

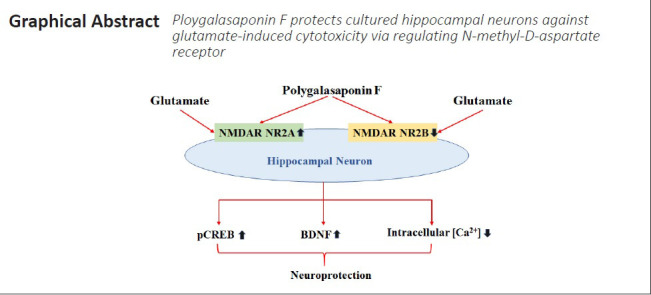
Key Words: BDNF, Ca2+ homeostasis, excitotoxicity, glutamate, hippocampal neurons, pCREB, polygalasaponin F, neuroprotection, NR2A, NR2B
Abstract
Excess extracellular glutamate leads to excitotoxicity, which induces neuronal death through the overactivation of N-methyl-D-aspartate receptors (NMDARs). Excitotoxicity is thought to be closely related to various acute and chronic neurological disorders, such as stroke and Alzheimer’s disease. Polygalasaponin F (PGSF) is a triterpenoid saponin monomer that can be isolated from Polygala japonica, and has been reported to protect cells against apoptosis. To investigate the mechanisms underlying the neuroprotective effects of PGSF against glutamate-induced cytotoxicity, PGSF-pretreated hippocampal neurons were exposed to glutamate for 24 hours. The results demonstrated that PGSF inhibited glutamate-induced hippocampal neuron death in a concentration-dependent manner and reduced glutamate-induced Ca2+ overload in the cultured neurons. In addition, PGSF partially blocked the excess activity of NMDARs, inhibited both the downregulation of NMDAR subunit NR2A expression and the upregulation of NMDAR subunit NR2B expression, and upregulated the expression of phosphorylated cyclic adenosine monophosphate-responsive element-binding protein and brain-derived neurotrophic factor. These findings suggest that PGSF protects cultured hippocampal neurons against glutamate-induced cytotoxicity by regulating NMDARs. The study was approved by the Institutional Animal Care Committee of Nanchang University (approval No. 2017-0006) on December 29, 2017.
Chinese Library Classification No. R453; R741; R285
Introduction
Excessively high concentrations of extracellular glutamate can induce excitotoxicity through the overactivation of glutamate receptors (mainly N-methyl-D-aspartate receptors [NMDARs]), leading to a series of abnormal events, such as intracellular Ca2+ overload, oxidative stress, and mitochondrial dysfunction, ultimately causing neuronal death (Mehta et al., 2013). Glutamate excitotoxicity-induced neuronal death is widely observed in many neurological disorders, including ischemia, trauma, epilepsy, amyotrophic lateral sclerosis, and neurodegenerative disorders such as Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease. Furthermore, glutamate excitotoxicity-induced neuronal death is one of the major pathogenic mechanisms underlying these neurological disorders (Lau and Tymianski, 2010; Mehta et al., 2013; Huberfeld and Vecht, 2016; Dorsett et al., 2017; Jiang et al., 2019; Chu et al., 2020). NMDARs are essential for many normal brain functions, such as synaptic plasticity, learning, and memory (Bliss and Collingridge, 1993). The inhibition of NMDAR activity in vivo can also cause apoptosis in developing neurons (Ikonomidou et al., 1999). Thus, blocking excitotoxicity while preserving the physiological functions of NMDARs may be a promising strategy for treating glutamate-induced excitotoxicity (Xia et al., 2010).
It has been reported that the location of NMDARs determines whether they are coupled to pro-death or pro-survival signals (Hardingham and Bading, 2010). A single NMDAR is composed of heteromeric subunits, including two NR1 and two NR2 subunits. The NR2 subunits can be NR2A–NR2D or NR3 (Huo et al., 2015). The NR1 subunits form an ion channel, while the NR2 subunits modulate the activity of the ion channel (Liu et al., 2017). Different NR2-containing NMDARs (NR1/NR2A or NR1/NR2B) exhibit different biophysical properties and play antagonistic roles in synaptic transmission and excitotoxicity (Furukawa et al., 2005; Chen et al., 2008). In the adult brain, NR2B-containing NMDARs are located mainly at extrasynaptic sites, while NR2A-containing NMDARs are highly expressed at the synapse (Papouin et al., 2012). Activation of NR2A-containing (i.e., synaptic) NMDARs triggers pro-survival signals, whereas stimulation of NR2B-containing (i.e., extrasynaptic) NMDARs triggers pro-death signals (Rueda et al., 2016).
Polygala japonica is a traditional Chinese medical herb with expectorant, anti-inflammatory, antibacterial, and antidepressant effects (Sun et al., 2012; Chinese Pharmacopoeia Commission, 2015). Polygalasaponin F (PGSF), a triterpenoid saponin monomer isolated from P. japonica, has various effects on the nervous system. A previous study has demonstrated that PGSF has anti-inflammatory effects via inhibiting the release of the inflammatory cytokine tumor necrosis factor-α and nitric oxide in PC12 cells (Wei et al., 2014). PGSF also exerts a protective effect on PC12 cell survival in the presence of rotenone and sodium dithionite (Shi et al., 2013; Wu et al., 2014). In ischemic models, PGSF reduces the size of the infarct area in middle cerebral artery occlusion mice via an anti-apoptotic mechanism (Zhou et al., 2019).
