

SUMMARY
Tumor-initiating stem cells (TSCs) are critical for drug resistance and immune escape. However, the mutual regulations between TSC and tumor microenvironment (TME) remain unclear. Using DNA-label retaining, single-cell RNA sequencing (scRNA-seq), and other approaches, we investigated intestinal adenoma in response to chemoradiotherapy (CRT), thus identifying therapy-resistant TSCs (TrTSCs). We find bidirectional crosstalk between TSCs and TME using CellPhoneDB analysis. An intriguing finding is that TSCs shape TME into a landscape that favors TSCs for immunosuppression and propagation. Using adenoma-organoid co-cultures, niche-cell depletion, and lineaging tracing, we characterize a functional role of cyclooxygenase-2 (Cox-2)-dependent signaling, predominantly occurring between tumor-associated monocytes and macrophages (TAMMs) and TrTSCs. We show that TAMMs promote TrTSC proliferation through prostaglandin E2 (PGE2)-PTGER4(EP4) signaling, which enhances β-catenin activity via AKT phosphorylation. Thus, our study shows that the bidirectional crosstalk between TrTSC and TME results in a pro-tumorigenic and immunosuppressive contexture.
Graphical Abstract
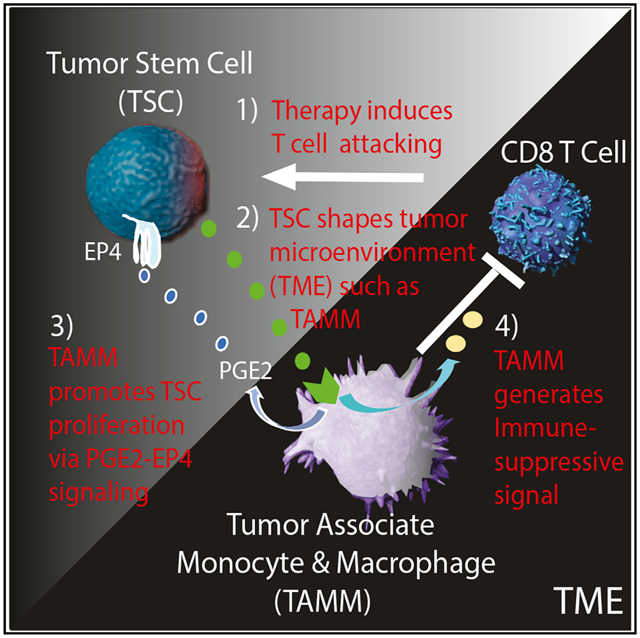
In brief
He et al. examined the nature of therapy-resistant tumor-initiating stem cells (TSCs) in murine intestinal adenoma using single-cell RNA sequencing (scRNA-seq) analysis and revealed a bidirectional crosstalk between TSC and its microenvironment, TME, in which TSC shapes TME, especially tumor-associated monocyte and macrophage (TAMM), into an immunosuppressive barrier and pro-tumorigenic niche.
INTRODUCTION
The roles of TME in supporting tumorigenesis are well documented (Arneth, 2019); however, the direct interaction between TME and tumor-initiating stem cells (TSCs) remains largely unaddressed (Clarke et al., 2006; Kreso and Dick, 2014; Li and Neaves, 2006). In this study, our intention was to characterize the diverse cellular components of intestinal adenoma and to dissect the TME-associated molecular signaling in response to chemoradiotherapy (CRT). We were interested in the following questions: (1) Do therapy-resistant TSCs (TrTSCs) exist in intestinal adenoma? (2) How do dynamic changes in TME, such as the innate immune component, regulate TrTSCs? and (3) How do TrTSCs influence innate and adaptive immune cells (Bonaventura et al., 2019; Melero et al., 2014; Murciano-Goroff et al., 2020)?
Intestinal tumorigenesis, including initial adenoma formation and subsequent carcinoma development, is a complex cascade of events. It often starts as local abnormal proliferation foci or abnormal tubular hyperplastic structures and then progresses from low-grade dysplasia (LGD) to high-grade dysplasia (HGD). It follows with invasion of the local region with immature features of tumor cells (adenocarcinoma in situ [ACIS]) and subsequent invasion into the muscular layer (invasive adenocarcinoma [InvAC]). Finally, metastasis occurs as cancer cells migrate away from the primary tumor, travel through the blood or lymphatic vessel, and form new tumors in distant organs (Fleming et al., 2012). At each step of tumorigenesis, various genetic mutations combine with abnormal changes in signaling to upregulate proto-oncogene activity and ablate tumor suppressor function (Fearon and Vogelstein, 1990; Knudson, 2001). These pathways include Wnt-β-catenin, mitogen-activated protein kinase (MAPK), PTEN (Phosphatase And Tensin Homolog)-controlled PI3Kca and AKT, transforming growth factor β (TGF-β), TP53, and other pathways (Fearon and Vogelstein, 1990; Jackstadt and Sansom, 2016).
TSCs are known to be critical in tumor initiation and progression (Clarke and Fuller, 2006; Dick, 2008). However, in response to CRT, it remains largely unclear whether and how the TME facilitates TSC survival from therapy challenges and supports subsequent tumor regrowth (Li and Neaves, 2006). The TME consists of tumor-associated stromal cells, endothelial cells, tissue-resident and recruited innate immune cells (i.e., tumor-associated monocytes and macrophages [TAMMs]), and adaptive immune T and B cells (Borovski et al., 2011; Quail and Joyce, 2017). TAMMs are the main components of myeloid-derived cells (MDCs) and functionally overlap with myeloid-derived suppressor cells (MDSC) in terms of T cell inhibition (Carvalho et al., 2018; Chen et al., 2018; Guadagno et al., 2018; Poh and Ernst, 2018).
MDCs have been known to produce a prominent proinflammatory eicosanoid, prostaglandin E2 (PGE2), which can also be produced by other cell types, including fibroblasts and late-stage malignant epithelial (EP) cells (EpCs) (Aoki et al., 2017; Harizi, 2013; Shao et al., 2006; Zelenay et al., 2015). PGE2 interacts with prostaglandin receptors (EPs) (Che et al., 2017; Miyoshi et al., 2017) to activate β-catenin signaling (Castellone et al., 2005; Evans, 2009; Hsu et al., 2017; Miyoshi et al., 2017), although the link between EPs and β-catenin is unclear.
Over the last few decades, many mouse models have been developed to study adenoma formation and adenocarcinoma development in the colorectum. The APCMin/+ model is the most used mouse model because it develops adenomas that mimic familial adenomatous polyposis (FAP) in humans (Moser et al., 1990; Su et al., 1992). There are many other mouse models that reflect different stages of tumorigenesis (Jackstadt and Sansom, 2016). Only recently have mouse models such as KPN (KrasG12D/+trP53−/− N1icd) and Villin-Cre:Apcfl/fl;R26-LSL-RasG12V;TP53KO been reported that mimic human colorectal cancer (CRC), deeming them useful for subtype and metastatic studies (Fumagalli et al., 2020; Jackstadt et al., 2019). However, these two models still come with caveats, including a rare rate of metastasis or the requirement of transplantation of organoids derived from the mutant mouse model.
Thus far, there has been a limited choice in a mouse CRC model considered suitable for a CRT response study, which requires many mice. Our study characterized the diverse cellular components of intestinal adenoma and the associated dynamic and complex signaling that takes place during CRT. We applied a variety of technologies, including DNA label retaining, single-cell RNA sequencing (scRNA-seq) analysis, flow cytometry assay, and electron microscopy (EM) technology to provide a comprehensive atlas of TME cellular components and signaling modules between TSCs and TME. We conducted in vitro and in vivo experiments to functionally characterize one of the identified signaling modules, the PGE2-EP4-mediated β-catenin axis. Finally, we examined the extent to which our findings in the mouse adenoma may provide insight into human CRCs.
RESULTS
CRT induces, whereas celecoxib-targeted therapy prevents, the progression of adenoma
CRT is used to treat tumors and cancer clinically. Celecoxib, a selective cyclooxygenase-2 (COX-2) inhibitor, is used to reduce inflammation in cancer patients who have received CRT, with an unexpected benefit of increasing overall survival in a limited number of CRC patients (Lin et al., 2006); however, the underlying mechanism and the potential consequences of these therapies are largely unclear. We used the APCMin/+ mouse model to study the response of tumor cells to CRT and celecoxib. Measuring the kinetics of adenoma formation in the APCMin/+ mouse model as shown in Figure 1A, we observed on average 25 tumors at 80 days, with a plateau of 55–65 adenomas around 100–120 days. These mice did not survive beyond 140–150 days. We initiated the therapy procedure in APCMin/+ mice between days 90 and 100 when adenomas were established and relatively stable (Figure 1A). We adapted a previously reported therapy regimen for human CRC patients (Lin et al., 2006). There were four therapy groups (Figure 1B): group 1, vehicle control; group 2 (CRT), CRT with 5 Gγ abdominal irradiation at 12 h prior to 7 days of therapy with capecitabine (a pro-drug of 5FU) and wait for 7 days before tissue harvest; group 3 (Cele), celecoxib for 14 days; and group 4 (Comb), combined CRT + celecoxib protocol.
Figure 1. CRT induced, but celecoxib prevented, adenoma progression and different responses of slow-cycling and active-cycling tumor cells to CRT.
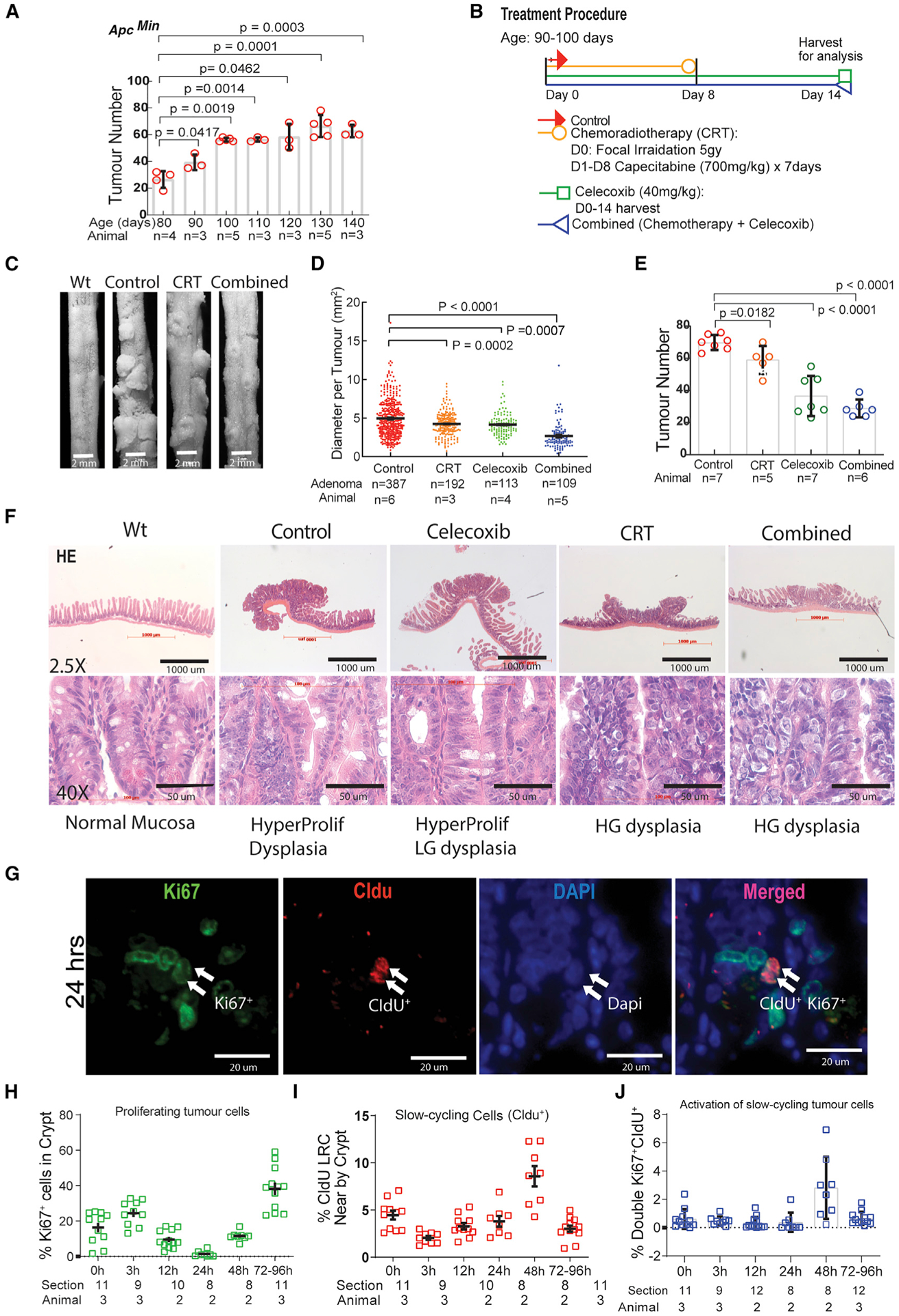
(A) Determining the rate and time frame of adenoma formation and development in ApcMin/+ mice.
(B) The procedures of CRT, celecoxib, and the combined therapies with the two.
(C) Images of tumors with different therapies.
(D and E) Measurement of adenoma size and number with different therapies.
(F) Pathohistological studies using H&E staining to monitor the progression or prevention of adenoma induced by CRT or celecoxib.
(G) Image showing division of slow-cycling cells.
(H–J) Measurement of percentage changes of active-cycling (H), slow-cycling (I), and activation of slow-cycling (J) tumor cells, respectively, in response to CRT. Ordinary one-way ANOVA multile comparisons with post-hoc t-tests (A,D,E), Means+/−SD. Data represent means with SD from a pool of section images with two to three independent experiments (H,I,J).
All groups were harvested at day 15. CRT reduced tumor diameter (Figures 1C and 1D) but had much less influence on tumor number (Figures 1C and 1E). Celecoxib had a similar reduction on tumor diameter but also significantly reduced tumor number. The Comb group showed reduced tumor number and dramatically reduced tumor diameter (Figures 1C–1E). We further examined the histological changes in adenomas following CRT and/or celecoxib therapy as revealed by hematoxylin and eosin (H&E) staining (Figure 1F). As Figure 1F shows (upper panel), when compared with the normal crypts and villi, morphological changes in the adenomas are consistent in size and quantity measurements (Figures 1C–1F). Pathophysiologically, normal intestinal mucosa showed columnar-to-cuboidal cells that were tightly packed with minimal branching and a straight luminal surface, whereas the adenomas in the control group showed hyperplasia (HyP) and LGD, with pseudostratified nuclei, minimal loss of polarity, occasional mitotic cells, and elongated nuclei that did not reach the luminal cell surface. Intriguingly, CRT-promoted adenoma progressed into the HGD with pathological features of stratified hyperchromatic nuclei reaching luminal border and prominent vesicular nucleoli nuclei, indicating increased euchromatin. The glands were back to back and mixed with either cribriform glands or irregularly branched crypts. The mitotic figures were prominent. However, celecoxib therapy prevented the adenoma progression from LGD to HGD. We noticed that, although the combined therapy led to the greatest reduction in tumor size, the tumors still maintained morphological HGD (Figure 1F, lower panel). This pathophysiological analysis indicates that CRT induces the progression of adenoma to the precancerous stage of HGD, whereas celecoxib maintains adenoma at the stage of HyP and LGD. This provides a dynamic window for us to study the response of TSC and TME to CRT and celecoxib.
Differential responses of active and slow-cycling tumor cells to CRT
Because CRT targets actively proliferating tumor cells, we hypothesized that slow-cycling tumor cells might be enriched in therapy-resistant or refractory cells, and these could contribute to tumor regrowth post-CRT. To test the response of proliferating and slow-cycling tumor cells to CRT, we differentially labeled active and slow-cycling cells as illustrated in Figure S1A (see details in STAR Methods). We then performed immunofluorescent (IF) analysis: chloro-deoxyuridine+ (CldU+) cells represented slow-cycling cells, Ki67+ cells represented proliferating cells, CldU+Ki67+ cells represented activated cells from the quiescent or slow-cycling state, and DAPI+ represented live cells (Figures 1G and S1B).
From 0 to 24 h, we observed a substantial reduction of proliferating cells as measured as percentage of Ki67+/DAPI+ to avoid counting dead cells. (Figure 1H). In comparison, we observed relatively stable populations of CldU+ and CldU+ Ki67+ cells (Figures 1I and 1J). From 24 to 96 h, we observed a biphasic change in the number of CldU+Ki67+ cells: an increase between 24 and 48 h, suggesting activation and division of slow-cycling cells (Figures 1G–1I), and a decline after 48 h, reflecting both a dilution of CldU during cell division and a reversion to quiescence (CldU+Ki67−) (Figures 1I and 1J). The remaining CldU+Ki67− cells at 96 h post-CRT most likely represent activated slow-cycling cells that have reverted to a quiescent state. Intriguingly, there were many more Ki67+ cells (~10%) than CldU+Ki67+ cells (3%) at 48 h (compare Figures 1H and 1J). This is most likely due to proliferation of progeny cells derived from the initial division of slow-cycling tumor cells post-CRT (Figure 1G).
In summary, these observations support our hypothesis that slow-cycling cells are resistant but proliferating cells are sensitive to CRT. In addition, slow-cycling cells not only survived CRT but also were activated at 24 h and proliferating and generating progeny at 24–48 h post-CRT. A portion of activated (CldU+Ki67+) cells reverted to quiescent state (CldU+Ki67−) after division, but most of the derived progenies underwent rapid proliferation (CldU−Ki67+). Thus, this temporal series of analyses imply that the slow-cycling tumor cells are indeed therapy refractory, serving as a potential resource for tumor regrowth post-CRT by enriching for TrTSCs.
