

Graphical abstract

Keywords: Dengue virus, NS2B-NS3-pro inhibitors, Peptidomimetics, HTS, High-throughput virtual screening (HTVS), Dengue-COVID-19 co-infection
Abbreviations: DENV, Dengue virus; DHF, Dengue hemorrhagic fever; DSS, Dengue shock syndrome; HCV, Hepatitis C Virus; HIV, Human Immuno-deficiency Virus; HTS, High-throughput screening; HTVS, High-throughput virtual screening; JEV, Japanese encephalitis virus; NMR, Nuclear magnetic resonance; ORF, Open reading frames; SARS, Severe acute respiratory syndrome-coronavirus; SI, Selectivity index; SLC, Split luciferase complementation; WNV, West Nile virus; YFV, Yellow fever virus
Abstract
Dengue remains a disease of significant concern, responsible for nearly half of all arthropod-borne disease cases across the globe. Due to the lack of potent and targeted therapeutics, palliative treatment and the adoption of preventive measures remain the only available options. Compounding the problem further, the failure of the only dengue vaccine, Dengvaxia®, also delivered a significant blow to any hopes for the treatment of dengue fever. However, the success of Human Immuno-deficiency Virus (HIV) and Hepatitis C Virus (HCV) protease inhibitors in the past have continued to encourage researchers to investigate other viral protease targets. Dengue virus (DENV) NS2B-NS3 protease is an attractive target partly due to its role in polyprotein processing and also for being the most conserved domain in the viral genome. During the early days of the COVID-19 pandemic, a few cases of Dengue-COVID 19 co-infection were reported. In this review, we compared the substrate-peptide residue preferences and the residues lining the sub-pockets of the proteases of these two viruses and analyzed the significance of this similarity. Also, we attempted to abridge the developments in anti-dengue drug discovery in the last six years (2015–2020), focusing on critical discoveries that influenced the research.
1. Introduction
Nearly 390 million people globally are at risk of developing the arthropod-borne viral disease dengue.1 Dengue virus (DENV)2 belongs to the genus flavivirus of the Flaviviridae family.3 The genus flavivirus also includes pathogenic viruses like West Nile virus (WNV), Yellow Fever virus (YFV), and Japanese Encephalitis virus (JEV).4 There are four distinct but closely related serotypes of dengue viruses: DENV 1, DENV 2, DENV 3, and DENV 4. Further, each serotype can be sub-classified into genotypes and strains.5 The virus is primarily transmitted by the arthropod vector Aedes aegypti and, to a lesser extent, by Aedes albopictus. 6 It is responsible for varying degrees of clinical manifestations in the infected individuals, including mild flu-like symptoms to severe symptoms such as dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). Failing to attend DHF and DSS cases immediately can prove to be fatal.7 Fortunately, less than 1% of dengue patients develop DHF and DSS.8 Based on the incidence of DHF and DSS in the case of dengue infection, the severity of the four serotypes is found to be in the following order (DENV 2 > DENV 1 > DENV 3 > DENV 4). DENV 4 is rarely fatal.9 Infection with one serotype does not necessarily offer cross-immunity to other serotypes. Further, any subsequent infection can develop into severe dengue, commonly referred to as “antibody-dependent enhancement.”10, 11 Over the years, dengue has severely affected populations in the tropical and subtropical regions of the world.12 Some studies investigating the prevalence of dengue have identified 128 countries at risk of dengue13 epidemics, among which Asia alone accounts for 70% of cases. The year 2019 recorded the highest ever reported dengue cases worldwide, with Afghanistan as the new entrant, witnessing dengue infection for the first time. In the year 2020, cases were on the rise in countries like Bangladesh, Brazil, Cook Islands, Ecuador, India, Indonesia, Maldives, Mauritania, Mayotte (Fr), Nepal, Singapore, Sri Lanka, Sudan, Thailand, Timor-Leste, and Yemen.14 On the vaccine front, the current status is not highly encouraging. The currently available DENV vaccine, Dengvaxia®, launched by Sanofi Pasteur in 2015, has only been approved in 20 countries due to concerns about vaccinating seronegative patients. Seropositive and seronegative patients responded differently to the vaccine; the vaccinated seronegative patients were vulnerable to severe dengue contracting the first natural DENV infection. Currently, only individuals in the age group of 9–45 years and with at least one reported previous DENV infection are eligible for vaccination.14, 15 Hence, these limitations aggressively demand a parallel attempt at drug development. As our group has previously published the progress made in the development of DENV protease inhibitors.16 In continuation of our previous work, in this review, along with a discussion on Dengue-COVID 19 co-infection, we have compiled the progress made in the last six years (2015–2020) toward the development of DENV protease inhibitors. We have broadly categorized our discussion into peptide inhibitors, small molecule inhibitors, inhibitors identified through a rational approach, inhibitors identified through modification of previously reported inhibitors, and drug repurposing. A section on patents and information on clinical trials has also been added. Wherever possible, we have discussed the evolution of potent molecules, reported mechanism of action of the inhibitors, and their interactions with the available crystal structures of DENV protease that demonstrated inhibitory activity. Further, the emergence of COVID-19 has stirred fresh concerns in tropical regions due to anticipated threats following a dengue co-infection. While the data on this topic is relatively premature and inconclusive, we have touched upon Dengue-COVID-19 co-infections and outcomes in patients. This has also inspired us to draw two comparisons, one between the substrate-peptide residue preferences of DENV NS2B-NS3 protease (hereafter referred to as DENV NS2B-NS3 pro) and SARS-CoV 3CLpro and the other between various residues lining the sub-pockets of DENV NS2B-NS3 pro and SARS-CoV 3CLpro.
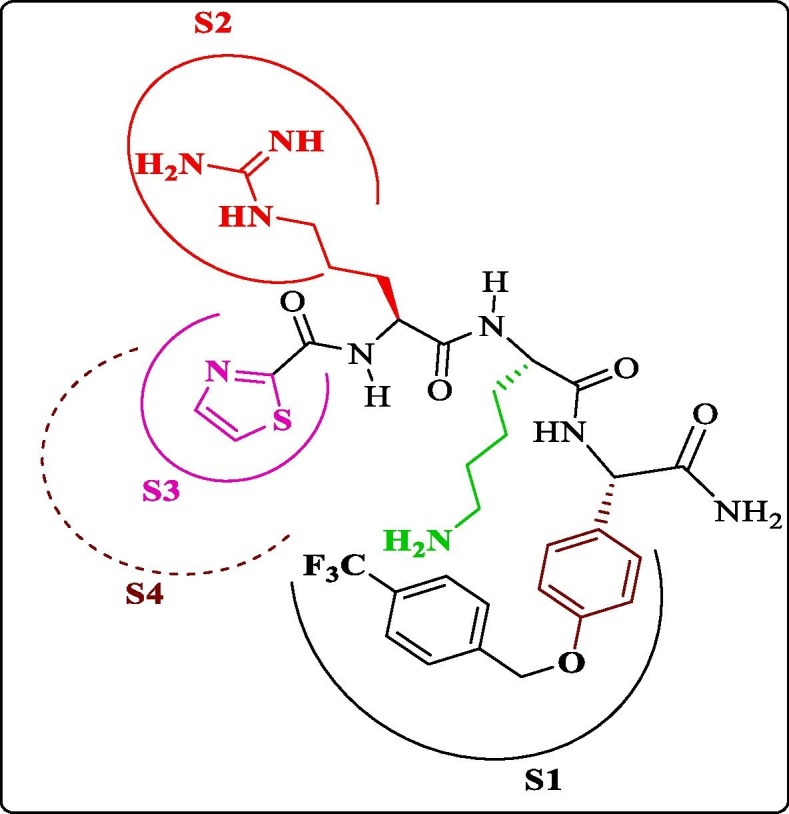
2. DENV genome and replication cycle
The DENV genome is approximately 11 kb long, single-stranded, positive-sense RNA, encoding a single polyprotein. The genome is processed into three structural proteins, namely, the capsid (C), envelope (E), and membrane (M) proteins and seven nonstructural proteins, i.e., NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 by the host proteases (Furin and signalase) and the two-component viral NS2B-NS3 protease ( Figure 1A ). 17, 18, 19 The DENV NS2B-NS3 pro (serine protease), which belongs to the trypsin superfamily, contains a catalytic triad in its active site formed by His51, Asp75, and Ser135 residues.20 The N-terminal domain of NS3 houses NS3 protease, which, together with 40 residues (hydrophilic) of NS2B,19 exhibits the serine protease activity.21 The lack of this NS2B-NS3 interaction renders the NS3 protease less active or inactive.19 Further, this NS2B-NS3 alignment forms the S2 and S3 sub-pockets in the protease active site. 22 Since viral replication and propagation depend on the activity of DENV NS2B-NS3 pro, the complex serves as an attractive target for the antiviral drug design against DENV.23 Figure 1B shows the basic steps involved in the replication cycle of the DENV. 18, 24, 25
Figure 1A.

Dengue virus genome.
Figure 1B.

