

Abstract
Purpose:
Mutations in the eyes shut homolog (EYS) gene are a frequent cause of autosomal recessive retinitis pigmentosa (arRP). This study used multi-modal retinal imaging to elucidate genotype-phenotype relationships in EYS-related RP (EYS-RP).
Design:
Cross-sectional study.
Method:
Multimodal retinal imaging and electrophysiologic testing was assessed for 16 patients with genetic confirmation of EYS-RP.
Results:
A total of 27 unique EYS variants were identified in 16 patients. Seven patients presented with an unusual crescent-shaped hyperautofluorescent (hyperAF) ring on fundus autofluorescence (FAF) imaging encompassing a large nasal-superior area of the posterior pole. Three patients had a typical circular or oval perifoveal hyperAF ring and six patients had no hyperAF ring. Spectral domain optical coherence tomography (SD-OCT) and en face OCT showed preserved ellipsoid zone and retinal thickness spatially corresponding to areas within the hyperAF rings. Eleven patients presented with a rod-cone dystrophy on full-field electroretinogram (ffERG), one patient presented with cone-rod dystrophy, and four patients did not undergo ERG testing. A significant spatial association was found between EYS variant position and autofluorescent phenotype, with variants occurring at a nucleotide position greater than GRch37 6:65300137 (c.5617C) being more associated with patients exhibiting autofluorescent rings at presentation.
Conclusions:
EYS-RP is a heterogeneous manifestation. Variants occurring in positions closer to the C-terminus of EYS are more common in patients presenting with autofluorescent rings on FAF imaging.
Keywords: Retinitis Pigmentosa, EYS, Autofluorescence
Introduction
Retinitis pigmentosa (RP) is an inherited and irreversible retinal degeneration that affects approximately 1 in 4,000 individuals in its nonsyndromic form.1–3 Over 70 mutated genes have been identified to cause nonsyndromic RP which includes several modes of inheritance, illustrating its genetic heterogeneity (https://sph.uth.edu/RETNET, accessed March 2018). RP is most commonly inherited in an autosomal recessive manner (arRP),3 of which, biallelic mutations in the eyes shut homolog (EYS) gene (OMIM 612424) is a known cause.4–6 Previous studies of arRP found EYS mutations in 5–33% of cases and amongst various ethnicities, suggesting a global presence.5, 7–16 The highest prevalence exists in the Japanese population, in which EYS mutations are estimated to be the most common cause of inherited retinal degenerations.17
Homozygosity mapping and positional cloning with comparative genomics led to the identification of EYS mutations as a cause of arRP.4, 6 Spanning 2 Mb of genomic DNA, EYS is the largest known retina-specific gene and encodes a product 3,165 amino acids in length.4, 18 EYS has been characterized most in Drosophila, in which the homologue is named spacemaker, or spam, which encodes a protein that plays a role in light-sensitive rhabdomere assembly.19 In humans, photoreceptor outer segments are the structural equivalent to insect light-sensitive rhabdomeres. Corroborating these findings is recent evidence that localized EYS to the outer segments of post-mortem human retina which suggests an important role in formation and structural integrity of photoreceptor architecture, specifically ciliary axoneme stability in rods and cones.20 Ablation of EYS with TALEN technology in zebrafish causes outer segment protein mislocalization and F-actin disruption, interestingly producing a cone-rod-like dystrophy.21
EYS appears to be relatively conserved across mammals, but deleted in many species including little brown bats, armadillos, sheep, and cattle. Smaller species such as rodents and guinea pigs have accumulated reading-frame disruptions and expression studies in mice suggest an absence of protein product.4 Thus, given the lack of animal models, studying disease phenotype in humans is increasingly important for understanding disease presentation and progression. To date, studies discerning clinical characteristics and retinal phenotype of EYS-associated RP (EYS-RP) patients are few in number,15, 22–25. In this cross-sectional study, we identify a heterogeneous phenotype on short wavelength fundus autofluorescence (FAF) and describe genetic correlates in a large cohort of EYS-RP patients.
Methods
Subjects.
This cross-sectional study was approved by the Edward S. Harkness Eye Institute and Columbia University Internal Review Boards (IRB), and adhered to the tenets of the Declaration of Helsinki. All data, imaging, and genetic information presented in this study are in accordance with HIPAA and not identifiable to individual patients. Clinical information, FAF imaging, spectral-domain (SD) and en face optical coherence tomography (OCT), and full-field electroretinogram (ffERGs) of sixteen unrelated patients seen between 2010 and 2017 at the Harkness Eye Institute electrophysiology clinic were retrospectively studied. All patients harbored at least two EYS variants. Patients with only one EYS variant were not included.
Genetic analyses.
For each patient, peripheral whole blood lymphocytes were collected and DNA was subsequently isolated for sequencing. Eight patients underwent clinical laboratory improved amendments (CLIA)-approved whole exome sequencing with Sanger sequencing confirmation at the Center of Personalized Genomic Medicine (PGM) of Columbia University Medical Center (CUMC) (New York, NY). Whole exome sequencing for P6 was performed in the laboratory of Dr. Rando Allikmets at CUMC (New York, NY). One patient received next generation sequencing of 31 genes known to cause inherited retinal dystrophies from Prevention Genetics (Marshfield, WI). The remaining patients received next generation sequencing of candidate genes known to cause inherited retinal dystrophies at the Casey Eye Institute Molecular Diagnostics Laboratory (Portland, OR). All identified variants were analyzed using Alamut software version 2.2 (Interactive Biosoftware, Rouen, France). Functional annotation of variants was carried out with ANNOVAR using pathogenicity scores of M-CAP, REVEL, Eigen, CADD, DANN, and SPIDEX.26–32 As a general guideline, pathogenic consequences are predicted for variants with scores over 0.025 for M-CAP, 0.5 for REVEL, 0.5 for Eigen, 20 for CADD, 0.97 for DANN, and more than 2 or less than −2 for SPIDEX psi z-score.
Retinal Imaging.
Following dilation, FAF and SD-OCT images were taken using a Spectralis HRA+OCT device (Heidelberg Engineering, Heidelberg, Germany). FAF images were taken for all patients in a 30-degree field with 1536 × 1536 pixel resolution, and a stimulus and barrier filter of 486nm and 521nm, respectively. For patients in which a hyperautofluorescent ring extended beyond the 30-degree field, a 55-degree field was acquired during follow-up with a wide-angle lens. SD-OCT scans were acquired using real-time registration of an infrared reflectance image in conjunction with an 870nm light source. All patients received high resolution scans of the fovea (horizontal cut, 9mm, ART, average of a minimum of 50 images). En face OCT of the central macula (6mm × 6mm) were acquired at least once for seven patients during follow-up (Zeiss AngioPlex OCT Angiography; Dublin, CA).
Full-field Electroretinogram (ffERG).
Twelve of 16 patients underwent ffERG testing for both eyes using a Diagnosys Espion Electrophysiology system (Diagnosys LLC, Lowell, MA, USA). Recordings were performed using Dawson, Trick, Lawson (DTL)-recording electrodes in accordance with the International Society for Clinical and Electrophysiology of Vision (ISCEV) standards in scotopic and photopic states.33, 34 In patients with 30 Hz-flicker ffERG recordings that were less than 5μV or predicted to be based on dilated fundus exam, Burian-Allen (BA) contact lens electrodes were used instead to measure this response. The recorded signal is subsequently passed through narrow band-passed filtering and undergoes computed averaging.35–37
Results
Clinical data.