Thus, PGSF appears to have protective effects against injuries of the nervous system. The present study aimed to investigate whether PGSF has a protective effect against glutamate excitotoxicity-induced neuronal death in primary cultured hippocampal neurons. We also attempted to address whether PGSF produces such a protective effect by regulating NR2A- and NR2B-containing NMDARs.
Materials and Methods
Animals
Female Wistar rats (weighing ~200 g, 6–8 weeks old, n = 24) and male C57BL/6 mice (weighing ~30 g, 4–6 weeks old, n = 12) were obtained from the Department of Experimental Animals, Institution of Life Science, Nanchang University (license No. SYXK (Gan) 2015-0002). Rats were grouped and housed at two animals per cage under temperature- and light-controlled conditions (22–23°C, 12-hour light/dark cycle), with food and water provided ad libitum. All experiments conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication 85-23, revised in 1985) and were approved by the Institutional Animal Care Committee of Nanchang University (approval No. 2017-0006) on December 29, 2017.
Primary culture of hippocampal neurons
Hippocampal neurons were obtained from embryonic day 17–19 Wistar rats (provided by the Department of Experimental Animals, Institution of Life Science, Nanchang University). The hippocampus was rapidly dissected from each embryo and minced into small pieces (less than 1 mm per side). These small pieces were collected, centrifuged at 800 × g for 1 minute, and digested with pre-warmed 0.05% trypsin (Gibco, Grand Island, NY, USA) for 10 minutes. To stop the digestion, 10% fetal bovine serum (Gibco) was added. Neuronal cells (i.e., the precipitation) were collected after centrifugation at 800 × g for 5 minutes, and were then dissolved in Neurobasal Medium (Gibco) containing 10% fetal bovine serum and 1% penicillin/streptomycin combination (Gibco). The cells were diluted to a final density of 1 × 106/mL and seeded onto culture dishes (Thermo Fisher Scientific, Waltham, MA, USA) pre-coated with poly-D-lysine (Sigma, St. Louis, MO, USA), and were then incubated at 37°C with 5% CO2. After 4 hours of incubation, the media was changed to Neurobasal Medium containing 2% B27, 1% penicillin/streptomycin combination, and 1% glutamine. Half of the media was changed every 3 days.
Cell viability
Hoechst 33342 and propidium iodide dye (Beyotime Biotechnology, Shanghai, China) double staining was used to determine cell death. The hippocampal neurons were seeded in 96-well plates and cultured for 6 days. To measure glutamate excitotoxicity, the cells were exposed to 20, 40, 60, 80, and 100 µM glutamate for 24 hours. In addition, to investigate the protective effects of PGSF on glutamate-induced excitotoxicity, PGSF (Pufei De Biotech Co., Chengdu, China) was dissolved in dimethyl sulfoxide (Sigma) to 1 mM stock concentration and diluted to a final concentration of 2, 4, 6, 8, or 10 µM in cell culture medium. The cells were pretreated with 2, 4, 6, 8, or 10 µM PGSF for 30 minutes, and then glutamate (Sigma) was added to the cells to a concentration of 100 µM. After being cultured for 24 hours, cell viability was observed. Furthermore, to determine the toxicity of PGSF, cells were treated with 2, 4, 6, 8, or 10 µM PGSF. Cell viability was then observed by adding Hoechst 33342 (10 µg/mL) and propidium iodide (5 µM) to the media of neuronal cells for 15 minutes. The cells were then imaged randomly under a fluorescence microscope (Zeiss, Oberkochen, Germany) and the stained cells were counted. Cell viability was calculated according to the following formula: Percentage viability (%) = (number of Hoechst 33342-stained cells – number of Hoechst 33342/propidium iodide double-stained cells)/number of Hoechst 33342-stained cells × 100.
Apoptosis assay
Hippocampal neuron apoptosis was determined using immunofluorescence staining of cleaved caspase-3. After the hippocampal neurons were seeded in cultured dishes pre-coated with poly-D- lysine in 24-well plates and cultured for 6 days, the cells were exposed to 100 µM glutamate, 100 µM glutamate + 10 µM PGSF, or 100 µM glutamate + 10 µM MK801 ((5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate; an NMDAR antagonist; Sigma) for 24 hours. For immunofluorescence, the cultured cells were washed twice with phosphate-buffered saline (PBS) for 5 minutes and fixed with 4% paraformaldehyde in PBS for 1 hour at room temperature. The fixed cells were then washed three times with PBS and blocked with 5% goat serum (Solarbio Life Science, Beijing, China) in PBS for 2 hours at room temperature. After incubation in the primary antibody (rabbit anti-cleaved caspase-3; 1:400, Cat# 9661; Cell Signaling, Danvers, MA, USA) at 4°C overnight, the cells were washed three times with PBS and incubated with the secondary antibody (donkey anti-rabbit, 1:500, Cat# Q22089; Life Technologies, Grand Island, NY, USA) for 2 hours at room temperature. Nuclei were visualized by staining with 4′,6-diamidino-2-phenylindole (Beyotime Biotechnology). The stained cells were then washed three times with PBS and imaged under a fluorescence microscope (Zeiss AxioImager A2, Oberkochen, Germany) using a 10× objective.
