

Abstract
The presenilin genes (PSEN1 and PSEN2) are mainly responsible for causing early-onset familial Alzheimer’s disease, harboring ~300 causative mutations, and representing ~90% of all mutations associated with a very aggressive disease form. Presenilin 1 is the catalytic core of the γ-secretase complex that conducts the intramembranous proteolytic excision of multiple transmembrane proteins like the amyloid precursor protein, Notch-1, N- and E-cadherin, LRP, Syndecan, Delta, Jagged, CD44, ErbB4, and Nectin1a. Presenilin 1 plays an essential role in neural progenitor maintenance, neurogenesis, neurite outgrowth, synaptic function, neuronal function, myelination, and plasticity. Therefore, an imbalance caused by mutations in presenilin 1/γ-secretase might cause aberrant signaling, synaptic dysfunction, memory impairment, and increased Aβ42/Aβ40 ratio, contributing to neurodegeneration during the initial stages of Alzheimer’s disease pathogenesis. This review focuses on the neuronal differentiation dysregulation mediated by PSEN1 mutations in Alzheimer’s disease. Furthermore, we emphasize the importance of Alzheimer’s disease-induced pluripotent stem cells models in analyzing PSEN1 mutations implication over the early stages of the Alzheimer’s disease pathogenesis throughout neuronal differentiation impairment.
Key Words: familial Alzheimer's disease, familial Alzheimer's disease-induced pluripotent stem cells models, induced pluripotent stem cells, neurogenesis, neuronal differentiation, Notch, presenilin 1, PSEN1 mutations, γ-secretase complex
Physiological Role of Presenilins
Presenilins (PS) are a family of highly conserved multi-pass transmembrane proteins located in different cellular structures such as endosomes, lysosomes, nuclear envelope, mitochondria, the trans-Golgi network, and the endoplasmic reticulum (Escamilla-Ayala et al., 2020). PS constitutes the catalytic core of the γ-secretase complex, known better for its proteolytic action on amyloid precursor protein (APP) resulting in amyloid beta-peptide (Aβ) accumulation, one of Alzheimer’s disease (AD) critical events. PS can be classified as γ-secretase-dependent and γ-secretase-independent. Most of the PS functions described up to date are γ-secretase-dependent mainly because, as described before, its primary role is to provide the complex’s catalytic core. The γ-secretase complex is a high molecular weight endoprotease that comprises four essential subunits: PS, Pen2, nicastrin, and Aph-1. Among these subunits, PS has shown to mediate most proteolytic events since the secretase activity is dramatically reduced when PS1 is deleted (Oikawa and Walter, 2019). It is noteworthy that some subunits have homologs and isoforms such as PS (PS1 and PS2) and Aph1 (Aph1a and Aph1b), respectively. These variations result in the in vivo assembly of at least six different complexes, though their functional differences are still poorly understood (Yonemura et al., 2016; Stanga et al., 2018).
Many studies have described that multiple γ-secretase complexes have different substrate specificities and have shown that exscind several Type-I transmembrane proteins. The best-studied substrates are APP and Notch for its fundamental implication in AD as well as in cell fate determination and development (Kopan and Ilagan, 2004; Stanga et al., 2018). Nevertheless, numerous other substrates have been identified, including Nectin1a, N- and E-cadherin, ErbB4, LRP, Jagged, CD44, Delta, and Syndecan. These proteins play essential roles in multiple processes, including neural progenitor maintenance, neurogenesis, neurite outgrowth regulation, synaptic function, neuronal function, myelination, and plasticity. Multiple of these processes are disrupted throughout AD pathogenesis, indicating that the complex usually serves as a mediator of diverse signaling pathways (Zoltowska and Berezovska, 2018; Güner and Lichtenthaler, 2020).
Search Strategy and Selection Criteria
Literature cited in this review published from 1998 to 2020 was searched on databases including Web of Science, PubMed and Google Scholar. The keywords used were: “PSEN1 mutations”, “Alzheimer’s Disease and PSEN1”, “PSEN1 and neuronal differentiation” “PSEN1 mutations and neurogenesis”, “Familiar Alzheimer’s Disease-induced pluripotent stem cell models”, and “γ-secretase complex and neurogenesis”.
Role of Presenilin 1 on Neural Differentiation
The PS1 protein is ~45–50 kDa in size and is widely expressed in various tissues, comprehending the brain, predominantly in the cell body and dendrites of neurons (Uhlén et al., 2015). Beyond its roles in the APP processing, evidence from in vivo research supports PS1 function during early neurogenesis, with pivotal functions in the proliferation and maintenance of neural progenitor cells (NPC), the appropriate migration of neurons in the cerebral cortex, and the spatiotemporal control of neuronal differentiation and new-born neuron survival capacity (for a detailed review see (Lazarov and Marr, 2010; Bonds et al., 2015). Homozygous PS1-null mice embryos result in perinatal lethality, displaying severe skeletal deformities, intracranial hemorrhages, and cerebral cavitation associated with massive neuronal loss (Donoviel et al., 1999). Additionally, several studies in PSEN1 cKO mice reported decreasing proliferation rates and enhanced differentiation capacity of NPCs (Bonds et al., 2015).
Using selected information where the implication of PS1 in the CNS is evidenced, we created a predicted protein-protein interaction network map with the software STRING (Szklarczyk et al., 2019). The predicted interactome network (Figure 1) emphasizes the implication of PS1 in NPC’s self-renewal and differentiation capacity, highlighting the biological processes of neuron generation in green color, with 17 members interacting and a false discovery rate (FDR) of 3.77E-12 (0.70 of confidence). The dark-blue color shows the canonical WNT signaling with five-member and an FDR of 2.06E-6. In yellow color, Notch signaling is shown with eight members, and an FDR of 2.24E-10. In pink color, the canonical WNT signaling regulation is shown with five members and an FDR of 6.01E-15. The neuronal differentiation is shown in red color with 15 members and an FDR of 5.03E-12. Finally, in light-blue color, the primary molecular function is the protein binding with 19 members and 9.96E-15 FDR.
Figure 1.
