

Abstract
The vertebrate inner ear is a labyrinthine sensory organ responsible for perceiving sound and body motion. While a great deal of research has been invested in understanding the auditory system, a growing body of work has begun to delineate the complex developmental program behind the apparatuses of the inner ear involved with vestibular function. These animal studies have helped identify genes involved in inner ear development and model syndromes known to include vestibular dysfunction, paving the way for generating treatments for people suffering from these disorders. This review will provide an overview of known inner ear anatomy and function and summarize the exciting discoveries behind inner ear development and the evolution of its vestibular apparatuses.
Keywords: balance, inner ear, otocysts, otoliths, saccule, semi‐circular canals, signaling pathways, utricle, vertigo, vestibular apparatus, vestibular system, vestibulocochlear nerve
The vertebrate inner ear is an important sensory organ for perceiving gravity and body motion. The aim of this review is to provide an overview of known inner ear anatomy and vestibular function and summarize the exciting discoveries achieved over the years through various animal studies to better understand inner ear development and the evolution of its vestibular apparatuses.

1. INTRODUCTION
Our sensory organs provide us with a way to interact with the world by detecting chemical or physical stimuli, either internally or within our environment, and transducing these signals along a neural circuit to the brain for processing and response determination (Straka, 2020; Straka et al., 2016). The inner ear is one of the most fascinating sensory organs because it is responsible for two key systems of perception: the auditory system for perceiving sound and the vestibular system for sensing balance and spatial orientation (Brister et al., 2020; Straka, 2020). Research into both systems is essential for understanding the diseases that impact inner ear function, but as 1–2 of every 1,000 children born in the United States suffer from a hearing impairment and roughly 2 billion older adults suffer from hearing loss, this has attracted a greater deal of research into the auditory system (Centers for Disease Control & Prevention, 2017; Hoffman et al., 2017; Schilder et al., 2019; Yamoah et al., 2020). However, the necessity for research into the vestibular system remains, given that approximately 3.3 million (5.2%) children between the ages of 2 and 17 in the United States suffer from some form of vestibular dysfunction, as characterized by delayed motor development or chronic dizziness and balance issues (Li et al., 2016). Head and neck injuries resulting in lesions to the inner ear and its internal circuit to the brain and congenital disorders whose symptoms do not present until later in life can also result in vestibular dysfunction, usually in the form of bouts of vertigo, the illusion of spinning or tilting, either of the environment or oneself (Gurr & Moffat, 2001; Kolev & Sergeeva, 2016; Yamoah et al., 2020). This illusory perception of motion confers physical risk, although sensory impairments associated with the inner ear also have an impact on mental health. Notably, disabilities related to inner ear dysfunction, particularly those that are acute, also contribute to a higher incidence of anxiety and depression (Yuan et al., 2015). Therefore, to combat health‐related issues caused by vestibular disorders, research into the development of the inner ear is essential for treating patients suffering from inner ear dysfunction and identifying preventative methods for birth defects associated with the vestibular system.
A growing body of work invested in comprehending the complex developmental program behind the vestibular apparatuses of the inner ear has provided an extensive foundation for understanding diseases involved in this system (Fattal et al., 2018; Toriello & Smith, 2013). In fact, recent research in animal models has already helped to identify the genetic factors behind some disorders that include vestibular dysfunction, such as Branchio‐oto‐renal syndrome, Pendred syndrome, Jervell and Lange‐Nielsen syndrome, Delpire‐McNeill syndrome, and Usher syndrome (Table 1; Ceruti et al., 2002; Kulkarni et al., 2012; McNeill et al., 2020; Stinckens et al., 2001; Weil et al., 1996). Therefore, the purpose of this review is to give an overview of what is known about inner ear anatomy and function and to summarize the exciting research that has already been conducted on the development and evolution of this intricate sensory organ.
TABLE 1.
Genes associated with vestibular dysfunction in human syndromes or disease phenotypes, as well as their phenotypes in animal disease models. OMIM reference numbers have been included where available
| Human gene name | Possible human behavioral or anatomical phenotypes | Animal model gene name and disease phenotypes | Citations |
|---|---|---|---|
| CADHERIN 23; CDH23 (605516) |
|
|
|
| CHLORIDE INTRACELLULAR CHANNEL 5; CLIC5 (607293) |
|
|
|
| CHROMODOMAIN HELICASE DNA‐BINDING PROTEIN 7; CHD7 (608892) |
|
|
|
| COAGULATION FACTOR C HOMOLOGY; COCH (603196) |
|
|
|
| ELONGATION FACTOR Tu GTP‐BINDING DOMAIN‐CONTAINING 2; EFTUD2 (603892) |
|
|
|
| EPHRIN RECEPTOR EPHA2; EPHA2 (176946) |
|
|
|
| EPHRIN RECEPTOR EPHB2; EPHB2 (600997) |
|
|
|
| EPITHELIAL SPLICING REGULATORY PROTEIN 1; ESRP1 (612959) |
|
|
|
| EYA TRANSCRIPTION COACTIVATOR AND PHOSPHATASE 1; EYA1 (601653) |
|
|
|
| FIBROBLAST GROWTH FACTOR 3; FGF3 (164950) |
|
|
|
| FIBROBLAST GROWTH FACTOR RECEPTOR 2; FGFR2 (176943) |
|
|
|
|
GAP JUNCTION PROTEIN, BETA‐2; GJB2 (121011) |
|
|
|
| GAP JUNCTION PROTEIN, BETA‐6; GJB6 (604418) |
|
|
|
| H6 FAMILY HOMEOBOX 2; HMX2 (600647) |
|
|
|
| H6 FAMILY HOMEOBOX 3; HMX3 (613380) |
|
|
|
| HOMEOBOX A1; HOXA1 (142955) |
|
|
|
| HOMEOBOX A2; HOXA2 (604685) |
|
|
|
| JAGGED 1; JAG1 (601920) |
|
|
|
|
LIM HOMEOBOX TRANSCRIPTION FACTOR 1, ALPHA; LMX1A (600298) |
|
|
|
| LOW DENSITY LIPOPROTEIN RECEPTOR‐RELATED PROTEIN 2; LRP2 (600073) |
|
|
|
| LYSINE‐SPECIFIC METHYLTRANSFERASE 2D; KMT2D (602113) |
|
|
|
| MYOSIN, HEAVY CHAIN 9, NONMUSCLE; MYH9 (160775) |
|
||
| MYOSIN VIIA; MYO7A (276903) |
|
|
|
| OTOGELIN; OTOG (604487) |
|
|
|
| PAIRED BOX 2; PAX2 (167409) |
|
|
|
| POTASSIUM CHANNEL, VOLTAGE‐GATED, KQT‐LIKE SUBUNIT SUBFAMILY, MEMBER 1; KCNQ1 (607542) |
|
|
|
| POU DOMAIN, CLASS 3, TRANSCRIPTION FACTOR 4; POU3F4 (300039) |
|
|
|
| PROTOCADHERIN 15; PCDH15 |
|
|
|
| RIBOSOMAL PROTEIN S26; RPS26 (603701) |
|
|
|
| SEMAPHORIN 3E; SEMA3E (608166) |
|
|
|
| SIX HOMEOBOX 1; SIX1 (601205) |
|
|
|
| SMALL NUCLEAR RIBONUCLEOPROTEIN POLYPEPTIDES B AND B1; SNRPB (182282) |
|
|
|
| SOLUTE CARRIER FAMILY 26, MEMBER 4; SLC26A4 (605646) |
|
|
|
| SOLUTE CARRIER FAMILY 12, MEMBER 2; SLC12A2 (600840) |
|
|
|
| SORTING NEXIN 10; SNX10 (614780) |
|
|
|
| SRY‐BOX 10; SOX10 (602229) |
|
||
|
TRANSCRIPTION FACTOR AP2‐ALPHA; TFAP2A (107580) |
|
|
|
| USH1 PROTEIN NETWORK COMPONENT HARMONIN; USH1C (605242) |
|
|
2. ANATOMY AND FUNCTION OF THE INNER EAR VESTIBULAR SYSTEM
The inner ear is a complex, labyrinthine structure situated within the temporal bone on each side of the head (Figure 1; Fekete & Wu, 2002a; Fritzsch et al., 2002). It is composed of a central vestibule connected to the cochlea on one side, which houses the auditory machinery of the ear, and three orthogonally situated semi‐circular canals on the other. A series of mosaic patches—composed of mechano‐receptive hair cells and their support cells housed within the central vestibule and the semi‐circular canals—represent the vestibular sensory organs of the ear.
FIGURE 1.

Schematic illustration of the adult human inner ear, with an emphasis on the vestibular apparatuses (not to scale)
The first of these vestibular organs, the semi‐circular canals, each terminate in a sac that houses an epithelium composed of hair cells and support cells that are sheathed by a gelatinous mass (Mescher, 2010). This gelatinous mass, known as cupula, extends from each sensory epithelia to the ampulla, a bulge situated directly across from an epithelium within a given canal. The hair cells within this epithelium are covered by stereocilia, mechanoreceptors that, when deflected, trigger the release of a chemical transmitter to the nerve terminals synapsed to their base (Barrett et al., 2012; Elliott et al., 2018). The deflection of these stereociliary bundles in the semi‐circular canals is triggered in a unique way during angular, or "rotational,” accelerations of the head. When the head turns, the inner ear moves with it, but the endolymph, a potassium‐rich fluid contained within the ear, remains inert. Because this fluid remains stationary, it exerts force against the cupula, deflecting the stereocilia within to sense the speed of rotation relative to the axis of the canal.
The second group of vestibular organs is known as the maculae, two sensory epithelia positioned such that one, the saccule, is oriented vertically within the vestibule, while the other, the utricle, is oriented horizontally (Chagnaud et al., 2017; Purves et al., 2001). As such, they detect motion in the vertical and horizontal planes, respectively, although their anatomy and function differ from that of the semi‐circular canals. Instead of detecting angular rotations, these sensory organs are responsible for detecting linear accelerations and the position of the head relative to gravity. Additionally, although the hair cells within the maculae are similarly covered by a heavy, gelatinous mass, it is embedded with small, calcium carbonate crystals known as otoliths or otoconia. This otolithic membrane moves according to the direction of gravity, such that when the head is tilted, the membrane shifts, deflecting the stereocilia of the underlying hair cells (Hain & Helminski, 2007).
Another important, although non‐sensory, component of the vestibular system is the endolymphatic duct, which extends dorsally from the utricular and saccular ducts housed within the central vestibule and through the vestibular aqueduct to connect with the endolymphatic sac, which is partially contained between the bony labyrinth surrounding the ear and the posterior cranial fossa (Kopecky et al., 2013; Lo et al., 1997). While endolymph is produced primarily by the dark cells of the vestibular labyrinth and stria vascularis of the cochlea, this endolymphatic duct and sac system is primarily responsible for regulating inner ear pressure by either decreasing it through the resorption of endolymph or by increasing it through the production of proteoglycans to attract and bind water (Kingma & van de Berg, 2016; Wackym et al., 1987).
3. OVERVIEW OF DEVELOPMENT OF THE VESTIBULAR STRUCTURES OF INNER EAR
While vestibular disorders can arise from physical trauma to the head or neck (Kolev & Sergeeva, 2016), they can also occur from the improper development of the vestibular structures previously described. As such, understanding the developmental program of the inner ear and identifying the genes involved in this process can be informative for elucidating the mechanisms of inner ear dysfunction, whether they appear at birth or manifest later in life (Fattal et al., 2018). This in turn allows for the identification of potential targets for gene therapy and the development of chemical therapeutic agents for preventative strategies and treatment of vestibular defects.
The aim of the following sections is to discuss some of the most important discoveries already made in animal models concerning the developmental program of the inner ear. It will outline the first step of inner ear development, the induction of the otic placode, which is the source of all inner ear cell types, and the subsequent formation of the otic vesicle, a closed‐off portion of the neural ectoderm that is subjected to signaling cues that pattern, or confer an identity on, the various otic cell types that will eventually arise in this sensory organ (Figure 2). These sections will also provide a summary description of how the vestibular structures are thereby formed, highlighting genes that have also been identified in human patients suffering from vestibular disorders, some of which can be found in Table 1.
FIGURE 2.
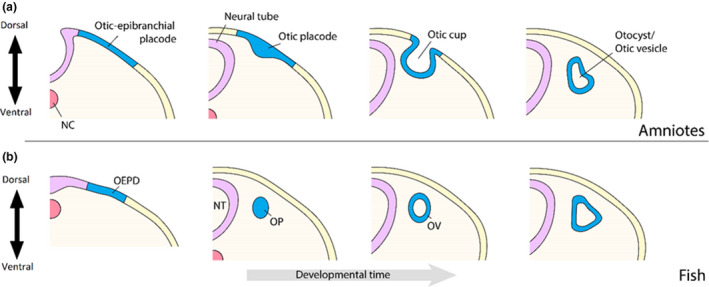
Early inner ear development. The inner ear is formed within the pre‐placodal region of the ectoderm that is first segregated into the otic/epibranchial precursor domain. A portion of this domain will then give rise to the otic placode, a thickening of the endoderm that invaginates in amniotes (a) or cavitates in fish (b) to form the otocyst/otic vesicle. This vesicle will be subjected to signaling cues that confer various otic identities on the vesicle and shape it into a labyrinth structure that houses both vestibular and auditory apparatuses
4. THE OTIC PLACODE AND FORMATION OF THE OTIC VESICLE
Important discoveries have already been made concerning the developmental plan of the inner ear leading up to the induction of an ectodermal thickening known as the otic placode. Studies in zebrafish have demonstrated that bone morphogenetic protein (BMP) signaling in the late blastula and early gastrula stages of development establish the competence of the pre‐placodal region (PPR) within the ectoderm at the neural plate border where the otic placode originates by inducing the expression of the inner ear marker foxi1 and other transcription factors (Figure 2; Kwon et al., 2010). It is also known that the PPR does not expand from the head into the embryonic trunk due to canonical WNT signaling, which instead promotes neural crest formation posterior to this placodal region (Litsiou et al., 2005). Subsequently, PPR specification is co‐operatively achieved by the dorsal expression of fibroblast growth factors (FGF) and platelet‐derived growth factors (PDGF), which act as BMP antagonists. Studies using the aquatic frog Xenopus have unveiled another key player for PPR specification, the retinoic acid (RA) signaling pathway, which both restricts the posterior domain of fgf8 expression in the PPR and induces the expression of the posteriorly restricted PPR genes tbx1 and ripply3 (Arima et al., 2005; Janesick et al, 2012; Shiotsugu et al., 2004).
Following PPR induction, this region is segregated into several domains fated for the placodal development of different sensory tissues, such as the otic/epibranchial precursor domain that gives rise to both the otic and epibranchial placodes (Figure 2; Freter et al., 2008; Ohyama et al., 2006; Schlosser & Ahrens, 2004). This entire domain is defined in zebrafish by the ectodermal expression of the Pax transcription factors, pax2a and pax8, although the subsequent gradient expression of pax2a within this region during somitogenesis further defines the individual placodal fates in zebrafish alone; lower levels favor an epibranchial fate, while higher levels direct otic fate as promoted by canonical WNT signaling (McCarroll et al., 2012; Ohyama et al., 2006). However, in Xenopus and chick, otic placodal fate relies instead on the expression of Gbx2 (Steventon et al., 2012). Interestingly, Pax2/8 double null mutant mice are able to develop an otocyst, although further morphogenesis of the inner ear does not proceed and sensorineural development is severely impaired (Bouchard et al., 2010). It is also important to note that while various cell fates are not specified until later patterning of the ear, studies in zebrafish demonstrate that foxi1, which enables the expression of pax8 at this time, provides otic cells with the competence to embark on a neuronal fate, and dlx3b/4b, which subsequently enable pax2a activation, then promote sensory fate while restricting neuronal fate (Hans et al., 2013; Hans et al., 2004). Other factors implicated in otic induction that are highly conserved across vertebrates include Fgf3 and Fgf8, with additional FGFs participating in this process in a species‐dependent manner (Alvarez et al., 2003; Freter et al., 2008; Ladher et al., 2005; Léger & Brand, 2002; Liu et al., 2003; Mansour et al., 1993; Maroon et al., 2002; Martin & Groves, 2006; Maulding et al., 2014; Padanad et al., 2012; Park & Saint‐Jeannet, 2008; Phillips et al., 2001; Wright & Mansour, 2003). While the loss of activity from any one FGF gene does not appear to prevent otic induction, impairing at least two members of this family of signaling molecules will result in a complete loss or deficiency of otic tissues.
Once the otic placode is induced, it invaginates into an otic cup in amniotes (Figure 2a) or cavitates in fish (Figure 2b) to then form the otic vesicle/otocyst (Figure 2; Kaufman, 1992; Maroon et al., 2002; Schlosser & Northcutt, 2000). While the molecular mechanisms behind its cavitation in fish are yet unknown, it is understood that the invagination of this vesicle in amniotes first requires an expansion at the basal aspect of the otic cells, followed by an apical constriction (Sai et al., 2014). Both stages are driven by the activation of myosin‐II, although it triggers the depolymerization of actin basally, in response to localized FGF signaling, and actomyosin contraction apically, which is a RhoA‐dependent process (Sai & Ladher, 2008; Sai et al., 2014).
5. AXIAL PATTERNING OF INNER EAR
During and after vesicle formation, otic cells will take on a neural, sensory, or non‐sensory cell fate. Cells destined to take on a neural or sensory fate originate in the neural–sensory‐competent domain (NSD) that originates within the anterior region of the developing vesicle (Fekete & Wu, 2002b). Cells within this region that express Neurog1, a key basic helix–loop–helix (bHLH) transcription factor in neuronal determination, are specified to become neuroblasts, which delaminate from the vesicle ventrally, form a transient amplifying population, and give rise to the afferent neurons of the vestibulocochlear (VIII) cranial ganglion, whereas cells that instead express Neurod1 are later fated to give rise to hair cells (Ma et al., 1999; Matei et al., 2005; Sapède et al., 2012). The posterior region of the otic vesicle gives rise to predominantly non‐sensory tissues, although the posterior crista is eventually situated within this area. When considering how the anteroposterior asymmetry of the ear is determined and differential cell fates are directed along this axis, the RA and FGF signaling pathways have been identified as key factors (Bok et al., 2011; Maier & Whitfield, 2014). Studies in zebrafish and chick show that the expression of fgf3 in the hindbrain confers an anterior identity within the otic placode, while RA signaling, induced by the expression of aldh1a3 in the head mesenchyme posteroventral to the placode, promotes the posterior expression of tbx1, a negative regulator of neurogenesis within the ear (Figure 3a; Bok et al., 2011; Maier & Whitfield, 2014; Radosevic, 2011; Radosevic et al., 2011; Raft et al., 2004).
FIGURE 3.