The lifecycle of the Dengue virus.
3. Dengue-COVID 19 co-infection
The first case of Dengue-COVID-19 co-infection was reported by Verduyn et al. in August 2020. This has since been a significant concern in tropical areas where flaviviruses and COVID-19 infections may coexist. Since the patient showed quite severe dengue infection with no history of prior dengue infection, Verduyn et al. hypothesized that Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection might have aggravated the severity of dengue. However, this contention needs further exploration as it was difficult to distinguish between the clinical features of Dengue and COVID-19. They reported that the patient showed some symptoms like prolonged fever, facial flushing skin, body-ache myalgia, arthralgia, erythema, retro-orbital eye pain, which were consistent with dengue. However, some of these overlap with the clinical features of COVID-19. Besides these, other symptoms, including thrombocytopenia and elevated liver enzymes, were also reported in both diseases.26 Following this report, several other reports of co-infection have emerged across the globe. Epelbein et al. reported a case of co-infection in a traveler returning from France.27 Further, Saddique et al. highlighted a case where similarity of symptoms was such that a proper distinction was only possible following a laboratory test.28 These observations hint at the difficulty in distinguishing the clinical manifestation arising from the co-infection, thus highlighting the need for exploring the co-infections further.
4. DENV NS2B-NS3 pro and SARS-CoV 3CLpro substrate-peptide residue preferences
The initial efforts in the search for DENV protease inhibitors were based on the residue sequences at various cleavage sites. Further, this approach was supported by the finding that NS2B-NS3 proteases of the Flavivirus family have a preference for substrates having dibasic residues (Lys or Arg) at the P1 and P2 site, and further residue preference at P1′site was a small amino acid (Gly, Ala, and Ser).29, 30 Further, Li et al. in 2005 performed functional substrate profiling of P1-P4 and P1′-P4′ for all the four serotypes of DENV protease complexes using tetrapeptide and octapeptide positional scanning peptides to explore the substrate specificity of DENV NS2B-NS3 pro serotypes 1–4.31 Table 1 presents the outcome of their work. SARS-CoV-2, which caused the COVID-19 pandemic, belongs to the family Coronaviridae and genus Betacoronoviruses. Coronaviruses are enveloped, single-stranded, positive-sense RNA viruses responsible for various diseases, including respiratory diseases in both animals and humans.32, 33, 34, 35 The genome of SARS-CoV-2 is ∼ 29.9 kb in length, and it has 14 open reading frames (ORF’s) encoding for 27 proteins.36 This viral genome encodes two proteases, besides Papain-like protease (PLpro), the Chymotrypsin-like cysteine protease, the 3C-like protease (3CLpro) is considered to be the main protease (also called Mpro) responsible for polyprotein processing. In addition, among the coronaviruses, the sequence for 3CLpro is highly conserved.37, 38 The fact that none of the human host cell proteases have such substrate specificity39, 40, 41 further strengthens the rationale behind the design and development of 3CL protease inhibitors. The catalytic dyad of SARS-CoV 3CLpro is composed of Cys145 and His41, which is different from the serine protease catalytic triad (Ser-His-Asp).42 Here, we compared the substrate-peptide residue preferences of both DENV NS2B-NS3 pro and the SARS-CoV 3CLpro of coronaviruses. Since these two proteases have already been established as potential targets,23, 43 it would be interesting to see the outcome of this comparison. In 2010, Chuk et al. presented the substrate-peptide residue preferences of SARS-CoV 3CLpro (Table 1 ). From the table, it is evident that Val > Phe > Thr residues were favored at P5 position in that order, while small hydrophobic residues like Cys and Val were more favored at P4 position, and positively charged residues with a tendency to form β-sheet structure were more favored (Arg > Val) at the P3 position. Further, at the P2 position, hydrophobic residue without β branch was most favored, and the order of residue preference was Leu > Met > Phe. Moreover, β-branch residues like Ile and Val were least preferred. At the P1 position, Gln was most favored, with Gln > His > Met as the order of preference for this position. For the prime side residue preferences, P1′ favored small residues; the order of preference was Ser > Ala > Cys > Gly. The P2′ and P3′ positions did not exhibit any specific residue preferences; however, it was observed that small residues like Ser, Gly, and Ala were more prone to cleavage by the protease than the larger residues. It was also observed that solvent-exposed sites like P5, P3, and P3′ favored positively charged residues suggesting that the electrostatic interactions being responsible for the catalytic activity of the 3CLpro. The P1 site was most selective as cleavage was only observed in the presence of Gln, Met, or His. Substituting all the favored residue on positions P5 to P1 resulted in a 2.8 fold higher activity than the wild-type substrate sequence.44 Table 1 shows the substrate-peptide residue preferences of DENV 1–4 NS2B-NS3pro and the SARS-CoV 3CLpro.31, 44 In summary, substrate residue specificity of DENV NS2B-NS3 pro is mainly seen for the P2–P1–P1′ regions. While P2–P1 had dibasic residue preference (Lys, Arg, except for Gln as P2 residue at the NS2B/NS3 cleavage point in DENV 1–4 pro), the P1′region had a preference for smaller sized residues (Ser, Gly). In the case of SARS-CoV 3CLpro, specificity was seen for P2, P1, and P1′ region,45 where it was observed that Gln was indispensable for the P1 site. Leu was favored most at P2 site, and similar to DENVpro, Ser was favored at P1.’
Table 1.
Substrate-peptide residue preferences of DENV NS2B-NS3 pro and SARS-CoV 3CLPro.
| Viral proteases | Natural-cleavage sites | Non-prime site residues |
Prime site residues |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P5 | P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ | P5′ | |||
| DENV1 | NS2A-NS2B | I | W | G | R | K | Cleavage point | S | W | P | L | N |
| NS2B-NS3 | K | K | K | Q | R | S | G | V | L | W | ||
| NS3-NS4A | A | A | G | R | R | S | V | S | G | D | ||
| NS4B-NS5 | G | G | G | R | R | G | T | G | A | Q | ||
| DENV2 | NS2A-NS2B | T | S | K | K | R | S | W | P | L | N | |
| NS2B-NS3 | V | K | K | Q | R | A | G | V | L | W | ||
| NS3-NS4A | A | A | G | R | K | S | L | T | L | N | ||
| NS4B-NS5 | T | N | T | R | R | G | T | G | N | I | ||
| DENV3 | NS2A-NS2B | T | L | K | R | R | S | W | P | L | N | |
| NS2B-NS3 | K | Q | T | Q | R | S | G | V | L | W | ||
| NS3-NS4A | A | A | G | R | K | S | I | A | L | D | ||
| NS4B-NS5 | G | T | G | K | R | G | T | G | S | Q | ||
| DENV4 | NS2A-NS2B | G | A | S | R | R | S | W | P | L | N | |
| NS2B-NS3 | V | K | T | Q | R | S | G | A | L | W | ||
| NS3-NS4A | A | S | G | R | K | S | I | T | L | D | ||
| NS4B-NS5 | Q | T | P | R | R | G | T | G | T | T | ||
| SARS-CoV 3CLpro | Wild-type autocleavage sequence | S | A | V | L | Q | S | G | F | R | K | |
| Most preferable sequence | V | C | R | L | Q | S | S | R | ||||
5. Residues lining the sub-pockets of DENV pro and SARS-CoV 3CL pro
As mentioned above, the substrate specificities for both the proteases have been well defined. In this section, the corresponding sub-pocket occupancies of the residues involved have been discussed. Table 2 summarizes the residues lining the corresponding S4, S3, S2, S1, and S1′ pockets of both DENVpro 31 and SARS-CoV-2 3CLPro.46, 47, 48, 49 The overlap of residues within the pockets is due to the common wall between the pockets. It is evident from the comparison that there is no exact overlap of residues lining the sub-pockets of both the proteases, except for Ser and Leu in the S1 pocket, Gln and His in the S2 pocket, and Arg and Asp in the S3 pocket. However, although the residues are different, residues with similar properties overlap the sub-pockets of both the proteases. The S1 sub-pocket residues (Tyr, Leu, Ile, Phe, and Met) lining both the proteases have hydrophobic side chains. Similarly, in the S2 pocket, residues (Arg, His) from both the proteases have positively charged basic side chains. In addition, residues with polar uncharged side chains (Gln, Asn, and Thr) line both the proteases. In the S3 sub-pocket, residues (Leu, Val, Met, and Tyr) lining both the proteases have hydrophobic side chains. It can also be seen that residues with positively charged basic side chains (Arg, Lys, and His) constitute the sub-pockets. Finally, the S4 sub-pocket of both the proteases are lined by residues with hydrophobic side chains (Val, Met, and Leu). Besides this, residues like Cys, Pro, and Gly lining different sub-pockets of both the proteases have tendencies to form various interactions, including hydrogen bond and hydrophobic interactions. While much needs to be explored before arriving at a conclusion, we believe that this similarity could be exploited in the future for developing dual inhibitors against both proteases.
Table 2.
Residue lining the sub-pockets of DENV NS2B-NS3 pro and SARS-CoV-2 3CLpro.
 |
Note: Residues with similar color share properties by virtue of their side chains.
6. Design and discovery of DENV NS2B-NS3 pro inhibitors
This section summarizes the research on inhibitors reported in the last six years (2015–20) and summarizes the predicted interactions of a few key residues. Most interactions were common among reported peptide inhibitors. Inhibitors other than peptides were also critically examined for their predicted interactions with the proteases. Besides, any common attributes from different chemical structures were also examined. Since we have already compared the substrate-peptide residue preferences and residues lining the proteases for DENV and SARS-CoV-2 in the aforementioned sections, the insights provided below could guide the development of potent dual inhibitors. However, experimental validations remain an ultimate tool for any in-silico favored results.
6.1. Peptide inhibitors
Anti-DENV drug development began with the design of substrate-based peptide inhibitors against DENV NS2B-NS3 pro. However, the relatively flat topography of the DENV protease active site posed numerous challenges in drug design.22 In addition, there was a significant possibility of developing compounds with compromised pharmacokinetic properties as the non-prime side favored positively charged residues which would ultimately impart a negative charge to the active site. Despite these apprehensions, attempts were made to design peptide inhibitors and peptidomimetics. Eventually, inhibitors with nanomolar affinities were obtained. The peptide inhibitors reported in the last six years have been discussed in the subsections, namely substrate-peptide analogs of non-prime side, peptide conjugates, and cyclic peptides.
6.1.1. Substrate-peptide analogs of non-prime side and Substrate-peptide conjugate from common tripeptide (Cap-Arg-Lys-Phg-NH2)
In this section, we discuss three inhibitors developed by modifying a common tripeptide with varying cap (Cap-Arg-Lys-Phg-NH2) reported by Behnam et al.50 The first modification was done by Weigel et al.. They observed that previous modifications of peptide hybrids did not optimize the S2 pocket, considering the target affinity.50, 51, 52 Hence, arginine mimetic moieties were screened to find a suitable fit. This work revolved around already established tripeptide Bz-Arg-Lys-Phg-NH2.50 They synthesized and performed in-vitro characterization of potent peptidic inhibitors of DENV pro by incorporating phenylalanine and phenylglycine derivatives as arginine-mimicking groups. Figure 2 shows the best two analogs, N-Benzoyl-capped tripeptides having (4-guanidino)-l-phenylalanine (1) and benzamidine (2) as arginine mimetic modification with inhibitory concentrations in the sub-micromolar range. Further, the cap modifications yielded the most promising compound 3 ( Figure 2 ) with an IC50 value of 0.21 µM and a Ki value of 139 nM. Antiviral activity of the best molecules and the reference compound (Bz-Arg-Lys-Phg-NH2) on Huh-7 cell line infected with DENV2 serotype showed a 58% reduction of viral titer for compound 4, which was highest in the series ( Figure 2 ). However, PAMPA studies discouraged further development due to poor membrane permeability of the 15 synthesized tripeptides, possibly due to the presence residues at P1 and P2 position having positively charged side chains amounting to high polarity; this was also considered the reason for low antiviral activity. Molecular docking studies of compound 3 ( Figure 2 ) against protease (PDB 3U1I) predicted that the basic residue of arginine mimetic analog occupied the S2 pocket and appeared to be involved in electrostatic interaction with Asp75 also other occupancies could be seen in the figure.53
Figure 2.

Weigel et al. (2015) tripeptide modification & predicted occupancy of the inhibitor (3) at subsites of protease.
The second modification-based study in this series was conducted by Behnam et al. (2015), who developed substrate-based peptide inhibitors and extended their work toward the development of potential dual inhibitors of DENV and WNV pro by synthesizing and performing an extensive biological evaluation of inhibitors containing benzyl ethers of 4-hydroxyphenylglycine as non-natural peptidic building blocks. Figure 3 outlines the critical inhibitors from their work and displays the best compound obtained at each modification step. Their work was centered around retro-peptide sequences (Bz-Arg-Lys-Nle-NH2), having good inhibitory activity against DENV2 pro.51 The replacement of Nle with Phg (Bz-Arg-Lys-l-Phg-NH2, IC50 = 3.32 µM) improved the inhibition profile.50 Thus, the same molecule used by Weigel et al. was optimized here in a three-step protocol, involving optimization of this sequence which ultimately settled with modifying the C-terminal 4-hydroxyphenylglycine of the tripeptide to the benzyloxy ether (Bz-Arg-Lys-(4-benzyloxy)-d-Phg-NH2) and having an IC50 value of 0.367 µM. Further, simultaneous C & N- terminal modification of the optimized sequence was performed, and N-terminal modification led to various caps being optimized to yield dual inhibitors with nanomolar inhibition. Finally, a few caps (with thiophene ring and thiazole ring) showed favorable results, and resulting compounds 5 and 6 (Figure 3) had an IC50 value of 109 nM and 99 nM respectively against DENV2 pro. The C-terminal optimization generated Bz-Arg-Lys-(4-CF3-benzyl ether)-d-Phg-NH2, (7, Figure 3) with an IC50 of 0.069 µM. The final fragment merging yielded compounds 8 and 9 ( Figure 3) with Ki values of 19 nM and 12 nM, respectively, against DENV2 pro. Several of these newly discovered protease inhibitors significantly reduced dengue and WNV titers in cell-based assays of virus replication. The most active analog, compound 10 ( Figure 3 ), showed an EC50 value of 3.4 μM against DENV-2 and 15.5 μM against WNV. In general, the compounds showed less inhibition in WNV, suggesting a subtle difference in active sites of DENV and WNV proteases. Once again, PAMPA studies revealed weak permeability, possibly due to the same reason. Docking studies of tripeptides against DENV3 pro (PDB 3U1I) showed that the tripeptides assumed a near cyclic confirmation inside the binding site of the protease. Figure 4 shows the predicted occupancy of different pockets by inhibitor 9. The occupancy was similar to that of the previous inhibitor and an identified, predicted key interaction was that of Alkyl side chain residue Val155 which exhibited a π-σ interaction with the phenyl ring of ether.54
Figure 3.

Behnam et al. (2015) modifications.
Figure 4.
Predicted pocket interaction of inhibitor (9) within DENV2 NS2B NS3 pro.
Sometimes, peptides are conjugated with small molecules for improved inhibitory activity against the targeted enzyme. One such attempt was made by Bastos et al. in 2015 while searching for a dual inhibitor against DENV and WNV NS2B-NS3 protease. They modified a molecule similar to that developed by Behnam et al. with a varying cap group.50 Thus, the third attempt to modify tripeptides led to the modification of compound 11, giving rise to compounds 12, 13 ( Figure 5 ), which turned out to be potent inhibitors of DENV NS2B-NS3 pro (IC50 = 0.46 ± 0.02 µM) and WNV pro (0.75 ± 0.04 µM), respectively. The Ki value of compound 12 was found to be 0.40 ± 0.03 µM. The docking of compound 12 against DENV pro predicted the occupancy of phenylglycine in the S1 pocket and arginine in the S2 pocket. The cap occupied the S3 and S4 pockets, and lysine was positioned between S1 and S2 pockets. The results showed four hydrogen bonding and twelve nonbonding interactions, besides this one of the predicted key interactions involved residue, Tyr215, engaged in a π-π stacking interaction55.
Figure 5.