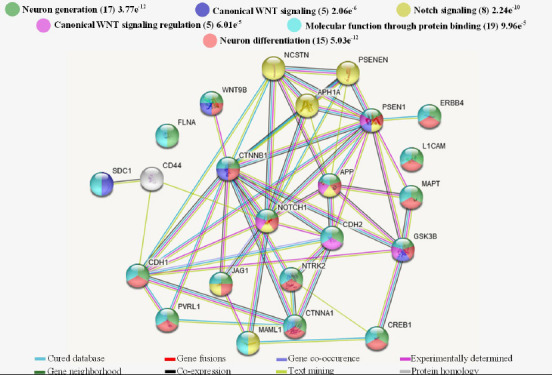
Interactome of polypeptides linked to PS1 concerning NPC proliferation and differentiation.
UniProtKB codes were submitted to the String program to create a predicted interaction network. Colored stripes illustrate evidence for each interaction. The proteins involved in neuron generation (17 members), the canonical Wnt signaling (5 members), the Notch signaling (8 members), regulation of the Wnt signaling (5 members), neuron differentiation (15 members), and which molecular function mediated by the protein binding (19 members) were highlighted in green, dark blue, red, yellow, pink, red and light-blue nodes respectively. All proteins interactions show statistical significance (P-value ≤ 0.05). APH1A: Gamma-secretase subunit APH-1A; APP: amyloid-beta precursor protein; CDH1: cadherin-1; CDH2: cadherin-2; CREB1: cyclic amp-responsive element-binding protein 1; CTNNA1: catenin alpha-1; CTNNB1: catenin beta-1; ERBB4: receptor tyrosine-protein kinase erbB-4; FLNA: filamin-A; GSK3B: glycogen synthase kinase-3 beta; JAG1: protein jagged-1; L1CAM: neural cell adhesion molecule L1; MAML1: mastermind-like protein 1; MAPT: microtubule-associated protein tau; NCSTN: nicastrin; NOTCH1: neurogenic locus notch homolog protein 1; NTRK2: BDNF/NT-3 growth factors receptor; PSEN1: presenilin-1; PSENEN: gamma-secretase subunit PEN-2; PVRL1: nectin-1; SCD1: syndecan-1; WNT9B: protein Wnt-9b.
Here, the best-known biological functions of PS1 are associated with the Notch signaling and neuronal differentiation regulation. Notch proteins are large signaling receptors involved in cell-fate determination and patterning during development. In the adult, Notch activity also is implicated in neural stem cell maintenance, neural differentiation, and regulating neurite outgrowth. Studies using knockouts and γ-secretase inhibitors established a relationship between Notch and PS, observing that the loss of PS activity resembled losses in Notch1 function. Notch signaling can be activated by PS1 throughout the Notch1 intracellular domain delivery and downstream signaling initiation (Bonds et al., 2015; Marathe and Alberi, 2015). Handler et al. (2000) suggested that Notch signaling impairment by PSEN1 inhibition results from the downregulated expression of Notch1 downstream target gene Hes5. The Notch1 signaling impairment and PSEN1 inhibition promote a massive premature neuronal differentiation resulting in a partial depletion of the NPC pools (Handler et al., 2000). Interestingly, reports have shown that Notch signaling also regulates dendrite development in new-born neurons. The conditional knockout of Notch1 in adult-born neurons reduced dendritic branching, while its overexpression significantly increased the dendrite arborization but inhibits neurite outgrowth (Bonds et al., 2015; Ding et al., 2016).
The PS1-induced Notch signaling pathway indirectly mediates the CREB-target genes expression (brain-derived neurotrophic factor and c-fos included), implicated in neuronal survival and long-lasting synaptic changes. This indirect activation appears to be related to the putative CREB-binding protein (CBP) promoter containing a binding site for the Notch-downstream transcription factor CBF1/RBP-Jk (Saura et al., 2004). Besides, Francis et al. (2006) reported a direct PS1-mediated CREB pathway activation that involves the P38 and p42/p44 MAPK and PI3-kinase pathways. In immature neurons, the signaling mediated by CREB is required for initial neuronal dendritic branching by directing dendrite formation and elongation (Bustos et al., 2017), as well to the response to neurotrophic factors promoting neural survival pathways that involve AMP, PKA, MAPK, and genes such as Bcl2 and Mcl1 (Walton and Dragunow, 2000; Landeira et al., 2016). Peculiarly, the PS-mediated disassembly of N-cadherin (CDH2) and E-cadherin (CDH1) also affects CREB signaling. A report showed that N-cadherin CTF bind to CREB-binding protein, promoting its degradation, which in turn, downregulates CREB transcription (Marambaud et al., 2002).
E-cadherin and N-cadherin are the best-known cadherins that mediate cell adhesion, with critical roles in the normal development and maintenance of cell-cell contacts. Cadherins bind to β-catenin to adhesion stabilization through the actin cytoskeleton (Marambaud et al., 2002). Reports showed that PS1 might have dual activity regard to cell-cell adhesion: (i) under cell-cell adhesion conditions, PS1 promotes the cadherin-catenin complex anchors to the cytoskeleton, promoting Ca2+-dependent cell aggregation (Baki et al., 2001), (ii) conversely when cell dissociation is needed, PS1 promotes adherents junctions disassembling (Marambaud et al., 2002). The PS1-dependent E-cadherin processing results in a remarkably cytosolic increase of the complex E-cad/CTF2 and soluble β-catenin, a Wnt signaling pathway decisive regulator (Parisiadou et al., 2004; Li et al., 2016). However, there are contradictory data related to PS1 implication in the β-catenin regulation. For example, Killick et al. (2001) reported that PS1 antagonizes the cytoplasmatic and nuclear β-catenin as a response to Wnt1 or Dvl; consequently, there is negative regulation of Wnt signaling-dependent transcriptional activation (Killick et al., 2001). In the absence of PS1, the β-catenin pools are significantly higher, which probably could be related to the hyperproliferative phenotype found in PSEN1 null fibroblast (Xia et al., 2001). In contrast, Uemura et al. (2003) suggested that PS1 exert a positive effect in β-catenin nuclear translocation regulation with a subsequent increase of cyclin D transcription, which in turn preceded SH-SY5Y cell differentiation through the β-catenin/TCF/LEF-1 pathway (Uemura et al., 2003; Bonds et al., 2015). Furthermore, another PS1 interacting protein is glycogen synthase kinase 3β (GSK3β), primarily characterized by Wnt signaling involvement. It has been shown that GSK3β phosphorylates PS1 on serine residue 397, regulating its stabilization. Besides, GSK3β influence PS1 ability to associate with β-catenin, improving the stability of the Complex (Duggan and McCarthy, 2016).
