 3. All the ligand molecules as A6, A2, and A7 revealed 0 violations in Lipinski rules, similar bioavailability, and a high rate of gastrointestinal absorption. Only the A2 ligand molecule has glycoprotein permeability. Toxicity prediction was analyzed to determine the compounds were whether toxic or not. Predicted results were shown in Table 4. Determination of carcinogenicity and cytotoxicity of A6, A2, A7 were predicted inactiveness (non-toxic).
Table 3.
ADME and drug-likeness parameters of the selected IPAs.
| ID | MW (g/mol) | nHBD | nHBA | Log S | GA | CPY | BBB | Pgp | BA | Log Po/w | SA | nLV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A6 | 373.51 | 1 | 3 | −6.41 | High | CPY2 D6 inhibitor | Yes | No | 0.55 | 5.24 | 3.46 | 0 |
| A2 | 343.25 | 1 | 3 | −5.17 | High | CYP2D6 inhibitor, CYP3A4 inhibitor | Yes | Yes | 0.55 | 2.95 | 3.82 | 0 |
| A7 | 373.51 | 1 | 3 | −6.41 | High | CYP2D6 inhibitor | Yes | No | 0.55 | 5.24 | 3.46 | 0 |
Key: Molecular weight (MW), Number of hydrogen bond donor (nHBD), Water solubility (Log S), gastrointestinal absorption (GI), CYP isoform inhibitor (CPY), blood-brain barrier permeant (BBB), P-glycoprotein substrate (Pgp), Bio-availability (B), consensus Log Po/w, Synthetic Accessibility (SA), Number of Lipinski violation (nLV).
Table 4.
Toxicity prediction of the selected IPAs.
| Compound | Carcinogenicity | Cytotoxicity |
|---|---|---|
| A6 | Inactive | Inactive |
| A2 | Inactive | Inactive |
| A7 | Inactive | Inactive |
4. Conclusion
As an extension of our previous work, this research adopted the in-silico approach in analyzing the binding profiling of some newly designed IPA compounds as potential hits of anti-TB candidates. The template scaffold (Ligand 26) was selected for the in-silico design strategy and ligand compounds (A1–A8) were designed which exhibited better binding affinities when compared with that of the scaffold template (6.8 kcal/mol) and isoniazid standard drug (6.00 kcal/mol). In addition, all docking results of designed ligands with the targeted protein showed binding affinities ranging from (−8.5 kcal/mol to −11 kcal/mol). The drug-likeness and pharmacokinetic profile prediction results for the selected ligands with higher binding affinities (A6, A2, and A7) showed zero violations of Lipinski rules with similar bioavailability, and high rate in gastrointestinal absorption, while toxicity parameters such as carcinogenicity and cytotoxicity were all predicted as non-toxic (inactiveness).
Ethical statement
Not applicable
CRediT authorship contribution statement
Mustapha Abdullahi: Conceptualization, Methodology, Data curation, Visualization, Investigation, Supervision, Writing - original draft. Niloy Das: Software, Visualization, Validation, Writing - review & editing. Shola Elijah Adeniji: Data curation, Formal analysis, Supervision. Alhassan Kabiru Usman: Investigation, Writing - review & editing. Ahmad Muhammad Sani: Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Duarte R., Aguiar A., Pinto M., Furtado I., Tiberi S., Lönnroth K. Different disease, same challenges: social determinants of tuberculosis and COVID-19. Pulmonology. 2021;27(4):338–344. doi: 10.1016/j.pulmoe.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visca D., Ong C.W.M., Tiberi S., Centis R., D’Ambrosio L., Chen B. Tuberculosis and COVID-19 interaction: a review of biological, clinical and public health effects. Pulmonology. 2021;27(2):151–165. doi: 10.1016/j.pulmoe.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdul-Hammed M., Adedotun I.O., Falade V.A., Adepoju A.J., Olasupo S.B., Akinboade M.W. Target-based drug discovery, ADMET profiling, and bioactivity studies of antibiotics as potential inhibitors of SARS-CoV-2 main protease (sMpro) VirusDisease. 2021;1–29 doi: 10.1007/s13337-021-00717-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pethe K., Bifani P., Jang J. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med. 2013;19(9):1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 5.Jadhavar P.S., Patel K.I., Dhameliya T.M., Saha N. Bioorganic Chemistry Benzimidazoquinazolines as new potent anti-TB chemotypes: design, synthesis, and biological evaluation. Bioorg Chem. 2020;99(March) doi: 10.1016/j.bioorg.2020.103774. [DOI] [PubMed] [Google Scholar]
