

Abstract
Cancer represents an evolutionary process through which growing malignant populations genetically diversify, leading to tumour progression, relapse and resistance to therapy. In addition to genetic diversity, the cell-to-cell variation that fuels evolutionary selection also manifests in cellular states, epigenetic profiles, spatial distributions and interactions with the microenvironment. Therefore, the study of cancer requires the integration of multiple heritable dimensions at the resolution of the single cell — the atomic unit of somatic evolution. In this Review, we discuss emerging analytic and experimental technologies for single-cell multi-omics that enable the capture and integration of multiple data modalities to inform the study of cancer evolution. These data show that cancer results from a complex interplay between genetic and non-genetic determinants of somatic evolution.
The evolutionary history of cancer includes malignant transformation, followed by progression to more aggressive and resistant forms1, ultimately leading to its devastating clinical impact2. Somatic mutations, including single nucleotide variants (SNV) and structural variations, are critical for cancer initiation and evolution1,3. Nonetheless, in recent years, pervasive somatic mutations have been identified across healthy tissues4–8, particularly those exposed to environmental carcinogens, such as skin and the oesophagus, which suggests that cancer often arises from pre-malignant clonal outgrowths. Importantly, somatic mutations in clonal outgrowths that do not progress, or even regress, overlap with recurrent drivers of cancer4–9. These data suggest that genetic mechanisms alone may be insufficient to drive malignant transformation10.
Somatic mutations.
Alterations in the DNA acquired post-conception (versus germline mutations) and able to be passed onto progeny of mutated cells. Somatic mutations can be detected by sequencing in otherwise histologically normal appearing tissue, are often associated with age and environmental exposures, and can manifest in cancer driver genes.
Clonal.
A population of cells with the same underlying genetic make-up (that is, with the same somatic mutations), which can beget subclonal populations that have acquired additional genetic aberrancies.
Drivers.
Somatic mutations that increase tumour cell fitness.
As malignant populations grow, cells undergo further genetic diversification that enables tumour progression, relapse and resistance to therapy3. However, clear genetic drivers of progression, metastasis and therapeutic resistance are identified in only a subset of tumours, pointing to non-genetic contributors of cancer progression11,12. Recent advances, particularly in single-cell technologies, have revealed intratumoural heterogeneity in cell states, epigenetic profiles, spatial dynamics and interactions with the tumour microenvironment. These axes of potentially heritable intratumoural variation may provide additional cues for cancer evolution. Thus, the integration of multiple layers of information for individual cancer cells, via single-cell multi-omics, is critically needed for a comprehensive understanding of the mechanisms of cancer evolution (FIG. 1).
Fig. 1 |. Single-cell multi-omics for deciphering clonal evolution in cancer.
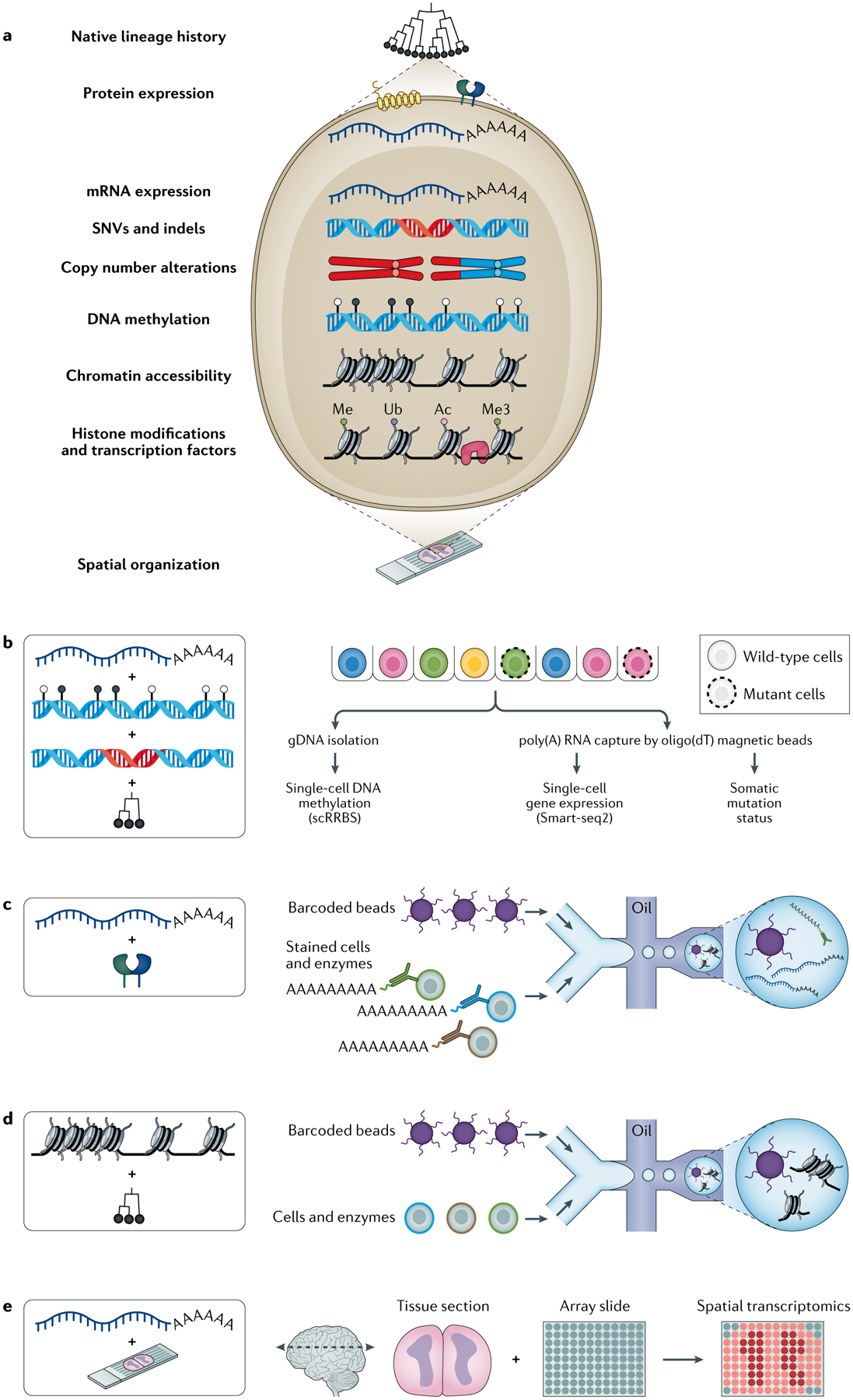
Analytic or experimental integrations of multiple data ‘omics’ modalities in single-cells advance our understanding of mechanisms of clonal evolution. a | Cancer cell representation with heritable traits that can be interrogated via multi-omics platforms. b | Extracting DNA methylation (DNAme) and transcriptomic information from the same cells experimentally has been achieved by modifying plate-based single-cell RNA sequencing (scRNA-seq) protocols (for example, Smart-seq2), in which both RNA and DNA are respectively isolated from the same cells for whole-transcriptome and DNAme data through bisulfite sequencing121,128,129. Heritable stochastic DNAme changes can then be exploited as native barcodes to directly infer the high-resolution phylogenetic history of tumour cells121. c | scRNA-seq with integration of protein expression measurements can be performed in parallel for the same cells122,123. DNA-barcoded antibodies, acting as synthetic transcripts, are used to convert the detection of proteins into a quantitative readout. This allows the immunophenotyping of cells to be integrated with an unbiased transcriptome analysis using existing single-cell sequencing approaches. d | High-sensitivity somatic genotyping in which, for instance, any mutation in mitochondrial DNA may serve as lineage markers. Interrogating these naturally occurring genetic barcodes within scATAC-seq97 (or scRNA-seq) provides high-resolution phylogenies coupled with cell state information. e | As an example of spatially aware platforms160,162–164,166,169, spatial transcriptomics165 utilizes molecular barcodes for the detection of mRNA molecules and maps them to their spatial positioning. gDNA, genomic DNA; indels, insertions or deletions; scRRBS, single-cell reduced-representation bisulfite sequencing; SNVs, single nucleotide variants.
Cell states.
A cell’s phenotype, as inferred by transcriptional or protein markers, that are often transitional (for example, intermediate states within a developmental system such as haematopoiesis or epithelium).
Single-cell multi-omics.
Analytic or experimental integrations of multiple data ‘omics’ modalities in single cells.
Herein, we review recent experimental and analytic innovations in single-cell technologies that integrate multiple dimensions of heritable information in individual tumour cells. This Review addresses both genetic and non-genetic routes of cancer evolution that can be uniquely interrogated through single-cell multi-omics advancements. As comprehensive discussions of genetic sources of cancer heterogeneity3 and the technical aspects of integrative single-cell analyses13 have been recently undertaken, we focus herein on nascent but compelling evidence that motivates the integration of multiple strata of information in single cancer cells to decipher tumour heterogeneity and evolution.
Genetic heterogeneity and lineage tracing
Inference of clonal architecture in bulk sequencing.
Genetic heterogeneity through the continuous acquisition of somatic mutations underlies clonal evolution in many cancers. The clonal architecture of these genetically heterogeneous populations has been previously inferred through bulk next-generation sequencing14. The integration of read depth and variant allele frequencies of somatic mutations in whole-exome or whole-genome sequencing (WGS) data can be used to infer tumour purity, ploidy and local copy number for each mutation and thus to determine the cancer cell fractions (CCFs) harbouring the mutations14. These data can resolve clonal and subclonal relationships to a limited extent (FIG. 2a). In aneuploid solid tumours, WGS can help time early versus late clonal somatic alterations by determining whether SNVs occurred before or after large amplification processes such as whole-genome duplication15.
Fig. 2 |. Phylogenetic inference for retrospective lineage tracing.