During neurogenesis, one of PS’s most relevant functions is the proper migration from neuronal cells’ birthplace to a final location for integration (Wang et al., 2017). Mice with a null PSEN1 die prematurely and display multiple abnormalities, including impairments in cell migration during neocortex development (Handler et al., 2000; Louvi et al., 2004; Buchsbaum and Cappello, 2019). Louvi et al. (2004) reported that loss of PS1 function appears to have a strong link with neuronal migration impairments, compromising radial, and tangential migration. Interestingly, the failure to migrate to an appropriate cortical position in the PSEN1 cKO mice correlates with the radial glia reduction and Cajal-Retzius neuron survival (Louvi et al., 2004; Barber and Pierani, 2016).
During the neuronal differentiation, PS1 associates with cytoskeleton components, including MAPT and filamin, regulating neurite outgrowth and stabilization (Zhang et al., 1998a; Pigino et al., 2001). It is unclear how PS1 might mediate this process. Additionally, despite the critical role of presenilins in brain development, their effect during adult neurogenesis remains unclear. In this line, Gadadhar et al. (2011) reported that in neurosphere cultures, downregulation of PS1 improves differentiation without altering their multipotentiality and decreases NPCs’ proliferation. PS1 depletion also downregulates β-catenin and EGFR, implicated in the neural stem cells self-renewal and proliferation regulation (Gadadhar et al., 2011). Also, Bonds et al. (2015) reported that PS1 downregulation reduces p-β-catenin and Notch intracellular cleavage fragments, promoting the cell cycle exit and differentiation. As the expression of β-catenin in NPC is decreased by PS1 downregulation, it may promote a reduction of proliferation-inducing genes (Bonds et al., 2015). In contrast, Dhaliwal et al. (2018) reported that PSEN1 ablation made no changes in maintenance, proliferation, and NPC differentiation. Interestingly, the retroviral-labeled presenilin-null adult neurons show typical electrophysiological properties (Dhaliwal et al., 2018).
To date, there are 149 different substrates identified for the γ-secretase complex. It is unknown if these proteins processing induces aberrant cleavages or generates toxic products, such as Aβ. However, the substrates’ normal biological functions involved in diverse signaling pathways are affected by the γ-secretase complex-mediated processing. Consequently, impaired signaling contributing to neurodegeneration during AD pathogenesis could result from a disruption in the γ-secretase complex (Güner and Lichtenthaler, 2020).
In summary, the reduction of PS1 levels in NPC favors a decreased proliferation rate and enhanced NPC differentiation. Simultaneously, it results in a dendritic branching and spine density reduction and decreased survival rate in newly generated neuronal cells. The predicted interactome network emphasizes the implication of PS1 in NPC self-renewal and differentiation capacity. Hence, the discovery of possible signaling pathways linked to familial Alzheimer’s disease (FAD), and in turn, a possible therapeutic strategy design could be improved by using these types of interactome analyses.
Implication of PSEN1 Mutations in Neural Differentiation
AD is a chronic neurodegenerative disorder characterized by an acute accumulation of neurofibrillary tangles and amyloid plaques in specific brain regions, such as the hippocampus and the cortex, leading to a progressive loss of neurons. Amyloid plaques are insoluble extracellular aggregations of Aβ protein, while intracellularly paired helical filaments composed of hyperphosphorylated tau (an abnormal microtubule-associated protein) accumulate in the form of neurofibrillary tangles. The Aβ protein is released after the sequential β- and γ-secretase-mediated proteolytic processing of APP (DeTure and Dickson, 2019).
It is well known that most of the AD cases correspond to the sporadic variant of the disorder; however, there is a genetic predisposition variant that involves three different genes originating the disorder. These AD-related genes are PSEN1 and 2, and APP (encoding for PS1, PS2, and APP proteins respectively) from which mutations in PSEN genes exhibit high penetrance causing the most FAD cases. The most commonly mutated gene and mostly associated with a severe form of the disease is PSEN1, with approximately 319 mutations reported in the Alzforum database (https://www.alzforum.org/mutations/psen-1) (Lanoiselée et al., 2017; Kabir et al., 2020).
The FAD-linked mutations in PSEN genes have been associated with the proteolytic processing of APP because of the γ-secretase-complex implication over the generation of Aβ peptide residues (37 to 49), increasing the self-aggregation Aβ42-residue type abundance, leading to nucleation, oligomerization, and neuropathogenicity, establishing a critical role for PS in AD pathogenesis (de Leeuw and Tackenberg, 2019).
There is growing evidence regarding PS1 and APP implication in different physiological processes in neural stem cells and NPC, including differentiation, proliferation, and survival (Figure 2). Consequently, an impaired function or regulation induced by FAD-linked PSEN1 mutations compromise these processes and cause aberrant signaling, synaptic dysfunction, memory impairment, increased Aβ42/Aβ40 ratio, aberrant control of neuronal differentiation, contributing to neurodegeneration, mostly in the frontotemporal region (Lazarov and Marr, 2010; Dhaliwal et al., 2018).
Figure 2.
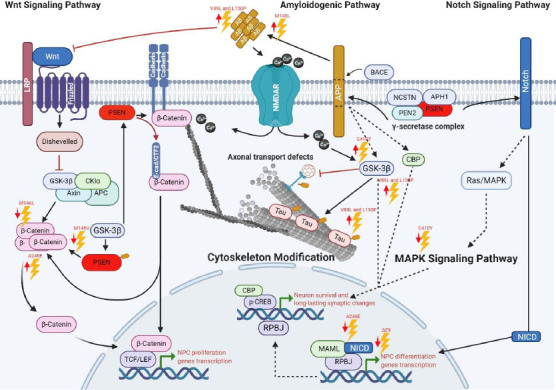
Major pathways disturbed by PSEN1 mutations.