Bastos et al. (2015) modifications.
The common observation about the occupancies of the residues of the potent molecules in different pockets is shown in Table 3 . It can be seen that they have almost a common pattern of occupancy. Arginine mimetic modification of Weigel et al. however, influences the position of Phg wherein it gets displaced to the S1′ pocket. The best inhibition (IC50 = 0.018 µM) of protease was witnessed when Phg occupied the S1 pocket. This tripeptide was so far, the best reported protease inhibitor identified. However, as with previous cases, pharmacokinetic challenges remained a major concern, for which the probable solutions have been discussed later on in this review.
Table 3.
Compounds arising from tripeptide modification and their predicted pocket occupancies.
| Tripeptide for modification50 | Potent compounds | IC50 (µM) | |
| Cap-Arg-Lys-Phg-NH2 | Compound 3 | IC50 = 0.21 µM | |
| Compound 9 | IC50 = 0.018 µM | ||
| Compound 12 | IC50 = 0.46 ± 0.02 µM | ||
| Residues of inhibitor | Predicted pocket occupancies | ||
| Compound 3* | Compound 9# | Compound 12 | |
| Phg/Phg modification# | S1′ | S1 | S1 |
| Arg/Arg-mimetic* | S2 | S2 | S2 |
| Lys | S1 | S1, S3, S4 | S1, S2 |
| Cap group (varying) | S3 | S3, S4 | S3, S4 |
6.1.1.1. Cyclic peptides against DENV NS2B-NS3 pro
The peptides discussed above targeted the non-prime side and had their own merits and demerits. Lin et al. (2017) observed that the currently available designs focused on the non-prime side of the DENV protease active site and almost always led to low lipophilic and nonspecific scaffolds. This prompted them to exploit the advantages of aprotinin (58 amino acids protein) for designing new cyclic peptides as aprotinin showed a high affinity for DENV2 pro (Ki = 26.9 nM). Their strategy of designing new cyclopeptides was based on the structure of aprotinin targeting the active site pockets S3-S4′ of the DENV pro. After analyzing aprotinin’s binding loop, they found that the prime side significantly modulated DENV pro binding affinity.56 Their designed cyclic peptides showed interaction with both sides of the active site. The design was based on the two loops of aprotinin, linked together with a glycine linker. The first loop, called the binding loop, was significant in binding to DENV pro, while the second loop was incorporated into the design to maintain binding loop structure and rigidity. Also, their previous work identified the key factors affecting binding affinity, which were hydrogen bonding contributed by P1 and P2′ residues, the P3′ and P4′ residue hydrophobic packing, and maintaining the aprotinin binding loop conformation intact in the designed cyclic peptide.56 Analogous to the structure of aprotinin, there was a need to optimize the binding loop from residue 12 to 18 (Pro13 to Ile18/Ile19) of aprotinin, the linker between the binding loop, and the second loop of aprotinin spanning from residue 35 to 39 (Tyr35/Gly36 to Arg39) in the newly designed cyclopeptides. Also, the linker glycine residues were either retained or were omitted entirely between Ile18/Ile19 and Tyr35/Gly36. Aprotinin’s disulfide bond was retained by keeping a disulfide bond between Cys14 of the first loop and Cys38 of the second loop. The length of the binding loop was optimized and compounds with and without linker (one or two Gly residues) were observed, the length of the second loop was optimized and eventually these modifications yielded cyclic peptides of varying lengths. Also, the preferences of different residues at a different position were understood and correlated with the obtained Ki values. The best inhibitor showed a Ki value of 2.9 µM against wild-type DENV3 pro. Also, the preference of residues for the P1 and P2′ positions was established. The key finding from this effort was that the preference between two basic amino acids Lys and Arg, was context-dependent at P1, and Arg was favored at P2′ position as it appeared to have more interactions with other residues through hydrogen bonding and van der Waal forces. The choice of optimal C-terminal residue was dependent on the length of the cyclic peptide since different lengths favored different residues at the C-terminal. However, in the native aprotinin, Arg39 was replaced with alanine. The flexibility was limited to alanine in most of the derivatives, and results were based on binding interactions. Similarly, the significance of P3 residue (N-terminal) was analyzed, and it was found that proline was more favored than a benzoyl cap at this position. However, the presence of proline was found to be context-dependent in the entire experiment as the removal of proline in some cyclic peptides did not change the activity significantly. Table 4 shows the binding affinity of cyclic peptides resulting from the incorporated changes in the optimization process. Although the most effective cyclic peptide obtained had a Ki value of 2.9 µM against wild-type DENV3 pro, this value was quite high when compared to the aprotinin inhibition constant of 29 nM against DENV2 pro. Hence, few strategies including, making the molecule more rigid and less bulky, improving the permeability and substituting cleavable peptide bonds between P1 and P1′ with non-cleavable bonds to ensure cyclic structure intact were recommended and further it was predicted that any combination of these set of changes could yield a more rigid molecule with good binding affinity.57
Table 4.
Lin et al. (2017) modifications and the resulting inhibition constants of various cyclic peptides. # Peptide bond, *Disulphide bond, Bz: Benzoyl capping.
| Cyclic peptides | (12–18) Binding loop residues (aprotinin) |
Glycine Linker | (35–39) Aprotinin’s second loop residues |
Ki (µM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ | 35 | 36 | 37 | 38 | 39 | ||||
| Length optimization | |||||||||||||||
| 1. | Pro | Cys* | Lys | Ala | Arg | Ile | Ile | Gly | Gly | Tyr | Gly | Gly | Cys* | Arg | 232.3 ± 69.9 |
| 2. | Pro | Cys* | Lys | Ala | Arg | Ile | Ile | Gly | Gly | Tyr | Gly | Gly | Cys* | Ala | 678.3 ± 244.1 |
| 3. | Pro | Cys* | Lys | Ala | Arg | Ile | – | Gly | – | Tyr | Gly | Gly | Cys* | Ala | 376.8 ± 204.1 |
| 4. | Pro | Cys* | Arg | Ala | Arg | Ile | Tyr | Gly | Gly | Cys* | Ala | 966.1 ± 328.8 | |||
| 5. | Pro | Cys* | Lys | Ala | Arg | Ile | Tyr | Gly | Gly | Cys* | Ala | 14.5 ± 6.7 | |||
| 6. | Pro | Cys* | Arg | Ala | Arg | Ile# | Tyr# | Gly | Gly | Cys* | Ala | 2.9 ± 0.8 | |||
| 7. | Pro | Cys* | Arg | Ala | Arg | Ile | Tyr | – | Gly | Cys* | Ala | 467.9 ± 68.7 | |||
| 8. | Pro | Cys* | Arg | Ala | Arg | Ile | – | Gly | Gly | Cys* | Ala | 780.3 ± 195.5 | |||
| 9. | Pro | Cys* | Arg | Ala | Arg | Ile | – | Gly | Gly | Cys* | Arg | 29.7 ± 5.8 | |||
| 10. | – | Cys* | Arg | Ala | Arg | Ile | Tyr | Gly | Gly | Cys* | Arg | 145.1 ± 27.9 | |||
| 11. | Pro | Cys* | Arg | Ala | Arg | Ile | – | – | Gly | Cys* | Ala | 101.2 ± 24.8 | |||
| Sequence optimization | |||||||||||||||
| Pro | Cys* | Arg | Ala | Arg | Ile# | Tyr# | Gly | Gly | Cys* | Ala | 2.9 ± 0.8 | ||||
| 12. | Bz | Cys* | Arg | Ala | Arg | Ile | Tyr | Gly | Gly | Cys* | Ala | 33.2 ± 6.5 | |||
| 13. | Pro | Cys* | Arg | Ala | Gln | Ile | Tyr | Gly | Gly | Cys* | Ala | No Binding | |||
| 14. | Pro | Cys* | Arg | Ala | Trp | Ile | Tyr | Gly | Gly | Cys* | Arg | 19.7 ± 8.8 | |||
| 15. | Pro | Cys* | Arg | Ala | Trp | Ile | Tyr | Gly | Gly | Cys* | Ala | 144.8 ± 54.4 | |||
| 16. | Pro | Cys* | Arg | Ala | Arg | Ile | Asp | Gly | Gly | Cys* | Ala | 69.3 ± 24.4 | |||
| 17. | Pro | Cys* | Arg | Val | Arg | Ile | Asp | Gly | Gly | Cys* | Ala | 35.8 ± 16.4 | |||
| 18. | Pro | Cys* | Arg | Val | Arg | Ile | Tyr | Gly | Gly | Cys* | Ala | 25.6 ± 9.9 | |||
Although nearly all the parameters were incorporated for optimization in the above study, the results were not significantly encouraging. It was suggested that reducing the molecule's flexibility could provide a more potent inhibitor as high flexibility was previously attributed to the low protease inhibition. In addition to improving the protease inhibitory profile, it is also essential to optimize the compound for antiviral activity. However, studies in the past made no such attempts. Takagi et al. (2017) synthesized 33 cyclic peptides, and for the first time, identified a novel cyclic peptide inhibitor with antiviral activity. Their work began with the modification of the lead compound from Xu et al. (eight-residue cyclic peptide, 14) ( Figure 6 ). After alanine scanning and studying the model by Xu et al., they learned that P1-Lys, P3- Lys, P2-Arg interactions were essential for the activity and, therefore, were retained in the new design. Also, since the environment around P1,’ P2′, and P4′ residues appeared hydrophobic, it was also believed that these positions could be substituted with hydrophobic groups and could result in hydrophobic interactions between the inhibitor and the enzyme. After optimizing the P1,’ P2,’ and P4′ positions, they established that at P1′-Ser was indispensable and P2′-Gly replacement with the hydrophobic group was valuable. It was also revealed that preferential replacement at this position was in the following order: l-homophenylalanine > l-phenylglycine > l-phenylalanine = d-homophenylalanine. Further, P4′ was fixed with d-phenylalanine. P3′-Ser yielded satisfactory results compared to P3′-Ala. However, the best protease inhibitory activity was observed when, d-proline replaced the flexible P4-Gly. The best cyclic peptide 15 ( Figure 6) had an IC50 value of 0.95 µM. Docking study of this molecule using DENV2 pro, attributed good protease inhibition to the β-turn like conformation of the cyclic peptide inside the protease. Markedly, Compound 15 was not an as effective antiviral agent as intended. The high hydrophilicity due to the side chains of the residues would have inhibited the membrane permeability of the cyclopeptide. Significant efforts were made to improve its permeability58, 59 while maintaining protease inhibition and antiviral efficiency. In the process, 11 cyclic peptides obtained were further subjected to antiviral assay. Finally, the optimization efforts yielded a cyclopeptide (P4′ d-Arg, P3′ l-Arg, P2′ l-Lys, P1′ l-Lys, P1 D-2NaI, P2 l-hPhe, P3 l-Phe, and P4 d-NaI), which showed promising antiviral activity with an EC50 value of 2.2 µM without significantly compromising the IC50 value (1.1 µM).60 The cyclic peptide was designed keeping the substrate-peptide residue preferences into consideration. The aromatic residues, introduced into the design improved the activity. Notably, the permeability issues were addressed without compromising the protease activity. Such attempts viz. introduction of amphipathic sequence motifs and the proper combination of arginine and hydrophobic aromatic residues could be applied to future cyclic molecules to improve the permeability issues and eventually obtain the desired potent antiviral agent.
Figure 6.

Cyclic peptides of Xu et al. (14) & Takaji et al. (15).
6.2. Small molecule inhibitors
Small molecules offer significant advantages over large peptides in terms of complexity, feasibility, and scalability. Small molecule inhibitors can introduce large conformational changes in DENV NS2B-NS3 pro.61 This section describes various attempts to develop non-peptide small molecule inhibitors. These have been sub-classified into inhibitors from structure-guided small molecule optimization, from natural sources, from High-throughput screening (HTS), HTVS, and a rational design approach. Further, in these subsections, the chemical class is also mentioned. In this section, we discuss various efforts made toward optimizing the potential inhibitors by recognizing the scaffolds involved in the protease inhibitory activity as well as gaining knowledge about protease residues predicted to be involved in the interaction with the inhibitor.
6.2.1. Inhibitor from structure-guided small molecule optimization (pyrazole ester derivative)
Koh et al. used a potent WNV protease inhibitor (19, Figure 7 , IC50 = 0.105 µM), a pyrazole ester derivative for DENV pro drug development. The compound was previously identified by Johnston et al. by performing HTS using the National Institute of Health compound library containing > 65,000 compounds.62 The structure-guided small molecule optimization effort improved compound potency and selectivity for DENV protease. Figure 7 shows how the modifications affected the IC50 values of compound 20 (IC50 = 0.5 ± 0.1 µM) and 21 (IC50 = 2.9 ± 0.5 µM). Protease inhibitor binding interactions were confirmed biophysically using nuclear magnetic resonance (NMR). The molecular weight of DENV2 pro treated with compound 19 and DENV pro in control (DMSO solution) by ESI-TOF MS indicated benzoylation of DENV2 pro by inhibitor. Finally, the group confirmed a covalent interaction between catalytic triad serine and inhibitor 19, as no change in molecular weight was observed when inhibitor 19 was treated with mutant DENV2 pro, which had Ala in place of Ser135 in the NS3 domain. The inhibitor appeared to have been occupying the active site. NMR analysis provided an insight into the protease inhibitor binding interactions. Further, they explored protease inhibitor molecular interaction by titrating inhibitor 19 against DENV2 pro. The chemical shift perturbation findings were in agreement with ESI-TOF MS results.
Figure 7.

Koh et al. (2015), structure-guided small molecule optimization.
Further, molecular docking of inhibitor 19 against DENV3 pro (PDB 3U1I) predicted the occupancy of inhibitor at different sites (Figure 8 ). Also, a few other interactions were predicted, including van der Waals and hydrogen bonding. Few interactions and positionings involved the identified key residues (Pro132 and Tyr161), the ester phenyl group occupied the P1 site, and the phenyl group appeared to be trapped between Pro132 and Tyr161, and a π-π interaction was predicted between the ester phenyl group and Tyr161. This work strongly suggested that this class of compounds inhibited flavivirus protease through targeted covalent modification of active site serine.63
Figure 8.

Predicted occupancy of the inhibitor (19) at different DENV protease sites.
6.2.2. Inhibitors from natural sources
6.2.2.1. Flavonoids, polyphenolic metabolites (PCA) and flavanones
Over the years, many natural sources have been explored for DENV pro inhibition. Encouraged by the role of bioflavonoids, flavonoids, and chalcone derivatives in DENV pro inhibition and newly identified allosteric sites in the DENV2 pro responsible for non-competitive inhibition,64, 65, 66 De Souse et al. (2015) evaluated six flavonoids (Agathisflavone, quercitrin, and isoquercitrin isolated from a natural source and three purchased from a commercial source) against DENV2 and DENV3 NS2B-NS3 pro. Agathisflavone (22, Figure 9 ) emerged as the best non-competitive inhibitor of both the proteases with IC50 values of 15.1 ± 2.2 µM and 17.5 ± 1.4 µM against DENV2 and DENV3 pro, respectively. Molecular docking studies with DENV3 pro (PDB 3U1I) predicted and identified an allosteric binding site, putative for other non-competitive inhibitors.67 Further, Ispita et al. (2019) performed in-silico screening and pharmacokinetic evaluation of 10 established phytoconstituents of Carica papaya. Molecular docking studies using the X-ray crystal structure of DENV2 pro (2FOM) identified two flavonoids, apigenin, and luteolin, with the best docking score (-7.7 kcal/mol). However, luteolin (23, Figure 9) was considered a more preferred lead owing to its predicted interaction with the residues of the catalytic triad. Luteolin was also predicted to be involved with two identified key residues (Asp75 and Gly153) via hydrophobic interactions.68 Recently, Dwivedi et al. studied the anti-DENV properties of Azadirachta indica. Virtual screening exercise on 49 reported bioflavonoids against DENV pro helped identify four potential DENV NS2B-NS3 pro inhibitors viz kaempferol-3-O-rutinoside (-9.55 kcal/mol), rutin (-9.32 kcal/mol), hyperoside (-7.87 kcal/mol), and epicatechenin (-7.62 kcal/mol). The interaction responsible for the predicted high docking score of kaempferol-3-O-rutinoside (24, Figure 9) were six hydrogen bonds between NS3 protease-kaempferol-3-O-rutinoside complex, involving key residues Asp75, Gly151, and Asn152, and other critical residues involved in hydrophobic interactions were His51, Lys73, Pro132, Ser135, Val154, and Tyr161. In addition, residues Gly151 and Gly153 also contributed toward glycine interactions. Cell viability and the in-vitro antiviral studies done on reference compound quercetin, kaempferol-3-O-rutinoside, and epicatechenin (25, Figure 9) showed kaempferol-3-O-rutinoside as a potent DENV inhibitor (77.7% at 100 µM) and epicatechenin (66.2% at 1000 µM) as a moderate inhibitor when compared to quercetin.69 In recent work, Farooq et al. (2020) adopted the molecular docking approach with the additional incorporation of antimicrobial and antioxidant activity and identified nine bioactive compounds from Carica papaya, of which three compounds viz. epigallocatechin (26, Figure 9), catechin (27, Figure 9), and protocatechuic acid (PCA, 28, Figure 9) showed good binding affinities with DENV NS2B-NS3 pro in the in-silico studies. These three compounds were predicted to show strong interaction with the residues of the catalytic triad (His51, Asp75, and Ser135) of DENV pro.70
Figure 9.