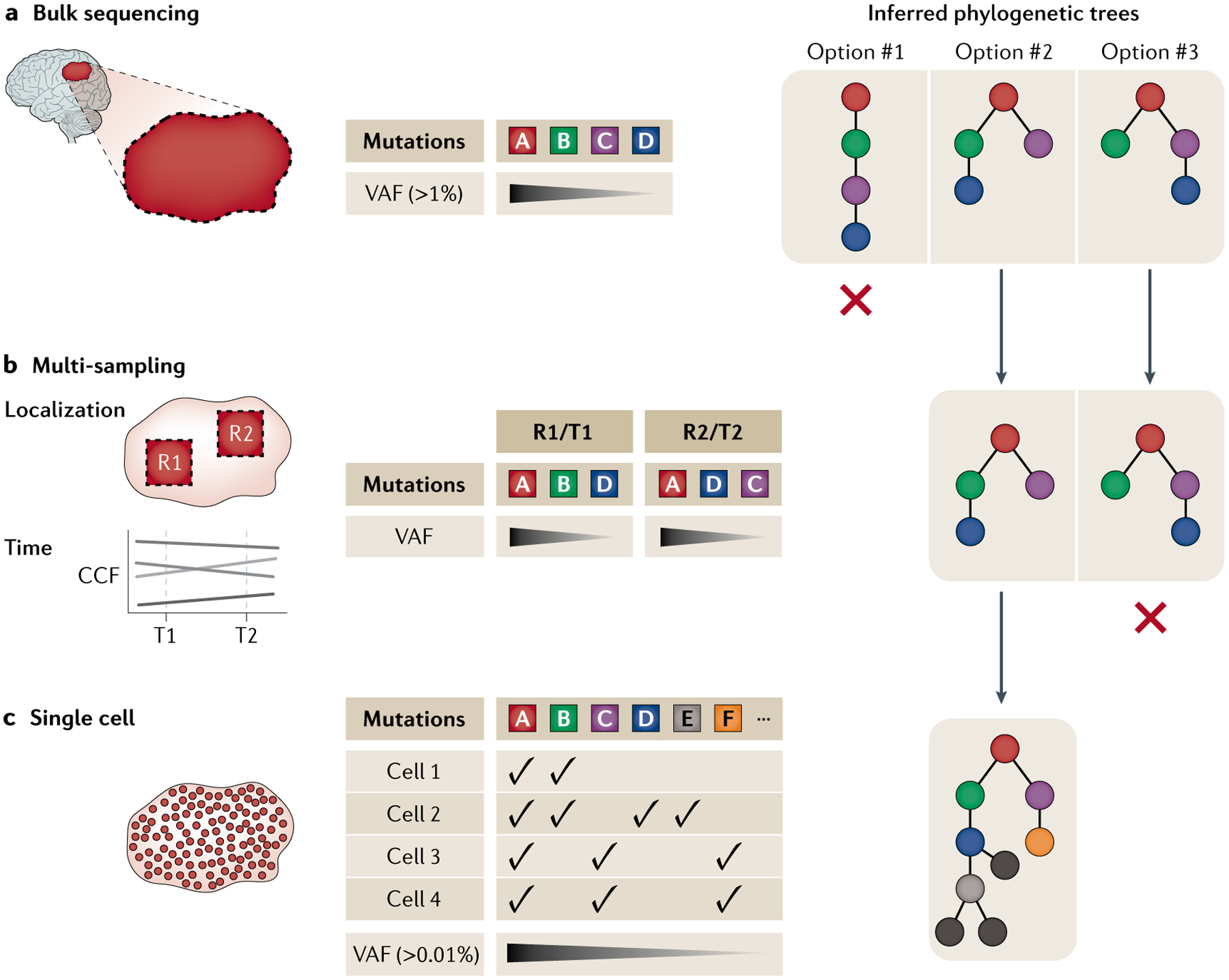
a | Bulk next-generation sequencing allows inference of clonal architecture phylogenetic trees of genetically heterogeneous populations. However, these data can resolve clonal and subclonal relationships to a limited extent by enabling the assessment of the order of acquisition of mutations (A–D) and are limited in their abilities to resolve the phylogenetic relationships of clones, especially at low cancer cell fractions (CCFs). b | Multi-sampling at different time points (T1, T2) during clonal evolution or at different regions (R1, R2) within a tumour (to assess for intratumoural clonal spatial composition) can provide higher-resolution phylogenetic relationships, even for subclones with low CCFs, owing to coordinated patterns of CCF fluctuations over time. c | Even though additional resolution is gained through multi-sampling, resolving phylogeny at single-cell resolution (by single-cell whole-genome sequencing or targeted sequencing) is required to derive the precise clonal dynamics and evolutionary history of a tumour. High-resolution trees pave the way for critical inferences derived directly from primary patient tumours for defining key parameters of somatic evolution. VAF, variant allele frequency.
Variant allele frequencies.
Frequencies of mutated alleles in the sequenced reads from next-generation sequencing. Variant allele frequencies reflect copy number, zygosity, tumour purity and the fraction of cancer cells that harbour the mutation (for example, clonal versus subclonal).
Tumour purity.
Per cent of a tumour mass composed of tumour cells, versus admixed non-neoplastic cells, such as tumour-infiltrating immune cells and stromal cells.
Cancer cell fractions.
(CCFs). Fractions of cells that harbour a given mutation.
When applied to large cohorts, these analyses provide a blueprint of the evolutionary sequence in which driver mutations arise. Thus, driver events can be separated into early, often ancestral mutations (for example, DNMT3A and TET2 mutations in myeloid neoplasms) versus late subclonal drivers that arise as a result of ongoing selection within the malignant population (for example, SF3B1, TP53 and NPM1 mutations in lymphoid and myeloid malignancies)14,16,17. Similarly, in a large cohort of renal cell carcinomas, tumours could be classified into evolutionary subtypes (for example, tumours with driver events in PBRM1, which subsequently acquire alterations in SETD2 or PI3K) that could be linked with prognostic information (for example, ‘PBRM1 → SETD2’ or ‘PBRM1 → PI3K’ subtypes with an attenuated disease course)18. Thus, timing analyses have charted different evolutionary trajectories or preferred mutational sequences in cancer, which suggest optimized paths and mutational interdependency17,19. While the presence of subclonal drivers predicts poor clinical outcome in patients with cancer14,17,20, early clonal drivers that develop years before diagnosis might present opportunities for early intervention. Nevertheless, methods that use bulk sequencing inference are fundamentally limited in their abilities to resolve the phylogenetic relationships of clones, especially at low CCFs21 (FIG. 2a).
Defining clonal dynamics through multi-sampling.
Cancer evolution is a dynamic process, whereby clones of different fitness may reshape the genetic make-up of the tumour in the face of challenges such as treatment with effective therapeutics. Multi-sampling at different time points during clonal evolution can provide higher-resolution phylogenetic relationships even for subclones with low CCFs due to coordinated patterns of CCF fluctuations over time22 (FIG. 2b). With a greater number of sampling time points, individual subclones can be identified at a CCF significantly different from other subclones, especially if they have distinct growth dynamics21. Furthermore, serial sequencing not only enhances clonal decomposition but also enables clone-specific fitness measurements21,23,24. Indeed, dense temporal sampling for either circulating leukaemia cells14,17,21,23,24 or circulating tumour DNA in solid malignancies, enables the measurement of clonal growth kinetics in relation to therapy through mathematical modelling25,26. For example, chronic lymphocytic leukaemia (CLL) relapsed after chemotherapy demonstrated an increased CCF with TP53 mutations compared with the pre-treatment tumour, suggesting a greater fitness advantage of the TP53-mutated subclone in the face of therapy17. By contrast, targeted CLL therapy provided a fitness advantage to clones harbouring point mutations in the target protein (BTK)23. In solid tumours, tracking circulating tumour DNA from patients with colorectal carcinomas treated with EGFR blockade identified the clonal dynamics of drug-resistant clone expansion upon treatment and a decline upon drug withdrawal25. Notably, mathematical modelling of clonal growth enables forward and backward extrapolation to predict future clonal fractions and to estimate the number of resistant cells at treatment initiation23. These data also pave the way for the algorithmic optimization of combination therapies based on continuous measurement of the therapy-specific fitness effects on different clones27.
Another form of multi-sampling is interrogating multiple regions within a tumour to assess for intratumoural clonal spatial composition. Analogous to temporal sampling, multiregional sampling of non-small-cell lung carcinomas helped to refine clonal relationships and, thus, to improve clinical stratification20. In a striking example of evolutionary selection, multiregional sampling revealed the convergent evolution of driver copy number alterations (CNAs), whereby the same driver CNA involved distinct parental alleles across the tumour20. Multiregional tumour sampling also enables comparison of the primary tumour with secondary spread to metastatic foci28, identifying drivers of metastases (for example, CNAs in MYC, YAP1 and MMP13 in brain-metastasized lung adenocarcinomas versus primary tumours29).
Copy number alterations.
(CNAs). Changes in the number of copies of a DNA segment due to deletions or gains in the genome.
While the breadth of genome-wide data enables inference for retrospective lineage tracing, a different, focused approach has leveraged the high mutability of microsatellites in mismatch repair-deficient colorectal carcinomas. Indeed, microsatellite sites have long been appreciated as a molecular clock, providing critical insights into clonal evolution30,31. For example, the molecular age of pre-malignant colorectal adenomas was comparable to that of adenocarcinomas, arguing against a stepwise progression from adenomas to adenocarcinomas30. In a more recent study, microsatellite sites were used for lineage tracing in multiregional analyses of only 20 poly-G repeat regions, challenging the ‘sequential progression model’, which posits that metastases to distant organs arise from lymph node metastases32. This study demonstrated that, in two-thirds of cases, both lymph node and distant metastases arose directly from the primary colonic adenocarcinomas32, with clear implications for interpreting the staging of biopsy samples. Building on this experience, a similar approach was undertaken targeting microsatellite sites to demonstrate that polyclonal seeding was more frequent in lymph node metastases than in distant metastases33. Thus, analyses of metastatic disease can further elucidate patterns of clonal spread34 and enable inferences of tumour evolution migration patterns35.
Retrospective lineage tracing.
Clonal architecture and/or phylogenetic reconstruction of primary samples through naturally accumulated heritable marks, such as copy number alterations, single nucleotide variants or DNA methylation, as opposed to prospective lineage tracing, in which lineage barcodes are artificially introduced into a model organism.
Molecular clock.
A method to deduce the temporal history (often in terms of number of divisions) of a cell or group of cells based on genetic or epigenetic changes that reflect time (or number of divisions).
Tracing genetic history in single cells.
Despite the additional resolution that multi-sampling provides to the clonal deconstruction of cancer evolution, resolving phylogeny at single-cell resolution is required to derive the precise clonal dynamics and evolutionary history of a tumour (FIG. 2c). Prospective lineage tracing through optical36–38 or sequencing barcodes39–42 has enabled in vitro or in vivo modelling of tumour evolution, including with methods capable of capturing additional cellular features such as single-cell gene expression data43–48 (see recent reviews49–51).
However, these methods are not applicable for retrospective lineage tracing in primary human tissues, where reliance on ‘native barcodes’ is key (FIGS 2,3). High-throughput, single-cell-targeted DNA sequencing, using a droplet-based microfluidics platform, for a panel of recurrent driver mutations has allowed the highly sensitive capture of somatic genotypes of thousands of cells to reveal subclonal shifts from diagnosis to relapse in acute myeloid leukaemia (AML)52. Another leading method of retrospective lineage tracing inference, applied directly to human cancers, is single-cell WGS (scWGS). scWGS enables the inference of CNAs that serve as natural barcodes through which the phylogenetic relationships between individual cells can be drawn53–55. Early single-cell sequencing studies of breast cancers resolved distinct clones within a tumour and demonstrated that metastatic foci were seeded from a single expanded clone54. Individual cells with non-recurrent complex CNAs provided a glimpse of the underlying genetic diversity that fuels clonal evolution54. Recently, high-throughput scWGS approaches have been introduced, enabling the sequencing of thousands of breast cancer cells and identifying early ancestral (for example, MCL1 and MYC) versus subclonal (for example, RAD18) amplifications56. scWGS is characterized by sparse coverage of SNVs, limiting the resolution of lineage reconstruction based on a relatively small number of large somatic CNAs. Furthermore, examples of convergent evolution in which the same CNAs are seen in both parental alleles within a single tumour, as noted above19,57, caution against classic phylogenetic reconstructions with the infinite sites assumption (that is, that mutations used for lineage tracing do not recur and are not lost). Thus, phylogenetic methods with ‘finite sites’ models may be better suited to address aneuploid tumours58 (the challenges of tumour phylogenetics have been reviewed elsewhere59). Other limitations include the introduction of technical errors from whole-genome amplifications such as allelic dropouts, non-uniform coverage, and PCR errors and recombinations49. Novel computational tools have been developed that adapt the classic phylogenetic methods for species evolution to address the technical noise of single-cell data60,61.