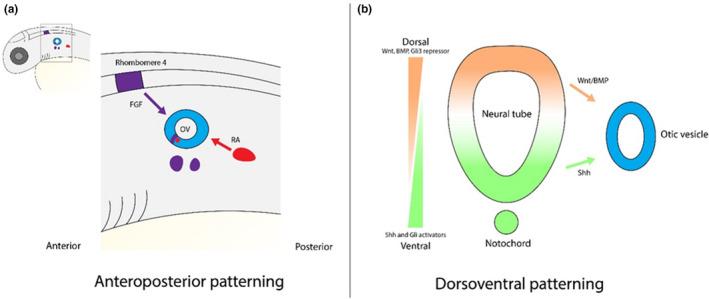
Axial patterning regulators of the inner ear. (a) The fibroblast growth factor (FGF) and retinoic acid (RA) signaling pathways are responsible for conferring anteroposterior identities during early inner ear development. Initially, fgf3 from the hindbrain confers anterior identities within the placode while RA, metabolized by aldh1a3 in the head mesenchyme posteroventral to the placode, promotes posterior identities. Once the otic vesicle forms, it continues to be subjected to FGF and RA signaling, particularly within the neuro‐sensory domain of the vesicle; at this time, FGFs are also expressed ventral to the ear. (b) Dorsoventral patterning of the ear is regulated by the Wnt, Bone morphogenetic protein (BMP), and sonic hedgehog (SHH) signaling pathways. SHH, secreted from the floor plate of the hindbrain and the notochord, is mediated by opposing gradients of the Gli3 repressor within the dorsal epithelium of the otic vesicle and Gli2/3 activators within the ventral domain. Wnt signaling is driven by the secretion of ligands from the dorsal hindbrain, whereas BMP signaling is driven by the secretion of ligands from the dorsal hindbrain and within the otic epithelium. Both Wnt and BMP have been implicated in restricting SHH signaling to the ventromedial region of the ear; ultimately, SHH acts as a ventralizing agent within the ear while WNT and BMP act as dorsalizing agents
Once the otic vesicle is formed, FGF and RA signaling continue to maintain anteroposterior patterning of the ear (Maier & Whitfield, 2014). Both within the anterior aspect of the vesicle and directly ventral to it, the expression of FGFs in zebrafish induces the expression of aldh1a3 within the posterior region of the NSD to create a localized source of RA signaling (Figure 3a). At this point, FGF and RA create a negative feedback loop that regulates the development of sensory hair cells. More precisely, high FGF levels of signaling in the anterior otic vesicle promote the expression of otx1b, which restricts neurogenesis and promotes the development of structural cells, with lower levels allowing for the development of sensory cells. Concurrently, RA signaling both restricts otx1b expression posteriorly to permit the development of sensory cells and instead promotes tbx1 expression to allow for the maturation of otic neuroblasts. However, the FGF and RA signaling pathways are not the only players in anteroposterior patterning and neurogenesis. Sonic hedgehog (SHH) signaling is also believed to be involved in this process, as murine Shh −/− mutants display reduced Neurog1 and Neurod1 expression, although the underlying mechanism by which this pathway regulates these genes is not yet well understood (Riccomagno et al., 2002).
Patterning of the inner ear is not limited to the anteroposterior axis. Sonic hedgehog (SHH), secreted from the floor plate and notochord, has been identified as a key regulator of dorsoventral identities, as mediated by opposing gradients of the Gli3 repressor within the dorsal domain of the otic epithelium and Gli2/3 activators ventrally (Figure 3b; Bok et al., 2007; Brown & Epstein, 2011). An absence of SHH signaling during inner ear development, as achieved in mouse embryos mutant in Smo, an essential transducer of this pathway, results in a wide range of morphological abnormalities later in otic development (Brown & Epstein, 2011). These abnormalities include missing ventral otic derivatives, such as the saccular macula and cochlear duct, and the malformation or absence of dorsal derivatives, such as the utricular macula, endolymphatic duct, and semi‐circular canals. Complimentary to these data, ectopic SHH signaling within and around the inner ear results in a complete absence of vestibular structures and a malformed cochlear duct (Riccomagno et al., 2002). Through these studies, it has been determined that SHH is required for specifying otic cells within the ventral region that would normally express the markers Otx1, Otx2, and Pax2, as Shh −/− mutants display reduced or absent expression of these genes. Ectopic SHH signaling has demonstrated that it also regulates cochlear formation through Brn4 and Tbx1 expression in the periotic mesenchyme, expanding the former ventrally and the latter dorsally, and through Pax2 expression in the otic epithelium, which is expanded from the ventromedial region to throughout the vesicle. However, SHH does not act alone in determining dorsoventral identities. The canonical WNT signaling pathway has also been implicated as a key player in specifying dorsal otic fates, with a loss of signaling during otic development resulting in missing vestibular structures, although due to the fact that some cells populating the cochlea are presumed to be exposed to WNT signals early on in otic development, a truncation of the cochlear duct is also observed (Riccomagno et al., 2005). As to how WNT specifics dorsal fates, this is accomplished through the secretion of the canonical WNT ligands Wnt1 and Wnt3a from the dorsal hindbrain (Figure 3b). In Wnt1 −/−; Wnt3a −/− mutants, there is a loss of Dlx5, Dlx6, and Gbx2 expression in the dorsal region. Conversely, increased WNT signaling, as achieved through lithium chloride treatment, results in the ventral expansion of these genes in wild‐type embryos and rescued expression in Wnt1 −/−; Wnt3a −/− mutants. When taking into consideration any possible cross talk between SHH and WNT at this stage of development, the observed dorsal expansion of the SHH effector Gli1 in Wnt1 −/−; Wnt3a −/− mutants suggests that WNT acts to restrict SHH signaling to the ventromedial region of the ear. However, the failed restriction of SHH signaling with lithium chloride treatment in these mutants implies that WNT does not act alone in this process.
More recently, the BMP pathway has been suggested to aid in dorsal specification, as seen with the loss of one or more semi‐circular canals with reduced signaling in chick embryos, as well as the restriction of SHH in the ear (Ohta & Schoenwolf, 2018; Ohta et al., 2016). BMP ligands expressed in the dorsal hindbrain and otic epithelium promote the expression of the dorsal markers Hmx3, through the non‐canonical activation of cAMP‐dependent Kinase A (PKA), and Dlx5, through the canonical Smad‐dependent pathway (Figure 3b; Ohta et al., 2016). Because the PKA phosphorylation of Gli results in the processing of Gli repressors, non‐canonical BMP signaling can negatively regulate SHH through the reduced expression of the SHH target gene Otx2 via the overexpression of the BMP ligand Bmp4. Additionally, loss of BMP signaling in the otic epithelium results in the expansion of both Otx2 and Pax2 expression. Together, this suggests that there is an intricate network of cross talk between these pathways, where SHH acts as a ventralizing agent that favors the development of the auditory structures of the ear while WNT and BMP act as dorsalizing agents that predominantly promote the development of the vestibular structures of the ear.
The final axis of the inner ear is the medial‐lateral, although little is known about how it is established. Research in chick embryos shows that some of the genes already discussed, such as Pax2 and Gbx2, have distinct dorsomedial expression patterns in the ear (Brigande et al., 2000; Choo et al., 2006). Interestingly, the Pax2 expression domain is thought to mark one side of the medial‐lateral boundary of the inner ear, on the other side of which we see the ventral‐lateral expression of SOHo (Brigande et al., 2000). In fact, the dorsal boundary between the expression pattern of these two genes marks the location of the endolymphatic duct’s outgrowth, and Pax2 −/− mutants display malformed endolymphatic ducts that are fused with the common crux, the branching point between the anterior and posterior semi‐circular canals, and which are often not properly restricted from the saccular compartment (Bouchard et al., 2010; Brigande et al., 2000; Burton et al., 2004). Gbx2 −/− mutants also display abnormalities of this channel, most commonly in the form of an absent endolymphatic duct (Choo et al., 2006). Less is known about how lateral structures of the ear are specified, but fate‐mapping studies in chick through the injection of fluorescent carbocyanine dyes into the cells of the rim of the otic cup suggest that the establishment of the medial‐lateral axis depends on the establishment of the other axes first (Brigande et al., 2000). This research suggests that while the otic vesicle/otocyst is forming, the lateral region of the otic vesicle that gives rise to the canals first originates in the dorsal region.
6. VESTIBULAR NEURONAL AND SENSORIAL DEVELOPMENT
While most genes involved in the specification of different cell fates appear once patterning of the otic vesicle is complete, neuronal specification occurs as early as the formation of the otic/epibranchial precursor domain, with the early expression of foxi1 imparting the competence for neuronal cell fate and dlx3b and dlx4b subsequently promoting sensory fate (Hans et al., 2004, 2013). In mouse embryos, once patterning of the otic vesicle is complete and the NSD is defined by FGF and RA signaling, it is within this region that we see the expression of essential neuronal and sensory markers. As previously described, Neurog1 and Neurod1 mark cells within the NSD are fated to become neuroblasts and hair cells, respectively, although mutations in either result in defects of both otic neurons and hair cells (Jahan, Pan, et al., 2010; Ma et al., 1999; Matei et al., 2005; Sapède et al., 2012). In particular, a loss of Neurog1 in mice results in an earlier hair cell cycle exit, resulting in the failed formation of sensory neurons and a truncated clonal expansion of in hair cell precursors, leading to the development of fewer hair cells overall (Matei et al., 2005). Loss of Neurod1 results in the formation of hair cells within the sensory ganglia of the inner ear, neuronal death, and the failed segregation of vestibular and cochlear afferents, leading to a projection of a single, mixed nerved to the cochlea (Jahan, Kersigo, et al., 2010; Jahan, Pan, et al., 2010; Macova et al., 2019). Other essential neuronal and sensory markers include Eya1, Six1, and Sox2 (Kiernan et al., 2005; Nichols et al., 2008; Xu et al., 1999; Zheng et al., 2003; Zou et al., 2008). Both EYA1 and SIX1 have been implicated in Branchio‐oto‐renal syndrome, which is usually characterized by hearing impairments and kidney and urinary tract malformations but can also include vestibular abnormalities, such as a truncated posterior semi‐circular canal or an enlarged endolymphatic sac and duct (Abdelhak et al., 1997; Ceruti et al., 2002; Melnick et al., 1976; Ruf et al., 2004). Studies in mice models have shown that loss of either otic Eya1 or Six1 results in the decreased expression of the sensorineural markers Neuorg1 and Neurod1 and that their co‐overexpression is capable of inducing neurogenesis in the ear (Ahmed, Xu, et al., 2012; Li, Zhang, et al., 2020). Sox2 has also been determined to be required for the expression of both Neurog1 and NeuroD1, engaging in an inhibitory feedback loop with these genes, and was initially assumed to control the number of neuroblasts that give rise to the afferent neurons of the VIII cranial ganglion (Evsen et al., 2013; Steevens et al., 2017). However, more recent work in mice utilizing the conditional deletion of Sox2 in otic tissues demonstrates that it does not appear to affect the initial delamination of neuroblasts, although the sensory epithelia of the inner ear ultimately fails to develop and early neurons perish (Dvorakova et al., 2020). It is possible that Sox2 functions in the developing ear by interacting with either Eya1 or Six1, as they have already been shown to co‐operatively promote the expression of other otic markers in cochlear cell cultures (Ahmed, Wong, et al., 2012). This includes Atoh1, an essential basic helix–loop–helix (bHLH) transcription factor associated with hair cell differentiation, which is normally suppressed by Neurod1 to prevent the premature or ectopic differentiation of hair cells (Chen et al., 2002; Jahan, Pan, et al., 2010; Kopecky et al., 2013; Pan et al., 2011). It is important to also note that Notch signaling plays an important role within the NSD for determining hair cell and to support cell fates (Pan et al., 2013; Petrovic et al., 2014). Initially, as directed through the Notch ligand Jag1, Notch‐mediated lateral induction induces Sox2 expression to specify the prosensory domain and prevent early hair cell differentiation. Once Atoh1 is expressed, it in turn induces Dl1, another Notch ligand. This creates a competition between Di1 and Jag1 for Notch, where the former favors the development of support cells and the latter the development of hair cells, creating a latticed pattern of the two cell types within this sensory domain.
Knowing that the NSD gives rise to the various sensory epithelia found in the mature inner ear, it is important to recognize that medial‐lateral patterning plays a role in how the neuroblasts in the pre‐segregated domain are determined to form ganglia of either the vestibular or auditory branches of the VIII cranial nerve. It has been discovered that otic cells that express Fgf3 within the anterior‐lateral region of this domain delaminate first and generate vestibular neurons (Bell et al., 2008; Koo et al., 2009). Meanwhile, cells that express Gata3 and Lmx1a within the medial region delaminate later and generate auditory neurons (Karis et al., 2001; Koo et al., 2009; Koundakjian et al., 2007; Lawoko‐Kerali et al., 2004). However, another possible player in the process is Myo7A, which encodes the unconventional myosin VIIA, a critical motor protein that in chick embryos is expressed in differentiating vestibular neurons before it is subsequently restricted to the vestibular afferents of the cristae (Nguyen et al., 2015). It is possible that Myo7A, therefore, acts in the innervation of these afferents or axonal migration and pathfinding, although most research into this gene has focused on the role it later plays in organizing the stereocilia bundles of vestibular and auditory hair cells and, therefore, its connection to Usher Syndrome type 1 in human patients (Ernest et al., 2000; Gibson et al., 1995; Nguyen et al., 2015; Nichols et al., 2008; Weil et al., 1996).
Further studies have demonstrated that NSD separation into the individual sensory epithelia throughout the ear requires restriction of Lmx1a expression to the non‐sensory in mice, with studies in Lmx1a/Lmx1b −/− mice demonstrating a severe disruption in auditory development that results in a complete loss of the organ of Corti, the sensory epithelium of the cochlea (Chizhikov et al., 2021; Nichols et al., 2008). As for the separation between more distinct regions of the initial sensory domain, Fgf10 appears to play an important role in the separation of the cristae, as mutants lack a posterior crista, only producing reduced and malformed anterior and lateral cristae (Pauley et al., 2003). Another player is Foxg1, whose null mutants often have fused anterior and lateral cristae and demonstrate severe abnormalities of both vestibular and auditory innervation (Hwang et al., 2009; Pauley et al., 2006). Otx1 also seems to play a role in cristae separation, as null mutants in mice and zebrafish lack a lateral crista, as well as a lateral canal (Fritzsch et al., 2001; Hammond & Whitfield, 2006; Morsli et al., 1999). However, these mutants also possess a fused utricle and saccule, which additionally implicates Otx1 in the proper segregation of the two maculae.
7. SEMI‐CIRCULAR CANAL DEVELOPMENT
The cells that give rise to the semi‐circular canals, which are responsible for detecting angular accelerations of the head, originate in a lateral region of the vesicle adjacent to where the presumptive cristae will form (Brigande et al., 2000; Chang, Brigande, et al., 2004). In fact, the cristae are likely responsible for inducing the formation of the canal ducts through the BMP and FGF signaling pathways, as Bmp4 in mice, chick, and zebrafish is essential for regulating Bmp2 and Dlx5, two key players in canal formation, and Fgf10‐/‐ mutants in chick lack all three canals (Chang, Brigande, et al., 2004; Chang et al., 2008; Hammond et al., 2009; Omata et al., 2007; Pauley et al., 2003; Wright & Mansour, 2003). Fgf3 has also been identified as another FGF involved in canal morphogenesis, as knockouts in mice occasionally lack a posterior canal (Mansour et al., 1993; Pauley et al., 2003).
Although there are morphological variations in how the canals are formed across vertebrates, the end result is three semi‐circular canals situated at right angles relative to one another. In zebrafish and Xenopus, we see an outgrowth of projections that bulge into the vesicle and fuse at a central point to form the pillars around which the three canals are formed (Figure 4a; Paterson, 1949; Waterman & Bell, 1984). In both of these species, glycosaminoglycan hyaluronan production is essential for these outgrowths, although other factors discovered in zebrafish have also been implicated in the formation of these projections, such as atropin2, atp1a1a.2, atp2b1a, cdh2, grhl2, fgf8, ncs1a, and otx1 (Asai et al., 2006; Babb‐Clendenon et al., 2006; Blasiole et al., 2006; Blasiole et al., 2005; Busch‐Nentwich et al., 2004; Cruz et al., 2009; Haddon & Lewis, 1991; Hammond & Whitfield, 2006; Han et al., 2011; Neuhauss et al., 1996). More recently, gpr126, an adhesion G protein‐coupled receptor gene expressed in the projections, has been identified as an important regulator of genes that encode extracellular matrix core proteins or are involved with modifying the extracellular matrix, such as chsy1, has3, hapln1a, hapln3, ugdh, and vcana (Geng et al., 2013). Changes to the extracellular matrix can understandably impact the proliferation or migration of cells within a given tissue, directly affecting tissue morphology, although they could contribute to signal transduction processes. Future research could, therefore, focus on determining which signaling pathways might direct or be directed by such changes to the extracellular matrix during semi‐circular canal morphogenesis.
FIGURE 4.
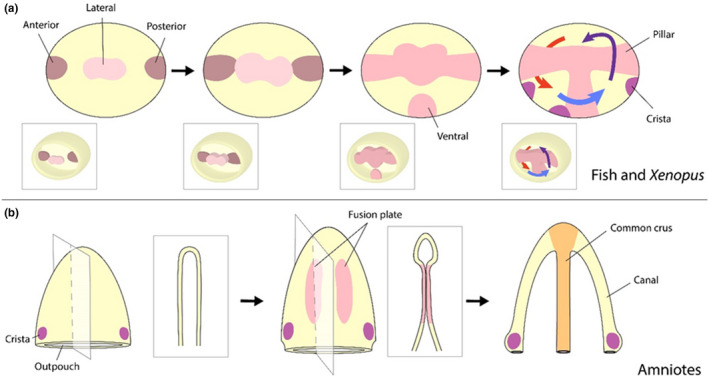
Semi‐circular canal morphogenesis. The cells that give rise to the canals originate in a lateral region adjacent to the presumptive cristae. However, there are variations between how these canals form in anamniotes, such as zebrafish and Xenopus, and amniotes, such as mouse and chick. (a) Canal formation in zebrafish and Xenopus begins with an outgrowth of projections that bulge inward into the otic vesicle. When these projections fuse, they become the pillars around which the canals are formed. (b) In amniotes, the canals are sculpted from two out‐pouches, one ventral (shown above) and one horizontal. Fusion plates are formed by opposing epithelium that extend toward one another. Following fusion, these plates are resorbed through apoptosis. In the above example, this process forms the anterior and posterior canals, with the common crux situated between them
In amniotes, morphogenesis of the semi‐circular canals begins with the formation of two out‐pouches, one vertical, situated dorsally, and one horizontal, which forms laterally around the middle of the otocyst (Bissonnette & Fekete, 1996; Fekete, 1999; Kopecky et al., 2012). The canonical WNT signaling pathway is believed to play an essential role leading up to this stage in promoting cell proliferation and repressing apoptosis, likely ensuring that a sufficient number of cells are available for the formation of these out‐pouches (Noda et al., 2012). With the ventral out‐pouch, opposing epithelial extend toward one another to form a fusion plate, which fuses and then is resorbed through apoptosis to create two canals along the rim, the anterior and posterior, which are connected centrally at the common crus (Figure 4b). The horizontal out‐pouch undergoes a similar process of fusion and resorption to give rise to the lateral canal. Despite the division between horizontal canal formation and that of the anterior and posterior canals in amniotes, Lmo4 in mouse embryos appears to be responsible for the outgrowth of both pouches by regulating cell proliferation and maintaining the expression of many players in canal morphogenesis, including Bmp4, Fgf10, Gata3, and Dlx5 (Deng et al., 2010). Here also the canonical WNT signaling pathway has been identified as an important factor behind semi‐circular morphogenesis for its role in inducing the expression of Netrin1, which induces fusion and resorption of the fusion plate by promoting periotic mesenchymal proliferation to push the opposing epithelial together and the detachment of the fusion plate from the basement membrane (Noda et al., 2012; Salminen et al., 2000).
8. PRODUCTION AND MAINTENANCE OF ENDOLYMPH
During its development, the otic vesicle is filled with endolymph, a fluid that is essential for sensory hair cell function and proper otolith or otoconia composition. With respect to hair cell function, the endolymph has a high concentration of potassium, which flows through the transducer channels of these cells with the deflection of their stereocilia and thereby triggers their depolarization (Battey, 2000; Bosher & Warren, 1968). Upon depolarization, the hair cells release neurotransmitters at their synapses with the afferent neurons of the vestibulocochlear nerve, the first step in triggering the perception of motion or sound. Some genes in human patients with auditory or vestibular impairments, such as those suffering from Jervell and Lang‐Nielsen syndrome and Delpire‐McNeill syndrome, have been implicated in controlling the composition or homeostasis of endolymph (Kulkarni et al., 2012; McNeill et al., 2020). Mutations in animal models often resulting in reduced endolymphatic volume and potential, commonly leading to a shrinkage or collapse of the membranous labyrinth and hair cell death (Blasiole et al., 2006; Casimiro et al., 2001; Cruz et al., 2009; Delpire et al., 1999; Dixon et al., 1999; Flagella et al., 1999). These genes include Slc12a2, which encodes the basal Na–K–1Cl co‐transporter that pumps potassium from the hair cells into the homologous epithelial “dark” cells surrounding the vestibular sensory epithelia, and both Kcnq1 and Kcne1, the subunits of KvLQT1, a channel on the apical side of dark cells responsible for returning potassium to the endolymph (Blasiole et al., 2006; Casimiro et al., 2001; Cruz et al., 2009; Delpire et al., 1999; Dixon et al., 1999; Flagella et al., 1999).