A comprehensive dissection of adenoma during CRT using scRNA-seq analysis
To test our hypothesis that slow-cycling tumor cells enrich for TrTSCs, to which TME may also play a protective role, we used scRNA-seq to analyze adenoma in ApcMin/+ mice to identify TrTSCs and the TME. We isolated and enriched viable single cells from EP and mesenchymal cell (MC) fractions of intestinal adenoma of ApcMin/+ mice at various time points during CRT and conducted scRNA-seq (see STAR Methods) (Figure 2A; Table S1). We then combined the scRNA-seq dataset of 79,801 total cells from various time points with and without CRT and performed Uniform Manifold Approximation and Projection (UMAP) analysis (Becht et al., 2018) (Figure 2B). According to the gene expression profile (Figures 2D and 2E), we categorized the UMAP (Figure 2B) into five subgroups: EpCs, TSCs, stromal cells, innate immune cells, and adaptive immune cells.
Figure 2. Analyses of therapy-resistant (Tr) cell population and TME cellular components using scRNA-seq approach.
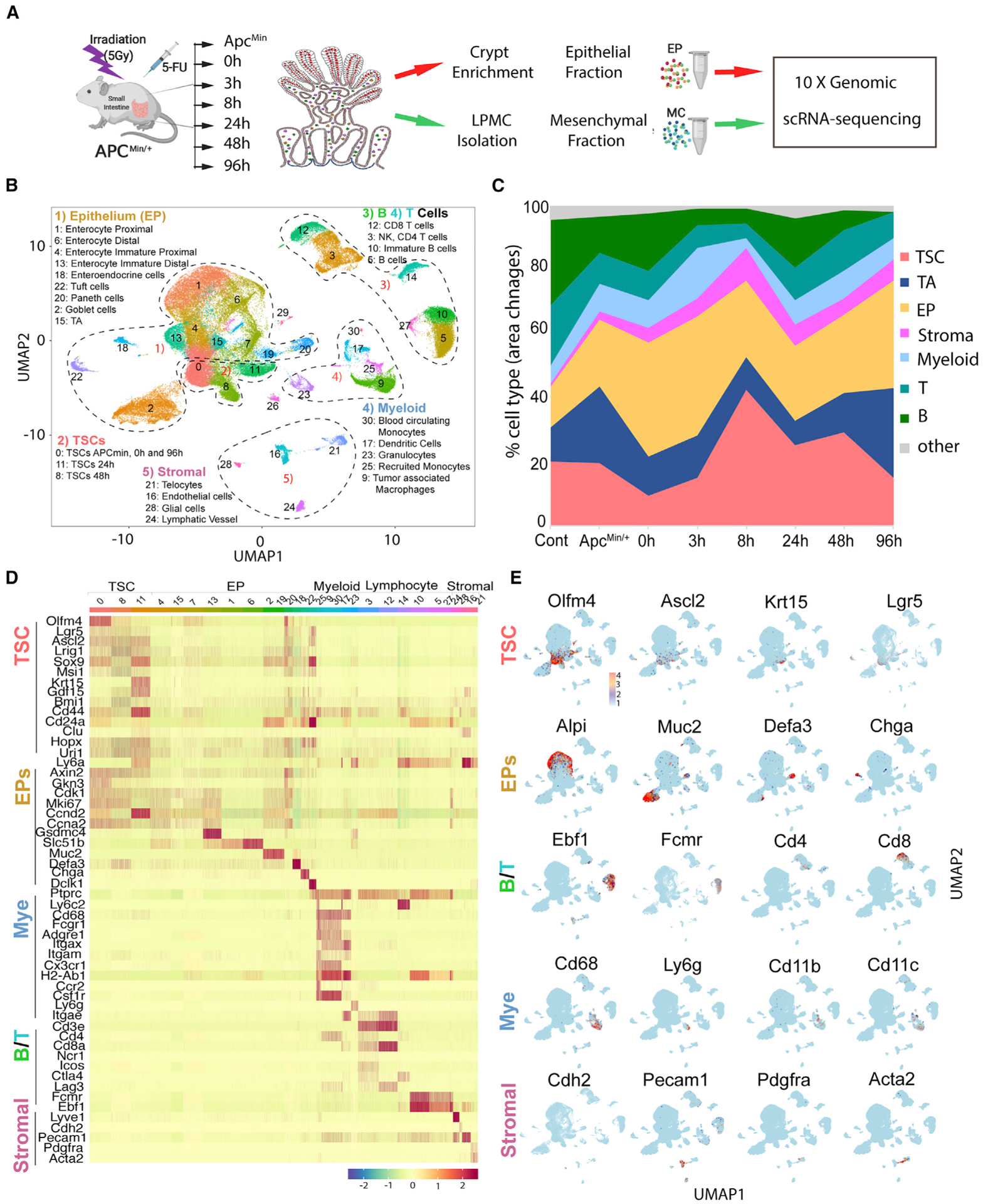
(A) The experimental procedure for single-cell harvest from adenoma, EpC versus MC separation, and scRNA-seq analysis.
(B) UMAP analysis of scRNA-seq data combining all cells isolated during CRT.
(C) Percentage of different cell types as indicated between WT control and adenoma (Apcmin), as well as during CRT.
(D) Heatmap of known genes for categorizing different cell groups.
(E) Expression pattern of some representative genes in UMAP.
We next analyzed the gene expression profiles of EpC and TSC groups (Figures 2D and S2). TSCs included three subclusters (SCs): C0, C8, and C11, representing TSCs at indicated time points of CRT. TSCs expressed genes found in ISCs, including Lgr5, Ascl2, Olfm4, Hoxp, Lrig1, and Bmi1, as well as surface marker genes, including CD44 and CD24a (Figures 2D, 2E, and S2). EpCs include transit-amplifying (TA) cells (C4, C7, C13, C15) (Figure S3A, Alpi; Figure S3B, mKi67), enteroendocrine (EE) cells (C18, ChgA), Tuft cells (C22, Dclk1), Goblet cells (C2, Muc2), Paneth cells (C19, 20, Defa3), and enterocytes (C1, C6, Alpi). Paneth cells transcriptomics include Lyz and other immune genes and thus more closely resemble innate immune cells compared with other EP lineages (Figures 2D, 2E, and S3A; Table S2).
The TME is composed of stromal cells and immune cells (Figures 2B–2E). Stromal cells include telocytes (C21, Pdgfra), myofibroblasts (the bridge between C16 and C21, Acta2), lymphatic (C24, Lyve1) and blood endothelial cells (C16, Pecam1), and glial cells (C28, Ncam1, and Gfap). Innate immune cells include circulating (C30, Ly6c2) and recruited monocytes (RMs; C25, Ccr2), dendritic cells (C17, CD11c), granulocytes (C23, Ly6g), and macrophages (C9, CD11b). Adaptive immune cells include regulatory T cells (C3, Cd4), cytotoxic T cells (C12, Cd8), natural killer (NK) cells (between C3 and C12, Ncr1), and B cells (C5, C10, C27, Ebf1) (Figures 2D and 2E; Table S2).
The percentage of each cell type within the entire cell population in scRNA-seq data was used to calculate the relative enrichment and dynamic change of each cell type (Popescu et al., 2019; Saunders et al., 2018). As Figure 2C shows, from 3 to 8 h post-CRT, TA cells were reduced most rapidly, followed by a large loss of EpCs. In contrast, the TSC percentage was relatively enriched. In comparing the percentage of immune cells, it revealed a decline of T and B cells but an increase in innate immune cells. The percentage of stromal cells remained relatively stable. MDCs were seen to increase in two waves: the first at 0–3 h and the second at 8–24 h; both were prior to TSC increase comparing with the rest of the tumor cells.
Identification of TrTSCs that survive from CRT, restore the TSC pool, and support tumor regrowth
To identify TrTSCs, we first examined changes in EpCs during CRT. Figure 3A shows a distribution of TSCs (C0, C8, C11), TA, and all five EP lineages (Figure 2). One day after radiotherapy and prior to chemotherapy, there was a decline in TA cells and a portion (cycling) of TSCs (Figure 3B, compare −12 h and 0 h). Between 3 and 8 h, there was a more dramatic reduction of EpCs, including differentiated cells. The lowest survival cell number was seen at 8 h post-CRT, which substantially enriched the TSC population (C11) as a result of a large loss of proliferating tumor cells (Figure 3B). We also observed a shift in the TSC cluster from 0 to 11, then to 8 (Figure 3B, 8–24 h), and finally from 8 to 0 (Figure 3B, 48–96 h). Compared with 48 h, there was a large increase in TA cells and all EP lineages at 96 h, suggesting that restored TSCs subsequently produced TA cells and all other lineage cells (Figure 3B, 96 h).
Figure 3. Identification of genes predominantly expressed in TrTSCs.
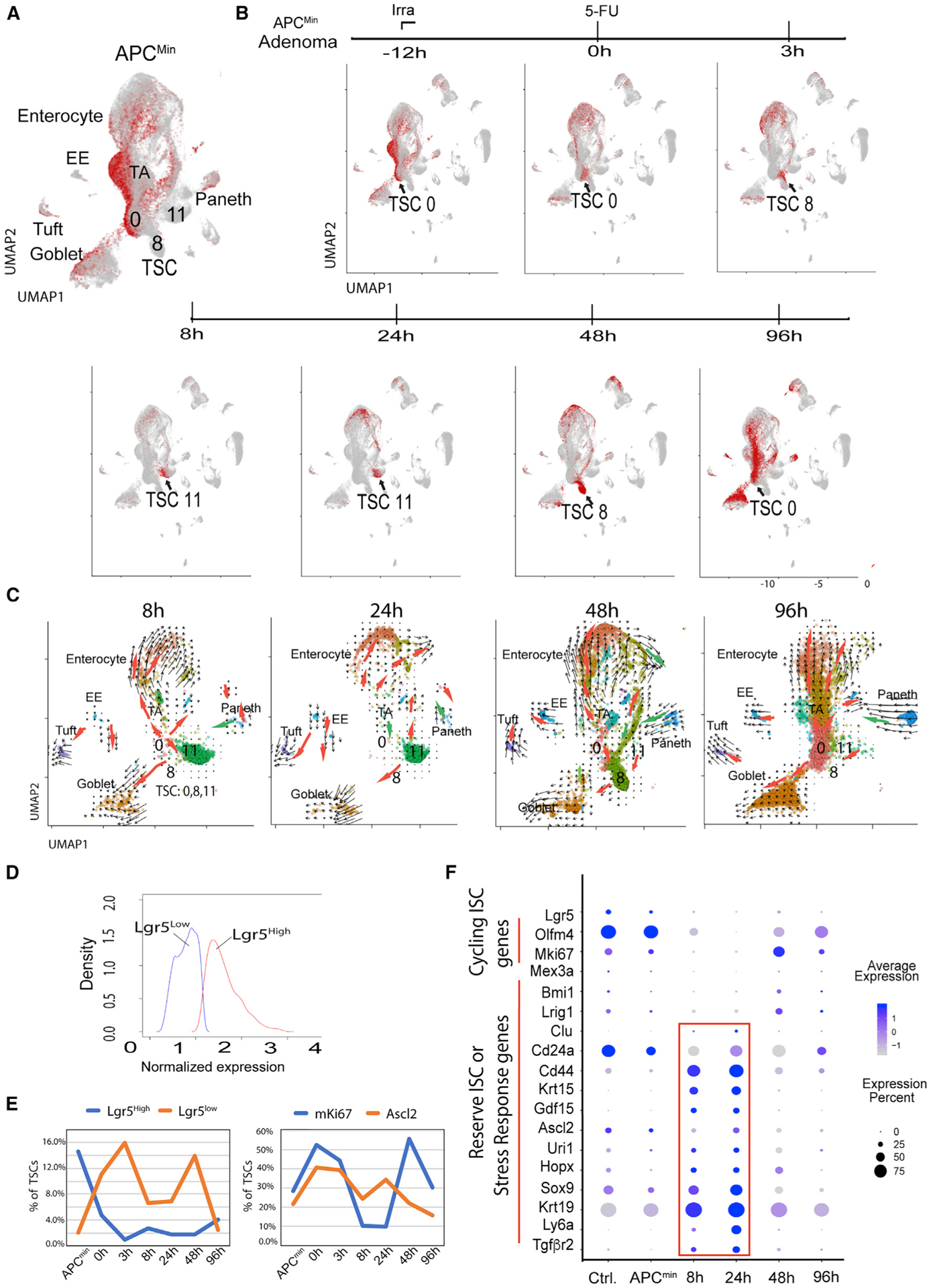
(A) TSC clusters 0, 8, and 11 and distribution of epithelial lineages produced by TSCs.
(B) Overall responses of different cell types to CRT and TrTSC population.
(C) Trajectory analysis of epithelial fraction of adenoma between 8 and 96 h after CRT.
(D and E) Determining Lgr5hi and Lgr5lo cell subsets and their corresponding changes during CRT.
(F) Dot blot analysis of a list of genes with information of cell numbers and gene levels.
To further verify whether the TrTSC population supports the regrowth of the tumor cells post-CRT, we conducted a trajectory analysis using the RNA velocity program (La Manno et al., 2018). Given that the lowest number of surviving tumor cells post-CRT is at 8 h (Figure 3B), we chose the 8- to 96-h window post-CRT to analyze the regeneration process in the EP fraction of adenoma (Figure 3B). At both 8 and 96 h, according to the arrows generated by the RNA velocity, the TSC population gave rise to Goblet cells, EE, and further to Tufts cells, enterocytes (Figures 2B–2E) via TA cells, and TA to Paneth progenitors and further to Paneth cells (Figure 3C, red arrows). Only a portion of Paneth cells seem to trend toward dedifferentiation from Paneth to TA cells (Figure 3C, green arrows). At 24–48 h, the trajectory was similar, apart from an increased trend toward reprogramming at 24 h. This increase was more prominent at 48 h, as indicated by green arrows involving multiple lineages.
In summary, we used scRNA-seq analysis to monitor the responses of different subtypes of tumor cells to CRT at various time points. Although a large portion of cycling stem and progenitor cells, as well as differentiated tumor cells, were eliminated, we found a specific TSC subset that survived CRT (Figure 3B, 8 h), restored the TSC pool (Figure 3B, 24–48 h), and supported robust tumor regrowth (Figure 3B, 96 h). Furthermore, results from the trajectory analysis indicate that the TrTSC population is derived from (1) surviving TSCs (Figure 3C, at 8–24 h) and (2) reprogramming from progenitor and differentiated cells (Figure 3C at 48 h). Intriguingly, it seems that surviving TSCs gave rise to progenitor and lineages (8–24 h) prior to being reprogrammed from progenitor and differentiated cells (48 h).
Identification of genes expressed in TrTSCs
Next, we identified genes expressed in TrTSCs. First, we pooled all TSC clusters 0, 8, and 11 together with wild-type (WT) ISCs as a control. The UMAP analysis classified TSCs into eight SCs in line with different CRT time points. We noticed that stem (S) SC1,2 representing TSCs in ApcMin/+ was different from the WT SSC0 control (Figure S3B). The TSC cluster dynamically changed from the initial position of SSC1,2 to the final position of SSC8 at 96 h (Figure S3B). The restored SCC8 is very close to but different from the initial SSC1,2. This is further supported by differential gene expression and Gene Ontology (GO) term analysis, which not only revealed several upregulated signaling pathways (including MAPK and TP53) but also an unexpected feature of co-existence of glycolysis and fatty acid metabolism in TSCs post-CRT (Figures S3D and S3E).
In examining the expression of known ISC genes, we observed that Lgr5+, Olfm4+, and mKi67+cells were largely reduced at 24 h (Figures 3D–3F and S2). We noticed that in terms of percentage, the Lgr5hi subset was predominant over the Lgr5lo subset in percentage without CRT. Post-CRT, however, the Lgr5hi subset dramatically declined (Figure 3E, at 3 h, left panel), while the Lgr5lo subset increased at 3–48 h; the declining Lgr5lo subset inversely correlated with the increasing Lgr5hi subset at 96 h, suggesting that the former may give rise to the latter during the restoration of the TSC pool (Figure 3E). In contrast, another ISC representative gene Ascl2+ TSCs had a similar pattern to the Lgr5lo subset during CRT (Figures 3E and 3F).
We next examined genes associated with reserve, revival, or regenerative (r) ISCs (Ayyaz et al., 2019; Bankaitis et al., 2018; Cheung et al., 2020; Li and Clevers, 2010). Those known rISC genes, including Krt15, Clu, Ly6a (Sca1), Uri1, Hopx, Sox9, and newly identified GDF15 and Tgfβr2, were significantly upregulated only during CRT (Figures 3F and S2). This represents a common feature among these rISC genes in response to stress (highlighted in red box in Figure 3F). The Asl2+Krt15+ double-positive (DP) TSCs were predominantly detected between 8 and 24 h post-CRT. Similarly, CD24+CD44+ DP cells were more enriched during the same time window (Figures 3F and S2). Unlike CD44 and Krt19, which increased during CRT but were more broadly expressed (Asfaha et al., 2015), the percentage of Bmi1-, Clu-, and Lrig1-expressing cells was relatively low, but indeed enriched in TrTSCs (Figures 3F and S2). In summary, Ascl2+Krt15+ together with other stress-response genes as described above are predominantly expressed in TrTSCs between 8 and 24 h after CRT, prior to the TSC expansion between 24 and 48 h (Figures 3D, 3F, S2, S3B, and S3C).
We found that Cdkn2c (P18, a G1-phase CDK inhibitor) could be detected in a small number of TSCs with or without therapy, whereas expression of Cdkn1c (p57, a G0-phase CDK inhibitor) was not detected (Figure S2). This observation suggests that TrTSCs may have been arrested at the G1 phase.
Identification of dynamic signaling modules between TME and TrTSC during CRT
Cell-cell communication mediated by ligand-receptor complexes is critical to coordinating diverse biological processes. The recently reported CellPhoneDB analytic tool is an integrated database resource with a statistical framework to identify which ligand-receptor pairs display significant interactions between two cell types from single-cell transcriptomics data (Efremova et al., 2020). We applied CellPhoneDB analysis to our scRNA-seq data to identify interacting ligand-receptor pairs between different TME cell types and TSCs during CRT: Figure 4A shows signaling modules for TME transition to TSCs, and Figure S4A displays signaling modules for TSCs transition to TME.
Figure 4. Analysis of TME-TSC signaling modules by CellPhoneDB and MDC subclusters (SCs).