Fig. 3 |. Interrogating native barcodes for retrospective lineage tracing.
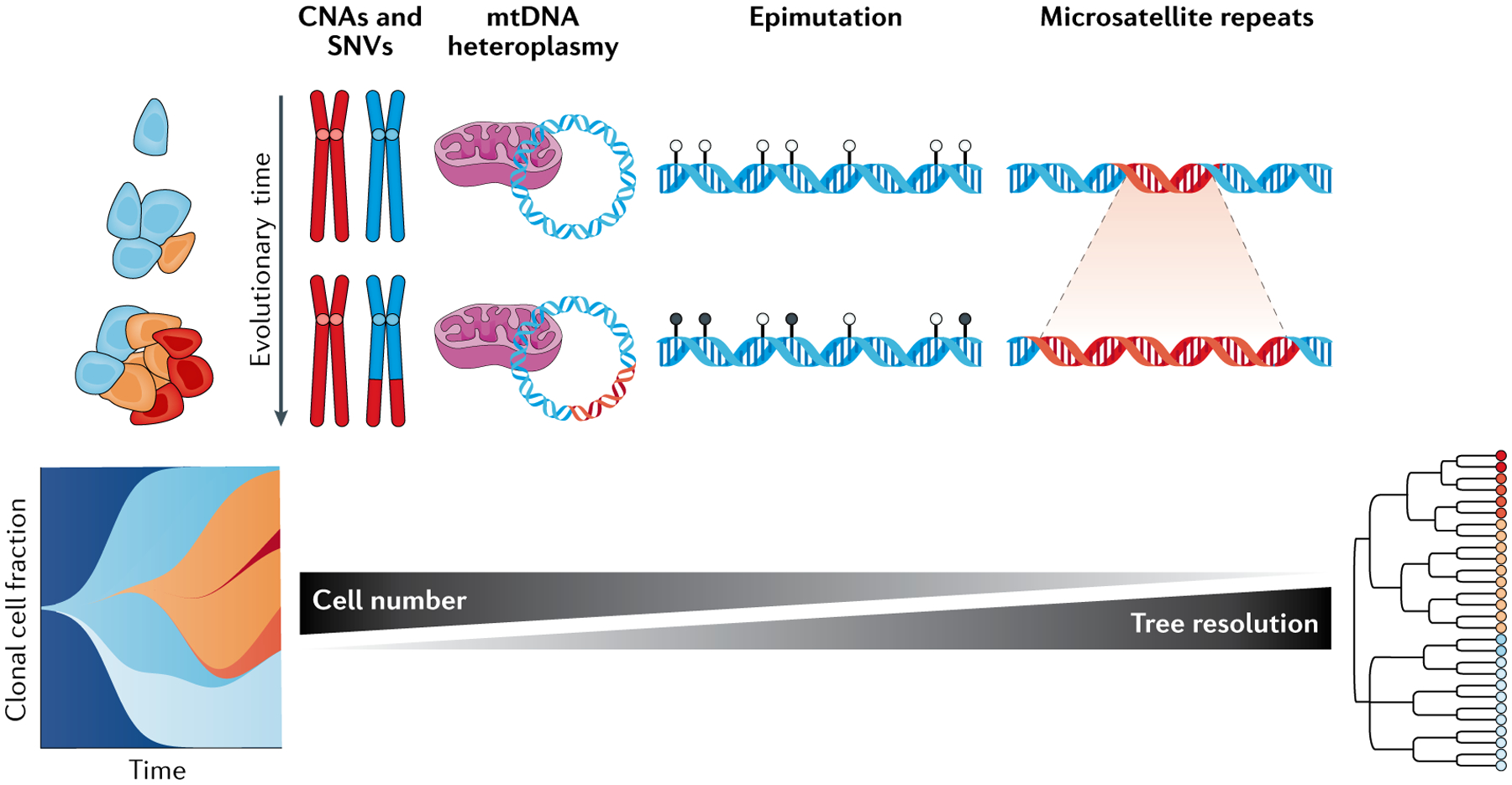
Clonal architecture and/or phylogenies can be reconstructed from primary samples through naturally accumulated heritable marks, that is, ‘native barcodes’, such as copy number alterations (CNAs), single nucleotide variants (SNVs), small insertions or deletions (indels) in microsatellite repeat regions, DNA methylation changes, and mutations in mitochondrial DNA (mtDNA). Emerging and potential multi-omics technologies for lineage inference display a trade-off between throughput and lineage inference resolution (black bars). For instance, single-cell retrospective lineage tracing inference methods using CNAs (from single-cell whole-genome sequencing or single-cell RNA sequencing datasets) provide a low resolution of the underlying genetic diversity that fuels clonal evolution but can be applied to high number of cells. High-sensitivity somatic genotyping of a large number of loci may enable clonal reconstruction and a high-resolution retrospective lineage tracing with methods that interrogate mtDNA or microsatellite sites. Finally, heritable stochastic DNA methylation changes can serve as a molecular clock and therefore be exploited as native barcodes to infer phylogenetic history.
Increasing DNA input through the use of single-cell-derived colonies (that is, populations of cells derived from a single clone) obviates the technical artefacts from whole-genome amplifications. The high-resolution lineage reconstruction of single-cell colony or organoid data has been applied to normal haematopoietic development54 and cancerous tissues62. The high-resolution trees paved the way for critical inferences derived directly from primary patient material of key parameters of somatic evolution such as determining the size and rate of growth of the self-renewing haematopoietic stem cell pool in a normal adult63. This study also began to link clonal dynamics with lineage fates, demonstrating that the human adult haematopoietic stem cells contributed to myeloid and B cells but not to T cells in the 140 sequenced single-cell clones. While this experiment was limited in throughput, it provided a model for retracing the genetic lineage history and fate decisions of normal and cancer stem cells in primary human samples, anticipating the integration of genetic data with other known axes of variation at transcriptional and epigenetic levels.
Heterogeneity of cell states in tumour evolution
Cell state plasticity as a mediator of cancer evolution.
Intratumoural genetic variability underlies tumour heterogeneity and provides a heritable diversity that drives clonal evolution across cancer. Nevertheless, genetic mechanisms alone may not capture the full spectrum of intratumoural heterogeneity. One such variability that has long been appreciated is the plasticity of cell states within a single tumour. A classic example is the epithelial–mesenchymal transition (EMT) in epithelial tissue-derived cancers, which is reminiscent of transitions between epithelial and mesenchymal cells during embryonic development64. EMT, a heterogeneous and dynamic disposition with intermediary or partial EMT meta-states, is governed by a complex network of transcription factors such as SNAI1, SNAI2, ZEB1, TWIST1 and other regulators64. Clinically, EMT is associated with varying invasive and metastatic potential65.
More broadly, cancers often recapitulate physiological developmental programmes and may thus be composed of stem-like and differentiated cell types, as has been observed in heamatopoietic66,67, brain68 and epithelial tumours69,70. These observations support a hierarchical model of cancer71. While still debated in the cancer field, in this model, cancer stem-like cells are at the apex of the tumour differentiation hierarchy and reflect traits of normal stem cells such as an extensive proliferative and self-renewal capacity. For example, AML cells with the most immature phenotype demonstrate robust repopulating capacities across leukaemia subtypes regardless of distinct somatic drivers67. Consistent with the suggestion that cancer initiates in cells with stem-like properties, a primitive stem-like programme has been shown to precede malignant transformation10. Specifically, activating an embryonic neural crest progenitor state in BRAF-mutated and TP53-mutated melanocytes resulted in the induction of melanoma10. These results indicate that somatic mutations cooperate with stem-like states for cancer initiation10. While these data provide compelling evidence for therapeutically targeting the most stem-like cells, tumour cell plasticity may complicate this strategy. Differentiated cancer cells demonstrated the capacity to de-differentiate into stem-like states66,69,70, providing evidence for a plastic bidirectional interchange between stem and differentiated cell states in malignant populations.
Stochastic cell state transitions can also serve as mediators of cancer resistance. In vitro and mouse models have identified transcriptional persister states in rare cells that enable survival through drug insult and facilitate the acquisition of full-blown genetically driven resistance to therapy11,12,72. Furthermore, lineage plasticity, such as the transformation of prostatic73 or lung adenocarcinoma74 into small cell carcinoma or of CLL into diffuse large B cell lymphomas in Richter transformation75, has long been appreciated as a non-genetic mechanism of therapy resistance. This form of lineage plasticity provides a path to therapy resistance via cell-identity reprogramming that eliminates dependencies on therapeutically targeted pathways. Collectively, these data show that cancer evolution is a result of a complex interplay between genetic diversity and cancer’s ability to toggle between cell states that jointly allow cancer populations to scan the fitness landscape for superior evolutionary trajectories and roadmaps to circumvent therapeutic barriers. We thus require methodologies that integrate cell state heterogeneity with a purely genetic cancer evolution model.
Heterogeneous transcriptional states at single-cell resolution.
As transcriptional state heterogeneity is a key ingredient in tumour initiation and progression11,12,72, single-cell RNA sequencing (scRNA-seq) of individual cells from primary tumours has emerged as a transformative technology. Studies across cancer types have revealed intratumoural cell state heterogeneity as the rule rather the exception. Heterogeneity in cell states is attributed not only to basic cellular processes, such as cell cycle, metabolic and hypoxia-induced stress states, but also to developmental programmes and clinically relevant resistance programmes68,76,77.
scRNA-seq provided high-resolution mapping of stem-like states78 and developmental hierarchies68 directly in patient samples. For example, in IDH1/2-mutated oligodendrogliomas, scRNA-seq identified stem-like cancer cells with activation of neural stem cell programmes at the top of the hierarchy that branches into two distinct cellular states resembling astrocytic and oligodendrocytic lineages68. Consistent with the cancer stem cell model79, the expression of cell cycle-related genes was highly enriched in stem-like cells. In single-cell analyses of melanoma, a stemness signature coincided with an AXL-high drug-resistance programme to RAF and MEK inhibitors76, directly co-mapping critical enabling properties, such as growth and resistance to therapy, to the same cells. scRNA-seq further enabled the detection of rare cells in patients who represent residual disease after therapy80. Comparison of scRNA-seq of pre-treatment and post-treatment multiple myeloma identified residual tumour cells (often comprising a small minority of the plasma cell population) based on the known malignant signatures of pre-treated myeloma80. Thus, scRNA-seq can distinguish rare tumour cells amid non-neoplastic cells for early disease or minimal residual disease detection and for the identification of targetable resistance programmes.