Dark cells, like the marginal cells of the cochlea, are also responsible for producing endolymph during otic development, although there appears to be some variation in the composition and function of endolymph‐producing cells surrounding the various sensory epithelia between species (Dohlman, 1965; Harada et al., 1989; Hommerich, 1990; Kimura et al., 1964; Villegas et al., 2001; Wangemann, 1995). However, one gene that has been found to be essential for the development and function of both dark and marginal cells is the estrogen‐related receptor beta, Nr3b2 (Chen & Nathans, 2007; Wangemann, 1995). In Nr3b2‐/‐ mouse mutants, these cells lack multiple ion channels and transporters and possess the characteristic narrowing of the membranous labyrinth of the semi‐circular canals normally found with improper endolymph composition or homeostasis (Chen & Nathans, 2007).
9. OTOCONIA (OTOLITH) FORMATION AND TETHERING
To enable the detection of gravity and linear accelerations, the maculae rely on a collection of calcium carbonite crystallites embedded in a gelatinous matrix of proteoglycans and proteins that overlay the hair cells (Ross & Pote, 1984). These bio‐mineral structures are called otoliths in fish and otoconia in most other vertebrates, and while there are structural differences between the two, there is a significant overlap in the genes responsible for otoconia and otolith formation. Some of these shared genes are involved in endolymph composition, which has a direct effect on the composition of these bio‐mineral structures, impacting their size and shape. These include the anion transporter Pendred, which is encoded by Slc26a4, and those encoding the enzyme carbonic anhydrase, which are together responsible for maintaining an alkaline endolymphatic pH, primarily through the production or transport of (Everett et al., 2001; Kido et al., 1991; Kim & Wangemann, 2010, 2011; Nakaya et al., 2007; Tsujikawa et al., 1993). Human patients with mutations in SLC26A4 suffer from the characteristic enlarged vestibular aqueduct associated with Pendred syndrome, which typically results in progressive hearing loss and episodic vertigo (Stinckens et al., 2001). Other genes encode proteins that are responsible for the mobilization of calcium, such as Otop1, which also participates in protein secretion, and Atp2b2, a calmodulin‐sensitive plasma membrane calcium‐ATPase that increases calcium concentration around the maculae (Blasiole et al., 2006; Hughes et al., 2007; Kim et al., 2010, 2011; Kozel et al., 1998). The loss of either of these genes in animal models results in the complete absence of otoliths or otoconia (Blasiole et al., 2006; Hughes et al., 2007; Kim & Wangemann, 2010, 2011; Kozel et al., 1998).
Other key players in otoconia or otolith formation are the proteins that make up the otolithic membrane, which are collectively referred to as otoconins, although there is some variation in the role each gene plays between species. These encode, for example: Oc90, which recruits calcium and other otoconial matrix components to facilitate crystal nucleation and growth; Otolin‐1, a collagen that interacts with Oc90 to act as a scaffold in the same processes; and keratan sulfate proteoglycans, which interact with both Oc90 and Otolin‐1, as well as other proteoglycans and collagens, to attract calcium (Deans et al., 2010; Killick & Richardson, 1997; Lu et al., 2010; Petko et al., 2008; Xu et al., 2010; Yang, Zhao, et al., 2011; Zhao et al., 2007). Null mutants in mice and zebrafish in these genes often result in lost or greatly reduced otoconia or otoliths (Deans et al., 2010; Killick & Richardson, 1997; Lu et al., 2010; Murayama et al., 2005; Petko et al., 2008; Xu et al., 2010; Yang, Zhao, et al., 2011; Zhao et al., 2007).
Another important step in otoconia or otolith formation is the initial tethering of these bio‐crystals to their underlying sensory epithelia, a task shared by various glycoproteins. Two such glycoproteins are otogelin, which is collagenous, and otogelin‐like, which is non‐collagenous, and which together are necessary for both the anchoring of the otolithic membrane to the maculae and the anchoring of the cupula to the crista of the semi‐circular canals (Bonnet et al., 2013; Cohen‐Salmon et al., 1997; Simmler, Cohen‐Salmon, et al., 2000; Simmler, Zwaenepoel, et al., 2000; Yariz et al., 2012). Other important factors for proper tethering include α‐Tectorin and β‐Tectorin, which each encode non‐collagenous glycoproteins that are expressed in the macular sensory epithelia (Legan et al, 1997; Rau et al., 1999). In mouse and zebrafish studies, null mutants of α‐Tectorin have scattered otoconia and reduced otolithic membrane, whereas β‐Tectorin have fewer or fused otoliths (Legan et al., 2000; Yang, Cheng, et al., 2011). Interestingly, analysis of the acellular membrane overlying the auditory sensory epithelium of the inner ear, the tectorial membrane, demonstrates that it also contains α‐Tectorin and β‐Tectorin (Goodyear & Richardson, 2002). Studies in mice demonstrate that a loss of α‐Tectorin results in a decoupling of the tectorial membrane from the underlying sensory epithelium, leading to impaired sound amplification and frequency‐dependent hearing loss (Legan et al., 2000). Oddly enough, Tectb −/− mice lead to an increased sharpness in cochlear frequency tuning, although there is a loss of sensitivity for frequencies below 20Hz, presumably from an enlargement of the tectorial membrane at the apical end of the cochlea (Russell et al., 2007).
10. OVERVIEW OF THE EVOLUTION OF THE VERTEBRATE INNER EAR
To understand the development of an organ, it is important to consider the changes that it has undergone over the course of its evolution. While we are still piecing together the evolutionary steps behind the development of the inner ear, the aim of this section is to touch upon some of the theories behind the origin of this sensory organ and how variations in its anatomy developed to suit the needs of different species.
11. EVOLUTION OF THE SPECIALIZED ENDORGANS OF THE INNER EAR
Although the “inner ear” is considered a solitary organ, it contains multiple apparatuses specialized in the detection of different stimuli. In general, the cochlea detects sound, the semi‐circular canals detect angular accelerations, and the maculae detect gravity and linear accelerations, raising the question of which apparatus came first. Up until the late 1800s, the inner ear was believed to serve a purely auditory function, with the semi‐circular canals and maculae supplementing the detection of sound by acting as a type of intensity coding system (Haeckel, 1877). However, lesions of the inner ear and later physiological observations revealed that the canals instead served a crucial purpose in detecting rotations of the head while the otoliths were involved with the perception of gravity (Breuer, 1873; Mach, 1873). Subsequently, it was discovered that while aquatic vertebrates possessed a structured labyrinth similar to that of land vertebrates, the anatomy of their inner ear did not immediately suggest the presence of a clearly analogous auditory apparatus to our own, which was the first indication that a specialized auditory system was a more recent acquisition (Retzius, 1881). While we now know that aquatic vertebrates are capable of detecting sound with their otolith organs, the evolution of a more intricate auditory system better capable of detecting airborne sounds would have been necessary for terrestrial tetrapods to avoid predators or track prey on land (Christensen, Christensen‐Dalsgaard, et al., 2015; Christensen, Lauridsen, et al., 2015; Christensen‐Dalsgaard et al., 2011; Clack, 2015; Lu & Xu, 2002; Lu et al., 2003, 2010).
Given its close proximity to the macular recess, the auditory sensory epithelium of the inner ear, known as the organ of Corti in mammals and the basilar papilla in amphibians, reptiles, and birds, is believed to have evolved from the otolith organs (Fritzsch & Wake, 1988). The evidence behind this theory is made apparent when considering the morphological similarities between the hair cells of each apparatus. Analyses of the sensory epithelia of the vestibular system reveal that two kinds of hair cells can be found in both the maculae and the ampullae, which are simply described as “type I” and “type II.” Type II hair cells occur in all vertebrates and are believed to be evolutionarily older, while type I hair cells are only found in birds, mammals, and reptiles (Wersall, 1956; Wersäll & Bagger‐Sjöbäck, 1974). Type I hair cells possess a flask‐shaped body, calyceal (“cup‐like”) afferent nerve endings that encompass the basolateral surface of one or more hair cells, and efferent synapses that contact the calyx rather than the hair cell itself; type II hair cells possess more cylindrical bodies, connect with multiple afferent bouton synapses, and contact their efferent synapses directly (Deans, 2013; Eatock et al., 1998; McInturff et al., 2018; Wersäll & Bagger‐Sjöbäck, 1974). Type II hair cells also have fewer stereocilia per bundle (Peterson et al., 1996; Wersäll & Bagger‐Sjöbäck, 1974). The resting potential of type I hair cells is also incredibly negative, and their primary current is a large and non‐activating K+ delayed inward rectifier, which allows these cells to produce faster voltage responses (Correia & Lang, 1990; Eatock & Songer, 2011; Meredith & Rennie, 2016; Rennie & Correia, 1994; Rusch & Eatock, 1996). The calyceal afferent synapses of type I cells also make these hair cells capable of much a faster, non‐quantal (“non‐vesicle”) form of transmissions that involve K+ accumulation in the synaptic cleft and ion channels in the synaptic membranes that face this cleft (Eatock, 2018). By comparison, type II hair cells have a more depolarized membrane potential and smaller activation range, although this is believed to increase their sensitivity (Correia et al., 1996). Both of these hair cells also possess ribbon synapses, a type of synapse that is unique to sensory tissues, differentiated from chemical synapses by a specialized organelle referred to as the “ribbon” that holds synaptic vesicles close to the active zone of the cell (Goldberg et al., 1990; Lysakowski & Goldberg, 1997; Matthews & Fuchs, 2010; Odermatt & Lagnado, 2009). This ribbon is thought to transport these vesicles to the site of fusion in an inconcurrent manner, allowing for the more sustained release of neurotransmitters (Odermatt & Lagnado, 2009).
The sensory epithelium of the auditory system is likewise composed of two types of hair cells: inner hair cells, which are the primary auditory receivers, and outer hair cells, which both mechanically amplify low‐intensity sound and are involved with sharpening frequency tuning curves (Aitkin, 1995; Ashmore, 1987, 2008; Dallos, 1996; Kössl & Vater, 1995; Ladhams & Pickles, 1996; Wever et al., 1971). The morphology of inner hair cells is that of a flask‐shaped body with a thick stereociliary bundle, like that of type I hair cells, while outer hair cells are more cylindrical and possess fewer stereocilia, like that of type II hair cells (Forge et al., 1991; Schwander et al., 2010; Slepecky, 1996). Both hair cells also possess ribbon synapses, although neither of these cell types possess the calyceal afferent synapses seen in the vestibular system (Francis et al., 2004; Khimich et al., 2005; Liberman, 1980; Liberman et al., 1990). As vertebrates transitioned from an aquatic to a terrestrial environment, the elongation of the limbs and neck allowed for a wider range and more abrupt motions of the head and body (Biewener, 1983; Ericsson et al., 2013; Kear et al., 2008; Young, 2013). However, this transition brought with it an increased risk of injury from falling. Although we still have much to learn about how calyceal synapses came to be, one of the driving forces behind their evolution solely within the vestibular system could be to provide faster reflexes in a terrestrial environment (Bierbower & Cooper, 2013; Korn & Faber, 2005).
Looking to the acellular membranes of the sensory epithelia of the inner ear, we can also glean something about the evolution of the various apparatuses of the inner ear. The tectorial membrane of the cochlea contains both α‐Tectorin and β‐Tectorin, which are utilized in the striolar region of the macula for otolith or otoconial tethering (Goodyear & Richardson, 2002). The otolithic membrane and tectorial membrane of the basilar papilla are also attached to their respective sensory epithelia by fibrous veils. These shared traits further support the notion that the auditory sensory epithelium evolved from the macular striola. However, an analysis of these acellular membranes also raises an interesting point about whether the macula or the ampulla predate one another. For example, one of the most prominent theories of otic evolution suggests that the inner ear originated from the lateral line system due to the close proximity of their placodes during development and the similar structure of the lateral line neuromasts to the ampullae that being of a group of hair cells embedded in a cupular membrane (Fritzsch et al., 1998; Popper et al., 1992; Wever, 1974). The cupular membrane of the inner ear is also less complex than that of the otolithic membrane, lacking either of the tectorins and being composed of randomly oriented filaments versus the densely and more aligned filaments of the otolithic membrane (Goodyear & Richardson, 2002). However, gravioreceptive sensors appear much earlier in history. Cnidarians, such as jellyfish, possess statocysts, small sensory vesicles that contain heavy crystal (“statolith”)‐bearing cells that perceive both the direction of gravity and water currents (Park et al., 2012; Schafer, 1878). The semi‐circular canals do not appear until much later with the arrival of the chordates, whose increasingly dynamic lifestyles required a more complicated gaze and posture stabilizing system (Higuchi et al., 2019; Mazan et al., 2000; Park et al., 2012). This, therefore, argues that the inner ear likely originated from a similar gravioreceptive sensor, from which additional, more specialized endorgans appeared to enable the refined detection of sound and motion (Duncan & Fritzsch, 2012; Wever, 1974).
Analysis of the sensory epithelia of the inner ear also provides us with clues about the development of its non‐sensory tissues (Fritzsch & Straka, 2014). For example, inductive signals from the sensory epithelium, such as Bmp4 and Fgf10, drive semi‐circular canal morphogenesis, demonstrating a reliance of non‐sensory development on inductive signals from sensory tissues and highlighting the fact that the evolution of the cristae predates that of canal formation (Chang, Brigande, et al., 2004; Chang et al., 2008; Pauley et al., 2003). In fact, no mutants bearing inner ear defects have been discovered in which the semi‐circular canals form normally when sensory development is perturbed (Anagnostopoulos, 2002; Chang, Cole, et al., 2004; Whitfield et al., 1996). Furthermore, it has even been demonstrated that the induction of hair cells is enough to generate vesicles in vitro, mirroring one of the initial steps of inner ear development (Koehler et al., 2013). Even within the sensory epithelium itself, the specification and organization of non‐sensory structures relies on the proper sensory development of their neighboring cells. For example, null mutant mice of Neurog1 demonstrate a substantial reduction in the number of hair cells in all sensory epithelia of the inner ear; meanwhile non‐sensory structures such as the cruciate eminence, a division that appears within the cristae of some species, are converted into hair cells (Harada, 1972, 1974; Ma et al., 2000; Matei et al., 2005). While there is still a great deal to learn concerning how otic sensory and non‐sensory tissues communicate with one another, the secret to how a simple vestibular organ developed into a more intricate labyrinth likely resides within its mechanosensory hair cells (Duncan & Fritzsch, 2012).
12. EVOLUTION OF THE SEMI‐CIRCULAR CANALS AND COMPLEX MOTOR BEHAVIORS
Although the labyrinthine nature of the inner ear is often considered a “vertebrate” invention, we now know the intricate structure of this sensory organ belongs to the chordates, as even early cyclostomata were found to have distinct semi‐circular canals (Wever, 1974). Therefore, by looking to the Agnatha, the sister group of jawed vertebrates, we can perhaps further understand how the inner ear more recently changed over the course of evolution to produce the characteristic three‐canal labyrinth of the gnathostomes and how these changes then enabled a more complex repertoire of motor behaviors.
Instead of the three orthogonally oriented canals found in gnathostomes, both ostracoderms, which are the extinct armored jawless fish of the Paleozoic, and lampreys possess only two canals, each terminating in an ampulla (Higuchi et al., 2019; Mazan et al., 2000). These canals are homologous to the anterior and posterior canals found in jawed vertebrates, suggesting that the lateral canal is a more recent acquisition. Hagfish, the only other modern representative of agnathans besides lampreys, instead possess one ring‐like semi‐circular canal predicted to have formed around a solitary epithelial projection, much in the same way the canal pillars form in zebrafish or Xenopus (Chagnaud et al., 2017; Higuchi et al., 2019; Paterson, 1949; Waterman & Bell, 1984). While this initially suggests that the one‐canal morphology is an ancestral trait, hagfish possess two ampullae located at opposite ends of their ring‐like canal. Following the “one ampulla per canal” theory, this instead proposes a two‐canal situation in our common ancestor, suggesting that a secondary fusion of these canals occurred in hagfish over the course of evolution (Goodrich, 1909). Even though we are uncertain about the true ancestral state, a recent comparative analysis between extant cyclostomes has reinforced what we know about the highly conserved molecular mechanisms behind inner ear development (Higuchi et al., 2019). For example, in lampreys, it has been discovered that Tbx1/10A is expressed in the posterior domain of the otic vesicle, where it plays a role in specifying the anteroposterior axis of the inner ear by suppressing genes involved in the neurogenesis and de‐epithelialization of vestibular ganglionic cells, much like its homolog in jawed vertebrates, TBX1 (Higuchi et al., 2019; Radosevic, 2011; Radosevic et al., 2011; Raft et al., 2004). Additionally, dorsoventral specification of the lamprey ear is reliant on Hedgehog signaling, much as it is in gnathostomes, with the expression of patched A originating in the ventral region of the vesicle (Higuchi et al., 2019). However, why cyclostomes failed to develop a lateral canal has yet to be determined. It was once assumed that the OTX family of transcription factors were responsible for lateral canal development, as mice mutant in Otx1 are missing this third canal, but the expression of OtxA within the ventral domain of cyclostomes suggests that these transcription factors alone cannot account for the evolution of this canal (Acampora et al., 1998; Higuchi et al., 2019; Morsli et al., 1999).
Beyond their use in comparative molecular analyses, studies of the inner ear of the Agnatha also provide valuable insight into the evolution of more complex motor behaviors. For example, one question that the Agnatha allows us to address is whether three orthogonally oriented semi‐circular canals are required for the detection of rotational accelerations on all three axes. To reiterate, lampreys possess only two canals, lacking the lateral canal found in gnathostomes responsible for detecting horizontal accelerations of the head, which would initially imply that they are unable to sense horizontal movement (Maklad et al., 2014). However, current analyses of their locomotion, a rhythmic undulation of the head and body on the horizontal plane, suggest that they would, in fact, require the perception of horizontal accelerations to move efficiently. Additionally, while the innervation of the lamprey eye is unique, they possess caudal and rostral recti muscles, implying that they should be capable of the vestibule–ocular reflex (i.e., stabilization of the eye) during horizontal movements of the head (Cohen, 1974; Maklad et al., 2014). In fact, a more in‐depth analysis of the electrophysiological response of their vestibulocochlear nerve shows that stimuli in all planes, including the horizontal, are capable of eliciting a response, even though the response from each of its two ampullae is somewhat modified from what is observed in higher vertebrates (Maklad et al., 2014). To summarize, this supports the idea that lampreys are able to perceive and react to stimuli on the horizontal plane, despite lacking a lateral canal, although the evolution of a lateral canal likely aided in the refined detection of stimuli on this plane.