The PSEN1 mutations impair the NSC capacity of self-renewal and differentiation in vitro and in vivo conditions. Among all mutations described here, the most frequently signaling pathways affected are Notch and Wnt. Interestingly, some mutations affecting GSK3β share downstream effectors with Notch/CREB signaling. Dotted arrows indicate a possible route, and upper red arrows indicate activity increasing, lower red arrows indicate a diminished activity. Created with BioRender.com. APC: Adenomatous polyposis coli protein; APH1: gamma-secretase subunit APH-1A; APP: amyloid precursor protein; Aβ: amyloid beta; BACE: beta-site APP cleaving enzyme; Ca: calcio; CBP: CREB-binding protein; CKlα: casein kinase Iα; CTF2: C terminal N-cadherin fragment; E-cad: E-Cadherin; LRP: lipoprotein receptor-related protein; MAML: mastermind-like protein; MAPK: mitogen-activated protein kinase; NCSTN: nicastrin; NICD: Notch intracellular domain; NMDAR: glutamate receptor ionotropic 3A; NPC: neural progenitor cells; PEN2: gamma-secretase subunit presenilin enhancer 2; PEN2: presenilin enhancer protein 2; PSEN: presenilin; RPBJ: recombining binding protein suppressor of hairless; TCF/LEF: T-cell factor/lymphoid enhancer factor; Wnt: Wingless-related integration site.
It has been suggested that PSEN1 mutations exert detrimental consequences on development because some of them cause very-early-onset AD (before de age of 30 when most aggressive) (DeTure and Dickson, 2019); therefore, many reports have emerged trying to elucidate the roles of PSEN1 mutations over neuronal differentiation (Table 1). Many of them have determined that FAD-linked mutations are mostly associated with dysfunctional mechanisms in the γ-secretase complex (Xia et al., 2015; Watanabe and Shen, 2017).
Table 1.
PSEN1 mutations linked to neuronal differentiation in Alzheimer’s disease
| Mutation | Model | Biological effect in neuronal differentiation | Codon change | Reference |
|---|---|---|---|---|
| P117L | Transgenic mice | Reduced number of NPC in the hippocampus | CCA to CTA | Wen et al. (2002) |
| Transgenic mice | Impairs new neuron production in the hippocampus by decreasing NPC survival | Wen et al. (2004) | ||
| Transgenic mice NPC | Decreased neurogenesis and promotes neuritic outgrowth in newly generated neurons | Eder-Colli et al. (2009) | ||
| M146V | Knockin mice | Impaired hippocampus-dependent associative learning correlated with reduced neurogenesis in the dentate gyrus | ATG to GTG | Wang et al. (2004) |
| Transgenic mice | Increased neuronal apoptosis by altering the stability of β-catenin | Zhang et al. (1998b) | ||
| ΔE9 | Transgenic mice NPC | Reduced self-renewal capacity and premature exit toward neuronal fates | Complex | Veeraraghavalu et al. (2010) |
| Diminished both levels of endogenous Notch/CBF-1 transcriptional activity and transcripts encoding Notch target genes | ||||
| Transgenic mice | Diminished constitutive proliferation and steady-state subventricular zone progenitor pool size | |||
| Non-transgenic mice hippocampal NPC | Did show no differences in proliferation and differentiation capacity compared to wild type | Veeraraghavalu and Sisodia (2013) | ||
| Transgenic mice | Reduced proliferation of NPC and their neuronal differentiation | Demars et al. (2010) | ||
| M146L | Non-transgenic mice hippocampal NPC | Did show no differences in NPC proliferation and differentiation capacity compared to wild type | ATG to CTG | Veeraraghavalu and Sisodia (2013) |
| Transgenic mice | Inhibition of Wnt signaling by increasing β-catenin phosphorylation and degradation | ATG to TTG | Kawamura et al. (2001) | |
| iPSC-derived NPC | Elevated Aβ42/Aβ40 ratios that affect their developmental potential and survival | Sproul et al. (2014) | ||
| A246E | iPSC-derived neurons | Impaired NPC proliferation and survival by premature differentiation | Yang et al. (2017) | |
| Decreased NICD levels | GCG to GAG | |||
| Transgenic mice | Elevated β-catenin levels | Chevallier et al. (2005) | ||
| Stimulated NPC proliferation | ||||
| PC12 cells and primary cultured hippocampal | Decreased survival signaling through the downregulation of Akt/PKB pathway | Weihl et al. (1999) | ||
| neurons | ||||
| C410Y | Decreased inactivation of GSK-3β, increasing the activity of GSK-3β, and | TGT to TAT | ||
| decreased levels of soluble β-catenin | ||||
| I143T | Transgenic mice | Increased neuronal apoptosis by altering the stability of β-catenin | ATT to ACT | Zhang et al. (1998b) |
| P264L | Primary cortical neurons from knock-in mice | Did not show increased neuron degeneration | CCG to CTG | Siman et al. (2000) |
| V89L | iPSC-derived neurons | Increased levels of active GSK3β | Ochalek et al. (2017) | |
| Increased Aβ42/Aβ40 ratio | ||||
| Increased tau phosphorylation | ||||
| L150P | Upregulation of APP synthesis and APP carboxy-terminal fragments cleavage | CTG to CCG | ||
| Y115C | iPSC-derived cortical | Did not show elevated Aβ42/Aβ40 ratio either tau phosphorylation | TAT to TGT | Moore et al. (2015) |
| neurons |
AKT/PKB: Protein kinase B; APP: amyloid precursor protein; Aβ: amyloid beta; CBF-1: centromere binding factor 1; GSK3β: glycogen synthase kinase 3 beta; iPSCs: induced pluripotent stem cells; NPC: neural progenitor cells.
Furthermore, in neurogenesis, this loss of function causes an aberrant ErbB-4 and E-cadherin-mediated signaling and the downregulation of Notch signaling (Handler et al., 2000; Demars et al., 2010).