Furthermore, not only can scRNA-seq measure cell states at a given time but emerging analytic technologies can also leverage scRNA-seq data to inform the likely dynamics of cell states. Various methods have emerged that reconstruct differentiation trajectories, collectively termed pseudotime projection analyses, based on the inference of similarity gradients in transcriptional data50,51. For example, one of the first of these techniques, Monocle, constructs minimum spanning trees to build backbones onto which single cells are mapped81. This perspective capitalizes on the fact that scRNA-seq is not dependent on sorting based on known cell surface markers and thus captures transitional states, which may be informative for cell state dynamics inference. Another distinct framework predicts future cell states by quantifying the ratio of unspliced to spliced mRNA within standard scRNA-seq to determine ‘RNA velocity’, that is, the rate of change in mRNA abundance82. As genes that are ‘turned on’ show a higher ratio than ‘turned off’ genes, RNA velocity can predict the transcriptional profile that the cell will assume, enabling prediction of the future cell state together with measurement of the current cell state. Both these approaches provide powerful measurements of cell state dynamics in both normal and cancerous tissues. Notably, these approaches are independent of genetic lineage tracing and thus offer the tantalizing prospect of linking cell state dynamics with genetic identity through single-cell multi-omics integration to elucidate the genetic underpinnings or decoupling of cell state dynamics in somatic evolution.
Coupling genetic lineage history with single-cell transcriptional states.
Retracing the lineage history of cancer cells with distinct phenotypic variations (for example, along developmental hierarchies or in treatment tolerance) requires the direct integration of genetic information with scRNA-seq data. For example, prospective barcoding through lentiviral transfection of patient-derived xenografts demonstrated that tumour cells with the same barcodes (that is, originating from the same cell) occupied distinct lineage-specific states, supporting the plasticity of glioblastoma cells77. However, methods that rely on artificial lineage barcodes cannot be applied to primary human samples, which necessitate the use of naturally occurring genetic barcodes of somatic mutations (FIGS 2,3).
Experimental methods that integrate DNA and RNA expression in single cells have been developed83–85. For example, G&T-seq can capture single-cell genomic and whole-transcriptome data by extracting and separating genomic DNA and mRNA for amplification and sequencing85. Another method, sci-L3, improves the throughput of the dual assay using combinatorial indexing84. These technologies share the same limitations of unimodal single-cell WGS, as discussed above, such as scalability, sparsity, PCR errors and allelic dropouts. The application of scWGS has limited value for largely euploid malignancies, for which a targeted sequencing approach may be more informative. Still, these methods provide the powerful ability to link genetic lineage history with the phenotypic states within the same cells for aneuploid tumours.
Furthermore, the rich genetic information available in the transcriptome itself has been leveraged to infer CNAs in large gains and losses by assessing imbalances in gene expression across chromosomes. This approach enabled the interrogation of the link between clonal identities and developmental states in these early landmark single-cell studies of aneuploid cancers68,76,78. In the setting of oligodendrogliomas, clonal divergence did not align with the predominant cell states, although skewed enrichment of subclones could be identified in distinct cell states68. In glioblastomas, specific copy number gains, such as those in EGFR, CDK4 and PDGFRA, respectively, underpin the biases in the transcriptional identities of tumour cells into astrocytic, neural-progenitor and oligodendrocyte-progenitor states77. Advanced analytic methods have since integrated SNPs for copy-neutral loss-of-heterozygosity events in conjunction with global gene expression levels for CNAs86,87. Application of these methods to multiple myeloma linked the subclonal deletion del(16) in the initial cancer cells with a transcriptional signature resembling relapsed extramedullary myeloma cells, demonstrating a potential link between aggressive behaviour and subclonal drivers in the original tumour86.
scRNA-seq data have also been interrogated for SNV drivers via the Smart-seq2 protocol, which captures full-length transcripts87. Mutation status in the oncohistone H3K27M was determined in 34% of tumour cells, with greater efficiency in the polyadenylated transcript H3F3A versus the non-polyadenylated HIST1H3B, reflecting the poly-A capture method of the Smart-seq2 platform87. Detection was markedly improved with the addition of H3F3A mutation locus-specific primers within the Smart-seq2 procedure (97% of 44 analysed cells). Similar methods using Smart-seq2 have been developed to link genetic alterations and transcriptional states in BCR–ABL-positive chronic myeloid leukaemia stem cells to uncover signatures of persistence in the setting of treatment with tyrosine kinase inhibitors88. Altogether, these studies demonstrate that clonal divergence maintains a complex relationship with cell-state heterogeneity, varying between tumour types. These results further highlight the need to integrate genetic identities with transcriptional states at the single-cell level at higher throughput.
Somatic genotyping in high-throughput single-cell RNA-seq.
The efficiency of somatic genotyping in the studies described above was low due to sparse coverage and the low throughput of scRNA-seq methods that profile the entire transcript (for example, Smart-seq2). Highly sensitive somatic genotyping through targeted amplification of both genomic DNA and cDNA enabled the integration of clonal structure with transcriptomic states in chronic myeloid neoplasms89. The added advantage of genomic DNA amplification is the genotyping of lowly or non-expressed mutations. While this method was built within the Smart-seq2 protocol, its throughput was improved to thousands of cells with the development of a 384-well format that generates 3´-biased libraries89. To measure genotype to phenotype relationships across tens of thousands of cells, the detection of SNVs has also been pursued in high-throughput droplet-based scRNA-seq (10x Genomics) by deeply sequencing the final library (~200,000 reads per cell versus the recommended minimum of ~20,000 reads per cell)90. This approach provided a framework to dissect subclonal transcriptional identities of AML such as that of GATA2-mutated subclones90.
Our group and others have further modified scRNA-seq platforms to enable the highly sensitive capture of somatic mutations in conjunction with whole transcriptomes in tens of thousands of single cells via targeted amplification of barcoded cDNA of the mutated locus of interest91,92 (FIG. 4a). Leveraging a nanowell-based scRNA-seq technology, van Galen et al. performed targeted somatic genotyping in scRNA-seq in the context of AML91. These data have demonstrated that AML transcriptional identities reflecting haematopoietic differentiation states closely correlated with the underlying genetic drivers (for example, FLT3 internal tandem duplications in the primitive progenitor cell state versus FLT3 Asn841Lys in differentiated cells within the same tumour)91, analogous to the close alignment of genotype and cell state in glioblastoma described above77. While the nanowell throughput enabled the transcriptomic profiling of thousands of cells, the efficiency of mutation locus capture was low for most targets owing to the low expression level of the targeted genes and the distance between the targeted locus and the transcript ends. However, the concurrent capture of whole-transcriptome information enabled the inference of mutation status based on transcriptional similarities between genotyped and non-genotyped cells through a Random forest classifier.
Fig. 4 |. Somatic mutations reshape differentiation topologies.
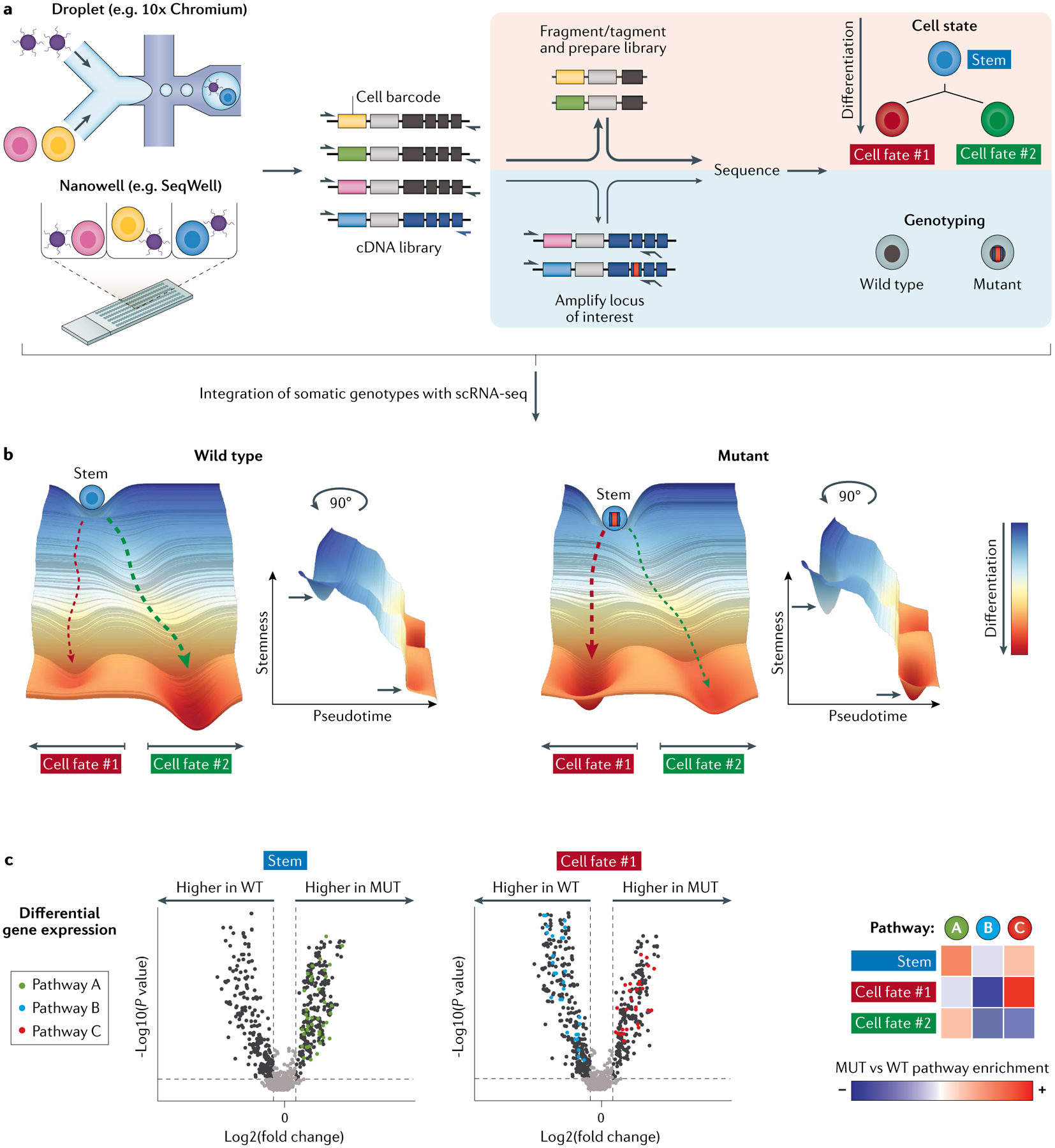
a | A schematic workflow of genotyping in single-cell RNA sequencing (scRNA-seq) by which cell state and somatic genotyping can be simultaneously captured for single cells. High-throughput digital scRNA-seq platforms (shown on the left) employ tagmentation or fragmentation for transcript-end biased cDNA short-read sequencing. Thus, loci harbouring somatic mutations are often lost. To overcome this limitation, these multi-omics techniques split the full-length cDNA for the targeted amplification of loci of interest on the one hand (shown on bottom panel with blue background) and for standard digital scRNA-seq on the other (shown on top panel with orange background). The two libraries are then intersected via shared cell barcodes analytically (not shown) to co-map somatic mutations and whole transcriptome data at single-cell resolution. b | The direct linking of somatic genotypes with whole transcriptome enables researchers to superimpose and chart two differentiation topologies within the same sample, namely the native wild type and the mutated one, thus turning the co-mingling of mutated and wild-type cells from a limitation to an advantage. The differentiation topologies (graphs) are built from scoring of stemness, pseudotime and differentiation states (that is, cell fate #1 and #2). c | By superimposing two differentiation topologies, we can identify the fitness impact of a somatic mutation within each cell state. Differential gene expression between mutant and wild-type cells can be identified (as shown by the volcano plots of differentially expressed genes). Pathway enrichment analysis of differentially regulated genes reveals activated or downregulated pathways (as shown by the annotated points on the volcano plots and heatmap, showing degree of enrichment). The differentially expressed genes and regulated pathways may vary as a function of cell state (that is, stem versus cell fate #1 versus cell fate #2). WT, wild type; MUT, mutant.
As an alternative approach, we developed Genotyping of Transcriptomes (GoT), which modifies droplet-based scRNA-seq (10x Genomics)92. GoT enabled highly sensitive genotyping in thousands of cells, resulting in a genotyping efficiency of ~90% for the main somatic mutation targets in the CALR driver gene. Nonetheless, the distance between the targeted locus and the transcript ends limited the sensitivity of genotyping for some targets, likely due to the inefficiency of large amplicons to cluster in standard short-read sequencing. To overcome this limitation, we introduced two complementary approaches, including long-read sequencing and circularization-GoT, in which sequential rounds of circularization and inverse PCR remove the intervening sequence between target locus and cell barcodes, producing a fragment that can be sequenced with standard short-read sequencing. We also noted that cells heterozygous for mutations may be erroneously assigned as wild type owing to partial sampling of the mRNA pool in scRNA-seq or as a result of transcriptional bursts. To overcome these potential biases, we undertook down-sampling of genotyping unique molecular identifier (UMI) counts for mutation calling and multivariate modelling with genotyping UMI. We also systematically examined the relationship of genotype-based findings to varying the minimum UMI count thresholds to assess the robustness of the findings. While GoT remains a challenge for lowly expressed genes, continued optimization of digital scRNA-seq platforms for enhanced overall RNA capture efficiencies will improve cDNA-based genotyping rates.
The ability of GoT to jointly capture cell state and genotype information at high-throughput showed that, in CALR-mutated myeloproliferative neoplasms, the predominant transcriptional identities of haematopoietic progenitor cells were uncoupled with the somatic mutation status; that is, mutant and wild-type cells were mingled throughout the haematopoietic differentiation tree, confirming that precision genotyping is necessary to distinguish mutant from wild-type cells92. Thus, in this context, GoT enables the overlay of the mutant differentiation trajectories onto the normal map of haematopoietic development within the same individual, eliminating potential biological confounders and technical batch effects (FIG. 4b). The direct comparison of mutant and wild-type differentiation landscapes identified that the mutant cell frequency increased with myeloid differentiation, suggesting a differential fitness advantage as a function of progenitor identity. To corroborate this result, we integrated pseudotime analysis (to provide cell state dynamics) with genetic mapping of the cells and demonstrated that mutated cells were biased towards a differentiated state compared with wild-type cells. Consistently, the mutation-related cell cycle gene expression increase was more pronounced in differentiated progenitors, such as megakaryocytic progenitors, compared with haematopoietic stem cells. Mutation-induced alterations in the unfolded protein response, NF-κB activity and JAK–STAT signalling all varied as a function of progenitor type and stage of maturation, demonstrating that the underlying cell identity constrains the impact of the driver mutation and that the resulting cancer cell phenotype is a function of the interaction between cell identity and the somatic mutation (FIG. 4c).
Such methods may be particularly helpful in studying early clonal expansions in human tissues93. Recently, clonal expansions within morphologically normal tissues, resulting in somatically acquired mosaicisms, were identified throughout the body4–6. These clonal expansions often harbour somatic mutations in known cancer drivers (for example, TP53 and NOTCH1), especially in tissues under environmental stress such as the skin, oesophagus and lung4,5,7,8. Similarly, within the haematopoietic system, recurrent drivers of myeloid malignancies, for example, DNMT3A and TET2 mutations, have been demonstrated often at low variant allele frequencies in individuals without haematological abnormalities94. This state, termed ‘clonal haematopoiesis’, nonetheless predisposes these individuals to an increased risk of developing blood cancers94,95 and cardiovascular disease96. However, a critical question remains as to the phenotypic consequences of these mutations that enable their clonal outgrowth. Thus, the application of multi-omics technologies to these otherwise normal-appearing tissues may help identify deviant transcriptional programmes that enable the earliest clonal growths in human tissue.
Expanding highly sensitive somatic genotyping to a larger number of loci may also enable clonal reconstruction and high-resolution retrospective lineage tracing coupled with cell identity and cell state information. For this purpose, any mutation, including those in mitochondrial DNA97–99 or microsatellite sites100, may serve as a lineage marker (FIGS 1d,2,3). The incorporation of these naturally occurring genetic barcodes into multi-omics single-cell sequencing may provide high-resolution phylogenies coupled with nuanced cell state data, ultimately allowing us to decipher the cell fate decisions of cancer cells.
Epigenetic plasticity in cancer evolution
Epigenetic profiles underlie cell states.
The cancer-enabling phenotypes described above, such as persistence, self-renewal and lineage plasticity, need to be inherited to contribute to the evolution of cancer to progression and resistance. However, these cell states have often been shown to be uncoupled from genetic identity. Accumulating data suggest that cell states may be encoded and propagated epigenetically12,101–103, consistent with epigenetic encoding of cell identity in normal developmental biology. Epigenetics encompasses additional layers of heritable changes104, including DNA methylation (DNAme), chromatin accessibility states and histone modifications, mediated largely through the propagation of key transcription factors105. As a whole, however, epigenetic modifications result in the highly coordinated regulation of transcriptional activity, which is important for normal development and tissue specification.
In cancer, the high prevalence of mutations in epigenetic modifiers, such as TET2, DNMT3A and ASXL1 in haematopoietic neoplasms16,106, and SWI/SNF (BAF) chromatin remodelling complexes in solid tumours107, points to the significance of epigenetics in mediating tumorigenesis. In addition, recent mechanistic explorations have pointed to epigenetics as underlying transcriptional signatures of persistence and resistance11,102. In glioblastomas, persistent tumour stem cells displayed a global reconfiguration of the repressive histone methylation (H3K27me3), with a focal increase in the active enhancer mark (H3K27ac) associated with Notch signalling and quiescence108. In oestrogen receptor-positive breast cancer, KDM5 histone demethylase expression underpinned transcriptional heterogeneity and drug resistance102. Thus, a growing body of evidence points to the significance of epigenetic encoding of cell states in cancer.
Corrupted epigenetic identities enable tumour plasticity.
Epigenetic identities are faithfully propagated from the first transformed cell, allowing inference of cell of origin109–111 akin to the faithful propagation of the genetic information of the ancestral cancer cell. Nonetheless, the epigenome also parallels the genome in undergoing diversification in the growing malignant population. Thus, just as stochastic errors in the genome result in genetic intratumoural heterogeneity, stochastic aberrations in the epigenome generate epigenetic intratumoural diversity. This understanding has emerged from measurement of DNAme at the level of the single DNA fragment. These studies, based on bulk bisulfite sequencing, have shown thousands of loci that exhibit a ‘noisy’ stochastic pattern of DNAme changes, representing large heritable cell-to-cell variation in the epigenome of somatic tissues, both normal and cancerous112.
Building on these foundational studies112, the examination of primary cancer tissues revealed that tumour cells exhibit DNAme diversification in parallel to genetic evolution. Sampling of multiple regions within the primary tumour, metastases and pre-malignant outgrowths in prostatic adenocarcinomas revealed that lineage tracing via DNAme heterogeneity closely mirrored the phylogenetic relationships built based on copy number genetic diversity113. Similarly, in colorectal cancer models, DNAme analysis of single-cell-derived colon cancer organoids revealed a marked DNAme heterogeneity that was propagated stably, in parallel with genetic diversification62. Large-scale analysis of DNAme in primary CLL samples confirmed greater intratumoural DNAme heterogeneity than normal B cells and was predictive of adverse outcome114.
Multiple, coordinated epigenetic layers regulate gene expression and cell identity, and early data suggest that epigenetic diversification extends beyond the level of DNAme. For example, corrupted coordination between layers of the epigenome resulted in co-mapping to the same genomic loci of typically mutually exclusive activating and repressive histone modifications in CLL, likely reflecting cell-to-cell diversity in histone modification and associated with greater transcriptional diversity115.
Importantly, recent evidence supports the notion that selection may operate on epigenetic diversity, akin to the well-established genetic diversity model of clonal evolution116,117. In IDH-mutant gliomas, stochastic hypermethylation of the CTCF insulator protein-binding motifs resulted in the loss of insulation between enhancers and genes, which could then be hijacked as a mechanism for oncogenic gene activation116. Conversely, aberrantly restrictive states, through promoter hypermethylation or polycomb-mediated repression, can inhibit the induction of differentiation programmes, arresting the cancer cells at a proliferative state, as was shown in EZH2-mutated B cell lymphomas117–119. Finally, corrupted epigenetic encoding through the process of epimutation (for example, stochastic DNAme changes) can lower the barriers for transition between cell states117 (FIG. 5). This phenomenon may underlie the increased plasticity of malignancies, undermining differentiation hierarchies and enabling processes such as de-differentiation into stem-like states114,117. Such plastic differentiation topologies have been shown through mathematical modelling to amplify positive selection and therefore the evolutionary capacity of cancer120. Taken together, epigenetic information emerges as a central heritable encoding of critical cell states and may thus provide an additional field of operation for the evolutionary process of diversification and selection115 (TABLE 1).
Fig. 5 |. An integrative model of cancer progression.