Given that three unique canals are not required for sensing motion in a three‐dimensional space, this raises the question of how other changes in otic anatomy translate to differences in behavior. Studies have shown that the “radius of curvature” of a canal, and therefore its overall size, plays a significant role in the agility of a species (Spoor et al., 2007). For example, it is found that primates and other mammals, such as galagos and tarsiers, possess a large canal radius relative to body mass, enabling them to move faster and with less circumspection than other mammals, such as extant sloths, which are well known for their slow locomotion (Billet et al., 2013; Spoor et al., 2007). This correlation extends beyond mammals, such that when comparing fish with land‐based vertebrates of the same size, a much greater curvature to body mass ratio is found, corresponding to a greater sensitivity to angular accelerations (Jones & Spells, 1963). However, these species all possess an orthogonal three‐canal system. When considering a species such as the hagfish, which have only a single vertical canal, it is interesting to see what other anatomical compensations need to be made to allow for efficient movement (Higuchi et al., 2019; McVean, 2009). Hagfish also make a fascinating research subject for this topic of study because they do not possess functional eyes, or do they have a conventional “neck,” which means the degree to which their solitary canal needs to measure angular accelerations is not reliant on their ability to stabilize their gaze when they are in motion (McVean, 2009). However, electrophysiological analyses indicate that hagfish can still detect rotational accelerations and on more than one axis, although with less sensitivity, particularly on the horizontal plane. One reason their single canal detects accelerations on multiple axes could be that, unlike other fish, the internal radius of their canal is much larger than its radius of curvature. However, this anatomical difference might actually exist to compensate for the lack of cupula overlying the ring‐like crista of the hagfish ear, although it was originally thought that the cupula serves to stabilize and protect the hair cell bundles within the ampullae (Dohlman, 1980). During an acceleration, this cupula is displaced, which deflects the embedded hair cells within and triggers a response. It is, therefore, hypothesized that a canal’s radius of curvature must be large enough to reduce the time required for the cupula to return to its resting position and, therefore, allow the embedded hair cells to detect the next acceleration. In hagfish, this requirement for a large radius of curvature might simply not exist due to the lack of cupula (McVean, 1991). Altogether, while these studies have given us an idea of how inner ear anatomy affects the motor capabilities of a species, there is still a great deal we can learn about how the species with a rudimentary inner ear system, or no inner ear at all, perceive and respond to motions of the head and body.
13. CONCLUDING REMARKS
A great deal of research has been invested in understanding how the inner ear develops and how its developmental program has evolved over the course of vertebrate history. This has aided the medical field considerably by providing insights into how mutant variants of human genes in animal models impact both their independent cellular function and the overall function of the inner ear. However, while many genetic factors have already been identified and investigated, there are still gaps in our knowledge concerning all the players involved in otic development and how they interact with other unknown or known factors. Nevertheless, headway has already been made in restoring vestibular function through gene editing tools in some animal models of vestibular diseases. This includes the intraperitoneal injection of antisense oligonucleotides to block the defective cryptic splice site of Ush1c or the supplemental knock‐in of this gene with synthetic adeno‐associated viral vectors to rescue vestibular function in mice models of Usher syndrome (Delmaghani & El‐Amraoui, 2020; Lentz et al., 2013; Pan et al., 2017). While it remains to be seen how soon these techniques can be applied to human patients, continuing research into the developmental biology of the inner ear is essential in identifying new targets for this promising area of therapeutics.
CONFLICT OF INTEREST
All authors certify that they have no involvement in or affiliation with any organization or entity with financial or non‐financial interests concerning the subject matter discussed in this manuscript.
AUTHOR CONTRIBUTIONS
The body of this manuscript and Table 1 were written by Kacey Mackowetzky. Figure 1 was illustrated by Emily Mackowetzky. Figures 2, 3, 4 were illustrated by Kevin Yoon. The supervisory author is Andrew Waskiewicz, who edited the manuscript and provided funding for its publication. Funding for the publication of this review was provided in a grant from the National Science and Engineering Research Council of Canada.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Abdelhak, S., Kalatzis, V., Heilig, R., Compain, S., Samson, D., Vincent, C. et al. (1997) A human homologue of the Drosophila eyes absent gene underlies branchio‐oto‐renal (BOR) syndrome and identifies a novel gene family. Nature Genetics, 15(2), 157–164. 10.1038/ng0297-157 [DOI] [PubMed] [Google Scholar]
- Acampora, D., Avantaggiato, V., Tuorto, F., Barone, P., Reichert, H., Finkelstein, R. et al. (1998) Murine Otx1 and Drosophila otd genes share conserved genetic functions required in invertebrate and vertebrate brain development. Development, 125(9), 1691–1702. [DOI] [PubMed] [Google Scholar]
- Ahmed, M., Wong, E.Y., Sun, J., Xu, J., Wang, F. & Xu, P.X. (2012) Eya1‐Six1 interaction is sufficient to induce hair cell fate in the cochlea by activating Atoh1 expression in cooperation with Sox2. Developmental Cell, 22(2), 377–390. 10.1016/j.devcel.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, M., Xu, J. & Xu, P.‐X. (2012) EYA1 and SIX1 drive the neuronal developmental program in cooperation with the SWI/SNF chromatin‐remodeling complex and SOX2 in the mammalian inner ear. Development, 139(11), 1965–1977. 10.1242/dev.071670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, Z.M., Riazuddin, S., Ahmad, J., Bernstein, S.L., Guo, Y., Sabar, M.F. et al. (2003) PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Human Molecular Genetics, 12(24), 3215–3223. 10.1093/hmg/ddg358 [DOI] [PubMed] [Google Scholar]
- Aitkin, L. (1995) The auditory neurobiology of marsupials: a review. Hearing Research, 82(2), 257–266. [DOI] [PubMed] [Google Scholar]
- Aker, M., Rouvinski, A., Hashavia, S., Ta‐Shma, A., Shaag, A., Zenvirt, S. et al. (2012) An SNX10 mutation causes malignant osteopetrosis of infancy. Journal of medical genetics, 49(4), 221–226. [DOI] [PubMed] [Google Scholar]
- Alagramam, K.N., Murcia, C.L., Kwon, H.Y., Pawlowski, K.S., Wright, C.G. & Woychik, R.P. (2001) The mouse Ames waltzer hearing‐loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nature Genetics, 27(1), 99–102. 10.1038/83837 [DOI] [PubMed] [Google Scholar]
- Alasti, F., Sadeghi, A., Sanati, M.H., Farhadi, M., Stollar, E., Somers, T. et al. (2008) A mutation in HOXA2 is responsible for autosomal‐recessive microtia in an Iranian family. The American Journal of Human Genetics, 82(4), 982–991. 10.1016/j.ajhg.2008.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsmadi, O., Meyer, B.F., Alkuraya, F., Wakil, S., Alkayal, F., Al‐Saud, H. et al. (2009) Syndromic congenital sensorineural deafness, microtia and microdontia resulting from a novel homoallelic mutation in fibroblast growth factor 3 (FGF3). European Journal of Human Genetics, 17(1), 14–21. 10.1038/ejhg.2008.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez, Y., Alonso, M.T., Vendrell, V., Zelarayan, L.C., Chamero, P., Theil, T. et al. (2003) Requirements for FGF3 and FGF10 during inner ear formation. Development, 130(25), 6329–6338. 10.1242/dev.00881 [DOI] [PubMed] [Google Scholar]
- Anagnostopoulos, A.V. (2002) A compendium of mouse knockouts with inner ear defects. TRENDS in Genetics, 18(10), S21–S38. 10.1016/S0168-9525(02)02753-1 [DOI] [PubMed] [Google Scholar]
- Arima, K., Shiotsugu, J., Niu, R., Khandpur, R., Martinez, M., Shin, Y. et al. (2005) Global analysis of RAR‐responsive genes in the Xenopus neurula using cDNA microarrays. Developmental Dynamics, 232(2), 414–431. 10.1002/dvdy.20231 [DOI] [PubMed] [Google Scholar]
- Asai, Y., Chan, D.K., Starr, C.J., Kappler, J.A., Kollmar, R. & Hudspeth, A.J. (2006) Mutation of the atrophin2 gene in the zebrafish disrupts signaling by fibroblast growth factor during development of the inner ear. Proceedings of the National Academy of Sciences, 103(24), 9069–9074. 10.1073/pnas.0603453103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore, J.F. (1987) A fast motile response in guinea‐pig outer hair cells: the cellular basis of the cochlear amplifier. Journal of Physiology, 388, 323–347. 10.1113/jphysiol.1987.sp016617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore, J. (2008) Cochlear outer hair cell motility. Physiological Reviews, 88(1), 173–210. 10.1152/physrev.00044.2006 [DOI] [PubMed] [Google Scholar]
- Babb‐Clendenon, S., Shen, Y.‐C., Liu, Q., Turner, K.E., Mills, M.S., Cook, G.W. et al. (2006) Cadherin‐2 participates in the morphogenesis of the zebrafish inner ear. Journal of Cell Science, 119(24), 5169–5177. 10.1242/jcs.03299 [DOI] [PubMed] [Google Scholar]
- Barrett, K., Barman, S., Boitano, S. & Brooks, H. (2012) Chapter 10. Hearing & equilibrium. In Ganong’s review of medical physiology. 24th edition. New York: McGraw‐Hill. [Google Scholar]
- Battey, J.F.Jr (2000) A genetic approach to understanding inner ear function. The Journal of clinical investigation, 106(12), 1431–1432. 10.1172/JCI11763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, D., Streit, A., Gorospe, I., Varela‐Nieto, I., Alsina, B. & Giraldez, F. (2008) Spatial and temporal segregation of auditory and vestibular neurons in the otic placode. Developmental Biology, 322(1), 109–120. 10.1016/j.ydbio.2008.07.011 [DOI] [PubMed] [Google Scholar]
- Bergstrom, D.E., Gagnon, L.H. & Eicher, E.M. (1999) Genetic and physical mapping of the dreher locus on mouse chromosome 1. Genomics, 59(3), 291–299. 10.1006/geno.1999.5873 [DOI] [PubMed] [Google Scholar]
- Bierbower, S.M. & Cooper, R.L. (2013) The mechanistic action of carbon dioxide on a neural circuit and NMJ communication. Journal of Experimental Zoology Part A: Ecological Genetics and Physiology, 319(6), 340–354. 10.1002/jez.1798 [DOI] [PubMed] [Google Scholar]
- Biewener, A.A. (1983) Allometry of quadrupedal locomotion: the scaling of duty factor, bone curvature and limb orientation to body size. Journal of Experimental Biology, 105(1), 147–171. 10.1242/jeb.105.1.147 [DOI] [PubMed] [Google Scholar]
- Billet, G., Germain, D., Ruf, I., de Muizon, C. & Hautier, L. (2013) The inner ear of Megatherium and the evolution of the vestibular system in sloths. Journal of Anatomy, 223(6), 557–567. 10.1111/joa.12114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonnette, J.P. & Fekete, D.M. (1996) Standard atlas of the gross anatomy of the developing inner ear of the chicken. The Journal of Comparative Neurology, 368(4), 620–630. 10.1002/(sici)1096-9861(19960513)368:4<620:Aid-cne12>3.0.Co;2-l [DOI] [PubMed] [Google Scholar]
- Blasiole, B., Canfield, V.A., Vollrath, M.A., Huss, D., Mohideen, M.A., Dickman, J.D. et al. (2006) Separate Na, K‐ATPase genes are required for otolith formation and semicircular canal development in zebrafish. Developmental Biology, 294(1), 148–160. 10.1016/j.ydbio.2006.02.034 [DOI] [PubMed] [Google Scholar]
- Blasiole, B., Kabbani, N., Boehmler, W., Thisse, B., Thisse, C., Canfield, V. et al. (2005) Neuronal calcium sensor‐1 gene ncs‐1a is essential for semicircular canal formation in zebrafish inner ear. Journal of Neurobiology, 64(3), 285–297. 10.1002/neu.20138 [DOI] [PubMed] [Google Scholar]
- Bok, J., Dolson, D.K., Hill, P., Rüther, U., Epstein, D.J. & Wu, D.K. (2007) Opposing gradients of Gli repressor and activators mediate Shh signaling along the dorsoventral axis of the inner ear. Development, 134(9), 1713–1722. 10.1242/dev.000760 [DOI] [PubMed] [Google Scholar]
- Bok, J., Raft, S., Kong, K.A., Koo, S.K., Dräger, U.C. & Wu, D.K. (2011) Transient retinoic acid signaling confers anterior‐posterior polarity to the inner ear. Proceedings of the National Academy of Sciences of the United States of America, 108(1), 161–166. 10.1073/pnas.1010547108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolz, H., von Brederlow, B., Ramírez, A., Bryda, E.C., Kutsche, K., Nothwang, H.G. et al. (2001) Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nature Genetics, 27(1), 108–112. 10.1038/83667 [DOI] [PubMed] [Google Scholar]
- Bonnet, C., Louha, M., Loundon, N., Michalski, N., Verpy, E., Smagghe, L. et al. (2013) Biallelic nonsense mutations in the otogelin‐like gene (OTOGL) in a child affected by mild to moderate hearing impairment. Gene, 527(2), 537–540. 10.1016/j.gene.2013.06.044 [DOI] [PubMed] [Google Scholar]
- Bosher, S.K. & Warren, R.L. (1968) Observations on the electrochemistry of the cochlear endolymph of the rat: a quantitative study of its electrical potential and ionic composition as determined by means of flame spectrophotometry. Proceedings of the Royal Society of London. Series B. Biological Sciences, 171(1023), 227–247. 10.1098/rspb.1968.0066 [DOI] [PubMed] [Google Scholar]
- Bosman, E.A., Penn, A.C., Ambrose, J.C., Kettleborough, R., Stemple, D.L. & Steel, K.P. (2005) Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Human Molecular Genetics, 14(22), 3463–3476. 10.1093/hmg/ddi375 [DOI] [PubMed] [Google Scholar]
- Bosman, E.A., Quint, E., Fuchs, H., de Angelis, M.H. & Steel, K.P. (2009) Catweasel mice: a novel role for Six1 in sensory patch development and a model for branchio‐oto‐renal syndrome. Developmental Biology, 328(2), 285–296. 10.1016/j.ydbio.2009.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard, M., de Caprona, D., Busslinger, M., Xu, P. & Fritzsch, B. (2010) Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Developmental Biology, 10(1), 89. 10.1186/1471-213X-10-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer, J. (1873) Über die Bogengänge des Labyrinths. Vorläufige Mitteilung . Anz kk Ges d Aerzte Wien D, 7, 15–18. [Google Scholar]
- Brigande, J.V., Kiernan, A.E., Gao, X., Iten, L.E. & Fekete, D.M. (2000) Molecular genetics of pattern formation in the inner ear: do compartment boundaries play a role? Proceedings of the National Academy of Sciences of the United States of America, 97(22), 11700–11706. 10.1073/pnas.97.22.11700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister, E., Agarwal, A. & Richter, C.‐P. (2020) 2.02 ‐ The sensory organ of hearing. In Fritzsch, B. (Ed.) The senses: A comprehensive reference, 2nd edition. Oxford: Elsevier, pp. 18–31. [Google Scholar]
- Brown, A.S. & Epstein, D.J. (2011) Otic ablation of smoothened reveals direct and indirect requirements for Hedgehog signaling in inner ear development. Development (Cambridge, England), 138(18), 3967–3976. 10.1242/dev.066126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, Q., Cole, L.K., Mulheisen, M., Chang, W. & Wu, D.K. (2004) The role of Pax2 in mouse inner ear development. Developmental Biology, 272(1), 161–175. 10.1016/j.ydbio.2004.04.024 [DOI] [PubMed] [Google Scholar]
- Busch‐Nentwich, E., Söllner, C., Roehl, H. & Nicolson, T. (2004) The deafness gene dfna5 is crucial for ugdh expression and HA production in the developing ear in zebrafish. Development, 131(4), 943–951. 10.1242/dev.00961 [DOI] [PubMed] [Google Scholar]
- Casimiro, M.C., Knollmann, B.C., Ebert, S.N., Vary, J.C., Greene, A.E., Franz, M.R. et al. (2001) Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange– Nielsen Syndrome. Proceedings of the National Academy of Sciences of the United States of America, 98(5), 2526–2531. 10.1073/pnas.041398998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2017) 2017 summary of diagnostics among infants not passing hearing screening. Retrieved from https://www.cdc.gov/ncbddd/hearingloss/2017‐data/06‐diagnostics.html [Google Scholar]
- Ceruti, S., Stinckens, C., Cremers, C.W. & Casselman, J.W. (2002) Temporal bone anomalies in the branchio‐oto‐renal syndrome: detailed computed tomographic and magnetic resonance imaging findings. Otol Neurotol, 23(2), 200–207. 10.1097/00129492-200203000-00016 [DOI] [PubMed] [Google Scholar]
- Chagnaud, B.P., Engelmann, J., Fritzsch, B., Glover, J.C. & Straka, H. (2017) Sensing external and self‐motion with hair cells: A Comparison of the Lateral Line and Vestibular Systems from a Developmental and Evolutionary Perspective. Brain, behavior and evolution, 90(2), 98–116. 10.1159/000456646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, W., Brigande, J.V., Fekete, D.M. & Wu, D.K. (2004) The development of semicircular canals in the inner ear: role of FGFs in sensory cristae. Development, 131(17), 4201–4211. 10.1242/dev.01292 [DOI] [PubMed] [Google Scholar]
- Chang, W., Cole, L., Cantos, R. & Wu, D.K. (2004) Molecular genetics of vestibular organ development. In Tam, P.P.L., Nelson, W.J. & Rossant, J. (Eds.) The vestibular system. New York, NY: Springer, pp. 11–56. [Google Scholar]
- Chang, W., Lin, Z., Kulessa, H., Hebert, J., Hogan, B.L.M. & Wu, D.K. (2008) Bmp4 is essential for the formation of the vestibular apparatus that detects angular head movements. PLoS genetics, 4(4), e1000050. 10.1371/journal.pgen.1000050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang‐Chien, J., Yen, Y.C., Chien, K.H., Li, S.Y., Hsu, T.C. & Yang, J.J. (2014) The connexin 30.3 of zebrafish homologue of human connexin 26 may play similar role in the inner ear. Hearing Research, 313, 55–66. 10.1016/j.heares.2014.04.010 [DOI] [PubMed] [Google Scholar]
- Chen, J. & Nathans, J. (2007) Estrogen‐related receptor beta/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Developmental Cell, 13(3), 325–337. 10.1016/j.devcel.2007.07.011 [DOI] [PubMed] [Google Scholar]
- Chen, P., Johnson, J.E., Zoghbi, H.Y. & Segil, N. (2002) The role of Math1 in inner ear development: uncoupling the establishment of the sensory primordium from hair cell fate determination. Development, 129(10), 2495–2505. 10.1242/dev.129.10.2495 [DOI] [PubMed] [Google Scholar]
- Chizhikov, V.V., Iskusnykh, I.Y., Fattakhov, N. & Fritzsch, B. (2021) Lmx1a and Lmx1b are redundantly required for the development of multiple components of the mammalian auditory system. Neuroscience, 452, 247–264. 10.1016/j.neuroscience.2020.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo, D., Ward, J., Reece, A., Dou, H., Lin, Z. & Greinwald, J. (2006) Molecular mechanisms underlying inner ear patterning defects in kreisler mutants. Developmental Biology, 289(2), 308–317. 10.1016/j.ydbio.2005.10.007 [DOI] [PubMed] [Google Scholar]
- Christensen, C.B., Christensen‐Dalsgaard, J. & Madsen, P.T. (2015) Hearing of the African lungfish (Protopterus annectens) suggests underwater pressure detection and rudimentary aerial hearing in early tetrapods. Journal of Experimental Biology, 218(Pt 3), 381–387. 10.1242/jeb.116012 [DOI] [PubMed] [Google Scholar]