DENV NS2B-NS3 pro inhibitors from natural sources.
In 2019, Lim et al. isolated six compounds from Ganoderma lucidium Var. antler. Molecular docking studies revealed, Hesperitin, a bioflavonone (29, Figure 9), as a DENV2 pro inhibitor among the six identified compounds. Also, docking affinities of hesperitin were near the reference ligand, 4-hydroxypanduratin A. Further HOMO-LUMO calculations also indicated good ligand-protease interaction. Hesperitin appeared to interact with the residues of the catalytic triad (His51, Asp75, and Ser135) and oxyanion hole through hydrogen bonding, van der Waal, and hydrophobic interactions.71
6.2.2.2. Terpenoids
Dwivedi et al. (2016) explored the in-silico inhibitory potential of five triterpenoids obtained from neem plant against DENV NS2B-NS3 pro. Using the crystal structure of DENV NS2B-NS3 pro (PDB 2VBC) for molecular docking, they reported the binding affinities of three triterpenoids viz. Nimbin, desacetylnimbin, and desacetylsalannin to be 5.56, –5.24, and –3.43 kcal/mol, respectively. The high binding affinity of Nimbin (30, Figure 10 ) was attributed to four predicted hydrogen bonds of Nimbin with the enzyme, which included three bonds with the residues of the catalytic triad (His51, Asp75, Ser135) and one with Asn152 residue. In addition, a hydrophobic interaction with six other residues, including the key residues Val154, Pro132, and Gly153, also contributed toward binding affinity.72 Bharadwaj et al. (2019) explored Ganoderma lucidium for anti-DENV infection treatment. Before this, G. lucidium was reported to have a pool of bioactive compounds, including polysaccharides and triterpenes.73 Several medicinal properties were attributed to G. lucidium, including antiviral activity against HIV.74 Structure-based virtual screening of 22 triterpenoids, known for their antiviral activities against crystal structure DENV2 pro (PDB 2FOM), identified four triterpenoids with a docking score higher than the reference inhibitor. The triterpenoid Ganodermanontriol (31, Figure 10), with a docking score of −6.291 kcal/mol, showed three hydrogen bonding interactions, including key residues Lys73 and Asn167. Besides this, residues involved in hydrophobic interactions included Val154 and Val155. Residues His51, Asn152, and Asn167 were predicted to be involved in polar interaction, while Gly153 was involved in glycine interaction. In addition, positive charge interaction with Lys73 and Lys74, and negative charge interaction with Asp75 residues also contributed to the effective binding. The in-vitro DENV inhibition assay of this compound showed a 40% reduction in viral titer at 50 µM concentration.75
Figure 10.

DENV NS2B-NS3 pro inhibitors from natural sources.
6.2.2.3. Curcumin and its analogs
In a previous HTS study, Balasubramanian et al. identified curcumin (unpublished report) as a DENV NS2B-NS3 pro inhibitor.76 However, poor plasma availability of curcumin, owing to retro-aldol conversion to ferulic acid at the plasma pH 77, 78 prompted them to synthesize four analogs. Although DENV pro inhibitory assays showed modest activity for the analogs (IC50 between 36.23 and 60.98 µM), inhibition of DENV replication in the replicon-based assay was promising. A cyclohexanone analog (32, Figure 11 ) of curcumin showed an EC50 value of 8.07 ± 1.52 µM, whereas EC50 for plaque assay was 2.34 ± 0.21 µM. Compared to curcumin, a cyclopentanone (33, Figure 11) analog with a selectivity index (SI) of 16.27 was the most active and less toxic.79 In another effort to synthesize curcumin analog, Zamri et al. (2019) reported a benzenesulphonyl curcumin analog (34, Figure 11), ((3E,5E)-3,5-bis(4-methoxybenzylidene)-1-(phenylsulfonyl)piperidin-4-one), and in-silico studies revealed promising DENV NS2B-NS3 pro inhibitory activity attributed to the predicted interactions with His51, Asp75, and Ser135 of the catalytic triad. MD simulations showed binding free energy of −61.01 kcal/mol when compared to −59.78 Kcal/mol of the positive control, Panduratin A.80
Figure 11.

DENV NS2B-NS3 pro inhibitors from natural sources.
6.2.2.4. Miscellaneous
To identify anti-dengue agents from natural products, Lee et al. (2016) reported the anti-dengue activity of peridinin (35, Figure 12 ), a carotene-like substance, for the first time. Peridinin, along with eight other compounds, was obtained from the ethanolic extract of Formosan zoanthid, Palythoamutuki. Their in-vitro potential for inhibiting DENV2 replication was assessed. Peridinin inhibited DENV2 in-vitro replication with an EC50 of 4.5 ± 0.46 µM, while EC50 reported for the other three serotypes ranged between 5.84 and 7.62 µM. Further, Peridinin proved to be an effective DENV2 pro inhibitor with an EC50 of 8.5 ± 0.41 µM.81 Tataringa et al. made the first attempt to perform an in-silico study of 11 compounds from the GC–MS analyzed, Romanian Allium cepa oil. All the 11 compounds were docked to the seven selected protein targets of DENV, including DENV NS2B-NS3 pro. Hexadecanoic acid (36, Figure 112) emerged as the best compound with the best binding affinity against almost all the protein targets, including the protease target. It was predicted to form two hydrogen bonds with the protease target.82
Figure 12.

DENV NS2B-NS3 pro inhibitors from natural sources.
However, flavonoids are known to yield false-positive activities in biochemical assays. We observed that none of the original studies have discussed assay interference properties of compounds obtained from natural sources. Upon analyzing all the compounds among those compounds obtained from natural resources, we found that seven out of total of 15 compounds were found to have PAINS groups. Of these, four compounds had catechol moiety while three others had ene-one-ene configuration. Considering these findings, we believe that although promising, the data obtained after studying natural compounds, especially those from the flavonoid class cannot be relied upon completely due to false findings.
6.2.3. Inhibitors from HTS
6.2.3.1. Benzothiazole and benzimidazole derivatives
HTS remains an effective tool for screening compounds to identify lead compounds from a library of the molecules. Wu et al. (2015), to identify new chemical scaffolds for inhibition of the DENV2-3 pro, performed HTS using an in-house library of approximately
250 compounds and identified a lead molecule containing diarylthio-ether (37, Figure 13 ). Modifications to improve the affinity resulted in the synthesis of seven analogs. Significant modifications included thiophene ring replacement with alkyl chain and other heteroaromatic ring systems. Further modifications saw the amide bond replacement with ester and amino groups. However, substitution with alkyl chain, ester, and amine groups led to the loss of activity. As shown in Figure 13, subsequent replacements led to the identification of compound 38 ( Figure 13 ), which gave the best IC50 (3.6 ± 0.11 µM) against DENV2 pro and with a non-competitive mode of inhibition. Docking studies of compound 38 predicted the interaction of one –OH group with the side chain of Asn152 and with the backbone of Lys74 while that of the second –OH group with Lys73. However, a dimethoxy substitution, compound 40 ( Figure 13 ), was found to be inactive. Further replacement of CF3-Ph- by OH-Napthyl group and “S” by “O” yielded compound 39 ( Figure 13 ), which had an IC50 of 4.2 ± 0.44 µM against DENV2 pro. Results of the viral replication assay showed low EC50 values for most compounds. A remarkable finding was that the dimethoxy derivative, compound 40, showed a good reduction in viral titer against DENV2, with an EC50 value of 2.5 ± 0.11 µM. However, it was inactive in the protease assay. In contrast, the findings for compound 38 were the opposite. Compound 38 was not active at a concentration below 3 µM in the antiviral assay. Further, it was hypothesized that compound 40 was a prodrug, which gets demethylated to the dihydroxy form to exhibit the action.83
Figure 13.

Schematic representation of Wu et al. (2015) modification of lead obtained through HTS.
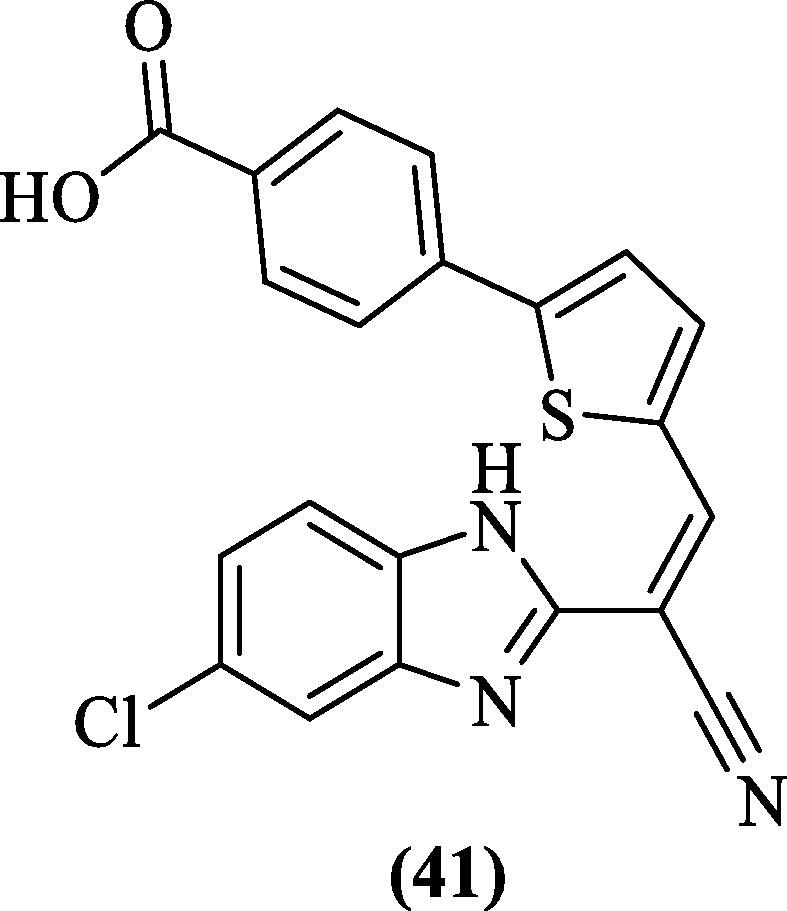
In yet another work, Raut et al. (2015) randomly screened ∼ 1000 small molecules from an ‘in-house’ library to identify potential dengue protease inhibitors. A benzimidazole derivative, compound 41 ( Figure 14 ), emerged as the most potent inhibitor of DENV2 pro (IC50 = 5.95 μM). Enzyme kinetic studies indicated a mixed-type inhibition; this was effective against all other DENV serotypes and reduced the viral titer of DENV-1, 2, 3, 4 by 50, 83, 75, and 73% respectively in a cell-based inhibition assay. Docking analysis predicted inhibitor binding to an allosteric site near the active site.84
Figure 14.
DENV NS2B-NS3 pro inhibitors obtained from HTS.
6.2.3.2. Polyphenolic and catechol scaffolds
Balasubramanian et al. (2016) also employed the HTS approach to screen approximately 1,20,000 compounds against DENV2 pro and identified compounds with polyphenolic and catechol scaffolds. Of the eight thoroughly investigated compounds, three compounds (42, 43, and 44, Figure 15 ) were potent inhibitors of all four serotypes. Their DENV 1–4 pro inhibitory concentrations (IC50) were found to be in the range of 2.94–4.06 μM, 0.64–1.15 μM, and 0.13–0.77 μM, respectively, while the EC50 values obtained from DENV2 replication inhibition assay using BHK21 cells for compounds 42 and 43 were 6.9 ± 0.6 µM and 2.29 ± 0.3 µM, respectively. All three compounds were competitive inhibitors of DENV2 pro, and molecular docking studies proposed active site binding of all the three inhibitors.76
Figure 15.

DENV NS2B-NS3 pro inhibitors obtained from HTS.
6.2.4. Inhibitors identified through HTVS
HTVS has become an essential part of drug discovery efforts, saving time and providing an opportunity for screening molecules rationally. HTVS, over the years, has helped researchers identify potential HITs and, eventually, potential lead compounds. In the last decade, many antivirals have emerged as a result of this in-silico approach.
6.2.4.1. Piperazine-naphthyridine derivative and isoindoline dione derivatives
Li et al. (2015), through their virtual screening campaign using a library of 5 million compounds obtained from four different commercial sources, attempted structure-guided discovery of a small non-peptide as an inhibitor of DENV2 pro. Fourteen compounds based on docking scores were identified and subjected to biological screening. The top HIT compound 45 (Figure 16 ) showed an EC50 of 5.0 µM in BHK21 cells in the protease inhibition assay. Docking study of compound 45 further supported their pharmacophore-based design concept, with a binding free energy of −10.65 kcal/mol. The result was in agreement with the interactions seen in the known crystal structure (PDB 3U1I). Also, compound 45 was predicted to occupy the pockets of the binding site, and the benzyl ring of the compound appeared to occupy the hydrophobic region between Pro132 and Val155. Further lone pair of “N” on the adjacent atom was predicted to interact with the hydroxyl group of Tyr161, and an additional π-π interaction could also be seen also it appeared that an additional water molecule linked the amino group of the inhibitor with Met184 and Gly153 to provide stabilization to the inhibitor 45–protease complex.85
Figure 16.