Calcium imaging
The intracellular free Ca2+ was measured using the Ca2+ indicator Fluo-4 AM (Molecular Probes, Eugene, OR, USA). Hippocampal neurons were seeded on glass slides in 24-well plates and cultured for 6 days. The cells were then incubated with Fluo-4 AM (5 μM) and Pluronic F-127 (0.05%) (Molecular Probes) in HEPES-buffered solution (NaCl 135 mM, KCl 5 mM, MgSO41 mM, CaCl22 mM, HEPES 20 mM, glucose 5 mM, lactic acid 10 mM, and Na-pyruvate 1 mM; pH 7.4) for 30 minutes at room temperature in the dark to stain the cells. The cells were then treated with glutamate or PGSF + glutamate for 4 hours. Next, intracellular Ca2+ concentrations were determined using fluorescence intensity, which was measured with a 488 nm excitation and 535 nm emission using a confocal microscope (Olympus, Tokyo, Japan). For the Ca2+-free experiments, the neuronal cells were incubated with Fluo-4 AM (5 μM) and Pluronic F-127 (0.05%) in Ca2+-free HEPES-buffered solution (NaCl 135 mM, KCl 5 mM, MgSO41 mM, EGTA 1 mM, HEPES 20 mM, glucose 5 mM, lactic acid 10 mM, and Na-pyruvate 1 mM; pH 7.4) for 30 minutes at room temperature in the dark. Intracellular Ca2+ concentrations were measured and imaged using a confocal microscope (Olympus). The images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA; version: 1.53c).
Slice electrophysiology
To obtain brain slices, C57BL/6 mice were anesthetized by ethyl ether (Xilong Scientific Co., Shantou, China; Song et al., 2017) and sacrificed for use in the whole-cell patch-clamp technique. Their brains were then transferred to ice-cold carbogenated (95% O2/5% CO2) artificial cerebrospinal fluid solution (ACSF) containing (in mM): NaCl, 124; NaHCO3, 26; D-(+)glucose, 10; KCl, 2.5; CaCl2, 2.5; MgSO4·7H2 O, 2; and NaH2 PO4·2H2 O, 1.25. Brain slices (300 µm thickness) were prepared and transferred to warmed carbogenated ACSF solution (32°C) for 10 minutes. During the recordings, slices were immersed in ACSF, which flowed continuously at a rate of 2 mL/min at 32°C. Hippocampal neurons of brain slices were patch-clamped using glass pipettes (2.7–4.0 MΩ; Vital Sense Scientific Instruments Co., Ltd., Wuhan, China) under a microscope (Nikon, Tokyo, Japan). Voltage-clamp recordings were recorded using an Axon Instruments MultiClamp 700B amplifier (Molecular Devices, San Jose, CA, USA), and currents were digitized using an Axon Instruments 1550A digitizer controlled with the Axon Instruments pClamp 10.5 software (Axon Instruments, Foster City, CA, USA) on a computer. To measure the NMDAR current, an excitatory postsynaptic current (EPSC) was evoked at +40 mV to remove the magnesium block from the NMDAR. The gamma-aminobutyric acid antagonist bicuculline (20 µM; MedChemExpress, Monmouth Junction, NJ, USA) was added to block gamma-aminobutyric acid currents. In addition, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 µM; MedChemExpress) was added during the experiment to block α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid currents. The internal medium consisted of (in mM): cesium methanesulfonate, 125; CsCl, 5; HEPES, 10; ethylene glycol tetraacetic acid, 0.2; MgCl2, 1; Mg-ATP, 4; Na-GTP, 0.3; phosphocreatine, 10; and QX314, 5 (pH 7.40, 285 mOsm). The peak amplitude of the NMDAR currents was selected for data analysis. The inhibitory effects were calculated as follows: Inhibitory effects (%) = (1 – NMDAR-mediated currents (drug treatment)/NMDAR-mediated currents (control)) × 100.
Western blot assay
The cultured hippocampal neurons were seeded in six-well plates and cultured for 6 days. After being treated with glutamate (100 μM) or PGSF (2–10 μM) + glutamate (100 μM), the total proteins of the hippocampal neurons were harvested in cell lysis buffer containing protease and phosphatase inhibitors (Beyotime Biotechnology). Protein concentrations were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis with standard proteins. The sample proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were then blocked with 5% nonfat milk at room temperature for 1 hour and probed with primary antibodies (rabbit anti-NR2A, 1:100, Cat# 4205S, Cell Signaling; rabbit anti-NR2B, 1:100, Cat# 4207S, Cell Signaling; rabbit anti-pCREB, 1:100, Cat# 9198S, Cell Signaling; rabbit anti-BDNF, 1:100, Cat# 47808S, Cell Signaling; rabbit anti-glyceraldehyde-3-phosphate dehydrogenase [GAPDH], 1:100, Cat# 2118, Cell Signaling) at 4°C overnight. Next, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:3000, Cat# D601016-0005, Sangon Biotech, Shanghai, China) for 2 hours at room temperature. The blots were visualized using an eECL Western Blot Kit (Cat# CW0048S, CWBio, Beijing, China), and band intensities were analyzed using Image Lab software (Bio-Rad, Hercules, CA, USA; version: 5.2.1).