- 6.Dhameliya T.M., Patel K.I., Tiwari R., Vagolu S.K., Panda D., Sriram D. Synthesis, and biological evaluation of benzo [d] imidazole-2-carboxamides as new anti-TB agents. Bioorg Chem. 2021;107:104538. doi: 10.1016/j.bioorg.2020.104538. [DOI] [PubMed] [Google Scholar]
- 7.Wang A., Lv K., Li L., Liu H., Tao Z., Wang B. Design, synthesis and biological activity of N-(2-phenoxy)ethyl imidazo[1,2-a]pyridine-3-carboxamides as new antitubercular agents. Eur J Med Chem. 2019;178:715–725. doi: 10.1016/j.ejmech.2019.06.038. [DOI] [PubMed] [Google Scholar]
- 8.Ko Y., Choi I. Putative 3D structure of QcrB from Mycobacterium tuberculosis cytochrome bc1 complex, a novel drug-target for new series of antituberculosis agent Q203. Bull Korean Chem Soc. 2016;37(5):725–731. doi: 10.1002/bkcs.10765. [DOI] [Google Scholar]
- 9.Pan Z., Wang Y., Gu X., Wang J., Cheng M. Refined homology model of cytochrome Bcc complex B subunit for virtual screening of potential anti-tuberculosis agents. J Biomol Struct Dyn. 2020;38(16):4733–4745. doi: 10.1080/07391102.2019.1688196. [DOI] [PubMed] [Google Scholar]
- 10.Abdullahi M, Shallangwa GA, Ibrahim MT, et al. QSAR studies on some C-14urea tetrandrine compounds as potent anti-cancer agents against leukemia cell line (K562). J Turkish Chem Soc, Section A: Chem. 2018;5(3). 10.18596/jotcsa.457618.
- 11.Abdullahi M., Uzairu A., Shallangwa G.A., Mamza P., Arthur D.E., Ibrahim M.T. In-silico modelling studies on some C14-urea-tetrandrine derivatives as potent anti-cancer agents against prostate (PC3) cell line. J King Saud Univ – Sci. 2020;32(1):770–779. doi: 10.1016/j.jksus.2019.01.008. [DOI] [Google Scholar]
- 12.Abdullahi M., Elijah S. In-silico molecular docking and ADME / pharmacokinetic prediction studies of some novel carboxamide derivatives as anti-tubercular agents. Chemistry Africa. 2020;3(4):989–1000. doi: 10.1007/s42250-020-00162-3. [DOI] [Google Scholar]
- 13.Abdullahi M., Shallangwa G.A., Uzairu A. In silico QSAR and molecular docking simulation of some novel aryl sulfonamide derivatives as inhibitors of H5N1 influenza A virus subtype. Beni-Suef Univ J Basic Appl Sci. 2020;2(9):1–12. doi: 10.1186/s43088-019-0023-y. [DOI] [Google Scholar]
- 14.Oduselu GO, Ajani OO, Ajamma YU, Brors B, Adebiyi E. Homology modelling and molecular docking studies of selected substituted benzo[d]imidazol-1-yl)methyl)benzimidamide scaffolds on plasmodium falciparum adenylosuccinate lyase receptor. Bioinform Biol Insights. 2019;13. 10.1177/1177932219865533. [DOI] [PMC free article] [PubMed]
- 15.Mora Lagares L., Minovski N., Caballero Alfonso A.Y., Benfenati E., Wellens S., Culot M. Homology modeling of the human p-glycoprotein (Abcb1) and insights into ligand binding through molecular docking studies. Int J Mol Sci. 2020;21(11):4058. doi: 10.3390/ijms21114058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abdullahi M., Adeniji S.E., Arthur D.E., Haruna A. Homology modeling and molecular docking simulation of some novel imidazo[1,2-a]pyridine-3-carboxamide (IPA) series as inhibitors of Mycobacterium tuberculosis. J Genet Eng Biotechnol. 2021;19(1):1–13. doi: 10.1186/s43141-020-00102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdullahi M., Uzairu A., Shallangwa G.A., Mamza P., Arthur D.E., Ibrahim M.T. In-silico modelling studies on some C 14 -urea-tetrandrine derivatives as potent anti-cancer agents against prostate (PC3) cell line. J King Saud Univ – Science. 2020;32(1):770–779. doi: 10.1016/j.jksus.2019.01.008. [DOI] [Google Scholar]
- 18.Daggupati T., Pamanji R., Yeguvapalli S. In silico screening and identification of potential gsk3β inhibitors. J Recept Signal Transd. 2018;38(4):279–289. doi: 10.1080/10799893.2018.1478854. [DOI] [PubMed] [Google Scholar]
- 19.Peele K.A., Potla C., Srihansa T. Informatics in medicine unlocked molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: a computational study. Inf Med Unlocked. 2020;19(March) doi: 10.1016/j.imu.2020.100345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prottoy N.I., Sarkar B., Ullah A., Hossain S., Boby A.S., Araf Y. Molecular docking and pharmacological property analysis of antidiabetic agents from medicinal plants of bangladesh against type II diabetes: a computational approach. PharmaTutor. 2019;7(9):6–15. [Google Scholar]