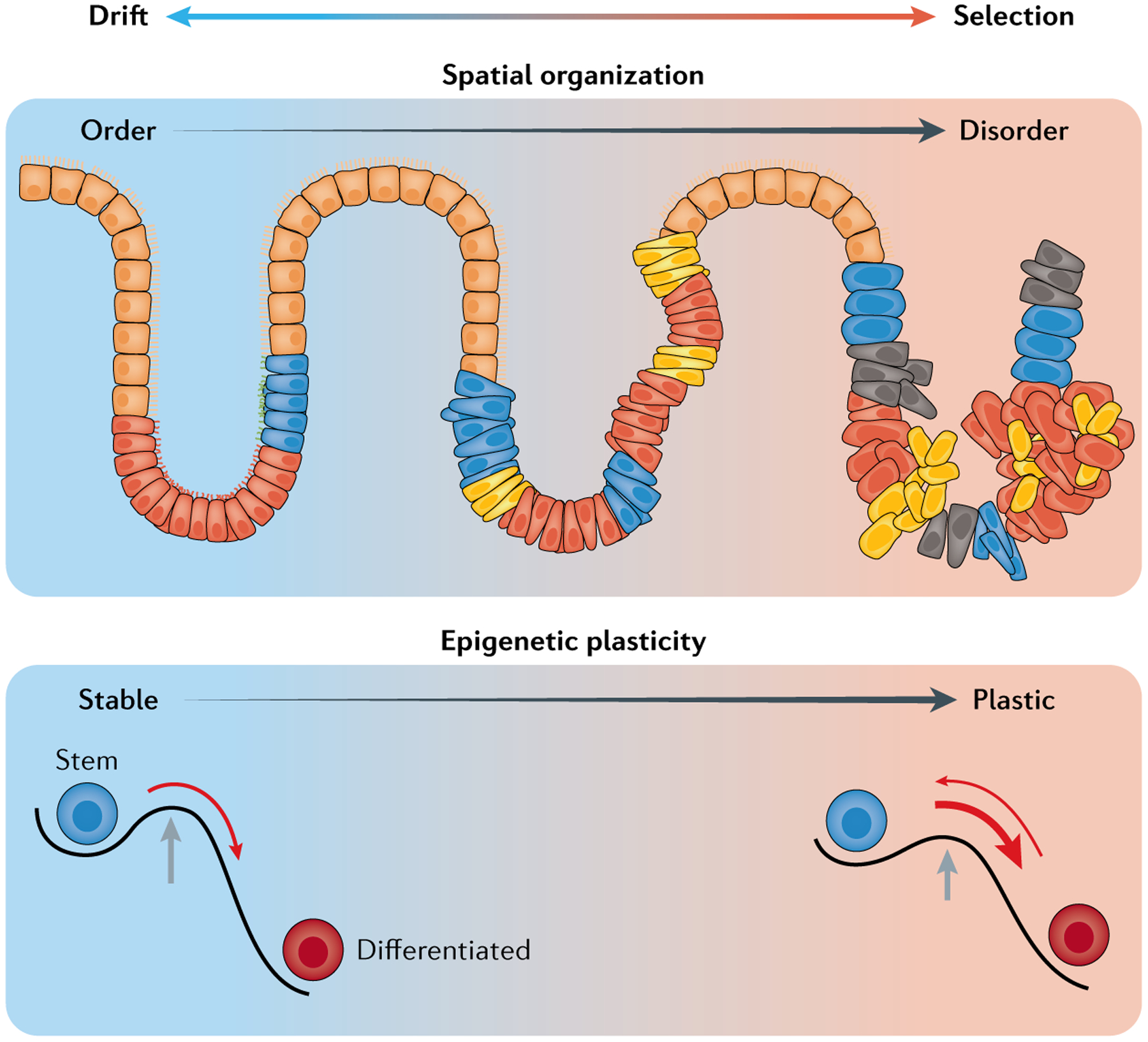
Single-cell multi-omics profiling of malignancies and clonal expansions in normal tissues may help unravel the underlying model through which rogue somatic evolutionary processes are suppressed in the multi cellular human host. Emerging data suggest that genetic constraints (that is, the time needed for the accumulation of multiple driver events)173 may need to act in concert with other mechanisms to suppress somatic evolution. One such mechanism may involve spatial constraints, as in the case of the colonic crypts that reduce the effective population size by splitting the overall population of colonic stem cells to isolated habitats, thereby favouring drift over selection36,174 (top panel). Greater spatial mixing with malignant progression can thus serve to amplify the selection and development of resistance to therapy142. Another such mechanism may be the complex differentiation hierarchies that suppress positive selection120. Thus, the patterns of de-differentiation related to the relaxation of epigenetic identity barriers in cancer may have the opposite effect, serving to amplify positive selection117,120 (bottom panel). Of note, these differentiation hierarchies may be encoded extrinsically in highly organized tissues, such as epithelial organs via cytokine gradients175, or intrinsically in less spatially defined tissues, such as the bone marrow, through complex, deep epigenetic hierarchies176.
Table 1 |.
The role of epigenetics in tumour evolution
| Epigenetic defects in cancer | Mechanism of tumour evolution | Examples |
|---|---|---|
| Disrupted epigenetic modifiers | Somatic mutations in epigenetic modifiers | DNMT3A, TET2 and AXL1 mutations in haematological malignancies105; SWI/SNF (BAF) mutations in solid tumours107 |
| Somatic mutations in epigenetic substrates | Histone H3F3A and HIST1H3B K27M mutations in paediatric high-grade gliomas177,178 | |
| Dysregulated expression of epigenetic modifiers | KDM5 overexpression linked with drug resistance in breast cancer102 | |
| Epigenetic diversification and/or plasticity | Changes in epimutation rate | Increased epimutation in CLL versus normal B cells121; AML subsets defined by epimutation rates179 |
| Suppression of differentiation | EZH2-activating mutations in DLBCL inhibit differentiation programmes118,119 | |
| Discoordination of epigenetic layers | Discoherence of DNA methylation and histone modifications in CLL115 | |
| Global hypomethylation linked with genomic instability | MMR-positive CRC displays de novo methylating defect whereas MMR-deficient CRC does not, suggesting two modes of chromosomal instability180 | |
| Positive selection of epigenetic disruption | Hypermethylation of CTCF insulator protein binding motifs results in loss of insulation between enhancers and genes116 |
AML, acute myeloid leukaemia; CLL, chronic lymphocytic leukaemia; CRC, colorectal cancer; DLBCL, diffuse large B cell lymphoma; MMR, mismatch repair.
Epimutation.
Heritable stochastic errors in epigenetic marks (best described in DNA methylation).
Multi-omics technology links genetic, epigenetic and transcriptional information in single cells.
Novel analytical and experimental methods are under development to capture the single-cell epigenome alongside cell state readouts such as transcriptional and protein expression phenotypes121. scRNA-seq with integration of protein expression122,123 and single-nuclei chromatin accessibility assays (ATAC-seq) have been performed in parallel on the same cancer samples (FIG. 1c), with analytic linking of transcriptional states with dysregulation of the regulatory networks as inferred from chromatin accessibility124. In mixed lineage biphenotypic leukaemias (with markers of both myeloid and lymphoid differentiation in the same leukaemia), mapping tumour cells to the map of normal haematopoietic differentiation with both mRNA and ATAC-seq data enabled causal linking of transcription factor activation with its downstream targets (for example, RUNX1 upregulating CD69)124. Further application of such analytically or experimentally joined dual high-throughput scRNA-seq and ATAC-seq protocols (for example, sci-CAR)125 to cancer specimens will likely help discern precise cis-regulatory sites and target genes and identify the key regulatory networks that govern clonal evolution.
The experimental extraction of DNAme and transcriptomic information from the same cells has been achieved by modifying plate-based scRNA-seq protocols, for example, Smart-seq2, in which both RNA and DNA are respectively isolated from the same cells for whole-transcriptome and DNAme data (through bisulfite sequencing)85,121,126–129. One method, scTrio-seq was applied to multiregional sampling of hepatocellular and colorectal carcinomas to retrace the genetic lineage histories of cancer cells in the context of transcriptional and methylation states126,127. In colorectal carcinomas, methylation profiles were closely linked with genetic lineages as defined by CNAs127. While transcriptional programmes were negatively correlated with the methylation of promoters, as expected, transcriptional states were not consistently linked with subclonal genetic identities.
Linking DNAme and whole-transcriptome data in single CLL cells revealed that the cell-to-cell methylation variation was closely associated with gene expression changes121. While the limitation of sparsity in single-cell genomics data also extends to single-cell bisulfite sequencing data (resulting in a capture rate of ~10% of the targeted methylome per cell), global changes in DNAme provide valuable insights into clonal evolution such as the ability to measure epimutation rates. Notably, heritable DNAme epimutations also serve as a molecular clock130–134 and can therefore be exploited as native barcodes to directly infer the high-resolution phylogenetic history of tumour cells in primary CLL samples (FIGS 1b,3). Further integration of somatic genotyping in this multi-omics procedure was applied to the subclonal SF3B1 mutation, which revealed that mutated cells congregated within one clade in the DNAme-based phylogenetic tree with distinct transcriptional output, providing orthogonal validation to tree inferences and enabling the estimation of nodal age at SF3B1 mutation acquisition121. Finally, the multi-omics projection of transcriptional information directly onto the lineage tree revealed the activation of distinct pathways in different clades with therapeutic exposure121. While the application of joint single-cell epigenetic multi-omics platforms135,136 to cancer is still nascent, these data highlight the promise of single-cell integrations of various heritable, yet plastic, dimensions of cancer cells for tracking — and ultimately predicting — clonal evolution.
Spatial dynamics and the microenvironment
Spatial dynamics as a heritable source of tumour heterogeneity.
Spatial localization represents another heritable dimension in tumour cell evolution associated with fitness, as tumour cells tend to co-localize with their parent cells in spatially constrained growth19,20,54,137. In certain cases, for example, cells at the leading edge of a solid tumour mass are in the active cell cycle (‘boundary-bound’ growth), pointing to spatial constraints as an important heritable trait of fitness related to metastatic potential and survival138,139. Recent data suggested that the spatial distribution of tumour cells is associated not only with fitness but also with clonal evolution140. The multiregional sampling of single glands (composed of <10,000 cells) within the same tumour further revealed a high degree of clonal mixing in colorectal carcinomas in striking contrast to pre-malignant adenomas of comparable size, which demonstrated segregation of clonal identities140 (FIG. 5). Consistent with these data, mathematical simulations identified the clonal mixing of tumour cells to be an early event of colorectal carcinomas141. Thus, analogous to the ongoing corruption of epigenetic profiles of cancer cells, the break-down of tissue architecture may be a critical feature of clonal evolution, as even minute cell dispersals increase tumour fitness and its ability to overcome therapeutic challenges142. Notably, while leukaemias, or more broadly blood cancers, are less strictly bound by spatial constraints, the disorganization of the bone marrow is a long-appreciated feature of myeloid neoplasms143 and, thus, the distortion of tissue architecture is a shared feature of progression between solid cancers and blood malignancies.
Spatial embedding of the evolutionary process also results in varying microenvironmental interactions for the growing malignant population. Across all cancer types, the tumour ecosystem, including the immune cells, endothelial cells and stromal cells, has been demonstrated to be a critical determinant of transformation, progression and response to therapy144–146. In a recent fascinating example, non-small-cell lung carcinomas were found to modulate neoantigen expression for immune escape through either promoter methylation or CNAs in a manner highly correlated with specific immune microenvironments147. Similarly, tumour-to-immune cell interactions were observed in multiregional sampling of high-grade ovarian carcinoma148, demonstrating that tumour-associated T cells negatively correlated with clonal heterogeneity, selecting for clones with immune escape mechanisms148. These investigations have shown that heritable evolutionary changes (at either the genome or epigenome level) vary within the same malignant population as a function of the local interactions with the non-malignant cellular neighbours. Specifically, these studies suggest that the microenvironment is exerting selective pressures on subclones and actively moulding cancer evolution.
The tumour microenvironment at single-cell resolution.
While the above-described bulk multi-omics analyses of primary cancer samples have provided strong support for the impact of the microenvironment on the evolutionary process, the cellular complexity of the microenvironment requires single-cell analysis to provide a high-resolution map of its interactions with the tumour. In head and neck squamous cell carcinomas, through scRNA-seq, interactions between tumour cells with a partial EMT programme and the stromal compartment were identified149. The complexity of the microenvironment has also been recently demonstrated with scRNA-seq, showing biases in the immune microenvironment that support immune evasion such as through expansion of T cells with exhaustion programmes76. Indeed, single-cell omics profiling has been transformative for the fields of immunology and immuno-oncology150,151; however, an in-depth discussion of immune cell biology is beyond the scope of this review and has been reviewed recently elsewhere150,151.
Nonetheless, as scRNA-seq is typically performed on fully dissociated tissue, it does not preserve the architecture of the tumour cells and microenvironment. Cell-to-cell interactions may be nonetheless inferred from scRNA-seq by correlating the expression of known ligands and receptors152,153. While analytic tools have also been developed to predict the spatial arrangement of highly structured tissue architectures154,155, highly disordered cancer tissues may prove more challenging for inferences based on scRNA-seq of dissociated samples.
Examining tumour microenvironments with spatially aware single-cell technologies.
Spatially aware single-cell platforms may thus be particularly transformative in the study of the cancer microenvironment. For protein detection, multiplexed labelling with metal isotopes (for example, imaging mass cytometry (IMC)156 and multiplexed ion beam imaging (MIBI)157) or fluorophores (for example, multiplex immunofluorescence (MxIF)158 and cyclic immunofluorescence (CycIF)159) enable the detection of dozens of markers at cellular or subcellular resolution. The application of these methods to cancer samples revealed significant intratumoural heterogeneity, including spatially dependent diverging signalling pathways158. The detection of mRNA with immunofluorescence (for example, multiplexed error robust-fluorescence in situ hybridization (MERFISH)160 and sequential FISH (seqFISH)161,162) and in situ RNA sequencing of amplified cDNA (FISSEQ and STARmap)163,164 have increased the targets to hundreds of genes. Spatial transcriptomic platforms that utilize molecular barcodes for the detection of mRNA molecules have drastically increased the dimensionality165,166 (FIG. 1e). These data can be analytically integrated with high-throughput scRNA-seq platforms to inform each dataset, whereby the dissociated cells from the scRNA-seq data may be remapped to their spatial positioning based on spatially patterned gene expressions167,168. The multiplexing of protein and gene expression was achieved through extension of the IMC platform by integration of in situ hybridization of targeted mRNA molecules such that RNA to protein expression correlations could be determined169. For example, in breast cancer, HER2 gene and protein expressions were highly correlated, whereas CK19 showed poor correlation between protein and corresponding gene levels. The same method identified the T cell-recruiting cytokine CXCL10 as expressed in tumour cells associated with T cells, providing insights into the tumour–immune cell interface. Computational tools have been developed in parallel to quantify contributions of gene expression or protein expression from cell-intrinsic versus environmental factors170, with respect to both spatial positioning171 and cell-to-cell interactions172. Application of these spatially aware platforms, integrated with high-throughput single-cell multi-omics, promises to enhance our understanding of the cell-to-cell interactions and spatial constraints that enable the fitness optimization and evolution of cancer cells.
Conclusions and perspectives
Cancer evolution encompasses a complex interplay of genetic, cell state, epigenetic, spatial and microenvironmental factors. Recently, novel multi-omics technologies have begun to integrate across these genetic and non-genetic determinants of tumour evolution at the critical resolution of the single cell — the fundamental evolutionary unit.
These methods pave the way to address central questions on cancer evolution through the study of human tissue. For example, to define the factors that drive the malignant transformation versus involution of clonally expanded cells requires the ability to genotype cells for driver mutations as well as to gather information about their transcriptional and epigenetic states. Another open field of investigation in human malignancies is the assessment of lineage fate decisions of cancer stem cells that culminate from the interactions of somatic mutations with cell states. These cell states may in turn be determined by intrinsic epigenetic underpinnings and/or extrinsic cues from the environment, emphasizing the need for multi-omics single-cell data integration across modalities.
As a final perspective, single-cell multi-omics profiling of malignancies and clonal expansions in normal tissues may help unravel the underlying model through which rogue somatic evolutionary processes are suppressed in the multicellular human host (FIG. 5). Emerging data that show the pervasiveness of somatic driver mutations in normal tissue4–6,94 suggest that genetic constraints (that is, the time needed for the accumulation of multiple driver events)173 may need to act in concert with other mechanisms to suppress somatic evolution. One such mechanism may involve spatial constraints, as in the case of the colonic crypts that reduce the effective population size by splitting the overall population of colonic stem cells to isolated habitats, thereby favouring drift over selection36,174. Greater spatial mixing with malignant progression can thus serve to amplify selection and the development of resistance to therapy142. Another such mechanism may be the complex differentiation hierarchies; this notion is supported by mathematical modelling of evolutionary dynamics, showing that evolutionary graphs that reflect differentiation hierarchies have an organization that suppresses positive selection120. By contrast, the patterns of de-differentiation related to the relaxation of epigenetic identity barriers in cancer may have the opposite effect, leading to evolutionary graphs that serve to amplify positive selection117,120. Of note, these differentiation hierarchies may be encoded extrinsically in highly organized tissues, such as epithelial organs via cytokine gradients175, or intrinsically in less spatially defined tissues, such as the bone marrow, through complex, deep epigenetic hierarchies176. As clonal outgrowths represent a first critical step towards circumventing this barrier to somatic evolution, they tend to affect cytokine-related mechanisms in epithelial tissues (for example, mutations related to Notch and WNT signalling) and epigenetic mechanisms in haematopoietic tissue (for example, DNAme modifier mutations). Growing clonal populations can then serve as a superior substrate for an effective evolutionary process, ultimately leading to the selection of malignant clones.
In other words, we envision that the application of single-cell multi-omics to somatically evolving human tissue will provide critical clues as to the basic system properties that discourage the trillions of somatic cells from rescinding the multi-cellular contract and optimizing their fitness at the expense of the host. Such integrative analysis, empowered by the simultaneous interrogation of the multi-faceted axes of diversity that fuel somatic evolution, may thus advance this exciting new frontier in evolutionary biology at the intersection between multi-cellular species evolution and asexual reproduction of somatic cells more akin to unicellular lifeforms.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Greaves M & Maley CC Clonal evolution in cancer. Nature 481, 306–313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duffy TP Portraits of an illness. Trans. Am. Clin. Climatol. Assoc 120, 209–225 (2009). [PMC free article] [PubMed] [Google Scholar]
- 3.Turajlic S, Sottoriva A, Graham T & Swanton C Resolving genetic heterogeneity in cancer. Nat. Rev. Genet 20, 404–416 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Martincorena I et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yizhak K et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364, eaaw0726 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yokoyama A et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Martincorena I et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated the ubiquitous nature and positive selection of somatic mutations in cancer driver genes in normal skin tissue.
- 8.Yoshida K et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teixeira VH et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat. Med 25, 517–525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaufman CK et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 351, aad2197 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work showed that somatic drivers of melanoma, when superimposed on a progenitor cell state, induced malignant melanoma, highlighting the significance of cell state for tumorigenesis.
- 11.Shaffer SM et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified a transient transcriptional state in melanoma cells that leads to stable drug resistance, underscoring cell state heterogeneity as a key mediator of tumour evolution.
- 12.Hata AN et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med 22, 262–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stuart T & Satija R Integrative single-cell analysis. Nat. Rev. Genet 20, 257–272 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Landau DA et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerstung M et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papaemmanuil E et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med 374, 2209–2221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landau DA et al. Mutations driving CLL and their evolution in progression and relapse. Nature 526, 525–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turajlic S et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx renal. Cell 173, 581–594.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turajlic S et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell 173, 595–610.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jamal-Hanjani M et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Gruber M et al. Growth dynamics in naturally progressing chronic lymphocytic leukaemia. Nature 570, 474–479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrated the ability to define patterns of subclonal growth rates of CLL through dense temporal sequencing.
- 22.Leshchiner I et al. Comprehensive analysis of tumour initiation, spatial and temporal progression under multiple lines of treatment. Preprint at bioRxiv 10.1101/508127 (2018). [DOI] [Google Scholar]
- 23.Burger JA et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat. Commun 7, 11589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landau DA et al. The evolutionary landscape of chronic lymphocytic leukemia treated with ibrutinib targeted therapy. Nat. Commun 8, 2185 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siravegna G et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med 21, 795–801 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abbosh C et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446–451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolan PO et al. Genotype-fitness maps of EGFR-mutant lung adenocarcinoma chart the evolutionary landscape of resistance for combination therapy optimization. Cell Syst. 10, 52–65.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wedge DC et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet 50, 682–692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shih DJH et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat. Genet 52, 371–377 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsao JL et al. Genetic reconstruction of individual colorectal tumor histories. Proc. Natl Acad. Sci. USA 97, 1236–1241 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsao JL, Davis SD, Baker SM, Liskay RM & Shibata D Intestinal stem cell division and genetic diversity. A computer and experimental analysis. Am. J. Pathol 151, 573–579 (1997). [PMC free article] [PubMed] [Google Scholar]
- 32.Naxerova K et al. Hypermutable DNA chronicles the evolution of human colon cancer. Proc. Natl Acad. Sci. USA 111, E1889–E1898 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reiter JG et al. Lymph node metastases develop through a wider evolutionary bottleneck than distant metastases. Nat. Genet 52, 692–700 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerlinger M et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med 366, 883–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El-Kebir M, Satas G & Raphael BJ Inferring parsimonious migration histories for metastatic cancers. Nat. Genet 50, 718–726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snippert HJ et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Sutherland KD et al. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19, 754–764 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Quintana E et al. Efficient tumour formation by single human melanoma cells. Nature 456, 593–598 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhang HE et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med 21, 440–448 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Eirew P et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518, 422–426 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen LV et al. DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nat. Commun 5, 5871 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hwang B et al. Lineage tracing using a Cas9-deaminase barcoding system targeting endogenous L1 elements. Nat. Commun 10, 1234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frieda KL et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spanjaard B et al. Simultaneous lineage tracing and cell-type identification using CRISPR-Cas9-induced genetic scars. Nat. Biotechnol 36, 469–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raj B et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol 36, 442–450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKenna A et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alemany A, Florescu M, Baron CS, Peterson-Maduro J & van Oudenaarden A Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Kalhor R et al. Developmental barcoding of whole mouse via homing CRISPR. Science 361, eaat9804 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woodworth MB, Girskis KM & Walsh CA Building a lineage from single cells: genetic techniques for cell lineage tracking. Nat. Rev. Genet 18, 230–244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kester L & van Oudenaarden A Single-cell transcriptomics meets lineage tracing. Cell Stem Cell 23, 166–179 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Baron CS & van Oudenaarden A Unravelling cellular relationships during development and regeneration using genetic lineage tracing. Nat. Rev. Mol. Cell Biol 20, 753–765 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Pellegrino M et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 28, 1345–1352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith MA et al. E-scape: interactive visualization of single-cell phylogenetics and cancer evolution. Nat. Methods 14, 549–550 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Navin N et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim C et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 173, 879–893.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laks E et al. Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing. Cell 179, 1207–1221.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuipers J, Jahn K, Raphael BJ & Beerenwinkel N Single-cell sequencing data reveal widespread recurrence and loss of mutational hits in the life histories of tumors. Genome Res. 27, 1885–1894 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zafar H, Tzen A, Navin N, Chen K & Nakhleh L SiFit: inferring tumor trees from single-cell sequencing data under finite-sites models. Genome Biol. 18, 178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwartz R & Schaffer AA The evolution of tumour phylogenetics: principles and practice. Nat. Rev. Genet 18, 213–229 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jahn K, Kuipers J & Beerenwinkel N Tree inference for single-cell data. Genome Biol. 17, 86 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davis A & Navin NE Computing tumor trees from single cells. Genome Biol. 17, 113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roerink SF et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556, 457–462 (2018). [DOI] [PubMed] [Google Scholar]; This study utilized clonal organoids of colorectal carcinoma to chart mutational and DNAme lineage trees within tumours.