Statistical analysis
All results are expressed as the mean ± standard error of the mean (SEM) and were analyzed using GraphPad Prism software (Version 7.04, GraphPad Inc., San Diego, CA, USA). One-way analysis of variance with Dunnett’s post hoc test was used for multiple comparisons. The two-tailed Student’s t-test was used for two-sample comparisons. P < 0.05 was considered statistically significant.
Results
PGSF enhances the viability of cultured neurons treated with glutamate
We used cultured hippocampal cells to investigate the neuroprotective effects of PGSF because hippocampal neurons are vulnerable to both ischemia and neurodegenerative disorders (Pulsinelli et al., 1982; Lu et al., 2014). We first cultured the hippocampal neurons for 6 days, and then investigated the concentration-dependent effects of glutamate on the viability of such neurons. Five different concentrations of glutamate (20, 40, 60, 80, or 100 µM) were used to treat the neurons for 24 hours (Figure 1A). The viability of the neurons under each concentration of glutamate was measured. As shown in Figure 1B, there was a decrease in the survival percentage of hippocampal cells with an increase in glutamate concentration (F(5, 57) = 1.27).
Figure 1.
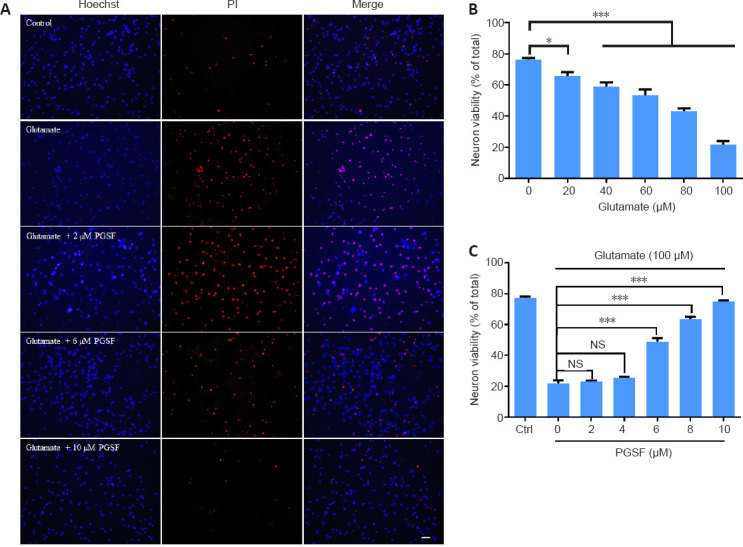
PGSF inhibits glutamate-induced death of cultured hippocampal neurons.
Cells were treated for 24 hours with glutamate alone or in the presence of PGSF (2, 6, 8, or 10 µM), and neuronal death was detected by Hoechst 33342 (blue) and PI (red) staining. (A) Representative images of neurons under control conditions and after treatment with glutamate (100 µM) alone or in the presence of PGSF. Cells stained with Hoechst 33342 only are surviving cells, and those co-stained with Hoechst 33342 and PI are dead cells. Scale bar: 20 μM. (B) Quantitative analysis of neuronal viability after treatment with different concentrations of glutamate. The experiment was repeated four times. (C) Quantitative analysis of neuronal viability in cultured cells treated with 100 µM glutamate in the presence of different doses of PGSF. The experiment was repeated nine times. Data are expressed as the mean ± SEM. *P < 0.05, ***P < 0.001 (one-way analysis of variance followed by Dunnett’s post hoc test). Original data for Figure 1 are shown in Additional file 2. NS: Not significant; PGSF: polygalasaponin F; PI: propidium iodide.
We next investigated the protective effects of PGSF on cultured neurons exposed to 100 µM glutamate. Hippocampal cells were treated with 100 µM glutamate, 2 µM PGSF + 100 µM glutamate, 4 µM PGSF + 100 µM glutamate, 6 µM PGSF + 100 µM glutamate, 8 µM PGSF + 100 µM glutamate, or 10 µM PGSF + 100 µM. As shown in Figure 1C, although 2 µM and 4 µM PGSF had no protective effects against glutamate-induced cell death (F(6, 50) = 3.04), 6, 8, and 10 µM PGSF significantly enhanced neuronal viability in a concentration-dependent manner (cell viability: 48.88 ± 2.39% under 6 µM PGSF; 63.61 ± 1.32% under 8 µM PGSF; and 74.83 ± 0.85% under 10 µM PGSF). The toxicity of PGSF was also investigated; the concentrations of PGSF used in the present experiment had no toxicity to neuronal cells (data not shown).