DENV pro inhibitors obtained from HTVS.
Timiri et al. (2015) performed HTVS using the ZINC 8 database against 2FOM to identify an inhibitor of DENV2 pro. After analyzing the top hundred molecules manually, they synthesized phthalimide-sulphonamide derivatives. Twenty derivatives of 4-(1, 3-dioxo-2, 3-dihydro-1H-isoindol-2-yl) benzene- 1-sulphonamide were synthesized and screened for DENV2 pro inhibitory activity. Compound 46 ( Figure 16 ) inhibited protease with an IC50 value of 48.2 µM. The docking score of this compound was −5.12 kcal/mol, and the predicted critical interactions shown by this compound were π-π stacking interactions between ring A and Tyr200, A hydrogen bonding interaction between “O” atom of ring A and “N” atom of imidazole ring in Hip 90. Another hydrogen bonding interaction was observed between the “N” atom attached to sulphonamide and the “O” atom of the carbonyl group of Asn191. They also observed hydrophobic interactions of ring A with Asp168, Phe169, Pro171, Tyr189, Gly190 residues, hydrophobic interaction between 4-ethyl phenyl group attached to sulphonamide and Trp89 and Val111, and a π-cation interaction between electron cloud of ring B and cationic “N” of imidazole ring in Hip90. MD simulations revealed that NS2B-NS3 pro was more stable when bound with inhibitor (46). 86
6.2.4.2. Indole, Thioguanine and oxadiazole-Aryloxazinone derivatives
Further, Pelliccia et al. (2017) adopted a virtual screening approach to identify allosteric inhibitors against DENV NS2B-NS3 pro. Based on their findings, they designed and synthesized inhibitors and evaluated their inhibitory potential by performing the cell-based and enzyme-based assay against DENV NS2B-NS3 pro. Among the synthesized compounds, the EC50 values of compound 47 ( Figure 17 ) in the cell-based and enzyme-based assay was found to be 6.7 ± 0.78 µM and 4.7 ± 0.3 µM, respectively. Further, 47 inhibited DENV replication in Huh-7 cells with an EC50 of 4.60 ± 0.03 µM. Docking studies of this compound predicted the hydrogen bond between the carbonyl portion of the ester and the Asp152 side chain. Also, the ester moiety appeared to be positioned inside the binding pocket. Further, hydrogen bond was predicted between indole, NH and residue Asn167. Residues Val146, Leu149, and Leu76 were predicted to form hydrophobic interaction with the indole ring, and the methoxyphenyl moiety also showed hydrophobic interactions with Trp83 and a π-cation interaction with Lys74.87
Figure 17.

DENV pro inhibitors obtained from HTVS.
Hariono et al. (2019) identified a HIT against DENV2 pro by virtual screening a library from the National Cancer Institute database. The experimental IC50 of this HIT was found to be 62 µM against DENV2 pro. Modification of this HIT (having thioguanine scaffold) resulted in a more potent compound (48, Figure 17), having an IC50 value of 0.38 µM. Further, from the molecular modeling studies, residues Ser135, Tyr161, and Gly153 were predicted to form hydrogen bonding interactions with the inhibitor (48). Few other residues, i.e., Pro132, Gly151, His51, Asp75, and Asn152, appeared to have hydrophobic interactions.88 Recently, Bhowmick et al. (2020) performed a multi-step virtual screening of the Asinex database (containing 590,859 compounds) against DENV2 pro using crystal structure 2FOM. The best molecule identified (49, Figure 17) had a docking score of −7.619 kcal/mol, and the binding free energy of this molecule calculated from MD simulation trajectories, using the MM/GBSA approach was found to be −60.666 kcal/mol. These metrics were superior to the standard inhibitor (MB21). Further, residues Lys73, Gly153, and Asn167 were predicted to form four hydrogen bonds with the inhibitor (49). Residues Lys74 and Leu76 were involved in hydrophobic interactions with the inhibitor, and the only π-cation interaction observed was between Lys74 and inhibitor.89
6.3. Inhibitors identified through a rational approach
6.3.1. Piperidinyl and piperidone derivatives
Inspired by their previous studies on natural products and their role as antiviral agents against DENV2, Gan et al. (2017) reported two piperidinyl compounds (50 and 51, Figure 18 ) showing antiviral activity against DENV2 with an EC50 value of 3.2 and 2.4 µM, respectively. Both the compounds were synthesized from nicotinic acid. After the cell-based assay, protease inhibition was analyzed. However, they were weaker protease inhibitors, as reflected by their IC50 values of 213.7 µM (50) and 257.4 µM (51).90
Figure 18.

Potent inhibitors of Gan et al. (2017).
The 3,5-bis(arylidene)-4-piperidone derivatives have been previously reported for their antiviral,91 anticancer,92 antioxidant,93 and anticholinesterase94 activities. The α, β-unsaturated ketone analogs from Boesenbergia rotunda were reported to be potent inhibitors of DENV2 serine pro.64 In addition, analogs of 4-hydroxypanduratin were also reported as DENV pro inhibitors.95 Osman et al. (2017) attempted to exploit the benefits of α, β-unsaturated ketones. They synthesized ten compounds by incorporating a piperidone ring to the α, β-unsaturated ketone system and evaluated their inhibitory activity against DENV2 pro. They performed in-silico studies and correlated the in-silico and in-vitro results. Docking studies using DENV2 pro (PDB 2FOM) identified two inhibitors (52 and 53, Figure 19 ) with good docking scores. These molecules also showed good in-vitro affinities in agreement with the in-silico binding scores. Nitro derivatives of 3, 5-bis (arylidene)-4-piperidones (52, 53) had IC50 values of 15.22 and 16.23 µmol/L, respectively, as compared to the standard panduratin A, which had an IC50 value of 57.28 µmol/L. In molecular docking studies, both compounds appeared to occupy the active site. Compound 52 was predicted to form five hydrogen bonds with the His51, Pro132, Ser135, Gly153, and Arg54 residues. A π-π stacking interaction was also observed between the phenyl ring and His51. Compound 53 was predicted to formed six hydrogen bonds with His51, Pro132, Ser135, Gly153, Arg54, and Trp50 residues.96
Figure 19.

Best reported inhibitors of Osman et al. (2017).
6.3.2. Thiosemicarbazones and 4-benzyloxyphenylglycine derivatives
Considering the biological properties offered by thiosemicarbazones and reports of their role as antiviral agents, Padampriya et al. (2018) synthesized six thiosemicarbazone-derived phenyl-acetyl derivatives and analyzed their in-vitro antiviral activity against the DENV2 serotype. Compound 54 (Figure 20 ) showed antiviral activity. Its docking study against DENV2 pro (PDB 2FOM) revealed occupancy at the active site where the inhibitor formed four hydrogen bonds with residues Ser135, Gly151, Pro132, and Asp75.97 In recent work, Kühl et al. (2020) reported 55 non-peptidic small molecule derivatives of 4-benzyloxyphenylglycine as DENV2 pro inhibitors. In their work, the core 4-benzyloxyphenylglycine was coupled to a lipophilic alkyl chain via an amide bond. The carboxyl portion was further connected via another amide bond to a phenyl group with further variations in basic residues. Variants were produced by fragment merging using biochemical assays for categorizing and combining most active residues. Compound 55 ( Figure 20 ) (the l-isomer), a phenyl guanidine variant, yielded upper-nanomolar DENV2 pro-HeLa EC50 (0.49 ± 0.08 µM) activity. The d-isomer of 55 was found to have 20-fold low potency compared to l-isomer, partly indicating a different binding mode. An added hexanoic acid cap at N-terminal improved the activity of 55 over other substituents. Modeling studies performed against DENV3 pro (PDB 3U1I) revealed that the positively charged phenylguanidine moiety was positioned in the S1 pocket-forming an ionic interaction with the negatively charged aspartate residue. The S2 pocket was occupied by (4-Benzyloxy)-l-phenylglycine, and the phenyl ring of the phenylglycine formed π − π-stacking interaction with catalytic histidine. The hexanoic acid contributed to lipophilic interactions and occupied the S3 pocket.98
Figure 20.

Potent inhibitors of Padampriya et al. (54) and Kühl et al. (55).
6.4. Inhibitors reported by modification of previously reported inhibitors
Weng et al. modified their previously reported most potent linear dipeptide DENV2 pro inhibitor.99 They synthesized a series of 16 newly fused bicyclic compounds of pyrrolidino[1,2-c] imidazolidinone derivatives and evaluated their inhibitory activity against DENV2NS2B-NS3 pro and wild-type DENV-2 virus. The C-3 stereochemistry of the fused ring system played a crucial role in determining the activity of the fused ring derivatives. The linear dipeptide (56) and a non-peptidic fused ring derivative (57, Figure 21 ) showed a similar activity profile. Both had an IC50 value of 1.2 ± 0.4 µM and had almost similar EC50 values of 38.7 ± 5.4 µM and 39.4 ± 6.2 µM, respectively. Also, the 3-beta isomer (58, Figure 21) was inactive; thus, the importance of absolute configuration at this position could be understood. Meanwhile, their CC50 values indicated that they had no cytotoxic effect in the tested concentration range. The 3-alpha isomer of the fused ring scaffold was studied with the help of molecular docking against the chain-A of the X-ray crystal structure of DENV2 pro (PDB 2FOM) to understand the interactions which could be attributed to the protease inhibition activity. It was predicted that ring A (p-Nitro phenyl group) occupied the cleft of Tyr150, Ser135, and Pro132. A hydrogen bond was predicted between the oxygen atom of the nitro group of ring A and Ser135 of the catalytic triad. Further, fused ring B, N-4 nitrogen atom appeared to be involved in hydrogen bond formation with imidazole of His51 by donating its lone pair. Ring C was close to Gly133, Ile36, and Val52. The two hydrogen bonds formed with the residues of the catalytic triad thus indicated stable binding of the inhibitor with the protein and eventually inhibiting the catalytic activity of the serine protease. Although the EC50 values were not very promising, this study could provide a new approach for discovering non-peptidic DENV pro inhibitors in the future.100
Figure 21.

Weng et al. (2017) modification.
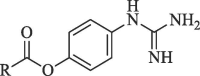
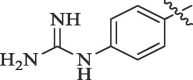
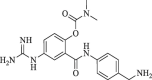
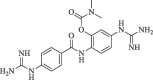
In yet another modification effort, Sunderman et al. (2019) modified a previously reported dibasic 4-guanidinobenzoate, which inhibited DENV2 pro in the low micro-molar range.101 Their objective was to study the SAR and improve the drug-likeness of lead compounds ( figure 22 ). Earlier Aryl-4-guanidinobenzoates were known for the formation of reversible or pseudo-irreversible covalent bonds.102 Also, the replacement of the ester group with amide and the introduction of carbamates between two aromatic rings was associated with inhibition of serine protease activity.103 These changes were incorporated in the lead structure, and 19 analogs of the lead were synthesized and evaluated for DENV2 and WNV pro inhibition. These results have been summarized in (Table 5, Table 6, Table 7 ). The 4-guanidinobenzoate modifications (Table 5) showed that the activity decreased when the aromatic ring was replaced with the aliphatic chain. The 4-guanidinophenol modifications (Table 6) also did not yield encouraging results. However, they were more effective when compared to the 4-guanidinobenzoate modifications. The introduction of the carbamate group between the aromatic groups also resulted in less active compounds (Table 7) as they showed weak inhibitory activity. SAR studies on these compounds established the importance of the electrophilic ester group and 4-guanidinobenzoate for protease inhibition. Also, it indicated that 4-guanidinophenol modification was more acceptable than 4-guanidinobenzoate modification. Binding mode analysis for the lead compound showed covalent interaction with DENV pro. However, stability in the basic buffer remained a significant concern.104
Figure 22.