Some studies have shown that PSEN1 P117L mutation triggers a decreased neurogenesis by impairing the NPC survival in the dentate gyrus (Wen et al., 2004). In contrast, overexpression of wild type PSEN1 in the dentate gyrus did not affect but contributed to neurogenesis (Wen et al., 2002). This finding suggests that this mutation promotes enhanced apoptosis, reduced survival of new neurons, or a neuron-glia switch during fate specification. Similarly, Eder-Colli et al. (2009) reported that the in vitro neurogenesis of murine embryonic NPC was impaired by the FAD-linked PSEN1 P117L, whereas new neurons had enhanced neuritic outgrowth (Eder-Colli et al., 2009).
In comparison to wild-type mice, knock-in mice harboring PSEN1 M146V mutation showed an associative learning impairment, consistent with a dentate gyrus neurogenesis reduction. Suggesting that memory deficits related to AD may be due to neurogenesis impairment (Wang et al., 2004).
Veeraraghavalu et al. (2010) demonstrated that in transgenic mice harboring PSEN1 ΔE9 mutation, the Notch signaling is altered through the endogenous transcriptional activity of Notch/CBF1 and by the reduction of transcripts encoding Notch-target genes in NPC from the SVZ. Moreover, they showed that in vitro SVZ NPC cultures derived from mice harboring PSEN1 ΔE9 mutation presented a limited capacity for self-renewal and premature differentiation into neurons (Veeraraghavalu et al., 2010). Otherwise, Veeraraghavalu and Sisodia (2013) reported non-transgenic mouse adult hippocampal NPC carrying either PSEN1 ΔE9 or M146L mutation and showed no dissimilarities in their capacity to differentiate and proliferate compared to wild type. These suggest that impairments in the proliferation and neuronal differentiation capacity in PSEN1 mutant mice are due to the expression of the PSEN1 mutation by different cells rather than NPC, but residing in the same neurogenic cell niche (Veeraraghavalu and Sisodia, 2013).
Besides, Demars et al. (2010), in a study using APPswe/PS1 PSEN1 ΔE9 animal model, suggested that FAD mutations induce early and severely NPC intracellular changes along with altered neurogenic microenvironment in both the hippocampus and SVZ, leading to impaired neurogenesis. They also proposed that these impairments modify the hippocampal and olfactory function, contributing to enhancing the neuronal vulnerability in AD (Demars et al., 2010).
Previously it was suggested that PSEN1 A246E and C410Y mutations affect GSK3β activity (Weihl et al., 1999). Moreover, the GSK3β suppression leads to an enhanced stabilization of β-catenin and TCF/LEF activation, which is essential for neural stem cells self-renewal and NPC proliferation. Hence, the dysregulated activity of GSK3β underlies alterations in tau phosphorylation, neuronal differentiation, neuronal survival, and activation of transcription of neurogenesis-related target genes and proteins (Otto et al., 2016).
Chevallier et al. (2005) reported that mice carrying the PSEN1 A246E mutation showed an increase in β-catenin levels and stimulated hippocampal NPC proliferation without survival or differentiation capacity repercussion, suggesting that this mutation influences NPC cell growth by atypical β-catenin signaling.
Similarly, another study showed that PSEN1 M146L mutation inhibits Wnt signaling by increasing β-catenin phosphorylation and degradation in vitro and in vivo (Kawamura et al., 2001). Therefore, a downregulated β-catenin signaling increases neuronal vulnerability to apoptosis and specific developmental brain defects, whereas restoration of regular β-catenin activity leads to NPC’s pool expansion and neurogenesis restoration (Zhang et al., 2011).
Moreover, Wnt signaling’s loss may induce susceptivity to Aβ-mediated apoptosis (De Ferrari et al., 2003). Thus, PSEN1 M146V, C410Y, and I143T mutations promote apoptosis through β-catenin nuclear accumulation and the early-onset AD (Zhang et al., 1998b). In contrast, PSEN1 P264L mutant mice did not display any massive neuronal loss, indicating that not all PSEN1-linked mutations lead to neurodegeneration (Siman et al., 2000).
Despite PSEN1 implication in several cellular processes, the current transgenic models fail to recapitulate early postnatally events in determined cortical areas and NPC’s pools (Lazarov and Marr, 2010). Additionally, the limited access to AD-affected human tissue made it intricate to study early molecular events during AD pathogenesis.
Modeling Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells
Several research groups reported the generation of human neurons from induced pluripotent stem cells (iPSCs) obtained from FAD patients that share the same genetic background (Yagi et al., 2011). Accordingly, several studies demonstrated that iPSC-derived neurons from AD patients exhibit many AD pathology traits. These studies support the implementation of iPSCs as a platform for a new generation of in vitro AD models focused on the study of molecular mechanisms, novel biomarkers identification, and novel drugs evaluation (D’Avanzo et al., 2015; Raja et al., 2016; Hernández-Sapiéns et al., 2020; Li et al., 2020).
For example, a remarkable report by Sproul et al. (2014) studied for the first time NPCs derived from FAD-iPSC carrying the PSEN1 A246E or M146L mutations. They demonstrated the impact of these mutations in the Aβ42/Aβ40 ratios compared with control cells. They concluded that high Aβ42:Aβ40 ratios affect NPC’s differentiation capacity and survival (Sproul et al., 2014). Similarly, Yang et al. (2017), using the same cell system, showed that the PSEN1 mutations induce premature differentiation of NPC, possibly through the upregulation of genes related to neuron development, neuron projection, and neuron maturation. Furthermore, they observed a downregulation in genes mainly associated with cell cycle, which could explain the diminished NPC self-renewal and elevated apoptosis. Interestingly, they also found a significant reduction in NICD levels, consistent with a study by Borghese et al. (2010), who demonstrated that inhibiting Notch signaling results in neuronal differentiation enhancement (Borghese et al., 2010; Yang et al., 2017).
Ochalek et al. (2017), using iPSC-derived neurons carrying PSEN1 V89L and L150P mutations, observed a highly active GSK3β, indicating a direct role in Tau hyperphosphorylation (Ochalek et al., 2017). Since Wnt regulates NPC self-renewal by inactivating GSK3β and stabilizing β-catenin, it is expected that an imbalance in the signaling pathway contributes to the dysfunction in NPC maintenance and differentiation capacity.