A total of 16 unrelated patients with two or more expected pathogenic mutations in EYS were included in this analysis. Fifteen patients were diagnosed with arRP based on symptoms, family history, ocular exam, retinal imaging, and/or a rod-cone sequence of degeneration on ffERG (Fig. 1). One patient was diagnosed with cone-rod dystrophy based on presenting symptoms and ffERG (P7). Demographic and genetic information are presented in Table 1. Clinical characteristics of patients in this study are shown in Table 2. The average age at presentation was 51 years (range, 28 to 83), affecting 12 males and 4 females. Best-corrected visual acuity ranged from 20/20 to light perception. Refractive error was recorded for 20 eyes, of which 17 were myopic and three hyperopic. Six patients had a history of prior cataract extraction and seven additional patients showed evidence of opacification of at least one lens. Intraretinal pigment migration was observed in 14 of 16 cases.
Figure 1. EYS-associated Retinitis Pigmentosa.

Digital color fundus photos of the right and left macula (top row) show attenuated arterioles, pale discs, and sparse intraretinal pigment migration (arrows) in a patient with EYS-RP (P8). Short wavelength FAF of both eyes (second row) reveals the typical phenotype of RP with a circular perifoveal hyperautofluorescent ring (arrows) and peripheral RPE atrophy. Note the ring is symmetric and round. High resolution SD-OCT imaging through the right (third row) and left (fourth row) fovea shows characteristic peripheral thinning of the outer retinal laminae and shortening of the ellipsoid zone line which is spared in the central macula. EYS, eyes shut homolog; RP, retinitis pigmentosa; RPE, retinal pigment epithelium; FAF, fundus autofluorescence.
Table 1. Genotype of EYS Patients.
Age, gender, and identified EYS variants for all patients included in this cross-sectional study.
| ID | Age | Gender | Genotype | |
|---|---|---|---|---|
| P1 | 58 | M | EYS | c.9383_9387delAATTA (p.Lys3128Serfs*5) c.6571+5G>A (p.?) |
| P2 | 46 | M | EYS | c.8012T>A (p.Leu2671*)† c.6416G>A (p.Cys2139Tyr)† c.1641_1644delTCAG (p.Ser547Argfs*62) |
| P3 | 66 | M | EYS | c.1155T>A (p.Cys385*) homozygous c.8648_8655delCATGCAGA (p.Thr2883Lysfs*4) |
| P4 | 33 | F | EYS | c.3555C>G (p.Cys1185Trp) c.8111T>G (p.Leu2704*) |
| P5 | 83 | M | EYS | c.9286_9295delGTAAATATCG (p.Val3096Leufs*28) homozygous |
| P6 | 44 | M | EYS | c.8413dupA (p.Thr2805Asnfs) c.3443+1G>T (p.?) |
| P7 | 65 | M | EYS | c.8111T>G (p.Leu2704*) c.9317_9336del20 (p.Thr3106Lysfs*13) |
| P8 | 37 | M | EYS | c.1961dupA (p.Asn654Lysfs*5) c.5645–1197-c.5927+3169 |
| P9 | 61 | F | EYS | c.6714delT (p.Ile2239Serfs*17) c.9299_9302delCTCA (p.Thr3100Lysfs*26) |
| P10 | 28 | M | EYS | c.7578+1G>A (p.?) homozygous |
| P11 | 58 | M | EYS | c.1645G>T (p.Glu549*) c.2992+1G>A (p.?) |
| P12 | 54 | M | EYS | c.6794delC (p.Pro2265Glnfs*46) homozygous |
| P13 | 30 | F | EYS | c.3443+1G>T (p.?) homozygous c.3250A>C (p.Thr1084Pro) homozygous c.4402G>C (p.Asp1468His) homozygous |
| P14 | 45 | F | EYS | c.963_979delAAAAGGATCTTCCAGCC (p.Pro321Profs) c.6794delC (p.Pro2265Glnfs*46) |
| P15 | 70 | M | EYS | c.1308C>A (p.Cys436*) c.5928delG (p.Arg1976Serfs*11) |
| P16 | 30 | M | EYS | c.4120C>T (p.Arg1374*) homozygous |
Footnote:
, in cis
Table 2. Clinical Characteristics.
Patient demographics and ocular exam findings for 16 EYS-RP patients.
| ID | Age | Sex | Age at Diagnosis | Visual Acuity (OD, OS) | Refraction (OD/OS) | Lens (OD/OS) | Pigment | CME | |
|---|---|---|---|---|---|---|---|---|---|
| Crescent Ring | P1 | 58 | M | 50 | 20/70, 20/60 | +2.75, −1.50 × 86 +1.75, −1.00 × 119 |
−/− | + | + |
| P2 | 46 | M | 46 | 20/20, 20/25 | −1.50, −1.75 × 175 −0.75, −2.25 × 002 |
−/− | − | − | |
| P3 | 66 | M | 65 | 20/20, 20/800 | NR | PCIOL OU | + | − | |
| P4 | 33 | F | 31 | 20/20, 20/20 | −3.75, −1.50 × 11 −3.50, −1.50 × 9 |
−/− | + | − | |
| P5 | 83 | M | 65 | 20/100, 20/80 | −4.50, −1.50 × 75 −5.25, −0.75 × 75 |
ACIOL OU | + | − | |
| P6 | 44 | M | NR | 20/125, 20/100 | −1.25, −0.75 × 80 −1.25, −0.75 × 80 |
NSC/− | + | − | |
| P7 | 65 | M | 65 | 20/50, 20/60 | −3.50, −1.00 × 174 −2.75, −0.75 × 4 |
NSC OU | − | − | |
| Typical Ring | P8 | 37 | M | 30 | 20/20, 20/20 | −3.75, +1.75 × 112 −3.25, +1.00 × 86 |
PSC/PSC | + | − |
| P9 | 61 | F | NR | 20/50, 20/25 | NR | PCIOL OU | + | + | |
| P10 | 28 | M | 27 | 20/20, 20/25 | NR | PSC/− | + | − | |
| No Ring | P11 | 58 | M | 45 | 20/20, 20/25 | −1.25, −0.75 × 180 +0.50, −2.00 × 170 |
PCIOL OU | + | − |
| P12 | 54 | M | 28 | 20/LP, 20/LP | NR | PCIOL OU | + | − | |
| P13 | 30 | F | 30 | 20/60, 20/60 | −3.00, -- −3.75, −0.25 × 75 |
PSC/PSC | + | + | |
| P14 | 45 | F | 24 | 20/70, 20/80 | −1.75, −1.00 × 87 −2.00, −0.25 × 16 |
NSC/NSC | + | − | |
| P15 | 70 | M | NR | 20/70, 20/150 | NR | PCIOL OU | + | − | |
| P16 | 30 | M | 10 | 20/25, 20/20 | NR | PSC/PSC | + | − |
Footnotes: BCVA, best-corrected visual acuity; OD, right eye; OS, left eye; CME, cystoid macular edema; NR, not recorded; PCIOL, posterior chamber intraocular lens; ACIOL, anterior chamber intraocular lens; PSC, posterior subcapsular cataract; NSC, nuclear sclerotic cataract.
Fundus Autofluorescence.
Short-wavelength FAF imaging with a 30-degree field was performed in 16 patients (31 eyes) (Fig. 2). Images of the right eye for one patient (P12) was unattainable due to difficulty with fixation. In 10 patients, a distinct hyperautofluorescent ring could be observed, with seven patients (P1–7) (13 eyes) exhibiting a unique crescent-shaped boundary whose edge encroached on the central macula, generally advancing most from the temporal and inferior directions. One patient (P3), showed a crescent-shaped ring in the right eye and a typical RP ring in the left eye. FAF images were acquired with a 55 degree field of view in patients with crescent-shaped rings, confirming a larger proportion of the nasal and superior retina being encompassed by the crescent ring, in reference to the fovea (Fig. 3). Of the seven patients with crescent-shaped rings, wide-field imaging of four patients (P1, P2, P4, and P6) revealed an area of retina nasal to the optic disc which was located within the hyperautofluorescent boundary. Crescent-shaped rings were symmetrical between eyes, except in previously mentioned P3. Typical RP rings were observed in both eyes of three patients (P8, P9, and P10). The remaining patients (P11–16) did not have an easily-identifiable crescent-shaped or typical RP ring. Areas between the inner and outer ring borders were hyperautofluorescent on FAF with outer ring boundaries generally appearing more delineated, as seen in previous studies.38 Inner ring boundaries were easily identifiable in seven of ten patients exhibiting rings. Generally, extensive RPE atrophy could be appreciated up to the vascular arcades. Coalescing lacunar or punched out-appearing RPE atrophy was seen in six patients. A hypoautofluorescent bulls-eye-like pattern of maculopathy was present in six patients (P6, P7, P9, P11, P14, and P15) and extensive foveal or macular RPE atrophy was seen in four patients (P5, P7, P12, P15).