(Lead compound).
Table 5.
Modification of 4-guanidinobenzoate part by Sunderman et al. (2019).
 |
Percentage inhibition of NS2B-NS3 pro |
|
|---|---|---|
| R | DENV1 | WNV# |
 (Lead compound) (Lead compound)
|
98.1 | 29.8 |
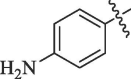 |
45.4 | No inhibition |
 |
4.2 | No inhibition |
 |
4.6 | No inhibition |
| Camostat | 19.6 | 12.6 |
50 µM substrate concentration and 50 µM compound concentration.
50 µM substrate concentration and 25 µM compound concentration.
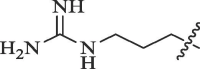
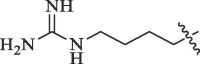
Table 6.
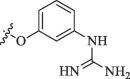
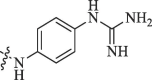
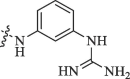
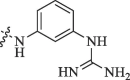
Modification of 4-guanidinophenol part by Sunderman et al. (2019).
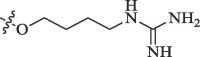
 |
Percentage inhibition of NS2B-NS3 pro |
|
|---|---|---|
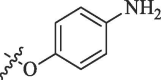
| R | DENV* | WNV# |
 |
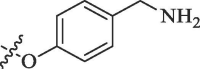
51.3 | 17.5 |
 |
23.2 | 17.9 |
 |
36.8 | No inhibition |
 |
56.8 | No inhibition |
 |
49.0 | 17.1 |
 |
8.8 | No inhibition |
 |
40.1 | 36.5 |
50 µM substrate concentration and 50 µM compound concentration.
50 µM substrate concentration and 25 µM compound concentration.
Table 7.
Carbamate analogs and their modification by Sunderman et al. (2019).
| Compound | Percentage inhibition of NS2B-NS3 pro |
|
|---|---|---|
| DENV* | WNV# | |
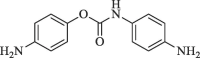 |
33.7 | 72.8 |
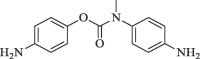 |
29.8 | 68.0 |
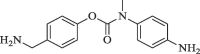 |
22.9 | 59.3 |
| 31.3 | 62.9 | |
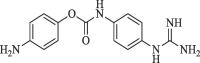 |
52.0 | 61.9 |
|
60.8 | 68.8 |
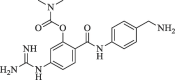 |
74.4 | 73.3 |
 |
56.0 | 76.1 |
 |
20.2 | 23.9 |
50 µM substrate concentration and 50 µM compound concentration.
50 µM substrate concentration and 25 µM compound concentration.
These two modifications yielded successful results. Although the results were not highly encouraging, the insights from these studies certainly improve our understanding of the dos and don’ts in the drug design process.
6.5. Inhibitor reported by investigation of antiviral activities of previously reported inhibitors
Chu et al. (2015) investigated the antiviral activities of 15 small molecule and peptide-based NS2B-NS3 pro inhibitors on dengue serotype 2-infected HuH-7 human hepatocarcinoma cells. Experimental results revealed an anthraquinone (ARDP0006) as the most potent inhibitor, which reduced dengue viral titer by>1 log PFU/mL at 1 µM concentration in their cell-based assays involving HuH-7 and K562 cell lines. It was also non-cytotoxic at 1 µM concentration over three days of incubation on HuH-7 cells using the Alamar Blue cellular toxicity assay. Table 8 summarizes the work done by this group. The key conclusion of their study was that, although a molecule was an effective DENV2 pro inhibitor, the potential to be a successful drug candidate lay in its ability to penetrate the cells chosen for the study and reduce viral titer. Ideally, DENV2 pro inhibitors should be amenable to modifications that enhance the permeability, which is indicated by their PAMPA values. This molecule could be exploited for drug development in the future.105
Table 8.
Result of antiviral activity of previously reported inhibitors, Chu et al. (2015).
| Sl. No. | Structure | Supplied by | % purity |
Ki/EC50/IC50 (Reported) |
Cellular toxicity |
Antiviral activity |
Reason for no antiviral activity |
Permeability (P) (10-6 cmS-1) |
|---|---|---|---|---|---|---|---|---|
| 1 | Ac-RTSKKR-CONH2 | GL Biochem (China) |
>95% | Ki = 12.14 µM | X | X | Inability to permeate & disadvantage of Peptide nature of the drug | 0 Not detectible |
| 2 |  |
Maybridge (UK) | >95% | Ki = 44 ± 5 µM | X | X | Positive charge on guanidine and basic moiety limitations | 0 Not detectible |
| 3 | BZ-Ala-Lys-Arg-Arg-H | Mimotopes (Australia) |
62% | Ki = 5.3 µM | X | X | Very high charge On compound |
0 Not detectible |
| 4 |  |
Otava (Ukraine) |
>95% | Ki = 17.0 ± 4.3 µM IC50 = 67.0 ± 7 µM |
X | X | High IC50 value | 4.12 ± 0.45 |
| 5 |
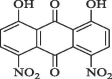 ARDP0006 |
Sigma-Aldrich (USA) |
>95% |
EC50 = 4.2 ± 1.9 µM |
X | Highly permeable | 4.45 ± 0.6 | |
| 6 | 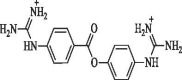 |
In-house | >95% | Ki = 2.0 µM | X | X | Poor permeability owing to high polarity | 0 Not detectible |
| 7 | 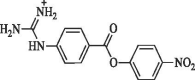 |
Cambridge Screening Library (USA) |
>95% | NR | X | X | Lacked membrane permeability | 0 Not detectible |
| 8 | 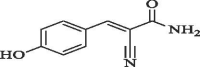 |
Maybridge (UK) | >95% | Ki = 35.7 µM | X | X | Unknown reason |
8.40 ± 0.15 |
| 9 |  |
Cambridge Screening Library (USA) |
>95% | Ki = 15 ± 3 µM | X | X | Lacked membrane permeability | 0 Not detectible |
| 10 |  |
Cambridge Screening Library (USA) |
>95% | IC50 = 27.0 ± 1.3 µM | X | X | Positively charged nature of compound | 0 Not detectible |
| 11 |  |
GL Biochem (China) |
>95% | Ki = 15.6 ± 1.1 µM | X | X | Highly cationic nature of compound | 0 Not detectible |
| 12 |  |
GL Biochem (China) |
>95% | Ki = 1.4 ± 0.1 µM | X | X | Highly cationic nature of compound | 0 Not detectible |
| 13 | Alexidine hydrochloride | Sigma-Aldrich (USA) |
>95% | Ki = 41 ± 3 µM | X | X | Cationic guanidine moieties | 0 Not detectible |
| 14 | Ivermectin | Sigma-Aldrich (USA) |
>95% | Ki = 79 ± 21 µM | --- | --- | --- | |
| 15 | Selamectin | Sigma-Aldrich (USA) |
>95% | Ki = 63 ± 18 µM | ‘ --- | --- | --- |
6.6. Inhibitors from drug repurposing (Nelfinavir, Policresulen, and Erythrosin B)
Drug refocusing as a concept has gained significant popularity with the emergence of the COVID-19 pandemic. Similar approaches have also been used for DENV drug discovery. Bhakat et al. (2015) first reported drug repurposing efforts where they analyzed nine peptidomimetic FDA-approved HIV/HCV inhibitors by molecular docking and using a cell-based antiviral assay. Docking of the HIV/HCV inhibitors was done using the closed conformation of DENV NS2B-NS3 pro (PDB 3U1I), and the results predicted that the HIV/HCV inhibitors were able to bind the active sites of DENV NS2B-NS3 pro. The results were further refined using MD-based MM/GBSA rescoring, and it was observed that van der Waals interactions were the key driving forces for the inhibitor-enzyme interactions in the active site. Nelfinavir (Decahydroisoquinoline scaffold derivative, 59, Figure 23 ) showed hydrogen bonding interactions with Met84 and Gly153, a π-π stacking interaction with Tyr161, and a side chain hydrogen bond interaction with Thr83 also contributed to the binding.
Figure 23.