These and other studies demonstrated the importance of iPSC technology for developing in vitro disease models that allow us to study the earliest molecular events underlying the pathology. The information obtained from in vitro AD models can provide the basis for identifying new diagnostic biomarkers, new therapeutic strategies, and the development of “precision medicine”. Additionally, the use of patient-specific iPSC-derived neurons represents a breakthrough strategy for studying the effects of FAD mutations in multiple physiological processes, such as NPC proliferation and differentiation.
Conclusions and Future Perspectives
The PSEN1 protein function and its association with familial AD have been widely studied. Their biological roles’ complexity is becoming increasingly evident because of their implicated cellular processes. Several studies attributed a critical role of PS1 in the neurogenesis process, controlling crucial aspects of the NPC self-renewal and differentiation and in new-born neuron generation. Its implication in dendritic morphogenesis and its subsequent stabilization is remarkable.
To date, FAD is associated with approximately 274 mutations linked to PSEN1. Most of them are missense mutations. Interestingly, the effects of PS1 mutation and their contribution to FAD were first associated with a γ-secretase gain of function due to its ability to influence the APP cleavage and modify the Aβ42/Aβ40 ratio. Afterward, another hypothesis suggested a function inhibition by β-catenin absence and Notch intracellular domain and their downstream target genes. Therefore, the biochemical impairment of PS1 is critical in FAD development. In addition to Aβ production, PS1 mutation may alter neurogenesis directly and indirectly, such as NPC self-renewal and differentiation impairment through different signaling pathways, contributing considerably to cognitive impairment.
Despite the relevance of AD research in biomedicine, the molecular pathways leading to neurogenesis impairment and neurodegeneration are still unknown. Moreover, many knowledge gaps remain because of the limited access to AD-affected human tissue and the limited reliability of current transgenic and in vitro AD models. The iPSC technology has revolutionized AD in vitro models to overcome these gaps by maintaining donors’ genetic information. The analysis of neurogenesis using FAD iPSC-derived NPC offers a unique chance to analyze their biology in a pathological adult environment and the cellular mechanisms regulated by the γ-secretase complex. Thus, an examination of regulated intramembrane proteolysis and its subsequent cellular processes in human neural cells carrying FAD-related PSEN mutations could provide essential clues to a comprehensive understanding of how these mutations cause AD and how to design novel therapeutic strategies.
Acknowledgments:
We would like to thank MD Muhamad Festok from Yale University, United States of America for his valuable effort in editing the English language of the manuscript.
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was supported by the Consejo Nacional de Ciencia y Tecnología Scholarship 711893 (to MAH) and 711874 (to EER).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Funding: This work was supported by the Consejo Nacional de Ciencia y Tecnología Scholarship 711893 (to MAH) and 711874 (to EER).
References
- 1.Baki L, Marambaud P, Efthimiopoulos S, Georgakopoulos A, Wen P, Cui W, Shioi J, Koo E, Ozawa M, Friedrich VL, Robakis NK. Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc Natl Acad Sci U S A. 2001;98:2381–2386. doi: 10.1073/pnas.041603398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barber M, Pierani A. Tangential migration of glutamatergic neurons and cortical patterning during development: lessons from Cajal-Retzius cells. Dev Neurobiol. 2016;76:847–881. doi: 10.1002/dneu.22363. [DOI] [PubMed] [Google Scholar]
- 3.Bonds JA, Kuttner-Hirshler Y, Bartolotti N, Tobin MK, Pizzi M, Marr R, Lazarov O. Presenilin-1 dependent neurogenesis regulates hippocampal learning and memory. PLoS One. 2015;10:e0131266. doi: 10.1371/journal.pone.0131266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borghese L, Dolezalova D, Opitz T, Haupt S, Leinhaas A, Steinfarz B, Koch P, Edenhofer F, Hampl A, Brüstle O. Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells. 2010;28:955–964. doi: 10.1002/stem.408. [DOI] [PubMed] [Google Scholar]
- 5.Buchsbaum IY, Cappello S. Neuronal migration in the CNS during development and disease: insights from in vivo and in vitro models. Development. 2019;146:dev163766. doi: 10.1242/dev.163766. [DOI] [PubMed] [Google Scholar]
- 6.Bustos FJ, Jury N, Martinez P, Ampuero E, Campos M, Abarzúa S, Jaramillo K, Ibing S, Mardones MD, Haensgen H, Kzhyshkowska J, Tevy MF, Neve R, Sanhueza M, Varela-Nallar L, Montecino M, van Zundert B. NMDA receptor subunit composition controls dendritogenesis of hippocampal neurons through CAMKII, CREB-P, and H3K27ac. J Cell Physiol. 2017;232:3677–3692. doi: 10.1002/jcp.25843. [DOI] [PubMed] [Google Scholar]
- 7.Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol. 2005;167:151–159. doi: 10.1016/S0002-9440(10)62962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Avanzo C, Aronson J, Kim YH, Choi SH, Tanzi RE, Kim DY. Alzheimer’s in 3D culture: challenges and perspectives. Bioessays. 2015;37:1139–1148. doi: 10.1002/bies.201500063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Ferrari GV, Chacón MA, Barría MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- 10.de Leeuw S, Tackenberg C. Alzheimer’s in a dish – induced pluripotent stem cell-based disease modeling. Transl Neurodegener. 