Figure 2. Heterogeneous phenotype of EYS-RP on FAF.
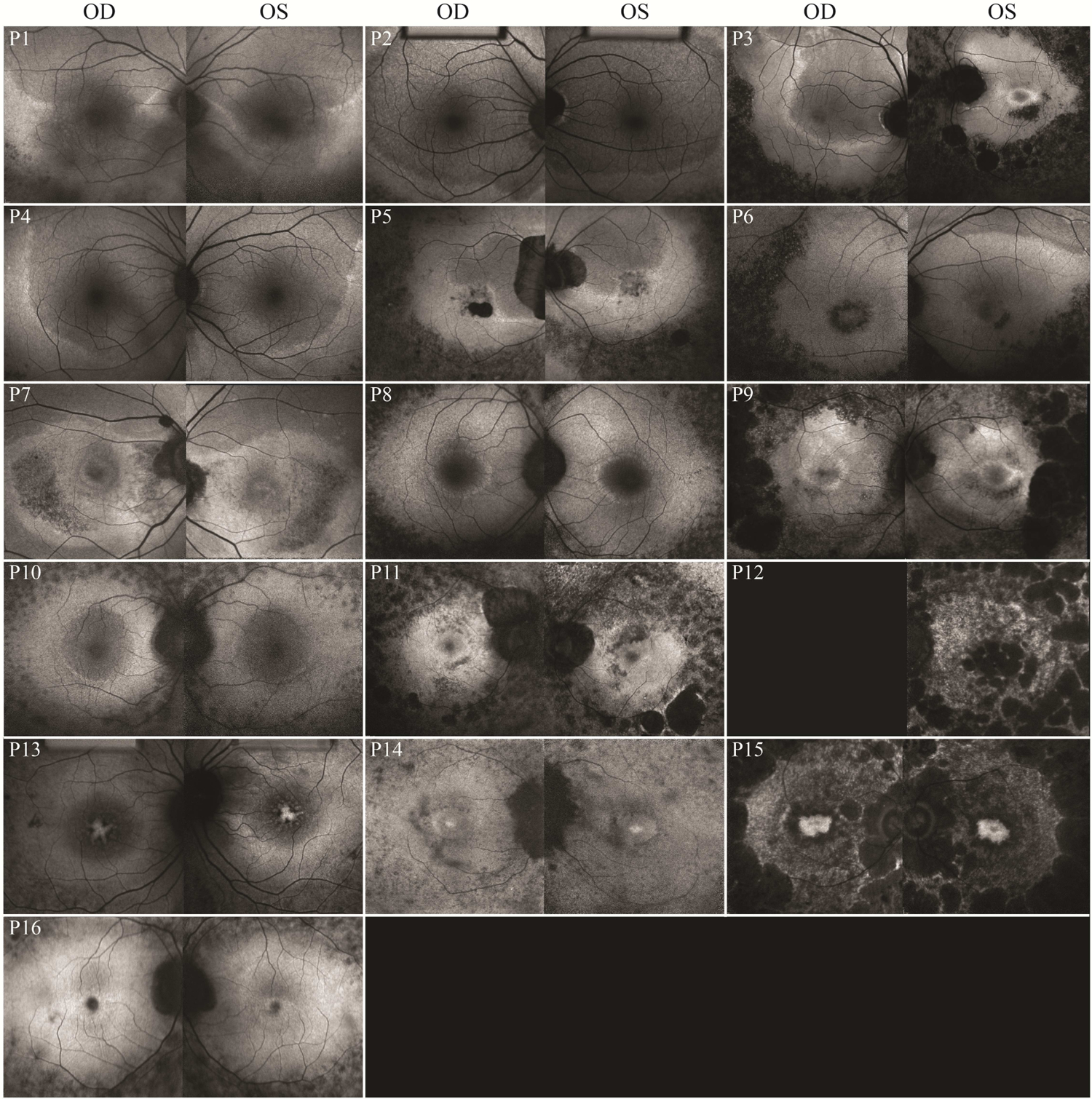
Short wavelength FAF imaging of each patient (30 degree field). Seven patients (P1–7) had atypical, larger crescent-shaped FAF rings. EYS, eyes shut homolog; RP, retinitis pigmentosa; FAF, fundus autofluorescence.
Figure 3. Wide-field FAF imaging of an unusual EYS-RP phenotype with FAF ring boundaries appearing crescent-like.
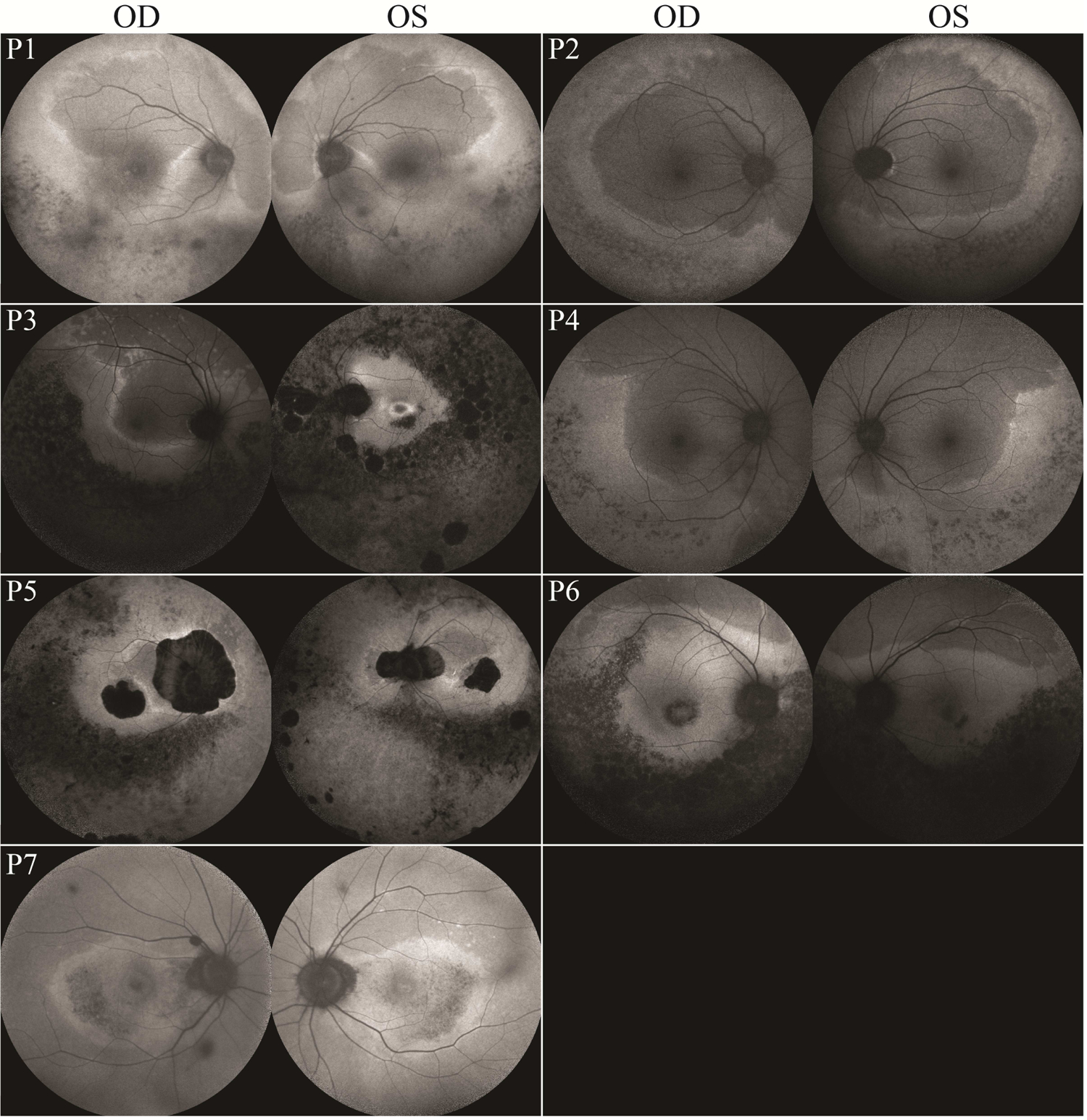
Short wavelength FAF imaging of the right and left eyes of each patient exhibiting an atypical FAF ring phenotype (55 degree field). Note the varying levels of macular involvement amongst patients and the asymmetric presentation of P3. EYS, eyes shut homolog; RP, retinitis pigmentosa; FAF, fundus autofluorescence.