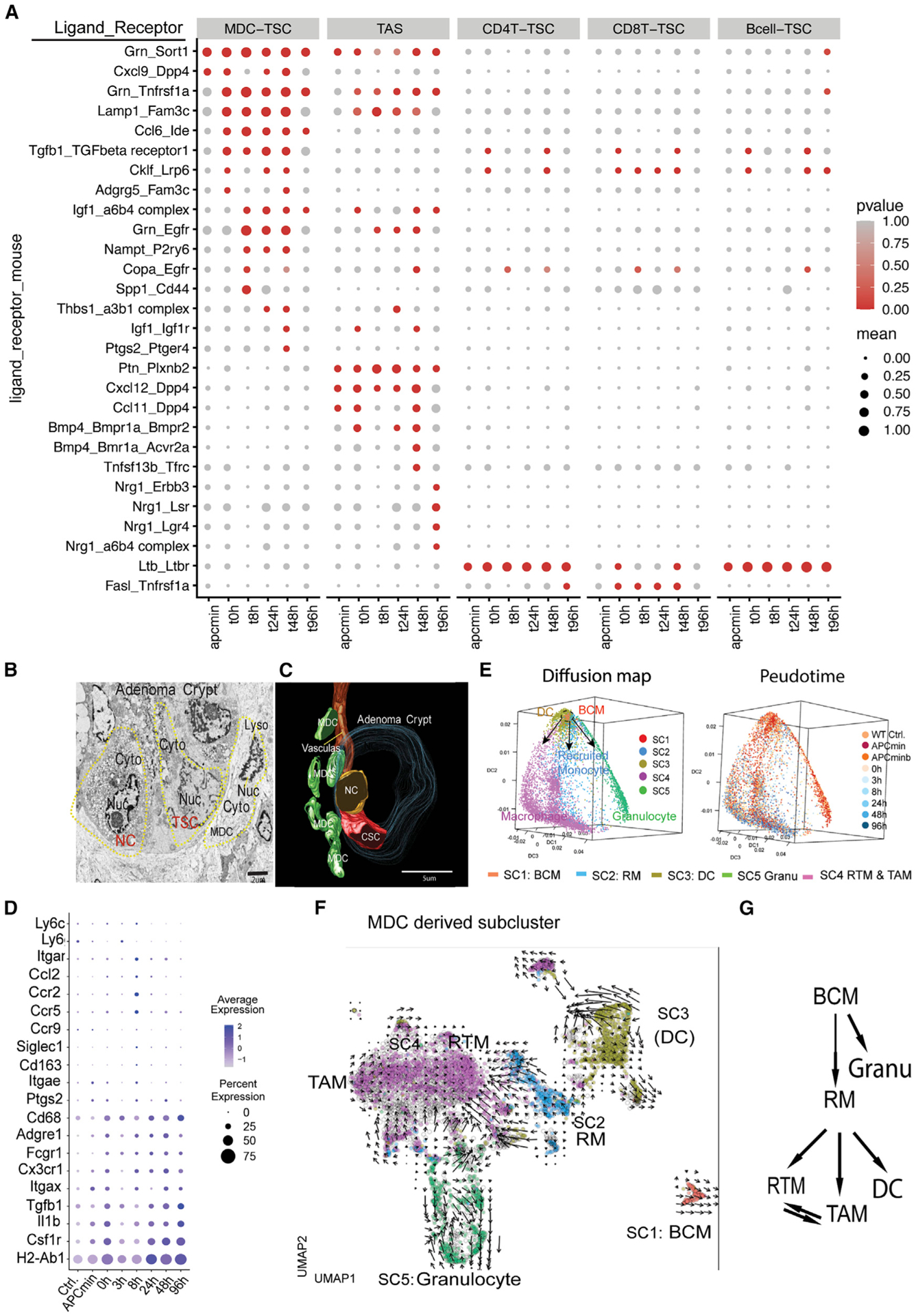
(A) Signaling modules indicated by ligand-receptor pairing between TME cellular components and TSCs identified using CellPhoneDB.
(B and C) Recruiting of TAMMs to the TSC niche revealed by TEM (B) and 3D-SEM (C).
(D) Dot plot showing representative gene expression within MDCs during CRT.
(E) Diffusion map and time change related to SCs in MDCs with the genes in each SC listing in Table S1.
(F) Trajectory analysis of SCs of MDCs using RNA velocity program.
(G) Lineage relationship between different MDC subpopulations indicated by combining diffusion map and RNA velocity analyses.
We unexpectedly found that TSCs directed multiple TME cell types to send immunosuppressive signals, which in turn protected TSCs from CD8 T cell attack. For example, CD8 T cells provide an apoptosis-inductive signal to TSCs via FASL-TNFR (Caulfield and Lathem, 2014) signaling in response to CRT (Figure 4A). Inversely, TSCs provide macrophage migration inhibitory factor (MIF) signal transition to multiple TME cells, which in turn (Figures S4A and S4B) generate immunosuppressive signals via CD74 receptor to suppress CD8 T cells (Figueiredo et al., 2018). There is also a predominant RPS19 (ribosome protein S19)-C5AR1 (complement C5a receptor 1) interaction between TSC-MDC, which in turn can upregulate immunosuppressive signaling (Figures S4A and S4B). Interrupting the RPS19-C5AR1 interaction was reported to increase the infiltration by CD8 T cells in breast cancer (Markiewski et al., 2017).
Figure S5 illustrates sequential events in response to CRT: TrTSCs survived, while cycling stem cells were eliminated between 0 and 8 h, followed with upregulation of stress-response genes and the related signaling in TrTSCs (8–24 h) (Figure 3F), TSC expansion at 24–48 h, and restoration at 96 h (Figures 3B andS4 S5). During these processes, dynamic signals were seen from various TME cell types regulating TSCs (Figures 4A and S4C). At the initial stage of CRT (0–8 h), CRT caused tumor cell apoptosis, which induced an inflammatory response as reflected by Granulin (GRN)-Sortilin (Sort1, a TNFR family) signaling from MDCs and TASs transition to TSCs. GRN-Sort1 signaling plays an anti-inflammation role in wound healing (Rhost et al., 2018). Between 8 and 24 h post-CRT, upregulations of TGF-β1 and Osteopontin (OPN encoded by Spp1) signals, respectively, from MDCs (Figure 4A) coincide with upregulations of corresponding TGF-βR and CD44 receptors in TrTSCs (Figure 3F), respectively (Figure 4A). Interestingly, both TGF-β1 and OPN-CD44 signals were reported to be involved in controlling immune checkpoint gene expression (Klement et al., 2018; Miao et al., 2019). During the TSC expansion period (24–48 h after CRT), MDCs seemed to play a critical role because there were multiple signals between MDCs and TSCs, including CCR5-CCL1/5, insulin growth factor 1 (IGF1)-IGF1 receptor (IGF1R), and PGE2 (reflected by Ptgs2)-Ptger4 (Prostaglandin E Receptor EP4) (EP4). CCRL1,5/CCR5 signaling has the potential to recruit additional MDCs to the TSC site (Walens et al., 2019). Both IGF1 and PGE2 signaling may contribute to TSC expansion via MAPK Figure S2(Figure S5) and EP4 pathways, respectively (Choi et al., 2008; Greenhough et al., 2009). PGE2 correlates with the upregulation of Pgst2 (COX-2), an enzyme essential for PGE2 synthesis (Greenhough et al., 2009). The identified PGE2-EP4 interaction time coincided with the TSC expansion time window at 24–48 h after CRT, consistent with other observations using different approaches (please see below). In line with our observation from immunostaining that MDCs became distant from slow-cycling (CldU+) cells at 96 h after CRT (Figure S1B), here, CellPhoneDB analysis also showed that a declining MDC-derived signaling and an increasing TAS-dependent signaling was a trend at 96 h after CRT. For example, NGR1 (Neuregulin 1, an EGF ligand) was shown to be involved in a protein complex with multiple receptors between TASs and TSCs to facilitate Wnt and EGF signaling (Figures 4A and S5).
Dynamic recruitment of MDCs to the slow-cycling cells in response to CRT
CellPhoneDB analysis showed complex signaling from many TME cell types and suggested a dual role of MDCs in both suppressing cytotoxic CD8 T cells and prompting TSC survival and expansion during the acute response to CRT. We parallelly investigated the interaction between MDCs and TrTSCs using the IF assay. We examined the spatial relationship between CD68+ MDCs and proliferating (Ki67+, green), as well as slow-cycling (CldU+, red), cells in response to CRT (Figure S1B). At 0 h, CD68+ MDCs were relatively distant from crypts and localized in the centralized region of adenoma (Figure S1B, 1st column). At 3–24 h after CRT, these cells moved closer, some were adjacent to slow-cycling cells (Figure S1B, 2nd and 3rd columns) but separated from CldU+ (slow-cycling) cells at 96 h after CRT (Figure S1B, 4th column). We also compared the pairing of CD68+ MDCs/CldU+ cells to that of CD3+ T/CldU+ cells (Figures S1B and S1C). Intriguingly, an increased CD68+ MDCs/CldU+ cell pairing inversely correlated with a decreased CD3+ T/CldU+ cell pairing between 0 and 48 h after CRT (Figures S1D and S1E). Taken together, this suggests a potentially dual role of CD68+ MDCs in regulating slow-cycling tumor cells and inhibiting T cells following CRT.
To examine the nature of cells in the area to which CD68+ MDCs were recruited, we used EM to analyze the cellular ultra-structure of adenoma (Figures 4B and S6A). Under transmission EM, stem cells were clearly distinguishable from differentiated cells, with distinct features including a large nucleus/cytoplasm ratio (reflecting an undifferentiated state) and evenly distributed and less-condensed ribosomes (reflecting a low translational rate). Surviving stem-like cells (TSC) were further distinguished from neighboring necroptotic cells (NCs), because the NC had increased cell volumes, swollen organelles, plasma membrane permeabilization, and cellular collapse as observed starting at 3 h after CRT (Figure 4B).
Consistent with the results of IF assay (Figure S1B), we noted that macrophage-like cells were recruited to sites adjacent to stem-like cells. Macrophages have an irregularly shaped nucleus and cell membrane, a few mitochondria, and multiple lysosomes in the cytoplasm (Figure 4B). We further used three-dimensional segmentation electron microscopy (3D-SEM) technology to reconstitute 3D structures of the related crypt region of adenoma to examine the spatial relationships of these cell types. A reconstitution of 212 slices of SEM sections revealed recruitment of MDCs (in green) from blood vessels to the sites adjacent to stem-like cells (in red) (Figures 4C and S6A; Videos S1 and S2). In conclusion, we further confirmed with transmission electronic microscopy (TEM) and SEM technology that, in response to CRT, MDCs were recruited to the site of stem-like cells.
Using these diverse approaches of IF and TEM/SEM, we showed that slow-cycling tumor cells enrich for TrTSCs. MDCs were dynamically recruited to the site of potentially slow-cycling TrTSCs following CRT, suggesting a role for them to regulate TrTSCs under stress.
Dynamics of MDC subpopulations revealed by scRNA-seq analysis
We selected the MDC clusters from scRNA-seq data for further UMAP analysis. Based on the report (Mildner et al., 2017) that categorized stages and lineages of MDCs with associated surface markers and transcription factors (Figure 4D), we further classified MDCs into five SCs (Figure 4E; Table S3): blood-circulating monocytes (BCMs, SC1), RMs (SC2) that largely overlap MDSCs (myeloid-derived suppressor cells), dendritic cluster (DC, SC3), resident tissue macrophages (RTMs, SC4) that largely overlap with tumor-associated macrophages (TAMs), and granulocytes (SC5) (Table S3).
Next, we used the diffusion map approach, which reflects a stochastically and continual computational lineage modeling, to analyze MDCs in conjunction with the corresponding time course of CRT (Figure 4E). In response to CRT, the dominant population changed from BCMs (0–3 h) to RM (3–24 h) and further to TAM and RTM (48–96 h). In addition, we conducted RNA velocity analysis (Figure 4F) and further showed that RM (SC2) gave rise to DC (SC3) and TAMs/RTMs (SC4). Within SC4, TAMs further converted to RTMs (Figure 4G).
Inhibition of the PGE2 pathway reduces MDCs’ ability to promote adenoma-organoid growth
Moving forward, we selected a critical ligand-receptor pair to conduct a functional study. TrTSC expansion occurred at 24–48 h after CRT, during which the interaction between PGE2 and EP4 was prominent (Figure 4A). Although Ptgs2 expression remains predominant in MDCs in adenoma (Figure S7A), expression of Ptger4 was very broad but became relatively enriched in TSCs at 24–48 h after CRT, largely as a result of elimination of other EP4+ tumor cells (Figure S7A). These observations led us to hypothesize that MDCs are a main resource of PGE2, which contributes to promoting TSC expansion via EP4-mediated signaling after CRT.
To test this hypothesis, we utilized an ex vivo 3D organoid culture system to assess whether MDCs support organoid growth (Sato et al., 2009; Wang et al., 2013). We used microbeads (Method) to enrich monocyte components of MDCs (M-MDCs, Ly6c+Ly6gdimCD11b+CD11clo) from the intestine of APCMin/+ mice following the previously reported method (Chun et al., 2015; Tavazoie et al., 2018). We then co-cultured M-MDCs with adenoma-derived organoids from the same mice for 8 days in consideration of the short lifespan of macrophage in culture. Attachment of macrophage-like cells to the cultured organoids was verified using scanning EM (Figures S6A and S6B). We found that M-MDCs, which most likely converted to TAMs during the culture with organoids, stimulated the budding of crypts and overall organoid growth (Figure 5A). To quantify the overall effect of M-MDCs on adenoma-derived organoid growth, we measured organoid area and number in a projected image. We observed a significant increase in the organoid mass (= No. × area) (Figure 5B). To test whether the effect was dependent on the COX-2-PGE2-EP4 pathway, we used a COX-2 inhibitor, celecoxib, or the EP4 inhibitor, AH23848. Although showing much less inhibition on the organoid growth in the control (no M-MDC) (Figure 5B, left panel), we observed substantial inhibition on M-MDC-enhanced organoid growth by adding celecoxib or AH23848 individually or in combination. Of note, two independent M-MDC added experiments showed similar trends of significant inhibition using either celecoxib or AH23848 with a difference of lower M-MDC viability in exp2 (76%) compared with that in exp1 (86%) (Figures 5A and 5B, right panel).
Figure 5. Roles of TAMMs in supporting organoid culture in vitro and tumor growth in vivo.

(A and B) Images of growth changes in the co-culture of adenoma-derived organoids with or without M-MDCs and with or without adding EP4 and COX-2 inhibitors (A), and the quantification of organoid mass (= No. × area) under different conditions. This quantification was based on two independent experiments each with multiple replicates. Each experiment was normalized to the mean of the crypt-only group, and then two experiments were subject to statistical analysis. Two-way ANOVA with post hoc test was used to compare group means. (B). For normalized mass data: we fit a two-way ANOVA model with all data from two experiments. Then, post-hoc t-tests were performed to test specific comparisons of interest. P-values were adjusted using the sidak method. The same experiment was repeated two times at different times and data are Mean+/− SEM.
(C and D) IF assay of crypt budding and p-ßcatS552 detection (C), and western blot analyses of the p-ßcatS552 protein level in organoid culture with or without adding inhibitors (D).
(E and F) Flow cytometry analysis of RMs (E) and TAMs (F) and inhibition of Cox-2 reduced both RMs and TAMs. T-tests with Means +/− SD.
(G) Dot plot showing gene expression associated with RM and TAMs.
(H) Adenoma growth was substantially reduced by Clodrosome compared with Encapsome.
(I–K) Clodrosome caused significant depletion of CD11bintCX3CR1+ cells, which was shown to be Ly6c−MHCII+, thus fitting the definition of TAMs (B). H-K, T-tests with Welch’s correction Means +/−SD.
We next examined the downstream effect of the COX-2-PGE2-EP4 pathway using anti-phosphorylated β-catS552 (anti-p-β-catS552) antibody. This is because PGE2-EP signaling activates AKT, which in turn enhances β-catenin signaling as reflected by p-β-catS552 (He et al., 2007). The specificity of anti-p-β-catS552 antibody was verified in the β-catenin KO mouse model (Figure S6C). We found that adding EP4 and COX-2 inhibitors reduced the number of p-β-catS552-expressing cells analyzed with the IF assay and reduced the crypt-budding event (Figure 5C). We also performed western blot analysis on the co-cultured organoids and found that adding celecoxib or AH23848 indeed reduced the protein level of p-β-catS552 (Figure 5D). Given the known p-β-catS552 as a readout of coordinated AKT and β-catenin signaling in promoting stem cell expansion and tumor initiation (He et al., 2007; Perry et al., 2011, 2020), our result shows that inhibition of the PGE2 pathway indeed suppresses TSC expansion and the subsequent crypt budding, both of which are critical to tumor growth.
Inhibition or depletion of TAMMs reduced tumors
To verify the function of M-MDCs in supporting tumorigenesis in vivo, we first used flow cytometry to distinguish subsets of M-MDCs, including RM (Figure S7B) and TAM (Figure S7C). We then examined the effect of CRT and celecoxib on these MDCs subsets (Figure 1B and 7 on and 7 off procedure). PGE2 is known to promote differentiation of RMs to TAMs (Liu and Cao, 2016; Rong et al., 2016). Although CRT alone reduced RMs (which enriched proliferating progenitor cells) without affecting TAMs measured 7 days after CRT, celecoxib reduced both RMs and more significantly TAMs. The combination therapies significantly reduced both RMs and TAMs compared with that of control (Figures 5E and 5F). Furthermore, the dot blot based on scRNA-seq data confirmed the conversion of RM (Ly6c2+MHCII−) to TAM (Ly6c2−MHCII+) during CRT (Figure 5G). It also revealed two waves for TAM formation; both TAM peaks were just prior to the recruitment of MDC to TSC (Figures S1B and S4A).
