

Abstract
The incidence of zoonotic diseases is increasing worldwide, which makes identifying parasites likely to become zoonotic and hosts likely to harbour zoonotic parasites a critical concern. Prior work indicates that there is a higher risk of zoonotic spillover accruing from closely related hosts and from hosts that are infected with a high phylogenetic diversity of parasites. This suggests that host and parasite evolutionary history may be important drivers of spillover, but identifying whether host–parasite associations are more strongly structured by the host, parasite or both requires co-phylogenetic analyses that combine host–parasite association data with host and parasite phylogenies. Here, we use host–parasite datasets containing associations between helminth taxa and free-range mammals in combination with phylogenetic models to explore whether host, parasite, or both host and parasite evolutionary history influences host–parasite associations. We find that host phylogenetic history is most important for driving patterns of helminth-mammal association, indicating that zoonoses are most likely to come from a host's close relatives. More broadly, our results suggest that co-phylogenetic analyses across broad taxonomic scales can provide a novel perspective for surveying potential emerging infectious diseases.
This article is part of the theme issue ‘Infectious disease macroecology: parasite diversity and dynamics across the globe’.
Keywords: host–parasite, network, mammals, helminths, co-phylogenetics, zoonoses
1. Background
Zoonoses, parasites transmitted between humans and animal taxa, are a rising threat to public health and wildlife conservation. With the increase in the emergence of zoonotic infectious diseases, research to predict and survey potentially zoonotic host–parasite interactions is critical [1–4]. Recent studies suggest that host shifts are more likely to occur between closely related hosts, indicating, for example, that close relatives of humans are most likely to cause a spillover event [5–9]. Spillover also probably depends on parasite characteristics, with parasites with broad host ranges more likely to cause a zoonotic infection [10,11]. Analyses that combine association data with host and parasite phylogenies can help us to identify how host–parasite interactions are shaped by both host and parasite evolutionary history, thereby allowing us to more accurately identify potential emerging infectious diseases.
We explore four alternative hypotheses for how host and parasite evolutionary history could influence host–parasite associations. The first predicts that these associations are random with respect to both the host and parasite phylogenies; in this scenario, host–parasite interactions are driven by abiotic or biotic factors that are not phylogenetically structured. For instance, the geographical separation appears to be the primary determinant of interactions between hosts and parasites in several systems (e.g. nematodes and stick insects [12]; parasites of primates [5]; parasites of carnivores [8]). The second hypothesis predicts that host–parasite associations are driven by codivergence, such that closely related hosts are infected by closely related parasites (and thus closely related parasites infect closely related hosts). This pattern has been observed in pocket gophers with chewing lice [13,14], although strict cospeciation appears to be rare, as most parasites infect multiple hosts, and most hosts are infected by multiple parasites [15,16]. The last two hypotheses predict that the pattern of host–parasite association is largely driven by either host or parasite evolution alone. For instance, if the pattern was driven by host evolution, closely related hosts would be infected by similar parasites, regardless of the relatedness of the parasites, as has been shown for rabies viruses in North American bats [17]. Alternatively, if the pattern was driven by parasite evolution, closely related parasites would infect similar hosts, regardless of the relatedness of those hosts. Although this hypothesis has been rarely indicated in the literature, it is reasonable to assume that closely related parasites would infect similar hosts as a result of the evolution of similar transmission modes and immune evasion strategies. While we look at these hypotheses separately, they are not independent of one another and more than one of these hypotheses could be true at the same time.
Determining which of these hypotheses provides the best fit with the data would be useful for identifying potential zoonotic threats. For example, if neither host nor parasite evolution shapes host–parasite interactions, then determining the ecological factors, abiotic and biotic, that drive these interactions becomes the critical step in identifying potential sources of emerging infectious diseases. By contrast, if host–parasite associations are driven by codivergence, then the parasites most likely to spillover will be those that both infect the close relatives of the focal host and are closely related to parasites already infecting that host. If host evolution alone plays the dominant role, then the surveillance strategy is similar, but we must pay attention to all the parasites of the focal host's close relatives, regardless of whether they are related to the host's known parasites. For example, the current strategy for identifying novel human zoonoses is to survey all of the parasites of Primates, essentially assuming that any of their parasites have high potential for spilling over into humans [18–20]. On the other hand, if parasite evolution alone plays the dominant role, surveying for potential zoonoses should focus on parasite clades with species known to infect humans; practically, this is more challenging because we cannot narrow surveillance efforts to particular host taxa, potentially greatly enlarging the taxonomic, geographical, and ecological range of the survey.
Here, we use a host–parasite database (global mammal parasite database, GMPD [21]) and a museum-verified dataset (Nearctic dataset [22]) containing associations between helminth taxa and free-range mammals in combination with reconstructed host and parasite molecular phylogenies [23,24] to assess evidence for each of the above mechanisms in shaping the pattern of helminth-mammal associations. Food-borne zoonotic helminths are increasing in prevalence and distribution [25]. For instance, Oesophagostoma bifurcum (the causative agent of oesophagostomiasis) and Trichenella spp. (the causative agent of trichinellosis) have increased in prevalence [26,27]. We conducted co-phylogenetic analyses using multiple methods [28–32]. Our results suggest that host phylogenetic history plays the dominant role in driving patterns of helminth-mammal association, indicating that zoonoses are most likely to come from a host's close relatives.
2. Methods
The data for this analysis comprises: (i) an incidence matrix of host–parasite interactions; (ii) a host phylogeny; and (iii) a parasite phylogeny. We use two different mammal-helminth datasets for our analyses: the first comes from the GMPD [21] and the second from a dataset of museum-verified Nearctic host–parasite associations [24]. The GMPD contains over 10 000 records of helminths in the phyla Acanthocephala, Nematoda and Platyhelminthes infecting hosts in the orders Artiodactyla (even-toed ungulates), Carnivora, Perissodactyla (odd-toed ungulates) and Primates. This dataset omits rodents and includes associations derived from the literature, and it is possible that some parasites may have been misidentified in the original publications. Parasite misidentification could lead to an inaccurate interpretation of parasite sharing. For example, a parasite that infects two hosts, but which was misidentified in one host could lead us to underestimate parasite sharing, whereas single-host parasites which were classified as the same species in two separate hosts would lead us to overestimate parasite sharing. To evaluate the sensitivity of our analyses to such errors, we also analyse data from the Nearctic dataset (which was restricted to voucher-verified museum specimens), for which we have more confidence in correct parasite identification, although cryptic parasite diversity (that is, evolutionary distinctiveness that is not manifested in observable phenotypes) could still pose a challenge to taxonomists in some instances. The Nearctic dataset contains a similar phylogenetic breadth of helminths infecting mammals in the orders Artiodactyla, Carnivora, Didelphimorphia (opossums), Eulipotyphla (moles and shrews), Lagomorpha (rabbits) and Rodentia. Host–parasite association data comes from the H. W. Manter Parasitology Collection, the former United States National Parasitology Collection, the Museum of Southwestern Biology and the Canadian Museum of Nature. We also analysed a smaller dataset containing only the Primate-helminth associations in the GMPD. The use of the three different datasets allows us to address the sensitivity of our results to potential sampling biases that may be present in any one dataset.