DENV NS2B-NS3 pro inhibitors identified using the drug repurposing approach.
Interestingly the docking score of Nelfinavir (-8.9 kcal/mol) complimented the antiviral cell-based assay results. Nelfinavir showed the best antiviral activity against DENV-2 with EC50 of 3.5 ± 0.4 µM and a SI of 4.6.106 Further, Wu et al. (2015), from their in-house library of old drugs (∼1000 compounds), identified a topical hemostatic and antiseptic 2-hydroxy-3,5-bis [(4-hydroxy- 2-methyl-5-sulfophenyl) methyl]-4-methyl-benzene-sulfonic acid (Policresulen) as a potent inhibitor of DENV2 pro with an IC50 value of 0.48 μg/mL. Policresulen acted as a competitive inhibitor of the protease and slightly affected protease stability. Also, policresulen inhibited DENV2 replication in BHK-21 cells giving an IC50 value of 4.99 μg/mL. Furthermore, IC50 for cytotoxicity to BHK-21 cells was found to be 459.45 μg/mL. Policresulen is a mixture of various analogs.107 Compound 60 (Figure 23) represents the major component of policresulen, and for a better understanding of the binding interaction, it was docked with the structural NS2B-NS3 pro model (PDB 2M9Q). The study predicted that in ring A, sulfo group oxygen appeared to form a hydrogen bond with the Gln106 side chain in the P1 region of the protease. The phenol group formed hydrogen bonds with the main chain carbonyl of Gln114. Ring B formed hydrophobic interactions with Ile109, Ile115, and Val 131 in the hydrophobic P2 region of the protease. An electrostatic interaction was also seen between the sulfo group and His130. In the ring C, the oxygen of the sulfo group formed a hydrogen bond with side chain residues of Thr132 and Arg133 in the positive P3 region of the protease.108 Recently, Li et al. reported Erythrosin B (EB) (61, Figure 23), an FDA- approved food additive, as a potent inhibitor of flaviviruses, including DENV pro with low IC50 value. They used their previously developed split luciferase complementation (SLC) based HTS assay to identify molecules blocking the interaction between NS2B and NS3 of DENV2 pro.109 EB non-competitively inhibited the interaction between DENV2 NS2B and NS3 with an IC50-SLC value of 15 µM. The antiviral efficacy of EB was evaluated using viral plaque reduction assay, and EC50 determined against DENV2 was 1.2 µM. Since inhibitors targeting NS2B-NS3 interaction sites are potentially broad-spectrum inhibitors for flaviviruses,109, 110 EB’s potential to inhibit other flaviviruses was also investigated, and results were encouraging. Viral titer reduction was found to be dose-dependent in other flaviviruses.111
6.7. Clinical trials and patents
We searched for dengue fever in the clinical trials database (https://clinicaltrials.gov) and found only 19 records reflecting drug trials. A summary of the patents available for dengue protease inhibitors is provided in Table 9 ; none of these records reflected any protease inhibitors.
Table 9.
Summary of Patents related to Dengue Protease Inhibitors.
| Title | Inventors | Patent number | Related publication (if any) |
|---|---|---|---|
| Small molecule inhibitors of DENV proteases | Viswanathan Usha, Watowich Stanley J |
US2015141521A1 US9408813B2 |
Viswanathan et al. 2014112 |
| Small molecule inhibitors of dengue and WNV proteases | Gilbertson Scott, Tomlinson Suzanne M, Watowich Stanley J |
US2013035284A1 US8778876B2 |
Tomlinson et al. 200925 |
| Dengue protease substrates and inhibitors | Harris Jennifer, Li Jun, Tumanut Christine |
WO2006014232A2 WO2006014232A3 |
Li et al. 200531 |
| Flavivirus protease inhibitors | Manzano Mark, Padmanabhan Radhakrishnan, TeramotoTadahisa |
WO2014066502A2 WO2014066502A3 |
Balasubramanian et al. 201676 |
| Dengue and WNV protease inhibitors | Harisha Attimogae Shivamurthy, Nagarajan Kuppuswamy, Padmanabhan Radhakrishnan, Rao Kothapalli Sundarraja, Shashiprabha, Shridhara Kanakamajalu |
WO2014164667A1 | Kálai et al. 201193 |
| Use of the acetic acid compound 2-[1-[[(1r)-1-[3-[2-(7-chloroquinolin-2-yl) etenyl] phenyl]-3-[2-(2-hydroxypropan-2-) phenyl] propyl] sulfanylmethyl] cyclopropyl] as alosteric inhibitor of the DENV NS2B/NS3 protease | Aldo Segura Cabrera, Carlos Armando García Pérez, Juan Santiago, Salas Benito, Mario Alberto, Rodríguez Pérez |
MX2015013285A | NA |
7. Conclusion
Despite best efforts by various research groups over the years, none of the DENV protease inhibitors has entered clinical trials. The failure of the only vaccine developed has further increased the demand for the search for a potent drug. The structure and function of DENV protease is well understood. Based on the efforts made by researchers in the industry and academia, it is well understood that aiming only to develop inhibitors with sub-micromolar or nanomolar affinities will not serve the purpose. The fundamental challenge in developing an effective drug lies in dealing with the toxicity, stability, and permeability of DENV protease inhibitors. Improving the permeability of the peptide inhibitors while maintaining the protease inhibitory profile requires a balanced selection of functional groups to be incorporated in the design. One such attempt was made by Takagi et al. while designing the cyclic peptides, where they found the introduction of aromatic residues in the design improved the inhibitory profile. Also, in general, the peptide inhibitors showed marked improvement in protease inhibition by aromatization of N & C-terminals. An insight into the protease inhibitor interaction provided by the different research groups in their work lead us to identify certain key residues which could be targeted using a rational design approach. Since these residues of DENV protease appear to be involved in a variety of interactions as shown in Table 10 (hydrogen bonding interactions, hydrophobic interactions, π-π interaction, π-cation interaction, glycine interaction, polar interaction, π-σ interaction) and since the chemical scaffolds with which they interacted belonged to diverse chemical classes, this advantage could be leveraged in drug design approach, both for the designing the small molecule inhibitors as well as peptide inhibitors. Importantly the identified key residues showed interaction with more than half of the reported inhibitors. Table 10 shows the key residues of the proteases which interacted with the inhibitors having a wide range of chemical scaffolds in their architecture. It can be seen that the residues of the catalytic triad and residues Asn152, Pro132, Lys73 were commonly engaged in hydrogen bonding and hydrophobic interactions. In contrast, certain residues like Tyr161 (π-π interaction), His51 (π-π interaction), Gly153 (Glycine interaction), Lys74 (π-cation interaction), Val155 (π-σ interaction) showed some characteristic interactions confined to these residues. Exploring the occupancies of S1,’ S1, S2, S3, and S4 pockets by different scaffolds (Table 10) and the residues interacting with these pockets could help us develop novel scaffolds (peptides and small molecules). At the same time, a rational attempt could be made to address the pharmacokinetic issues of the tripeptide inhibitors by proper substitution of basic residues at P1 and P2 position of tripeptide molecule with different heterocyclic/aromatic/cyclic/bicyclic rings, which were predicted to occupy different positions in the S1 and S2 pockets of different reported inhibitors. Table 10 also provides few hydrophobic scaffolds as possible substitutes of the tripeptide cap group, often appearing to occupy the S3 and S4 sub-pockets of the protease. Hence the scaffolds occupying the S3 and S4 sub-pockets could prove to be good substitutes or other possible substitutes of the cap of the tripeptides, and their utility in enhancing the pharmacokinetic properties could be further tested. Each scaffold with its substation offers a definite property to the molecule, which is finally responsible for the inhibitory activity. Table 10 summarizes scaffolds present in various compounds. Since we are far from obtaining a small molecule inhibitor with a nanomolar affinity, any combination of scaffolds with moderate inhibition could be rationally selected for further development. While assembling active portions from these scaffolds could yield active molecules, their synthetic feasibility should also be considered. Table 11 presents a summary of potent molecules from different sections. DENV and COVID-19 co-infection has raised serious concerns. The emergence of co-infections inspired us to compare substrate-peptide residue preferences and the residues lining the sub-pockets of both the proteases. To the best of our knowledge, this review is the first attempt to compare the similarities and differences between both proteases. Viral propagation in both the families of viruses is dependent on the activity of the proteases involved. Thus, we believe that our insights on similarities in the residue preferences of both the proteases and the analysis on residues lining the sub-pockets of both the proteases could help new researchers strategize their new drug discovery efforts and guide established researchers to steer their research in this direction. This approach should especially spur the development of peptidomimetics which have shown a significantly potent inhibitory activity in the past. However, this topic needs further exploration since the insights on SARS-CoV-2 3CLpro are relatively fresh. Our group is currently making efforts toward screening the reported DENV NS2B-NS3 pro inhibitors and other libraries in the database against SARS-CoV-2 3CLpro, and we have got encouraging results. The results will be published in the followup manuscript.
Table 10.
A summary of key residues identified as having interaction with DENV pro.
 |
Note: Similar color residues share similar properties by virtue of their side chains.
Table 11.
Few selected potent molecules from different classes.
| Chemical class/molecule | Compound No. | IC50/ EC50 | Activity (inhibition) |
|---|---|---|---|
| Peptides | |||
| Tripeptide Cap-Arg-Lys-Phg-NH2 |
9 | IC50 = 0.018 µM | DENV2 pro |
| Cyclic peptide | 15 | IC50 = 0.95 µM. | DENV2 pro |
| Small molecules | |||
| Pyrazole ester derivative | 20 | IC50 = 0.5 ± 0.1 µM | DENV2 pro |
| Peridinin | 35 | EC50 = 4.5 ± 0.46 µM | DENV2 replication |
| From HTS | 38 | IC50 = 3.6 ± 0.11 µM | DENV2 pro |
| 43 | EC50 = 2.29 ± 0.3 µM | DENV2 replication | |
| From HTVS | 47 | EC50 = 4.60 ± 0.03 µM | DENV2 replication |
| Rational approach | |||
| Piperidinyl derivative | 51 | EC50 = 2.4 µM | Antiviral against DENV2 |
| 4-benzyloxyphenylglycine derivatives | 55 | EC50 = 0.49 ± 0.08 µM | DENV2 pro HeLa EC50 |
| Drug repurposing | |||
| Nelfinavir | 59 | EC50 = 3.5 ± 0.4 µM | Antiviral against DENV2 |
| Policresulen | 60 | IC50 = 0.48 μg/mL | DENV2 pro |
| Erythrosin B | 61 | EC50 = 1.2 µM | Antiviral against DENV2 |
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors would like to thank the funding agency, DST-SERB, Govt. of India, for providing financial support through their project (EMR /2016/005711) dated 7th August 2017 and Birla Institute of Technology, Mesra, Ranchi, India for providing the necessary infrastructural facilities.
References
- 1.Bhatt S., Gething P.W., Brady O.J., et al. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashburn PM, Craig CF. Experimental investigations regarding the etiology of dengue fever. 1907. J Infect Dis. 2004;189(9):1747-1783; discussion 1744. http://doi.org/10.1086/383418. [DOI] [PubMed]
- 3.Lescar J., Luo D., Xu T., et al. Towards the design of antiviral inhibitors against flaviviruses: The case for the multifunctional NS3 protein from Dengue virus as a target. Antiviral Res. 2008;80:94–101. doi: 10.1016/j.antiviral.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Stevens A.J., Gahan M.E., Mahalingam S., Keller P.A. The medicinal chemistry of dengue fever. J Med Chem. 2009;52:7911–7926. doi: 10.1021/jm900652e. [DOI] [PubMed] [Google Scholar]
- 5.Nitsche C., Holloway S., Schirmeister T., Klein C.D. Biochemistry and medicinal chemistry of the dengue virus protease. Chem Rev. 2014;114:11348–11381. doi: 10.1021/cr500233q. [DOI] [PubMed] [Google Scholar]
- 6.Kautner I., Robinson M.J., Kuhnle U. Dengue virus infection: Epidemiology, pathogenesis, clinical presentation, diagnosis, and prevention. J Pediatr. 1997;131(4):516–524. doi: 10.1016/S0022-3476(97)70054-4. [DOI] [PubMed] [Google Scholar]
- 7.Simmons C.P., Farrar J.J., Van Vinh C.a.N., Wills B. Current concepts: Dengue. N Engl J Med. 2012;366:1423–1432. doi: 10.1056/NEJMra1110265. [DOI] [PubMed] [Google Scholar]
- 8.Simmons C.P., Farrar J.J., van Vinh Chau N., Wills B. Barriers to preclinical investigations of anti-dengue immunity and dengue pathogenesis. Nat Rev Microbiol. 2012;366:1423–1432. doi: 10.1056/NEJMra1110265. [DOI] [PubMed] [Google Scholar]
- 9.Kyle J.L., Harris E. Global spread and persistence of dengue. Annu Rev Microbiol. 2008;62:71–92. doi: 10.1146/annurev.micro.62.081307.163005. [DOI] [PubMed] [Google Scholar]
- 10.Guzman M.G., Alvarez M., Halstead S.B. Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: An historical perspective and role of antibody-dependent enhancement of infection. Arch Virol. 2013;158:1445–1459. doi: 10.1007/s00705-013-1645-3. [DOI] [PubMed] [Google Scholar]
- 11.Halstead S.B. Neutralization and Antibody-Dependent Enhancement of Dengue Viruses. Adv Virus Res. 2003;60:421–467. doi: 10.1016/S0065-3527(03)60011-4. [DOI] [PubMed] [Google Scholar]
- 12.WHO. Global Strategy for Dengue Prevention and Control.; 2012. doi:/entity/denguecontrol/9789241504034/en/index.html.
- 13.Brady O.J., Gething P.W., Bhatt S., et al. Refining the Global Spatial Limits of Dengue Virus Transmission by Evidence-Based Consensus. PLoS Negl Trop Dis. 2012;6 doi: 10.1371/journal.pntd.0001760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.World Health Organisation. Dengue and severe dengue. WHO Fact Sheet. Accessed January 5, 2021. https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue.
- 15.Guy B., Noriega F., Ochiai R.L., et al. A recombinant live attenuated tetravalent vaccine for the prevention of dengue. Expert Rev Vaccines. 2017;16:671–683. doi: 10.1080/14760584.2017.1335201. [DOI] [PubMed] [Google Scholar]
- 16.Timiri A.K., Sinha B.N., Jayaprakash V. Progress and prospects on DENV protease inhibitors. Eur J Med Chem. 2016;117:125–143. doi: 10.1016/j.ejmech.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Chambers T.J., Hahn C.S., Galler R., Rice C.M. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- 18.Melino S., Paci M. Progress for dengue virus diseases: Towards the NS2B-NS3pro inhibition for a therapeutic-based approach. FEBS J. 2007;274:2986–3002. doi: 10.1111/j.1742-4658.2007.05831.x. [DOI] [PubMed] [Google Scholar]
- 19.Yusof R., Clum S., Wetzel M., Murthy H.M.K., Padmanabhan R. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J Biol Chem. 2000;275:9963–9969. doi: 10.1074/jbc.275.14.9963. [DOI] [PubMed] [Google Scholar]
- 20.Bazan J.F., Fletterick R.J. Detection of a trypsin-like serine protease domain in flaviviruses and pestviruses. Virology. 1989;171:637–639. doi: 10.1016/0042-6822(89)90639-9. [DOI] [PubMed] [Google Scholar]
- 21.Falgout B., Pethel M., Zhang Y.M., Lai C.J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol. 1991;65:2467–2475. doi: 10.1128/jvi.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erbel P., Schiering N., D’Arcy A., et al. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat Struct Mol Biol. 2006;13:372–373. doi: 10.1038/nsmb1073. [DOI] [PubMed] [Google Scholar]
- 23.Phong W.Y., Moreland N.J., Lim S.P., Wen D., Paradkar P.N., Vasudevan S.G. Dengue protease activity: The structural integrity and interaction of NS2B with NS3 protease and its potential as a drug target. Biosci Rep. 2011;31:399–409. doi: 10.1042/BSR20100142. [DOI] [PubMed] [Google Scholar]
- 24.Mukhopadhyay S., Kuhn R.J., Rossmann M.G. A structural perspective of the Flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 25.Tomlinson S.M., Malmstrom R.D., Russo A., Mueller N., Pang Y.P., Watowich S.J. Structure-based discovery of dengue virus protease inhibitors. Antiviral Res. 2009;82:110–114. doi: 10.1016/j.antiviral.2009.02.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verduyn M., Allou N., Gazaille V., et al. Co-infection of dengue and covid-19: A case report. PLoS Negl Trop Dis. 2020;14:1–5. doi: 10.1371/journal.pntd.0008476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epelboin L., Blondé R., Nacher M., Combe P., Collet L. COVID-19 and dengue co-infection in a returning traveller. J Travel Med. 2021;27:1–2. doi: 10.1093/JTM/TAAA114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saddique A., Rana M.S., Alam M.M., et al. Emergence of co-infection of COVID-19 and dengue: A serious public health threat. J Infect. 2020;81:e16–e18. doi: 10.1016/j.jinf.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chambers T.J., Weir R.C., Grakoui A., et al. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc Natl Acad Sci U S A. 1990;87:8898–8902. doi: 10.1073/pnas.87.22.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preugschat F., Yao C.W., Strauss J.H. In vitro processing of dengue virus type 2 nonstructural proteins NS2A, NS2B, and NS3. J Virol. 1990;64:4364–4374. doi: 10.1128/jvi.64.9.4364-4374.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J., Lim S.P., Beer D., et al. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J Biol Chem. 2005;280:28766–28774. doi: 10.1074/jbc.M500588200. [DOI] [PubMed] [Google Scholar]
- 32.Woo P.C.Y., Lau S.K.P., Huang Y., Yuen K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp Biol Med. 2009;234:1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- 33.Chan J.F.W., Li K.S.M., To K.K.W., Cheng V.C.C., Chen H., Yuen K.Y. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J Infect. 2012;65:477–489. doi: 10.1016/j.jinf.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan J.F.W., Lau S.K.P., Woo P.C.Y. The emerging novel Middle East respiratory syndrome coronavirus: The “knowns” and “unknowns”. J Formos Med Assoc. 2013;112:372–381. doi: 10.1016/j.jfma.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorbalenya A.E., Baker S.C., Baric R.S., et al. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu A., Peng Y., Huang B., et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe. 2020;27:325–328. doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang H., Xie W., Xue X., et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L. Lin d, Sun X, curth U, drosten c, Sauerhering L, Becker S, Rox K and Hilgenfeld R: Crystal structure of SARS-coV-2 main protease provides a basis for design of improved α‑ketoamide inhibitors. Science (80-). 2020;368:409-412. [DOI] [PMC free article] [PubMed]