Given that adding M-MDCs increased adenoma-derived organoid growth (Figures 5A and 5B) and reduced TAMMs (Figures 5E and 5F) correlated with decreased tumorigenesis in vivo (Figures 1A–1E), we further tested whether direct depletion of macrophages could affect tumorigenesis. Because Clodrosome was shown to deplete macrophages (Mok et al., 2014), we examined the effect of Clodrosome in the APCMin/+ model and, using Encapsome as a control, analyzed 7–9 days after three treatments. We first observed that adenomas shrank dramatically and that the total numbers of adenomas were significantly reduced (~50%) by the Clodrosome compared with Encapsome treatments (Figure 5H). Using flow cytometry, we found that a population of CD11bloCX3CR1+ was largely reduced (60%) in the Clodrosome-treated group compared with the Encapsome-treated control group (Figures 5I–5K). Further examining the surface markers, we confirmed that the Clodrosome-sensitive population was predominantly (90%) MHCII+ and LyC6− (Figures S7D and S7E), fitting the definition of TAMs. Thus, depletion of TAMs using Clodrosome significantly reduced adenoma number in ApcMin/+ mice.
CD68+ TAMMs promote TSC expansion and tumorigenesis in vivo
We next investigated a role of TAMMs in regulating TSCs in vivo. First, we observed co-staining of Krt15 with CldU+ slow-cycling cells after CRT using the IF assay (Figure 6A). Although the incompatibility of anti-Bmi1 with anti-CldU antibodies affected our ability to perform the co-staining of Bmi1 and CldU, we detected that both Krt15+ and Bmi1+ TrTSCs were enriched in the TSC cluster at 8–24 h after CRT (Figure 6B). These observations suggest that Bmi1 is enriched in slow-cycling TrTSCs. We therefore used the Bmi1-CreER mouse model to conduct the clonal formation and linage tracing assay. We obtained the adenoma model by crossing Bmi1-CreER with R26-LSL-GFP and Apcmin/+ mouse lines, thus enabling us to study Bmi1-driven adenoma clones (Figure 6C). We observed that Bmi1-CreER-derived GFP+ adenoma clones (Figure 6D) include all four epithelia lineages (Figure 6E). We detected association of CD68+ TAMMs with Bmi1-GFP+ single cell (Figure 6F, left panel) and with Bmi1-GFP+ clones in adenoma (Figure 6F, right panel). We then observed a trend in which inhibition of Cox-2 reduced the number of Bmi1-GFP+ cells at the single-cell level (Figure 6G) and reduced the number of Bmi1-CreER-derived GFP+ crypts in APCMin/+ mice (Figure 6H). In contrast, CRT only slightly affected the numbers of Bmi1-GFP+ TSCs (Figure 6G) and Bmi1-Cre-derived tumor clones as measured 7-days after CRT (Figure 6H).
Figure 6. Testing a role of PGE2 signaling in promoting TSCs propagation and tumorigenesis using lineage tracing assay.
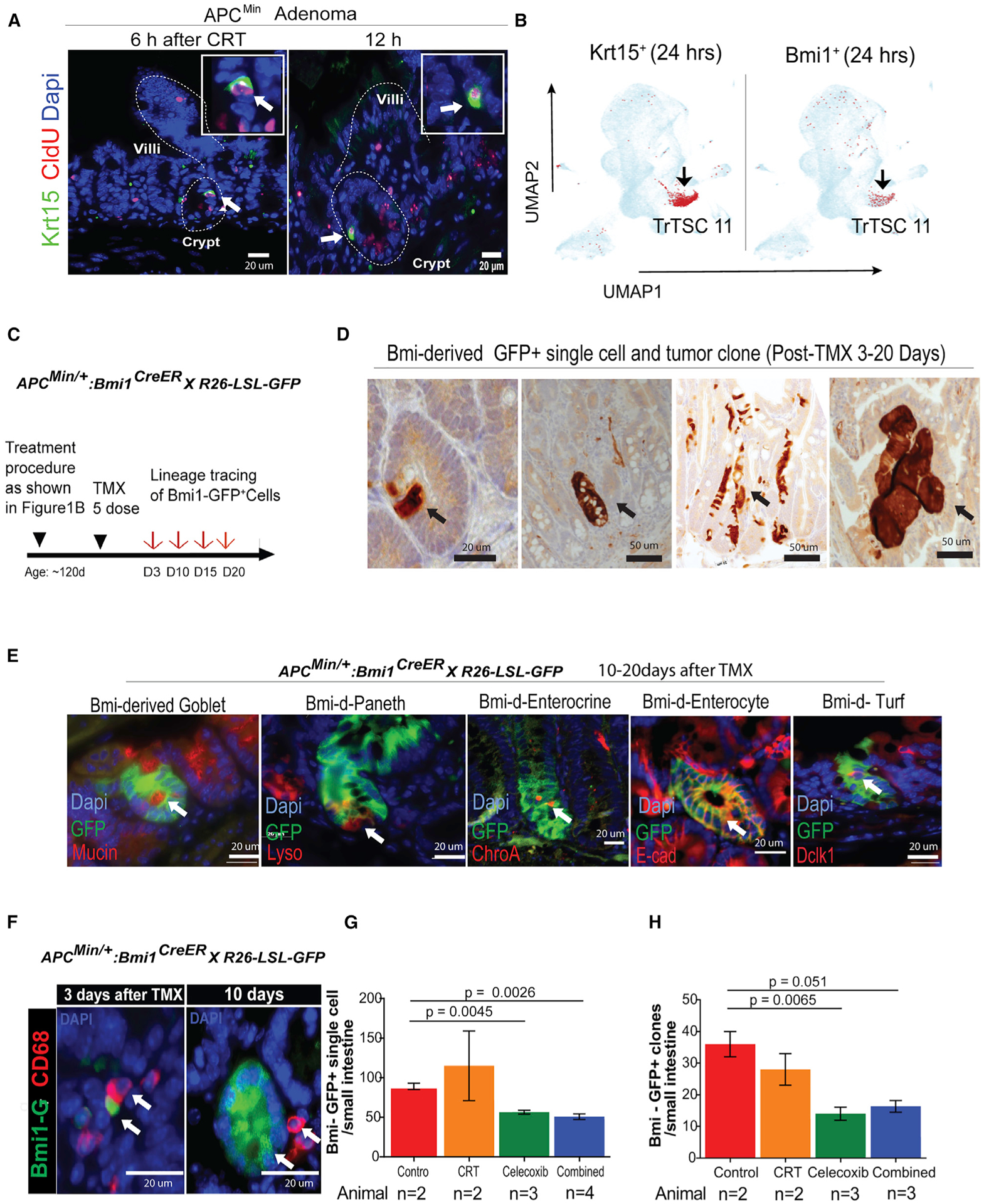
(A) IF co-staining of TrCSC markers Krt15 (green) with CldU+ (red) slow-cycling cells.
(B) Predominant expression of Krt15 or Bmi1 in TSC C11 at 24 h after CRT.
(C) The procedure for marking Bmi1-Cre-derived single TSCs.
(D) IH tracing Bmi1-derived tumor clones (brown).
(E) Lineage tracing showing that Bmi1-CreER-derived clones include all four epithelial lineages in intestine.
(F) After TMX induction, association of CD68+ with Bmi1+ (green) single cells and Bmi1-derived tumor clones.
(G) Percentage of Bmi1+ single cells per small intestine with different therapies (*n = 2–4 mice).
(H) Quantification of Bmi1-Cre-derived tumor clones per small intestine with different therapies (*n = 2–3 mice).
*Because of the COVID-19 pandemic, the mouse experiment to increase the control group of animal number in the Bmi1-CreER:R26LSL-GFP:ApcMin/+-induced lineage assay was affected. T-tests, Means+/−SD.
A potential role of TAMMs in promoting TSC/CSC proliferation in human CRCs
Finally, we determined to what extent our finding made in the murine adenoma model study may be relevant to human CRC. Up to 35 patients’ sections out of 59 samples of human CRC patients were qualified for conducting H&E and IF assays. We first categorized the stages of adenoma formation, progression, and cancer development according to the pathological feature of each patient’s CRC section (Figures 7A and S8). Using Krt15+Ascl2+ DP cells as TrTSC markers, we examined the association of CD68+ MDCs with TSCs/CSCs at different stages of tumor and cancer development.
Figure 7. Examination of TrTSC/CSC markers and the association of MDCs with TSC/CSC at different adenoma progression and cancer stages in sections from human CRC patients using IH/IF assay.
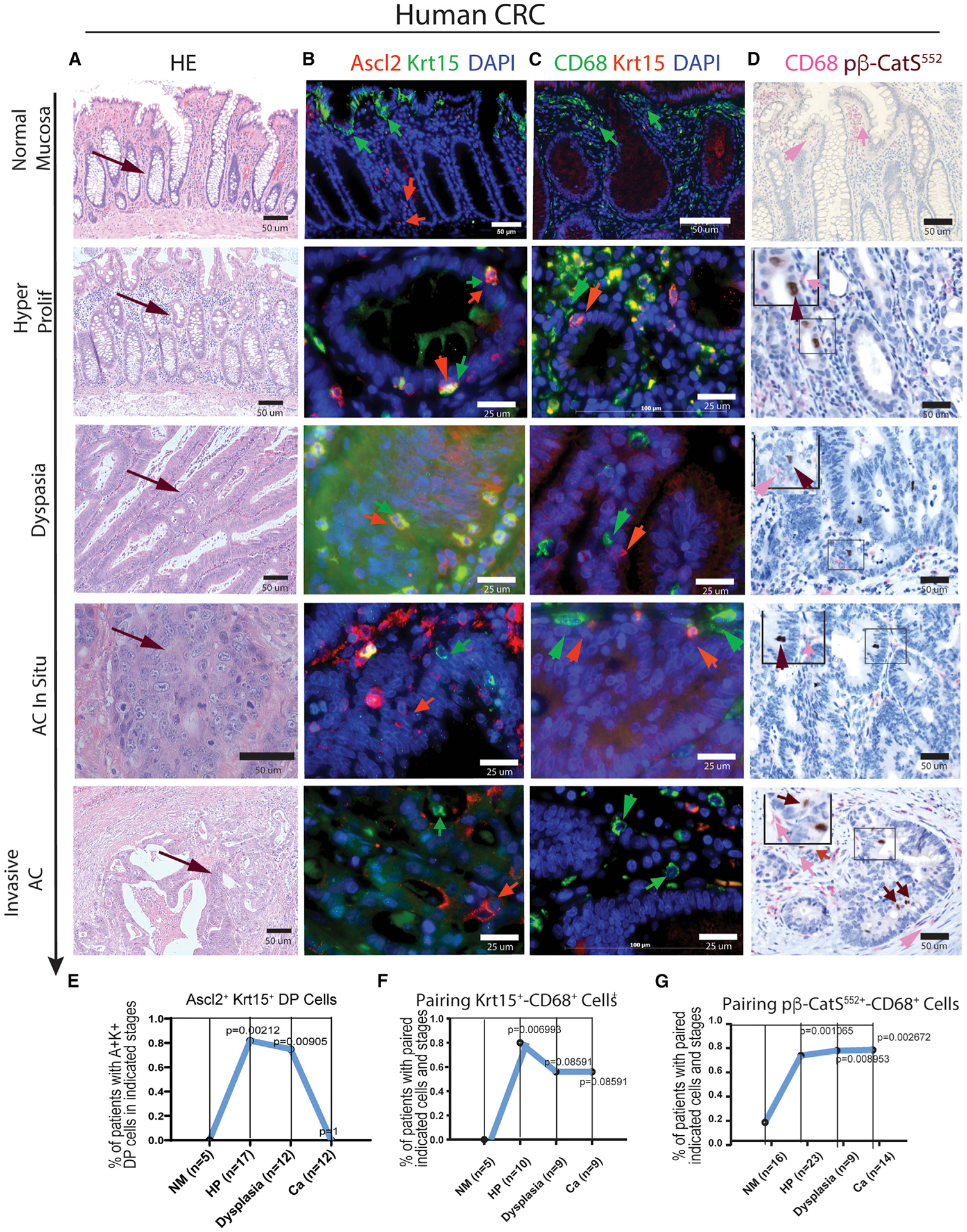
(A–D) H&E staining shows the pathological feature of adenoma progression and cancer stages in sections of human CRC (A). With the corresponding sections, the following panels show (B) IF co-staining of markers Krt15 and Ascl2, (C) IF co-staining of CD68 and Krt15, and (D) IHC co-staining of p-β-catS552 and CD68.
(E–G) Statistical analysis of the significance of correlation for detection of Krt15+Ascl2+ TrTSCs (E), association of CD68+ MDCs with Krt15+ TSCs (F), or association of CD68+ MDCs with p-β-catS552 TSCs/CSCs in the corresponding stages in human CRCs (G). Fisher’s exact test was used to test whether the percentage of double-positive samples in HM differed from the ones on the other stages.
In the regions near the boundary of the CRC domain and in what morphologically seems to be “normal mucosa” (Figure 7A, top panel), Ascl2+ or Krt15+ EpCs were detected in the lower part or in the tip region of crypt, whereas Krt15+Ascl2+ DP EpCs were rarely seen in crypts (Figure 7B, top panel). In contrast, the Krt15+Ascl2+ DP EpCs were frequently detected in the adenoma stage (Figures 7B and 7E). However, Krt15+Ascl2+ DP EpCs declined in cancer (Figures 7B and 7E), partly because of down-regulation of Krt15 in Ascl2+ EpCs in the cancer stage (Table S4).
Next, we examined the association of CD68+ cells with Krt15+ EpCs (Figure 7C). Of note, there is an antibody incompatibility between CD68 and Ascl2. In the “normal mucosa” region (Figure 7A, top panel), a cluster of CD68+ cells was located beneath the Krt15+ crypt-tip EpCs. Paring of CD68+ and Krt15+ EpCs was rare in the lower part of crypts (Figure 7C, top panel) but was significantly more frequent in the HyP stage than in either HDG or cancerous stages (Figures 7C and 7F; Table S4).
To determine whether MDCs play a role in promoting TSC proliferation, we used anti-p-β-catS552 antibody. This is because p-β-catS552-marked TSCs/CSCs are often located at the apical side of crypts, indicating the active dividing state (He et al., 2007; Perry et al., 2020; McKinley et al., 2018). Pairing of CD68+ MDCs with nuclear-localized p-β-catS552+ EpCs was rare in “normal mucosa” (Figures 7D and 7G) but much more frequent in both adenoma and cancerous stages (Figures 7D and 7G; Table S4). This observation is consistent with the result of adenoma-derived organoid culture, in which p-β-catS552 + cells activity and apical position correlated with the crypt budding event (Figure 5C); thus, both ex vivo and in vivo observations support the role of MDCs in promoting TSCs/CSCs activation and proliferation not just in murine adenoma but also in human CRC patients.
DISCUSSION
Using the classical Apcmin/+ adenoma mouse model, we have systematically investigated intestinal adenoma cells under the stress of CRT and identified TrTSCs. We observed a bidirectional regulation between TSC and TME. Although TME, especially TAMMs, generated immunosuppressive signals against cytotoxic CD8 T cells, unexpectedly, it was TrTSCs that shape TME into an immunosuppressive barrier. Considering the limitation of this adenoma model, we investigated CRC sections of human CRC patients and found that carcinoma development coexisted with multiple stages of adenoma progression, which illustrates the continuity of carcinogenesis (Figures 7, S8A, and S9A). This observation offered us an opportunity to test the extent to which our findings in the murine model could provide an insight into the corresponding stages in human CRC.
Identification of TrTSCs in adenoma
Normally, cycling ISCs maintain homeostatic epithelium but are sensitive to stress (Barker et al., 2007); it is rISCs that survive the stress and support EP regeneration (Bankaitis et al., 2018; Karmakar et al., 2020; Li and Clevers, 2010). Akin to this scenario (Li and Neaves, 2006), we show in adenoma that cycling TSCs expressing Lgr5hi, Olfm4, and mKi67 were largely eliminated by CRT, whereas TrTSCs expressing Lgr5neg-lo and Ascl2 (Figures 3D, 3E, and S9B) survived CRT and upregulated several rISC representative or stress-responsible genes (Figure 3F). These rISC genes are also expressed in various progenitor cells (Karmakar et al., 2020; Yousefi et al., 2017), which, however, were largely eliminated by CRT (Figure S2). Therefore, TrTSC is defined from the functional aspect: it not only survives CRT but also restores the TSC pool and supports tumor regrowth. The existence of TrTSCs was clearly shown at 8–24 h after CRT as indicated by the trajectory analysis (Figures 3A, 3B, and S2); TrTSCs could also be derived from the dedifferentiated tumor cells as shown at 48 h after CRT (Figures 3B, S3A, S9B, and S9B′).
Bidirectional and dynamic crosstalk between TSCs and TME
To investigate direct interactions between TSCs and TME, we identified key ligand-receptor pairs and the associated signaling modules using the CellPhoneDB program. CD8 T cells were shown to potentially induce apoptosis of TSCs via CRT-induced Fas ligand (FASL)-TNFRSF1 signaling (Figure 4A) (Caulfield and Lathem, 2014). Intriguingly, it was TSCs secreted MIF via TME widely expressed CD74 (Figueiredo et al., 2018) to shape the TME into an immunosuppressive barrier (Figure S4B) (Müller et al., 2020). In addition, a unique RPS19-C5AR1 (Markiewski et al., 2017) interaction between TSCs and TAMMs also carries a potential to generate immunosuppressive signals (Figures S4A and S4B). Thus, compared with other TME cell types, TAMMs played a predominant role in forming an immune barrier against CD8 T cells, consistent with the initial increase in MDCs and the decline in T cells, which correlated with the survival of TrTSCs (Figures S1A and S1B). Inversely, TAMMs also promoted TSC proliferation at 24–48 h (see below for details). At 96 h, much of MDC-TSC signaling declined because of separation of MDCs from TSCs, and stroma-derived signaling became dominant to facilitate TSCs restoration to homeostasis (Figures S5 and S9).
TAMMs promote TSC propagation
Both macrophage and Treg cells were reported to inhibit CD8 T cells in the tumor neighborhood (Schürch et al., 2020; Taniguchi et al., 2020). Here we show that TAMMs are a key TME component in forming an immune barrier against CD8 T cells and in promoting TSC expansion. Mechanistically, the PGE2-EP4 signaling between TAMM-TSC promoted the proliferation of TrTSCs via AKT enhanced β-catenin signaling (Figures 5A–5D) (He et al., 2007). This conclusion is supported by various approaches (Figures 4, 5, 6, and S9C). Moreover, CRT-induced recruitment of TAMMs to the TrTSC niche was dynamic and clearly coincides with the initial protection of TSCs at 0–8 h and the subsequent expansion of TrTSCs at 24–48 h (Figure S9B).