The host phylogeny comes from the Phylogenetic Atlas of Mammal Macroecology's mammal phylogeny (PHYLACINE [23,33]) trimmed to include only the hosts found in each dataset. The parasite phylogeny comes from a novel molecular phylogeny of parasitic helminths built from sequences of parasite genes (CO1, 18S and 28S) found in GenBank (see [24] for details on phylogeny construction). Subsetting the GMPD to include only hosts and parasites for which phylogenetic information is available yielded 971 associations among 197 mammal species and 248 helminth species. Subsetting the Nearctic dataset resulted in 379 associations among 69 mammal species and 90 helminth species. The Primate dataset included 113 associations among 58 primate species and 31 helminth species.
We combined the phylogenies and host–parasite association datasets and conducted co-phylogenetic analyses using several different methodological approaches. We first applied the method of Hommola et al. [29], which tests for a correlation between the pairwise phylogenetic distance between all pairs of hosts and all pairs of parasites in the dataset. That is, the method looks at pairs of host–parasite associations and calculates the phylogenetic distance between the hosts in the pair and between the parasites in the pair: the host distance will be zero for pairs representing two parasites infecting the same host; similarly, the parasite distance will be zero for pairs representing a parasite infecting two hosts. After computing these branch lengths for all pairs, the method then estimates the correlation between the host distances and the parasite distances over all host–parasite association pairs. A high correlation means that, when two hosts are far apart on the tree, the parasites that infect them also tend to be far apart; similarly, when hosts are closely related, their parasites tend to be closely related as well. A low correlation would mean that there is no relationship between the distance between hosts and between parasites. We compute this correlation in our three datasets (in R, following Hadfield et al. [32]), and then compare the observed correlation against randomly permuted data.
The association data is randomly permuted in three ways. Legendre et al. [28] proposed a permutation such that each parasite infects the same number of hosts, but the identity of those hosts is randomly determined (referenced as L1). An alternative is to permute the matrix of associations such that each host is infected by the same number of parasites, but the identity of those parasites is randomly determined (referenced as L2). Hommola et al. [29] proposed a third possibility, where only the total number of host–parasite associations is preserved, and those associations are randomly determined (referenced as H). Under all perturbations, the test statistic is computed to produce a null distribution against which the observed value can be compared. As pointed out by Hadfield et al. [32], comparing the value of the test statistic against the null distributions generated by different types of permutation provides slightly different information. In particular, the first permutation (L1) is sensitive to host–parasite coevolution, structure in host evolutionary interactions (which occur if related hosts are infected by similar parasites, irrespective of the parasite phylogeny), structure in parasite evolutionary interactions (which occur if related parasites infect similar hosts, irrespective of the host phylogeny), and phylogenetic signal in the parasite species richness within hosts (because the permutation alters the number of parasites infecting each host). The second permutation (L2) tests for coevolution, structure in host and parasite evolutionary interactions, and for phylogenetic signal in the host range of parasites (because the permutation alters the number of hosts each parasite infects). The third permutation (H) tests for coevolution, structure in host and parasite evolutionary interactions, and for phylogenetic signal in both parasite species richness and host range. This method can test whether host–parasite associations are random or shaped by phylogenetic codivergence, but it cannot provide information about whether host or parasite evolution is more important; if closely related hosts share similar, but unrelated, parasites (or the opposite), the observed correlation would still probably be zero, as it would in the case where host and parasite associations are random.
The second method we use is the ParaFitGlobal method of Legendre et al., which evaluates the evidence for coevolution between parasites and hosts [28]. This method works by testing for congruence between host and parasite phylogenetic trees, that is, it tests whether hosts and their parasites have equivalent positions in their respective trees. Perfect congruence would signal tight codiversification of specialist parasites with their hosts, whereas no congruence would signal that host–parasite associations are formed randomly with respect to the evolutionary history of each species. As such, the null hypothesis that this method is testing is that the evolution of hosts and parasites are independent. This method was one of the first developed that could account for the fact that many parasites can infect more than one host, and that hosts are often infected by many parasites. ParaFitGlobal computes a ‘fourth corner’ statistic [24] based on the product of matrices describing (i) the presence/absence of each host–parasite association; (ii) the parasite phylogenetic tree; and (iii) the host phylogenetic tree. To determine whether this statistic has a value that is different from that expected by chance, the presence/absence data is randomly permuted using the same set of permutations described above (in R, following Hadfield et al. [32]).
The third and fourth methods, in contrast with the first two, provide additional information that can help us to determine whether the pattern of host–parasite association is more strongly structured by host evolutionary history, parasite evolutionary history or coevolution. The third method, from Ives & Godfray [30], transforms the branch lengths of the host and parasite phylogenies, thereby adjusting the covariance between any two tips of either tree, to maximize the fit of an evolutionary model to the observed host–parasite association data. By contrast, ParaFitGlobal holds the branch lengths, and thus covariance, constant [28]. The covariance in the Ives and Godfray method is specified by an Ornstein-Uhlenbeck process with a parameter d that determines the strength of the phylogenetic signal, changing the value of d essentially transforms the underlying branch lengths [34]. The Ornstein-Uhlenbeck model is often described as a model for stabilizing selection: a deterministic tendency towards an ‘optimal’ value for a trait evolving along a phylogeny [35]. If d = 0, there is no phylogenetic covariance between tips, which can be described as a star phylogeny, whereas d = 1 implies no stabilizing selection and a covariance that is described by Brownian motion (BM) evolution. A value of 0 < d < 1 implies some amount of stabilizing selection, and d > 1 suggests disruptive selection. Because there are two phylogenies, the method assumes that there is some value, dh, that best describes the covariance between host species and a separate parameter, dp, that describes the covariance between parasite species. The method estimates the values of dh and dp that minimize the mean square error (MSE) between the model-predicted host–parasite associations and the observed host–parasite associations. Comparing the values of dh and dp give a sense of how much phylogenetic signal in parasite–hosts associations is owing to the host versus the parasite, and comparing these values against either d = 0 or d = 1 gives a sense of the overall phylogenetic signal in the data.
We use the Ives and Godfray method for the Nearctic and Primate dataset, fitting simplified versions of the model that either fix dh = dp = 0 (star phylogeny) or dh = dp = 1 (BM). Then, we fit the full model that fits dh and dp to the data. Unfortunately, the complete helminth GMPD dataset was too large for the full method to work: both the code provided in the electronic supplementary material of Ives & Godfray [30] and in the R package picante [36] that implements the method generate matrices that are so large that they consume all available computer memory. However, Hadfield et al. [32] provide code for computing a statistic that is proportional to MSE under the assumption that dh = dp = 1 (BM evolution); their code does not generate large matrices and is thus possible to apply all three datasets. This statistic is very similar to that of Legendre et al. [28]. It is also possible to compute the MSE under the assumption that dh = dp = 0 (star phylogeny) for all three models. We were therefore able to compute these statistics for all three datasets and use the same set of permutations described above to evaluate whether their values are different from a null distribution based on randomly permuted host–parasite associations. However, it was not possible to find the maximum-likelihood values of dh and dp.