PGSF inhibits glutamate-induced apoptosis in neuronal cells
Apoptotic cell death is executed by the activation of procaspase-3. We therefore used cleaved caspase-3, an activated form of caspase-3, to examine neuronal cell death (Park et al., 2010). Neuronal cells were treated for 24 hours with glutamate (100 µM), glutamate (100 µM) + PGSF (10 µM), or glutamate (100 µM) + MK801 (10 µM). Cellular apoptosis was then calculated by counting the cleaved caspase-3-positive cells. As shown in Figure 2A, more cleaved caspase-3-positive cells were observed under glutamate treatment, whereas numbers returned to control levels when PGSF or MK801 was co-applied with glutamate. MK801 is a non-selective antagonist of NMDARs, and was used here as a positive control. Figure 2B shows the quantification of the neuronal apoptosis ratio under the control, glutamate, glutamate + PGSF, and glutamate + MK801 conditions (F(3, 24) = 0.772). Thus, PGSF was able to protect neurons against glutamate excitotoxicity-induced apoptotic death.
Figure 2.
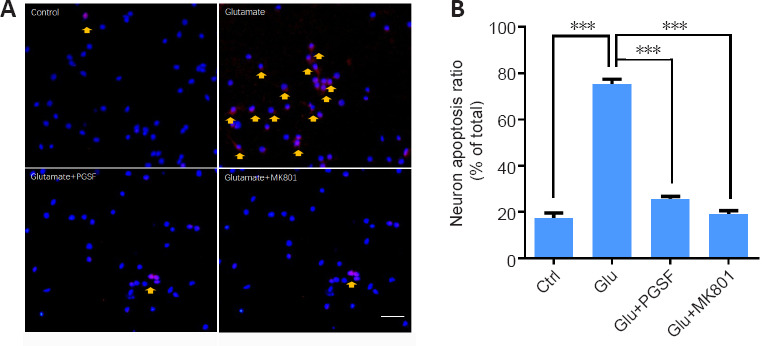
PGSF inhibits glutamate-induced apoptosis in cultured hippocampal neurons.
Cultured neurons were treated for 24 hours with glutamate (100 µM) alone or in the presence of PGSF (10 µM) or MK801 (10 µM), and neuronal apoptosis was detected using cleaved caspase-3 staining. (A) Representative images of neurons under control conditions and after treatment with glutamate, glutamate + PGSF, or glutamate + MK801. There were many more cleaved caspase-3-positive neurons in the glutamate-only group than in the other groups. Neurons displaying signs of apoptosis are indicated by arrows. Scale bar: 20 µm. (B) Quantitative analysis of the neuronal apoptosis ratio under control conditions and after treatment with glutamate, glutamate + PGSF, or glutamate + MK801. Data are shown as the mean ± SEM. The experiment was repeated four times. ***P < 0.001 (one-way analysis of variance followed by Dunnett’s post hoc test). Original data for Figure 2 are shown in Additional file 3. Ctrl: Control; Glu: glutamate; MK801: (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate, an N-methyl-D-aspartate receptor antagonist; PGSF: polygalasaponin F.
PGSF inhibits the overload of cytosolic Ca2+ induced by glutamate excitotoxicity
Glutamate excitotoxicity-induced cytosolic Ca2+ overload is highly associated with neuronal death (Xiong et al., 2004). We therefore examined whether the neuroprotective effects of PGSF were associated with the inhibition of cytosolic Ca2+ overload. Cultured hippocampal cells were incubated with glutamate (100 µM), glutamate (100 µM) + PGSF (10 µM), or glutamate (100 µM) + MK801 (10 µM), and Ca2+ signals were measured using Flu-4 AM fluorescent probes at 4 hours post-incubation. Cytosolic Ca2+ in the cultured cells increased markedly upon treatment with glutamate (Figure 3A and B), and this Ca2+ overload was suppressed when PGSF was co-applied (Figure 3C). A similar suppressive effect was observed when the non-selective NMDAR antagonist MK801 was co-administered (Figure 3D). The increased cytosolic Ca2+ that was induced by glutamate exposure came from extracellular Ca2+ because such an effect did not occur in the absence of extracellular Ca2+ (Figure 3E). The quantitative analysis of these results revealed that PGSF inhibited Ca2+ influx (F(4, 106) = 4.12; Figure 3F, which is in accordance with the results of previous studies demonstrating that Ca2+ overload after excitotoxic stimuli comes from the influx of extracellular Ca2+ (Berdichevsky et al., 1983; Lai et al., 2014).
Figure 3.

PGSF inhibits glutamate-induced Ca2+ overload in cultured hippocampal neurons.