A limitation in using the APCmin adenoma model and an insight into human CRCs
By examining pathohistological sections representing up to 35 out of 59 cohort human CRC patients (Figures 7A and S8A), we observed that Krt15+Ascl2+ DB EpCs were rare in the “normal mucosa” region; furthermore, these cells were mainly detected in the adenoma and declined in the adenocarcinoma stages (Figures 7B and 7E). This comparison indicates a limitation in using the APCmin/+ mouse model in terms of its relevance to human adenocarcinoma. Similarly, we observed that co-localization of CD68+ TAMMs with Krt15+ EpCs occurred primarily in the adenoma stage, but not in the adenocarcinoma stage (Figure 7F). However, when p-β-catS552 was used, a different CSC marker that reflects PGE2-EP4 signaling to enhance Akt and β-catenin activities (He et al., 2007; Perry et al., 2011, 2020b), the association of CD68+ TAMMs with TSC/CSCs persisted in the carcinoma stage of human CRC patients (Figures 7D and 7G). Furthermore, we have recently reported that p-β-catS552 functionally empowers CSC immune escape via upregulation of multiple immune checkpoint genes (Perry et al., 2020b). This provides insight for understanding the putative role TAMMs play in both protecting TSC/CSC from cytotoxic T cells and supporting TSC/CSC propagation. Future studies are required using CRC or patient-derived xenograft (PDX) animal models to verify these findings, including the PGE2-EP4 and P-β-CatS552 axis, as well as putative MIF-CD74- and RPS19-C5AR1-mediated immunosuppressive signaling.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Linheng Li (lil@stowers.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at https://www.stowers.org/research/publications/libpb-1315 upon publication.
All software and packages applied are publicly available and listed in the key resources table. The specific analysis parameters used are explained in the method section.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Ly6c antibody [ER-MP20] | Abcam | ab15627;RRID:AB_302004 |
| InVivoMAb anti-mouse Ly6G/Ly6C (Gr-1), 1mg | BioXCell | BE0075;RRID:AB_10312146 |
| Anti-Rabbit p-β-Catenin (Ser552) Monoclonal Antibody | Zymed custom antibody | Affinity Pure Rb Ab, 0.5mg/ml |
| Anti-Rabbit β-actin polyclonal Antibody | Abcam | Cat# ab8227 RRID: AB_2305186 |
| Anti-Mouse MHC Class II Monoclonal Antibody (AF6-120.1), APC | Thermo Fisher | Cat# 17-5320-82;RRID:AB_2573212 |
| SP-1 Chromogranin A (Porcine) Antibody | Immunostar | 20086;RRID:AB_572226 |
| FcR Blocking Reagent, mouse | Miltenyi Biotec | 130-092-575;RRID:AB_2892833 |
| Ly-6G (Gr-1) Monoclonal Antibody (1A8-Ly6g), PE-eFluor 610, eBioscience | ThermoFisher | 61-9668-82;RRID:AB_2574679 |
| Ly-6C Monoclonal Antibody (HK1.4), Alexa Fluor 488, eBioscience | ThermoFisher | 53-5932-82;RRID:AB_2574427 |
| CD11b Monoclonal Antibody (M1/70), APC-eFluor 780, eBioscience | ThermoFisher | 47-0112-82;RRID:AB_1603193 |
| Lysozyme EC 3.2.1.17 | Agilent | A009902-2;RRID:AB_578661 |
| Prostaglandin E Receptor EP4 Antibody | VWR/Bioss | bs-8538R |
| Anti-F4/80 antibody [SP115] (ab111101), Rabbit | Abcam | ab111101;RRID:AB_10859466 |
| Anti-CD68 antibody (ab125212), Rabbit | Abcam | ab125212:RRID:AB_10975465 |
| Anti-CD11b antibody [EPR1344] (ab133357), rabbit | Abcam | ab133357;RRID:AB_2650514 |
| CD44-Biotin, mouse | Miltenyi Biotec | 130-110-082;RRID:AB_2661032 |
| CD24-Biotin, mouse | Miltenyi Biotec | 130-102-128;RRID:AB_2656584 |
| Anti-CD68 antibody [C68/684] | Abcam | ab201340;RRID:AB_2747829 |
| Anti-CD11b antibody [M1/70] | Abcam | ab8878;RRID:AB_306831 |
| Anti-CD11b antibody [EPR1344] (ab133357), rabbit | Abcam | ab133357;RRID:AB_2650514 |
| Anti-Rabbit Krt15 Polyclonal Antibody | Novus | Cat# NBP1-85602 RRID: AB_11037933 |
| Anti-Mouse Ascl2 Monoclonal Antibody | LSBio | Cat# LS-C126891;RRID:AB_10831114 |
| CD9 | Abcam | ab92726;RRID:AB_10561589 |
| Bmi1 antibody | Abcam | ab14389;RRID:AB_2065390 |
| Annexin A2 | Abcam | ab178677;RRID:AB_1140698 |
| Annexin A1 | Abcam | ab214486;RRID:AB_722804 |
| Ascl2 antibody | Biorbyt | orb155740;RRID:AB_10865613 |
| E-Cadherin (24E10) Rabbit mAb | Cell Signaling Technology | 3195;RRID:AB_2291471 |
| PD-1 Monoclonal Antibody (7A11B1) | ThermoFisher | MA5-15780;RRID:AB_11152225 |
| Beta-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | 3700S;RRID:AB_2242334 |
| Mouse TrueBlot ULTRA:Anti-Mouse Ig HRP | Rockland | 18-8817-33;RRID:AB_2610851 |
| CST alpha-tubulin Antibody | Cell Signaling Technology | 2144;RRID:AB_2210548 |
| Bmi1 (D20B7) XP® Rabbit mAb | Cell Signaling Technology | 6964;RRID:AB_10828713 |
| Purified Mouse Anti-Human CD63 Clone H5C6 | Fisher/BD Biosciences | 556019;RRID:AB_396297 |
| Krt15 antibody (Cytokeratin 15) | Fisher/Novus Biologicals | NBP2-50461AF488; RRID:AB_1084834 |
| BrdU antibody | Fisher/Novus Biologicals | NB600-720;RRID:AB_10002707 |
| S100A10 Antibody | VWR/ProteinTech | 11250-1-AP;RRID:AB_2269906 |
| Donkey Anti-Rabbit IgG H&L (Alexa Fluor® 594) |
Abcam | ab150076;RRID:AB_2782993 |
| ASCL2 Antibody (C terminus) LS-C126891 | LifeSpan Biosciences | LS-C126891-100;RRID:AB_10831114 |
| Cytokeratin 15 Antibody | Novus Biologicals | NBP1-85602;RRID:AB_11037933 |
| MHC Class II I-Ab Monoclonal Antibody (AF6-120.1), APC, eBioscience | ThermoFisher | 17-5320-82;RRID:AB_2573212 |
| Mouse IgG2a kappa Isotype Control (eBM2a), APC, eBioscience | ThermoFisher | 17-4724-81;RRID:AB_470188 |
| CD45 Monoclonal Antibody (30-F11), PE-Cyanine5, eBioscience | ThermoFisher | 15-0451-83;RRID:AB_468753 |
| Rat IgG2b kappa Isotype Control (eB149/10H5), PE-Cyanine5, eBioscience | ThermoFisher | 15-4031-82;RRID:AB_470133 |
| CD11b Monoclonal Antibody (M1/70), PE, eBioscience | ThermoFisher | 12-0112-83;RRID:AB_2734870 |
| Rat IgG2b kappa Isotype Control (eB149/10H5), PE, eBioscience | ThermoFisher | 12-4031-82;RRID:AB_470042 |
| CD11c Monoclonal Antibody (N418), PE-Cyanine7, eBioscience | ThermoFisher | 25-0114-82;RRID:AB_469590 |
| Armenian Hamster IgG Isotype Control (eBio299Arm), PE-Cyanine7, eBioscience | ThermoFisher | 25-4888-82;RRID:AB_470204 |
| FITC anti-mouse CX3CR1 Antibody | VWR/Biolegend | 149020;RRID:AB_2565703 |
| FITC Mouse IgG2a, κ Isotype Ctrl Antibody | VWR/Biolegend | 400208;RRID:AB_2884007 |
| Annexin V-mFluor Violet 540 conjugate | AAT Bioquest | 20080 |
| EP4 antibody | Abcam | ab217966 |
| CD3 | Abcam | ab16669;RRID:AB_443425 |
| Anti-BrdU antibody [BU1/75 (ICR1)] | Abcam | ab6326;RRID:AB_305426 |
| CD68 Antibody | Abcam | ab201340;RRID:AB_991703 |
| CD19 Antibody | Abcam | ab203615;RRID:AB_1140606 |
| Ascl2 antibody, 100 μg | Biorbyt | orb155740 |
| Cytokeratin 15 Antibody | Fisher/Novus Biologicals | NBP2-53308-100ug |
| TCF21 Antibody | Fisher/Novus Biologicals | NBP1-88637;RRID:AB_11008184 |
| Rat monoclonal [YTS169.4] to CD8 | Abcam | ab22378;RRID:AB_447033 |
| Anti-Sox9 antibody | Sigma/Millipore | AB5535;RRID:AB_2239761 |
| RM-9106-S1/Ab Ki-67 0.5mL/EA | VWR | MISC-CLINICAL |
| Mouse monoclonal [2B3] to RUNX3 | Abcam | ab135248 |
| Rabbit monoclonal [EP3251] to Pax2 | Abcam | ab79389;RRID:AB_1603338 |
| ASCL2 Antibody (C terminus) LS-C126891 | LifeSpan Biosciences | LS-C126891-100;RRID:AB_10831114 |
| Cytokeratin 15 Antibody | Novus Biologicals | NBP1-85602;RRID:AB_11037933 |
| Cox2 (D5H4) XP Rabbit mAb | Cell Signaling Technology | 12282S;RRID:AB_2571729 |
| Purified Mouse Anti-E-Cadherin | Fisher/BD Biosciences | 3700S |
| PTGER4/EP4 Antibody | Novus Biologicals | NBP1-84833;RRID:AB_11019807 |
| p23/PTGES3 Antibody (JJ3) | Novus Biologicals | NB300-576;RRID:AB_10000921 |
| HSP90 beta Antibody | Novus Biologicals | NBP1-77563;RRID:AB_11011699 |
| Anti-HSP90aa1 | Novus Biologicals | NB120-2928;RRID:AB_790272 |
| B7-H1/PD-L1/CD274 Antibody (M - 0.1 MG) | Fisher/Novus Biologicals | NBP1-432620 |
| Anti-Prostaglandin E2 antibody | Abcam | ab2318;RRID:AB_302974 |
| Cleaved Caspace-3 (Asp175) Antibody, 100 μl (10 western blots) | Cell Signaling Technology | 9661S;RRID:AB_2341188 |
| BrdU/CldU antibody | Fisher/Novus Biologicals | NB500-169;RRID:AB_10002608 |
| Cox2 Antibody | Cell Signaling Technology | 4842;RRID:AB_2084968 |
| ANTI-CD44 ANTIGEN ISOFORM 4 | Fisher | PIPA521419 |
| CD44v6 Monoclonal Antibody (9A4) | Fisher | MA1-81995;RRID:AB_928369 |
| Phospho-Akt (Ser473) (D9E) XP® | Cell Signaling Technology | 4060S;RRID:AB_2315049 |
| Thermo Scientific Lab Vision Ki-67, Rabbit Monoclonal Antibody | ThermoFisher | RM-9106-S1;RRID:AB_149792 |
| Anti-RIP3 antibody | Abcam | ab56164;RRID:AB_2178667 |
| Anti-CD8 antibody [YTS169.4] | Abcam | ab22378;RRID:AB_447033 |
| Annexin V-mFluor Violet 540 conjugate | AAT Bioquest | 20082 |
| CD24 Monoclonal Antibody (M1/69), eFluor 450, eBioscience | ThermoFisher/eBioscience | 48-0242-82;RRID:AB_1311169 |
| MHC Class II I-Ab Monoclonal Antibody (AF6-120.1), APC, eBioscience | ThermoFisher/eBioscience | 17-5320-82;RRID:AB_2573212 |
| Anti-CD44 antibody [T2–F4] | Abcam | ab40983;RRID:AB_726520 |
| Thermo Scientific Lab Vision Ki-67, Rabbit Monoclonal Antibody | ThermoFisher | RM-9106-S1;RRID:AB_149792 |
| CldU antibody | Novus Biologicals | NB500-169;RRID:AB_10002608 |
| Anti-Mouse CD24 FITC 50ug | eBioscience | 11-0241-81;RRID:AB_464984 |
| Lysozyme EC 3.2.1.17 | Dako | A009902-2:RRID:AB_2341231 |
| Anti-GRP78/BiP (ET-21) antibody produced in rabbit | Sigma | G9043-200UL; RRID:AB_2279879 |
| Mouse ALCAM Phycoerythrin Affinity Purified PAb, Goat IgG (CD166 PE) | R&D Systems | FAB1172P;RRID:AB_2242595 |
| CD44 P7 | BioLegend | 103030;RRID:AB_830787 |
| Anti-Mouse CD24 eFluor® 450 (Pacific Blue® replacement) 100 ug (CD24 eF450) | eBioscience | 48-0242-82;RRID:AB_1311169 |
| goat anti-rabbit IgG, F(ab’)2-APC-Cy7 | Santa Cruz Biotechnology | sc-3847; RRID:AB_649105 |
| Anti-GRP78/BiP (ET-21) antibody produced in rabbit | Sigma | G9043-200UL;RRID:AB_2279879 |
| MTA1 (D40D1) XP® Rabbit mAb 100ul (10 western blots) | Cell Signaling Technology | 5647S;RRID:AB_10705601 |
| BrdU antibody | VWR | 95019-028 |
| CD44 | Fisher | MA1-81995;RRID:AB_928369 |
| Biological samples | ||
| Human familial adenomatous polyposis (FAP) paraffin-embedded tissue sections | University of Kansas Medical Center | Pathology & Laboratory UKMC, KC, KS 66160 |
| Human colorectal cancer (CRC) paraffin-embedded tissue sections | University of Kansas Medical Center | Pathology & Laboratory UKMC, KC, KS 66160 |
| Chemicals, peptides, and recombinant proteins | ||
| EP2 antagonist | Cayman Chemical | 14050 |
| M-CSF, Macrophage-Colony Stimulating Factor Protein, Recombinant human | Millipore | GF053 |
| EP4 antagonist | Sigma | A8227-25MG |
| Human Recombinant M-CSF, Size: 100 μg | StemCell Technologies | 78057 |
| DNase I | Fisher/Worthington | LS006361 |
| Liberase TM Research Grade | Sigma | 5401119001 |
| Celecoxib | LC Laboratories | C-1502 |
| Capecitabine, 10 g | LC Laboratories | C-2799 |
| JAG-1 (188–204), Jagged-1 (188–204), Notch Ligand” | Anaspec | AS-61298 |
| Recombinant Murine Noggin, 20 ug | PeproTech | 250-38 |
| Recombinant Murine EGF, 100 ug | PeproTech | 315-09 |
| Recombinant Mouse R-Spondin 1, CF, 250 ug | R&D Systems | 3474-RS-250 |
| Recombinant Murine Wnt-3a, 2 ug | R&D Systems | 1324-WN-002 |
| Stemolecule Thiazovivin (ROCK inhibitor), 1 mg | Reprocell | 04-0017 |
| Stemolecule CHIR99021, 2 mg | Reprocell | 04-0004 |
| IntestiCult Organoid Growth Medium (Mouse) | StemCell Technologies, Inc. | 06005 |
| Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix, Phenol Red-free, LDEV-free, 10 mL | VWR | 356231 |
| PF3644022, 10 mg | Sigma | H6278-50MG |
| 4-HYDROXYTAMOXIFEN | VWR | 103302-538 |
| Collagenase 1A, 100mg | Sigma | C2674-100MG |
| Hyaluronidase, 100mg | Sigma | H3506-100MG |
| Evans Blue, 10 g | Sigma | E2129-10G |
| MATRIGEL MATRIX NO PHENOL 10ML | VWR/Corning | 356237 |
| Advanced DMEM/F-12 | ThermoFisher | 12634-010 |
| E4 pipette | Rainin | 17014490 |
| B-27® Serum-Free Supplement (50X), Liquid, 10 ml | ThermoFisher | 17504-044 |
| N-2 Supplement (100X), Liquid, 5 ml | ThermoFisher | 17502-048 |
| N-ACETYL-L-CYSTEINE BIOREAGENT CELL CUL& | Sigma | A9165-5G |
| Y-27632 | Sigma | Y0503-1MG |
| Stemolecule Thiazovivin 1 mg | Stemgent | 04-0017 |
| Recombinant Human R-Spondin 1, CF 25 μg | R&D Systems | 4645-RS-025/CF |
| Basement Membrane Matrix, Growth Factor Reduced (GFR), Phenol Red-free, 10ml *LDEV-Free | BD Biosciences | 356231 |
| SOLUTION BSS W/O CA+ MG+ PH-RD 1L | VWR | 12001-980 |
| Critical commercial assays | ||
| Myeloid-Derived Suppressor Cell Isolation Kit | Miltenyi Biotec | Cat# 130-094-538 |
| MACS separation using LS columns | Miltenyi Biotec | Cat# 130-042-401 |
| 10X Chromium Single Cell 3′ Library & Gel Bead Kit (v2) | 10x Genomics | Cat# PN-120267 |
| TruSeq Stranded mRNA Prep Kit | illumine | Cat# 20020594 |
| Bioanalyzer RNA Analysis | Agilent Technologies | Cat# 5067-1511 |
| Complementary DNA synthesis; High-capacity RNA to cDNA | Life Technologies | Cat# 4387406 |
| Propidium Iodide Solution | Miltenyi Biotec | 130-093-233 |
| MACS BSA Stock Solution | Miltenyi Biotec | 130-091-376 |
| Dead Cell Removal Kit | Miltenyi Biotec | 130-090-101 |
| autoMACS Rinsing Solution | Miltenyi Biotec | 130-091-222 |
| Anti-Mouse IgG1 MicroBeads | Miltenyi Biotec | 130-047-102;RRID:AB_244355 |
| CD45 MicroBeads, mouse | Miltenyi Biotec | 130-052-301;RRID:AB_2877061 |
| CD31 MicroBeads, mouse | Miltenyi Biotec | 130-097-418;RRID:AB_2814657 |
| Anti-Ter-119 MicroBeads, mouse | Miltenyi Biotec | 130-049-901 |
| Miltenyi Biotec | Miltenyi Biotec | 130-042-201 |
| Dead Cell Removal Kit | Miltenyi Biotec | 130-090-101 |
| RBC lysis buffer | Sigma | 11814389001 |
| Annexin V-mFluor Violet 540 conjugate | AAT Bioquest | 20082 |
| CD11b MicroBeads | Miltenyi Biotec | 130-049-601 |
| CD11c MicroBeads | Miltenyi Biotec | 130-108-338 |
| Trypan blue solution 0.4%, 100ml | ThermoFisher | 15250061 |
| KnockOut Serum Replacement | ThermoFisher | 10828028 |
| Restore PLUS Western Blot Stripping Buffer | ThermoFisher | 46430 |
| Deposited data | ||
| 10X single cell (sc) RNA-seq data | Gene Expression Omnibus (GEO: GSE136256) | |
| Experimental models: Organisms/strains | ||
| C57BL/6 | Jackson Laboratories | 000664;RRID:IMSR_JAX:000664 |
| C57BL/6J-ApcMin/J (Apc-Min) | Jackson Laboratories | 002020;RRID:IMSR_JAX:002020 |
| BALB/cJ (BALB/c) | Jackson Laboratories | 000651;RRID:IMSR_JAX:000651 |
| B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J (R26-YFP) | Jackson Laboratories | 006148;RRID:IMSR_JAX:006148 |
| B6;129-Bmi1tm1(cre/ERT)Mrc/J (Bmi1-CreER) | Jackson Laboratories | 010531;RRID:IMSR_JAX:010531 |
| B6.129P2-Lgr5tm1(cre/ERT2)Cle/J (LGR5KI) | Jackson Laboratories | 008875;RRID:IMSR_JAX:008875 |
| Software and algorithms | ||
| FlowJo Software v 7.0 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| ImageJ | OpenSource | https://imagej.nih.gov/ij/download.html |
| GraphPad Prism v 8.1.2 | Prism | https://www.graphpad.com/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal therapy procedure
All mice were maintained under pathogen-free conditions, and all procedures performed in this study were approved by the Institutional Animal Care and Use Committee at the Stowers Institute for Medical Research. APCMin/+ mice (both male and female) were treated starting at days 90–100. Mouse models by crossing Bmi1-CreER mice with R26-LSL-GFP and Apcmin/+ mouse lines (both male and female) were used for tumor clone study. Four experimental groups were conducted in this study: Control group: mice were treated via gavage with 40mMol/l citrate buffer and 5% gum Arabic for 7 days. Chemoradiotherapy group: mice were abdominally irradiated (one-time 5Gy at 12hr before day 0) prior to gavage administration of capecitabine (700 mg/kg BW) for 7 days. Celecoxib therapy group: mice were administrated with celecoxib (100 ppm [parts per million] celecoxib mixed in 1 kg food) right after focal irradiation and continuing throughout the experimental procedure until harvested for analysis. Combined therapy group: mice were treated with both celecoxib and chemoradiotherapy. Antibiotics (Baytril, 0.277 mg/ml) were administered in the drinking water 3 days prior to irradiation and for 21 days post irradiation. Mice were analyzed between day 14 and day 21 post CRT apart from some cases that required a longer period.