The fourth method we explore is from Krasnov et al. [31], which first estimates the modularity of the network formed by host–parasite associations: in a host–parasite network with high modularity, one would find clusters of hosts and parasites that interact mainly with one another, and not with other clusters of hosts and parasites. Modules are computed using the cluster_walktrap function in the R package igraph [37]. It then estimates whether the hosts and parasites that belong to the same module tend to be closely related by calculating the correlation between co-membership in a module and phylogenetic distance. Essentially, the method evaluates whether, across all hosts (and parasites), the pairwise phylogenetic distance between hosts (or parasites) within a single module is less than the pairwise phylogenetic distance between hosts (or parasites) in separate modules. If so, then there is evidence for phylogenetic structuring of host–parasite associations. Again, we use permutations of the host–parasite association data to determine whether the observed correlations between co-membership and phylogenetic distance are different to that expected from randomly constructed host–parasite association networks.
3. Results
(a) . Hommola method
Using the Hommola method, the null hypothesis is that there is no correlation between the phylogenetic distance between any pair of hosts and the parasites that infect them.
A high correlation, on the other hand, indicates that the phylogenetic distance between a pair of hosts is mirrored in the parasites that infect them, suggesting a pattern of codiversification. We find that there is only a low correlation between the pairwise phylogenetic distances among all pairs of hosts and all pairs of parasites in the GMPD (r < 0.001), and this low correlation is similar to what is observed in randomly permuted datasets (table 1) Similarly, there is no biologically meaningful correlation between shared branch lengths in the Nearctic dataset. This indicates a lack of support for codiversification structuring host–parasite associations in these datasets. However, there is a significant positive correlation in the L1 permutation test of the Primate dataset, suggesting that there is a detectable phylogenetic signal in the pattern of host–parasite associations among Primates and their parasites that is more strongly influenced by the host phylogeny, although the statistical relationship is also weak (table 1).
Table 1.
Significance of the observed values of the Hommola et al. [29] test statistic based on three different bootstrap permutation methods. (L1: where each parasite infects the same number of hosts, but the identity of those hosts is randomly determined; L2: where each host is infected by the same number of parasites, but the identity of those parasites is randomly determined and H: where the total number of host–parasite associations is preserved, but associations are randomly determined.)
| dataset | test statistic | L1 p-value | L2 p-value | H p-value |
|---|---|---|---|---|
| GMPD | 0.00 | 0.353 | 0.365 | 0.334 |
| Primates only (GMPD) | 0.06 | 0.001 | 0.115 | 0.071 |
| Nearctic | −0.00 | 0.365 | 0.383 | 0.336 |
(b) . Legendre method
Using ParaFitGlobal, the null hypothesis is that host and parasite evolution are independent. We see evidence that would support either coevolution, host evolutionary interactions (closely related hosts infected by similar, but unrelated, parasites), parasite evolutionary interactions (closely related parasites infected similar, but unrelated, hosts), or phylogenetic signal in parasite species richness (number of parasite species that infect a host), or host range (number of host species each parasite infects) in all three datasets. The observed value of the test statistic (which has no direct interpretation) is more extreme than the values observed for almost all of the bootstrap permutations using any method of permutation (table 2). Thus, there appears to be strong evidence for the phylogenetic signal in the pattern of host–parasite associations, though using this method, we are not able to determine whether that is primarily a result of evolutionary signal coming through host or parasite evolutionary history.
Table 2.
Significance of the observed values of the Legendre et al. [28] test statistic based on three different bootstrap permutation methods. (L1: where each parasite infects the same number of hosts, but the identity of those hosts is randomly determined; L2: where each host is infected by the same number of parasites, but the identity of those parasites is randomly determined and H: where the total number of host–parasite associations is preserved, but associations are randomly determined.)
| dataset | test statistic | L1 p-value | L2 p-value | H p-value |
|---|---|---|---|---|
| GMPD | 637.82 | <0.001 | <0.001 | <0.001 |
| Primates only (GMPD) | 6.70 | 0.004 | <0.001 | 0.001 |
| Nearctic | 12.31 | <0.001 | <0.001 | <0.001 |
(c) . Ives and Godfray method
Using the method of Ives & Godfray [30], we compare the fit of three models to the data: a non-phylogenetic model (dh = dp = 0), a BM model(dh = dp = 1) and a model that transforms the branch lengths of the phylogeny to maximize the fit of the model (dh, dp flexible). We found that for the Nearctic dataset, the best-fitting evolutionary model was a star phylogeny (dh = dp = 0). This can be seen both in the MSE estimates of the different models and from the estimates of dh and dp in the full method (table 3). For the Primate dataset, however, the best-fitting evolutionary model estimated dh and dp, although the best-fitting estimates for both dh and dp are small. However, the value of dh is significantly larger than that of dp, suggesting a larger role for the host phylogeny in shaping host–parasite associations among Primates and their parasites (table 3).
Table 3.
MSE estimates for each model fitted to the Primate (from the GMPD) and the Nearctic datasets. (Estimates for dh and dp included in parentheses.)
| dataset | MSE full (dh, dp estimated) | MSE star (dh = dp = 0) | MSE BM (dh = dp = 1) |
|---|---|---|---|
| Primates only (GMPD) | 0.056 (dh = 4.9 × 10−7, dp = 8.5 × 10−27) | 0.059 (dh = dp = 0) | 1.92 (dh = dp = 1) |
| Nearctic | 0.059 (dh = 3.9 × 10−95, dp = 2.5 × 10−14) | 0.057 (dh = dp = 0) | 30.49 (dh = dp = 1) |
Using the permutation test again allows us to test the null hypothesis that the pattern of host–parasite associations is independent of either the host or parasite evolutionary history. Interestingly, however, the permutation test results indicate that the fit of the BM model to the real data in all three datasets is often significantly better than the fit of that model to randomly permuted data (table 4). This suggests that, despite the relatively poor fit of the BM model to the Nearctic and Primate datasets, there is still phylogenetic signal in the pattern of host–parasite associations. In particular, the fit to the real data is much better than the fit to data that has been permuted such that each parasite infects the same number of hosts, but the identity of those hosts is shuffled (L1), compared to permutations that preserve the number of parasites infecting each host (L2) or fully permuted data. Given the interpretations mentioned before, this is evidence for phylogenetic signal in the parasite richness of each host, further indicating a stronger role for host evolution in shaping host–parasite interactions.
Table 4.
Signal estimates for BM model for each dataset based on three different bootstrap permutation methods. (L1: where each parasite infects the same number of hosts, but the identity of those hosts is randomly determined; L2: where each host is infected by the same number of parasites, but the identity of those parasites is randomly determined; and H: where the total number of host–parasite associations is preserved, but associations are randomly determined.)
| dataset | model | test statistic | L1 p-value | L2 p-value | H p-value |
|---|---|---|---|---|---|
| GMPD | BM | 339667.56 | 0.009 | 0.002 | 0.361 |
| Primates only (GMPD) | BM | 3453.45 | 0.006 | <0.001 | 0.047 |
| Nearctic | BM | 189337.53 | 0.016 | 0.100 | 0.158 |
(d) . Krasnov method
Using the method in Krasnov et al. [31], we test the null hypothesis that membership in a host–parasite module is unrelated to the phylogenetic distance between the hosts and parasites within the network module. If there is phylogenetic structuring in host–parasite associations, the pairwise phylogenetic distance between hosts (or parasites) within a module is less than the pairwise phylogenetic distance between separate modules. We detected 38 distinct host–parasite clusters in the GMPD, with an overall modularity score of 0.63 (figure 1); this is significantly higher than the modularity of randomly permuted datasets that varied the identity (but not number) of hosts each parasite infected (p < 0.001) or datasets that varied the identity (but not number) of parasite each host was infected by (p < 0.001). There are several highly connected modules comprising dense networks of closely interacting hosts and parasites, surrounded by many small, often disconnected modules. There are five modules with 20 or more interacting hosts and parasites, the largest of which contains 154 hosts and parasites. This module contains 62% of the Carnivore hosts, as well as large fractions of parasites from many of the parasite classes (e.g. 62% of the Platyhelminthes and 59% of the Acanthocephala). The second largest module (77 species) contains 65% of the Artiodactyla (even-toed ungulates) and their parasites. The third largest module (46 species) contains 57% of the Primates and their parasites. The fourth largest module (37 species) contains all of the Perissodactyla (odd-toed ungulates) and their parasites. The modules therefore appear to be highly structured by host phylogeny.
Figure 1.
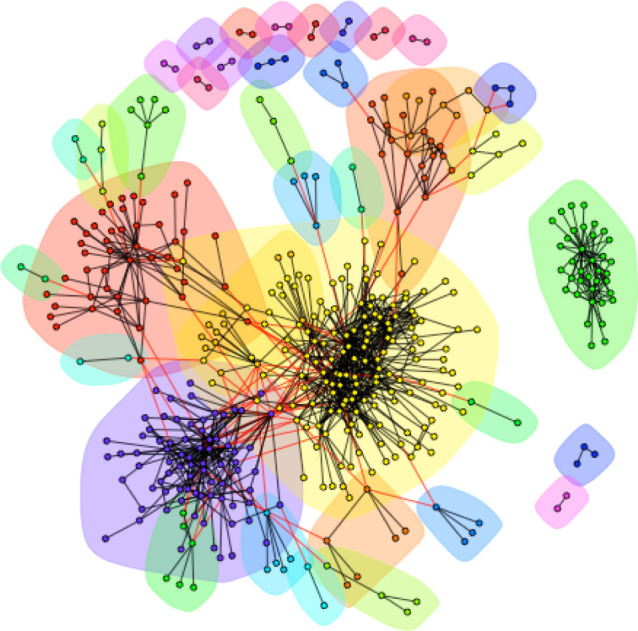
Host–parasite network from the GMPD includes 38 distinct modules. Closely interacting modules of hosts and parasites are clustered by colour: nodes of the same colour belong to the same module. Modules are arranged such that the more connections between modules, the closer they are in space. Black vertices show host–parasite interactions within a module; red vertices show host–parasite connections between modules.
For the Nearctic dataset, we detected 17 distinct host–parasite clusters, with an overall modularity score of 0.5 (figure 2). Similar to the GMPD dataset, this is significantly higher than the modularity of randomly permuted datasets that varied the identity (but not number) of hosts each parasite infected (p < 0.001) or datasets that varied the identity (but not number) of parasite each host was infected by (p < 0.001). There are also several large modules in the Nearctic dataset, although the overall pattern reveals more connections between modules than was evident in the GMPD data. Some of these modules appear to be more structured by the parasites. For example, the largest network contains all of the Artiodactyla in the dataset. The next largest network contains 46% of the Carnivores in the dataset and includes several species of tapeworm (Taenia spp.) that have carnivores as definitive hosts, suggesting a role for parasite phylogeny or codivergence in structuring some modules. Carnivores are also common in the third largest network (which also contains marsupials and rodents). The overall higher interconnectedness of the network, and lower modularity, appears to be driven by the inclusion of rodents in this dataset, as rodents appear in 11 of the 17 modules.
Figure 2.
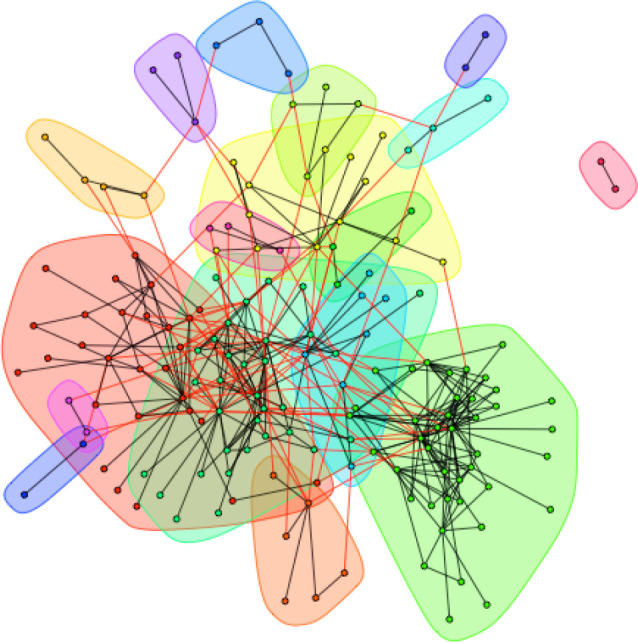
Host–parasite network from the Nearctic dataset includes 17 distinct modules. The high degree of overlap between modules near the centre of the figure and the large number of red vertices indicates a much higher degree of host–parasite sharing among modules in this dataset compared to the GMPD data. Closely interacting modules of hosts and parasites are clustered by colour: nodes of the same colour belong to the same module. Modules are arranged such that the more connections between modules, the closer they are in space. Black vertices show host–parasite interactions within a module; red vertices show host–parasite connections between modules.
For the Primate dataset, we detected 20 distinct host–parasite clusters, with an overall modularity score of 0.53 (figure 3); interestingly, this is significantly higher than the modularity of randomly permuted datasets that varied the identity (but not number) of hosts each parasite infected (p = 0.04), but not datasets that varied the identity (but not number) of parasite each host was infected by (p = 0.82). There are many very small modules, comprising a single-host–parasite pair. The largest module (of 23 species) contains 18 species of Great Apes (including both Pan species), lesser apes, and Old and New World monkeys.
Figure 3.
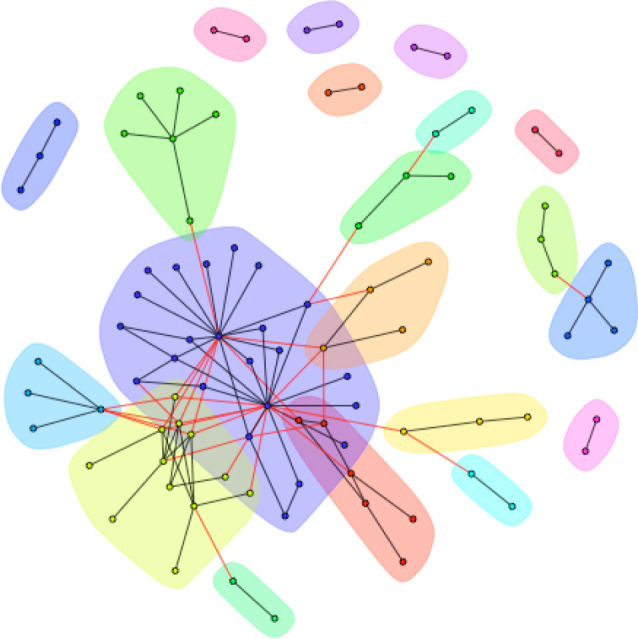
Host–parasite network from the Primate subset of the GMPD includes 20 distinct modules. Closely interacting modules of hosts and parasites are clustered by colour: nodes of the same colour belong to the same module. Modules are arranged such that the more connections between modules, the closer they are in space. Black vertices show host–parasite interactions within a module; red vertices show host–parasite connections between modules.