Cells were incubated for 4 hours with glutamate (100 µM) alone or in the presence of PGSF (10 µM) or MK801 (10 µM), or under Ca2+-free conditions. Intracellular concentrations of Ca2+ were visualized by staining with Fluo-4 AM. (A–E) Representative images of neurons under the different experimental conditions. Compared with the neurons in the other groups, fluorescence intensity was markedly stronger in the neurons treated with glutamate alone. Scale bar: 50 µm. (F) Quantitative analysis of the relative fluorescence intensity of neurons under the different experimental conditions. Data are shown as the mean ± SEM. The experiment was repeated four times. **P < 0.01 (one-way analysis of variance followed by Dunnett’s post hoc test). Original data for Figure 3 are shown in Additional file 4. AU: Arbitrary unit; Glu: glutamate; MK801: (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate, an N-methyl-D-aspartate receptor antagonist; PGSF: polygalasaponin F.
PGSF suppresses NMDAR-mediated currents in cultured hippocampal neurons
Calcium imaging revealed that PGSF inhibited the influx of Ca2+, suggesting that PGSF might block NMDARs. To investigate this concept, we used a whole-cell patch-clamp technique to record the NMDAR-mediated currents in cultured hippocampal neurons treated with PGSF. Bicuculline and DNQX were co-administered to suppress the gamma-aminobutyric acid inhibitory postsynaptic current and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor-mediated EPSC, respectively. Our data revealed that PGSF (10 μM) blocked the NMDAR-mediated EPSC by 47.7 ± 34.03% (Figure 4A and B). Furthermore, as a positive control, MK801 (10 μM) blocked the NMDAR-mediated EPSC by 89.54 ± 1.32% (Figure 4B and C), which was consistent with the results of a previous study demonstrating that MK801 blocks almost 100% of NMDAR-mediated synaptic currents (Xia et al., 2010). This result suggests that PGSF might be a relatively mild NMDAR antagonist compared with MK801.
Figure 4.
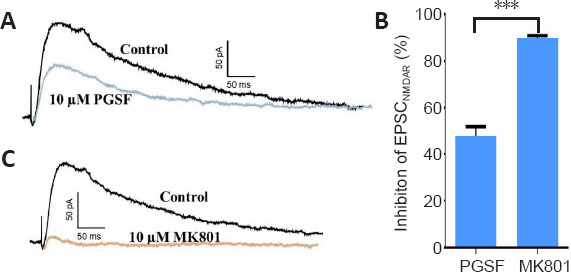
PGSF suppresses NMDAR-mediated currents in cultured hippocampal neurons.
(A) PGSF (10 µM) partially blocked the NMDAR-mediated component of EPSCs. (B) MK801 (10 µM), as a positive control, almost completely blocked the NMDAR-mediated component of EPSCs. (C) Quantitative analysis of the inhibitory effects of PGSF and MK801 on NMDAR-mediated currents. Data are shown as the mean ± SEM. The experiment was repeated four times. ***P < 0.001 (two-tailed Student’s t-test). Original data for Figure 4 are shown in Additional file 5. EPSC: Excitatory postsynaptic current; MK801: (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate, an N-methyl-D-aspartate receptor antagonist; NMDAR: N-methyl-D-aspartate receptor; PGSF: polygalasaponin F.
PGSF reverses the glutamate-induced regulation of NR2A and NR2B expression
The NR2A and NR2B subunits of NMDARs play different roles in neuronal survival and death (Lai et al., 2014). The results described in the previous sections indicate that PGSF prevents neuronal cells from glutamate excitotoxicity by partially blocking NMDARs. We therefore investigated the effects of PGSF on the expression of NMDARs in cultured hippocampal neurons. Because NMDARs in the forebrain are composed of subunits that include NR2A and NR2B (Ishii et al., 1993), we used western blot assays to detect the effects of PGSF on the expression of NR2A and NR2B. As shown in Figure 5, treatment with glutamate (100 µM) suppressed NR2A expression (F(4, 15) = 0.58, P < 0.001, Figure 5A) and enhanced NR2B expression (F(4, 15) = 2.98, P < 0.001, Figure 5B) in a time-dependent manner, with effects appearing 10 minutes after glutamate application and reaching a maximum at 30 minutes after application. When glutamate (100 µM) was co-applied with PGSF (10 µM) or MK801 (10 µM, as a positive control), the glutamate-induced suppression of NR2A expression (F(3, 12) = 0.74, P < 0.001, Figure 5C) and enhancement of NR2B expression (F(3, 12) = 3.17, P < 0.001, Figure 5D) were reversed to control levels.
Figure 5.
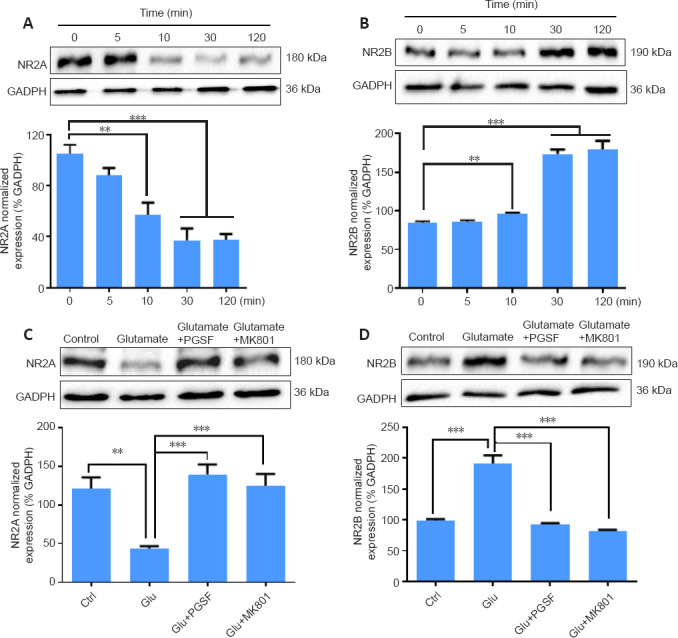
PGSF reverses the glutamate-induced regulation of NR2A and NR2B expression in cultured hippocampal neurons.