- Christensen, C.B., Lauridsen, H., Christensen‐Dalsgaard, J., Pedersen, M. & Madsen, P.T. (2015) Better than fish on land? Hearing across metamorphosis in salamanders. Proceedings of the Royal Society B: Biological Sciences, 282(1802), 10.1098/rspb.2014.1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen‐Dalsgaard, J., Brandt, C., Wilson, M., Wahlberg, M. & Madsen, P.T. (2011) Hearing in the African lungfish (Protopterus annectens): pre‐adaptation to pressure hearing in tetrapods? Biology Letters, 7(1), 139–141. 10.1098/rsbl.2010.0636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clack, J.A. (2015) Evolutionary biology: the origin of terrestrial hearing. Nature, 519(7542), 168–169. 10.1038/519168a [DOI] [PubMed] [Google Scholar]
- Cohen, B. (1974) The vestibulo‐ocular reflex arc. In Kornhuber, H.H. (Ed.) Vestibular System Part 1: Basic Mechanisms. Berlin, Heidelberg: Springer, pp. 477–540. [Google Scholar]
- Cohen‐Salmon, M., El‐Amraoui, A., Leibovici, M. & Petit, C. (1997) Otogelin: a glycoprotein specific to the acellular membranes of the inner ear. Proceedings of the National Academy of Sciences of the United States of America, 94(26), 14450–14455. 10.1073/pnas.94.26.14450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia, M.J. & Lang, D.G. (1990) An electrophysiological comparison of solitary type I and type II vestibular hair cells. Neuroscience letters, 116(1–2), 106–111. 10.1016/0304-3940(90)90394-O [DOI] [PubMed] [Google Scholar]
- Correia, M.J., Ricci, A.J. & Rennie, K.J. (1996) Filtering properties of vestibular hair cells: an update. Annals of the New York Academy of Sciences, 781, 138–149. 10.1111/j.1749-6632.1996.tb15698.x [DOI] [PubMed] [Google Scholar]
- Cowan, C.A., Yokoyama, N., Bianchi, L.M., Henkemeyer, M. & Fritzsch, B. (2000) EphB2 guides axons at the midline and is necessary for normal vestibular function. Neuron, 26(2), 417–430. 10.1016/S0896-6273(00)81174-5 [DOI] [PubMed] [Google Scholar]
- Cremers, C. & Huygen, P. (1984) Clinical features of female heterozygotes in the X‐linked mixed deafness syndrome (with perilymphatic gusher during stapes surgery). International Journal of Pediatric Otorhinolaryngology, 6, 179–185. 10.1016/S0165-5876(83)80118-9 [DOI] [PubMed] [Google Scholar]
- Crovetto, M.A., Whyte, J., Sarasola, E., Rodriguez, J.A. & García‐Barcina, M.J. (2012) Absence of COCH gene mutations in patients with superior semicircular canal dehiscence. American Journal of Medical Genetics Part A, 158(1), 251–253. 10.1002/ajmg.a.34377 [DOI] [PubMed] [Google Scholar]
- Cruz, S., Shiao, J.‐C., Liao, B.‐K., Huang, C.‐J. & Hwang, P.‐P. (2009) Plasma membrane calcium ATPase required for semicircular canal formation and otolith growth in the zebrafish inner ear. Journal of Experimental Biology, 212(5), 639–647. 10.1242/jeb.022798 [DOI] [PubMed] [Google Scholar]
- Dallos, P. (1996) Overview: cochlear neurobiology. The cochlea, 1–43. [Google Scholar]
- Deans, M.R. (2013) A balance of form and function: Planar polarity and development of the vestibular maculae. Seminars in Cell & Developmental Biology, 24(5), 490–498. 10.1016/j.semcdb.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans, M.R., Peterson, J.M. & Wong, G.W. (2010) Mammalian Otolin: a multimeric glycoprotein specific to the inner ear that interacts with otoconial matrix protein Otoconin‐90 and Cerebellin‐1. PLoS One, 5(9), e12765. 10.1371/journal.pone.0012765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmaghani, S. & El‐Amraoui, A. (2020) Inner ear gene therapies take off: current promises and future challenges. Journal of clinical medicine, 9(7), 2309. 10.3390/jcm9072309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire, E., Lu, J., England, R., Dull, C. & Thorne, T. (1999) Deafness and imbalance associated with inactivation of the secretory Na‐K‐2Cl co‐transporter. Nature Genetics, 22(2), 192–195. 10.1038/9713 [DOI] [PubMed] [Google Scholar]
- Deng, M., Pan, L., Xie, X. & Gan, L. (2010) Requirement for Lmo4 in the vestibular morphogenesis of mouse inner ear. Developmental Biology, 338(1), 38–49. 10.1016/j.ydbio.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Palma, F., Holme, R.H., Bryda, E.C., Belyantseva, I.A., Pellegrino, R., Kachar, B. et al. (2001) Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nature Genetics, 27(1), 103–107. 10.1038/83660 [DOI] [PubMed] [Google Scholar]
- Dixon, M.J., Gazzard, J., Chaudhry, S.S., Sampson, N., Schulte, B.A. & Steel, K.P. (1999) Mutation of the Na‐K‐Cl Co‐transporter gene Slc12a2 results in deafness in mice. Human Molecular Genetics, 8(8), 1579–1584. 10.1093/hmg/8.8.1579 [DOI] [PubMed] [Google Scholar]
- Dodson, K.M., Blanton, S.H., Welch, K.O., Norris, V.W., Nuzzo, R.L., Wegelin, J.A. et al. (2011) Vestibular dysfunction in DFNB1 deafness. American Journal of Medical Genetics Part A, 155(5), 993–1000. 10.1002/ajmg.a.33828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman, G.F. (1965) The mechanism of secretion and absorption of endolymph in the vestibular appartus. Acta Oto‐Laryngologica, 59(2–6), 275–288. 10.3109/00016486509124562 [DOI] [Google Scholar]
- Dohlman, G.F. (1980) Critical review of the concept of cupula functon. Acta Oto‐Laryngologica, 90(sup376), 2–30. 10.3109/00016488009108886 [DOI] [PubMed] [Google Scholar]
- Dror, A.A., Politi, Y., Shahin, H., Lenz, D.R., Dossena, S., Nofziger, C. et al. (2010) Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. Journal of Biological Chemistry, 285(28), 21724–21735. 10.1074/jbc.M110.120188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, J.S. & Fritzsch, B. (2012) Evolution of sound and balance perception: innovations that aggregate single hair cells into the ear and transform a gravistatic sensor into the organ of corti. The Anatomical Record, 295(11), 1760–1774. 10.1002/ar.22573 [DOI] [PubMed] [Google Scholar]
- Dvorakova, M., Macova, I., Bohuslavova, R., Anderova, M., Fritzsch, B. & Pavlinkova, G. (2020) Early ear neuronal development, but not olfactory or lens development, can proceed without SOX2. Developmental Biology, 457(1), 43–56. 10.1016/j.ydbio.2019.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eatock, R.A. (2018) Specializations for fast signaling in the amniote vestibular inner ear. Integrative and Comparative Biology, 58(2), 341–350. 10.1093/icb/icy069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eatock, R.A., Rüsch, A., Lysakowski, A. & Saeki, M. (1998) Hair cells in mammalian utricles. Otolaryngology—Head and Neck Surgery, 119(3), 172–181. 10.1016/S0194-5998(98)70052-X [DOI] [PubMed] [Google Scholar]
- Eatock, R.A. & Songer, J.E. (2011) Vestibular hair cells and afferents: two channels for head motion signals. Annual Review of Neuroscience, 34, 501–534. 10.1146/annurev-neuro-061010-113710 [DOI] [PubMed] [Google Scholar]
- Eccles, M.R. & Schimmenti, L.A. (1999) Renal‐coloboma syndrome: a multi‐system developmental disorder caused by PAX2 mutations. Clinical Genetics, 56(1), 1–9. 10.1034/j.1399-0004.1999.560101.x [DOI] [PubMed] [Google Scholar]
- Elliott, K.L., Fritzsch, B. & Duncan, J.S. (2018) Evolutionary and developmental biology provide insights into the regeneration of organ of corti hair cells. Frontiers in Cellular Neuroscience, 12, 252. 10.3389/fncel.2018.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmaleh‐Bergès, M., Baumann, C., Noël‐Pétroff, N., Sekkal, A., Couloigner, V., Devriendt, K. et al. (2013) Spectrum of temporal bone abnormalities in patients with Waardenburg syndrome and SOX10 mutations. AJNR. American Journal of Neuroradiology, 34(6), 1257–1263. 10.3174/ajnr.A3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson, R., Knight, R. & Johanson, Z. (2013) Evolution and development of the vertebrate neck. Journal of anatomy, 222(1), 67–78. 10.1111/j.1469-7580.2012.01530.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkman, L., McEvilly, R.J., Luo, L., Ryan, A.K., Hooshmand, F., O'connell, S.M. et al. (1996) Role of transcription factors a Brn‐3.1 and Brn‐3.2 in auditory and visual system development. Nature, 381(6583), 603–606. [DOI] [PubMed] [Google Scholar]
- Ernest, S., Rauch, G.‐J., Haffter, P., Geisler, R., Petit, C. & Nicolson, T. (2000) Mariner is defective in myosin VIIA: a zebrafish model for human hereditary deafness. Human Molecular Genetics, 9(14), 2189–2196. 10.1093/hmg/9.14.2189 [DOI] [PubMed] [Google Scholar]
- Everett, L.A., Belyantseva, I.A., Noben‐Trauth, K., Cantos, R., Chen, A., Thakkar, S.I. et al. (2001) Targeted disruption of mouse Pds provides insight about the inner‐ear defects encountered in Pendred syndrome. Human Molecular Genetics, 10(2), 153–161. 10.1093/hmg/10.2.153 [DOI] [PubMed] [Google Scholar]
- Evsen, L., Sugahara, S., Uchikawa, M., Kondoh, H. & Wu, D.K. (2013) Progression of neurogenesis in the inner ear requires inhibition of Sox2 transcription by Neurogenin1 and Neurod1. The Journal of Neuroscience, 33(9), 3879–3890. 10.1523/jneurosci.4030-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattal, D., Hansen, M. & Fritzsch, B. (2018). Aging‐related balance impairment and hearing loss. In Rizzo, M., Anderson, S. & Fritzsch, B. (Eds.) The Wiley Handbook on the Aging Mind and Brain. Hoboken, NJ: John Wiley & Sons Ltd, pp. 315–336. [Google Scholar]
- Favor, J., Sandulache, R., Neuhäuser‐Klaus, A., Pretsch, W., Chatterjee, B., Senft, E. et al. (1996) The mouse Pax21Neu mutation is identical to a human PAX2 mutation in a family with renal‐coloboma syndrome and results in developmental defects of the brain, ear, eye, and kidney. Proceedings of the National Academy of Sciences of the United States of America, 93(24), 13870–13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete, D.M. (1999) Development of the vertebrate ear: insights from knockouts and mutants. Trends in Neurosciences, 22(6), 263–269. 10.1016/s0166-2236(98)01366-6 [DOI] [PubMed] [Google Scholar]
- Fekete, D. & Wu, D. (2002) Development and evolution of hearing and balance‐Introduction. Hoboken, NJ, USA: John Wiley & Sons Inc. [Google Scholar]
- Fekete, D.M. & Wu, D.K. (2002) Revisiting cell fate specification in the inner ear. Current Opinion in Neurobiology, 12(1), 35–42. 10.1016/s0959-4388(02)00287-8 [DOI] [PubMed] [Google Scholar]
- Feng, Y. & Xu, Q. (2010) Pivotal role of hmx2 and hmx3 in zebrafish inner ear and lateral line development. Developmental Biology, 339(2), 507–518. 10.1016/j.ydbio.2009.12.028 [DOI] [PubMed] [Google Scholar]
- Flagella, M., Clarke, L.L., Miller, M.L., Erway, L.C., Giannella, R.A., Andringa, A. et al. (1999) Mice lacking the basolateral Na‐K‐2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. Journal of Biological Chemistry, 274(38), 26946–26955. 10.1074/jbc.274.38.26946 [DOI] [PubMed] [Google Scholar]
- Forge, A., Davies, S. & Zajic, G. (1991) Assessment of ultrastructure in isolated cochlear hair cells using a procedure for rapid freezing before freeze‐fracture and deep‐etching. Journal of Neurocytology, 20(6), 471–484. 10.1007/BF01252275 [DOI] [PubMed] [Google Scholar]
- Francis, H.W., Rivas, A., Lehar, M. & Ryugo, D.K. (2004) Two types of afferent terminals innervate cochlear inner hair cells in C57BL/6J mice. Brain Research, 1016(2), 182–194. 10.1016/j.brainres.2004.05.016 [DOI] [PubMed] [Google Scholar]
- Freter, S., Muta, Y., Mak, S.‐S., Rinkwitz, S. & Ladher, R.K. (2008) Progressive restriction of otic fate: the role of FGF and Wnt in resolving inner ear potential. Development, 135(20), 3415–3424. 10.1242/dev.026674 [DOI] [PubMed] [Google Scholar]
- Fritzsch, B., Barald, K.F. & Lomax, M.I. (1998) Early embryology of the vertebrate ear. In Rubel, E.W., Popper, A.N. & Fay, R.R. (Eds.) Development of the auditory system. New York, NY: Springer, pp. 80–145. [Google Scholar]
- Fritzsch, B., Beisel, K.W., Jones, K., Fariñas, I., Maklad, A., Lee, J. et al. (2002) Development and evolution of inner ear sensory epithelia and their innervation. Journal of Neurobiology, 53(2), 143–156. 10.1002/neu.10098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch, B., Signore, M. & Simeone, A. (2001) Otx1 null mutant mice show partial segregation of sensory epithelia comparable to lamprey ears. Development Genes and Evolution, 211(8–9), 388–396. 10.1007/s004270100166 [DOI] [PubMed] [Google Scholar]
- Fritzsch, B. & Straka, H. (2014) Evolution of vertebrate mechanosensory hair cells and inner ears: toward identifying stimuli that select mutation driven altered morphologies. Journal of Comparative Physiology A Neuroethology, Sensory, Neural, and Behavioral Physiology, 200(1), 5–18. 10.1007/s00359-013-0865-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch, B. & Wake, M. (1988) The inner ear of gymnophione amphibians and its nerve supply: a comparative study of regressive events in a complex sensory system (Amphibia, Gymnophiona). Zoomorphology, 108(4), 201–217. 10.1007/BF00312221 [DOI] [Google Scholar]
- Gagnon, L.H., Longo‐Guess, C.M., Berryman, M., Shin, J.‐B., Saylor, K.W., Yu, H. et al. (2006) The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. The Journal of Neuroscience, 26(40), 10188–10198. 10.1523/jneurosci.2166-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng, F.‐S., Abbas, L., Baxendale, S., Holdsworth, C.J., Swanson, A.G., Slanchev, K. et al. (2013) Semicircular canal morphogenesis in the zebrafish inner ear requires the function of gpr126 (lauscher), an adhesion class G protein‐coupled receptor gene. Development, 140(21), 4362–4374. 10.1242/dev.098061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, F., Walsh, J., Mburu, P., Varela, A., Brown, K.A., Antonio, M. et al. (1995) A type VII myosin encoded by the mouse deafness gene shaker‐1. Nature, 374(6517), 62–64. 10.1038/374062a0 [DOI] [PubMed] [Google Scholar]
- Goldberg, J.M., Lysakowski, A. & Fernández, C. (1990) Morphophysiological and ultrastructural studies in the mammalian cristae ampullares. Hearing Research, 49(1–3), 89–102. 10.1016/0378-5955(90)90097-9 [DOI] [PubMed] [Google Scholar]
- Goodrich, E.S. (1909). Cyclostomes and fishes. A Treatise on Zoology, 518. Retrieved from https://ci.nii.ac.jp/naid/20000758058/en/ [Google Scholar]
- Goodyear, R.J. & Richardson, G.P. (2002) Extracellular matrices associated with the apical surfaces of sensory epithelia in the inner ear: molecular and structural diversity. Journal of Neurobiology, 53(2), 212–227. 10.1002/neu.10097 [DOI] [PubMed] [Google Scholar]
- Gordon, C.T., Petit, F., Oufadem, M., Decaestecker, C., Jourdain, A.‐S., Andrieux, J. et al. (2012) EFTUD2 haploinsufficiency leads to syndromic oesophageal atresia. Journal of Medical Genetics, 49(12), 737–746. [DOI] [PubMed] [Google Scholar]
- Gurr, B. & Moffat, N. (2001) Psychological consequences of vertigo and the effectiveness of vestibular rehabilitation for brain injury patients. Brain Injury, 15(5), 387–400. 10.1080/02699050010005904 [DOI] [PubMed] [Google Scholar]
- Haddon, C.M. & Lewis, J.H. (1991) Hyaluronan as a propellant for epithelial movement: the development of semicircular canals in the inner ear of Xenopus. Development, 112(2), 541–550.Retrieved from https://dev.biologists.org/content/develop/112/2/541.full.pdf [DOI] [PubMed] [Google Scholar]
- Haeckel, E. (1877) Anthropogenie: oder, Entwickelungsgeschichte des menschen, Keimes‐und stammesgeschichte. W. Engelmann. [Google Scholar]
- Hain, T.C. & Helminski, J.O. (2007) Anatomy and physiology of the normal vestibular system. Vestibular Rehabilitation, 1(1), 2. [Google Scholar]
- Hammond, K.L., Loynes, H.E., Mowbray, C., Runke, G., Hammerschmidt, M., Mullins, M.C. et al. (2009) A late role for bmp2b in the morphogenesis of semicircular canal ducts in the zebrafish inner ear. PLoS One, 4(2), e4368. 10.1371/journal.pone.0004368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, K.L. & Whitfield, T.T. (2006) The developing lamprey ear closely resembles the zebrafish otic vesicle: otx1 expression can account for all major patterning differences. Development, 133(7), 1347–1357. 10.1242/dev.02306 [DOI] [PubMed] [Google Scholar]
- Han, Y., Mu, Y., Li, X., Xu, P., Tong, J., Liu, Z. et al. (2011) Grhl2 deficiency impairs otic development and hearing ability in a zebrafish model of the progressive dominant hearing loss DFNA28. Human Molecular Genetics, 20(16), 3213–3226. [DOI] [PubMed] [Google Scholar]
- Hans, S., Irmscher, A. & Brand, M. (2013) Zebrafish Foxi1 provides a neuronal ground state during inner ear induction preceding the Dlx3b/4b‐regulated sensory lineage. Development, 140(9), 1936–1945. 10.1242/dev.087718 [DOI] [PubMed] [Google Scholar]
- Hans, S., Liu, D. & Westerfield, M. (2004) Pax8 and Pax2a function synergistically in otic specification, downstream of the Foxi1 and Dlx3b transcription factors. Development, 131(20), 5091–5102. 10.1242/dev.01346 [DOI] [PubMed] [Google Scholar]
- Harada, Y. (1972) Surface structure of semicircular ampullae. Equilibrium Research, 31(suppl‐4), 53–58. 10.3757/jser.31.suppl-4_53 [DOI] [Google Scholar]
- Harada, Y. (1974) The shape of the vertical canal crista in various animals. Equilibrium Research, 33(1), 82–85. 10.3757/jser.33.82 [DOI] [Google Scholar]
- Harada, Y., Tagashira, N. & Hirakawa, K. (1989) Morphological study of the cells in the Planum Semilunatum. Acta Oto‐Laryngologica, 108(sup468), 17–21. 10.3109/00016488909139015 [DOI] [PubMed] [Google Scholar]
- Hennekam, R. & Goldschmeding, R. (1998) Complete absence of rib ossification, micrognathia and ear anomalies: extreme expression of cerebro‐costo‐mandibular syndrome? European Journal of Human Genetics, 6(1), 71–74. 10.1038/sj.ejhg.5200154 [DOI] [PubMed] [Google Scholar]
- Higuchi, S., Sugahara, F., Pascual‐Anaya, J., Takagi, W., Oisi, Y. & Kuratani, S. (2019) Inner ear development in cyclostomes and evolution of the vertebrate semicircular canals. Nature, 565(7739), 347–350. 10.1038/s41586-018-0782-y [DOI] [PubMed] [Google Scholar]
- Hoffman, H.J., Dobie, R.A., Losonczy, K.G., Themann, C.L. & Flamme, G.A. (2017) Declining prevalence of hearing loss in US adults aged 20 to 69 years. JAMA Otolaryngology‐Head & Neck Surgery, 143(3), 274–285. 10.1001/jamaoto.2016.3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holme, R.H. & Steel, K.P. (2002) Stereocilia defects in waltzer (Cdh23), shaker1 (Myo7a) and double waltzer/shaker1 mutant mice. Hearing Research, 169(1–2), 13–23. 10.1016/s0378-5955(02)00334-9 [DOI] [PubMed] [Google Scholar]
- Hommerich, C.P. (1990) Ototoxicity of loop diuretics. Morphological and electrophysiological examinations in animal experiments. Advances in Oto‐Rhino‐Laryngology, 44, 92–164. [PubMed] [Google Scholar]
- Hughes, I., Saito, M., Schlesinger, P.H. & Ornitz, D.M. (2007) Otopetrin 1 activation by purinergic nucleotides regulates intracellular calcium. Proceedings of the National Academy of Sciences of the United States of America, 104(29), 12023–12028. 10.1073/pnas.0705182104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, C.H., Simeone, A., Lai, E. & Wu, D.K. (2009) Foxg1 is required for proper separation and formation of sensory cristae during inner ear development. Developmental Dynamics: An Official Publication of the American Association of Anatomists, 238(11), 2725–2734. [DOI] [PubMed] [Google Scholar]
- Igawa, H.H., Nishizawa, N., Sugihara, T. & Inuyama, Y. (2000) Inner ear abnormalities in Kabuki make‐up syndrome: report of three cases. American Journal of Medical Genetics, 92(2), 87–89. [DOI] [PubMed] [Google Scholar]
- Jahan, I., Kersigo, J., Pan, N. & Fritzsch, B. (2010) Neurod1 regulates survival and formation of connections in mouse ear and brain. Cell and Tissue Research, 341(1), 95–110. 10.1007/s00441-010-0984-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahan, I., Pan, N., Kersigo, J. & Fritzsch, B. (2010) Neurod1 suppresses hair cell differentiation in ear ganglia and regulates hair cell subtype development in the cochlea. PLoS One, 5(7), e11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janesick, A., Shiotsugu, J., Taketani, M. & Blumberg, B. (2012) RIPPLY3 is a retinoic acid‐inducible repressor required for setting the borders of the pre‐placodal ectoderm. Development, 139(6), 1213–1224. 10.1242/dev.071456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, G.M. & Spells, K.E. (1963) A theoretical and comparative study of the functional dependence of the semicircular canal upon its physical dimensions. Proceedings of the Royal Society of London. Series B: Biological Sciences, 157, 403–419. 10.1098/rspb.1963.0019 [DOI] [PubMed] [Google Scholar]
- Jones, S.M., Robertson, N.G., Given, S., Giersch, A.B.S., Liberman, M.C. & Morton, C.C. (2011) Hearing and vestibular deficits in the Coch(−/−) null mouse model: comparison to the Coch(G88E/G88E) mouse and to DFNA9 hearing and balance disorder. Hearing Research, 272(1–2), 42–48. 10.1016/j.heares.2010.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans, M.C.J., Hoefsloot, L.H., van der Donk, K.P., Admiraal, R.J., Magee, A., van de Laar, I. et al. (2008) Familial CHARGE syndrome and the CHD7 gene: A recurrent missense mutation, intrafamilial recurrence and variability. American Journal of Medical Genetics Part A, 146A(1), 43–50. 10.1002/ajmg.a.31921 [DOI] [PubMed] [Google Scholar]
- Kantarci, H., Edlund, R.K., Groves, A.K. & Riley, B.B. (2015) Tfap2a promotes specification and maturation of neurons in the inner ear through modulation of Bmp, Fgf and notch signaling. PLoS Genetics, 11(3), e1005037. 10.1371/journal.pgen.1005037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci, S., Al‐Gazali, L., Hill, R.S., Donnai, D., Black, G.C., Bieth, E. et al. (2007) Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai‐Barrow and facio‐oculo‐acoustico‐renal syndromes. Nature Genetics, 39(8), 957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karis, A., Pata, I., van Doorninck, J.H., Grosveld, F., de Zeeuw, C.I., de Caprona, D. et al. (2001) Transcription factor GATA‐3 alters pathway selection of olivocochlear neurons and affects morphogenesis of the ear. Journal of Comparative Neurology, 429(4), 615–630. 10.1002/1096-9861(20010122)429:4<615:AID-CNE8>3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
- Katayama, K.‐I., Imai, F., Suto, F. & Yoshida, Y. (2013) Deletion of Sema3a or plexinA1/plexinA3 causes defects in sensory afferent projections of. Statoacoustic Ganglion Neurons, 8(8), e72512. 10.1371/journal.pone.0072512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, M. (1992) The atlas of mouse development. Academic Press. [Google Scholar]
- Kear, B.P., Lee, M.S., Gerdtz, W.R. & Flannery, T.F. (2008) Evolution of hind limb proportions in kangaroos (Marsupialia: Macropodoidea). In Sargis, E.J. & Dagosto, M. (Eds.) Mammalian evolutionary morphology. Dordrecht, the Netherlands: Springer, pp. 25–35. [Google Scholar]
- Khetarpal, U., Schuknecht, H.F., Gacek, R.R. & Holmes, L.B. (1991) Autosomal dominant sensorineural hearing loss: pedigrees, audiologic findings, and temporal bone findings in two kindreds. Archives of Otolaryngology‐Head & Neck Surgery, 117(9), 1032–1042. 10.1001/archotol.1991.01870210104022 [DOI] [PubMed] [Google Scholar]
- Khimich, D., Nouvian, R., Pujol, R., Tom Dieck, S., Egner, A., Gundelfinger, E.D. et al. (2005) Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature, 434(7035), 889–894. 10.1038/nature03418 [DOI] [PubMed] [Google Scholar]
- Kido, T., Sekitani, T., Yamashita, H., Endo, S., Masumitsu, Y. & Shimogori, H. (1991) Effects of carbonic anhydrase inhibitor on the otolithic organs of developing chick embryos. American Journal of Otolaryngology, 12(4), 191–195. 10.1016/0196-0709(91)90119-z [DOI] [PubMed] [Google Scholar]
- Kiernan, A.E., Pelling, A.L., Leung, K.K., Tang, A.S., Bell, D.M., Tease, C. et al. (2005) Sox2 is required for sensory organ development in the mammalian inner ear. Nature, 434(7036), 1031–1035. 10.1038/nature03487 [DOI] [PubMed] [Google Scholar]
- Killick, R. & Richardson, G.P. (1997) Antibodies to the sulphated, high molecular mass mouse tectorin stain hair bundles and the olfactory mucus layer. Hearing Research, 103(1–2), 131–141. 10.1016/s0378-5955(96)00174-8. [DOI] [PubMed] [Google Scholar]
- Kim, E., Hyrc, K.L., Speck, J., Lundberg, Y.W., Salles, F.T., Kachar, B. et al. (2010) Regulation of cellular calcium in vestibular supporting cells by otopetrin 1. Journal of Neurophysiology, 104(6), 3439–3450. 10.1152/jn.00525.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, E., Hyrc, K.L., Speck, J., Salles, F.T., Lundberg, Y.W., Goldberg, M.P. et al. (2011) Missense mutations in Otopetrin 1 affect subcellular localization and inhibition of purinergic signaling in vestibular supporting cells. Molecular and Cellular Neurosciences, 46(3), 655–661. 10.1016/j.mcn.2011.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H.M. & Wangemann, P. (2010) Failure of fluid absorption in the endolymphatic sac initiates cochlear enlargement that leads to deafness in mice lacking pendrin expression. PLoS One, 5(11), e14041. 10.1371/journal.pone.0014041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H.M. & Wangemann, P. (2011) Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PLoS One, 6(3), e17949. 10.1371/journal.pone.0017949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, R., Lundquist, P.G. & Wersaell, J. (1964) Secretory epithelial linings in the Ampullae of the guinea pig labyrinth. Acta Oto‐Laryngologica, 57, 517–530. 10.3109/00016486409137114 [DOI] [PubMed] [Google Scholar]
- Kingma, H. & van de Berg, R. (2016) Chapter 1 ‐ Anatomy, physiology, and physics of the peripheral vestibular system. In Furman, J.M. & Lempert, T. (Eds.) Handbook of Clinical Neurology. 137, Elsevier, pp. 1–16. [DOI] [PubMed] [Google Scholar]
- Koehler, K.R., Mikosz, A.M., Molosh, A.I., Patel, D. & Hashino, E. (2013) Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature, 500(7461), 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev, O.I. & Sergeeva, M. (2016) Vestibular disorders following different types of head and neck trauma. Functional Neurology, 31(2), 75–80. 10.11138/fneur/2016.31.2.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo, S.K., Hill, J.K., Hwang, C.H., Lin, Z.S., Millen, K.J. & Wu, D.K. (2009) Lmx1a maintains proper neurogenic, sensory, and non‐sensory domains in the mammalian inner ear. Developmental Biology, 333(1), 14–25. 10.1016/j.ydbio.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecky, B.J., Jahan, I. & Fritzsch, B. (2013) Correct timing of proliferation and differentiation is necessary for normal inner ear development and auditory hair cell viability. Developmental Dynamics: An Official Publication of the American Association of Anatomists, 242(2), 132–147. 10.1002/dvdy.23910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecky, B., Johnson, S., Schmitz, H., Santi, P. & Fritzsch, B. (2012) Scanning thin‐sheet laser imaging microscopy elucidates details on mouse ear development. Developmental Dynamics: An Official Publication of the American Association of Anatomists, 241(3), 465–480. 10.1002/dvdy.23736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn, H. & Faber, D.S. (2005) The Mauthner cell half a century later: a neurobiological model for decision‐making? Neuron, 47(1), 13–28. 10.1016/j.neuron.2005.05.019 [DOI] [PubMed] [Google Scholar]
- Kössl, M. & Vater, M. (1995) Cochlear structure and function in bats. In Popper, A.N. & Fay, R.R. (Eds.) Hearing by bats. New York, NY: Springer, pp. 191–234. [Google Scholar]
- Koundakjian, E.J., Appler, J.L. & Goodrich, L.V. (2007) Auditory neurons make stereotyped wiring decisions before maturation of their targets. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 27(51), 14078–14088. 10.1523/jneurosci.3765-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozel, P.J., Friedman, R.A., Erway, L.C., Yamoah, E.N., Liu, L.H., Riddle, T. et al. (1998) Balance and hearing deficits in mice with a null mutation in the gene encoding plasma membrane Ca2+‐ATPase isoform 2. Journal of Biological Chemistry, 273(30), 18693–18696. 10.1074/jbc.273.30.18693 [DOI] [PubMed] [Google Scholar]
- Kozlowski, D.J., Whitfield, T.T., Hukriede, N.A., Lam, W.K. & Weinberg, E.S. (2005) The zebrafish dog‐eared mutation disrupts eya1, a gene required for cell survival and differentiation in the inner ear and lateral line. Developmental Biology, 277(1), 27–41. [DOI] [PubMed] [Google Scholar]
- Kulkarni, A.M., Rajput, K., Raglan, E., Abrams, D. & Bitner‐Glindzicz, M. (2012) Is gross motor delay secondary to bilateral vestibular hypofunction in Jervell and Lange‐Nielsen syndrome? Audiological Medicine, 10(2), 93–98. 10.3109/1651386x.2012.686165 [DOI] [Google Scholar]
- Kwon, H.‐J., Bhat, N., Sweet, E.M., Cornell, R.A. & Riley, B.B. (2010) Identification of early requirements for preplacodal ectoderm and sensory organ development. PLoS genetics, 6(9), e1001133. 10.1371/journal.pgen.1001133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladhams, A. & Pickles, J. (1996) Morphology of the monotreme organ of Corti and macula lagena. Journal of Comparative Neurology, 366(2), 335–347. [DOI] [PubMed] [Google Scholar]
- Ladher, R.K., Wright, T.J., Moon, A.M., Mansour, S.L. & Schoenwolf, G.C. (2005) FGF8 initiates inner ear induction in chick and mouse. Genes & Development, 19(5), 603–613. 10.1101/gad.1273605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalwani, A.K., Linthicum, F.H., Wilcox, E.R., Moore, J.K., Walters, F.C., San Agustin, T.B. et al. (1997) A five‐generation family with late‐onset progressive hereditary hearing impairment due to cochleosaccular degeneration. Audiology and Neurotology, 2(3), 139–154. [DOI] [PubMed] [Google Scholar]
- Lalwani, A.K., Luxford, W.M., Mhatre, A.N., Attaie, A., Wilcox, E.R. & Castelein, C.M. (1999) A new locus for nonsyndromic hereditary hearing impairment, DFNA17, maps to chromosome 22 and represents a gene for cochleosaccular degeneration. American Journal of Human Genetics, 64(1), 318. 10.1086/302216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawoko‐Kerali, G., Rivolta, M.N., Lawlor, P., Cacciabue‐Rivolta, D.I., Langton‐Hewer, C., van Doorninck, J.H. et al. (2004) GATA3 and NeuroD distinguish auditory and vestibular neurons during development of the mammalian inner ear. Mechanisms of Development, 121(3), 287–299. 10.1016/j.mod.2003.12.006 [DOI] [PubMed] [Google Scholar]
- Le Caignec, C., Lefevre, M., Schott, J., Chaventre, A., Gayet, M., Calais, C. et al. (2002) Familial deafness, congenital heart defects, and posterior embryotoxon caused by cysteine substitution in the first epidermal‐growth‐factor–like domain of Jagged 1. The American Journal of Human Genetics, 71(1), 180–186. 10.1086/341327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legan, P.K., Lukashkina, V.A., Goodyear, R.J., Kössi, M., Russell, I.J. & Richardson, G.P. (2000) A targeted deletion in alpha‐tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron, 28(1), 273–285. 10.1016/s0896-6273(00)00102-1 [DOI] [PubMed] [Google Scholar]
- Legan, P.K., Rau, A., Keen, J.N. & Richardson, G.P. (1997) The mouse tectorins. Modular matrix proteins of the inner ear homologous to components of the sperm‐egg adhesion system. Journal of Biological Chemistry, 272(13), 8791–8801. 10.1074/jbc.272.13.8791 [DOI] [PubMed] [Google Scholar]
- Léger, S. & Brand, M. (2002) Fgf8 and Fgf3 are required for zebrafish ear placode induction, maintenance and inner ear patterning. Mechanisms of Development, 119(1), 91–108. 10.1016/s0925-4773(02)00343-x [DOI] [PubMed] [Google Scholar]
- Lentz, J.J., Jodelka, F.M., Hinrich, A.J., McCaffrey, K.E., Farris, H.E., Spalitta, M.J. et al. (2013) Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nature Medicine, 19(3), 345–350. 10.1038/nm.3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz, J., Pan, F., Ng, S.S., Deininger, P. & Keats, B. (2007) Ush1c216A knock‐in mouse survives Katrina. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 616(1–2), 139–144. 10.1016/j.mrfmmm.2006.11.006 [DOI] [PubMed] [Google Scholar]
- Li, C.‐M., Hoffman, H.J., Ward, B.K., Cohen, H.S. & Rine, R.M. (2016) Epidemiology of dizziness and balance problems in children in the United States: a population‐based study. The Journal of Pediatrics, 171, 240–247.e243. 10.1016/j.jpeds.2015.12.002 [DOI] [PubMed] [Google Scholar]
- Li, J., Zhang, T., Ramakrishnan, A., Fritzsch, B., Xu, J., Wong, E.Y.M. et al. (2020) Dynamic changes in cis‐regulatory occupancy by Six1 and its cooperative interactions with distinct cofactors drive lineage‐specific gene expression programs during progressive differentiation of the auditory sensory epithelium. Nucleic Acids Research, 48(6), 2880–2896. 10.1093/nar/gkaa012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M., Nishio, S.‐Y., Naruse, C., Riddell, M., Sapski, S., Katsuno, T. et al. (2020) Digenic inheritance of mutations in EPHA2 and SLC26A4 in Pendred syndrome. Nature Communications, 11(1), 1343. 10.1038/s41467-020-15198-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X., Oghi, K.A., Zhang, J., Krones, A., Bush, K.T., Glass, C.K. et al. (2003) Eya protein phosphatase activity regulates Six1‐Dach‐Eya transcriptional effects in mammalian organogenesis. Nature, 426(6964), 247–254. 10.1038/nature02083 [DOI] [PubMed] [Google Scholar]
- Liberman, M.C. (1980) Morphological differences among radial afferent fibers in the cat cochlea: an electron‐microscopic study of serial sections. Hearing Research, 3(1), 45–63. 10.1016/0378-5955(80)90007-6 [DOI] [PubMed] [Google Scholar]
- Liberman, M.C., Dodds, L.W. & Pierce, S. (1990) Afferent and efferent innervation of the cat cochlea: quantitative analysis with light and electron microscopy. The Journal of Comparative Neurology, 301(3), 443–460. 10.1002/cne.903010309 [DOI] [PubMed] [Google Scholar]
- Litsiou, A., Hanson, S. & Streit, A. (2005) A balance of FGF, BMP and WNT signalling positions the future placode territory in the head. Development, 132(18), 4051. 10.1242/dev.01964 [DOI] [PubMed] [Google Scholar]
- Liu, D., Chu, H., Maves, L., Yan, Y.‐L., Morcos, P.A., Postlethwait, J.H. et al. (2003) Fgf3 and Fgf8 dependent and independent transcription factors are required for otic placode specification. Development, 130(10), 2213–2224. 10.1242/dev.00445 [DOI] [PubMed] [Google Scholar]
- Lo, W.W., Daniels, D.L., Chakeres, D.W., Linthicum, F.H.Jr, Ulmer, J.L., Mark, L.P. et al. (1997) The endolymphatic duct and sac. AJNR. American Journal of Neuroradiology, 18(5), 881–887. [PMC free article] [PubMed] [Google Scholar]
- Lu, W., Zhou, D., Freeman, J.J., Thalmann, I., Ornitz, D.M. & Thalmann, R. (2010) In vitro effects of recombinant otoconin 90 upon calcite crystal growth. Significance of tertiary structure. Hear Res, 268(1–2), 172–183. 10.1016/j.heares.2010.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Z. & Xu, Z. (2002) Effects of saccular otolith removal on hearing sensitivity of the sleeper goby (Dormitator latifrons). Journal of Comparative Physiology A, 188(8), 595–602. [DOI] [PubMed] [Google Scholar]
- Lu, Z., Xu, Z. & Buchser, W. (2003) Acoustic response properties of lagenar nerve fibers in the sleeper goby, Dormitator latifrons. Journal of Comparative Physiology A, 189(12), 889–905. [DOI] [PubMed] [Google Scholar]
- Lu, Z., Xu, Z. & Buchser, W.J. (2010) Frequency coding of particle motion by saccular afferents of a teleost fish. Journal of Experimental Biology, 213(9), 1591–1601. 10.1242/jeb.038836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lufkin, T., Dierich, A., LeMeur, M., Mark, M. & Chambon, P. (1991) Disruption of the Hox‐1.6 homeobox gene results in defects in a region corresponding to its rostral domain of expression. Cell, 66(6), 1105–1119. [DOI] [PubMed] [Google Scholar]
- Lysakowski, A. & Goldberg, J.M. (1997) A regional ultrastructural analysis of the cellular and synaptic architecture in the chinchilla cristae ampullares. The Journal of Comparative Neurology, 389(3), 419–443. 10.1002/(sici)1096-9861(19971222)389:3<419:aid-cne5>3.0.co;2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Q., Anderson, D.J. & Fritzsch, B. (2000) Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. Journal of the Association for Research in Otolaryngology, 1(2), 129–143. 10.1007/s101620010017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Q., Fode, C., Guillemot, F. & Anderson, D.J. (1999) Neurogenin1 and neurogenin2 control two distinct waves of neurogenesis in developing dorsal root ganglia. Genes & Development, 13(13), 1717–1728. 10.1101/gad.13.13.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach, E. (1873) Physikalische versuche über den Gleichgewichtssinn des Menschen. Aus der kk Hof‐und Staatsdruckerei. [Google Scholar]
- Macova, I., Pysanenko, K., Chumak, T., Dvorakova, M., Bohuslavova, R., Syka, J. et al. (2019) Neurod1 Is essential for the primary tonotopic organization and related auditory information processing in the midbrain. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 39(6), 984–1004. 10.1523/jneurosci.2557-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, E.C. & Whitfield, T.T. (2014) RA and FGF signalling are required in the zebrafish otic vesicle to pattern and maintain ventral otic identities. PLoS Genetics, 10(12), e1004858. 10.1371/journal.pgen.1004858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maklad, A., Reed, C., Johnson, N.S. & Fritzsch, B. (2014) Anatomy of the lamprey ear: morphological evidence for occurrence of horizontal semicircular ducts in the labyrinth of Petromyzon marinus . Journal of anatomy, 224(4), 432–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolis, E.N., Yandavi, N., Nadol, J.B.Jr, Eavey, R.D., McKenna, M., Rosenbaum, S. et al. (1996) A gene for non‐syndromic autosomal dominant progressive postlingual sensorineural hearing loss maps to chromosome 14q12–13. Human Molecular Genetics, 5(7), 1047–1050. [DOI] [PubMed] [Google Scholar]
- Mansour, S.L., Goddard, J.M. & Capecchi, M.R. (1993) Mice homozygous for a targeted disruption of the proto‐oncogene int‐2 have developmental defects in the tail and inner ear. Development, 117(1), 13–28.Retrieved from https://dev.biologists.org/content/develop/117/1/13.full.pdf [DOI] [PubMed] [Google Scholar]
- Maroon, H., Walshe, J., Mahmood, R., Kiefer, P., Dickson, C. & Mason, I. (2002) Fgf3 and Fgf8 are required together for formation of the otic placode and vesicle. Development, 129(9), 2099–2108. [DOI] [PubMed] [Google Scholar]
- Martin, D.M., Sheldon, S. & Gorski, J.L. (2001) CHARGE association with choanal atresia and inner ear hypoplasia in a child with a de novo chromosome translocation t (2; 7)(p14; q21. 11). American Journal of Medical Genetics, 99(2), 115–119. [DOI] [PubMed] [Google Scholar]
- Martin, K. & Groves, A.K. (2006) Competence of cranial ectoderm to respond to Fgf signaling suggests a two‐step model of otic placode induction. Development, 133(5), 877–887. [DOI] [PubMed] [Google Scholar]
- Matei, V., Pauley, S., Kaing, S., Rowitch, D., Beisel, K., Morris, K. et al. (2005) Smaller inner ear sensory epithelia in Neurog1 null mice are related to earlier hair cell cycle exit. Developmental Dynamics, 234(3), 633–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, G. & Fuchs, P. (2010) The diverse roles of ribbon synapses in sensory neurotransmission. Nature reviews. Neuroscience, 11(12), 812–822. 10.1038/nrn2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulding, K., Padanad, M.S., Dong, J. & Riley, B.B. (2014) Mesodermal Fgf10b cooperates with other fibroblast growth factors during induction of otic and epibranchial placodes in zebrafish. Developmental Dynamics, 243(10), 1275–1285. 10.1002/dvdy.24119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazan, S., Jaillard, D., Baratte, B. & Janvier, P. (2000) Otx1 gene‐controlled morphogenesis of the horizontal semicircular canal and the origin of the gnathostome characteristics. Evolution & Development, 2(4), 186–193. 10.1046/j.1525-142x.2000.00062.x [DOI] [PubMed] [Google Scholar]