Optical Coherence Tomography.
All 16 patients showed evidence of thinning of the outer retinal laminae on SD-OCT imaging. Generally, a preserved ellipsoid zone (EZ)-line spatially corresponded to the outer boundary of the hyperautofluorescent rings seen on FAF. Thus, patients with a crescent ring showed preservation of the EZ layer over a larger area of the retina. Cystoid macular edema was present in three patients at presentation (P1, P9, and P13). En face OCT was performed in seven patients to assess retinal thickness and the inner segment/outer segment (IS/OS) junction (Fig. 4). The thickest areas of the retina localized to within the hyperautofluorescent ring. En face sections approximating the EZ also show relatively preserved IS/OS junction in areas corresponding to within the ring, however auto-segmentation did not consistently select EZ throughout the entire 6mm × 6mm frame for each patient. The three patients with crescent-shaped FAF rings who underwent en face OCT imaging had a higher proportion of preserved retinal thickness and EZ, judged qualitatively.
Figure 4. En face OCT of EYS-RP patients.
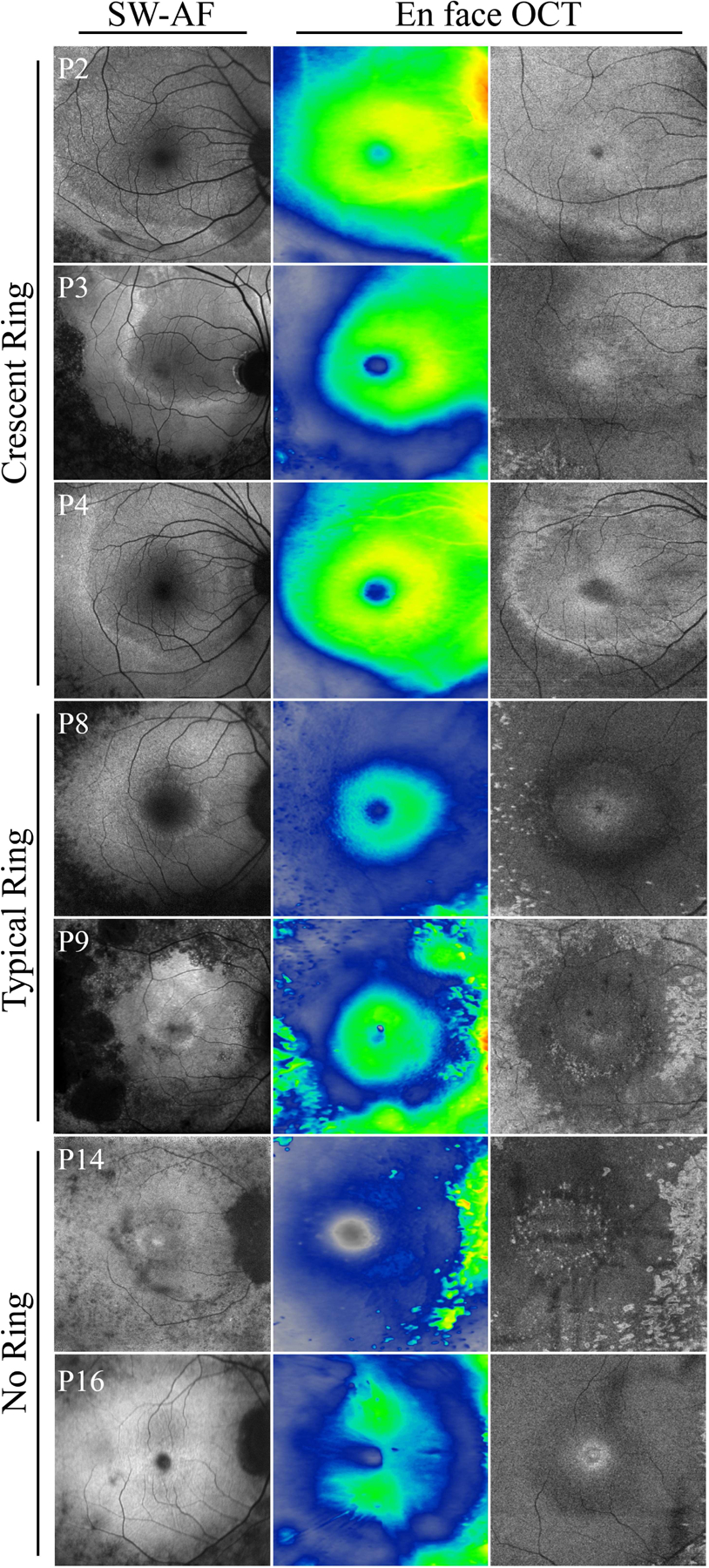
Short wavelength FAF images (first column) aligned with retinal thickness heat maps (second column) and en face OCT at the level of the IS/OS junction (third column) in EYS-RP. EYS patients with a larger ring exhibited a smaller area of thinned retina and EZ loss. Preserved retinal thickness and spared EZ spatially corresponded to the shape of the ring. In patients with no ring (P13 and P16), retinal thickness corresponded most to the nerve fiber layer and greater EZ loss was noted. EYS, eyes shut homolog; RP, retinitis pigmentosa; FAF, fundus autofluorescence; IS/OS, inner segment/outer segment; EZ, ellipsoid zone.
Full-field electroretinography (ffERG).
Twelve of 16 patients underwent ffERG evaluation (Table 3). Eleven patients showed a rod-cone sequence of degeneration and one patient (P7) was diagnosed with a cone-rod dystrophy. Scotopic rod-specific B-wave was extinguished in 16 of 24 eyes and photopic 30 Hz-flicker cone response was less than or equal to one microvolt in 6 of 24 eyes. Three of six patients with the crescent-ring phenotype in both eyes had an average 30 Hz-flicker response greater than 5μV, compared to 0 of 5 patients with no crescent ring phenotype evident in both eyes. Four of seven patients (57%) with crescent rings had recordable scotopic rod-specific B waves in both eyes, compared to patients with typical or absent rings all showing extinguished waveforms bilaterally. Two of seven patients with crescent rings required BA contact lenses to obtain recordable waveforms compared to all patients with either a typical or absent ring.
Table 3. Full-field electroretinography of EYS-RP patients.
Full-field electroretinogram 30 Hz-flicker and scotopic rod-specific B-wave amplitudes for 16 EYS-RP patients.
| ID | 30Hz-flicker OD (μv) | 30Hz-flicker OS (μv) | Scotopic Rod-specific B-wave OD (μv) | Scotopic Rod-specific B-wave OS (μv) | |
|---|---|---|---|---|---|
| Crescent Ring | P1 | 4.4dtl | 5.1dtl | 10.0 | 16.8 |
| P2 | 18.6dtl | 16.4dtl | 42.3 | 53.7 | |
| P3 | 2.2ba* | 10.3ba* | ext. | ext. | |
| P4 | 29.1dtl | 31.5dtl | 54.7 | 67.8 | |
| P5 | 5.0dtl | 4.9dtl | ext. | ext. | |
| P6 | 1.0ba | 4.3ba | ext. | ext. | |
| P7 | 30.6dtl | 34.0dtl | 86.3 | 109.7 | |
| Typical Ring | P8 | 0.8ba | 1.5ba | ext. | ext. |
| P9 | 0.6ba | 0.9ba | ext. | ext. | |
| P10 | 5.0ba | 3.8ba | ext. | ext. | |
| No Ring | P11 | -- | -- | -- | -- |
| P12 | -- | -- | -- | -- | |
| P13 | 0.5ba | 0.4ba | ext. | ext. | |
| P14 | -- | -- | -- | -- | |
| P15 | -- | -- | -- | -- | |
| P16 | 1.4ba | 1.5ba | ext. | ext. |
Footnotes: ba, Burian-Allen contact lens; dtl, Dawson, Trick, and Litzkow (DTL) electrodes;
, Crescent ring in OD and typical ring in OS; ext., extinguished; --, not performed.
Genetic analysis.
Variants in EYS were detected in all patients in the cohort (Table 1). In total, 27 unique variants were identified of which 12 have not been reported in the Reference Sequence (RefSeq) database (https://www.ncbi.nlm.nih.gov/refseq, accessed July 2017). Pathogenicity of each EYS variant was assessed with predictive programs which are summarized in Table 4. The most predominant coding effect amongst the variants were frameshifts (42.2%), followed by nonsense (23.7%) and non-coding variants (18.4%). All non-coding variants were in canonical splice acceptor and donor sites. Most frameshift variants were caused by small deletions ranging from single to 20 bps, all resulting in premature STOP codons and in several cases, the resulting mRNA is predicted to be targeted for nonsense mediated decay. Duplications were found in two patients including a variant spanning a 7,545 bp region of intron 28. The missense variants detected were mostly predicted deleterious except two of three homozygous variants identified in P13: c.3250A>C (p.Thr1084Pro) is of uncertain significance, and also c.4402G>C (p.Asp1468His) based on scores of M-CAP (0.06), REVEL (0.33), Eigen (−0.0124), CADD (24.5) and DANN (0.994).
Table 4: Detailed analysis of EYS variants.
Summary of EYS variants identified in the patient cohort, including functional analyses.
| dbSNP | Chr | GRCh37 | Nucleotide Change | Protein Variant | Count in Cohort | Allele Frequency | M-CAP | REVEL | Eigen | CADD | DANN | ClinVar Significance |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| -- | 6 | 66115144 | c.963_979delAAAAGGATCTTCCAGCC | p.Pro321Profs | 1 | |||||||
| rs143994166 | 6 | 66112400 | c.1155T>A | p.Cys385Ter | 2 | 0.0007 | −0.3334 | 28.2 | 0.983 | Likely pathogenic | ||
| -- | 6 | 66063502 | c.1308C>A | p.Cys436Ter | 1 | 0.000004073 | −0.0979 | 35 | 0.974 | |||
| rs752504462 | 6 | 66044995 | c.1641_1644delTCAG | p.Ser547Argfs | 1 | 0.000004084 | ||||||
| -- | 6 | 66044994 | c.1645G>T | p.Glu549Ter | 1 | −0.2863 | 25.8 | 0.965 | ||||
| rs749103801 | 6 | 66005817 | c.1961_1962insA | p.Asn654Lysfs | 1 | 0.000006599 | ||||||
| -- | 6 | 65596589 | c.2992+1G>A | 1 | 0.6634 | 23.3 | 0.931 | |||||
| rs778646190 | 6 | 65523464 | c.3250A>C | p.Thr1084Pro | 2 | 0.00006855 | 0.04 | 0.18 | −1.1326 | 0.322 | Uncertain | |
| rs373441420 | 6 | 65523270 | c.3443+1G>T | 3 | 0.00002119 | 0.846 | 23.4 | 0.976 | Pathogenic | |||
| -- | 6 | 65336027 | c.3555C>G | p.Cys1185Trp | 1 | 0.84 | 0.85 | 0.6131 | 26.4 | 0.99 | ||
| rs928803207 | 6 | 65301640 | c.4120C>T | p.Arg1374Ter | 2 | 0.00001347 | −0.0156 | 35 | 0.997 | |||
| rs778752557 | 6 | 65301358 | c.4402G>C | p.Asp1468His | 2 | 0.00008771 | 0.06 | 0.33 | −0.0124 | 24.5 | 0.994 | Uncertain |
| -- | 6 | 65150442 | c.5645-1197-c.5927+3169 | 1 | ||||||||
| -- | 6 | 65098733 | c.5928delG | p.Arg1976Serfs | 1 | |||||||
| rs749909863 | 6 | 64940493 | c.6416G>A | p.Cys2139Tyr | 1 | 0.0001 | 0.35 | 0.76 | 0.6828 | 29.5 | 0.997 | Uncertain | Pathogenic |
| -- | 6 | 64791744 | c.6571+5G>A | 1 | 0.00001486 | 1.3819 | 14.39 | 0.786 | ||||
| rs752953889 | 6 | 64776242 | c.6714delT | p.Ile2239Serfs | 1 | 0.00003948 | Likely pathogenic | |||||
| rs758109813 | 6 | 64709008 | c.6794delC | p.Pro2265Glnfs | 3 | 0.0002 | Pathogenic | |||||
| -- | 6 | 64498950 | c.7578+1G>A | 2 | 0.0000133 | 1.0605 | 25.9 | 0.996 | ||||
| rs527236076 | 6 | 64472413 | c.8012T>A | p.Leu2671Ter | 1 | 0.00001987 | 0.09 | 0.3156 | 37 | 0.984 | Likely pathogenic | |
| rs779983752 | 6 | 64436534 | c.8111T>G | p.Leu2704Ter | 2 | 0.00004052 | 0.6781 | 47 | 0.987 | |||
| -- | 6 | 64431514 | c.8413dupA | p.Thr2805Asnfs | 1 | |||||||
| rs528919874 | 6 | 64431272 | c.8648_8655delCATGCAGA | p.Thr2883Lysfs | 1 | 0.0008 | ||||||
| rs770748359 | 6 | 64430632 | c.9286_9295delGTAAATATCG | p.Val3096Leufs | 2 | 0.0002 | Pathogenic | |||||
| rs769824975 | 6 | 64430625 | c.9299_9302delCTCA | p.Thr3100Lysfs | 1 | 0.00004015 | ||||||
| -- | 6 | 64430591 | c.9317_9336delCCAATTTTGTTGGCAAAATT | p.Thr3106Lysfs | 1 | 0.00000672 | ||||||
| -- | 6 | 64430540 | c.9383_9387delAATTA | p.Lys3128Argfs | 1 |
All reference sequences are NM_001142800.1 HGVS.
Approximately 71.1% of variants occurred in the conserved domains of EYS, including 12 truncating variants in the Laminin G and 15 in the epidermal growth factor (EGF)-like domains. Nearly half (47.4%) of the variants harbored by the cohort were spatially distributed across the Laminin G domains toward the carboxy (−COOH) end of the protein (Fig 5). Of the 18 variants clustered within this portion of the protein, termed LamG/C-terminal region, 11 (61%) were found in patients presenting with a crescent ring; whereas only the c.5928delG (p.Arg1976Serfs*11) variant in P15 and the c.6794delC (p.Pro2265Glnfs*46) variant, homozygous in P12 and compound heterozygous in P14, were associated with advanced-disease patients (no rings). An examination of the spatial distribution of variants along the protein within each phenotypic group revealed that 69% and 83.3% of variants in the crescent and typical ring group, respectively, cluster toward the carboxy one-third end of the protein, while 75.0% of variants in the no ring group cluster in the amino (−NH2) one-third end of the protein. A Fisher’s Exact contingency test was used to assess the significance of this observation. Variants with a nucleotide position greater than GRch37 6:65300137 (c.5617C), which marks the beginning of the first Laminin G domain, were classified as falling within the defined “LamG/C-terminal” end of the protein (Fig. 5). A significant spatial association was found individually between ‘crescent’ versus ‘no ring’ phenotypes (p = 0.02), ‘typical’ versus ‘no ring’ phenotypes (p =0.02) and collectively between ‘crescent’/’typical’ rings together versus ‘no ring’ (p = 0.004). A statistical difference was not found between the ‘crescent’ and ‘typical’ ring phenotypes (p > 0.05).
Figure 5. Schematic representation of EYS protein domains and the distribution of variants with respect to FAF phenotype.
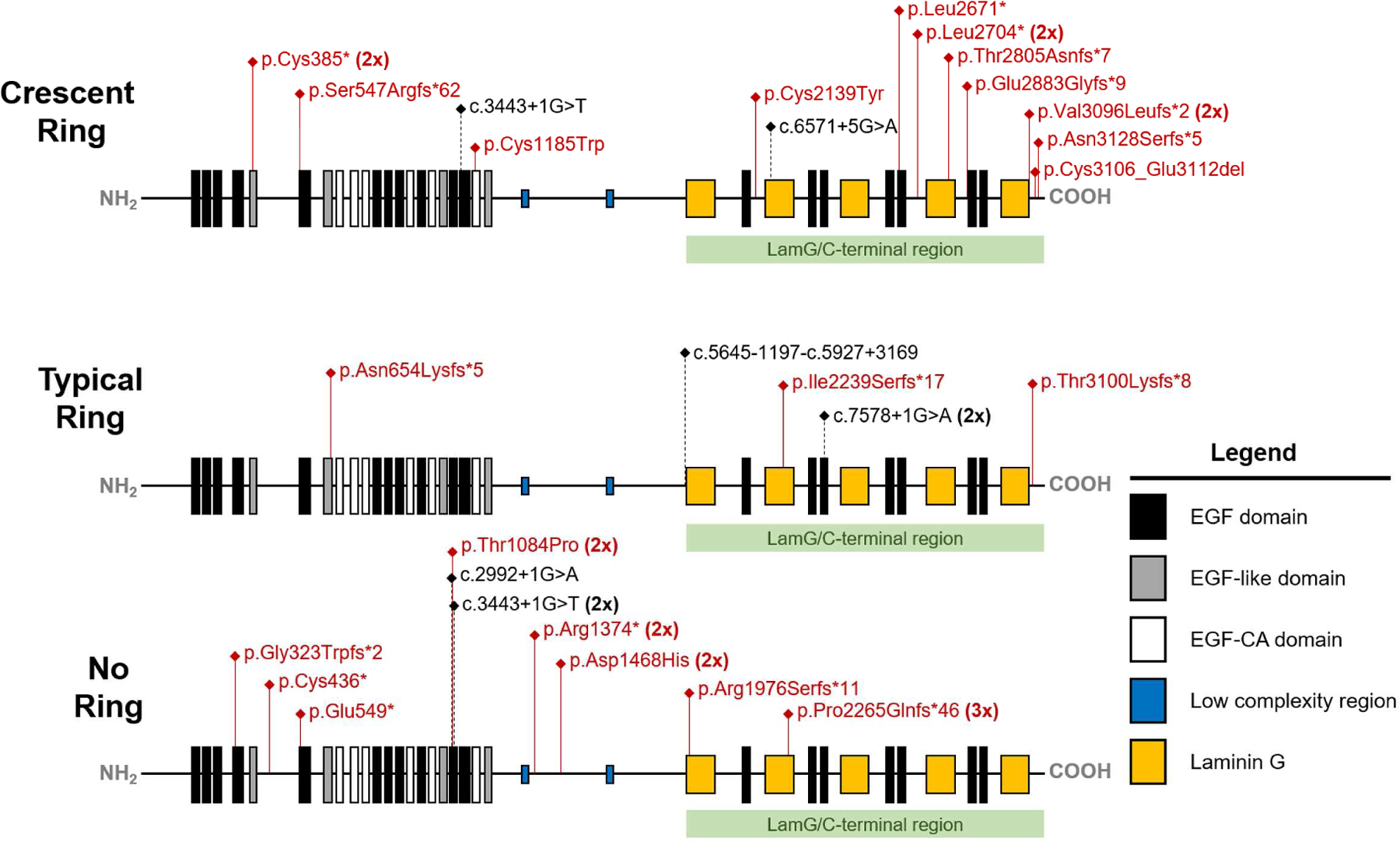
Variants identified in the coding region are listed in red with the putative position of non-coding variants listed in black and indicated by black dotted lines. Patients in the crescent and typical ring groups harbored variants which clustered predominantly in the Laminin G domains toward the carboxy (−COOH) end of the protein. Patients that exhibited advanced disease (no ring) harbored variants which clustered predominantly in the amino (−NH2) end of the protein amongst the epidermal growth factor (EGF) domains. EYS, eyes shut homolog; FAF, fundus autofluorescence.
Selected Case Studies
EYS-RP Masquerading as a Potential Drug Toxicity (P7).
A 65-year-old man presented by referral for an electrodiagnostics evaluation. He complained of bilateral floaters in both eyes, more in the right, and decreased visual acuity bilaterally. The progression was described to be slow but the patient was unable to describe the exact duration of symptoms. Past medical history included skin melanoma that was surgically removed four months prior and HIV infection controlled with multiple-drug therapy that included abacavir, lamivudine, efavirenz, and tenofovir. Past ocular history and family history were unremarkable. Referral to the Harkness Eye Institute electrophysiology clinic was arranged to assess for inflammatory conditions, retinal drug toxicity, and cancer-associated retinopathy. Visual acuity was best corrected to 20/50 and 20/60 in the right and left eyes, respectively. The anterior segments were within normal limits on ocular exam and mild nuclear sclerosis of the lens could be seen bilaterally. Intraocular pressures were 17 in the right and 18 in the left, respectively. Dilated fundus exam revealed an optic nerve with a healthy-appearing rim, clear vitreous, and bull’s eye maculopathy in both eyes. No vascular attenuation or pigment migration was appreciated in the periphery. SD-OCT revealed extensive thinning of the outer retinal laminae within the macula, with a relatively spared foveal ellipsoid zone in both eyes. FAF imaging showed an amorphous crescent-shaped ring that extended slightly superior and inferior to the optic disc; the rings were symmetric between eyes. ffERG showed photopic cone responses affected to a greater extent than scotopic responses and was non-progressive over two months, thus most consistent with a cone-rod dystrophy. Whole exome sequencing identified two novel variants in EYS which are predicted to be pathogenic; namely c.8111T>G (p.Leu2704*) and c.9317_9336del20 (p.Thr3106Lysfs*13). Family members were not available to complete segregation analyses. The patient was recommended to maintain his current drug regimen and follow-up at regular but infrequent intervals, as EYS-RP was the most likely diagnosis.
Delayed Diagnosis of EYS-RP in P13.
A 30-year-old woman presented by referral to the electrophysiology clinic of the Harkness Eye Institute for evaluation of macular edema. Systemic past medical history was unremarkable, however the patient’s past ocular history was notable for suspected uveitis with presumed secondary macular edema for which she received an intravitreal steroid injection from the referring ophthalmologist. This lead to subsequent steroid-induced glaucoma requiring valve surgery. She reported night blindness since the age of 19 years old. There was no family history of blinding disorders in her family. Vision could be best corrected to 20/60 bilaterally. The anterior segment appeared quiet on slit lamp exam with posterior subcapsular cataracts observed in both lenses. On dilated fundus exam, intraretinal pigment migration could be seen in the inferior periphery and macular changes spatially corresponding to cystoid macular edema on SD-OCT. Optic disc margins were mildly indistinct with no disc edema. There were vitreous cells that appeared more prevalent in the right eye. Peripheral thinning of the outer retinal layers and extensive cystoid macular edema causing schisis of the inner retina was appreciated on SD-OCT of both eyes. FAF imaging showed patchy areas of RPE atrophy and macular changes spatially corresponding to CME seen on fundoscopy and SD-OCT. ffERG was performed and showed completely extinguished maximal and rod-specific scotopic b-wave responses. Photopic 30 Hz-flicker was less than 1μV bilaterally using BA contact lens electrodes. An inherited retinal dystrophy was suspected and next generation sequencing of DNA collected from peripheral lymphocytes was performed for common genes known to cause inherited retinal dystrophies. Three variants were identified in EYS, all of which were homozygous: c.3443+1G>T, p.(?); c.3250A>C, p.(Thr1084Pro); c.4402G>C, p.(Asp1468His). All three variants were heterozygous in the patient’s non-affected daughter and mother. Of the three, the c.3443+1G>T, p.(?), variant is most likely the causal one. EYS-RP was diagnosed and the patient was treated with acetazolamide to manage the CME which decreased after three months of follow-up.
Discussion
Previous investigations have described EYS-RP as a relatively homogenous and severe phenotype amongst affected individuals, similar to other autosomal recessive RP phenotypes.7, 14, 15 The present cohort was seen at a single-center, and our cross-sectional analysis strongly suggests high clinical and genetic heterogeneity amongst patients. Our analysis suggests a relatively variable presentation, with patients exhibiting particular FAF phenotypes that potentially relate to the location of their variants along EYS. In this study, a spared ellipsoid zone was observed within the inner and outer borders of typical and crescent-shaped hyperautofluoresent rings, as expected. In early RP, the peripheral EZ is less visible and the edge of the EZ roughly approximates the visual field as the disease progresses.39, 40 Several studies have further described EZ changes in RP and used it as a reliable measure to monitor disease progression,41–45 including in a longitudinal progression study for EYS-RP.46 Future studies would be useful for assessing differences in the visual function and progression rates of patients with and without the crescent-shaped ring phenotype.
In two patients, panel-based and whole exome sequencing guided the clinical management. In one case (P7), confirming the diagnosis of EYS-RP meant that medications for HIV did not require adjustment and a non-progressive ffERG supported these recommendations. Lack of intraretinal pigment on exam and the unusual crescent-shaped ring may have contributed to an incomplete diagnosis. Additionally, the ffERG appeared most consistent with a cone-rod dystrophy in conjunction with the patient’s symptoms. Interestingly, recent data shows expression of EYS in both rods and cones, and ablation of EYS in zebrafish with TALEN technology yields a phenotype similar to cone-rod dystrophy.21 Few other cases of cone-rod dystrophy in EYS-patients are documented in the literature.47, 48 In the second case (P13), macular edema was initially ascribed to be secondary to uveitis, leading to intravitreal steroid injection by the referring provider and a complication of subsequent glaucoma. Early recognition of cystoid macular edema secondary to RP is important as CME may respond to less toxic, oral or topical carbonic anhydrase inhibitors as a first-line therapy.49, 50
The phenotypic stratification proposed is further strengthened by the observation that patients with crescent-shaped and typical rings are associated with variants in the distal portion, or C-terminal one-third of the EYS protein. Conversely, patients with no FAF ring, predominantly harbor variants in the amino-terminus of the protein. Given the relatively large size of the EYS protein (~2Mbs), we speculate that variants near the distal portion of the EYS protein result in less alteration of protein function, because the effect is not null, but rather impairs EYS binding via the affected Laminin G domains within its interactome. In a recent study by McGuigan and colleagues, autofluorescent imaging was performed on six EYS-RP patients,24 of which two showed preserved melanization on NIR-AF in a pattern similar to what we describe as a crescent-shape on short-wavelength autofluorescence. Genotype-phenotype correlations were not attempted in this study, but these two patients had four variants all occurring at nucleotide positions greater than GRch37 6:65300137 (c.5617C), thus falling within the defined “LamG/C-terminal” end of the protein.
There are limitations to our study. Segregation analysis was performed in 5 patients. EYS variants were interpreted as homozygous in four of the remaining 11 patients. Low availability of family members to confirm the phase of variants in the remaining seven patients was a contributing factor. Additionally, ffERGs were performed with either BA contact lenses or DTL-recording electrodes, and one patient manifested with a different ring subtype in each eye (P3) which hindered statistical comparison of electric responses between groups.
Further genetic analyses are needed of larger cohorts to confirm genotype-phenotype correlations presented in this study, as it is difficult to parse the contribution of a particular allele to disease phenotype in small cohorts of patients harboring mostly compound heterozygous variants. Genetic modifiers, in addition to the position of the variant within the gene, likely explain the variability of FAF phenotype and disease severity amongst patients. FAF imaging is useful in determining extent of disease involvement and, when used in conjunction with other modalities, may hasten the diagnosis of EYS-RP in cases that present atypically. Longitudinal studies and functional testing will be useful for comparing the progression rates of patients with differing ring types on FAF imaging.
Supplementary Material
{kind=link}
Acknowledgements
A) FUNDING/SUPPORT
Supported, in part, by grants from National Eye Institute, NIH [P30EY019007, R01EY018213, R01EY024698, R01EY026682, R21AG050437, R24EY019861], National Cancer Institute Core [5P30CA013696], the Research to Prevent Blindness (RPB) Physician-Scientist Award, unrestricted funds from RPB, New York, NY, USA. J.D.S is supported by the RPB Medical Student Eye Research Fellowship. S.H.T. is a member of the RD-CURE Consortium and is supported by the Tistou and Charlotte Kerstan Foundation and the Schneeweiss Stem Cell Fund, New York State [C029572]. The funding sources had no role in the study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
B) FINANCIAL DISCLOSURES
K.B.F. is a consultant for Opotvue, Optos, Heidelberg Engineering, Genentech, and Spark Therapeutics. He receives research support from Genentech/Roche. The remaining authors have no disclosures. None of the authors declare a conflict of interest.
C) OTHER ACKNOWLEDGEMENTS
None
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conference: Presented at the Annual Academy of Ophthalmology meeting, 2017
DATA AVAILABILITY
All collected raw data and analyses are included in this manuscript.