(A, B) Glutamate downregulated the expression of NR2A subunits and upregulated the expression of NR2B subunits in a time-dependent manner. (C, D) The glutamate-induced downregulation of NR2A and upregulation of NR2B was reversed by co-administration of PGSF. Data are shown as the mean ± SEM. The experiment was repeated four times. **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Dunnett’s post hoc test). Original data for Figure 5 are shown in Additional file 6. Ctrl: Control; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; Glu: glutamate; MK801: (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate, an N-methyl-D-aspartate receptor antagonist; PGSF: polygalasaponin F.
PGSF reverses glutamate-induced suppression of pro-survival signaling molecules
It has been reported that the activation of synaptic NR2A-containing NMDARs upregulates the levels of pCREB (a transcription factor) (Hardingham et al., 2001) and BDNF (a survival-promoting protein) (Hardingham et al., 2002; Shi et al., 2021), whereas the activation of extrasynaptic NR2B-containing NMDARs produces an opposite effect (Hardingham et al., 2002). We thus investigated whether PGSF was able to block glutamate-induced changes in pCREB and BDNF in cultured hippocampal neurons. As shown in Figure 6, treatment with glutamate (100 µM) significantly reduced the expression of pCREB (F(3, 8) = 0.78, P < 0.001, Figure 6A) and BDNF (F(3, 16) = 0.67, P < 0.001, Figure 6B); however, these suppressive effects were reversed in the presence of PGSF (10 µM) or MK801 (10 µM; Figure 6A and B).
Figure 6.
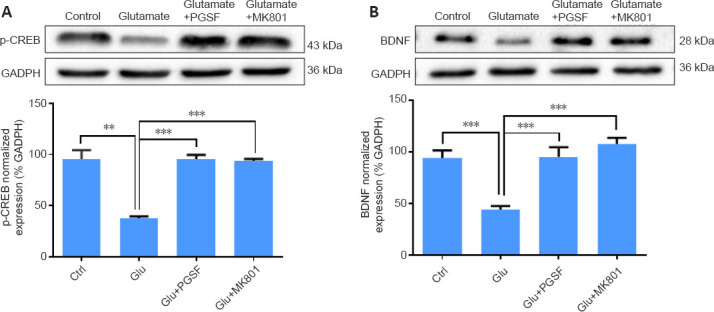
PGSF reverses the glutamate-induced suppression of pCREB and BDNF in cultured hippocampal neurons.
Glutamate (100 µM; 1 hour) suppressed the expression of pCREB and BDNF; however, such suppressive effects were not observed in the presence of PGSF (10 µM) or MK801 (10 µM). Data are shown as the mean ± SEM. The experiment was repeated three times. **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Dunnett’s post hoc test). Original data for Figure 6 are shown in Additional file 7. BDNF: Brain-derived neurotrophic factor; Ctrl: control; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; Glu: glutamate; MK801: (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate, an N-methyl-D-aspartate receptor antagonist; pCREB: phosphorylated cyclic adenosine monophosphate-responsive element binding protein; PGSF: polygalasaponin F.
Discussion
The present study demonstrated the protective effects of PGSF against glutamate-induced excitotoxicity in hippocampal neurons. Our results suggest that PGSF probably acts in combination with NMDARs, blocks the overload of Ca2+ influx, and rescues pCREB levels and BDNF expression, thus reversing glutamate-induced excitotoxicity.
In the present investigation, we demonstrated that PGSF reversed the glutamate-induced downregulation of a pro-survival signal. Ca2+ entry through synaptic NMDARs activates the transcription factor CREB (Hardingham et al., 1997), and CREB-dependent gene expression is causally linked with neuroprotective activity against apoptosis and excitotoxicity (Mantamadiotis et al., 2002). BDNF, one of the targets of Ca2+/CREB signaling, acts as a pro-survival signal and rescues neurons from excitotoxicity (Lee et al., 2005; Zhang et al., 2009). The activation of synaptic NR2A-containing NMDARs increases pCREB and BDNF levels, whereas the stimulation of extrasynaptic NR2B-containing NMDARs attenuates pro-survival signaling (Hardingham et al., 2002).