Human subjects
De-identified patient specimens were used in this study. Adult human familial adenomatous polyposis (FAP) and colorectal cancer (CRC) paraffin-embedded tissue sections were provided by the Department of Pathology & Laboratory Medicine at University of Kansas Medical Center.
METHOD DETAILS
Dual labeling of active-proliferating and slow-cycling tumor cells
APCMin/+ mice were labeled via intraperitoneal (IP) injection of chloro-deoxyuridine (CldU) (10mg/kg bodyweight [BW]), iodo-deoxyuridine (IdU) (10mg/kg BW), or bromodeoxyuridine (BrdU) (10mg/kg BW). For labeling of slow-cycling cells, mice were intraperitoneally injected with CldU (10mg/kg BW) twice a day, for 5 consecutive days, followed by a chase period (5 days) to enrich label retaining cells (LRCs). For labeling of proliferating cells, mKi67 was used.
Collection and fixation of normal and adenoma intestinal tissues
Mice were euthanized with CO2 followed by cervical dislocation. A ventral incision was made to collect the intestine. Opened intestine was washed with PBS. The intestine was placed on 30–40ml of zinc formalin, fixed for 20–24hrs, and then washed 5 times with water. The intestine was placed on 30–40ml of 70% ethanol and was rolled starting from the duodenum. The intestinal roll was stabilized with 27 ½ needle and placed back in 70% ethanol. The roll was prepared for histological processing (embedded in paraffin and cut into sections).
Isolation of epithelial single cell single cell
Intestine was dissected and washed 5 times with ice-cold HBSS w/o Ca2+Mg2. Minced intestinal adenoma tissues were directly treated with 30mM EDTA as described in a previous report (Wang et al., 2013). Samples were shaken vigorously by hand for 3–5min (~180 shakes/minute). Supernatant was removed, and crypts were resuspended with TryPLE Express containing 500uM NAC, 10uM Y-27632 and incubated for 6–8min in a 37°C water bath while being stirred occasionally by a P1000 pipette. Dissociated crypts were transferred to ice-cold DMEM/F12 media with 500uM N-Acetyl-L-cysteine, 10uM Y-27632 dihydrochloride ROCK inhibitor and shaken for 30 s to promote the dissociation and pass cell strainer 40um nylon filter the suspension. Single cell suspension was resuspended pellet in 3ml of ice-cold DMEM/F12 media with 500uM N-Acetyl-L-cysteine, 10uM Y-27632 dihydrochloride ROCK inhibitor and then passed through 20um syringe filter to remove debris. 2,000–3,000 epithelial cells/samples were counted, resuspended with HBSS without Ca2+Mg2+ + 0.4% BSA, and prepared for 10XGenomic scRNA sequencing.
Isolation of lamina propria mesenchymal single cell
After removing epithelial cells, the intestinal mesenchymal tissue was subjected to digestion with Liberase (0.6mg/ml) in HBSS buffer with Ca2+Mg2+ by incubation 37°C 20min with occasional gentle shaking(2X/min); DNaseI (2,000unit/ml) was then added, and tissue was further dissociated into single cells using Gentle MACS Dissociator on intestine program1. Single cells were suspended using 16G needle and filtered through 70 μm cell strainer to enrich lamina propria mesenchymal single cells. The cell pellet was resuspended in the medium (HBSS with Ca2+Mg2+ 3% FBS and 10uM Y27632). Then, 2,000–3,000 LPMC/samples were counted, resuspended with HBSS without Ca2+Mg2+ + 0.4% BSA, and prepared for 10XGenomic scRNA sequencing.
3D Ex vivo organoid co-culture with MDCs
MDCs were isolated from spleen, intestine adenoma of APCMin/+ mice and bone marrow of C57BL/6 from P7 using a Mouse MDSC Isolation Kit (Miltenyi Biotec, 130-094-538) according to the manufacturer’s instructions. Crypt isolation was performed according to the protocol from StemCell Technologies, Inc. (DOCMENT #28223 Version 2.0.1). Crypts (500/well) were then embedded in Matrigel (Corning, growth factor reduced, cat #354230) with MDCs (500 cells/well) at the ratio of 1:1, and cultured in IntestiCult organoid growth medium (StemCell Technologies, Inc., cat #06005) supplemented with 100 μg/ml Penicillin-Streptomycin, 5ng/ml granulocyte–macrophage colony-stimulating factor (Sigma-Aldrich, GF004), 2500 UI/ml macrophage colony-stimulating factor (StemCell Technologies, Inc., cat #78057). COX2 inhibitor (Celecoxib, > 99%) and EP4 inhibitor (AH23848 Hemicalcium salt, ≥ 90%) were purchased from LC Laboratories (cat #c-1502) and Sigma-Aldrich (cat #A8227) respectively, and dissolved in DMSO (Corning, cat #25–950-CQC) as 10mM in stock. Organoids co-cultured with MDSCs were treated with COX2 inhibitor (5–20 μM) and EP4 inhibitor (10–30 μM) from day2 to day14. Changes in organoid morphology after drug therapy were visualized with microscope AxioObserver.Z1with Incubator XLmulti S1.
10x Chromium single-cell RNA-seq library construction (v2)
Dissociated cells, having been sorted in HBSS without Ca2+Mg2+ + 0.4% BSA, were assessed for concentration and viability via a Nexcelom Cellometer Auto T4. Cells deemed to be at least 60% viable were loaded on a Chromium Single Cell Controller (10x Genomics, Pleasanton, CA), based on live cell concentration. Libraries were prepared using the Chromium Single Cell 3′ Library & Gel Bead Kit v2 (10x Genomics) according to manufacturer’s directions. Resulting short fragment libraries were checked for quality and quantity using an Agilent 2100 Bioanalyzer and Invitrogen Qubit Fluorometer. Libraries were pooled in groups, at equal molar concentrations. With cells captured estimated at ~570–18,800 cells per sample, libraries were sequenced to a depth necessary to achieve 13,500–175,000 mean reads per cell - ~40–340M reads each - on an Illumina HiSeq 2500 instrument using Rapid SBS v2 chemistry with the following paired read lengths: 26 bp Read1, 8 bp I7 Index and 98 bp Read2.
Flow Cytometry
Cells were harvested from Intestine LPMC of APCmin/+ mice in HBSS. Red blood cells were lysed using a 0.16 M ammonium chloride solution, and the cells were filtered with 70 μm strainers to generate single cell suspensions. For RM and TAM identification, cells were stained with antibodies against CD45, CD11b, CD11c, Ly6c, CX3CR1, MHCII. For MDSCs analysis, cells were stained with antibodies CD11b, Ly6c, Ly6g. All the antibodies were purchased from BD Biosciences, Biolegend and eBioscience. Antibody staining was performed at 4°C for 30min, and then stained with the viability dyes 7-Aminoactinomycin D (7-AAD, 0.1 mg/ml) to exclude dead cells. Cell analyses were performed on MACSQuant (Miltenyi Biotec) and ZE5 Cell Analyzer (Bio-Red). Data analysis was performed using FlowJo software V7.0.
Macrophage depletion
Macrophage depletion was described previously. Mice were treated with Clodrosome via IP (50mg/kg BW every other day, total four dosages) for depleting macrophages or administered with Encapsome (equal vol.) as control. Intestinal TAMs were analyzed at 7days, 8days, 9days, 10days post Clodrosome and Encapsome therapy.
Western blot analysis
Intestinal adenomas were isolated from APCMin/+ mice at indicated time points post therapy and minced into small pieces before lysis in 1 X Laemmli sample buffer (30mM Tris-HCl, pH6.8, 10% glycerol, 1%SDS, 0.005% bromophenol blue plus 355mM 2-mercaptoethanol). Tumor pieces in 1 X Laemmli sample buffer were boiled for 20min followed by full-speed centrifuge for 5min at RT. Intestine organoids were washed with cold PBS to remove Matrigel and lysis in 1 X Laemmli sample buffer. Lysates were centrifuged at 14000 g for 15min. Protein concentrations were determined using Bio-Rad protein assay reagent. Proteins in supernatant were loaded onto SDS-PAGE gel and separated by electrophoresis and transferred onto nitrocellulose membrane. Then, nitrocellulose membrane was blocked in 5% non-fat milk at RT for 2hrs and further incubated overnight in 5% non-fat milk with indicated primary antibodies (COX-2, Cell Signaling Technology, 4842S, 1:5,000; cleaved Caspase3, Cell Signaling Technology, 9661S, 1:5,000; EP2, Abcam, ab124419, 1:2,500; phospho-AKT, Cell signaling Technology, 9271S, 1: 20,000;. RIPK3, Cell Signaling Technology, 13526S, 1:5,000; phospho-beta-catenin, homemade, 1:5,000; actin, Abcam, ab8227, 1:10,000). After three washes in (TBST (Tris-buffered saline [TBS] and Tween 20), the membrane was incubated with horseradish peroxidase (HRP) conjugated secondary antibody at RT for 1hr followed by three washes in TBST. Chemiluminescent substrate (Pierce, ECL Western Blotting Substrate, cat 32106) was used to visualize targeted proteins on the membrane.
Histology, Immunohistochemical (IHC) and Immunofluorescent (IF) assays
Immunofluorescence staining was performed on the tissue based on previously established protocol. In short, paraffin-embedded samples were deparaffinized, the tissue rehydrated with clear rite (3X, 5min each) and then hydrated with an alcohol gradient (2× 100% ethanol, 2X 95%, 1X 705, and 3X distilled H2O, and 1XDPBS, 5min each) and blocked using a 3-serum blocker (2% mouse serum + 10% goat serum + 10% donkey serum in PBS) for 30min to 1hr. Antigen retrieval was performed in a microwave oven (BioGenex, EZ Retriever™) for 10min at 95°C in Citrate buffer. Tissue was stained with primary antibody in the antibody diluent overnight at 4°C. Samples were then washed with DPBS (3X, 5min each) and stained in secondary antibody (in antibody diluent) for 1hr at RT (in the dark). Slides were again washed with DPBS (3X, 5min each), then DAPI was stained, and coverslip was placed on each sample using anti-fade mounting media. Images were acquired on the upright Zeiss microscope.
For whole mount staining, intestine organoids were collected in ice-cold PBS to remove Matrigel and fixed in 4% PFA. Organoids were subsequently permeabilized in PBS 0.1% Tween 20 (30min at RT) before resuspension in blocking solution containing 0.2% normal donkey serum/PBS. For IHC and IF staining, sections were incubated after antigen retrieval with primary antibody overnight at 4°C. Primary antibodies: rabbit anti-p-β-catenin (homemade, 1:150) and mouse anti-E-cadherin (1:200, CST). Alexa Fluor 568 or Alexa Fluor 488 conjugate anti-rabbit and anti-mouse IgGs (1:200 dilution) were used to develop signals at RT for 1hr. DAPI was used to stain the nucleus. Coverslip was placed on each sample using anti-fade mounting media. Images were acquired on Nikon 3PO confocal microscope with 40x magnification.
Transmission electronic microscopy (TEM) and Serial Block-face Electron Microscopy (SEM) and Modeling
For TEM analysis, the intestinal tumor samples were prefixed with 2.5% paraformaldehyde and 2% glutaraldehyde in 50 mM sodium cacodylate containing 1% sucrose (PH7.4). The tissues were post fixed in 2% OsO4. After dehydration with a graded ethanol series, samples were infiltrated and embedded in Epon resin (EMS, Fort Washington, PA). Ultrathin (60–70nm nm) sections were collected on copper grids, stained with 2% uranyl acetate and 1% lead citrate. Sections were photographed using a FEI transmission electron microscope at 80kV.
For serial block-face electron microscopy (Denk and Horstmann, 2004), the intestine samples were fixed as described above. Samples were rinsed and then incubated with 1% uranium acetate overnight, treated with lead aspartate solution in 60°C oven for 30min. After rinsing, the samples were infiltrated and embedded in Epon. Block face imaging was conducted using a Gatan 3View 2XP inside a Zeiss Gemini SEM (Carl Zeiss). 3D models were made using a IMOD image-processing package (Kremer et al., 1996).
QUANTIFICATION AND STATISTICAL ANALYSIS
Tumor size quantification
The whole intestine images were acquired by a Leica stereoscope with INFINITY3–3UR scientific digital camera. All tumors were identified and circled manually with “Freehand selections” in ImageJ, and then sizes were quantified with ROI manager.
Organoid measurement method
Images for crypt area quantification were acquired with Nikon Eclipse TI equipped with a Yokagawa CSU W1 spinning disk and a Hamamatsu Flash 4.0 camera. 3D DAPI images were acquired via a 4× 0.2 NA air objective with 405 nm excitation and a standard DAPI emission filter. Images were collected with 50-micron spacing in the z dimension. Crypt segmentation was performed in Fiji (Schindelin et al., 2012) by first maximum projecting whole well images and then Gaussian blurring with a standard deviation of 4 pixels. Next, we subtracted a 100-pixel radius rolling background. Next, we thresholded the image at 2.5% of the maximum DAPI intensity and filled the holes in the resulting objects. Finally, we filtered out objects with an area less than 750 pixels to avoid noise and contaminants. Some organoids were very close to each other and were not successfully separated by the above algorithm. Those organoid masks were separated by drawing a dark line between them. Finally, the areas of the organoids were measured as the number of pixels contained within each mask.
Single cell RNA-seq and 10X data preprocessing
Cell QC was performed to reach 70%–90% viability with 2000–5000 cell range, through RNA QC, cDNA QC and then library QC. cDNA libraries generated from single cells were sequenced as paired-end reads on Illumina HiSeq 2500 machine. Raw sequencing data were processed using 10x Genomics Cell Ranger pipeline v3. Reads were demultiplexed into Fastq file format using Cellranger mkfastq. Genome index was built by Cellranger mkref using mouse genome mm10, ensembl 91 gene model. Data were aligned by STAR aligner, and cell counts tables were generated using Cellranger count function with default parameters.
Single cell RNA-seq and data analysis
Cellranger’s raw gene count matrices were further analyzed using the Seurat (v2.3.3) R package, following standard protocols. Cells with less than 200 detected genes and 500 UMIs were excluded from downstream analysis. Gene expression data were log-normalized to a scale factor of 10000, and then regressed on the number of UMIs. Principle component analysis (PCA) was done using only the highly variable genes, and 25 to 50 principal components (PCs) were used for clustering analysis at a resolution of 0.6 to identify distinct clusters of cells, based on PCElbowPlot results. UMAP-plot was used to visualize the clustering results. Known and de novo markers were used to classify cells into Lymphocyte, Epithelium, Stromal, Myeloid and TSC populations. Trajectory analysis was performed using R package DiffusionMap.