REFERENCES
- 1.Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci 1993;34(5):1659–76. [PubMed] [Google Scholar]
- 2.Hamel C Retinitis pigmentosa. Orphanet J Rare Dis 2006;1:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006;368(9549):1795–809. [DOI] [PubMed] [Google Scholar]
- 4.Abd El-Aziz MM, Barragan I, O’Driscoll CA, et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet 2008;40(11):1285–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abd El-Aziz MM, O’Driscoll CA, Kaye RS, et al. Identification of novel mutations in the ortholog of Drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 2010;51(8):4266–72. [DOI] [PubMed] [Google Scholar]
- 6.Collin RW, Littink KW, Klevering BJ, et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet 2008;83(5):594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandah-Rozenfeld D, Littink KW, Ben-Yosef T, et al. Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Invest Ophthalmol Vis Sci 2010;51(9):4387–94. [DOI] [PubMed] [Google Scholar]
- 8.Barragan I, Borrego S, Pieras JI, et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum Mutat 2010;31(11):E1772–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beryozkin A, Zelinger L, Bandah-Rozenfeld D, et al. Identification of mutations causing inherited retinal degenerations in the israeli and palestinian populations using homozygosity mapping. Invest Ophthalmol Vis Sci 2014;55(2):1149–60. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Liu X, Sheng X, et al. Targeted next-generation sequencing reveals novel EYS mutations in Chinese families with autosomal recessive retinitis pigmentosa. Sci Rep 2015;5:8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Y, Huang L, Sundaresan P, et al. Whole-exome Sequencing Analysis Identifies Mutations in the EYS Gene in Retinitis Pigmentosa in the Indian Population. Sci Rep 2016;6:19432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosono K, Ishigami C, Takahashi M, et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS One 2012;7(2):e31036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Zhang J, Li C, et al. Identification of a novel homozygous nonsense mutation in EYS in a Chinese family with autosomal recessive retinitis pigmentosa. BMC Med Genet 2010;11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwanami M, Oshikawa M, Nishida T, Nakadomari S, Kato S. High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 2012;53(2):1033–40. [DOI] [PubMed] [Google Scholar]
- 15.Littink KW, van den Born LI, Koenekoop RK, et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology 2010;117(10):2026–33, 2033 e1–7. [DOI] [PubMed] [Google Scholar]
- 16.Oishi M, Oishi A, Gotoh N, et al. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Invest Ophthalmol Vis Sci 2014;55(11):7369–75. [DOI] [PubMed] [Google Scholar]
- 17.Arai Y, Maeda A, Hirami Y, et al. Retinitis Pigmentosa with EYS Mutations Is the Most Prevalent Inherited Retinal Dystrophy in Japanese Populations. J Ophthalmol 2015;2015:819760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan MI, Collin RW, Arimadyo K, et al. Missense mutations at homologous positions in the fourth and fifth laminin A G-like domains of eyes shut homolog cause autosomal recessive retinitis pigmentosa. Mol Vis 2010;16:2753–9. [PMC free article] [PubMed] [Google Scholar]
- 19.Zelhof AC, Hardy RW, Becker A, Zuker CS. Transforming the architecture of compound eyes. Nature 2006;443(7112):696–9. [DOI] [PubMed] [Google Scholar]
- 20.Alfano G, Kruczek PM, Shah AZ, et al. EYS Is a Protein Associated with the Ciliary Axoneme in Rods and Cones. PLoS One 2016;11(11):e0166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Z, Hu X, Liu F, et al. Ablation of EYS in zebrafish causes mislocalisation of outer segment proteins, F-actin disruption and cone-rod dystrophy. Sci Rep 2017;7:46098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Audo I, Sahel JA, Mohand-Said S, et al. EYS is a major gene for rod-cone dystrophies in France. Hum Mutat 2010;31(5):E1406–35. [DOI] [PubMed] [Google Scholar]
- 23.Gu S, Tian Y, Chen X, Zhao C. Targeted next-generation sequencing extends the phenotypic and mutational spectrums for EYS mutations. Mol Vis 2016;22:646–57. [PMC free article] [PubMed] [Google Scholar]
- 24.McGuigan DB, Heon E, Cideciyan AV, et al. EYS Mutations Causing Autosomal Recessive Retinitis Pigmentosa: Changes of Retinal Structure and Function with Disease Progression. Genes (Basel) 2017;8(7):178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suto K, Hosono K, Takahashi M, et al. Clinical phenotype in ten unrelated Japanese patients with mutations in the EYS gene. Ophthalmic Genet 2014;35(1):25–34. [DOI] [PubMed] [Google Scholar]
- 26.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet 2016;99(4):877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 2016;48(12):1581–1586. [DOI] [PubMed] [Google Scholar]
- 28.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet 2016;48(2):214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46(3):310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015;31(5):761–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015;347(6218):1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCulloch DL, Marmor MF, Brigell MG, et al. Erratum to: ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 2015;131(1);131:81–3. [DOI] [PubMed] [Google Scholar]
- 34.McCulloch DL, Marmor MF, Brigell MG, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 2015;130(1):1–12. [DOI] [PubMed] [Google Scholar]
- 35.Andreasson SO, Sandberg MA, Berson EL. Narrow-band filtering for monitoring low-amplitude cone electroretinograms in retinitis pigmentosa. Am J Ophthalmol 1988;105:500–3. [DOI] [PubMed] [Google Scholar]
- 36.Berson EL. Long-term visual prognoses in patients with retinitis pigmentosa: the Ludwig von Sallmann lecture. Exp Eye Res 2007;85(1):7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sieving PA, Arnold EB, Jamison J, Liepa A, Coats C. Submicrovolt flicker electroretinogram: cycle-by-cycle recording of multiple harmonics with statistical estimation of measurement uncertainty. Invest Ophthalmol Vis Sci 1998;39(8):1462–9. [PubMed] [Google Scholar]
- 38.Schuerch K, Marsiglia M, Lee W, Tsang SH, Sparrow JR. Multimodal imaging of disease-associated pigmentary changes in retinitis pigmentosa. Retina 2016; 36(Suppl 1):S147–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hood DC, Ramachandran R, Holopigian K, Lazow M, Birch DG, Greenstein VC. Method for deriving visual field boundaries from OCT scans of patients with retinitis pigmentosa. Biomed Opt Express 2011;2(5):1106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lima LH, Burke T, Greenstein VC, et al. Progressive constriction of the hyperautofluorescent ring in retinitis pigmentosa. Am J Ophthalmol 2012;153(4):718–27, 727 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birch DG, Locke KG, Wen Y, Locke KI, Hoffman DR, Hood DC. Spectral-domain optical coherence tomography measures of outer segment layer progression in patients with X-linked retinitis pigmentosa. JAMA Ophthalmol 2013;131(9):1143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai CX, Locke KG, Ramachandran R, Birch DG, Hood DC. A comparison of progressive loss of the ellipsoid zone (EZ) band in autosomal dominant and x-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 2014;55(11):7417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hariri AH, Zhang HY, Ho A, et al. Quantification of Ellipsoid Zone Changes in Retinitis Pigmentosa Using en Face Spectral Domain-Optical Coherence Tomography. JAMA Ophthalmol 2016;134(6):628–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith TB, Parker M, Steinkamp PN, et al. Structure-Function Modeling of Optical Coherence Tomography and Standard Automated Perimetry in the Retina of Patients with Autosomal Dominant Retinitis Pigmentosa. PLoS One 2016;11(2):e0148022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun LW, Johnson RD, Langlo CS, et al. Assessing Photoreceptor Structure in Retinitis Pigmentosa and Usher Syndrome. Invest Ophthalmol Vis Sci 2016;57(6):2428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyata M, Ogino K, Gotoh N, et al. Inner segment ellipsoid band length is a prognostic factor in retinitis pigmentosa associated with EYS mutations: 5-year observation of retinal structure. Eye (Lond) 2016;30:1588–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katagiri S, Akahori M, Hayashi T, et al. Autosomal recessive cone-rod dystrophy associated with compound heterozygous mutations in the EYS gene. Doc Ophthalmol 2014;128:211–7. [DOI] [PubMed] [Google Scholar]
- 48.Littink KW, Koenekoop RK, van den Born LI, et al. Homozygosity mapping in patients with cone-rod dystrophy: novel mutations and clinical characterizations. Invest Ophthalmol Vis Sci 2010;51(11):5943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakthavatchalam M, Lai FHP, Rong SS, Ng DS, Brelen ME. Treatment of cystoid macular edema secondary to retinitis pigmentosa: a systematic review. Surv Ophthalmol 2017; pii: S0039–6257(17)30068–1. [DOI] [PubMed] [Google Scholar]
- 50.Mrejen S, Audo I, Bonnel S, Sahel JA. Retinitis Pigmentosa and Other Dystrophies. Dev Ophthalmol 2017;58:191–201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.