- McCarroll, M.N., Lewis, Z.R., Culbertson, M.D., Martin, B.L., Kimelman, D. & Nechiporuk, A.V. (2012) Graded levels of Pax2a and Pax8 regulate cell differentiation during sensory placode formation. Development, 139(15), 2740–2750. 10.1242/dev.076075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarren, A., Jayabose, S., Ozkaynak, M.F., Tugal, O. & Sandoval, C. (2007) Cleft palate, bilateral external auditory canal atresia, and other midline defects associated with Diamond‐Blackfan anemia: case report. Journal of Pediatric Hematology/Oncology, 29(5), 338–340. [DOI] [PubMed] [Google Scholar]
- McInturff, S., Burns, J.C. & Kelley, M.W. (2018) Characterization of spatial and temporal development of Type I and Type II hair cells in the mouse utricle using new cell‐type‐specific markers. Biology Open, 7(11), bio038083. 10.1242/bio.038083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill, A., Iovino, E., Mansard, L., Vache, C., Baux, D., Bedoukian, E. et al. (2020) SLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect. Brain, 143(8), 2380–2387. 10.1093/brain/awaa176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, A. (1991) The semicircular canals of the hagfish Myxine glutinosa . Journal of Zoology, 224(2), 213–222. 10.1111/j.1469-7998.1991.tb04800.x [DOI] [Google Scholar]
- McVean, A. (2009) The semicircular canals of the hagfish Myxine glutinosa . Journal of Zoology, 224, 213–222. 10.1111/j.1469-7998.1991.tb04800.x [DOI] [Google Scholar]
- Melnick, M., Bixler, D., Nance, W.E., Silk, K. & Yune, H. (1976) Familial branchio‐oto‐renal dysplasia: a new addition to the branchial arch syndromes. Clinical Genetics, 9(1), 25–34. 10.1111/j.1399-0004.1976.tb01546.x [DOI] [PubMed] [Google Scholar]
- Meredith, F.L. & Rennie, K.J. (2016) Channeling your inner ear potassium: K+ channels in vestibular hair cells. Hearing Research, 338, 40–51. 10.1016/j.heares.2016.01.015 [DOI] [PubMed] [Google Scholar]
- Mescher, A. (2010) Chapter 23. The eye and ear: special sense organs. In Weitz, M., Kearns, B. & Boyle, P. (Eds.) Junqueira’s basic histology: text & atlas, 12th edition. New York, NY: McGraw‐Hill Education/Medical. [Google Scholar]
- Miller, N.D., Nance, M.A., Wohler, E.S., Hoover‐Fong, J.E., Lisi, E., Thomas, G.H. et al. (2009) Molecular (SNP) analyses of overlapping hemizygous deletions of 10q25. 3 to 10qter in four patients: evidence for HMX2 and HMX3 as candidate genes in hearing and vestibular function. American Journal of Medical Genetics Part A, 149(4), 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millonig, J.H., Millen, K.J. & Hatten, M.E. (2000) The mouse Dreher gene Lmx1a controls formation of the roof plate in the vertebrate CNS. Nature, 403(6771), 764–769. [DOI] [PubMed] [Google Scholar]
- Morsli, H., Tuorto, F., Choo, D., Postiglione, M.P., Simeone, A. & Wu, D.K. (1999) Otx1 and Otx2 activities are required for the normal development of the mouse inner ear. Development, 126(11), 2335–2343. 10.1242/dev.126.11.2335 [DOI] [PubMed] [Google Scholar]
- Murayama, E., Herbomel, P., Kawakami, A., Takeda, H. & Nagasawa, H. (2005) Otolith matrix proteins OMP‐1 and Otolin‐1 are necessary for normal otolith growth and their correct anchoring onto the sensory maculae. Mechanisms of Development, 122(6), 791–803. 10.1016/j.mod.2005.03.002 [DOI] [PubMed] [Google Scholar]
- Mutai, H., Wasano, K., Momozawa, Y., Kamatani, Y., Miya, F., Masuda, S. et al. (2020) Variants encoding a restricted carboxy‐terminal domain of SLC12A2 cause hereditary hearing loss in humans. PLoS genetics, 16(4), e1008643. 10.1371/journal.pgen.1008643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya, K., Harbidge, D.G., Wangemann, P., Schultz, B.D., Green, E.D., Wall, S.M. et al. (2007) Lack of pendrin HCO3‐ transport elevates vestibular endolymphatic [Ca2+] by inhibition of acid‐sensitive TRPV5 and TRPV6 channels. American Journal of Physiology. Renal Physiology, 292(5), F1314–F1321. 10.1152/ajprenal.00432.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba, A., Abe, S., Shinkawa, H., Kimberling, W.J. & Usami, S.‐I. (2001) Genetic features of hearing loss associated with ear anomalies: PDS and EYA1 mutation analysis. Journal of Human Genetics, 46(9), 518–521. [DOI] [PubMed] [Google Scholar]
- Neuhauss, S.C., Solnica‐Krezel, L., Schier, A.F., Zwartkruis, F., Stemple, D.L., Malicki, J. et al. (1996) Mutations affecting craniofacial development in zebrafish. Development, 123(1), 357–367.Retrieved from https://dev.biologists.org/content/develop/123/1/357.full.pdf [DOI] [PubMed] [Google Scholar]
- Nguyen, K., Hall, A.L. & Jones, J.M. (2015) Expression of myosin VIIA in the developing chick inner ear neurons. Gene Expression Patterns, 19(1–2), 36–44. 10.1016/j.gep.2015.07.001 [DOI] [PubMed] [Google Scholar]
- Nichols, D.H., Pauley, S., Jahan, I., Beisel, K.W., Millen, K.J. & Fritzsch, B. (2008) Lmx1a is required for segregation of sensory epithelia and normal ear histogenesis and morphogenesis. Cell and Tissue Research, 334(3), 339–358. 10.1007/s00441-008-0709-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda, T., Oki, S., Kitajima, K., Harada, T., Komune, S. & Meno, C. (2012) Restriction of Wnt signaling in the dorsal otocyst determines semicircular canal formation in the mouse embryo. Developmental Biology, 362(1), 83–93. 10.1016/j.ydbio.2011.11.019 [DOI] [PubMed] [Google Scholar]
- Odermatt, B. & Lagnado, L. (2009) Ribbon synapses. In Squire, L.R. (Ed.) Encyclopedia of Neuroscience. Oxford: Academic Press, pp. 373–381. [Google Scholar]
- Ohta, S. & Schoenwolf, G.C. (2018) Hearing crosstalk: the molecular conversation orchestrating inner ear dorsoventral patterning. Wiley Interdisciplinary Reviews Developmental Biology, 7(1). 10.1002/wdev.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, S., Wang, B., Mansour, S.L. & Schoenwolf, G.C. (2016) BMP regulates regional gene expression in the dorsal otocyst through canonical and non‐canonical intracellular pathways. Development, 143(12), 2228–2237. 10.1242/dev.137133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama, T., Mohamed, O.A., Taketo, M.M., Dufort, D. & Groves, A.K. (2006) Wnt signals mediate a fate decision between otic placode and epidermis. Development, 133(5), 865. 10.1242/dev.02271 [DOI] [PubMed] [Google Scholar]
- Omata, Y., Nojima, Y., Nakayama, S., Okamoto, H., Nakamura, H., Funahashi, J. et al. (2007) Role of bone morphogenetic protein 4 in zebrafish semicircular canal development. Development, Growth & Differentiation, 49(9), 711–719. 10.1111/j.1440-169X.2007.00964.x [DOI] [PubMed] [Google Scholar]
- Padanad, M.S., Bhat, N., Guo, B. & Riley, B.B. (2012) Conditions that influence the response to Fgf during otic placode induction. Developmental Biology, 364(1), 1–10. 10.1016/j.ydbio.2012.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, B., Askew, C., Galvin, A., Heman‐Ackah, S., Asai, Y., Indzhykulian, A.A. et al. (2017) Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nature biotechnology, 35(3), 264–272. 10.1038/nbt.3801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, N., Jahan, I., Kersigo, J., Kopecky, B., Santi, P., Johnson, S. et al. (2011) Conditional deletion of Atoh1 using Pax2‐Cre results in viable mice without differentiated cochlear hair cells that have lost most of the organ of Corti. Hearing Research, 275(1–2), 66–80. 10.1016/j.heares.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, W., Jin, Y., Chen, J., Rottier, R.J., Steel, K.P. & Kiernan, A.E. (2013) Ectopic expression of activated notch or SOX2 reveals similar and unique roles in the development of the sensory cell progenitors in the mammalian inner ear. The Journal of Neuroscience, 33(41), 16146–16157. 10.1523/jneurosci.3150-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, B.‐Y. & Saint‐Jeannet, J.‐P. (2008) Hindbrain‐derived Wnt and Fgf signals cooperate to specify the otic placode in Xenopus. Developmental Biology, 324(1), 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, E., Hwang, D.‐S., Lee, J.‐S., Song, J.‐I., Seo, T.‐K. & Won, Y.‐J. (2012) Estimation of divergence times in cnidarian evolution based on mitochondrial protein‐coding genes and the fossil record. Molecular Phylogenetics and Evolution, 62(1), 329–345. 10.1016/j.ympev.2011.10.008 [DOI] [PubMed] [Google Scholar]
- Park, W.‐J., Meyers, G.A., Li, X., Theda, C., Day, D., Oriow, S.J. et al. (1995) Novel FGFR2 mutations in Crouzon and Jackson‐Weiss syndromes show allelic heterogeneity and phenotypic variability. Human Molecular Genetics, 4(7), 1229–1233. [DOI] [PubMed] [Google Scholar]
- Pasqualetti, M., Neun, R., Davenne, M. & Rijli, F.M. (2001) Retinoic acid rescues inner ear defects in Hoxa1 deficient mice. Nature Genetics, 29(1), 34–39. [DOI] [PubMed] [Google Scholar]
- Paterson, N.F. (1949) The development of the inner ear of Xenopus laevis. Proceedings of the Zoological Society of London, 119(2), 269–291. 10.1111/j.1096-3642.1949.tb00878.x [DOI] [Google Scholar]
- Pauley, S., Lai, E. & Fritzsch, B. (2006) Foxg1 is required for morphogenesis and histogenesis of the mammalian inner ear. Developmental Dynamics, 235(9), 2470–2482. 10.1002/dvdy.20839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauley, S., Wright, T.J., Pirvola, U., Ornitz, D., Beisel, K. & Fritzsch, B. (2003) Expression and function of FGF10 in mammalian inner ear development. Developmental Dynamics, 227(2), 203–215. 10.1002/dvdy.10297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, E., Cotton, J. & Grant, J. (1996) Structural variation in ciliary bundles of the posterior semicircular canal: quantitative anatomy and computational analysis. Annals of the New York Academy of Sciences, 781(1), 85–102. [DOI] [PubMed] [Google Scholar]
- Petko, J.A., Millimaki, B.B., Canfield, V.A., Riley, B.B. & Levenson, R. (2008) Otoc1: a novel otoconin‐90 ortholog required for otolith mineralization in zebrafish. Dev Neurobiol, 68(2), 209–222. 10.1002/dneu.20587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic, J., Formosa‐Jordan, P., Luna‐Escalante, J.C., Abelló, G., Ibañes, M., Neves, J. et al. (2014) Ligand‐dependent Notch signaling strength orchestrates lateral induction and lateral inhibition in the developing inner ear. Development, 141(11), 2313–2324. 10.1242/dev.108100 [DOI] [PubMed] [Google Scholar]
- Phillips, B.T., Bolding, K. & Riley, B.B. (2001) Zebrafish fgf3 and fgf8 encode redundant functions required for otic placode induction. Developmental Biology, 235(2), 351–365. 10.1006/dbio.2001.0297 [DOI] [PubMed] [Google Scholar]
- Phillips, J.B., Blanco‐Sanchez, B., Lentz, J.J., Tallafuss, A., Khanobdee, K., Sampath, S. et al. (2011) Harmonin (Ush1c) is required in zebrafish Müller glial cells for photoreceptor synaptic development and function. Disease Models & Mechanisms, 4(6), 786–800. 10.1242/dmm.006429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popper, A.N., Platt, C. & Edds, P.L. (1992) Evolution of the vertebrate inner ear: an overview of ideas. The Evolutionary Biology of Hearing, 49–57. [Google Scholar]
- Purves, D., Augustine, G., Fitzpatrick, D., Katz, L., LaMantia, A., McNamara, J. et al. (2001) The otolith organs: the utricle and sacculus. Neuroscience. Retrieved from: https://www.ncbi.nlm.nih.gov/books/NBK10792/ [Google Scholar]
- Quintero‐Rivera, F., Robson, C.D., Reiss, R.E., Levine, D., Benson, C.B., Mulliken, J.B. et al. (2006) Intracranial anomalies detected by imaging studies in 30 patients with Apert syndrome. American Journal of Medical Genetics Part A, 140(12), 1337. [DOI] [PubMed] [Google Scholar]
- Radosevic, M. (2011) Spatial control of inner ear neurogenesis by retinoic acid, Tbx1 and her genes. Universitat Pompeu Fabra. [Google Scholar]
- Radosevic, M., Robert‐Moreno, À., Coolen, M., Bally‐Cuif, L. & Alsina, B. (2011) Her9 represses neurogenic fate downstream of Tbx1 and retinoic acid signaling in the inner ear. Development, 138(3), 397–408. 10.1242/dev.056093 [DOI] [PubMed] [Google Scholar]
- Raft, S., Andrade, L.R., Shao, D., Akiyama, H., Henkemeyer, M. & Wu, D.K. (2014) Ephrin‐B2 governs morphogenesis of endolymphatic sac and duct epithelia in the mouse inner ear. Developmental Biology, 390(1), 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raft, S., Nowotschin, S., Liao, J. & Morrow, B.E. (2004) Suppression of neural fate and control of inner ear morphogenesis by Tbx1. Development, 131(8), 1801–1812. [DOI] [PubMed] [Google Scholar]
- Raphael, Y., Kobayashi, K.N., Dootz, G.A., Beyer, L.A., Dolan, D.F. & Burmeister, M. (2001) Severe vestibular and auditory impairment in three alleles of Ames waltzer (av) mice. Hearing Research, 151(1–2), 237–249. 10.1016/s0378-5955(00)00233-1 [DOI] [PubMed] [Google Scholar]
- Rau, A., Legan, P.K. & Richardson, G.P. (1999) Tectorin mRNA expression is spatially and temporally restricted during mouse inner ear development. The Journal of Comparative Neurology, 405(2), 271–280. [PubMed] [Google Scholar]
- Rennie, K.J. & Correia, M.J. (1994) Potassium currents in mammalian and avian isolated type I semicircular canal hair cells. Journal of Neurophysiology, 71(1), 317–329. 10.1152/jn.1994.71.1.317 [DOI] [PubMed] [Google Scholar]
- Retzius, G. (1881) Das Gehörorgan der Wirbelthiere: Morphologisch‐Histologische Studien. 1, Das Gehörorgan der Fische und Amphibien. Samson & Wallin in Commission. [Google Scholar]
- Riccomagno, M.M., Martinu, L., Mulheisen, M., Wu, D.K. & Epstein, D.J. (2002) Specification of the mammalian cochlea is dependent on Sonic hedgehog. Genes & Development, 16(18), 2365–2378. 10.1101/gad.1013302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccomagno, M.M., Takada, S. & Epstein, D.J. (2005) Wnt‐dependent regulation of inner ear morphogenesis is balanced by the opposing and supporting roles of Shh. Genes & Development, 19(13), 1612–1623. 10.1101/gad.1303905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, N.G., Cremers, C.W., Huygen, P.L., Ikezono, T., Krastins, B., Kremer, H. et al. (2006) Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Human Molecular Genetics, 15(7), 1071–1085. 10.1093/hmg/ddl022 [DOI] [PubMed] [Google Scholar]
- Rohacek, A.M., Bebee, T.W., Tilton, R.K., Radens, C.M., McDermott‐Roe, C., Peart, N. et al. (2017) ESRP1 mutations cause hearing loss due to defects in alternative splicing that disrupt cochlear development. Developmental Cell, 43(3), 318–331.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, M.D. & Pote, K.G. (1984) Some properties of otoconia. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 304(1121), 445–452.Retrieved from http://www.jstor.org/stable/2396113 [DOI] [PubMed] [Google Scholar]
- Ruf, R.G., Xu, P.X., Silvius, D., Otto, E.A., Beekmann, F., Muerb, U.T. et al. (2004) SIX1 mutations cause branchio‐oto‐renal syndrome by disruption of EYA1‐SIX1‐DNA complexes. Proceedings of the National Academy of Sciences of the United States of America, 101(21), 8090–8095. 10.1073/pnas.0308475101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch, A. & Eatock, R.A. (1996) A delayed rectifier conductance in type I hair cells of the mouse utricle. Journal of Neurophysiology, 76(2), 995–1004. 10.1152/jn.1996.76.2.995 [DOI] [PubMed] [Google Scholar]
- Russell, I.J., Legan, P.K., Lukashkina, V.A., Lukashkin, A.N., Goodyear, R.J. & Richardson, G.P. (2007) Sharpened cochlear tuning in a mouse with a genetically modified tectorial membrane. Nature Neuroscience, 10(2), 215–223. 10.1038/nn1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sai, X. & Ladher, R.K. (2008) FGF signaling regulates cytoskeletal remodeling during epithelial morphogenesis. Current Biology, 18(13), 976–981. 10.1016/j.cub.2008.05.049 [DOI] [PubMed] [Google Scholar]
- Sai, X., Yonemura, S. & Ladher, R.K. (2014) Junctionally restricted RhoA activity is necessary for apical constriction during phase 2 inner ear placode invagination. Developmental Biology, 394(2), 206–216. 10.1016/j.ydbio.2014.08.022 [DOI] [PubMed] [Google Scholar]
- Salminen, M., Meyer, B.I., Bober, E. & Gruss, P. (2000) Netrin 1 is required for semicircular canal formation in the mouse inner ear. Development, 127(1), 13–22.Retrieved from https://dev.biologists.org/content/develop/127/1/13.full.pdf [DOI] [PubMed] [Google Scholar]
- Sanyanusin, P., McNoe, L.A., Sullivan, M.J., Weaver, R.G. & Eccles, M.R. (1995) Mutation of PAX2 in two siblings with renal‐coloboma syndrome. Human Molecular Genetics, 4(11), 2183–2184. 10.1093/hmg/4.11.2183 [DOI] [PubMed] [Google Scholar]
- Sapède, D., Dyballa, S. & Pujades, C. (2012) Cell lineage analysis reveals three different progenitor pools for neurosensory elements in the otic vesicle. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 32(46), 16424–16434. 10.1523/JNEUROSCI.3686-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, E.A. (1878) Observations on the nervous system of Aurelia aurita . Philosophical Transactions of the Royal Society of London, 169, 563–575. [Google Scholar]
- Schilder, A.G.M., Su, M.P., Blackshaw, H., Lustig, L., Staecker, H., Lenarz, T. et al. (2019) Hearing protection, restoration, and regeneration: an overview of emerging therapeutics for inner ear and central hearing disorders. Otology & Neurotology, 40(5). Retrieved from https://journals.lww.com/otology‐neurotology/Fulltext/2019/06000/Hearing_Protection,_Restoration,_and_Regeneration_.2.aspx [DOI] [PubMed] [Google Scholar]
- Schlosser, G. & Ahrens, K. (2004) Molecular anatomy of placode development in Xenopus laevis. Developmental Biology, 271(2), 439–466. 10.1016/j.ydbio.2004.04.013 [DOI] [PubMed] [Google Scholar]
- Schlosser, G. & Northcutt, R.G. (2000) Development of neurogenic placodes in Xenopus laevis. The Journal of Comparative Neurology, 418(2), 121–146 [PubMed] [Google Scholar]
- Schorle, H., Meier, P., Buchert, M., Jaenisch, R. & Mitchell, P.J. (1996) Transcription factor AP‐2 essential for cranial closure and craniofacial development. Nature, 381(6579), 235–238. 10.1038/381235a0 [DOI] [PubMed] [Google Scholar]
- Schraders, M., Ruiz‐Palmero, L., Kalay, E., Oostrik, J., del Castillo, F.J., Sezgin, O. et al. (2012) Mutations of the gene encoding otogelin are a cause of autosomal‐recessive nonsyndromic moderate hearing impairment. The American Journal of Human Genetics, 91(5), 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwander, M., Kachar, B. & Müller, U. (2010) The cell biology of hearing. Journal of Cell Biology, 190(1), 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seco, C.Z., Oonk, A.M.M., Domínguez‐Ruiz, M., Draaisma, J.M.T., Gandía, M., Oostrik, J. et al. (2015) Progressive hearing loss and vestibular dysfunction caused by a homozygous nonsense mutation in CLIC5. European Journal of Human Genetics, 23(2), 189–194. 10.1038/ejhg.2014.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi, A., Ceruti, S., Trevisi, P., Gualandi, F., Busi, M., Donati, I. et al. (2011) LAMM syndrome with middle ear dysplasia associated with compound heterozygosity for FGF3 mutations. American Journal of Medical Genetics Part A, 155(5), 1096–1101. [DOI] [PubMed] [Google Scholar]
- Shine, I. & Watson, J. (1967) A new syndrome of sex‐linked congenital conductive deafness. In: Unpublished.