Calcium influx is an essential mediator of glutamate excitotoxicity, and NMDARs are the primary source of toxic Ca2+ entry (Choi and Rothman, 1990). The overactivation of NMDARs mediates the influx of a large amount of extracellular Ca2+ into neurons, which then triggers downstream neurotoxic cascades, including CREB shut-off and BDNF inactivation (Hardingham and Bading, 2010). The intracellular Ca2+ overload causes metabolic failure and oxidative stress, leading to the apoptosis or necrosis of neurons (Fei et al., 2020). Attenuating the increased levels of intracellular Ca2+ has been reported as an important strategy for preventing neuronal death (Choi, 1987). In the present study, we revealed that PGSF inhibited the Ca2+ influx that was induced by high concentrations of extracellular glutamate. It is highly possible that PGSF reduces the glutamate-induced apoptosis of cells by blocking Ca2+ influx.
Our results also revealed that PGSF was able to partially block NMDAR-mediated currents. According to the model by Hardingham and Bading (Hardingham and Bading, 2010), both the activation and the location of NMDARs affect the fate of neurons. Equivalent amounts of Ca2+ that flow through synaptic and extrasynaptic NMDARs trigger completely opposite downstream events. The entry of Ca2+ through synaptic NMDARs tends to render neurons more resistant to oxidative stress and apoptosis, while Ca2+ influx via extrasynaptic NMDARs preferentially initiates cellular death by triggering gene transcription that leads to oxidative stress and neuronal apoptosis (Hardingham et al., 2002; Hardingham and Bading, 2010). In the hippocampus of rats, NR2A-containing NMDARs located at synaptic sites are associated with cell survival, whereas NR2B-containing NMDARs located at extrasynaptic sites are linked to cell death (Lai et al., 2011; Martel et al., 2012). In the present study, PGSF reversed the glutamate-induced downregulation of NR2A expression and upregulation of NR2B expression. This may be an important route by which PGSF produces its neuroprotective effects.
NMDARs promote neuronal survival via the activation of protein kinase B (Akt) (Lafon-Cazal et al., 2002). In general, the stimulation of NR2A-containing NMDARs reduces neuronal death, while the stimulation of NR2B-containing NMDARs tends to promote neuronal death (Lai et al., 2014). Calcium travels through NR2A-containing NMDARs and activates the pro-survival protein phosphatidylinositol 3-kinase (PI3K). Active PI3K binds to the pleckstrin homology domain of Akt, facilitating the activation of Akt (Stephens et al., 1998). It has been reported that NMDAR-triggered activation of the PI3K/Akt pathway protects neurons from hypoxic and excitotoxic death (Soriano et al., 2006), whereas inhibition of the PI3K/Akt pathway exacerbates ischemic neuronal death (Endo et al., 2006). The present study did not provide evidence that PGSF exerts its neuroprotection via the PI3K/Akt pathway. However, Xie et al. (2020) reported that PGSF inhibits oxygen/glucose deprivation-induced neuronal apoptosis by activating the PI3K/Akt pathway. It is thus possible that the NR2A-containing NMDAR/PI3K/Akt signaling pathway may be a mechanism underlying the neuroprotective effects of PGSF.
Sun et al. (2012) reported that PGSF induces long-term potentiation in the hippocampus of adult rats via NMDAR activation. In their study, PGSF was administered intracerebroventricularly, and the long-term potentiation of population spikes was recorded from the dentate gyrus of the hippocampus in anesthetized rats. It is therefore hard to identify the locations and specific sites of PGSF action. The discrepancy between the effects of PGSF in the study by Sun et al. (2012) and those in the present study may have been caused by the different protocols that were used.
In the current study, we did not conduct any experiments examining whether PGSF was able to reach the hippocampus. However, Zhou et al. (2019) reported that PGSF reduces the size of the infarct areas (including the hippocampus) in rats with middle cerebral artery occlusion, suggesting that PGSF is able to reach the hippocampus in vivo. It is widely accepted that moderate NMDAR activity is beneficial for neurons (Paoletti et al., 2013). Moreover, drugs that inhibit NMDARs often exhibit protective effects against excitatory damage (Ikonomidou and Turski, 2002). However, many drugs that inhibit NMDARs fail to pass clinical trials because of their severe side effects, such as hyperlocomotion and cognitive deficits. For example, MK801 can induce schizophrenia symptoms (Micale et al., 2013). In contrast, a mild NMDAR antagonist that can block 27.1% of NMDAR-mediated EPSCs has been reported to have an excellent clinical safety profile because it selectively inhibits neuronal death while maintaining the normal function of NMDARs; it is used clinically for the treatment of neurodegenerative diseases (Xia et al., 2010; Saxton et al., 2012; Wang et al., 2015). The present study demonstrated that, although MK801 almost entirely blocked NMDAR-mediated EPSCs, PGSF only partially suppressed NMDAR-mediated EPSCs. These findings suggest that PGSF is a potential NMDAR antagonist that may be a promising therapeutic agent for protection against glutamate excitotoxicity.
Additional files:
Additional file 1: Open peer review reports 1 (94.2KB, pdf) and 2 (89.5KB, pdf) .
Footnotes
P-Reviewers: Amantea D, Armato U; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Gardner B, Yu J, Song LP; T-Editor: Jia Y Research Article
Funding:This work was supported by the National Natural Science Foundation of China, