

Abstract
DNA-templated molecular (dye) aggregates are a novel class of materials that have garnered attention in a broad range of areas including light harvesting, sensing, and computing. Using DNA to template dye aggregation is attractive due to the relative ease with which DNA nanostructures can be assembled in solution, the diverse array of nanostructures that can be assembled, and the ability to precisely position dyes to within a few Angstroms of one another. These factors, combined with the programmability of DNA, raise the prospect of designer materials custom tailored for specific applications. Although considerable progress has been made in characterizing the optical properties and associated electronic structures of these materials, less is known about their excited-state dynamics. For example, little is known about how the excited-state lifetime, a parameter essential to many applications, is influenced by structural factors, such as the number of dyes within the aggregate and their spatial arrangement. In this work, we use a combination of transient absorption spectroscopy and global target analysis to measure excited-state lifetimes in a series of DNA-templated cyanine dye aggregates. Specifically, we investigate six distinct dimer, trimer, and tetramer aggregates—based on the ubiquitous cyanine dye Cy5—templated using both duplex and Holliday junction DNA nanostructures. We find that these DNA-templated Cy5 aggregates all exhibit significantly reduced excited-state lifetimes, some by more than 2 orders of magnitude, and observe considerable variation among the lifetimes. We attribute the reduced excited-state lifetimes to enhanced nonradiative decay and proceed to discuss various structural factors, including exciton delocalization, dye separation, and DNA heterogeneity, that may contribute to the observed reduction and variability of excited-state lifetimes. Guided by insights from structural modeling, we find that the reduced lifetimes and enhanced nonradiative decay are most strongly correlated with the distance between the dyes. These results inform potential tradeoffs between dye separation, excitonic coupling strength, and excited-state lifetime that motivate deeper mechanistic understanding, potentially via further dye and dye template design.
Introduction
Molecular (dye) aggregates, which can form delocalized collective excitations called excitons, are central to a number of applications including natural and artificial light harvesting,1,2 organic optoelectronics,3 sensing,4−6 and nanoscale computing.7−12 The role of dye aggregates in some of these applications is to efficiently absorb light and transfer it to a reaction center, charge–transfer interface, or across an excitonic wire.1−4,10 Any loss along the way reduces the overall efficiency of the energy-transfer process. Conversely, dye aggregates have found a role in other applications such as sensing5,6 or photothermal therapy13,14 for their ability to quench emitted light, that is, quickly convert light energy into heat. Here, the contrast between the highly emissive monomer dyes and weakly emissive dye aggregates is essential. Thus, developing an understanding of the nonradiative processes that lead to quenching in dye aggregates can both improve the overall efficiency of energy conversion and computing applications as well as potentially enhance the overall contrast in sensing applications.
Since the discovery of dye aggregates in the 1930s by Scheibe and Jelley,15,16 a considerable amount of research has been dedicated to better understanding their unique electronic structure. Kasha, following Davydov’s work on molecular crystals,17,18 highlighted that many dye aggregate optical properties, such as new and shifted absorption bands, can be explained by a relatively simple model based on the mutual orientation of the transition dipole moments (TDMs) of constituent dyes. Kasha’s description of dye aggregation can, in many cases, accurately predict the energy-level splitting and redistribution of oscillator strength concomitant with dye aggregation. Kasha’s model, for example, predicts that when dyes pack with their TDMs in an end-to-end (J-aggregate) or stacked (H-aggregate) configuration, the absorption spectrum is redshifted or blueshifted, respectively, compared to the individual dye. The energy shift is largely related to whether transitions from the ground state to the lowest- or highest-energy excitonic states are allowed. In a J-aggregate, transitions to the lowest- and highest-energy excitonic states are optically allowed and forbidden, respectively, while the opposite is true for H-aggregates. It is worthwhile to note that more recently, the theoretical treatment of the electronic structure of dye aggregates has been further refined to include vibronic19 and charge-transfer20,21 coupling, the latter of which becomes important at small intermolecular distances.
While great progress has been made in understanding the electronic structure of dye aggregates, challenges remain in understanding their dynamical properties. Excited-state lifetimes, which are essential to any application utilizing dye aggregates, are a dynamical property controlled by both radiative and nonradiative decay processes.22 As noted above, optical transitions between the ground and lowest-energy excited states of J- and H-aggregates are allowed and forbidden, respectively. Given that emission is the inverse of absorption and involves the same electronic states, insights into the radiative decay rate can be gained from the static electronic structure. For example, the TDMs of dyes in a perfect J-aggregate sum together constructively, which enhances the radiative rate compared with the monomer. For a perfect H-aggregate, in contrast, the TDMs of the dyes destructively interfere, causing the radiative rate to become zero. This is the reason why, assuming nonradiative decay rates have not changed, it is generally suggested that J- and H-aggregates are emissive and dark, respectively. Although the effect of aggregation on the radiative rate is fairly well understood, in practice, several studies have shown that nonradiative decay processes can play a more predominant role in dye aggregate photophysics, ultimately governing their excited-state lifetimes.14,23−25 Clearly, developing a deeper understanding of excited-state lifetimes in dye aggregates, particularly the role of nonradiative decay processes, is of utmost importance.
There are a variety of ways to form dye aggregates. Dye aggregates can be formed spontaneously in concentrated solutions, well above the solubility limit, as was originally demonstrated by Scheibe and Jelley.15,16 Some control of the aggregate character in solutions of spontaneously aggregating dyes can be achieved through alterations of the solvent conditions such as the ionic strength and composition,26 solvent polarity, and temperature. Dye substituents can be modified to affect aggregate packing as well.27 However, strict control of the number of dyes in an aggregate is not possible for spontaneously formed aggregates larger than a dimer. Natural systems use proteins to overcome this limitation, which has resulted in sophisticated supramolecular assemblies for light harvesting.1,28 The examples from nature demonstrate that high specificity with regard to the number of interacting dyes, dye placement, and orientation is possible. Although considerable progress has been made in predicting how proteins fold,29−34 to date artificial protein-templated dye aggregates are yet to be demonstrated, likely due to complexities arising from adding a dye to the protein structure. Using DNA to template dye aggregates represents an alternative, promising approach. In contrast to amino acids, nucleic acids have relatively simple pairing rules, that is, Watson–Crick base pairing yet can form a diverse array of structures including duplexes, Holliday junctions, and higher-order structures.35−39 Many early, pioneering studies used duplex DNA to template dye aggregates via physisorption.40−42 More recently, an even higher level of precision and versatility has been achieved by covalently tethering dyes to DNA.24,43−48 Such precision and versatility enable the controlled assembly of dye aggregates with distinct packing geometries24,47,49 and dye aggregate networks with a specific number of constituent dyes,47,49,50 facilitating the study of structure–function relationships and design of aggregates with properties custom tailored and optimized for specific applications.
Cyanines are ubiquitous dyes, by far the most commonly employed family of dyes in covalently tethered DNA templating.46,49,51−54 Of these dyes, the pentamethine dye Cy5 (Scheme 1) is particularly attractive due to its strong absorption [i.e., TDM of ∼16 D (ref (55)) and associated peak extinction coefficient of ∼250,000 M–1 cm–1] in the visible spectral range (λmax ∼ 650 nm). According to Kasha’s molecular exciton model,56,57 the excitonic hopping parameter, J, sometimes referred to as the excitonic coupling strength, is directly proportional to the square of the TDM. Thus, a large TDM may lead to a large J. Indeed, our recent work has shown that J can be as large as 800 cm–1 (100 meV) in certain DNA-templated Cy5 aggregates.49,58 Additionally, Cy5 is quite photostable. Cy5 has a relatively high fluorescence quantum yield of ∼0.3,25,59 indicating that radiative decay is able to effectively compete with nonradiative decay processes. This results in a fairly long ∼1.5 ns lifetime for a Cy5 monomer, which we showed previously was dramatically reduced in dimer and tetramer aggregates due to accelerated nonradiative decay.25 The physical origin of this accelerated nonradiative decay, however, remains elusive. Furthermore, the generality of this effect and whether it depends on the packing configuration, such as the number of dyes and the distance and orientation between them, are yet to be explored.
Scheme 1. Cy5, a Common Fluorescence Labeling Dye Composed of a Pentamethine Bridge Flanked by Two Dimethyl Indolenine Rings, Shown Tethered to an Oligonucleotide Backbone via Two Propyl Linkers Covalently Bonded to the Deoxyribose and Phosphate Groups of the 5′ and 3′ Ends of the Sequence, Respectively.

In this work, we use a combination of femtosecond transient absorption (TA) spectroscopy and global target analysis (GTA) to examine the excited-state dynamics of a series of DNA-templated Cy5 aggregates. First, we examine the excited-state dynamics of duplex dimer and Holliday-junction tetramer structures formed using two distinct oligonucleotide strands. Next, we examine the excited-state dynamics of two different dimers, a trimer, and a tetramer using DNA Holliday junctions formed from four distinct oligonucleotide strands. In all cases, considerable reduction of the excited-state lifetime in the aggregates compared with the monomer is observed and, in addition, considerable variability is observed in the lifetimes of the aggregate structures. Pump-wavelength-dependent femtosecond TA measurements indicate that certain solutions exhibit an appreciable extent of heterogeneity, particularly with respect to aggregate structures. We proceed to discuss the structural factors influencing nonradiative decay in the DNA-templated aggregates, such as exciton delocalization and dye separation. Finally, we discuss factors contributing to the variability of the excited-state lifetimes, including potential sources of heterogeneity.
Methods
DNA-Dye Construct Preparation
Oligonucleotides labeled with Cy5 via dual phosphoramidite linkers and purified by dual high-performance liquid chromatography were obtained as lyophilized powders from Integrated DNA Technologies (IDT; Coralville, IA). Unlabeled oligonucleotides purified by desalting were also obtained from IDT. Stock solutions of labeled and unlabeled oligonucleotides were prepared at a concentration of 100 μM by hydrating with water obtained from a Barnstead Nanopure water purification system (ThermoFisher Scientific, Waltham, MA). Stock oligonucleotide solution concentrations were determined according to Beer’s law using the extinction coefficient provided by the vendor (at 260 nm) and the absorbance measured at 260 nm via a NanoDrop One UV–Vis spectrophotometer (ThermoFisher Scientific, Waltham, MA), which reports absorbance scaled to a path length of 1 cm. Solutions of DNA and DNA-dye constructs (i.e., labeled and unlabeled DNA duplexes and Holliday Junctions) were prepared by combining equimolar amounts of stock oligonucleotide solutions with an aqueous TAE buffer solution prepared at 10 times its typical concentration, that is, 10×. MgCl2 was added for selected DNA and DNA-dye constructs. The volumes of the stock oligonucleotide solutions, TAE buffer solution, and water were chosen so that the final concentration of the DNA or DNA-dye construct was in the range of 1–5 μM. The volumes were also chosen to achieve a final buffer concentration of 1× TAE and the final desired concentration of MgCl2, which ranged from 0 to 100 mM. The resulting solutions were vortex mixed for 30 s, then incubated overnight in a dark container at room temperature.
Steady-State Absorption Spectroscopy
Steady-state spectra were collected using either a Cary 60 or 5000 UV–Vis spectrophotometer (Agilent Technologies, Santa Clara, CA). Spectra were typically collected from 200 to 800 nm in 1 nm steps. Solutions were contained in a 2 mm quartz cuvette (Starna Cells, Atascadero, CA) and had a maximum absorbance of ca. 0.3 in the range of 500–700 nm.
Femtosecond TA Spectroscopy
All TA spectra were collected using a custom-built TA spectrometer. At the head of the system, a Ti:sapphire-based laser oscillator (Coherent, Santa Clara, CA) produces a train of femtosecond pulses at a rate of 80 MHz, centered at ∼800 nm and with an energy of ∼4 nJ/pulse. The output of the laser oscillator seeds a Ti:sapphire-based regenerative amplifier (Coherent, Santa Clara, CA), which produces an amplified train of ∼40 fs pulses at a rate of 1 kHz, centered at ∼800 nm and with an energy of ∼3 mJ/pulse. The output of the laser amplifier was used to drive an optical parametric amplifier (Coherent, Santa Clara, CA) to generate the pump beam, which varied from ∼560 to ∼700 nm. The probe beam was generated by focusing a small fraction of the laser amplifier output onto a 2 mm thick sapphire window (Newlight Photonics, Toronto, ON, Canada), which produced a white-light continuum spanning ∼420 to 750 nm (Section S1). The pump and probe beams were spatially overlapped at the sample position, and their relative time delay was controlled by varying the pump beam path distance via a mechanical delay stage (Aerotech, Pittsburgh, PA). TA spectra were collected by routing the probe beam to a spectrograph, which included a monochromator and an sCMOS array detector (Andor, Belfast, Northern Ireland). For all measurements, the relative orientation of the linearly polarized pump and probe beams was set to 55°, that is, the so-called “magic” angle. Pump and probe beam diameters were estimated to be ca. 215 and 140 μm, respectively, by measuring the beam power in the presence and absence of a 50 μm diameter high-energy pinhole (Newport, Santa Clara, CA) situated in the focal plane of the probe beam. Pump fluences ranged from 13 to 40 μJ/cm2 and are reported where appropriate. Solutions were contained in a 2 mm quartz cuvette (Starna Cells, Atascadero, CA) and stirred with magnetic stirrer bars (Starna Cells, Atascadero, CA) rotated over the course of the measurement with a magnetic stirring apparatus (Ultrafast Systems, Sarasota, FL). The pump pulse duration was determined to be ∼180 fs by performing an autocorrelation on a 2 mm quartz cuvette filled with distilled water situated at the sample position (Section S2).
Global and Target Analysis of TA
Prior to GTA, the femtosecond TA data were corrected for chirp in the probe continuum. The chirp in the probe continuum was modeled with a polynomial function, and the respective wavelength-dependent time offset correction factors were subtracted from the data. GTA of the corrected data was facilitated with the Java-based graphical user interface Glotaran60 and performed using the TIMP statistical package, which is part of the R computational platform.
Steady-State Optical Modeling
The optical properties of selected DNA-dye constructs were modeled using in-house software based on the Kühn–Renger–May method (version 13.7).47,49,58 The software simultaneously fits absorbance and circular dichroism data by constructing and diagonalizing a Holstein Hamiltonian19 that includes a single dominant vibronic mode. The fitting procedure begins with a user input dye configuration and performs a stochastic search by randomly perturbing the dye configuration and checking the fit quality. The output of the optical modeling is a set of vectors representing the spatial orientation and separation of the TDMs of the dyes in the aggregate. In the case of Cy5, the TDM is taken to be along the long axis of the pentamethine bridge (Scheme 1).
Results
Construct Preparation and Steady-State Optical Characterization
To examine the impact of DNA-dye configuration on the excited-state dynamics of Cy5 aggregates, we chose to study the two types of DNA-Cy5 constructs presented in Figure 1.
Figure 1.

(a) Type 1 DNA-dye constructs formed by mixing two complementary Cy5-labeled oligonucleotide strands in an aqueous buffer solution in the absence (left) and presence (right) of added MgCl2. Duplex DNA is the primary nanostructure in the absence of added MgCl2, whereas DNA Holliday junctions are formed when MgCl2 is added. There is one Cy5 dye per strand, and therefore, the duplex and Holliday junction template the aggregation of a Cy5 dimer and tetramer, respectively. (b) Normalized absorption spectra of the duplex dimer and “mobile” Holliday junction tetramer. The lower part of panel (b) depicts schematic illustrations of the duplex dimer and Holliday junction tetramer, where cyan circles indicate the approximate location of Cy5 dyes. (c) Type 2 DNA-dye constructs formed by mixing four mutually complementary oligonucleotide strands in an aqueous buffer solution with MgCl2 added. The solutions primarily consist of “immobile” Holliday junctions. Cy5 dyes are covalently tethered to two, three, or four positions within the Holliday junction. (d) Normalized absorption spectra of the two dimer species (adjacent and transverse), trimer, and tetramer formed via “immobile” Holliday junctions. The lower part of panel (d) depicts schematic illustrations of the dimer, trimer, and tetramer structures, where cyan circles indicate the approximate location of Cy5 dyes. The schematic depictions of the dye-labeled duplex and Holliday junction structures are not meant to imply specific geometric configurations of the dye or DNA backbone.
Cannon et al. showed that a range of DNA-Cy5 dye aggregate nanostructures are possible from a basis set of either two or four oligonucleotide strands.47,49Figure 1a shows the two DNA-dye aggregate nanostructures resulting from a basis set of two oligonucleotide strands. In this case, which we label the “type 1” set of DNA-dye constructs, it is possible to form a Cy5 dimer via duplex DNA along with a Cy5 tetramer via a DNA Holliday junction. Specifically, Cannon et al. showed that in the absence of added MgCl2, duplex DNA was the primary DNA nanostructure in the solution. However, through gel electrophoresis studies, Cannon et al. showed that adding MgCl2 to the solution promoted the association of four strands to form a “mobile” DNA Holliday junction tetramer. Furthermore, the Holliday junction is only observed when the two basis strands are labeled with Cy5, indicating that the DNA-dye construct may be additionally stabilized by the mutual attraction of the four dyes. Since two and four dyes are aggregated via DNA duplexes and Holliday junctions, we label these aggregates as the duplex dimer and type 1 tetramer structures, respectively. In the case of the duplex dimer and type 1 tetramer structures, the excitonically coupled absorption spectra exhibit redshifted and blueshifted spectral features consistent with J- and H-aggregation, respectively (Figure 1b). For the remainder of the paper, we refer to the solutions where primarily duplex dimers and Holliday junction tetramers are present as the no-salt type 1 and high-salt type 1 solutions, respectively.
Expanding the basis set to four oligonucleotides provides access to an even larger number of DNA-dye aggregate configurations (Figure 1c). Here, a single “immobile” DNA Holliday junction is targeted. In this case, the Holliday junction is targeted to minimize the possibility that lower-order DNA nanostructures, such as duplexes, might form and contribute to heterogeneity in the solution. We label these aggregates as the “type 2” set of DNA-dye constructs. In the type 2 set of DNA-dye constructs, the number of dyes in the aggregate can be varied between two, three, and four. Thus, the effect of the extent of exciton delocalization on excited-state lifetimes can be examined. Furthermore, two different dimer configurations are possible—an adjacent and transverse dimer. Cannon et al. showed that the adjacent dimer exhibited optical signatures of a J-aggregate packing arrangement, while the transverse dimer, trimer, and tetramer all exhibited signatures of H-aggregate packing.49 These signatures are evident in the absorption spectra, as shown in Figure 1d. Thus, the so-called type 2 set of DNA nanostructures provide access to a range of configurations, including different possible extents of exciton delocalization, proximal positions of the dyes, and packing arrangements.
Excited-State Dynamics of Type 1 DNA-Cy5 Constructs
We begin by examining the excited-state dynamics of the no-salt type 1 solution, which is composed largely of duplex dimer structures. We performed spectrally resolved femtosecond TA measurements exciting the solutions at specific wavelengths using a narrowband pump and tracking the ensuing excited-state dynamics across the visible spectrum using a continuum probe (see, e.g., Section S1). Here, we excite the solution at 675 nm (Figure 2a) which was selected to coincide with the main absorption band at ∼666 nm. The resulting TA surface for the no-salt type 1 solution shown in Figure 2a plots the evolution of the TA spectrum. Several positive and negative TA signals (plotted in units of ΔT/T) are evident, all of which decay on a rapid ca. 10 ps timescale. Figure 2b displays spectral slices that more clearly highlight the rapidly decaying positive and negative signals. At the earliest time delay shown of ∼1 ps, two prominent positive TA signals are apparent at 603 and 672 nm that generally align with the peaks appearing at ca. 600 and 666 nm in the steady-state absorption spectrum. As such, we assign these positive TA signals to ground-state bleach (GSB) features of the duplex dimer structure. In addition, several negative TA signals are observed at ca. 445, 493, and 638 nm, which we assign to excited-state absorption (ESA) features. At the longest time delay shown of ∼150 ps, all the signals have decayed except for a weak positive signal at ca. 655 nm, which we assign to the GSB feature associated with a small subpopulation of monomers.
Figure 2.
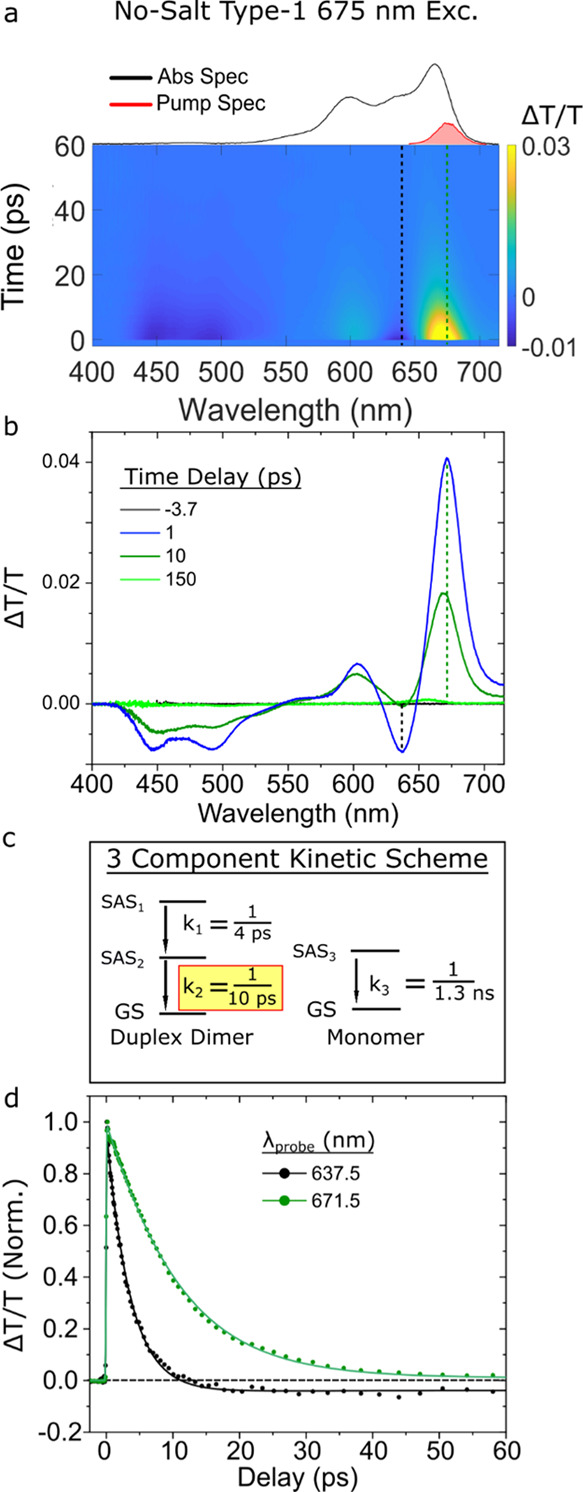
Femtosecond TA spectrum of the no-salt type 1 solution, predominantly composed of duplex J-dimer structures, collected with a pump wavelength of 675 nm at a fluence of 18 μJ/cm2. (a) Surface plot. The black and green vertical dashed lines correspond to individual kinetics traces plotted in panel (d). The absorption and pump spectra are plotted above the TA surface plot. The scale bar is indicated beside the plot. (b) Selected TA spectra. Time delays are indicated in the legend. (c) Kinetic scheme used to model the TA spectrum via GTA. Rate constants (expressed as inverse time constants) associated with conversion between different components exhibiting distinct species-associated spectra (SAS) are shown. The rate constant associated with the excited-state lifetime of the duplex dimer structure is highlighted in yellow. (d) Selected TA kinetics at probe wavelengths of 638 and 672 nm are shown, with the kinetics normalized to their maximum amplitude near the time origin of the measurement. Data are shown as circles, while fits are shown as solid lines.
To further investigate the excited-state dynamics of the duplex dimer structure, we performed a GTA of the TA of the no-salt type 1 solution. The kinetic scheme used to model the data is shown in Figure 2c. A more detailed mathematical and physical justification of the three-component kinetic scheme, including the species-associated spectra (SAS) for each component, can be found in Section S3 of Supporting Information. The kinetic scheme includes two components associated with the duplex dimer structure and a third component to account for a small subpopulation of monomers. With the exception of the small subpopulation of monomers, the no-salt type 1 solution is not expected to exhibit significant heterogeneity, particularly at the 675 nm excitation wavelength chosen for the TA measurement.25 Thus, we conclude that the two components in the kinetic scheme assigned to the duplex dimer structure decay in sequence. Figure 2d displays normalized kinetics traces and associated fits at probe wavelengths of 672 and 638 nm, which correspond to the decay of the most prominent GSB and ESA features, respectively, of the duplex dimer (see, e.g., Figure 2a). The GTA returned k1 and k2 rate constants, associated with the sequential decay of the duplex dimer, of ca. 1/3.6 and 1/10 ps–1, respectively, while the third rate constant, k3, associated with monomers, was fixed to the excited-state decay rate of the monomer, that is, 1/1.3 ns–1 (Section S4). Figure 2d shows that the ESA feature exhibits a decay component on a timescale much faster than the GSB feature, which the GTA derives to be 4 ps. In contrast, the GTA derives a time constant of 10 ps for the more slowly decaying GSB feature. The latter result is in excellent agreement with the 11 ps lifetime previously measured using a degenerate, narrowband pump–probe configuration.25
Next, we examined the excited-state dynamics of the high-salt type 1 solution, primarily composed of type 1 tetramer structures. In order to preferentially excite the type 1 tetramer structures, that is, to optically bias a population of excited states specifically associated with these structures, the solution was pumped at 555 nm, near where the type 1 tetramer structures absorb the strongest. The resulting TA surface plot is shown below the steady-state absorption spectrum in Figure 3a. A single positive TA signal is apparent in the surface plot at ca. 561 nm, which is situated between two negative TA signals, one at the shorter wavelength that is weak and broad, and another at the longer wavelength that is more intense and narrow. Notably, compared with the duplex dimer structure, the signal persists on a longer several tens of picosecond timescale. Figure 3b shows selected TA spectra taken from the TA surface plot. At an early time delay (1 ps), an intense and narrow positive TA band peaking at ca. 561 nm is observed, which aligns with the main absorption band of the high-salt type 1 solution. Given that the high-salt type 1 solution is composed largely of type 1 tetramer structures, we assign the positive TA band to the GSB of the type 1 tetramer structures. As noted above, the GSB of the type 1 tetramer structure is situated between two negative TA signals. Figure 3b shows that these signals, which we assign to ESA features, peak at ca. 467 and 608 nm. Notably, the 1 ps TA spectrum is essentially featureless between ca. 650–715 nm. This observation indicates that duplex dimer structures, which exhibit a pronounced GSB feature at ca. 671 nm (Figure 2b), contribute negligibly, if at all, to the observed TA. At intermediate time delays (10–30 ps), the TA spectrum appears to decay uniformly. This result is consistent with the interpretation that only a single aggregate structure, the type 1 tetramer, contributes to the TA signal. At long time delays (150 ps), the TA signal has completely recovered to baseline, and no monomer signal is evident. Thus, the selected TA spectra indicate that the 555 nm pump preferentially excites type 1 tetramer structures in the high-salt type 1 solution.
Figure 3.

Femtosecond TA spectra of the high-salt type 1 solution, primarily composed of type 1 tetramer structures, collected with pump wavelengths of 555 and 660 nm at pump fluences of 26 and 18 μJ/cm2, respectively. (a,e) Surface plots. Vertical dashed lines indicate kinetics traces that are plotted in panels (d,h), respectively. (b,f) Selected TA spectra for each pump wavelength. Time delays are indicated in the legend. (c,g) Kinetic schemes used for GTA. The rate constants associated with specific components are reported as inverse time constants. In the case of panel (g), all rate constants were fixed except for the rate constant associated with the fourth component (or SAS4). (d,h) Selected TA kinetics, normalized to their maximum amplitude near the time origin of the measurement. Data are shown as circles, while fits are shown as solid lines. Panel (d) displays TA kinetics at a probe wavelength of 561 nm, which corresponds to the GSB of the type 1 tetramer structure; the inset of panel (d) displays TA kinetics taken at probe wavelengths of 561 and 583 nm corresponding to the GSB and ESA, respectively, of the type 1 tetramer structure. Panel (h) displays TA kinetics at a probe wavelength of 561 nm, which corresponds primarily to the GSB of the type 1 tetramer structure, and at probe wavelengths of 635 and 667 nm, which correspond primarily to the ESA and GSB, respectively, of the duplex dimer structure.
To further investigate the excited-state dynamics of the type 1 tetramer structures, we performed a GTA of the TA of the high-salt type 1 solution excited at 555 nm. The two-component kinetic scheme used to model the data is shown in Figure 3c. A more detailed mathematical and physical justification of the two-component kinetic scheme, including the SAS for each component, can be found in Section S5. Figure 3d displays a normalized kinetics trace at 561 nm, and an overlay of normalized kinetics traces at 561 and 583 nm is shown in the inset. The kinetics traces are largely similar, with slight deviations evident on the sub-10 ps timescale. Two rate constants k1 and k2 with values of 1/2.3 and 1/30 ps–1, respectively, are derived from the GTA. While k1 could have several physical origins, including solvent dynamics and vibrational cooling, the value of ca. 1/30 ps–1 is clearly associated with the full recovery of the GSB and decay of the ESA features. As such, we assign k2 to the excited-state decay rate of the type 1 tetramer structures. The value of ca. 30 ps derived by the GTA is in good agreement with the lifetime of ca. 35 ps previously measured via degenerate, narrowband pump–probe.25
Having measured the “pure” spectrotemporal signals—that is, the spectra and rate constants—associated with the duplex dimer and type 1 tetramer structures, we next proceeded to simultaneously excite both populations in order to produce a heterogeneous TA data set. This was accomplished by exciting the high-salt type 1 solution at 660 nm, where both the duplex dimer and type 1 tetramer exhibit appreciable absorption (Figure 1b). As expected, both the TA surface plot and selected TA spectra appear to be a sum of the signals associated with the duplex dimer and type 1 tetramer structures (Figure 3e,f). This is perhaps most visible in the 1 ps TA spectrum, which exhibits positive and negative TA signals at ca. 667 and 635 nm, respectively, clearly reminiscent of the GSB and ESA features associated with the duplex dimer structure. As was observed in the low-salt type 1 solution excited at 675 nm, the ESA feature peaking at ca. 635 nm decays considerably by ca. 10 ps. In addition, a prominent positive TA feature is observed at ca. 561 nm, which is reminiscent of the GSB feature of the type 1 tetramer structure. Consistent with our expectations based on the ca. 10 and 30 ps lifetimes measured for the duplex dimer and type 1 tetramer structures, respectively, the positive TA signal at 667 nm decays faster than the positive TA signal at 561 nm. Clearly, the positive TA signals at 667 and 561 nm are attributable to the GSB features of the duplex dimer and type 1 tetramer structures, respectively. Thus, we have successfully achieved a heterogeneous TA data set where two aggregate structures are simultaneously excited.
To test the ability of GTA to decompose a TA data set into “pure” spectra and rate constants of the constituent components, we then performed a GTA on the heterogeneous TA data set including signatures of both duplex dimers and type 1 tetramers. A five-component kinetic scheme with both sequential and parallel pathways, as shown in Figure 3g, was used to model the data. A more detailed mathematical and physical justification of the five-component kinetic scheme, including the SAS for each component, can be found in Section S6. An essential aspect of the model is that it includes multiple parallel decay pathways to account for the heterogeneous nature of the solution and corresponding TA data set. Guided by the rate constants measured for the duplex dimer and type 1 tetramer detailed above, k1–k3 and k5 attributable to the duplex dimer, type 1 tetramer, and monomer were fixed to the values of k1 = 1/4 ps–1, k2 = 1/10 ps–1, k3 = 1/32 ps–1, and k5 = 1/1.3 ns–1, respectively. Only one component was used for the type 1 tetramer due to the small change in the amplitude between its two components (see, e.g., Figure 3c and Section S5). An additional component, not associated with any of the structures discussed thus far, was revealed by the GTA. The rate constant associated with this component, k4, was derived to be 1/160 ps–1. Additional details about this component, including a tentative physical assignment, can be found below. Figure 3h displays normalized kinetics traces at probe wavelengths of 561, 635, and 667 nm, which correspond primarily to the GSB of the type 1 tetramer, the ESA of the duplex dimer, and the GSB of the duplex dimer, respectively. Remarkably, the kinetics traces displayed in Figure 3h appear largely to represent a linear combination of the kinetics traces displayed in Figures 2d and 3d, i.e., those associated with the duplex dimer and type 1 tetramer structures, respectively, with the exception of a more pronounced long-lived signal readily attributable to a larger fraction of monomers photoexcited in the high-salt type 2 solution. Fits of the kinetics traces, derived from the five-component GTA, are also shown in Figure 3h.
We next proceed to compare the “pure” SAS derived for the duplex dimer and type 1 tetramer with the corresponding SAS derived from the heterogeneous TA data set. Figure 4 displays the “pure” SAS derived for the duplex dimer and type 1 tetramer along with the corresponding SAS derived from the heterogeneous TA data set. The five-component kinetic scheme used to model the heterogeneous TA data set is shown in Figure 4a above the SAS plots, with arrows pointing from each component to its corresponding SAS. To facilitate comparison, the SAS are normalized to the strongest GSB feature in each plot.
Figure 4.

(a) Five-component kinetic scheme used to model the high-salt type 1 solution pumped at 660 nm via a GTA (reproduced from Figure 3g). Four subpopulations—duplex dimers, type 1 tetramers, adjacent dimers, and monomers—decay in parallel in the model. The duplex dimers additionally exhibit a sequential decay in the model. (b) “Pure” SAS derived for the duplex dimer from the no-salt type-1 solution pumped at 675 nm (solid lines) and SAS from the heterogeneous data set, i.e., the high-salt type-1 solution pumped at 660 nm, attributed to the duplex dimer (dotted lines). SAS1 are plotted in black, and SAS2 are plotted in orange. (c) “Pure” SAS derived for the type 1 tetramer from the high-salt type-1 solution pumped at 555 nm (solid line) and the SAS from the heterogeneous data set pumped at 660 nm, attributed to the type 1 tetramer (dotted line). (d) SAS from the heterogeneous data set attributed to the fourth component, which we assign to a small subpopulation of adjacent dimer-like structures present in the high-salt type-1 solution.
The “pure” SAS derived for the duplex dimer and type 1 tetramer are in overall good agreement with the corresponding SAS derived from the heterogeneous TA data set. Figure 4b shows that the “pure” SAS1 for the duplex dimer and the corresponding SAS derived from the heterogeneous TA data set are nearly indistinguishable. The most notable difference appears in the comparison of the “pure” SAS2 for the duplex dimer with the corresponding SAS derived from the heterogeneous TA data set. Compared with the “pure” SAS2 for the duplex dimer, the SAS from the heterogeneous data set exhibits an additional positive amplitude at ca. 560 nm, marked with an asterisk. Given that the additional intensity is located in the vicinity of the main GSB for the type 1 tetramer that appears at ca. 560 nm, we attribute the additional intensity to component mixing between SAS2 and SAS3 due to their similar lifetimes (10 and 32 ps, respectively) and spectral overlap. Figure 4c shows that the “pure” SAS of the type 1 tetramer and the corresponding SAS derived from the heterogeneous TA data set are also largely indistinguishable. Here, we see overall good agreement between the SAS except for the spectral region between 650 and 700 nm, marked with two asterisks. Specifically, while the “pure” SAS have essentially zero amplitude in this spectral region, the SAS derived from the heterogeneous data set exhibit positive and negative TA features. We attribute these features to a combination of the component mixing between SAS3 and shorter timescale components along with the dynamic pump scatter present at intermediate time delays. Lastly, Figure 4d shows the SAS associated with the fourth component in the model, which, given the similarity of its SAS and rate constant with the type 2 adjacent dimer discussed in the following section, we tentatively attribute to an additional adjacent dimer-like structure present in the high-salt type 1 solution.
Overall, the results indicate that GTA is largely able to disentangle the SAS and rate constants associated with different aggregate structures in a heterogeneous solution and even uncover a previously unidentified aggregate structure. However, when lifetimes of species are similar and their spectra overlap, certain limitations such as component mixing can arise. These are important points of relevance to the next section.
Excited-State Dynamics of Type 2 DNA-Cy5 Constructs
With the excited-state dynamics of the type 1 DNA-dye constructs characterized, we next proceeded to characterize the excited-state dynamics of the type 2 DNA-dye constructs.
Dimers
We begin by examining the excited-state dynamics of the transverse dimer solution pumped at 600 nm near where the Holliday junction transverse dimer structures absorb most strongly. Figure 5a displays the associated TA surface plot, which is characterized by a broad GSB band with its most intense feature peaking at ca. 600 nm and a weaker ESA band at a shorter wavelength peaking at ca. 495 nm. Relatively uniform decay of these spectral bands is observed in the TA surface. Figure 5b displays selected TA spectra, which show how these features evolve over time. Comparing the TA spectrum at 1 ps, as shown in Figure 5b, to the absorption spectrum of the transverse dimer, the shoulder, intense bleach, and secondary bleach at 560, 600, and 635 nm, respectively, all align well with features in the steady-state absorption spectrum. The TA spectrum at early delay times (1 ps) largely resembles the TA spectra at intermediate (100 ps) and longer delay (400 ps). However, the ratio of the intensities of primary and secondary GSB features at 600 and 635 nm, respectively, decreases at longer delay times, which we attribute to long-lived monomer contribution to the TA. The close resemblance between steady-state and transient spectra, the relatively uniform TA decay, and the results of pump-wavelength-dependent TA (as described below) are all consistent with the interpretation that the transverse dimer solution exhibits little aggregate structural heterogeneity and that the TA data, as shown in Figure 5a,b, primarily represent the excited-state dynamics of transverse dimer structures.
Figure 5.

Femtosecond TA spectra of the type 2 transverse and adjacent dimer solutions pumped at 600 and 675 nm, respectively, with pump fluences of 13 and 15 μJ/cm2, respectively. (a,e) TA surface plots of the transverse and adjacent dimer solutions. Vertical dashed lines indicate selected kinetics traces plotted in panels (d,h). (b,f) Selected TA spectra. (c,g) Kinetic schemes used for GTA. The rate constants associated with specific components and SAS are reported as inverse time constants. In both analyses, the rate constant associated with the monomer was fixed to 1/1.3 ns–1. (d,h) Normalized kinetics traces taken at different probe wavelengths. Data are shown as circles, while fits are shown as solid lines. In panel (d), kinetics traces are displayed for probe wavelengths of 494 and 601 nm, which correspond to ESA and GSB bands, respectively. In panel (h), kinetics traces are displayed for probe wavelengths of 640 and 670 nm, which correspond to ESA and overlapping ESA and GSB bands, respectively.
To extract the lifetime of the transverse dimer structure, we performed GTA on the TA data. The four-component kinetic scheme used to model the data is shown in Figure 5c. A more detailed mathematical and physical justification of the four-component kinetic scheme, including the SAS for each component, can be found in Section S7 of Supporting Information. The kinetic scheme includes sequential kinetics associated with the transverse dimer structures and the parallel decay of a small subpopulation of monomers. With the fourth rate constant, k4, associated with monomer decay fixed to a value of 1/1.3 ns–1 (a value used in all subsequent analyses), we derived k1, k2, and k3 values of 1/2.2, 1/24, and 1/202 ps–1, respectively. We attribute k1 and k2 to excited-state dynamics that do not result in decay to the ground state as the primary difference between SAS are small spectral shifts and changes to ESA bands (Section S7). We attribute the third and final rate constant associated with the transverse dimer structures, that is, k3, to excited-state decay to the ground state. Figure 5d plots normalized kinetics traces at 601 and 494 nm along with fits derived from the GTA. The trace at 494 nm initially decreases in amplitude more rapidly than the trace at 601 nm, which reflects changes in the excited-state electronic structure associated with the short-wavelength ESA band that take place at a rate of 1/24 ps–1 (i.e., k2). In contrast, the 601 nm trace, which follows the decay of the main GSB feature, largely reflects the 1/202 ps–1 excited-state decay rate of the transverse dimer structures.
We next characterize the excited-state dynamics of the adjacent dimer solution. Specifically, we performed TA with a 675 nm pump wavelength, which corresponds to the strongest absorption band of the adjacent dimer solution. Figure 5e displays the TA surface. Two GSB features are visible, with the strongest and weakest features at 671 and 608 nm, respectively. An ESA feature at ca. 641 nm also appears to overlap the two GSB features. A second ESA feature is observed at shorter wavelengths at 499 nm. While the 608 and 671 nm GSB features align reasonably well with the maxima observed in the steady-state absorption spectrum, overall the TA spectra do not strongly resemble the steady-state absorption spectrum, possibly indicating that the adjacent dimer solution is heterogeneous. Excitation wavelength-dependent TA measurements (described below) confirm that the adjacent dimer solution may exhibit appreciable heterogeneity. Despite the heterogeneity, the redshifted absorption maximum of the adjacent dimer solution (Figure 1d) suggests that J-aggregates are the predominant species. By exciting at 675 nm, J-aggregates are thus selectively excited over H-aggregates or monomers. Selected TA spectra are plotted in Figure 5f. Similar to what we saw with the duplex dimer (Figure 3a–d), the two GSB features and the ESA at short wavelengths of the adjacent dimer appear to decay together, while the ESA at ca. 641 nm exhibits a more rapid decay. For example, at 1 ps delay, the amplitudes of both ESA features are similar, but after 100 ps, the ESA at 635 nm has decayed appreciably such that the overall TA signal has become positive in that region, while the ESA at ca. 500 nm is still negative.
To extract the lifetimes of the adjacent dimer structures, we performed a GTA, according to a four-component kinetic scheme (Figure 5g,h). Additional details regarding the mathematical and physical justification of the four-component kinetic scheme are shown in Section S8 of Supporting Information. We attribute k1 and k2 with values of 1/6.0 and 1/40 ps–1, respectively, to two rate constants associated with a single population of short-lived adjacent dimer structures. As we saw above for the duplex and transverse dimer structures, we assign k1 (1/6.0 ps–1) to an excited-state process that results in a change of the electronic structure, as primarily a reduction in the amplitude of the ESA band at 641 nm is observed between SAS1 and SAS2 (Section S8). We attribute k2 (1/40 ps–1) to the excited-state decay rate of the short-lived adjacent dimer structures. Based on the distinct spectral features of SAS3 as compared with SAS1 and SAS2 (Section S8), we attribute k3 (1/242 ps–1) to the excited-state decay rate of a subpopulation of long-lived adjacent dimer structures. Thus, two different dimer structures with excited-state lifetimes of ca. 40 and 240 ps are observed in the adjacent dimer solution.
To further characterize the heterogeneity of the transverse and adjacent dimer solutions, we examined the pump-wavelength dependence of their TA. While Figure 5 displays the TA of the transverse dimer solution pumped at 600 nm, Figure 6 shows the TA of the transverse dimer solution pumped at 660 nm, which was chosen to selectively excite monomer and J-aggregate subpopulations. Compared with the TA surface shown in Figure 5a, the TA surface shown in Figure 6a exhibits more pronounced GSB and ESA features at ca. 660 and 500 nm, respectively. Compared with Figure 5b, less uniform decay of the TA spectra is observed in Figure 6b. For example, the GSB at 605 nm decays more rapidly than the GSB at 660 nm, and the 400 ps TA spectrum largely resembles that of the monomer. We then pumped the adjacent dimer solution at 600 nm to potentially selectively excite H-aggregate subpopulations. Compared with Figure 5e where the solution was pumped at 675 nm, the TA surface, as shown in Figure 6d, exhibits additional GSB and ESA features at ca. 605 and 500 nm, respectively. Selected TA spectra, as shown in Figure 6e, indicate that the GSB features at ca. 605 and 660 nm decay nearly synchronously for approximately 100 ps, but at later times, the TA spectra resemble the monomer. Thus, it is clear from the pump-wavelength-dependent TA results, as shown in Figures 5 and 6, that both the transverse and adjacent dimer solutions exhibit some level of heterogeneity.
Figure 6.

Femtosecond TA spectra of the type 2 transverse and adjacent dimer solutions pumped at 660 and 600 nm, respectively, with pump fluences of 40 and 15 μJ/cm2, respectively. (a,d) TA surface plots of the transverse and adjacent dimer solutions. (b,e) Selected TA spectra. (c,f) 100 ps time delay, TA spectra along with the scaled basis spectra used to model the data and the resultant fit. Data are shown in black; transverse dimer, adjacent dimer, and monomer basis spectra are shown in purple, green, and blue, respectively; and the fit is shown in red.
To further characterize the heterogeneity in the transverse and adjacent dimer solutions, we modeled their TA spectra using the SAS (or “pure” spectra) of the monomers and transverse and long-lived adjacent dimer structures derived from the GTA. The rationale for using these basis spectra is the following: (i) a small subpopulation of monomers is known to be present in the solutions25 and (ii) the presence of long-lived adjacent dimer structures in the no-salt type 1 solution composed primarily of duplex dimer solution suggests such aggregates may be present in other solutions. Figure 6c displays the results of modeling the 100 ps timescale TA spectrum of the transverse dimer solution excited at 660 nm as a linear combination of these basis spectra. The model matches the data quite well and indicates that mostly monomers and transverse dimer structures are present in the solution, possibly also with a small amount of long-lived adjacent dimer structures. Due to the presence of a strong GSB feature at 600 nm in the TA spectrum of the adjacent dimer solution, which closely matches the primary GSB feature of the transverse dimer structures, it is sensible to conclude that transverse dimer structures may be contributing to the TA signal as well. Thus, the transverse dimer structure was included as a basis spectrum in the analysis of the TA spectrum of the adjacent dimer solution. Figure 6f displays the results of modeling the 100 ps timescale TA spectrum as a linear combination of monomer, adjacent dimer, and transverse dimer basis spectra. Again, the model matches the data well and indicates that an appreciable population of transverse dimer structures is present in the adjacent dimer solution. Based on these results, we thus conclude that with respect to aggregate structures, the transverse and adjacent dimer solutions exhibit limited and appreciable heterogeneity, respectively.
Trimer
Having characterized the excited-state dynamics of the transverse and adjacent dimer solutions, we next turn our attention to the excited-state dynamics of the trimer solution. Figure 7 displays the TA of the trimer solution pumped near its most intense absorption band at 560 nm. The TA surface and selected TA spectra, as shown in Figure 7a,b, exhibit two GSB features peaking at ca. 574 and 680 nm. Two ESA features are also observed at ca. 469 and 655 nm. Between early (1 ps) and intermediate (30 ps) time delays, the maximum of the primary GSB feature shifts toward longer wavelengths and broadens slightly. At the same time, the ESA maximum at 469 nm shifts to shorter wavelengths. At longer time delays (150 ps), the primary GSB feature has redshifted further, peaking at 582 nm, while the weak ESA observed at ca. 655 nm has decayed considerably. The significant spectral shifts observed in the TA data suggest that the trimer solution may be heterogeneous and that pumping the solution at 560 nm excites multiple aggregate structures.
Figure 7.
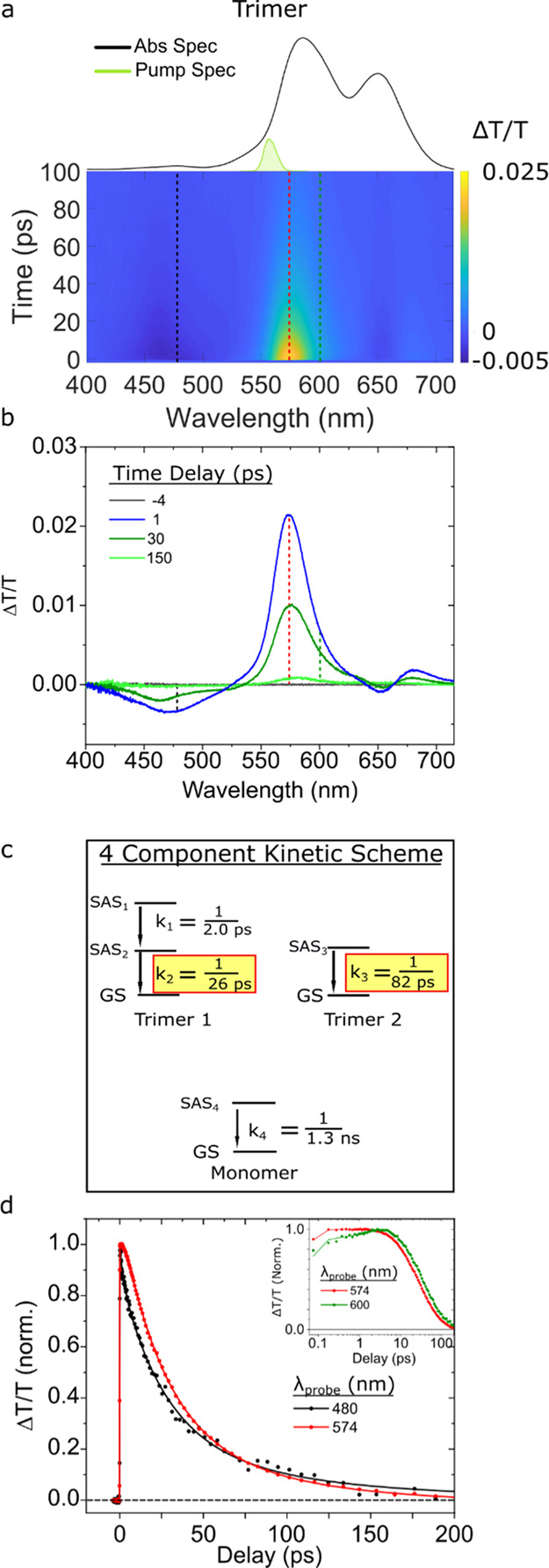
Femtosecond TA spectra of the type 2 trimer solution pumped at 560 nm with a pump fluence of 29 μJ/cm2. (a) TA surface plot. Vertical dashed lines indicate selected kinetics traces plotted in panel (d). (b) Selected TA spectra. (c) Kinetic scheme used for GTA. (d) Normalized kinetics traces and their fits based on the kinetic model from panel (c) taken at probe wavelengths of 480 and 574 nm (black and red, respectively). Data are shown as circles, while fits are shown as solid lines. To better visualize the initial kinetics, the inset of panel (d) is plotted on a logarithmic time axis for kinetics traces taken at probe wavelengths of 574 and 600 nm (red and green, respectively).
To extract the lifetime of the trimer structure(s) and investigate the possibility of multiple aggregate subpopulations, we subsequently performed GTA. The four-component kinetic scheme used to model the data is shown in Figure 7c. Additional mathematical and physical justification is provided in Section S9 of Supporting Information. We assign the k1 and k2 rate constants with values of 1/2.0 and 1/26 ps–1, respectively, to a single population of short-lived trimer structures. As we observed above for the dimer structures, k1 is assigned to changes in the electronic structure taking place at the excited state, and k2 is assigned to excited-state decay to the ground state. We assign the k3 rate constant, which had a value of 1/82 ps–1, to the parallel excited-state decay of a second long-lived trimer structure. As can be seen in the selected TA spectra displayed in Figure 7b, the GSB of the long-lived trimer structure is redshifted compared with that of the short-lived trimer structure. Figure 7d displays selected kinetics traces and associated fits taken at probe wavelengths of 574 and 600 nm. As can be seen more clearly in the inset, the traces at 574 and 600 nm have greater contributions from the short- and long-lived trimer structures, respectively. Thus, Figure 7d provides evidence for different relaxation rates of two trimer subpopulations, which decay with lifetimes of 26 and 82 ps.
To further characterize the heterogeneity in the trimer solution, we performed pump-wavelength-dependent TA. Two TA surfaces for the trimer solution excited at 560 and 660 nm are shown in Section S10. The two TA surfaces show very little resemblance as is evident by the presence of drastically different GSB features. These results suggest that the trimer solution exhibits a considerable degree of heterogeneity. A Gaussian fitting analysis of the steady-state absorption spectrum of the trimer solution further supports this interpretation (Section S11). The analysis indicates that the absorption band at 589 nm is well described by two Gaussian line shapes centered at 578 and 603 nm. The largest amplitude Gaussian line shape centered at 578 nm can be explained by a predominant population of trimer structures—the most prominent GSB feature of the trimer structures appears in the vicinity of ca. 575 nm (Figure S9.5), which fits the expectation that the most prominent blueshifted steady-state absorption band of the trimer structure appears intermediate between that of the analogous dimer and tetramer structures with bands at ca. 603 and 563 nm, respectively (see, e.g., Figure 1d). The Gaussian line shape centered at 603 nm could arise from a subpopulation of transverse dimer structures, which, as was just noted, absorbs most strongly in this spectral region. Additionally, the absorption band of the trimer solution that peaks at 650 nm is well described by two Gaussian line shapes centered at 647 and 671 nm. The Gaussian line shape centered at 647 nm coincides well with the monomer absorption band at ca. 650 nm. The Gaussian line shape centered at 671 nm, on the other hand, coincides well with the most prominent GSB feature of the adjacent dimer solution that peaks at 669 nm (Figure 5e,f), which we take to be approximately representative of the most intense steady-state absorption band of the adjacent dimer structures. To further explore the heterogeneity of the trimer solution, we modeled the 100 ps TA spectra of the solution excited at 660 nm, which appears to excite most subpopulations, with the same basis spectra as used above for the transverse and adjacent dimer solutions (Section S12). The model fits the data well and indicates that, in addition to two trimer structures evidenced by GTA, the trimer solution likely contains additional subpopulations of monomers, adjacent dimers, and transverse dimer-like structures, which is further consistent with Gaussian fitting analysis.
Tetramer
Lastly, we performed TA measurements on the tetramer solution. We first determined that the solution is mostly homogeneous except for a small subpopulation of monomers (see Section S13). As such, exciting the solution at 560 nm, which corresponds to the most intense absorption feature in the steady-state absorption spectrum, selectively excites the tetramer structure.
The TA surfaces and spectra for the type 2 tetramer structure (Figure 8a,b) are very similar to those collected for the type 1 tetramer structure selectively excited in the high-salt type 1 solution (Figure 3a,b). Specifically, both TA spectra exhibit an intense, narrow GSB feature peaking at ca. 565 nm, which coincides with the most intense band in the steady-state absorption spectrum. The GSB feature also straddles two less intense ESA features peaking at ca. 572 and 614 nm. To quantify the lifetime of the type 2 tetramer structure, we performed GTA according to a kinetic two-component scheme (see, e.g., Figure 8c and Section S14). As we saw for the other structures, we assign k1 to a change in the electronic structure taking place at the excited state, and k2 is assigned to the excited-state decay of the type 2 tetramer structure. Figure 8d displays kinetics traces and fits at probe wavelengths of 565 and 614 nm. The synchronous decay of these traces to baseline is consistent with the decay of a single population. The semilogarithmic plot in the inset of Figure 8d highlights the early-time component associated with changes in the electronic structure (i.e., k1). Based on analysis, we derived k1 and k2 rate constants with values of 1/1.3 and 1/40 ps–1, respectively.
Figure 8.
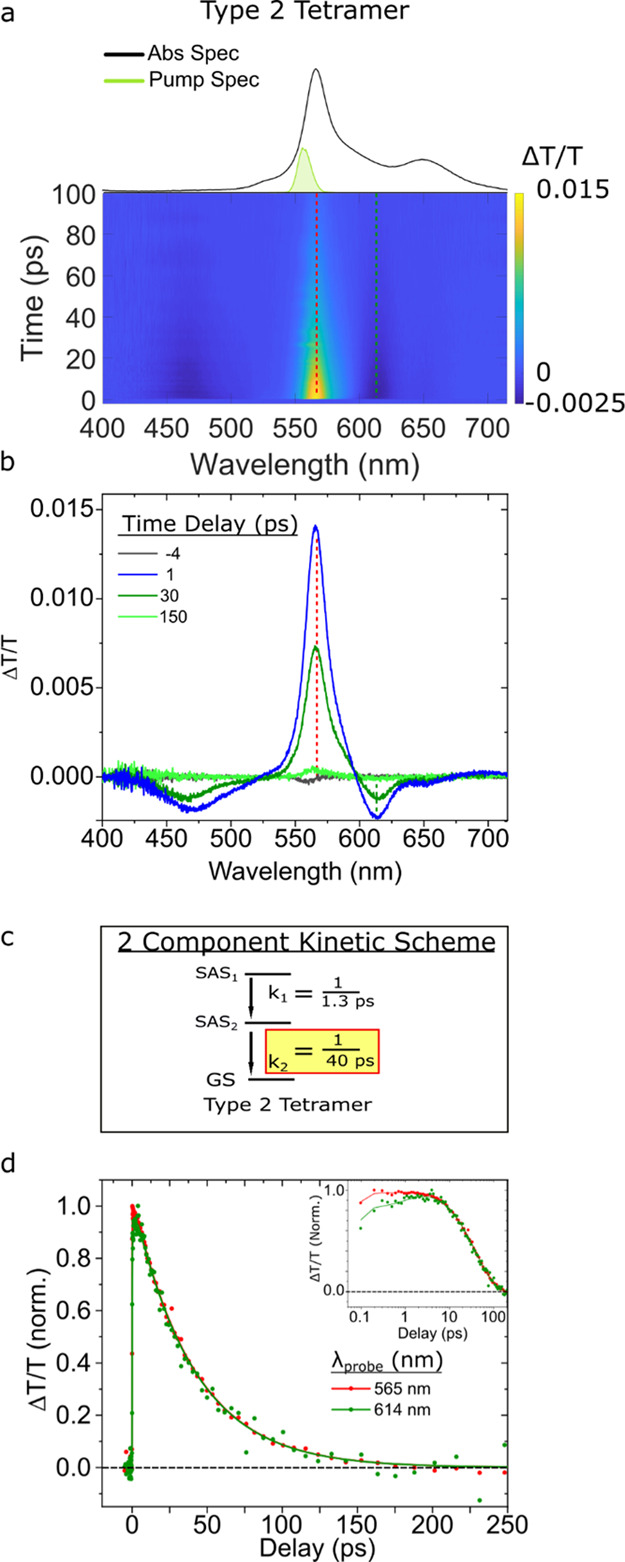
Femtosecond TA spectra of the type 2 tetramer solution pumped at 560 nm with a pump fluence of 14 μJ/cm2. (a) TA surface plots. Vertical dashed lines indicate selected kinetics traces plotted in panel (d). (b) Selected TA spectra. (c) Kinetic scheme used for GTA. (d) Normalized selected kinetics traces and their fits based on the kinetic models from panel (c) taken at probe wavelengths of 565 and 614 nm (red and green, respectively). Data are shown as circles, while fits are shown as solid lines. To better visualize the initial TA response of the solution, the inset of panel (d) is plotted on a logarithmic time axis for the kinetics traces taken at probe wavelengths of 565 and 614 nm.
Overview
In summary, we find that the excited-state lifetimes of all Cy5 aggregate configurations are significantly shortened compared with the monomer (Table 1).
Table 1. Excited-State Lifetimes of Type 1 and Type 2 DNA-Cy5 Constructs.
| type | construct | lifetime (ps) |
|---|---|---|
| N/A | monomer | 1300 |
| 1 | duplex dimer | 10 |
| 1 | tetramer | 30 |
| 2 | transverse dimer | 202 |
| 2 | short-lived adjacent dimer | 40 |
| 2 | long-lived adjacent dimer | 242 |
| 2 | short-lived trimer | 26 |
| 2 | long-lived trimer | 82 |
| 2 | tetramer | 40 |
The type 1 aggregate lifetimes are some of the shortest as the duplex dimer and type 1 tetramer exhibit lifetimes of ca. 10 and 30 ps, respectively. Conversely, the lifetimes of the type 2 aggregates exhibit a wider range of variability. Consistent with the type 1 aggregates, we found the primary adjacent dimer population, the two trimers, and the type 2 tetramer to exhibit lifetimes ranging from ca. 30 to 80 ps. In contrast, we found the transverse dimer and second adjacent dimer to exhibit considerably longer lifetimes of ca. 200 and 240 ps, respectively. In addition to measuring the lifetimes, we discovered evidence of heterogeneity in all of the aggregate solutions with the duplex dimer, transverse dimer, and type 2 tetramer solutions exhibiting relatively little heterogeneity and the adjacent dimer and trimer and solutions exhibiting more considerable heterogeneity. In the discussion section, we consider various mechanisms that may explain the origin of the enhanced nonradiative decay in the aggregates and the appreciable variability in their lifetimes.
Discussion
A primary goal of this study, and the focus of the first subsection, is to evaluate the relative impact of several structural factors—such as packing type, coupling strength, exciton delocalization, and dye separation—on excited-state lifetimes in DNA-templated Cy5 aggregates. In the second subsection, we proceed to discuss another factor contributing to the variability of excited-state lifetimes—heterogeneity—and the potential role of the DNA template.
Structural Factors Impacting Excited-State Lifetime
We first consider the possible relationship between packing type and excited-state lifetime. According to Kasha’s molecular exciton theory, dyes that pack as H-aggregates (J-aggregates) are expected to exhibit decreased (increased) radiative rates due to the out-of-phase (in-phase) interactions of their TDMs.57 As a result, assuming no change in the nonradiative decay rate, the H-aggregates (J-aggregates) should exhibit longer (shorter) lifetimes compared with the dye monomer. Instead, for the broad set of H- and J-aggregates examined in the present work (Figure 1), we find no such correlation between packing type and lifetime (Table 1). Specifically, for the transverse dimer, trimer, and tetramer structures, which adopted an H-aggregate packing type, we observe drastically reduced, rather than increased, lifetimes compared with the monomer. In the case of the duplex J-dimer and adjacent dimer structures, we also observe drastically reduced lifetimes compared with the monomer; lifetimes reduced to an extent inconsistent with enhanced radiative decay (i.e., superradiance) expected for a dimer aggregate.25 These results indicate that enhanced nonradiative decay is operative in both H- and J-aggregates. Thus, we conclude that there is no correlation between packing type and lifetime and that nonradiative decay is the primary decay pathway in the DNA-templated dye aggregates.
We next discuss the impact of coupling strength on excited-state lifetime. In a classic study, Sundström and Gillbro observed dramatically reduced lifetimes in aggregates of diethylthiadicarbocyanine iodide (DTDCI), a Cy5-like pentamethine dye.23 These authors attributed the reduced lifetimes observed in the aggregates, including a covalently bound dimer, to accelerated nonradiative decay. They proposed that accelerated nonradiative decay may be due to more strongly coupled dyes, based on predictions using a Fermi’s golden rule expression that related the nonradiative decay rate to coupling strength. In the case of our aggregates, specifically the duplex dimer, transverse dimer, and type 2 tetramer, we can determine the coupling strength, which we refer to as the excitonic hopping parameter (J). By modeling the optical properties of the different aggregate structures, which is described in greater detail below and in Section S15, we derived J to be 48 and 73 meV for the duplex and transverse dimer, respectively. For the type 2 tetramer, we derived a maximum value of 136 meV (i.e., Jmax), which represents J for the most strongly coupled dye pair, and an average value of 68 meV (i.e., Javg), which was taken as the average of J over all dye pairs. Table 2 lists these values along with their respective lifetimes.
Table 2. Excitonic Hopping Parameter and Excited-State Lifetimes of Selected Type 1 and Type 2 DNA-Dye Constructs.
| DNA-dye construct | Jmax (meV) | Javg (meV) | lifetime (ps) |
|---|---|---|---|
| duplex dimer | 48 | 11 | |
| transverse dimer | 73 | 200 | |
| type 2 tetramer | 136 | 68 | 40 |
In contrast to the expectation that stronger electronic coupling leads to accelerated nonradiative decay and reduced lifetimes, we find that the measured lifetimes are larger for those Cy5 aggregates with larger J. For example, the duplex dimer with the smallest J value exhibits the shortest lifetime. In contrast, the transverse dimer and type 2 tetramer with larger J values exhibit larger lifetimes. From this comparison, we conclude that increasing values of the excitonic hopping parameter do not result in enhanced nonradiative decay and shorter lifetimes.
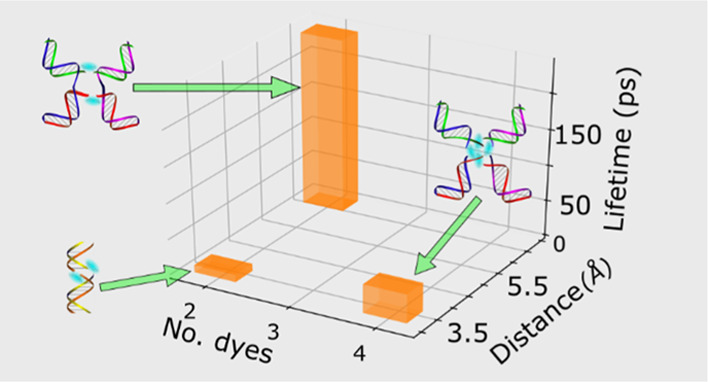
The extent of exciton delocalization (i.e., the number of dyes over which an excitation is collectively shared) is another factor that may impact aggregate lifetime. Theoretical work has indicated, for example, that nonradiative decay rates can decrease considerably with increasing extent of exciton delocalization.61,62 A central element of such a model is that larger aggregates are expected to exhibit smaller nuclear displacements upon photoexcitation, which results in reduced nonradiative decay rates. There is experimental precedent for this theoretical prediction. As noted above, Sundström and Gillbro observed drastically reduced lifetimes for aggregates of DTDCI, whose monomer lifetime is 1.5 ns. Specifically, these authors measured dimer and trimer aggregate lifetimes of 19 and 50 ps, respectively, which is consistent with the interpretation that an increasing extent of exciton delocalization may suppress nonradiative decay rates. This correlation is not just specific to DTDCI. Das and Kamat, for example, found that dimer and trimer aggregates of the dye thionine exhibited lifetimes of 40 and 60 ps, respectively, as compared with a lifetime of 420 ps for the monomer.63 Consistent with these observations, we find that the 40 ps lifetime of the tetramer is nearly four times as large as the lifetime of the shortest-lived dimer, the duplex dimer, which had a lifetime of 10 ps (Table 1). Additionally, we find that the shortest-lived trimer, with a lifetime of 26 ps, is intermediate between that of the duplex dimer and the tetramer. We chose to focus on the shortest-lived aggregates for this comparison because, as we propose below, we believe that an additional factor, dye separation, plays a primary role in influencing aggregate excited-state lifetime.
Finally, we consider the impact of dye separation on excited-state lifetime. This potential relationship is motivated by Liang et al.’s pioneering work examining lifetimes and fluorescence emission in dimers of squaraine dyes, a family of dyes structurally very similar to cyanines,64 bridged by a varying number of methylene groups. These authors observed the shortest lifetimes and least fluorescence emission for the most closely spaced dimers and found that lifetime and fluorescence emission increased progressively as the distance between the dyes became greater. In the case of our DNA-templated Cy5 aggregates, insights into dye separation can be gleaned by simulating the optical properties of the materials, namely, the absorbance and circular dichroism spectra, via an approach based on Kühn–Renger–May (KRM) theory.47,49,58Figure 9 displays the orientation of the TDMs that result from modeling the optical properties of the duplex dimer, the transverse dimer, and type 2 tetramer, highlighting the separation between TDMs in these aggregates.
Figure 9.
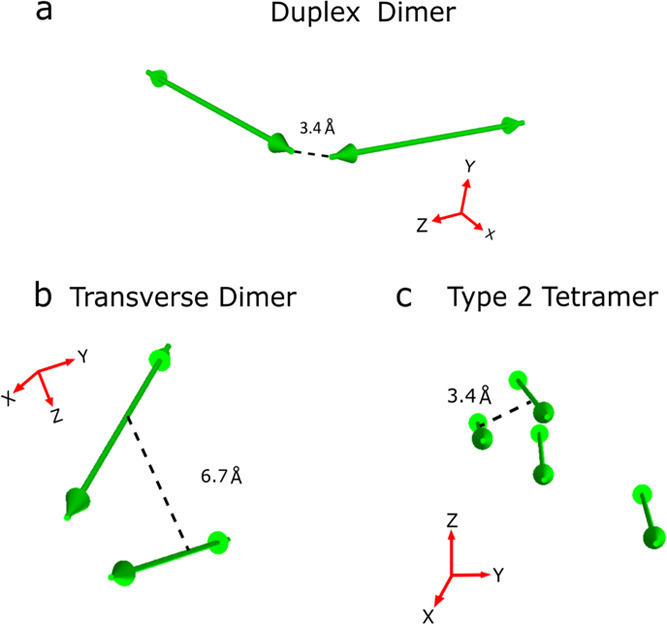
Orientation of TDM vectors for (a) duplex dimer, (b) transverse dimer, and (c) type 2 tetramer derived by modeling the optical properties of the materials via an approach based on KRM theory. The TDM vector orientations were derived in Section S15. Green double-headed arrows correspond to TDM vectors, which are taken to lie along the long axis of the Cy5 dye. Dashed lines indicate the critical approach distance derived by modeling; see the main text for further details. The critical approach distance for the duplex dimer (i.e., J-aggregate) was taken to be the end-to-end distance, while the critical approach distance for the transverse dimer and type 2 tetramer (i.e., H-aggregates) was taken to be the center-to-center distance.
Because of their fundamentally different packing configurations (see, e.g., Figures 1 and 9), a direct comparison of the inter-dye separations of the J-like duplex dimer structure and the H-like transverse dimer and type 2 tetramer structures is not possible. To facilitate comparison, we thus define the end-to-end and face-to-face dye separation observed in the J-like and H-like aggregate structures, respectively, as critical approach distances (Figure 9). Viewed in this manner, the modeling derives a dye separation distance of 3.4 Å for the duplex dimer, while for the transverse dimer and type 2 tetramer, dye separation distances of 6.7 and 3.4 Å, respectively, are derived. For the structures with the shortest dye separation, that is, the duplex dimer and type 2 tetramer, we measure the shortest lifetimes of 10 and 40 ps, respectively. For the transverse dimer, in contrast, where the distance is appreciably larger, we observe a much longer lifetime of 202 ps. Thus, consistent with the work of Liang et al.,64 we observe a correlation between lifetime and dye separation. The reason for this correlation may be that at small separation distances, the dyes exhibit appreciable wavefunction overlap. Furthermore, time-dependent changes in this wavefunction overlap may promote strong nonadiabatic coupling between the ground and excited states and thus facilitate nonradiative decay.14,25 We posit such wavefunction overlap may occur between the indolenine end groups and the bridging pentamethine chains for the duplex dimer and tetramer, respectively.
Heterogeneity and Variability of Lifetimes
Another factor contributing to the observed variability of excited-state lifetimes in the DNA-Cy5 aggregates may be heterogeneity. Heterogeneity can manifest itself as multiple DNA structures present in the solution, even though the number and type of basis DNA strands are identical. This can manifest in the form of static heterogeneity, where the DNA strands assemble and relax into multiple distinct DNA structures (or conformers). Additionally, dynamic heterogeneity may exist, wherein each structure fluctuates locally at the base pair level on a hundreds of μs timescale. This latter process is known colloquially as DNA “breathing”;65 it enables DNA replication and repair and hence life on earth.
In a prototypical example of static heterogeneity, Cannon et al. reported “type 1” solutions of DNA-templated Cy5 aggregates (Figure 1a,b) that consist of a mixture of duplex DNA and DNA Holliday junctions.47 Specifically, they showed that DNA-templated Cy5 aggregates, constructed of DNA strands capable of forming both duplex DNA and DNA Holliday junctions (see, e.g., Construct Preparation and Steady-State Optical Characterization in Results), can indeed consist of a mixture of duplex DNA and DNA Holliday junctions by varying the amount of MgCl2 present in the solution (Figure 10). The authors also showed that the interconversion between these structures is a slow process taking place on the order of minutes, which is consistent with the macroscopic structural changes necessary for this interconversion. The static heterogeneity present in the solution is evidenced in the TA results presented in Figures 3 and 4—the TA of the high-salt type 1 solution is well described by a model consisting of a mixture of duplex dimer and Holliday junction tetramer subpopulations. In addition, Figures 3 and 4 show that two more components are required to model the data—a component associated with a small subpopulation of monomers, plus another component with optical properties and dynamics consistent with the long-lived adjacent dimer subpopulation (see, e.g., Figure 5e–h and Section S6). We propose here that dynamic heterogeneity, or DNA “breathing”, may be the origin of these small subpopulations. Thus, leveraging the model originally developed by Cannon et al. that describes the solution as a mixture of duplex DNA and DNA Holliday junctions,47 we propose to expand the model to include two additional structures to explain the presence of these two additional subpopulations (see, e.g., Figure 10). Specifically, we propose that restricted and extended DNA “bubbles”, or local pockets in the secondary structure that arise from DNA breathing, give rise to a small subpopulation of long-lived adjacent dimers and monomers, respectively. The notion that DNA breathing might mediate interconversion between the short-lived duplex dimer and more longer-lived adjacent dimer and monomer configurations is wholly consistent with the correlation between dye separation and aggregate lifetime identified above. Additionally, in recent work by Marcus, von Hippel, and co-workers in which they performed single molecule studies of Cy3–Cy5 pairs attached to single-strand-duplex DNA junctions, they demonstrated the tendency for single-stranded DNA segments to adopt configurations that could be grouped into three sets, or macrostates, which they described as compact, partially extended, and fully extended.66 The restricted and extended bubble configurations we propose to explain the additional aggregate and monomer subpopulations observed here may be consistent with this interpretation of discrete, thermally accessible, transient dye–dye configurations.
Figure 10.

Proposed model of the high-salt type 1 solution, which consists of a mixture of duplex DNA and DNA Holliday junction structures. The part of the model shown in the orange box is a classic example of static heterogeneity. The interconversion between duplex DNA and DNA Holliday junctions is a macroscopic structural change that takes place on the timescale of minutes, thus establishing a steady-state mixture of components. We propose to expand this depiction of the high-salt type 1 solution with the addition of the structures shown in the gray box, which account for dynamic heterogeneity. Dynamic heterogeneity is present in these solutions via DNA “breathing”, a local structural change that takes place on a hundreds of μs timescale. These structural changes are proposed to explain the presence of small subpopulations of long-lived adjacent dimers and monomers in the solution, as exemplified by the structures labeled “restricted bubble” and “extended bubble”, respectively.
As another example of static heterogeneity, Mass et al. proposed a model for the “type 2” solutions of DNA-templated dye aggregates (Figure 1c,d) whereby the Holliday junction nanostructures can relax into multiple distinct configurations.58,67,68 Such configurations include an open configuration and two distinct isomers of a stacked configuration. Critically, these distinct DNA configurations impact the dye aggregate configurations that are accessed. For example, Figure 11 shows a range of possible DNA and dye aggregation configurations possible for the adjacent dimer, transverse dimer, trimer, and tetramer solutions.
Figure 11.
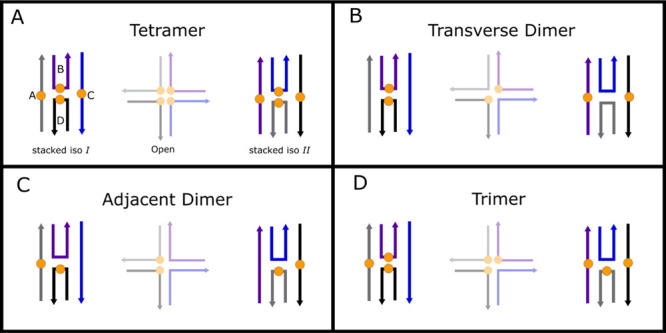
Proposed model for the composition of the type 2 solutions, which includes the (a) tetramer, (b) transverse dimer, (c) adjacent dimer, and (d) trimer solutions. The schematic shows three possible configurations of the DNA template, which impact the different dye aggregate configurations that are possible. Due to the particular solution conditions, the predominant DNA configurations are the two stacked iso I and II configurations. A third, open configuration, which is present in a low amount in these solutions, is shown in reduced opacity.
To determine whether the open or stacked DNA configurations were predominant, we first performed denaturation measurements. The results, as shown in Table 3 and Section S16, indicate that the stacked configurations are the predominant configurations in the adjacent dimer, transverse dimer, trimer, and tetramer solutions. All of these solutions, which were prepared with 15 mM MgCl2 added, are found to exhibit a denaturation temperature on the order of ∼60 °C, which is similar to that measured for a solution of unlabeled Holliday junctions with 15 mM MgCl2 added. In contrast, a much smaller denaturation temperature of ∼48 °C is measured for the unlabeled Holliday junctions prepared in a solution with 100 mM NaCl added. Given that the unlabeled Holliday junctions with 15 mM MgCl2 and 100 mM NaCl added are known to primarily adopt stacked and open configurations, respectively, we therefore conclude that the two stacked configurations, iso I and II, are the predominant DNA configurations in the “type 2” DNA-templated Cy5 aggregate solutions.
Table 3. Denaturation Temperatures of Type 2 DNA-Dye Constructs.
| DNA-dye construct | Tm (°C) |
|---|---|
| unlabeled (100 mM NaCl added) | 48.5 |
| unlabeled (15 mM MgCl2 added) | 60.5 |
| monomer | 59.1 |
| adjacent dimer | 59.0 |
| transverse dimer | 60.9 |
| trimer | 60.5 |
| tetramer | 62.4 |
Next, we consider the plausibility of the model proposed in Figure 11, which indicates that two stacked DNA configurations are possible. Focusing on the tetramer and transverse dimer structures, the model predicts that these structures may adopt either one or two distinct dye aggregate configurations, respectively. In the case of the tetramer solution, for example, iso I and II are essentially equivalent in the model (Figure 11a); that is, the four dyes are positioned close to one another in a similar manner, such that we might expect the same aggregate configuration for the iso I and II DNA configurations. In the case of the transverse dimer solution, two distinct aggregate configurations appear in the model (Figure 11b). Namely, one configuration, iso I, where the dyes are closely spaced, and a second configuration, iso II, where the dyes are much farther apart. The dyes are spaced so far apart, in fact, that we might expect them to not interact and to behave independently (i.e., as uncoupled monomers). Indeed, this interpretation is consistent with the disproportionately large subpopulation of monomers observed in the transverse dimer solution (Figure 6). Critically, the model predicts little aggregate heterogeneity for both the tetramer and transverse dimer structures. Consistent with this expectation, pump-wavelength-dependent TA measurements of the tetramer and transverse dimer solutions (see, e.g., Figures 5 and 6 and Section S13) indicated that these solutions exhibit little aggregate heterogeneity. Additional insights may be gleaned from Table 3. For example, we find that the denaturation temperatures of the tetramer and transverse dimer solutions are greater than that of the unlabeled Holliday junctions, which indicates that dyes in these structures may act to stabilize the overall DNA-dye nanostructure, presumably via dye–dye interactions. Thus, we conclude that the tetramer and transverse dimer solutions exhibit little aggregate heterogeneity, a result consistent with the model, and that stabilizing dye–dye interactions may contribute to further suppression of aggregate heterogeneity.
Now, we turn our attention to the adjacent dimer solution. For the adjacent dimer solution, the model predicts that, compared with the transverse dimer, the dyes are further separated and that, unlike for the tetramer, the dyes can be arranged in one of the two ways to form an aggregate (Figure 11c). In the case of the adjacent dimer solution, we observed a denaturation temperature that was less than that of the unlabeled Holliday junction (Table 3). The lower denaturation temperature of the adjacent dimer solution may be explained by weaker dye–dye interactions, that is, an overall destabilizing effect on the DNA-dye construct, which may, in part, be related to the larger separation of the dyes predicted by the model. The adjacent dimer solution also exhibited strong TA excitation wavelength dependence (Figure 6), suggesting appreciable heterogeneity. Although we found that the iso I and II configurations did not strongly impact aggregate heterogeneity for the tetramer and transverse dimers, apparently aggregate heterogeneity may occur in the case of the adjacent dimers due to the more distant spacing between dyes combined with differences in the flanking base pairs in the iso I and II configurations (Section S17). For example, Cunningham et al. showed that variations in the flanking base pairs can have a profound impact on aggregate packing type.24 Specifically, these authors showed that Cy3 dimers templated using duplex DNA exhibited H- and J-aggregate packing types when flanked with AT and GC base pairs, respectively. Consistent with this picture, a simple analysis of the TA of the DNA-templated Cy5 adjacent dimer aggregates indicated that the solution consisted of not only adjacent dimers, which tend to form J-aggregates with redshifted absorption spectra, but also a large proportion of transverse dimers, which tend to form H-aggregates with blueshifted absorption spectra (Figure 6). In further support of this interpretation, the absorption spectrum of the adjacent dimer solution (Figure 1d) exhibits obvious signatures of redshifted features attributable to adjacent dimers (i.e., J-aggregates) along with a strong absorption band at 600 nm that is consistent with the presence of an appreciable fraction of transverse dimers (i.e., H-aggregates). Lastly, it is critical to mention that the location of dyes both on the center and on the periphery or only on the center of the Holliday junction in the case of the adjacent and transverse dimer structures, respectively, may be another factor contributing to the differing degrees of heterogeneity observed in these solutions (Section S17). We conclude that the model may also explain the lower denaturation temperature and considerable heterogeneity observed for the adjacent dimer solution.
Regarding the trimer solution, the model suggests an even more complicated picture (Figure 11d). First, it is important to note that evidence indeed exists for the presence of “true” trimer configurations where the exciton is delocalized about all three dyes; for example, via the prominent H-aggregate absorption band that exhibits a blueshift intermediate between that of the transverse dimer and tetramer (Figure 1d). Furthermore, exciting the short-wavelength part of the blueshifted H-aggregate absorption band indeed appears to selectively excite “true” trimer configurations, which is evident by the mapping of the absorption spectrum onto the TA results in a similar manner to that observed for the tetramer (see, e.g., Figures 7 and 8). However, the model also predicts two trimer configurations that are not symmetric. On the one hand, the model predicts that the iso I configuration of the trimer could, in some sense, be considered a combination of iso I of the adjacent and transverse dimers. Viewed in this manner, we might expect to observe a subpopulation of these dimers in the trimer solution. Indeed, both the additional structures in the prominent H-aggregate absorption band at long wavelengths (Figure 1d and Section S11) along with the prominent GSB feature appearing at 600 nm in the femtosecond TA spectra for the trimer solution excited at 660 nm (Section S10) are readily explained by invoking the presence of an additional subpopulation of transverse dimers. Similar to the iso I configuration, iso II of the trimer can be viewed as a combination of the iso I and II of the adjacent dimer. In the paragraph above, we argued that the distant spacing of the dyes in these configurations may explain the lower denaturation temperature and more extensive heterogeneity observed in that solution. In the case of the trimer solution, the addition of a third dye may further act to destabilize the DNA-dye nanostructure. However, Table 3 indicates that the denaturation temperature of the trimer solution is very similar to that of the unlabeled Holliday junction. To explain this seeming anomaly, it is possible that transverse dimers present in the solution, via iso I, may act to stabilize the structure and thereby increase the denaturation temperature of the solution to approach that of the unlabeled Holliday junction. Coincidentally, this interpretation may also explain the prominence of spectral features attributable to transverse dimers in the trimer solution (Sections S10 and S12); that is, the increased stabilization that the transverse dimer imparts may also increase the fraction of iso I configurations in the solution.
Finally, an additional source of heterogeneity may arise from the “semi-mobility” of the type 2 DNA templates. Whereas, “mobile” Holliday junctions can undergo full branch migration (Figure 10, left panel) due to their fully symmetric nucleotide sequences (i.e., the two basis sequences are the reverse complement of one another), semi-mobile Holliday junctions are only able to undergo limited branch migration, the extent of which is determined by the length of the symmetric (i.e., reverse complementary) domains on the basis sequences at the center of the junction.69 For the type 2 DNA template, the bases adjacent to the dye sites allow for branch migration of a single base which permits the Holliday junction to alternately shorten and lengthen the vertical and horizontal arms, respectively, by one base. This process is illustrated for the type 2 sequences in Section S18. For the transverse dimer, the horizontal displacement would increase the dye separation and possibly promote a subpopulation of monomers. The effect would be similar for the trimer and tetramer but would instead promote smaller aggregates rather than monomers. Conversely, branch migration for the adjacent dimer would cause the dyes to move together as a unit and remain dimerized, but the changes in the DNA backbone may change the dye packing and lead to a distinct dimer subpopulation. Thus, both the stacked isomer and branch migration models may potentially contribute to the observed heterogeneity of the type 2 DNA templates.
In summary, the extensive heterogeneity observed in the trimer solution may well be explained by this model of static heterogeneity, which, as described above, suggests that many distinct dye aggregate configurations are possible. Clearly, the choice of the number of dyes and their relative position on the DNA template impacts the extent of heterogeneity, which, in the absence of mitigation strategies, may impact studies and applications that require homogeneous assemblies of materials.
Conclusions
In conclusion, we observed drastically reduced excited-state lifetimes across a broad set of DNA-templated Cy5 aggregates. For the type 1 aggregates, we measured lifetimes of ∼10 and ∼30 ps for the duplex dimer and type 1 tetramer, respectively, consistent with prior work.25 The type 2 aggregates also exhibited reduced lifetimes but with a broader range of timescales. Specifically, we measured lifetimes of ∼202 and ∼40 ps for the transverse dimer and tetramer, respectively, and lifetimes ranging from ∼40–242 and ∼26–82 ps for the adjacent dimer and trimer, respectively. The reduced lifetimes result from significantly enhanced nonradiative decay rates concomitant with aggregation. By comparing the orientation of dyes of selected structures with their respective lifetimes, we observed a correlation between dye separation and lifetime whereby close spacing between dyes resulted in the shortest lifetimes. We also observed a weak correlation of lifetime with exciton delocalization. These results suggest that DNA-templated dye aggregates may indeed represent designer materials that can be custom tailored for a broad set of specific applications. For applications where short lifetimes are advantageous, such as in sensing and photodynamic therapy where either large emission contrast or extensive heat dissipation are desired, our results suggest that tuning materials for short dye separation is desirable. For applications where long lifetimes are important, on the other hand, large dye separation may be required. If both short dye separation and long lifetime are important, that is, to achieve aggregates with large coupling strength and long-lived excited states, additional mechanistic insights may be needed to optimize these multiple parameters simultaneously; otherwise, there may be a tradeoff among dye separation, coupling strength, and lifetime.
We also provided significant insights into potential sources of heterogeneity in DNA-templated dye aggregates that may explain the broad set of measured lifetimes. Varying extents of heterogeneity in the different solutions, ranging from limited to extensive, were highlighted by pump-wavelength-dependent TA combined with global analyses via parallel decay models. For example, the duplex dimer and Holliday junction-based transverse dimer and tetramer solutions appeared to exhibit limited heterogeneity, whereas the Holliday junction-based adjacent dimer and trimer solutions appeared to exhibit more extensive heterogeneity. We consistently observed a subset of aggregates with longer lifetimes in most solutions, which we proposed to arise from transient local structural fluctuations of the DNA template via “breathing”. The local structural fluctuations of the DNA may lead to large dye separation and correspondingly longer lifetimes, with a limiting case being two non-interacting dye monomers separated by a large distance. Another potential source of heterogeneity we identified was associated largely with the type 2 aggregates. Specifically, we found that the DNA Holliday junctions used to template the type 2 aggregates adopted a stacked configuration and that the stacked configuration could exist in one of the two structural forms, iso I or II. The transverse dimer and tetramer aggregates had only one type of aggregate structure within this model, which was consistent with the general lack of heterogeneity observed in these solutions along with their higher denaturation temperatures. On the other hand, the adjacent dimer and trimer aggregates had two types of aggregate structures within this model, which was consistent with their more extensive heterogeneity and lower denaturation temperatures. Moving forward, these materials may need to be further modified, for example, by modifying the dye and/or DNA template to promote stronger dye and DNA interactions, respectively, to mitigate these potential sources of heterogeneity.
Acknowledgments
Research at Boise State University, including data collection, analysis, and interpretation and manuscript preparation, was supported by the National Science Foundation (NSF) through the Integrated NSF Support Promoting Interdisciplinary Research and Education (INSPIRE) via award no. 1648655. Specific equipment was supported by additional funding sources. The femtosecond laser amplifier and optical parametric amplifier were supported by the National Science Foundation NSF MRI via award no. 0923541. The femtosecond TA spectrometer was supported by a Department of Energy, Idaho National Laboratory, Laboratory Directed Research and Development project through blanket master contract no. 154754 between Battelle Energy Alliance and Boise State University, Release 15. The circular dichroism spectrometer was made available through the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering through the Established Program to Stimulate Competitive Research (EPSCoR) via award no. DE-SC0020089. The time-correlated single-photon counting spectrometer was supported by the Department of the Navy, Office of Naval Research (ONR) via ONR award no. N00014-19-1-2615.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.1c04517.
White-light probe spectrum, pump pulse duration, details of global target analyses, pump-wavelength dependence analyses, Gaussian analysis of the trimer solution absorption spectrum, four basis spectrum fit of TA of the trimer solution excited at 660 nm, analysis of heterogeneity of type 2 tetramer, details of the KRM modeling, DNA melting curves, Holliday junction stacked isomer differences, “semi-mobility” of the type 2 Holliday junction DNA template, supplemental methods, and supplemental references (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mirkovic T.; Ostroumov E. E.; Anna J. M.; van Grondelle R.; Govindjee; Scholes G. D. Light Absorption and Energy Transfer in the Antenna Complexes of Photosynthetic Organisms. Chem. Rev. 2017, 117, 249–293. 10.1021/acs.chemrev.6b00002. [DOI] [PubMed] [Google Scholar]
- Brixner T.; Hildner R.; Köhler J.; Lambert C.; Würthner F. Exciton Transport in Molecular Aggregates – From Natural Antennas to Synthetic Chromophore Systems. Adv. Energy Mater. 2017, 7, 1700236. 10.1002/aenm.201700236. [DOI] [Google Scholar]
- Ostroverkhova O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. 10.1021/acs.chemrev.6b00127. [DOI] [PubMed] [Google Scholar]
- Zang L.; Che Y.; Moore J. S. One-Dimensional Self-Assembly of Planar π-Conjugated Molecules: Adaptable Building Blocks for Organic Nanodevices. Acc. Chem. Res. 2008, 41, 1596–1608. 10.1021/ar800030w. [DOI] [PubMed] [Google Scholar]
- Arunkumar E.; Ajayaghosh A.; Daub J. Selective Calcium Ion Sensing with a Bichromophoric Squaraine Foldamer. J. Am. Chem. Soc. 2005, 127, 3156–3164. 10.1021/ja045760e. [DOI] [PubMed] [Google Scholar]
- Okamoto A. ECHO Probes: A Concept of Fluorescence Control for Practical Nucleic Acid Sensing. Chem. Soc. Rev. 2011, 40, 5815–5828. 10.1039/c1cs15025a. [DOI] [PubMed] [Google Scholar]
- Yurke B.; Kuang W. Passive Linear Nanoscale Optical and Molecular Electronics Device Synthesis from Nanoparticles. Phys. Rev. A 2010, 81, 033814. 10.1103/physreva.81.033814. [DOI] [Google Scholar]
- Graugnard E.; Kellis D. L.; Bui H.; Barnes S.; Kuang W.; Lee J.; Hughes W. L.; Knowlton W. B.; Yurke B. DNA-Controlled Excitonic Switches. Nano Lett. 2012, 12, 2117–2122. 10.1021/nl3004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laboda C.; Duschl H.; Dwyer C. L. DNA-Enabled Integrated Molecular Systems for Computation and Sensing. Acc. Chem. Res. 2014, 47, 1816–1824. 10.1021/ar500054u. [DOI] [PubMed] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Davis P. H.; Lee J.; Kuang W.; Hughes W. L.; Graugnard E.; Yurke B.; Knowlton W. B. Excitonic AND Logic Gates on DNA Brick Nanobreadboards. ACS Photonics 2015, 2, 398–404. 10.1021/ph500444d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya N. P. D.; Rappoport D.; Tabor D. P.; Aspuru-Guzik A. Excitonics: A Set of Gates for Molecular Exciton Processing and Signaling. ACS Nano 2018, 12, 6410–6420. 10.1021/acsnano.8b00584. [DOI] [PubMed] [Google Scholar]
- Kellis D. L.; Sarter C.; Cannon B. L.; Davis P. H.; Graugnard E.; Lee J.; Pensack R. D.; Kolmar T.; Jäschke A.; Yurke B.; et al. An All-Optical Excitonic Switch Operated in the Liquid and Solid Phases. ACS Nano 2019, 13, 2986–2994. 10.1021/acsnano.8b07504. [DOI] [PubMed] [Google Scholar]
- Hu W.; Miao X.; Tao H.; Baev A.; Ren C.; Fan Q.; He T.; Huang W.; Prasad P. N. Manipulating Nonradiative Decay Channel by Intermolecular Charge Transfer for Exceptionally Improved Photothermal Conversion. ACS Nano 2019, 13, 12006–12014. 10.1021/acsnano.9b06208. [DOI] [PubMed] [Google Scholar]
- Dean J. C.; Oblinsky D. G.; Rafiq S.; Scholes G. D. Methylene Blue Exciton States Steer Nonradiative Relaxation: Ultrafast Spectroscopy of Methylene Blue Dimer. J. Phys. Chem. B 2016, 120, 440–454. 10.1021/acs.jpcb.5b11847. [DOI] [PubMed] [Google Scholar]
- Scheibe G. Über die Veränderlichkeit der Absorptionsspektren in Lösungen und die Nebenvalenzen als ihre Ursache. ÜberAngew. Chemie 1937, 50, 212–219. 10.1002/ange.19370501103. [DOI] [Google Scholar]
- Jelley E. E. Molecular, Nematic and Crystal States of I: I-Diethyl--Cyanine Chloride. Nature 1937, 139, 631. 10.1038/139631b0. [DOI] [Google Scholar]
- Davydov A. S. Theory of Absorption Spectra of Molecular Crystals. Zh. Eksp. Teor. Fiz. 1948, 18, 210–218. [Google Scholar]
- Davydov A. S. The Theory of Molecular Excitons. Phys.-Usp. 1964, 7, 145–178. 10.1070/pu1964v007n02abeh003659. [DOI] [Google Scholar]
- Kühn O.; Renger T.; May V. Theory of Exciton-Vibrational Dynamics in Molecular Dimers. Chem. Phys. 1996, 204, 99–114. 10.1016/0301-0104(95)00448-3. [DOI] [Google Scholar]
- Hestand N. J.; Spano F. C. Molecular Aggregate Photophysics beyond the Kasha Model: Novel Design Principles for Organic Materials. Acc. Chem. Res. 2017, 50, 341–350. 10.1021/acs.accounts.6b00576. [DOI] [PubMed] [Google Scholar]
- Hestand N. J.; Spano F. C. Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer. Chem. Rev. 2018, 118, 7069–7163. 10.1021/acs.chemrev.7b00581. [DOI] [PubMed] [Google Scholar]
- Turro N. J.; Scaiano J. C.; Ramamurthy V.. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- Sundström V.; Gillbro T. Excited State Dynamics and Photophysics of Aggregated Dye Chromophores in Solution. J. Chem. Phys. 1985, 83, 2733–2743. 10.1063/1.449275. [DOI] [Google Scholar]
- Cunningham P. D.; Kim Y. C.; Díaz S. A.; Buckhout-White S.; Mathur D.; Medintz I. L.; Melinger J. S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. 10.1021/acs.jpcb.8b02134. [DOI] [PubMed] [Google Scholar]
- Huff J. S.; Davis P. H.; Christy A.; Kellis D. L.; Kandadai N.; Toa Z. S. D.; Scholes G. D.; Yurke B.; Knowlton W. B.; Pensack R. D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett. 2019, 10, 2386–2392. 10.1021/acs.jpclett.9b00404. [DOI] [PubMed] [Google Scholar]
- Heyne B. Self-Assembly of Organic Dyes in Supramolecular Aggregates. Photochem. Photobiol. Sci. 2016, 15, 1103–1114. 10.1039/c6pp00221h. [DOI] [PubMed] [Google Scholar]
- Haedler A. T.; Kreger K.; Issac A.; Wittmann B.; Kivala M.; Hammer N.; Köhler J.; Schmidt H.-W.; Hildner R. Long-Range Energy Transport in Single Supramolecular Nanofibres at Room Temperature. Nature 2015, 523, 196–199. 10.1038/nature14570. [DOI] [PubMed] [Google Scholar]
- Scholes G. D.; Fleming G. R.; Olaya-Castro A.; van Grondelle R. Lessons from Nature about Solar Light Harvesting. Nat. Chem. 2011, 3, 763–774. 10.1038/nchem.1145. [DOI] [PubMed] [Google Scholar]
- Snow C. D.; Nguyen H.; Pande V. S.; Gruebele M. Absolute Comparison of Simulated and Experimental Protein-Folding Dynamics. Nature 2002, 420, 102–106. 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- Baker D.; Sali A. Protein Structure Prediction and Structural Genomics. Science 2001, 294, 93–96. 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Dill K. A.; MacCallum J. L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- Yeates T. O. Geometric Principles for Designing Highly Symmetric Self-Assembling Protein Nanomaterials. Annu. Rev. Biophys. 2017, 46, 23–42. 10.1146/annurev-biophys-070816-033928. [DOI] [PubMed] [Google Scholar]
- Huang P.-S.; Boyken S. E.; Baker D. The Coming of Age of de Novo Protein Design. Nature 2016, 537, 320–327. 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- Bale J. B.; Gonen S.; Liu Y.; Sheffler W.; Ellis D.; Thomas C.; Cascio D.; Yeates T. O.; Gonen T.; King N. P.; et al. Accurate Design of Megadalton-Scale Two-Component Icosahedral Protein Complexes. Science 2016, 353, 389–394. 10.1126/science.aaf8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman N. C.; Kallenbach N. R. Design of Immobile Nucleic Acid Junctions. Biophys. J. 1983, 44, 201–209. 10.1016/s0006-3495(83)84292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winfree E.; Liu F.; Wenzler L. A.; Seeman N. C. Design and Self-Assembly of Two-Dimensional DNA Crystals. Nature 1998, 394, 539–544. 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Marblestone A. H.; Teerapittayanon S.; Vazquez A.; Church G. M.; Shih W. M. Rapid Prototyping of 3D DNA-Origami Shapes with caDNAno. Nucleic Acids Res. 2009, 37, 5001–5006. 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Wang D.; Yu L.; Huang C.-M.; Arya G.; Chang S.; Ke Y. Programmable Transformations of DNA Origami Made of Small Modular Dynamic Units. J. Am. Chem. Soc. 2021, 143, 2256–2263. 10.1021/jacs.0c10576. [DOI] [PubMed] [Google Scholar]
- Seifert J. L.; Connor R. E.; Kushon S. A.; Wang M.; Armitage B. A. Spontaneous Assembly of Helical Cyanine Dye Aggregates on DNA Nanotemplates. J. Am. Chem. Soc. 1999, 121, 2987–2995. 10.1021/ja984279j. [DOI] [Google Scholar]
- Wang M.; Silva G. L.; Armitage B. A. DNA-Templated Formation of a Helical Cyanine Dye J-Aggregate. J. Am. Chem. Soc. 2000, 122, 9977–9986. 10.1021/ja002184n. [DOI] [Google Scholar]
- Garoff R. A.; Litzinger E. A.; Connor R. E.; Fishman I.; Armitage B. A. Helical Aggregation of Cyanine Dyes on DNA Templates: Effect of Dye Structure on Formation of Homo- and Heteroaggregates. Langmuir 2002, 18, 6330–6337. 10.1021/la025742f. [DOI] [Google Scholar]
- Asanuma H.; Fujii T.; Kato T.; Kashida H. Coherent Interactions of Dyes Assembled on DNA. J. Photochem. Photobiol., C 2012, 13, 124–135. 10.1016/j.jphotochemrev.2012.04.002. [DOI] [Google Scholar]
- Cunningham P. D.; Khachatrian A.; Buckhout-White S.; Deschamps J. R.; Goldman E. R.; Medintz I. L.; Melinger J. S. Resonance Energy Transfer in DNA Duplexes Labeled with Localized Dyes. J. Phys. Chem. B 2014, 118, 14555–14565. 10.1021/jp5065006. [DOI] [PubMed] [Google Scholar]
- Melinger J. S.; Khachatrian A.; Ancona M. G.; Buckhout-White S.; Goldman E. R.; Spillmann C. M.; Medintz I. L.; Cunningham P. D. FRET from Multiple Pathways in Fluorophore-Labeled DNA. ACS Photonics 2016, 3, 659–669. 10.1021/acsphotonics.6b00006. [DOI] [Google Scholar]
- Markova L. I.; Malinovskii V. L.; Patsenker L. D.; Häner R. J- vs. H-Type Assembly: Pentamethine Cyanine (Cy5) as a near-IR Chiroptical Reporter. Chem. Commun. 2013, 49, 5298–5300. 10.1039/c3cc42103a. [DOI] [PubMed] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Patten L. K.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121, 6905–6916. 10.1021/acs.jpca.7b04344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma H.; Murayama K.; Kamiya Y.; Kashida H. The DNA Duplex as an Aqueous One-Dimensional Soft Crystal Scaffold for Photochemistry. Bull. Chem. Soc. Jpn. 2018, 91, 1739–1748. 10.1246/bcsj.20180278. [DOI] [Google Scholar]
- Cannon B. L.; Patten L. K.; Kellis D. L.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122, 2086–2095. 10.1021/acs.jpca.7b12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazuski R. J.; Díaz S. A.; Wood R. E.; Lloyd L. T.; Klein W. P.; Mathur D.; Melinger J. S.; Engel G. S.; Medintz I. L. Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency. J. Phys. Chem. Lett. 2020, 11, 4163–4172. 10.1021/acs.jpclett.0c01020. [DOI] [PubMed] [Google Scholar]
- Sohail S. H.; Otto J. P.; Cunningham P. D.; Kim Y. C.; Wood R. E.; Allodi M. A.; Higgins J. S.; Melinger J. S.; Engel G. S. DNA Scaffold Supports Long-Lived Vibronic Coherence in an Indodicarbocyanine (Cy5) Dimer. Chem. Sci. 2020, 11, 8546–8557. 10.1039/d0sc01127d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kringle L.; Sawaya N. P. D.; Widom J.; Adams C.; Raymer M. G.; Aspuru-Guzik A.; Marcus A. H. Temperature-Dependent Conformations of Exciton-Coupled Cy3 Dimers in Double-Stranded DNA. J. Chem. Phys. 2018, 148, 085101. 10.1063/1.5020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heussman D.; Kittell J.; Kringle L.; Tamimi A.; von Hippel P. H.; Marcus A. H. Measuring Local Conformations and Conformational Disorder of (Cy3)2 Dimer Labeled DNA Fork Junctions Using Absorbance, Circular Dichroism and Two-Dimensional Fluorescence Spectroscopy. Faraday Discuss. 2019, 216, 211–235. 10.1039/c8fd00245b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart S. M.; Chen W. J.; Banal J. L.; Bricker W. P.; Dodin A.; Markova L.; Vyborna Y.; Willard A. P.; Häner R.; Bathe M.; et al. Engineering Couplings for Exciton Transport Using Synthetic DNA Scaffolds. Chem 2021, 7, 752–773. 10.1016/j.chempr.2020.12.020. [DOI] [Google Scholar]
- Spiegel J. D.; Fulle S.; Kleinschmidt M.; Gohlke H.; Marian C. M. Failure of the IDA in FRET Systems at Close Inter-Dye Distances Is Moderated by Frequent Low Κ2 Values. J. Phys. Chem. B 2016, 120, 8845–8862. 10.1021/acs.jpcb.6b05754. [DOI] [PubMed] [Google Scholar]
- Kasha M. Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res. 1963, 20, 55–71. 10.2307/3571331. [DOI] [PubMed] [Google Scholar]
- Kasha M.; Rawls H. R.; Ashraf El-Bayoumi M. The Exciton Model in Molecular Spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. 10.1351/pac196511030371. [DOI] [Google Scholar]
- Mass O. A.; Wilson C. K.; Roy S. K.; Barclay M. S.; Patten L. K.; Terpetschnig E. A.; Lee J.; Pensack R. D.; Yurke B.; Knowlton W. B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124, 9636–9647. 10.1021/acs.jpcb.0c06480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhmer M.; Enderlein J. Fluorescence Spectroscopy of Single Molecules under Ambient Conditions: Methodology and Technology. ChemPhysChem 2003, 4, 792–808. 10.1002/cphc.200200565. [DOI] [PubMed] [Google Scholar]
- Snellenburg J. J.; Laptenok S.; Seger R.; Mullen K. M.; van Stokkum I. H. M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Software 2012, 49, 1. 10.18637/jss.v049.i03. [DOI] [Google Scholar]
- Scharf B.; Dinur U. Striking Dependence of the Rate of Electronic Radiationless Transitions on the Size of the Molecular System. Chem. Phys. Lett. 1984, 105, 78–82. 10.1016/0009-2614(84)80417-0. [DOI] [Google Scholar]
- Humeniuk A.; Mitrić R.; Bonačić-Koutecký V. Size Dependence of Non-Radiative Decay Rates in J-Aggregates. J. Phys. Chem. A 2020, 124, 10143–10151. 10.1021/acs.jpca.0c09074. [DOI] [PubMed] [Google Scholar]
- Das S.; Kamat P. V. Can H-Aggregates Serve as Light-Harvesting Antennae? Triplet–Triplet Energy Transfer between Excited Aggregates and Monomer Thionine in Aersol-OT Solutions. J. Phys. Chem. B 1999, 103, 209–215. 10.1021/jp983816j. [DOI] [Google Scholar]
- Liang K.; Farahat M. S.; Perlstein J.; Law K.-Y.; Whitten D. G. Exciton Interactions in Nonconjugated Squaraine Dimers. Mechanisms for Coupling and Consequences for Photophysics and Photochemistry. J. Am. Chem. Soc. 1997, 119, 830–831. 10.1021/ja962091z. [DOI] [Google Scholar]
- Von Hippel P. H.; Johnson N. P.; Marcus A. H. Fifty Years of DNA “Breathing”: Reflections on Old and New Approaches. Biopolymers 2013, 99, 923–954. 10.1002/bip.22347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israels B.; Albrecht C. S.; Dang A.; Barney M.; von Hippel P. H.; Marcus A. H. Sub-millisecond Conformational Transitions of Short Single-Stranded DNA Lattices by Photon Correlation Single-Molecule FRET. J. Phys. Chem. B 2021, 125, 9426–9440. 10.1021/acs.jpcb.1c04119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley D. M. J.; Norman D. G. The Holliday Junction Is Finally Seen with Crystal Clarity. Nat. Struct. Biol. 1999, 6, 897–899. 10.1038/13255. [DOI] [PubMed] [Google Scholar]
- Joo C.; McKinney S. A.; Lilley D. M. J.; Ha T. Exploring Rare Conformational Species and Ionic Effects in DNA Holliday Junctions Using Single-Molecule Spectroscopy. J. Mol. Biol. 2004, 341, 739–751. 10.1016/j.jmb.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Seeman N. C. Nucleic Acid Junctions and Lattices. J. Theor. Biol. 1982, 99, 237–247. 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.