RNA velocity
For all samples, the spliced/unspliced expression matrices were generated using command line tool “velocyto run10x” with default parameters and the same gene annotation file as Cellranger. The outputted loom files were then combined and loaded into R package Seurat to apply sctransform normalization analysis, dimension reduction, as well as clustering for velocity calculation. The RNA velocity plot was then drawn using R function “show.velocity.on.embedding.cor,” applying the same coordinates and color selection from the original UMAP for all cells.
CellPhoneDB analysis
CellPhoneDB (Efremova et al., 2020) was used to infer enriched ligand–receptor interactions using the single cell gene expression in different cell types. To create a database that could be used with mouse scRNA-seq data, we downloaded orthologs between human and mouse from Biomart, and only kept those with one-to-one relationship. We also added a few mouse-specific curated interaction pairs. A customized database was generated using ‘CellPhoneDB database generate’. The significant ligand-receptor interactions were performed using ‘CellPhoneDB method statistical analysis using our customized database and the default setting. The ones with P values < = 0.05 are considered to be significant.
Data repository
Raw Fastq/Bam files, TPM expression table, along with Cellranger raw count matrix can be retrieved from the GEO database with accession number GSE136256.
Automated Immunofluorescence Imaging Methods
Automated imaging of immunofluorescent sections stained as described above (“Immunohistochemical and immunofluorescent assay”) with Ki67 (labeled with Alexa Fluor 488 secondary), Cldu (labeled with Alexa Fluor 568), and DAPI staining. Images were acquired on an Olympus VS 120 Slide Scanner with a Hamamatsu ORCA-Flash 4.0 camera and a 40× 0.65 NA objective. Filter cubes were standard Olympus DAPI, FITC (for Alexa Fluor 488), and TRITC (for Alexa Fluor 568) cubes. Exposure times and illumination intensities were kept constant across time points and replicates.
Measurement and counting of immunofluorescent sections
Cell segmentation was performed as follows with custom written software utilizing the ImageJ (NIH, Bethesda, MD) program. First, images were down sampled by a factor of 3 from their original size, resulting in a pixel size of 0.66 μm. Next, the uniform background signal and camera offset were subtracted by subtracting the average intensity from a manually defined region near the tissue slice. Next, DAPI stained nuclei were detected using a “max not mask” approach. In this approach, the maximum intensity position above a specified threshold is identified and then masked with a circle of specified diameter. This process is repeated, each time finding maximum intensities outside the previously masked regions to identify all the nuclei. This methodology prevents overcounting of crowded nuclei (as is the case for intestinal crypts) due to the a priori specification of mask diameter corresponding approximately to the expected nuclear spacing. In our case, the intensity threshold was set to 25% of the maximum intensity in the DAPI channel and the mask diameter was 20 pixels (13.3 μm). After nuclear segmentation, intensities were measured for each nucleus in all channels, with a measurement diameter also equal to 20 pixels.
After measurement, it became clear that each sample differed in non-specific staining intensity. For Cldu, most of the cells in each sample were negative, allowing for subtraction of a majority negative signal intensity. This was determined by fitting the intensity histogram to a single Gaussian function and subtracting the center of that fit from the intensity distribution. For Ki67, the fraction of positive cells was much higher (sometimes approaching a majority), so we subtracted the 5th percentile of the intensity distribution from each image. Note that this approach may overestimate the non-specific staining level in each image, thereby leading to a slight underestimation of the number of positive cells in each image. The counted values appear to agree well with hand counting analysis of positive and double positive cells, validating our approach. The threshold value for Ki67 positive cells was set to 160 intensity units, while for Cldu the threshold value was set to 100 intensity units across all samples. These values were chosen based on manual inspection of the intensity distributions, comparison to hand counting analysis, and manual measurements of positive and negative cells. The absolute magnitude of these values did not dramatically affect the trends seen in Figure 3—changing them simply shifted those values at all time points to higher or lower fractions.
Technical replicates represented different intestinal tissue sections from the same animal while biological replicates represented samples from different animals (n = 2–3). Data points and statistics shown and analyzed were over all technical replicates. Outliers were removed based on both Dixon’s Q-test and the Grubb’s test. For CldU, a single 3hr replicate was identified as an outlier and for the co-occurrence analysis, two technical replicate outliers were removed from the 3hr time point, and one technical replicate was removed from the 48hr time point. P values were determined from a one-tailed t test comparison with the 0hr time point.
Statistical significance
Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software). All data are shown as the mean ± SEM. A Student’s t test was used to compare two sets of data. One-way ANOVA was used for multiple sample comparison. For ones showing significant overall difference among samples, post hoc t tests were performed to compare specific sample differences. Boxplot was generated using a R package ggplot2. A multiplicity adjusted significance threshold of p ≤ 0.05 was used throughout the study. Error bars indicate s.e.m. Sample sizes, experimental replicates, and specific statistical test used are described in the Figure Legends. For Figure 7, samples were visually inspected for the presence of adjacent or double positive cells by three independent researchers.
Supplementary Material
Highlights.
Therapy-resistant TSCs (TrTSCs) are slow-cycling, stress responsive and survive from CRT
TSC-TME crosstalk shapes an immunosuppressive and pro-tumorigenic contexture
TAMM, recruited by TrTSC, in turn supports TSC proliferation via PGE2-EP4 signaling
TAMM-TSC/CSC pairing persists from adenomas to cancer stages in human CRCs
ACKNOWLEDGMENTS
We thank X. Cao and L. Chen for scientific discussion. We thank R. Krumlauf, A. Sanchez, J. Perry, and M. Epp for comments and editing and K. Tannen for proofreading. We appreciate S. Tegegne, K. Zapien, Y. Wang, D. Tsuchiya, A. Venkatraman, Y. Li, Y. Wang, M. McClain, J. Teddy, and H. Marshall for technical assistance. This work was supported by Stowers Institute for Medical Research (SIMR-1004), NCI Cancer Center Support Grant P30 CA168524 to University of Kansas Cancer Center, and an NIH grant (U01DK085507) to L.L., a member of the Intestinal Stem Cell Consortium funded by NIDDK and NIAID. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109674.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aoki T, Frȍsen J, Fukuda M, Bando K, Shioi G, Tsuji K, Ollikainen E, Nozaki K, Laakkonen J, and Narumiya S (2017). Prostaglandin E2-EP2-NF-κB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci. Signal 10, eaah6037. [DOI] [PubMed] [Google Scholar]
- Arneth B (2019). Tumor Microenvironment. Medicina (Kaunas) 56, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asfaha S, Hayakawa Y, Muley A, Stokes S, Graham TA, Ericksen RE, Westphalen CB, von Burstin J, Mastracci TL, Worthley DL, et al. (2015). Krt19(+)/Lgr5(−) Cells Are Radioresistant Cancer-Initiating Stem Cells in the Colon and Intestine. Cell Stem Cell 16, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, et al. (2019). Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125. [DOI] [PubMed] [Google Scholar]
- Bankaitis ED, Ha A, Kuo CJ, and Magness ST (2018). Reserve Stem Cells in Intestinal Homeostasis and Injury. Gastroenterology 155, 1348–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, Caux C, and Depil S (2019). Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovski T, De Sousa E Melo F, Vermeulen L, and Medema JP (2011). Cancer stem cell niche: the place to be. Cancer Res. 71, 634–639. [DOI] [PubMed] [Google Scholar]
- Carvalho MI, Bianchini R, Fazekas-Singer J, Herrmann I, Flickinger I, Thalhammer JG, Pires I, Jensen-Jarolim E, and Queiroga FL (2018). Bidirectional Regulation of COX-2 Expression Between Cancer Cells and Macrophages. Anticancer Res. 38, 2811–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellone MD, Teramoto H, Williams BO, Druey KM, and Gutkind JS (2005). Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 310, 1504–1510. [DOI] [PubMed] [Google Scholar]
- Caulfield AJ, and Lathem WW (2014). Disruption of fas-fas ligand signaling, apoptosis, and innate immunity by bacterial pathogens. PLoS Pathog. 10, e1004252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che D, Zhang S, Jing Z, Shang L, Jin S, Liu F, Shen J, Li Y, Hu J, Meng Q, and Yu Y (2017). Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/β-catenin signalling pathway. Mol. Immunol 90, 197–210. [DOI] [PubMed] [Google Scholar]
- Chen J, Luo Y, Zhou Y, Qin S, Qiu Y, Cui R, Yu M, Qin J, and Zhong M (2018). Promotion of Tumor Growth by ADAMTS4 in Colorectal Cancer: Focused on Macrophages. Cell. Physiol. Biochem 46, 1693–1703. [DOI] [PubMed] [Google Scholar]
- Cheung P, Xiol J, Dill MT, Yuan WC, Panero R, Roper J, Osorio FG, Maglic D, Li Q, Gurung B, et al. (2020). Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell 27, 590–604.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Cho HY, Hoyt KR, Naegele JR, and Obrietan K (2008). IGF-1 receptor-mediated ERK/MAPK signaling couples status epilepticus to progenitor cell proliferation in the subgranular layer of the dentate gyrus. Glia 56, 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN, and Garrett WS (2015). CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 12, 244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MF, and Fuller M (2006). Stem cells and cancer: two faces of eve. Cell 124, 1111–1115. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, and Wahl GM (2006). Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 66, 9339–9344. [DOI] [PubMed] [Google Scholar]
- Denk W, and Horstmann H (2004). Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol 2, e329. 10.1371/journal.pbio.0020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick JE (2008). Stem cell concepts renew cancer research. Blood 112, 4793–4807. [DOI] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Evans T (2009). Fishing for a WNT-PGE2 link: beta-catenin is caught in the stem cell net-work. Cell Stem Cell 4, 280–282. [DOI] [PubMed] [Google Scholar]
- Fearon ER, and Vogelstein B (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767. [DOI] [PubMed] [Google Scholar]
- Figueiredo CR, Azevedo RA, Mousdell S, Resende-Lara PT, Ireland L, Santos A, Girola N, Cunha RLOR, Schmid MC, Polonelli L, et al. (2018). Blockade of MIF-CD74 Signalling on Macrophages and Dendritic Cells Restores the Antitumour Immune Response Against Metastatic Melanoma. Front. Immunol 9, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming M, Ravula S, Tatishchev SF, and Wang HL (2012). Colorectal carcinoma: Pathologic aspects. J. Gastrointest. Oncol 3, 153–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli A, Oost KC, Kester L, Morgner J, Bornes L, Bruens L, Spaargaren L, Azkanaz M, Schelfhorst T, Beerling E, et al. (2020). Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell 26, 569–578.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, and Kaidi A (2009). The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 30, 377–386. [DOI] [PubMed] [Google Scholar]
- Guadagno E, Presta I, Maisano D, Donato A, Pirrone CK, Cardillo G, Corrado SD, Mignogna C, Mancuso T, Donato G, et al. (2018). Role of Macrophages in Brain Tumor Growth and Progression. Int. J. Mol. Sci 19, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harizi H (2013). The immunobiology of prostanoid receptor signaling in connecting innate and adaptive immunity. BioMed Res. Int 2013, 683405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA, Dirisina R, Porter-Westpfahl KS, Hembree M, Johnson T, et al. (2007). PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat. Genet 39, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HH, Lin YM, Shen CY, Shibu MA, Li SY, Chang SH, Lin CC, Chen RJ, Viswanadha VP, Shih HN, and Huang CY (2017). Prostaglandin E2-Induced COX-2 Expressions via EP2 and EP4 Signaling Pathways in Human LoVo Colon Cancer Cells. Int. J. Mol. Sci 18, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackstadt R, and Sansom OJ (2016). Mouse models of intestinal cancer. J. Pathol 238, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackstadt R, van Hooff SR, Leach JD, Cortes-Lavaud X, Lohuis JO, Ridgway RA, Wouters VM, Roper J, Kendall TJ, Roxburgh CS, et al. (2019). Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 36, 319–336.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar S, Deng L, He XC, and Li L (2020). Intestinal epithelial regeneration: active versus reserve stem cells and plasticity mechanisms. Am. J. Physiol. Gastrointest. Liver Physiol 318, G796–G802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement JD, Paschall AV, Redd PS, Ibrahim ML, Lu C, Yang D, Celis E, Abrams SI, Ozato K, and Liu K (2018). An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Invest 128, 5549–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson AG (2001). Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 1, 157–162. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, and McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116, 71–76. 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kreso A, and Dick JE (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291. [DOI] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, and Clevers H (2010). Coexistence of quiescent and active adult stem cells in mammals. Science 327, 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, and Neaves WB (2006). Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 66, 4553–4557. [DOI] [PubMed] [Google Scholar]
- Lin EH, Curley SA, Crane CC, Feig B, Skibber J, Delcos M, Vadhan SR, Morris J, Ayers GD, Ross A, et al. (2006). Retrospective study of capecitabine and celecoxib in metastatic colorectal cancer: potential benefits and COX-2 as the common mediator in pain, toxicities and survival? Am. J. Clin. Oncol 29, 232–239. [DOI] [PubMed] [Google Scholar]
- Liu Y, and Cao X (2016). Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 30, 668–681. [DOI] [PubMed] [Google Scholar]
- Markiewski MM, Vadrevu SK, Sharma SK, Chintala NK, Ghouse S, Cho JH, Fairlie DP, Paterson Y, Astrinidis A, and Karbowniczek M (2017). The Ribosomal Protein S19 Suppresses Antitumor Immune Responses via the Complement C5a Receptor 1. J. Immunol 198, 2989–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Stuurman N, Royer LA, Schartner C, Castillo-Azofeifa D, Delling M, Klein OD, and Vale RD (2018). Cellular aspect ratio and cell division mechanics underlie the patterning of cell progeny in diverse mammalian epithelia. eLife 7, e36739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero I, Rouzaut A, Motz GT, and Coukos G (2014). T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 4, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, Singh B, Rosenblum MD, and Fuchs E (2019). Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells. Cell 177, 1172–1186.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schönheit J, Giladi A, David E, Lara-Astiaso D, LorenzoVivas E, Paul F, Chappell-Maor L, Priller J, Leutz A, et al. (2017). Genomic Characterization of Murine Monocytes Reveals C/EBPβ Transcription Factor Dependence of Ly6C− Cells. Immunity 46, 849–862.e7. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, VanDussen KL, Malvin NP, Ryu SH, Wang Y, Sonnek NM, Lai CW, and Stappenbeck TS (2017). Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J. 36, 5–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, Graeber T, West BL, Bollag G, and Ribas A (2014). Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 74, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, and Dove WF (1990). A dominant mutation that pre-disposes to multiple intestinal neoplasia in the mouse. Science 247, 322–324. [DOI] [PubMed] [Google Scholar]
- Müller L, Tunger A, Plesca I, Wehner R, Temme A, Westphal D, Meier F, Bachmann M, and Schmitz M (2020). Bidirectional Crosstalk Between Cancer Stem Cells and Immune Cell Subsets. Front. Immunol 11, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murciano-Goroff YR, Warner AB, and Wolchok JD (2020). The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 30, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JM, He XC, Sugimura R, Grindley JC, Haug JS, Ding S, and Li L (2011). Cooperation between both Wnt/beta-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewal and expansion. Genes Dev. 25, 1928–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JM, Tao F, Roy A, Lin T, He XC, Chen S, Lu X, Nemechek J, Ruan L, Yu X, et al. (2020). Overcoming Wnt-β-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat. Cell Biol 22, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh AR, and Ernst M (2018). Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu DM, Botting RA, Stephenson E, Green K, Webb S, Jardine L, Calderbank EF, Polanski K, Goh I, Efremova M, et al. (2019). Decoding human fetal liver haematopoiesis. Nature 574, 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2017). The Microenvironmental Landscape of Brain Tumors. Cancer Cell 31, 326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhost S, Hughes É, Harrison H, Rafnsdottir S, Jacobsson H, Gregersson P, Magnusson Y, Fitzpatrick P, Andersson D, Berger K, et al. (2018). Sortilin inhibition limits secretion-induced progranulin-dependent breast cancer progression and cancer stem cell expansion. Breast Cancer Res. 20, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, Huang B, Qiu S, Li X, He L, and Peng Y (2016). Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget 7, 83976–83986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, and Clevers H (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. [DOI] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, Zlobec I, Chu P, Black S, Demeter J, McIlwain DR, et al. (2020). Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 182, 1341–1359.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J, Sheng GG, Mifflin RC, Powell DW, and Sheng H (2006). Roles of myofibroblasts in prostaglandin E2-stimulated intestinal epithelial proliferation and angiogenesis. Cancer Res. 66, 846–855. [DOI] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, and Dove WF (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668–670. [DOI] [PubMed] [Google Scholar]
- Taniguchi S, Elhance A, Van Duzer A, Kumar S, Leitenberger JJ, and Oshimori N (2020). Tumor-initiating cells establish an IL-33-TGF-beta niche signaling loop to promote cancer progression. Science 369, eaay1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, Kurth I, Andreu-Agullo C, Derbyshire ML, Posada J, et al. (2018). LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 172, 825–840.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, and Alvarez JV (2019). CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. eLife 8, e43653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Scoville D, He XC, Mahe MM, Box A, Perry JM, Smith NR, Lei NY, Davies PS, Fuller MK, et al. (2013). Isolation and characterization of intestinal stem cells based on surface marker combinations and colony-formation assay. Gastroenterology 145, 383–395.e1-e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi M, Li L, and Lengner CJ (2017). Hierarchy and Plasticity in the Intestinal Stem Cell Compartment. Trends Cell Biol. 27, 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. (2015). Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at https://www.stowers.org/research/publications/libpb-1315 upon publication.