- 63.Lee-Six H et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nieto MA, Huang RY, Jackson RA & Thiery JP Emt: 2016. Cell 166, 21–45 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Pastushenko I et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018). [DOI] [PubMed] [Google Scholar]
- 66.McKenzie MD et al. Interconversion between tumorigenic and differentiated states in acute myeloid leukemia. Cell Stem Cell 25, 258–272.e9 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Bonnet D & Dick JE Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 3, 730–737 (1997). [DOI] [PubMed] [Google Scholar]
- 68.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrated distinct stem-like and differentiated cell states within oligodendrogliomas through scRNA-seq.
- 69.de Sousa e Melo F et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 543, 676–680 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Shimokawa M et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 545, 187–192 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Kreso A & Dick JE Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Bell CC et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun 10, 2723 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beltran H et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res 25, 6916–6924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oser MG, Niederst MJ, Sequist LV & Engelman JA Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 16, e165–e172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fabbri G et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J. Exp. Med 210, 2273–2288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Neftel C et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Batlle E & Clevers H Cancer stem cells revisited. Nat. Med 23, 1124–1134 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Ledergor G et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat. Med 24, 1867–1876 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han KY et al. SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res. 28, 75–87 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yin Y et al. High-throughput single-cell sequencing with linear amplification. Mol. Cell 76, 676–690.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Macaulay IC et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 12, 519–522 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Fan J et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 28, 1217–1227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Giustacchini A et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med 23, 692–702 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Rodriguez-Meira A et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol. Cell 73, 1292–1305.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a novel method to capture highly sensitive somatic genotyping in plate-based scRNA-seq by targeted amplification of both genomic DNA and cDNA.
- 90.Petti AA et al. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat. Commun 10, 3660 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Galen P et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell 176, 1265–1281.e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nam AS et al. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature 571, 355–360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrated that the transcriptional impact of somatic mutations in myeloid neoplasms varies as a function of cell state by developing a method for highly sensitive somatic genotyping in high-throughput scRNA-seq.
- 93.Izzo F et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet 52, 378–387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Desai P et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med 24, 1015–1023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jaiswal S, Natarajan P & Ebert BL Clonal hematopoiesis and atherosclerosis. N. Engl. J. Med 377, 1401–1402 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Ludwig LS et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176, 1325–1339.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work performed single-cell lineage tracing in human samples by interrogating mitochondrial DNA mutations within single-cell chromatin accessibility and RNA-seq data.
- 98.Stewart JB & Chinnery PF The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet 16, 530–542 (2015). [DOI] [PubMed] [Google Scholar]
- 99.Xu J et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. eLife 8, e45105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shlush LI et al. Cell lineage analysis of acute leukemia relapse uncovers the role of replication-rate heterogeneity and microsatellite instability. Blood 120, 603–612 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Latil M et al. Cell-type-specific chromatin states differentially prime squamous cell carcinoma tumor-initiating cells for epithelial to mesenchymal transition. Cell Stem Cell 20, 191–204.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hinohara K et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell 34, 939–953.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu VWC et al. Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell 167, 1310–1322.e17 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Catania S et al. Evolutionary persistence of DNA methylation for millions of years after ancient loss of a de novo methyltransferase. Cell 180, 263–277.e20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lappalainen T & Greally JM Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet 18, 441–451 (2017). [DOI] [PubMed] [Google Scholar]
- 106.Cancer Genome Atlas Research Network. et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.St Pierre R & Kadoch C Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr. Opin. Genet. Dev 42, 56–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liau BB et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 20, 233–246.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Beekman R et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat. Med 24, 868–880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin-Subero JI & Oakes CC Charting the dynamic epigenome during B-cell development. Semin. Cancer Biol 51, 139–148 (2018). [DOI] [PubMed] [Google Scholar]
- 111.Capper D et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Landan G et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet 44, 1207–1214 (2012). [DOI] [PubMed] [Google Scholar]
- 113.Brocks D et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 8, 798–806 (2014). [DOI] [PubMed] [Google Scholar]
- 114.Landau DA et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 26, 813–825 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pastore A et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat. Commun 10, 1874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Flavahan WA et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Flavahan WA, Gaskell E & Bernstein BE Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McCabe MT et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012). [DOI] [PubMed] [Google Scholar]
- 119.Beguelin W et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lieberman E, Hauert C & Nowak MA Evolutionary dynamics on graphs. Nature 433, 312–316 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Gaiti F et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 569, 576–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study performed single-cell multi-omics to capture whole transcriptomic, DNAme and somatic genotyping information from the same cells and performed lineage tracing through DNAme data onto which genetic and transcriptional identities were overlayed.
- 122.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group developed a method to capture targeted protein expression levels within droplet-based scRNA-seq platforms.
- 123.Peterson VM et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol 35, 936–939 (2017). [DOI] [PubMed] [Google Scholar]
- 124.Granja JM et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol 37, 1458–1465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hou Y et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bian S et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 362, 1060–1063 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Clark SJ et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun 9, 781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Angermueller C et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13, 229–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yatabe Y, Tavare S & Shibata D Investigating stem cells in human colon by using methylation patterns. Proc. Natl Acad. Sci. USA 98, 10839–10844 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shibata D Inferring human stem cell behaviour from epigenetic drift. J. Pathol 217, 199–205 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Siegmund KD, Marjoram P, Tavare S & Shibata D Many colorectal cancers are “flat” clonal expansions. Cell Cycle 8, 2187–2193 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shibata D Mutation and epigenetic molecular clocks in cancer. Carcinogenesis 32, 123–128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sottoriva A, Spiteri I, Shibata D, Curtis C & Tavare S Single-molecule genomic data delineate patient-specific tumor profiles and cancer stem cell organization. Cancer Res. 73, 41–49 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pott S Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife 6, e23203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Guo F et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 27, 967–988 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sottoriva A et al. A big bang model of human colorectal tumor growth. Nat. Genet 47, 209–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gong P, Wang Y, Liu G, Zhang J & Wang Z New insight into Ki67 expression at the invasive front in breast cancer. PLoS ONE 8, e54912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chkhaidze K et al. Spatially constrained tumour growth affects the patterns of clonal selection and neutral drift in cancer genomic data. PLoS Comput. Biol 15, e1007243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sun R et al. Between-region genetic divergence reflects the mode and tempo of tumor evolution. Nat. Genet 49, 1015–1024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ryser MD, Min BH, Siegmund KD & Shibata D Spatial mutation patterns as markers of early colorectal tumor cell mobility. Proc. Natl Acad. Sci. USA 115, 5774–5779 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Waclaw B et al. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 525, 261–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tricot G, De Wolf-Peeters C, Vlietinck R & Verwilghen RL Bone marrow histology in myelodysplastic syndromes. II. Prognostic value of abnormal localization of immature precursors in MDS. Br. J. Haematol 58, 217–225 (1984). [DOI] [PubMed] [Google Scholar]
- 144.Thorsson V et al. The immune landscape of cancer. Immunity 48, 812–830.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dong L et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 539, 304–308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kode A et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 506, 240–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rosenthal R et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang AW et al. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell 173, 1755–1769.e22 (2018). [DOI] [PubMed] [Google Scholar]
- 149.Puram SV et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen H, Ye F & Guo G Revolutionizing immunology with single-cell RNA sequencing. Cell Mol. Immunol 16, 242–249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Finotello F, Rieder D, Hackl H & Trajanoski Z Next-generation computational tools for interrogating cancer immunity. Nat. Rev. Genet 20, 724–746 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Vento-Tormo R et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kumar MP et al. Analysis of single-cell RNA-seq identifies cell-cell communication associated with tumor characteristics. Cell Rep. 25, 1458–1468.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nitzan M, Karaiskos N, Friedman N & Rajewsky N Gene expression cartography. Nature 576, 132–137 (2019). [DOI] [PubMed] [Google Scholar]; This work inferred spatial mapping of single-cells through scRNA-seq data obtained from dissociated cells.
- 155.Edsgard D, Johnsson P & Sandberg R Identification of spatial expression trends in single-cell gene expression data. Nat. Methods 15, 339–342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 157.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gerdes MJ et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl Acad. Sci. USA 110, 11982–11987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lin JR, Fallahi-Sichani M & Sorger PK Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun 6, 8390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shah S, Lubeck E, Zhou W & Cai L seqFISH accurately detects transcripts in single cells and reveals robust spatial organization in the hippocampus. Neuron 94, 752–758.e1 (2017). [DOI] [PubMed] [Google Scholar]
- 162.Eng C-HL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lee JH et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang X et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stahl PL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 166.Rodriques SG et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presented the Seurat algorithm for computationally mapping spatial locations of single cells from scRNA-seq derived from dissociated zebra embryos by integrating in situ hybridization data for landmark genes.
- 168.Achim K et al. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol 33, 503–509 (2015). [DOI] [PubMed] [Google Scholar]
- 169.Schulz D et al. Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. Cell Syst. 6, 25–36.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Svensson V, Teichmann SA & Stegle O SpatialDE: identification of spatially variable genes. Nat. Methods 15, 343–346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Schapiro D et al. histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods 14, 873–876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Arnol D, Schapiro D, Bodenmiller B, Saez-Rodriguez J & Stegle O Modeling cell-cell interactions from spatial molecular data with spatial variance component analysis. Cell Rep. 29, 202–211. e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tomasetti C, Marchionni L, Nowak MA, Parmigiani G & Vogelstein B Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl Acad. Sci. USA 112, 118–123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lopez-Garcia C, Klein AM, Simons BD & Winton DJ Intestinal stem cell replacement follows a pattern of neutral drift. Science 330, 822–825 (2010). [DOI] [PubMed] [Google Scholar]
- 175.Karin M & Clevers H Reparative inflammation takes charge of tissue regeneration. Nature 529, 307–315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cedar H & Bergman Y Epigenetics of haematopoietic cell development. Nat. Rev. Immunol 11, 478–488 (2011). [DOI] [PubMed] [Google Scholar]
- 177.Schwartzentruber J et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 178.Wu G et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet 44, 251–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li S et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med 22, 792–799 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lengauer C, Kinzler KW & Vogelstein B DNA methylation and genetic instability in colorectal cancer cells. Proc. Natl Acad. Sci. USA 94, 2545–2550 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]