- Shiotsugu, J., Katsuyama, Y., Arima, K., Baxter, A., Koide, T., Song, J. et al. (2004) Multiple points of interaction between retinoic acid and FGF signaling during embryonic axis formation. Development, 131(11), 2653–2667. 10.1242/dev.01129 [DOI] [PubMed] [Google Scholar]
- Simmler, M.‐C., Cohen‐Salmon, M., El‐Amraoui, A., Guillaud, L., Benichou, J.‐C., Petit, C. et al. (2000) Targeted disruption of otog results in deafness and severe imbalance. Nature Genetics, 24(2), 139–143. [DOI] [PubMed] [Google Scholar]
- Simmler, M.C., Zwaenepoel, I., Verpy, E., Guillaud, L., Elbaz, C., Petit, C. et al. (2000) Twister mutant mice are defective for otogelin, a component specific to inner ear acellular membranes. Mammalian Genome, 11(11), 960–966. [DOI] [PubMed] [Google Scholar]
- Slepecky, N.B. (1996) Structure of the mammalian cochlea. In Dallos, P., Popper, A.N. & Fay, R.R. (Eds.) The cochlea. New York, NY: Springer, pp. 44–129. [Google Scholar]
- Smith, R.J.H., Lee, E.C., Kimberling, W.J., Daiger, S.P., Pelias, M.Z., Keats, B.J.B. et al. (1992) Localization of two genes for usher syndrome type I to chromosome 11. Genomics, 14(4), 995–1002. 10.1016/s0888-7543(05)80122-3 [DOI] [PubMed] [Google Scholar]
- Smith, R.J.H., Pelias, M.Z., Daiger, S.P., Keats, B., Kimberling, W. & Hejtmancik, J.F. (1992) Clinical variability and genetic heterogeneity within the Acadian Usher population. American Journal of Medical Genetics, 43(6), 964–969. 10.1002/ajmg.1320430612 [DOI] [PubMed] [Google Scholar]
- Söllner, C., Rauch, G.J., Siemens, J., Geisler, R., Schuster, S.C., Müller, U. et al. (2004) Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature, 428(6986), 955–959. 10.1038/nature02484 [DOI] [PubMed] [Google Scholar]
- Song, M.H., Kwon, T.‐J., Kim, H.R., Jeon, J.H., Baek, J.‐I., Lee, W.‐S. et al. (2013) Mutational analysis of EYA1, SIX1 and SIX5 genes and strategies for management of hearing loss in patients with BOR/BO syndrome. PLoS One, 8(6), e67236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoor, F., Garland, T.Jr, Krovitz, G., Ryan, T.M., Silcox, M.T. & Walker, A. (2007) The primate semicircular canal system and locomotion. Proceedings of the National Academy of Sciences of the United States of America, 104(26), 10808–10812. 10.1073/pnas.0704250104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steevens, A.R., Sookiasian, D.L., Glatzer, J.C. & Kiernan, A.E. (2017) SOX2 is required for inner ear neurogenesis. Scientific Reports, 7(1), 4086. 10.1038/s41598-017-04315-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steventon, B., Mayor, R. & Streit, A. (2012) Mutual repression between Gbx2 and Otx2 in sensory placodes reveals a general mechanism for ectodermal patterning. Developmental Biology, 367(1), 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinckens, C., Huygen, P.L., Joosten, F.B., Van Camp, G., Otten, B. & Cremers, C.W. (2001) Fluctuant, progressive hearing loss associated with Menière like vertigo in three patients with the Pendred syndrome. International Journal of Pediatric Otorhinolaryngology, 61(3), 207–215. 10.1016/s0165-5876(01)00573-0 [DOI] [PubMed] [Google Scholar]
- Straka, H. (2020) 6.01 ‐ Overview: Vestibular System. In Fritzsch, B. (Ed.) The senses: A comprehensive reference, 2nd edition. Oxford: Elsevier, pp. 1–5. [Google Scholar]
- Straka, H., Zwergal, A. & Cullen, K.E. (2016) Vestibular animal models: contributions to understanding physiology and disease. Journal of Neurology, 263(1), 10–23. 10.1007/s00415-015-7909-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznajer, Y., Coldéa, C., Meire, F., Delpierre, I., Sekhara, T. & Touraine, R.L. (2008) A de novo SOX10 mutation causing severe type 4 Waardenburg syndrome without Hirschsprung disease. American Journal of Medical Genetics Part A, 146(8), 1038–1041. [DOI] [PubMed] [Google Scholar]
- Tekin, M., Hişmi, B.Ö., Fitoz, S., Özdağ, H., Cengiz, F.B., Sırmacı, A. et al. (2007) Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. The American Journal of Human Genetics, 80(2), 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekin, M., Öztürkmen Akay, H., Fitoz, S., Birnbaum, S., Cengiz, F., Sennaroğlu, L. et al. (2008) Homozygous FGF3 mutations result in congenital deafness with inner ear agenesis, microtia, and microdontia. Clinical Genetics, 73(6), 554–565. [DOI] [PubMed] [Google Scholar]
- Tekin, M., Sırmacı, A., Yüksel‐Konuk, B., Fitoz, S. & Sennaroğlu, L. (2009) A complex TFAP2A allele is associated with branchio‐oculo‐facial syndrome and inner ear malformation in a deaf child. American Journal of Medical Genetics Part A, 149(3), 427–430. [DOI] [PubMed] [Google Scholar]
- Tischfield, M.A., Bosley, T.M., Salih, M.A., Alorainy, I.A., Sener, E.C., Nester, M.J. et al. (2005) Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nature Genetics, 37(10), 1035–1037. 10.1038/ng1636 [DOI] [PubMed] [Google Scholar]
- Toriello, H. & Smith, S. (2013) Hereditary hearing loss and its syndromes. New York, NY: Oxford University Press. [Google Scholar]
- Tsai, H., Hardisty, R.E., Rhodes, C., Kiernan, A.E., Roby, P., Tymowska‐Lalanne, Z. et al. (2001) The mouse slalom mutant demonstrates a role for Jagged1 in neuroepithelial patterning in the organ of Corti. Human Molecular Genetics, 10(5), 507–512. [DOI] [PubMed] [Google Scholar]
- Tsujikawa, S., Yamashita, T., Tomoda, K., Iwai, H., Kumazawa, H., Cho, H. et al. (1993) Effects of acetazolamide on acid‐base balance in the endolymphatic sac of the guinea pig. Acta Oto‐Laryngologica Supplement, 500, 50–53. 10.3109/00016489309126179 [DOI] [PubMed] [Google Scholar]
- Tsukamoto, K., Suzuki, H., Harada, D., Namba, A., Abe, S. & Usami, S.‐I. (2003) Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. European Journal of Human Genetics, 11(12), 916–922. [DOI] [PubMed] [Google Scholar]
- Van de Laar, I., Dooijes, D., Hoefsloot, L., Simon, M., Hoogeboom, J. & Devriendt, K. (2007) Limb anomalies in patients with CHARGE syndrome: an expansion of the phenotype. American Journal of Medical Genetics Part A, 143A(22), 2712–2715. 10.1002/ajmg.a.32008 [DOI] [PubMed] [Google Scholar]
- Villegas, H., Merker, H.J., Helling, K., Clarke, A.H. & Scherer, H. (2001) Electron microscopic studies of ion‐ and H2O‐transporting epithelial cells in the horizontal ampulla of the pigeon. Histology and Histopathology, 16(4), 1161–1174. 10.14670/hh-16.1161 [DOI] [PubMed] [Google Scholar]
- Vissers, L.E., van Ravenswaaij, C.M., Admiraal, R., Hurst, J.A., de Vries, B.B., Janssen, I.M. et al. (2004) Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nature Genetics, 36(9), 955–957. [DOI] [PubMed] [Google Scholar]
- Wackym, P.A., Friberg, U., Bagger‐Sjöbäck, D., Linthicum, F.H., Friedmann, I. & Rask‐Andersen, H. (1987) Human endolymphatic sac: possible mechanisms of pressure regulation. The Journal of Laryngology & Otology, 101(8), 768–779. 10.1017/s0022215100102713 [DOI] [PubMed] [Google Scholar]
- Wada, T., Wakabayashi, Y., Takahashi, S., Ushiki, T., Kikkawa, Y., Yonekawa, H. et al. (2001) A point mutation in a cadherin Gene, Cdh23, causes deafness in a novel mutant, waltzer mouse niigata. Biochemical and Biophysical Research Communications, 283(1), 113–117. 10.1006/bbrc.2001.4724 [DOI] [PubMed] [Google Scholar]
- Wang, T.J., Huang, C.B., Tsai, F.J., Wu, J.Y., Lai, R.B. & Hsiao, M. (2002) Mutation in the FGFR2 gene in a Taiwanese patient with Beare‐Stevenson cutis gyrata syndrome. Clinical Genetics, 61(3), 218–221. [DOI] [PubMed] [Google Scholar]
- Wang, W., Chan, E.K., Baron, S., Van De Water, T. & Lufkin, T. (2001) Hmx2 homeobox gene control of murine vestibular morphogenesis. Development, 128(24), 5017–5029. [DOI] [PubMed] [Google Scholar]
- Wang, W., Grimmer, J.F., Van De Water, T.R. & Lufkin, T. (2004) Hmx2 and Hmx3 homeobox genes direct development of the murine inner ear and hypothalamus and can be functionally replaced by Drosophila Hmx. Developmental Cell, 7(3), 439–453. [DOI] [PubMed] [Google Scholar]
- Wangemann, P. (1995) Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hearing Research, 90(1–2), 149–157. 10.1016/0378-5955(95)00157-2 [DOI] [PubMed] [Google Scholar]
- Waterman, R.E. & Bell, D.H. (1984) Epithelial fusion during early semicircular canal formation in the embryonic zebrafish, Brachydanio rerio . The Anatomical Record, 210(1), 101–114. [DOI] [PubMed] [Google Scholar]
- Weil, D., Levy, G., Sahly, I., Levi‐Acobas, F., Blanchard, S., El‐Amraoui, A. et al. (1996) Human myosin VIIA responsible for the Usher 1B syndrome: a predicted membrane‐associated motor protein expressed in developing sensory epithelia. Proceedings of the National Academy of Sciences of the United States of America, 93(8), 3232–3237. 10.1073/pnas.93.8.3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wersall, J. (1956) Studies on the structure and innervation of the sensory epithelium of the cristae ampulares in the guinea pig; a light and electron microscopic investigation. Acta Oto‐Laryngologica Supplement, 126, 1–85. [PubMed] [Google Scholar]
- Wersäll, J. & Bagger‐Sjöbäck, D. (1974) Morphology of the vestibular sense organ. In Kornhuber, H.H. (Ed.) Vestibular System Part 1: Basic Mechanisms. Berlin, Heidelberg: Springer, pp. 123–170. [Google Scholar]
- Wesdorp, M., de Koning Gans, P.A., Schraders, M., Oostrik, J., Huynen, M.A., Venselaar, H. et al. (2018) Heterozygous missense variants of LMX1A lead to nonsyndromic hearing impairment and vestibular dysfunction. Human genetics, 137(5), 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wever, E.G. (1974) The evolution of vertebrate hearing. In Keidel, W.D. & Neff, W.D. (Eds.) Auditory System. Handbook of Sensory Physiology. 5, Berlin, Heidelberg: Springer, pp. 423–454. [Google Scholar]
- Wever, E.G., Mccormick, J.G., Palin, J. & Ridgway, S.H. (1971) The cochlea of the dolphin, Tursiops truncatus: hair cells and ganglion cells. Proceedings of the National Academy of Sciences of the United States of America, 68(12), 2908–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield, T.T., Granato, M., van Eeden, F.J., Schach, U., Brand, M., Furutani‐Seiki, M. et al. (1996) Mutations affecting development of the zebrafish inner ear and lateral line. Development, 123(1), 241–254.Retrieved from https://dev.biologists.org/content/develop/123/1/241.full.pdf [DOI] [PubMed] [Google Scholar]
- Winbo, A. & Rydberg, A. (2015) Vestibular dysfunction is a clinical feature of the Jervell and Lange‐Nielsen Syndrome. Scandinavian Cardiovascular Journal, 49(1), 7–13. 10.3109/14017431.2014.988172 [DOI] [PubMed] [Google Scholar]
- Wright, T.J. & Mansour, S.L. (2003) Fgf3 and Fgf10 are required for mouse otic placode induction. Development, 130(15), 3379–3390. 10.1242/dev.00555 [DOI] [PubMed] [Google Scholar]
- Xiang, M., Gan, L., Li, D., Chen, Z.‐Y., Zhou, L., O’Malley, B.W. et al. (1997) Essential role of POU–domain factor Brn‐3c in auditory and vestibular hair cell development. Proceedings of the National Academy of Sciences of the United States of America, 94(17), 9445–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P.‐X., Adams, J., Peters, H., Brown, M.C., Heaney, S. & Maas, R. (1999) Eya1‐deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nature Genetics, 23(1), 113–117. [DOI] [PubMed] [Google Scholar]
- Xu, Y., Zhang, H., Yang, H., Zhao, X., Lovas, S. & Lundberg, Y.W. (2010) Expression, functional, and structural analysis of proteins critical for otoconia development. Developmental Dynamics, 239(10), 2659–2673. 10.1002/dvdy.22405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, P.K., de Souza, T.A., Antiorio, A.T., Zanatto, D.A., Garcia‐Gomes, M.d.S.A., Alexandre‐Ribeiro, S.R. et al. (2019) Genetic and behavioral characterization of a Kmt2d mouse mutant, a new model for Kabuki Syndrome. Genes, Brain and Behavior, 18(8), e12568. [DOI] [PubMed] [Google Scholar]
- Yamoah, E.N., Li, M., Shah, A., Elliott, K.L., Cheah, K., Xu, P.‐X. et al. (2020) Using Sox2 to alleviate the hallmarks of age‐related hearing loss. Ageing Research Reviews, 59, 101042. 10.1016/j.arr.2020.101042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C.H., Cheng, C.H., Chen, G.D., Liao, W.H., Chen, Y.C., Huang, K.Y. et al. (2011) Zona pellucida domain‐containing protein β‐tectorin is crucial for zebrafish proper inner ear development. PLoS One, 6(8), e23078. 10.1371/journal.pone.0023078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., Zhao, X., Xu, Y., Wang, L., He, Q. & Lundberg, Y.W. (2011) Matrix recruitment and calcium sequestration for spatial specific otoconia development. PLoS One, 6(5), e20498. 10.1371/journal.pone.0020498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yariz, K.O., Duman, D., Zazo Seco, C., Dallman, J., Huang, M., Peters, T.A. et al. (2012) Mutations in OTOGL, encoding the inner ear protein otogelin‐like, cause moderate sensorineural hearing loss. American Journal of Human Genetics, 91(5), 872–882. 10.1016/j.ajhg.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, N.M. (2013) Macroevolutionary diversity of amniote limb proportions predicted by developmental interactions. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution, 320(7), 420–427. [DOI] [PubMed] [Google Scholar]
- Yuan, Q., Yu, L., Shi, D., Ke, X. & Zhang, H. (2015) Anxiety and depression among patients with different types of vestibular peripheral vertigo. Medicine, 94(5), e453. 10.1097/MD.0000000000000453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X., Yang, H., Yamoah, E.N. & Lundberg, Y.W. (2007) Gene targeting reveals the role of Oc90 as the essential organizer of the otoconial organic matrix. Developmental Biology, 304(2), 508–524. 10.1016/j.ydbio.2007.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, Q.Y., Yan, D., Ouyang, X.M., Du, L.L., Yu, H., Chang, B. et al. (2004) Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Human Molecular Genetics, 14(1), 103–111. 10.1093/hmg/ddi010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, W., Huang, L., Wei, Z.B., Silvius, D., Tang, B. & Xu, P.X. (2003) The role of Six1 in mammalian auditory system development. Development, 130(17), 3989–4000. 10.1242/dev.00628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, D., Erickson, C., Kim, E.‐H., Jin, D., Fritzsch, B. & Xu, P.‐X. (2008) Eya1 gene dosage critically affects the development of sensory epithelia in the mammalian inner ear. Human Molecular Genetics, 17(21), 3340–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.