- 41.Zhang L., Lin D., Kusov Y., et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J Med Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Z, Li Y, Loh YR, et al. NS2B-NS3 Protease from Zika Virus. Vol 354.; 2016. [DOI] [PubMed]
- 43.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorganic Med Chem Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chuck C.P., Chong L.T., Chen C., Chow H.F., Wan D.C.C., Wong K.B. Profiling of substrate specificity of SARS-CoV 3CLpro. PLoS ONE. 2010;5:1–7. doi: 10.1371/journal.pone.0013197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hegyi A., Ziebuhr J. Conservation of substrate specificities among coronavirus main proteases. J Gen Virol. 2002;83:595–599. doi: 10.1099/0022-1317-83-3-595. [DOI] [PubMed] [Google Scholar]
- 46.Yang H., Yang M., Ding Y., et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai W., Zhang B., Jiang X.M., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science (80-) 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gahlawat A., Kumar N., Kumar R., et al. Structure-Based Virtual Screening to Discover Potential Lead Molecules for the SARS-CoV-2 Main Protease. J Chem Inf Model. 2020;60:5781–5793. doi: 10.1021/acs.jcim.0c00546. [DOI] [PubMed] [Google Scholar]
- 49.Li Q., Kang C.B. Progress in developing inhibitors of sars-cov-2 3c-like protease. Microorganisms. 2020;8:1–18. doi: 10.3390/microorganisms8081250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Behnam M.A.M., Nitsche C., Vechi S.M., Klein C.D. C-terminal residue optimization and fragment merging: Discovery of a potent peptide-hybrid inhibitor of dengue protease. ACS Med Chem Lett. 2014;5:1037–1042. doi: 10.1021/ml500245v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nitsche C., Behnam M.A.M., Steuer C., Klein C.D. Retro peptide-hybrids as selective inhibitors of the Dengue virus NS2B-NS3 protease. Antiviral Res. 2012;94:72–79. doi: 10.1016/j.antiviral.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 52.Nitsche C., Schreier V.N., Behnam M.A.M., Kumar A., Bartenschlager R., Klein C.D. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J Med Chem. 2013;56:8389–8403. doi: 10.1021/jm400828u. [DOI] [PubMed] [Google Scholar]
- 53.Weigel L.F., Nitsche C., Graf D., Bartenschlager R., Klein C.D. Phenylalanine and Phenylglycine Analogues as Arginine Mimetics in Dengue Protease Inhibitors. J Med Chem. 2015;58:7719–7733. doi: 10.1021/acs.jmedchem.5b00612. [DOI] [PubMed] [Google Scholar]
- 54.Behnam M.A.M., Graf D., Bartenschlager R., Zlotos D.P., Klein C.D. Discovery of Nanomolar Dengue and West Nile Virus Protease Inhibitors Containing a 4-Benzyloxyphenylglycine Residue. J Med Chem. 2015;58:9354–9370. doi: 10.1021/acs.jmedchem.5b01441. [DOI] [PubMed] [Google Scholar]
- 55.Bastos Lima A., Behnam M.A.M., El Sherif Y., Nitsche C., Vechi S.M., Klein C.D. Dual inhibitors of the dengue and West Nile virus NS2B-NS3 proteases: Synthesis, biological evaluation and docking studies of novel peptide-hybrids. Bioorganic Med Chem. 2015;23:5748–5755. doi: 10.1016/j.bmc.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 56.Lin K.H., Nalivaika E.A., Prachanronarong K.L., Yilmaz N.K., Schiffer C.A. Dengue Protease Substrate Recognition: Binding of the Prime Side. ACS Infect Dis. 2016;2:734–743. doi: 10.1021/acsinfecdis.6b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin K.-H., Ali A., Rusere L., Soumana D.I., Kurt Yilmaz N., Schiffer C.A. Dengue Virus NS2B/NS3 Protease Inhibitors Exploiting the Prime Side. J Virol. 2017;91:1–10. doi: 10.1128/jvi.00045-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qian Z., Liu T., Liu Y.Y., et al. Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem Biol. 2013;8:423–431. doi: 10.1021/cb3005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qian Z., Larochelle J.R., Jiang B., et al. Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry. 2014;53:4034–4046. doi: 10.1021/bi5004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takagi Y., Matsui K., Nobori H., et al. Discovery of novel cyclic peptide inhibitors of dengue virus NS2B-NS3 protease with antiviral activity. Bioorganic Med Chem Lett. 2017;27:3586–3590. doi: 10.1016/j.bmcl.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 61.De La Cruz L., Nguyen T.H.D., Ozawa K., et al. Binding of low molecular weight inhibitors promotes large conformational changes in the dengue virus ns2b-ns3 protease: Fold analysis by pseudocontact shifts. J Am Chem Soc. 2011;133:19205–19215. doi: 10.1021/ja208435s. [DOI] [PubMed] [Google Scholar]
- 62.Johnston P.A., Phillips J., Shun T.Y., et al. HTS identifies novel and specific uncompetitive inhibitors of the two-component NS2B-NS3 proteinase of West Nile virus. Assay Drug Dev Technol. 2007;5:737–750. doi: 10.1089/adt.2007.101. [DOI] [PubMed] [Google Scholar]
- 63.Koh-Stenta X., Joy J., Wang S.F., et al. Identification of covalent active site inhibitors of dengue virus protease. Drug Des Devel Ther. 2015;9:6389–6399. doi: 10.2147/DDDT.S94207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kiat T.S., Pippen R., Yusof R., Ibrahim H., Khalid N., Rahman N.A. Inhibitory activity of cyclohexenyl chalcone derivatives and flavonoids of fingerroot, Boesenbergia rotunda (L.), towards dengue-2 virus NS3 protease. Bioorganic Med Chem Lett. 2006;16:3337–3340. doi: 10.1016/j.bmcl.2005.12.075. [DOI] [PubMed] [Google Scholar]
- 65.Yildiz M., Ghosh S., Bell J.A., Sherman W., Hardy J.A. Allosteric inhibition of the NS2B-NS3 protease from dengue virus. ACS Chem Biol. 2013;8:2744–2752. doi: 10.1021/cb400612h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mukhametov A., Newhouse E.I., Aziz N.A., Saito J.A., Alam M. Allosteric pocket of the dengue virus (serotype 2) NS2B/NS3 protease: In silico ligand screening and molecular dynamics studies of inhibition. J Mol Graph Model. 2014;52:103–113. doi: 10.1016/j.jmgm.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 67.De Sousa L.R.F., Wu H., Nebo L., et al. Flavonoids as noncompetitive inhibitors of Dengue virus NS2B-NS3 protease: Inhibition kinetics and docking studies. Bioorganic Med Chem. 2015;23:466–470. doi: 10.1016/j.bmc.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 68.Ghosh I, Talukdar P. Molecular docking and pharmacokinetics study for selected leaf phytochemicals from <i>Carica papaya<\i> Linn. against dengue virus protein, NS2B/NS3 protease. World Sci News. 2019;124:264–78.
- 69.Dwivedi V.D., Bharadwaj S., Afroz S., et al. Anti-dengue infectivity evaluation of bioflavonoid from Azadirachta indica by dengue virus serine protease inhibition. J Biomol Struct Dyn. 2020;1102 doi: 10.1080/07391102.2020.1734485. [DOI] [PubMed] [Google Scholar]
- 70.Farooq M.U., Munir B., Naeem S., et al. Exploration of Carica papaya bioactive compounds as potential inhibitors of dengue NS2B, NS3 and NS5 protease. Pak J Pharm Sci. 2020;33:355–360. doi: 10.36721/PJPS.2020.33.1.SUP.355-360.1. [DOI] [PubMed] [Google Scholar]
- 71.Lim W.Z., Cheng P.G., Abdulrahman A.Y., Teoh T.C. The identification of active compounds in Ganoderma lucidum var. antler extract inhibiting dengue virus serine protease and its computational studies. J Biomol Struct Dyn. 2020;38:4273–4288. doi: 10.1080/07391102.2019.1678523. [DOI] [PubMed] [Google Scholar]
- 72.Dwivedi V.D., Tripathi I.P., Mishra S.K. In silico evaluation of inhibitory potential of triterpenoids from azadirachta indica against therapeutic target of dengue virus, NS2B-NS3 protease. J Vector Borne Dis. 2016;53:156–161. [PubMed] [Google Scholar]
- 73.Chen S., Xu J., Liu C., et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat Commun. 2012;3:1–9. doi: 10.1038/ncomms1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.El-Mekkawy S., Meselhy M.R., Nakamura N., et al. Anti-HIV-1 and anti-HIV-1-protease substances from Ganoderma lucidum. Phytochemistry. 1998;49:1651–1657. doi: 10.1016/S0031-9422(98)00254-4. [DOI] [PubMed] [Google Scholar]
- 75.Bharadwaj S., Lee K.E., Dwivedi V.D., et al. Discovery of Ganoderma lucidum triterpenoids as potential inhibitors against Dengue virus NS2B-NS3 protease. Sci Rep. 2019;9:1–12. doi: 10.1038/s41598-019-55723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balasubramanian A., Manzano M., Teramoto T., Pilankatta R., Padmanabhan R. High-throughput screening for the identification of small-molecule inhibitors of the flaviviral protease. Antiviral Res. 2016;134:6–16. doi: 10.1016/j.antiviral.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anand P., Kunnumakkara A.B., Newman R.A., Aggarwal B.B. Bioavailability of curcumin: Problems and promises. Mol Pharm. 2007;4:807–818. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- 78.Wang Y.J., Pan M.H., Cheng A.L., et al. Stability of curcumin in buffer solutions and characterization of its degradation products. J Pharm Biomed Anal. 1997;15:1867–1876. doi: 10.1016/S0731-7085(96)02024-9. [DOI] [PubMed] [Google Scholar]
- 79.Balasubramanian A., Pilankatta R., Teramoto T., et al. Inhibition of dengue virus by curcuminoids. Antiviral Res. November 2018;2019:71–78. doi: 10.1016/j.antiviral.2018.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zamri A., Teruna H.Y., Rahmawati E.N., Frimayanti N., Ikhtiarudin I. Synthesis and in silico studies of a benzenesulfonyl curcumin analogue as a new anti dengue virus type 2 (DEN2) NS2B/NS3. Indones J Pharm. 2019;30:84–90. [Google Scholar]
- 81.Lee J.C., Chang F.R., Chen S.R., et al. Anti-dengue virus constituents from Formosan zoanthid Palythoa mutuki. Mar Drugs. 2016;14:1–10. doi: 10.3390/md14080151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tataringa G., Spac A., Sathyamurthy B., Zbancioc A.M. In silico studies on some dengue viral proteins with selected Allium cepa oil constituents from Romanian source. Farmacia. 2020;68:48–55. doi: 10.31925/farmacia.2020.1.8. [DOI] [Google Scholar]
- 83.Wu H., Bock S., Snitko M., et al. Novel dengue virus NS2B/NS3 protease inhibitors. Antimicrob Agents Chemother. 2015;59:1100–1109. doi: 10.1128/AAC.03543-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raut R., Beesetti H., Tyagi P., et al. A small molecule inhibitor of dengue virus type 2 protease inhibits the replication of all four dengue virus serotypes in cell culture. Virol J. 2015;12:1–7. doi: 10.1186/s12985-015-0248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li L., Basavannacharya C., Chan K.W.K., Shang L., Vasudevan S.G., Yin Z. Structure-guided discovery of a novel non-peptide inhibitor of dengue virus NS2B-NS3 protease. Chem Biol Drug Des. 2015;86:255–264. doi: 10.1111/cbdd.12500. [DOI] [PubMed] [Google Scholar]
- 86.Timiri A.K., Subasri S., Kesherwani M., et al. Synthesis and molecular modelling studies of novel sulphonamide derivatives as dengue virus 2 protease inhibitors. Bioorg Chem. 2015:74–82. doi: 10.1016/j.bioorg.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 87.Pelliccia S., Wu Y.H., Coluccia A., et al. Inhibition of dengue virus replication by novel inhibitors of RNA-dependent RNA polymerase and protease activities. J Enzyme Inhib Med Chem. 2017;32:1091–1101. doi: 10.1080/14756366.2017.1355791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hariono M., Choi S.B., Roslim R.F., et al. Thioguanine-based DENV-2 NS2B/NS3 protease inhibitors: Virtual screening, synthesis, biological evaluation and molecular modelling. PLoS ONE. 2019;14:1–21. doi: 10.1371/journal.pone.0210869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bhowmick S., Alissa S.A., Wabaidur S.M., Chikhale R.V., Islam M.A. Structure-guided screening of chemical database to identify NS3-NS2B inhibitors for effective therapeutic application in dengue infection. J Mol Recognit. 2020;33:1–18. doi: 10.1002/jmr.2838. [DOI] [PubMed] [Google Scholar]
- 90.Gan C.S., Lee Y.K., Heh C.H., Rahman N.A., Yusof R., Othman S. The synthetic molecules YK51 and YK73 attenuate replication of dengue virus serotype 2. Trop Biomed. 2017;34:270–283. [PubMed] [Google Scholar]
- 91.El-Subbagh H.I., Abu-Zaid S.M., Mahran M.A., Badria F.A., Al-Obaid A.M. Synthesis and biological evaluation of certain α, β-unsaturated ketones and their corresponding fused pyridines as antiviral and cytotoxic agents. J Med Chem. 2000;43:2915–2921. doi: 10.1021/jm000038m. [DOI] [PubMed] [Google Scholar]
- 92.Cheng D., Valente S., Castellano S., et al. Novel 3,5-bis(bromohydroxybenzylidene)piperidin-4-ones as coactivator-associated arginine methyltransferase 1 inhibitors: Enzyme selectivity and cellular activity. J Med Chem. 2011;54:4928–4932. doi: 10.1021/jm200453n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kálai T., Kuppusamy M.L., Balog M., et al. Synthesis of N-substituted 3,5-bis(arylidene)-4-piperidones with high antitumor and antioxidant activity. J Med Chem. 2011;54:5414–5421. doi: 10.1021/jm200353f. [DOI] [PubMed] [Google Scholar]
- 94.Basiri A., Xiao M., McCarthy A., Dutta D., Byrareddy S.N., Conda-Sheridan M. Design and synthesis of new piperidone grafted acetylcholinesterase inhibitors. Bioorganic Med Chem Lett. 2017;27:228–231. doi: 10.1016/j.bmcl.2016.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frimayanti N., Chee C.F., Zain S.M., Rahman N.A. Design of new competitive dengue Ns2b/Ns3 protease inhibitors-a computational approach. Int J Mol Sci. 2011;12:1089–1100. doi: 10.3390/ijms12021089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Osman H., Idris N.H., Kamarulzaman E.E., Wahab H.A., Hassan M.Z. 3,5-Bis(arylidene)-4-piperidones as potential dengue protease inhibitors. Acta Pharm Sin B. 2017;7:479–484. doi: 10.1016/j.apsb.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Padmapriya P., Gracy Fathima S., Ramanathan G., et al. Development of antiviral inhibitor against dengue 2 targeting Ns3 protein: In vitro and in silico significant studies. Acta Trop. 2018;188:1–8. doi: 10.1016/j.actatropica.2018.08.022. [DOI] [PubMed] [Google Scholar]
- 98.Kühl N., Graf D., Bock J., Behnam M.A.M., Leuthold M.M., Klein C.D. A New Class of Dengue and West Nile Virus Protease Inhibitors with Submicromolar Activity in Reporter Gene DENV-2 Protease and Viral Replication Assays. J Med Chem. 2020;63:8179–8197. doi: 10.1021/acs.jmedchem.0c00413. [DOI] [PubMed] [Google Scholar]
- 99.Zhou G.C., Weng Z., Shao X., et al. Discovery and SAR studies of methionine-proline anilides as dengue virus NS2B-NS3 protease inhibitors. Bioorganic Med Chem Lett. 2013;23:6549–6554. doi: 10.1016/j.bmcl.2013.10.071. [DOI] [PubMed] [Google Scholar]
- 100.Weng Z., Shao X., Graf D., et al. Identification of fused bicyclic derivatives of pyrrolidine and imidazolidinone as dengue virus-2 NS2B-NS3 protease inhibitors. Eur J Med Chem. December 2015;2017:751–759. doi: 10.1016/j.ejmech.2016.09.063. [DOI] [PubMed] [Google Scholar]
- 101.Knehans T., Schüller A., Doan D.N., et al. Structure-guided fragment-based in silico drug design of dengue protease inhibitors. J Comput Aided Mol Des. 2011;25:263–274. doi: 10.1007/s10822-011-9418-0. [DOI] [PubMed] [Google Scholar]
- 102.Beyler S.A., Zaneveld L.J.D. Antifertility activity of systemically administered proteinase (acrosin) inhibitors. Contraception. 1982;26:137–146. doi: 10.1016/0010-7824(82)90082-8. [DOI] [PubMed] [Google Scholar]
- 103.Bartolini M., Cavrini V., Andrisano V. Characterization of reversible and pseudo-irreversible acetylcholinesterase inhibitors by means of an immobilized enzyme reactor. J Chromatogr A. 2007;1144(1):102–110. doi: 10.1016/j.chroma.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 104.Sundermann T.R., Benzin C.V., Dražić T., Klein C.D. Synthesis and structure-activity relationships of small-molecular di-basic esters, amides and carbamates as flaviviral protease inhibitors. Eur J Med Chem. 2019;176:187–194. doi: 10.1016/j.ejmech.2019.05.025. [DOI] [PubMed] [Google Scholar]
- 105.Chu J.J.H., Lee R.C.H., Ang M.J.Y., et al. Antiviral activities of 15 dengue NS2B-NS3 protease inhibitors using a human cell-based viral quantification assay. Antiviral Res. 2015;118:68–74. doi: 10.1016/j.antiviral.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 106.Bhakat S., Delang L., Kaptein S., Neyts J., Leyssen P., Jayaprakash V. Reaching beyond HIV/HCV: Nelfinavir as a potential starting point for broad-spectrum protease inhibitors against dengue and chikungunya virus. RSC Adv. 2015;5:85938–85949. doi: 10.1039/c5ra14469h. [DOI] [Google Scholar]
- 107.AN, Ming; CHANG, Zhen; GAO, Lei; LI, Mei; CAO G. Determination of Four Related Substances in Policresulen Solution by HPLC. Chinese J Pharm Anal. 2004;24:536–37.
- 108.Wu D.W., Mao F., Ye Y., et al. Policresulen, a novel NS2B/NS3 protease inhibitor, effectively inhibits the replication of DENV2 virus in BHK-21 cells. Acta Pharmacol Sin. 2015;36:1126–1136. doi: 10.1038/aps.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li Z., Brecher M., Deng Y.Q., et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017;27(8):1046–1064. doi: 10.1038/cr.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li Z, Zhang J, Li H. Flavivirus NS2B/NS3 Protease: Structure, Function, and Inhibition. Elsevier Inc.; 2017. doi:10.1016/B978-0-12-809712-0.00007-1.
- 111.Li Z., Sakamuru S., Huang R., et al. Erythrosin B is a potent and broad-spectrum orthosteric inhibitor of the flavivirus NS2B-NS3 protease. Antiviral Res. December 2017;2018:217–225. doi: 10.1016/j.antiviral.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Viswanathan U., Tomlinson S.M., Fonner J.M., Mock S.A., Watowich S.J. Identification of a Novel Inhibitor of Dengue Virus Protease through Use of a Virtual Screening Drug Discovery Web Portal. J Chem Inf Model. 2014;54:2816–2825. doi: 10.1021/ci500531r. [DOI] [PubMed] [Google Scholar]