Looking across datasets, there is a consistent and significant negative correlation between co-membership in a module and phylogenetic distance for hosts, indicating that more closely related hosts are more likely to end up in the same module (table 5). However, parasite relatedness was never significantly related to membership in a module (table 6).
Table 5.
Correlation of host co-membership in a module with phylogenetic distance between hosts based on three different bootstrap permutation methods. (L1: where each parasite infects the same number of hosts, but the identity of those hosts is randomly determined; L2: where each host is infected by the same number of parasites, but the identity of those parasites is randomly determined and H: where the total number of host–parasite associations is preserved, but associations are randomly determined.)
| dataset | r | L1 p-value | L2 p-value | H p-value |
|---|---|---|---|---|
| GMPD | −0.49 | <0.001 | <0.001 | <0.001 |
| Primates only (GMPD) | −0.22 | <0.001 | <0.001 | <0.001 |
| Nearctic | −0.27 | <0.001 | <0.001 | <0.001 |
Table 6.
Correlation of parasite co-membership in a module with phylogenetic distance between parasites based on three different bootstrap permutation methods. (L1: where each parasite infects the same number of hosts, but the identity of those hosts is randomly determined; L2: where each host is infected by the same number of parasites, but the identity of those parasites is randomly determined and H: where the total number of host–parasite associations is preserved, but associations are randomly determined.)
| dataset | r | L1 p-value | L2 p-value | H p-value |
|---|---|---|---|---|
| GMPD | 0.03 | 0.79 | 0.83 | 0.86 |
| Primates only (GMPD) | 0.02 | 0.50 | 0.56 | 0.59 |
| Nearctic | −0.04 | 0.17 | 0.14 | 0.09 |
4. Discussion
We use host–parasite datasets containing associations between helminth taxa and free-range mammals in combination with reconstructed molecular phylogenies to explore phylogenetic patterns of host–parasite associations. We evaluate evidence for codivergence and examine whether parasite–host interactions are structured more by the phylogeny of hosts or parasites, using four co-phylogenetic analysis methods. Across three datasets that differ in their geographical extent and taxonomic breadth, we found fairly consistent evidence that host evolutionary history is the main determinant of helminth-mammal associations.
The test devised in Hommola et al. [29], which looks for correlations between the pairwise phylogenetic distances between pairs of hosts and pairs of parasites in each dataset, only found significant evidence for a correlation in the smallest dataset, that of primates and their helminth parasites (table 1). Conversely, both the ParaFitGlobal statistic [28] & the closely related test for BM evolution devised in Hadfield et al. [32], based on the MSE BM statistic of Ives & Godfray [30], provided strong evidence for non-randomness in the phylogenetic pattern of host–parasite associations across all three datasets. This difference between the methods is somewhat surprising, as other comparison studies have found that the Hommola statistic and ParaFitGlobal often perform quite similarly [29,32]. This may reflect differences in the sensitivity of each method to uncertainty in the underlying branch lengths: although all three methods make use of the phylogenies, ParaFitGlobal and MSE BM are calculated using the entire phylogeny, whereas the Hommola statistic draws on information contained in pairs of branches. Thus, an error in the length of any particular branch will probably have limited effect on ParaFitGlobal and MSE BM, but might have a much larger effect on the Hommola statistic, especially if the incorrect branch belongs to a host or parasite that is involved in many interactions. However, the ParaFitGlobal method does not account for phylogenetic independence (the data at more ancient nodes includes the same information as recent nodes along the same branches) which can exaggerate congruence (see [38,39]). Alternatively, it is possible that this difference simply reflects a strong influence of host evolutionary history only, which would appear as a lack of correlation between host and parasite pairwise phylogenetic distances, leading to a non-significant value for the Hommola et al. [29] statistic, but could be detected by the ParaFitGlobal statistic.
Another seemingly perplexing result was that the full Ives and Godfray method, applied to the smaller Nearctic and Primate datasets, found considerable support for the non-phylogenetic star phylogeny (table 3), even though permutation tests found evidence for phylogenetic constraint in the host–parasite associations (table 4). However, it is important to keep in mind that these analyses are quite different: the full Ives and Godfray method is comparing the fit of different evolutionary models to the observed host–parasite association dataset, whereas the permutation test is comparing the fit of a BM model to the observed data and randomly permuted data. It is not necessarily inconsistent for a non-phylogenetic model to provide the best fit to the observed data, but for the observed data to still contain more phylogenetic signal than would be found in randomized association data. Nevertheless, for the Primate dataset, where the full method provided a better fit than either BM or a star phylogeny, there was stronger evidence for phylogenetic signal coming through the host phylogeny than the parasite phylogeny.
The Krasnov method provided the most definitive test of whether host or parasite phylogeny appears to more strongly structure the host–parasite associations. All three datasets showed significant evidence of modularity (many more connections than expected between hosts and parasites within a module, and fewer than expected between hosts and parasite among modules); the observed modularity was similar to that observed in a dataset of fish parasites [40], and more than was observed in a dataset of rodents and fleas [31] and of another dataset of trophically transmitted parasites within an intertidal food web [41]. These modules were more strongly structured by host phylogeny than by parasite phylogeny, as evidenced by the strong positive correlation between co-membership in a phylogeny and phylogenetic distance (closely related hosts are more likely to co-occur in the same module than distantly related hosts [31,32,42]).
We thus conclude that there is support for evolution to play a role in structuring the interactions between helminths infecting mammals in all three datasets, although we do not see strong evidence for codivergence per se, or for cospeciation in particular. The most well-known evidence for cospeciation comes from studies of pocket gophers and their chewing lice; the strong evidence for cospeciation in this system has been attributed to the isolated lifestyle of the gophers, which acts as a sort of geographical isolation for the chewing lice [14,43,44]. While codivergence has been observed many times in both mammal (e.g. New world monkeys and protozoans [43]; mammals and fleas [32]) and non-mammal systems (e.g. doves and lice [45]; avian malaria [46–48]; blood alveolates and birds [49]; and parasitic fungi and plants [50]), cospeciation patterns among hosts and parasites are rare (reviewed in [38]). Increasingly, as we have found here, studies of mammal–parasite associations find that divergence patterns are explained, at least in part, by host evolutionary history (primates and parasites [5]; bats and rabies viruses [17]; mammals and fleas [31]; carnivores and parasites [8]). There is little to no evidence for host–parasite association patterns shaped by parasite evolutionary history; however, this is probably a result of not testing for the impact of parasite phylogeny owing to species extinction [51] and the lack of available molecular information on parasites to construct robust phylogenies [52].
The datasets used here have different strengths and weaknesses, from the perspective of detecting phylogenetic patterns in host–parasite association data. The GMPD is a very large dataset but it is taxonomically limited in its coverage of mammals; in particular, it is missing rodents. The presence of rodents in the geographically restricted Nearctic dataset may help to explain differences in the results between the GMPD and Nearctic datasets. In particular, the lower modularity and weaker correlation between co-occurrence and phylogenetic distance may be because of the inclusion of rodents, which are found in almost every module. Another potential complication with the Nearctic dataset is that it does not only contain definitive hosts, but also some intermediate hosts. For example, one of the smaller modules in the Nearctic dataset contains swift foxes (Vulpes velox), three jackrabbit prey species (Lepus californicus, Lepus townsendii and Lepus callotis) and two parasites (the cestode Mosgovoyia pectinata and the dog tapeworm Taenia serialis). Jackrabbits are known to be an intermediate host for T. serialis [53], whereas swift foxes are a definitive host [54]. Hosts that share a trophically transmitted parasite should definitely be found in the same module, but the potential for large phylogenetic distances between predators and prey may cause an overall weakening of the host phylogenetic signal.
It would be informative to expand upon the parasite phylogeny used here because it is less complete and more limited in taxonomic evenness than the phylogeny of mammal hosts. In particular, there are probably fewer closely related parasites within this phylogeny, potentially limiting our ability to detect phylogenetic signal within the parasite phylogeny. Only just over a third of taxa from the 10 best-sampled genera in the GMPD are included in the helminth phylogeny owing to the lack of molecular data. For example, in the GMPD, there are 21 species of Trichostrongylus, making it the third best-sampled parasite taxon; however, only two (9.5%) of these species are included in the phylogeny used here. Rounding out the genera within the helminth phylogeny presents a significant challenge, as helminths are both more diverse and ubiquitous than the mammal hosts they parasitize [55,56].
We find that host evolutionary history is the main determinant of host–parasite associations across all three datasets used here; however, we suggest that the relative importance of host evolutionary history is likely to vary across geographical scales [57]. The datasets used here cover rather large geographical areas (global and Nearctic distributions) and might better capture processes operating at biogeographical scales, including histories of postglacial range expansion that may have provided opportunities for spillover of parasites between previously allopatric hosts, and over deeper time, dynamics of host diversification and radiation [58,59]. At these large geographical and temporal scales, it is possible that relatively fast-evolving helminth lineages are little constrained in their host use by their own phylogenetic history [60,61]. It is possible that the signature of phylogenetic conservatism in host use on the helminth phylogeny would be more apparent at finer geographical scales and within more recently formed hosts assemblages [49,62–65]. Indeed, there is evidence that host movement can generate genetic structure in nematode parasites [66] and presumably, if host populations remain separated, we would expect parasite populations to become reproductively isolated. Such a scenario should generate a strong phylogenetic structure in host use—two closely related parasite lineages infecting the same host species. Why do we rarely detect such structuring in empirical datasets? One explanation is that the process of parasite speciation assumes host populations are in allopatry, and it is possible that secondary sympatry might even be inhibited by the parasites themselves (see apparent competition [67]). Further, because competitive exclusion tends to limit co-occurrence of closely related hosts at finer spatial scales—the Darwin-Hutchinson zone of Vamosi et al. [68] that defines the spatial scale small enough for individuals to interact, and taxonomic scale at which species are closely related enough to compete along similar niche axes—we rarely sample communities with recently diverged hosts that would be most likely to share closely related parasites.
5. Conclusion
Our study demonstrates that there is congruence between the helminth phylogeny and the phylogeny of their mammal hosts. These results align with previous work suggesting patterns of host–parasite association are shaped by host evolutionary history. Assessing the main determinants of host–parasite associations is a critical step in predicting emerging infectious diseases. Given that host evolutionary history shapes host–parasite associations, we recommend surveying all the parasites of the focal mammal's close relatives, regardless of whether they are closely related to the mammal's known parasites for identifying potential sources of zoonotic spillover. Additionally, we suggest that broad taxonomic scale co-phylogenetic analyses can provide novel insights on the evolutionary and ecological factors that shape host–parasite associations, which can complement trait-based analyses, and inform targeted surveillance of parasites that may spill into novel hosts.
Data accessibility
All datasets and code to fully replicate this study are available at: https://github.com/alainapb/Co_phylogenetic_hotspots.
Authors' contributions
All authors designed the study; C.E.C. performed co-phylogenetic analyses; all authors analysed the output. A.P.B. wrote the first draft of the manuscript. All authors contributed substantially to revisions.
Competing interests
We declare we have no competing interests.
Funding
We thank the members of the Macroecology of Infectious Disease Research Coordination Network (grant no. NSF-DEB 131223) for support in this work.
References
- 1.Cleaveland S, Laurenson MK, Taylor LH. 2001Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Phil. Trans. R. Soc. Lond. B 356, 991-999. ( 10.1098/rstb.2001.0889) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor LH, Latham SM, Woolhouse ME. 2001Risk factors for human disease emergence. Phil. Trans. R. Soc. Lond. B 356, 983-989. ( 10.1098/rstb.2001.0888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han BA, Kramer AM, Drake JM. 2016Global patterns of zoonotic disease in mammals. Trends Parasitol. 32, 565-577. ( 10.1016/j.pt.2016.04.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. 2017Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646-650. ( 10.1038/nature22975) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies TJ, Pedersen AB. 2008Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proc. R. Soc. B 275, 1695-1701. ( 10.1098/rspb.2008.0284) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramsden C, Holmes EC, Charleston MA. 2009Hantavirus evolution in relation to its rodent and insectivore hosts: no evidence for codivergence. Mol. Biol. Evol. 26, 143-153. ( 10.1093/molbev/msn234) [DOI] [PubMed] [Google Scholar]
- 7.Cooper N, Griffin R, Franz M, Omotayo M, Nunn CL. 2012Phylogenetic host specificity and understanding parasite sharing in primates. Ecol. Lett. 15, 1370-1377. ( 10.1111/j.1461-0248.2012.01858.x) [DOI] [PubMed] [Google Scholar]
- 8.Huang S, Bininda-Emonds OR, Stephens PR, Gittleman JL, Altizer S. 2014Phylogenetically related and ecologically similar carnivores harbour similar parasite assemblages. J. Anim. Ecol. 83, 671-680. ( 10.1111/1365-2656.12160) [DOI] [PubMed] [Google Scholar]
- 9.Longdon B, Brockhurst MA, Russell CA, Welch JJ, Jiggins FM. 2014The evolution and genetics of virus host shifts. PLoS Pathog. 10, e1004395. ( 10.1371/journal.ppat.1004395) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woolhouse MEJ, Gowtage-Sequeria S. 2005Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 11, 1842-1847. ( 10.3201/eid1112.050997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson CK, et al. 2015Spillover and pandemic properties of zoonotic viruses with high host plasticity. Sci. Rep. 5, e14830. ( 10.1038/srep14830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larose C, Schwander T. 2016Nematode endoparasites do not codiversify with their stick insect hosts. Ecol. Evol. 6, 5446-5458. ( 10.1002/ece3.2264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demastes JW, Hafner MS. 1993Cospeciation of pocket gophers (Geomys) and their chewing lice (Geomydoecus). J. Mammal. 74, 521-530. ( 10.2307/1382271) [DOI] [Google Scholar]
- 14.Demastes JW, Spradling TA, Hafner MS, Spies GR, Hafner DJ, Light JE. 2012Cophylogeny on a fine scale: Geomydoecus chewing lice and their pocket gopher hosts, Pappogeomys bulleri. J. Parasitol. 98, 262-270. ( 10.1645/GE-2904.1) [DOI] [PubMed] [Google Scholar]
- 15.Brooks DR. 1979Testing the context and extent of host-parasite coevolution. Syst. Biol. 28, 299-307. ( 10.1093/sysbio/28.3.299) [DOI] [Google Scholar]
- 16.Brooks DR, Hoberg EP, Boeger WA. 2015In the eye of the cyclops: the classic case of cospeciation and why paradigms are important. Comp. Parasitol. 82, 1-8. ( 10.1654/4724C.1) [DOI] [Google Scholar]
- 17.Streicker DG, Turmelle AS, Vonhof MJ, Kuzmin IV, McCracken GF, Rupprecht CE. 2010Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 329, 676-679. ( 10.1126/science.1188836) [DOI] [PubMed] [Google Scholar]
- 18.Huffman MA. 2013Preliminary survey of the distribution of four potentially zoonotic parasite species among primates in Sri Lanka. J. Natl Sci. Foundation Sri Lanka 41, 319-326. ( 10.4038/jnsfsr.v41i4.6246) [DOI] [Google Scholar]
- 19.Paige SB, Malavé C, Mbabazi E, Mayer J, Goldberg TL. 2015Uncovering zoonoses awareness in an emerging disease ‘hotspot’. Soc. Sci. Med. 129, 78-86. ( 10.1016/j.socscimed.2014.07.058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faust C, Dobson AP. 2015Primate malarias: diversity, distribution and insights for zoonotic Plasmodium. One Health 1, 66-75. ( 10.1016/j.onehlt.2015.10.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephens PR, et al. 2017Global mammal parasite database version 2.0. See 10.1002/ecy.1799. [DOI]
- 22.Botero-Cañola S. 2020Parasite geography: exploring drivers of parasite biodiversity distribution at species and community scales. Doctoral dissertation, The University of Nebraska-Lincoln, Lincoln, NE, USA. [Google Scholar]
- 23.Faurby S, Davis M, Pedersen RØ, Schowanek SD, Antonelli A, Svenning JC. 2018PHYLACINE 1.2: The phylogenetic atlas of mammal macroecology. Ecology 99, 2626. ( 10.1002/ecy.2443) [DOI] [PubMed] [Google Scholar]
- 24.Pfenning-Butterworth AC, Botero-Cañola S, Cressler CE. 2021On the evolution of host specificity: a case study in helminths. BioRxiv. ( 10.1101/2021.02.13.431093) [DOI]
- 25.Robinsn MW, Dalton JP. 2009Zoonotic helminth infections with particular emphasis on fasciolosis and other trematodiases. Phil. Trans. R. Soc. B 364, 2763-2776. ( 10.1098/rstb.2009.0089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarthy J, Moore TA. 2000Emerging helminth zoonoses. Int. J. Parasitol. 30, 1351-1360. ( 10.1016/S0020-7519(00)00122-3) [DOI] [PubMed] [Google Scholar]
- 27.Pozio E. 2007World distribution of Trichinella spp. infections in animals and humans. Vet. Parasitol. 149, 3-21. ( 10.1016/j.vetpar.2007.07.002) [DOI] [PubMed] [Google Scholar]
- 28.Legendre P, Desdevises Y, Bazin E. 2002A statistical test for host–parasite coevolution. Syst. Biol. 51, 217-234. ( 10.1080/10635150252899734) [DOI] [PubMed] [Google Scholar]
- 29.Hommola K, Smith JE, Qiu Y, Gilks WR. 2009A permutation test of host–parasite cospeciation. Mol. Biol. Evol. 26, 1457-1468. ( 10.1093/molbev/msp062) [DOI] [PubMed] [Google Scholar]
- 30.Ives AR, Godfray HCJ. 2006Phylogenetic analysis of trophic associations. Am. Nat. 168, E1-E14. ( 10.1086/505157) [DOI] [PubMed] [Google Scholar]
- 31.Krasnov BR, Fortuna MA, Mouillot D, Khokhlova IS, Shenbrot GI, Poulin R. 2012Phylogenetic signal in module composition and species connectivity in compartmentalized host-parasite networks. Am. Nat. 179, 501-511. ( 10.1086/664612) [DOI] [PubMed] [Google Scholar]
- 32.Hadfield JD, Krasnov BR, Poulin R, Nakagawa S. 2014A tale of two phylogenies: comparative analyses of ecological interactions. Am. Nat. 183, 174-187. ( 10.1086/674445) [DOI] [PubMed] [Google Scholar]
- 33.Faurby S, Svenning JC. 2015A species-level phylogeny of all extant and late quaternary extinct mammals using a novel heuristic-hierarchical Bayesian approach. Mol. Phylogenet. Evol. 84, 14-26. ( 10.1016/j.ympev.2014.11.001) [DOI] [PubMed] [Google Scholar]
- 34.Blomberg SP, Garland T Jr, Ives AR. 2003Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717-745. ( 10.1111/j.0014-3820.2003.tb00285.x) [DOI] [PubMed] [Google Scholar]
- 35.Hansen TF. 1997Stabilizing selection and the comparative analysis of adaptation. Evolution 51, 1341-1351. ( 10.1111/j.1558-5646.1997.tb01457.x) [DOI] [PubMed] [Google Scholar]
- 36.Kembel S, Cowan P, Helmus M, Cornwell W, Morlon H, Ackerly D, Blomberg C, Webb C. 2010Picante: R tools for integrating phylogenies and ecology. Bioinformatics26, 1463–1464. ( 10.1093/bioinformatics/btq166) [DOI] [PubMed]
- 37.Latapy M, Pons P. 2004Computing communities in large networks using random walks. arXiv cond-mat/0412368.
- 38.de Vienne DM, Refrégier G, López-Villavicencio M, Tellier A, Hood ME, Giraud T. 2013Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytol. 198, 347-385. ( 10.1111/nph.12150) [DOI] [PubMed] [Google Scholar]
- 39.Felsenstein J. 1985Phylogenies and the comparative method. Am. Nat. 125, 1-15. ( 10.1086/284325) [DOI] [Google Scholar]
- 40.Lima DP, Giacomini HC, Takemoto RM, Agostinho AA, Bini LM. 2012Patterns of interactions of a large fish–parasite network in a tropical floodplain. J. Anim. Ecol. 81, 905-913. ( 10.1111/j.1365-2656.2012.01967.x) [DOI] [PubMed] [Google Scholar]
- 41.Poulin R, Krasnov BR, Pilosof S, Thieltges DW. 2013Phylogeny determines the role of helminth parasites in intertidal food webs. J. Anim. Ecol. 82, 1265-1275. ( 10.1111/1365-2656.12101) [DOI] [PubMed] [Google Scholar]
- 42.Bellay S, de Oliveira EF, Almeida-Neto M, Junior DPL, Takemoto RM, Luque JL. 2013Developmental stage of parasites influences the structure of fish-parasite networks. PLoS ONE 8, e75710. ( 10.1371/journal.pone.0075710) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hafner MS, Nadler SA. 1990Cospeciation in host-parasite assemblages: comparative analysis of rates of evolution and timing of cospeciation events. Syst. Zool. 39, 192-204. ( 10.2307/2992181) [DOI] [Google Scholar]
- 44.Hafner MS, Page RD. 1995Molecular phylogenies and host-parasite cospeciation: gophers and lice as a model system. Phil. Trans. R. Soc. Lond. B 349, 77-83. ( 10.1098/rstb.1995.0093) [DOI] [PubMed] [Google Scholar]
- 45.Clayton DH, Bush SE, Goates BM, Johnson KP. 2003Host defense reinforces host–parasite cospeciation. Proc. Natl Acad. Sci. USA 100, 15 694-15 699. ( 10.1073/pnas.2533751100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ricklefs RE, Fallon SM. 2002Diversification and host switching in avian malaria parasites. Proc. R. Soc. Lond. B 269, 885-892. ( 10.1098/rspb.2001.1940) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alcala N, Jenkins T, Christe P, Vuilleumier S. 2017Host shift and cospeciation rate estimation from co-phylogenies. Ecol. Lett. 20, 1014-1024. ( 10.1111/ele.12799) [DOI] [PubMed] [Google Scholar]
- 48.Fecchio A, et al. 2018Host community similarity and geography shape the diversity and distribution of haemosporidian parasites in Amazonian birds. Ecography 41, 505-515. ( 10.1111/ecog.03058) [DOI] [Google Scholar]
- 49.Jenkins T, Thomas GH, Hellgren O, Owens IP. 2012Migratory behavior of birds affects their coevolutionary relationship with blood parasites. Evolution 66, 740-751. ( 10.1111/j.1558-5646.2011.01470.x) [DOI] [PubMed] [Google Scholar]
- 50.Peterson KR, Pfister DH, Bell CD. 2010Cophylogeny and biogeography of the fungal parasite Cyttaria and its host Nothofagus, southern beech. Mycologia 102, 1417-1425. ( 10.3852/10-048) [DOI] [PubMed] [Google Scholar]
- 51.Farrell M, Park A, Cressler C, Dallas T, Huang S, Mideo N, Morales-Castilla I, Davies J, Stephens P. 2021The ghosts of hosts past: impacts of host extinction on parasite specificity. Phil. Trans. R. Soc. B 376, 20200351. ( 10.1098/rstb.2020.0351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waxman D, Weinert LA, Welch JJ. 2014Inferring host range dynamics from comparative data: the protozoan parasites of new world monkeys. Am. Nat. 184, 65-74. ( 10.1086/676589) [DOI] [PubMed] [Google Scholar]
- 53.Ackert JE. 1915Experiments on Cysticerci of Taenia pisiformis, Bloch, and of Taenia serialis, Gervais. J. Parasitol. 1, 151-153. ( 10.2307/3271118) [DOI] [Google Scholar]
- 54.Criffield MA, Reichard MV, Hellgren EC, Leslie DM, Freel K. 2009Parasites of swift foxes (Vulpes velox) in the Oklahoma Panhandle. The Southwestern Naturalist 54, 492-498. ( 10.1894/MD-13.1) [DOI] [Google Scholar]
- 55.Burgin CJ, Colella JP, Kahn PL, Upham NS. 2018How many species of mammals are there? J. Mammal. 99, 1-14. ( 10.1093/jmammal/gyx147) [DOI] [Google Scholar]
- 56.Dobson A, Lafferty KD, Kuris AM, Hechinger RF, Jetz W. 2008Homage to Linnaeus: how many parasites? How many hosts? Proc. Natl Acad. Sci. USA 105(Suppl. 1), 11 482-11 489. ( 10.1073/pnas.0803232105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Majewska A, Huang T, Han B, Drake J. 2021Predictors of zoonotic potential in helminths. Phil. Trans. R. Soc. B 376, 20200356. ( 10.1098/rstb.2020.0356) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoberg EP. 1992Congruent and synchronic patterns in biogeography and speciation among seabirds, pinnipeds, and cestodes. J. Parasitol. 78, 601-615. ( 10.2307/3283535) [DOI] [PubMed] [Google Scholar]
- 59.Hoberg EP. 1995Historical biogeography and modes of speciation across high-latitude seas of the Holarctic: concepts for host–parasite coevolution among the Phocini (Phocidae) and Tetrabothriidae (Eucestoda). Can. J. Zool. 73, 45-57. ( 10.1139/z95-006) [DOI] [Google Scholar]
- 60.Toit ND, van Vuuren BJ, Matthee S, Matthee CA. 2013Biogeography and host-related factors trump parasite life history: limited congruence among the genetic structures of specific ectoparasitic lice and their rodent hosts. Mol. Ecol. 22, 5185-5204. ( 10.1111/mec.12459) [DOI] [PubMed] [Google Scholar]
- 61.Molnar RI, Bartelmes G, Dinkelacker I, Witte H, Sommer RJ. 2011Mutation rates and intraspecific divergence of the mitochondrial genome of Pristionchus pacificus. Mol. Biol. Evol. 28, 2317-2326. ( 10.1093/molbev/msr057) [DOI] [PubMed] [Google Scholar]
- 62.Huyse T, Volckaert FA. 2005Comparing host and parasite phylogenies: Gyrodactylus flatworms jumping from goby to goby. Syst. Biol. 54, 710-718. ( 10.1080/10635150500221036) [DOI] [PubMed] [Google Scholar]
- 63.Bruyndonckx N, Dubey S, Ruedi M, Christe P. 2009Molecular cophylogenetic relationships between European bats and their ectoparasitic mites (Acari, Spinturnicidae). Mol. Phylogenet. Evol. 51, 227-237. ( 10.1016/j.ympev.2009.02.005) [DOI] [PubMed] [Google Scholar]
- 64.Jorge F, Perera A, Poulin R, Roca V, Carretero MA. 2018Getting there and around: host range oscillations during colonization of the Canary Islands by the parasitic nematode Spauligodon. Mol. Ecol. 27, 533-549. ( 10.1111/mec.14458) [DOI] [PubMed] [Google Scholar]
- 65.Sweet AD, Johnson KP. 2018The role of parasite dispersal in shaping a host–parasite system at multiple evolutionary scales. Mol. Ecol. 27, 5104-5119. ( 10.1111/mec.14937) [DOI] [PubMed] [Google Scholar]
- 66.Blouin MS, Yowell CA, Courtney CH, Dame JB. 1995Host movement and the genetic structure of populations of parasitic nematodes. Genetics 141, 1007-1014. ( 10.1093/genetics/141.3.1007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holt RD, Bonsall MB. 2017Apparent competition. Ann. Rev. Ecol. Evol. Syst. 48, 447-471. ( 10.1146/annurev-ecolsys-110316-022628) [DOI] [Google Scholar]
- 68.Vamosi SM, Heard SB, Vamosi JC, Webb CO. 2009Emerging patterns in the comparative analysis of phylogenetic community structure. Mol. Ecol. 18, 572-592. ( 10.1111/j.1365-294X.2008.04001.x) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets and code to fully replicate this study are available at: https://github.com/alainapb/Co_phylogenetic_hotspots.