2019;8:21. doi: 10.1186/s40035-019-0161-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demars M, Hu Y-S, Gadadhar A, Lazarov O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J Neurosci Res. 2010;88:2103–2117. doi: 10.1002/jnr.22387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32–32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhaliwal J, Kannangara TS, Vaculik M, Xue Y, Kumar KL, Maione A, Béïque J-C, Shen J, Lagace DC. Adult hippocampal neurogenesis occurs in the absence of Presenilin 1 and Presenilin 2. Sci Rep. 2018;8:17931. doi: 10.1038/s41598-018-36363-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding XF, Gao X, Ding X-C, Fan M, Chen J. Postnatal dysregulation of Notch signal disrupts dendrite development of adult-born neurons in the hippocampus and contributes to memory impairment. Sci Rep. 2016;6:25780. doi: 10.1038/srep25780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duggan SP, McCarthy JV. Beyond γ-secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cell Signal. 2016;28:1–11. doi: 10.1016/j.cellsig.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Eder-Colli L, Abad-Estarlich N, Pannetier C, Vallet PG, Walzer C, Elder GA, Robakis NK, Bouras C, Savioz A. The presenilin-1 familial Alzheimer’s disease mutation P117L decreases neuronal differentiation of embryonic murine neural progenitor cells. Brain Res Bull. 2009;80:296–301. doi: 10.1016/j.brainresbull.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Escamilla-Ayala A, Wouters R, Sannerud R, Annaert W. Contribution of the Presenilins in the cell biology, structure and function of γ-secretase. Semin Cell Dev Biol. 2020;105:12–26. doi: 10.1016/j.semcdb.2020.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Francis YI, Stephanou A, Latchman DS. CREB-binding protein activation by presenilin 1 but not by its M146L mutant. Neuroreport. 2006;17:917–921. doi: 10.1097/01.wnr.0000220137.06542.a0. [DOI] [PubMed] [Google Scholar]
- 20.Gadadhar A, Marr R, Lazarov O. Presenilin-1 regulates neural progenitor cell differentiation in the adult brain. J Neurosci. 2011;31:2615. doi: 10.1523/JNEUROSCI.4767-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Güner G, Lichtenthaler SF. The substrate repertoire of γ-secretase/presenilin. Semin Cell Dev Biol. 2020;105:27–42. doi: 10.1016/j.semcdb.2020.05.019. [DOI] [PubMed] [Google Scholar]
- 22.Handler M, Yang X, Shen J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development. 2000;127:2593–2606. doi: 10.1242/dev.127.12.2593. [DOI] [PubMed] [Google Scholar]
- 23.Hernández-Sapiéns MA, Reza-Zaldívar EE, Cevallos RR, Márquez-Aguirre AL, Gazarian K, Canales-Aguirre AA. A three-dimensional Alzheimer’s disease cell culture model using iPSC-derived neurons carrying A246E mutation in PSEN1. Front Cell Neurosci. 2020;14:151. doi: 10.3389/fncel.2020.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabir MT, Uddin MS, Setu JR, Ashraf GM, Bin-Jumah MN, Abdel-Daim MM. Exploring the role of PSEN mutations in the pathogenesis of Alzheimer’s disease. Neurotox Res. 2020;38:833–849. doi: 10.1007/s12640-020-00232-x. [DOI] [PubMed] [Google Scholar]
- 25.Kawamura Y, Kikuchi A, Takada R, Takada S, Sudoh S, Shibamoto S, Yanagisawa K, Komano H. Inhibitory effect of a presenilin 1 mutation on the Wnt signalling pathway by enhancement of beta-catenin phosphorylation. Eur J Biochem. 2001;268:3036–3041. doi: 10.1046/j.1432-1327.2001.02197.x. [DOI] [PubMed] [Google Scholar]
- 26.Killick R, Pollard CC, Asuni AA, Mudher AK, Richardson JC, Rupniak HT, Sheppard PW, Varndell IM, Brion JP, Levey AI, Levy OA, Vestling M, Cowburn R, Lovestone S, Anderton BH. Presenilin 1 independently regulates beta-catenin stability and transcriptional activity. J Biol Chem. 2001;276:48554–48561. doi: 10.1074/jbc.M108332200. [DOI] [PubMed] [Google Scholar]
- 27.Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane. Nat Rev Mol Cell Biol. 2004;5:499–504. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- 28.Landeira BS, Santana TTdS, Araújo JAdM, Tabet EI, Tannous BA, Schroeder T, Costa MR. Activity-independent effects of CREB on neuronal survival and differentiation during mouse cerebral cortex development. Cereb Cortex. 2016;28:538–548. doi: 10.1093/cercor/bhw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanoiselée HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, Quillard-Muraine M, de la Sayette V, Boutoleau-Bretonniere C, Etcharry-Bouyx F, Chauviré V, Sarazin M, le Ber I, Epelbaum S, Jonveaux T, Rouaud O, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017;14:e1002270. doi: 10.1371/journal.pmed.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazarov O, Marr RA. Neurogenesis and Alzheimer’s disease: at the crossroads. Exp Neurol. 2010;223:267–281. doi: 10.1016/j.expneurol.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Kim HJ, Roh JH, Kim M, Koh W, Kim Y, Heo H, Chung J, Nakanishi M, Yoon T, Hong CP, Seo SW, Na DL, Song J. Pathological manifestation of the induced pluripotent stem cell-derived cortical neurons from an early-onset Alzheimer’s disease patient carrying a presenilin-1 mutation (S170F) Cell Proli. 2020;53:e12798. doi: 10.1111/cpr.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li P, Lin X, Zhang JR, Li Y, Lu J, Huang FC, Zheng CH, Xie JW, Wang JB, Huang CM. The expression of presenilin 1 enhances carcinogenesis and metastasis in gastric cancer. Oncotarget. 2016;7:10650–10662. doi: 10.18632/oncotarget.7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Louvi A, Sisodia SS, Grove EA. Presenilin 1 in migration and morphogenesis in the central nervous system. Development. 2004;131:3093. doi: 10.1242/dev.01191. [DOI] [PubMed] [Google Scholar]
- 34.Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/γ-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marathe S, Alberi L. Notch in memories: Points to remember. Hippocampus. 2015;25:1481–1488. doi: 10.1002/hipo.22426. [DOI] [PubMed] [Google Scholar]
- 36.Moore S, Evans Lewis DB, Andersson T, Portelius E, Smith J, Dias Tatyana B, Saurat N, McGlade A, Kirwan P, Blennow K, Hardy J, Zetterberg H, Livesey Frederick J. APP metabolism regulates Tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015;11:689–696. doi: 10.1016/j.celrep.2015.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochalek A, Mihalik B, Avci HX, Chandrasekaran A, Téglási A, Bock I, Giudice ML, Táncos Z, Molnár K, László L, Nielsen JE, Holst B, Freude K, Hyttel P, Kobolák J, Dinnyés A. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res Ther. 2017;9:90. doi: 10.1186/s13195-017-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oikawa N, Walter J. Presenilins and γ-secretase in membrane proteostasis. Cells. 2019;8:209. doi: 10.3390/cells8030209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otto GP, Sharma D, Williams RSB. Non-catalytic roles of presenilin throughout evolution. J Alzheimers Dis. 2016;52:1177–1187. doi: 10.3233/JAD-150940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parisiadou L, Fassa A, Fotinopoulou A, Bethani I, Efthimiopoulos S. Presenilin 1 and cadherins: stabilization of cell-cell adhesion and proteolysis-dependent regulation of transcription. Neurodegener Dis. 2004;1:184–191. doi: 10.1159/000080984. [DOI] [PubMed] [Google Scholar]
- 41.Pigino G, Pelsman A, Mori H, Busciglio J. Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci. 2001;21:834–842. doi: 10.1523/JNEUROSCI.21-03-00834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raja WK, Mungenast AE, Lin Y-T, Ko T, Abdurrob F, Seo J, Tsai LH. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS One. 2016;11:e0161969. doi: 10.1371/journal.pone.0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saura CA, Choi S-Y, Beglopoulos V, Malkani S, Zhang D, Rao BSS, Chattarji S, Kelleher RJ, III, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- 44.Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sproul AA, Jacob S, Pre D, Kim SH, Nestor MW, Navarro-Sobrino M, Santa-Maria I, Zimmer M, Aubry S, Steele JW, Kahler DJ, Dranovsky A, Arancio O, Crary JF, Gandy S, Noggle SA. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS One. 2014;9:e84547. doi: 10.1371/journal.pone.0084547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stanga S, Vrancx C, Tasiaux B, Marinangeli C, Karlström H, Kienlen-Campard P. Specificity of presenilin-1- and presenilin-2-dependent γ-secretases towards substrate processing. J Cell Mol Med. 2018;22:823–833. doi: 10.1111/jcmm.13364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uemura K, Kitagawa N, Kohno R, Kuzuya A, Kageyama T, Shibasaki H, Shimohama S. Presenilin 1 mediates retinoic acid-induced differentiation of SH-SY5Y cells through facilitation of Wnt signaling. J Neurosci Res. 2003;73:166–175. doi: 10.1002/jnr.10641. [DOI] [PubMed] [Google Scholar]
- 49.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 50.Veeraraghavalu K, Sisodia SS. Mutant presenilin 1 expression in excitatory neurons impairs enrichment-mediated phenotypes of adult hippocampal progenitor cells. Proc Natl Acad Sci U S A. 2013;110:9148. doi: 10.1073/pnas.1302106110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veeraraghavalu K, Choi SH, Zhang X, Sisodia SS. Presenilin 1 mutants impair the self-renewal and differentiation of adult murine subventricular zone-neuronal progenitors via cell-autonomous mechanisms involving Notch signaling. J Neurosci. 2010;30:6903–6915. doi: 10.1523/JNEUROSCI.0527-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walton MR, Dragunow M. Is CREB a key to neuronal survival. Trends Neurosci. 2000;23:48–53. doi: 10.1016/s0166-2236(99)01500-3. [DOI] [PubMed] [Google Scholar]
- 53.Wang R, Dineley KT, Sweatt JD, Zheng H. Presenilin 1 familial Alzheimer’s disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience. 2004;126:305–312. doi: 10.1016/j.neuroscience.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 54.Wang W, Moerman-Herzog AM, Slaton A, Barger SW. Presenilin 1 mutations influence processing and trafficking of the ApoE receptor apoER2. Neurobiol Aging. 2017;49:145–153. doi: 10.1016/j.neurobiolaging.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe H, Shen J. Dominant negative mechanism of Presenilin-1 mutations in FAD. Proc Natl Acad Sci U S A. 2017;114:12635–12637. doi: 10.1073/pnas.1717180114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci. 1999;19:5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wen PH, Shao X, Shao Z, Hof PR, Wisniewski T, Kelley K, Friedrich VL, Ho L, Pasinetti GM, Shioi J, Robakis NK, Elder GA. Overexpression of wild type but not an FAD mutant presenilin-1 promotes neurogenesis in the hippocampus of adult mice. Neurobiol Dis. 2002;10:8–19. doi: 10.1006/nbdi.2002.0490. [DOI] [PubMed] [Google Scholar]
- 58.Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, Haroutunian V, Elder GA. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol. 2004;188:224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 59.Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ., 3rd Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron. 2015;85:967–981. doi: 10.1016/j.neuron.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ, Koo EH, Wu X, Zheng H. Loss of presenilin 1 is associated with enhanced β-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:10863–10868. doi: 10.1073/pnas.191284198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- 62.Yang J, Zhao H, Ma Y, Shi G, Song J, Tang Y, Li S, Li T, Liu N, Tang F, Gu J, Zhang L, Zhang Z, Zhang X, Jin Y, Le W. Early pathogenic event of Alzheimer’s disease documented in iPSCs from patients with PSEN1 mutations. Oncotarget. 2017;8:7900–7913. doi: 10.18632/oncotarget.13776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yonemura Y, Futai E, Yagishita S, Kaether C, Ishiura S. Specific combinations of presenilins and Aph1s affect the substrate specificity and activity of γ-secretase. Biochem Biophys Res Commun. 2016;478:1751–1757. doi: 10.1016/j.bbrc.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 64.Zhang L, Yang X, Yang S, Zhang J. The Wnt/β-catenin signaling pathway in the adult neurogenesis. Eur J Neurosci. 2011;33:1–8. doi: 10.1111/j.1460-9568.2010.7483.x. [DOI] [PubMed] [Google Scholar]
- 65.Zhang W, Han SW, McKeel DW, Goate A, Wu JY. Interaction of presenilins with the filamin family of actin-binding proteins. J Neurosci. 1998a;18:914. doi: 10.1523/JNEUROSCI.18-03-00914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998b;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- 67.Zoltowska KM, Berezovska O. Dynamic nature of presenilin1/γ-secretase: implication for Alzheimer’s disease Pathogenesis. Mol Neurobiol. 2018;55:2275–2284. doi: 10.1007/s12035-017-0487-5. [DOI] [PMC free article] [PubMed] [Google Scholar]