All software and packages applied are publicly available and listed in the key resources table. The specific analysis parameters used are explained in the method section.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Ly6c antibody [ER-MP20] | Abcam | ab15627;RRID:AB_302004 |
| InVivoMAb anti-mouse Ly6G/Ly6C (Gr-1), 1mg | BioXCell | BE0075;RRID:AB_10312146 |
| Anti-Rabbit p-β-Catenin (Ser552) Monoclonal Antibody | Zymed custom antibody | Affinity Pure Rb Ab, 0.5mg/ml |
| Anti-Rabbit β-actin polyclonal Antibody | Abcam | Cat# ab8227 RRID: AB_2305186 |
| Anti-Mouse MHC Class II Monoclonal Antibody (AF6-120.1), APC | Thermo Fisher | Cat# 17-5320-82;RRID:AB_2573212 |
| SP-1 Chromogranin A (Porcine) Antibody | Immunostar | 20086;RRID:AB_572226 |
| FcR Blocking Reagent, mouse | Miltenyi Biotec | 130-092-575;RRID:AB_2892833 |
| Ly-6G (Gr-1) Monoclonal Antibody (1A8-Ly6g), PE-eFluor 610, eBioscience | ThermoFisher | 61-9668-82;RRID:AB_2574679 |
| Ly-6C Monoclonal Antibody (HK1.4), Alexa Fluor 488, eBioscience | ThermoFisher | 53-5932-82;RRID:AB_2574427 |
| CD11b Monoclonal Antibody (M1/70), APC-eFluor 780, eBioscience | ThermoFisher | 47-0112-82;RRID:AB_1603193 |
| Lysozyme EC 3.2.1.17 | Agilent | A009902-2;RRID:AB_578661 |
| Prostaglandin E Receptor EP4 Antibody | VWR/Bioss | bs-8538R |
| Anti-F4/80 antibody [SP115] (ab111101), Rabbit | Abcam | ab111101;RRID:AB_10859466 |
| Anti-CD68 antibody (ab125212), Rabbit | Abcam | ab125212:RRID:AB_10975465 |
| Anti-CD11b antibody [EPR1344] (ab133357), rabbit | Abcam | ab133357;RRID:AB_2650514 |
| CD44-Biotin, mouse | Miltenyi Biotec | 130-110-082;RRID:AB_2661032 |
| CD24-Biotin, mouse | Miltenyi Biotec | 130-102-128;RRID:AB_2656584 |
| Anti-CD68 antibody [C68/684] | Abcam | ab201340;RRID:AB_2747829 |
| Anti-CD11b antibody [M1/70] | Abcam | ab8878;RRID:AB_306831 |
| Anti-CD11b antibody [EPR1344] (ab133357), rabbit | Abcam | ab133357;RRID:AB_2650514 |
| Anti-Rabbit Krt15 Polyclonal Antibody | Novus | Cat# NBP1-85602 RRID: AB_11037933 |
| Anti-Mouse Ascl2 Monoclonal Antibody | LSBio | Cat# LS-C126891;RRID:AB_10831114 |
| CD9 | Abcam | ab92726;RRID:AB_10561589 |
| Bmi1 antibody | Abcam | ab14389;RRID:AB_2065390 |
| Annexin A2 | Abcam | ab178677;RRID:AB_1140698 |
| Annexin A1 | Abcam | ab214486;RRID:AB_722804 |
| Ascl2 antibody | Biorbyt | orb155740;RRID:AB_10865613 |
| E-Cadherin (24E10) Rabbit mAb | Cell Signaling Technology | 3195;RRID:AB_2291471 |
| PD-1 Monoclonal Antibody (7A11B1) | ThermoFisher | MA5-15780;RRID:AB_11152225 |
| Beta-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | 3700S;RRID:AB_2242334 |
| Mouse TrueBlot ULTRA:Anti-Mouse Ig HRP | Rockland | 18-8817-33;RRID:AB_2610851 |
| CST alpha-tubulin Antibody | Cell Signaling Technology | 2144;RRID:AB_2210548 |
| Bmi1 (D20B7) XP® Rabbit mAb | Cell Signaling Technology | 6964;RRID:AB_10828713 |
| Purified Mouse Anti-Human CD63 Clone H5C6 | Fisher/BD Biosciences | 556019;RRID:AB_396297 |
| Krt15 antibody (Cytokeratin 15) | Fisher/Novus Biologicals | NBP2-50461AF488; RRID:AB_1084834 |
| BrdU antibody | Fisher/Novus Biologicals | NB600-720;RRID:AB_10002707 |
| S100A10 Antibody | VWR/ProteinTech | 11250-1-AP;RRID:AB_2269906 |
| Donkey Anti-Rabbit IgG H&L (Alexa Fluor® 594) |
Abcam | ab150076;RRID:AB_2782993 |
| ASCL2 Antibody (C terminus) LS-C126891 | LifeSpan Biosciences | LS-C126891-100;RRID:AB_10831114 |
| Cytokeratin 15 Antibody | Novus Biologicals | NBP1-85602;RRID:AB_11037933 |
| MHC Class II I-Ab Monoclonal Antibody (AF6-120.1), APC, eBioscience | ThermoFisher | 17-5320-82;RRID:AB_2573212 |
| Mouse IgG2a kappa Isotype Control (eBM2a), APC, eBioscience | ThermoFisher | 17-4724-81;RRID:AB_470188 |
| CD45 Monoclonal Antibody (30-F11), PE-Cyanine5, eBioscience | ThermoFisher | 15-0451-83;RRID:AB_468753 |
| Rat IgG2b kappa Isotype Control (eB149/10H5), PE-Cyanine5, eBioscience | ThermoFisher | 15-4031-82;RRID:AB_470133 |
| CD11b Monoclonal Antibody (M1/70), PE, eBioscience | ThermoFisher | 12-0112-83;RRID:AB_2734870 |
| Rat IgG2b kappa Isotype Control (eB149/10H5), PE, eBioscience | ThermoFisher | 12-4031-82;RRID:AB_470042 |
| CD11c Monoclonal Antibody (N418), PE-Cyanine7, eBioscience | ThermoFisher | 25-0114-82;RRID:AB_469590 |
| Armenian Hamster IgG Isotype Control (eBio299Arm), PE-Cyanine7, eBioscience | ThermoFisher | 25-4888-82;RRID:AB_470204 |
| FITC anti-mouse CX3CR1 Antibody | VWR/Biolegend | 149020;RRID:AB_2565703 |
| FITC Mouse IgG2a, κ Isotype Ctrl Antibody | VWR/Biolegend | 400208;RRID:AB_2884007 |
| Annexin V-mFluor Violet 540 conjugate | AAT Bioquest | 20080 |
| EP4 antibody | Abcam | ab217966 |
| CD3 | Abcam | ab16669;RRID:AB_443425 |
| Anti-BrdU antibody [BU1/75 (ICR1)] | Abcam | ab6326;RRID:AB_305426 |
| CD68 Antibody | Abcam | ab201340;RRID:AB_991703 |
| CD19 Antibody | Abcam | ab203615;RRID:AB_1140606 |
| Ascl2 antibody, 100 μg | Biorbyt | orb155740 |
| Cytokeratin 15 Antibody | Fisher/Novus Biologicals | NBP2-53308-100ug |
| TCF21 Antibody | Fisher/Novus Biologicals | NBP1-88637;RRID:AB_11008184 |
| Rat monoclonal [YTS169.4] to CD8 | Abcam | ab22378;RRID:AB_447033 |
| Anti-Sox9 antibody | Sigma/Millipore | AB5535;RRID:AB_2239761 |
| RM-9106-S1/Ab Ki-67 0.5mL/EA | VWR | MISC-CLINICAL |
| Mouse monoclonal [2B3] to RUNX3 | Abcam | ab135248 |
| Rabbit monoclonal [EP3251] to Pax2 | Abcam | ab79389;RRID:AB_1603338 |
| ASCL2 Antibody (C terminus) LS-C126891 | LifeSpan Biosciences | LS-C126891-100;RRID:AB_10831114 |
| Cytokeratin 15 Antibody | Novus Biologicals | NBP1-85602;RRID:AB_11037933 |
| Cox2 (D5H4) XP Rabbit mAb | Cell Signaling Technology | 12282S;RRID:AB_2571729 |
| Purified Mouse Anti-E-Cadherin | Fisher/BD Biosciences | 3700S |
| PTGER4/EP4 Antibody | Novus Biologicals | NBP1-84833;RRID:AB_11019807 |
| p23/PTGES3 Antibody (JJ3) | Novus Biologicals | NB300-576;RRID:AB_10000921 |
| HSP90 beta Antibody | Novus Biologicals | NBP1-77563;RRID:AB_11011699 |
| Anti-HSP90aa1 | Novus Biologicals | NB120-2928;RRID:AB_790272 |
| B7-H1/PD-L1/CD274 Antibody (M - 0.1 MG) | Fisher/Novus Biologicals | NBP1-432620 |
| Anti-Prostaglandin E2 antibody | Abcam | ab2318;RRID:AB_302974 |
| Cleaved Caspace-3 (Asp175) Antibody, 100 μl (10 western blots) | Cell Signaling Technology | 9661S;RRID:AB_2341188 |
| BrdU/CldU antibody | Fisher/Novus Biologicals | NB500-169;RRID:AB_10002608 |
| Cox2 Antibody | Cell Signaling Technology | 4842;RRID:AB_2084968 |
| ANTI-CD44 ANTIGEN ISOFORM 4 | Fisher | PIPA521419 |
| CD44v6 Monoclonal Antibody (9A4) | Fisher | MA1-81995;RRID:AB_928369 |
| Phospho-Akt (Ser473) (D9E) XP® | Cell Signaling Technology | 4060S;RRID:AB_2315049 |
| Thermo Scientific Lab Vision Ki-67, Rabbit Monoclonal Antibody | ThermoFisher | RM-9106-S1;RRID:AB_149792 |
| Anti-RIP3 antibody | Abcam | ab56164;RRID:AB_2178667 |
| Anti-CD8 antibody [YTS169.4] | Abcam | ab22378;RRID:AB_447033 |
| Annexin V-mFluor Violet 540 conjugate | AAT Bioquest | 20082 |
| CD24 Monoclonal Antibody (M1/69), eFluor 450, eBioscience | ThermoFisher/eBioscience | 48-0242-82;RRID:AB_1311169 |
| MHC Class II I-Ab Monoclonal Antibody (AF6-120.1), APC, eBioscience | ThermoFisher/eBioscience | 17-5320-82;RRID:AB_2573212 |
| Anti-CD44 antibody [T2–F4] | Abcam | ab40983;RRID:AB_726520 |
| Thermo Scientific Lab Vision Ki-67, Rabbit Monoclonal Antibody | ThermoFisher | RM-9106-S1;RRID:AB_149792 |
| CldU antibody | Novus Biologicals | NB500-169;RRID:AB_10002608 |
| Anti-Mouse CD24 FITC 50ug | eBioscience | 11-0241-81;RRID:AB_464984 |
| Lysozyme EC 3.2.1.17 | Dako | A009902-2:RRID:AB_2341231 |
| Anti-GRP78/BiP (ET-21) antibody produced in rabbit | Sigma | G9043-200UL; RRID:AB_2279879 |
| Mouse ALCAM Phycoerythrin Affinity Purified PAb, Goat IgG (CD166 PE) | R&D Systems | FAB1172P;RRID:AB_2242595 |
| CD44 P7 | BioLegend | 103030;RRID:AB_830787 |
| Anti-Mouse CD24 eFluor® 450 (Pacific Blue® replacement) 100 ug (CD24 eF450) | eBioscience | 48-0242-82;RRID:AB_1311169 |
| goat anti-rabbit IgG, F(ab’)2-APC-Cy7 | Santa Cruz Biotechnology | sc-3847; RRID:AB_649105 |
| Anti-GRP78/BiP (ET-21) antibody produced in rabbit | Sigma | G9043-200UL;RRID:AB_2279879 |
| MTA1 (D40D1) XP® Rabbit mAb 100ul (10 western blots) | Cell Signaling Technology | 5647S;RRID:AB_10705601 |
| BrdU antibody | VWR | 95019-028 |
| CD44 | Fisher | MA1-81995;RRID:AB_928369 |
| Biological samples | ||
| Human familial adenomatous polyposis (FAP) paraffin-embedded tissue sections | University of Kansas Medical Center | Pathology & Laboratory UKMC, KC, KS 66160 |
| Human colorectal cancer (CRC) paraffin-embedded tissue sections | University of Kansas Medical Center | Pathology & Laboratory UKMC, KC, KS 66160 |
| Chemicals, peptides, and recombinant proteins | ||
| EP2 antagonist | Cayman Chemical | 14050 |
| M-CSF, Macrophage-Colony Stimulating Factor Protein, Recombinant human | Millipore | GF053 |
| EP4 antagonist | Sigma | A8227-25MG |
| Human Recombinant M-CSF, Size: 100 μg | StemCell Technologies | 78057 |
| DNase I | Fisher/Worthington | LS006361 |
| Liberase TM Research Grade | Sigma | 5401119001 |
| Celecoxib | LC Laboratories | C-1502 |
| Capecitabine, 10 g | LC Laboratories | C-2799 |
| JAG-1 (188–204), Jagged-1 (188–204), Notch Ligand” | Anaspec | AS-61298 |
| Recombinant Murine Noggin, 20 ug | PeproTech | 250-38 |
| Recombinant Murine EGF, 100 ug | PeproTech | 315-09 |
| Recombinant Mouse R-Spondin 1, CF, 250 ug | R&D Systems | 3474-RS-250 |
| Recombinant Murine Wnt-3a, 2 ug | R&D Systems | 1324-WN-002 |
| Stemolecule Thiazovivin (ROCK inhibitor), 1 mg | Reprocell | 04-0017 |
| Stemolecule CHIR99021, 2 mg | Reprocell | 04-0004 |
| IntestiCult Organoid Growth Medium (Mouse) | StemCell Technologies, Inc. | 06005 |
| Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix, Phenol Red-free, LDEV-free, 10 mL | VWR | 356231 |
| PF3644022, 10 mg | Sigma | H6278-50MG |
| 4-HYDROXYTAMOXIFEN | VWR | 103302-538 |
| Collagenase 1A, 100mg | Sigma | C2674-100MG |
| Hyaluronidase, 100mg | Sigma | H3506-100MG |
| Evans Blue, 10 g | Sigma | E2129-10G |
| MATRIGEL MATRIX NO PHENOL 10ML | VWR/Corning | 356237 |
| Advanced DMEM/F-12 | ThermoFisher | 12634-010 |
| E4 pipette | Rainin | 17014490 |
| B-27® Serum-Free Supplement (50X), Liquid, 10 ml | ThermoFisher | 17504-044 |
| N-2 Supplement (100X), Liquid, 5 ml | ThermoFisher | 17502-048 |
| N-ACETYL-L-CYSTEINE BIOREAGENT CELL CUL& | Sigma | A9165-5G |
| Y-27632 | Sigma | Y0503-1MG |
| Stemolecule Thiazovivin 1 mg | Stemgent | 04-0017 |
| Recombinant Human R-Spondin 1, CF 25 μg | R&D Systems | 4645-RS-025/CF |
| Basement Membrane Matrix, Growth Factor Reduced (GFR), Phenol Red-free, 10ml *LDEV-Free | BD Biosciences | 356231 |
| SOLUTION BSS W/O CA+ MG+ PH-RD 1L | VWR | 12001-980 |
| Critical commercial assays | ||
| Myeloid-Derived Suppressor Cell Isolation Kit | Miltenyi Biotec | Cat# 130-094-538 |
| MACS separation using LS columns | Miltenyi Biotec | Cat# 130-042-401 |
| 10X Chromium Single Cell 3′ Library & Gel Bead Kit (v2) | 10x Genomics | Cat# PN-120267 |
| TruSeq Stranded mRNA Prep Kit | illumine | Cat# 20020594 |
| Bioanalyzer RNA Analysis | Agilent Technologies | Cat# 5067-1511 |
| Complementary DNA synthesis; High-capacity RNA to cDNA | Life Technologies | Cat# 4387406 |
| Propidium Iodide Solution | Miltenyi Biotec | 130-093-233 |
| MACS BSA Stock Solution | Miltenyi Biotec | 130-091-376 |
| Dead Cell Removal Kit | Miltenyi Biotec | 130-090-101 |
| autoMACS Rinsing Solution | Miltenyi Biotec | 130-091-222 |
| Anti-Mouse IgG1 MicroBeads | Miltenyi Biotec | 130-047-102;RRID:AB_244355 |
| CD45 MicroBeads, mouse | Miltenyi Biotec | 130-052-301;RRID:AB_2877061 |
| CD31 MicroBeads, mouse | Miltenyi Biotec | 130-097-418;RRID:AB_2814657 |
| Anti-Ter-119 MicroBeads, mouse | Miltenyi Biotec | 130-049-901 |
| Miltenyi Biotec | Miltenyi Biotec | 130-042-201 |
| Dead Cell Removal Kit | Miltenyi Biotec | 130-090-101 |
| RBC lysis buffer | Sigma | 11814389001 |
| Annexin V-mFluor Violet 540 conjugate | AAT Bioquest | 20082 |
| CD11b MicroBeads | Miltenyi Biotec | 130-049-601 |
| CD11c MicroBeads | Miltenyi Biotec | 130-108-338 |
| Trypan blue solution 0.4%, 100ml | ThermoFisher | 15250061 |
| KnockOut Serum Replacement | ThermoFisher | 10828028 |
| Restore PLUS Western Blot Stripping Buffer | ThermoFisher | 46430 |
| Deposited data | ||
| 10X single cell (sc) RNA-seq data | Gene Expression Omnibus (GEO: GSE136256) | |
| Experimental models: Organisms/strains | ||
| C57BL/6 | Jackson Laboratories | 000664;RRID:IMSR_JAX:000664 |
| C57BL/6J-ApcMin/J (Apc-Min) | Jackson Laboratories | 002020;RRID:IMSR_JAX:002020 |
| BALB/cJ (BALB/c) | Jackson Laboratories | 000651;RRID:IMSR_JAX:000651 |
| B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J (R26-YFP) | Jackson Laboratories | 006148;RRID:IMSR_JAX:006148 |
| B6;129-Bmi1tm1(cre/ERT)Mrc/J (Bmi1-CreER) | Jackson Laboratories | 010531;RRID:IMSR_JAX:010531 |
| B6.129P2-Lgr5tm1(cre/ERT2)Cle/J (LGR5KI) | Jackson Laboratories | 008875;RRID:IMSR_JAX:008875 |
| Software and algorithms | ||
| FlowJo Software v 7.0 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| ImageJ | OpenSource | https://imagej.nih.gov/ij/download.html |
| GraphPad Prism v 8.1.2 | Prism | https://www.graphpad.com/ |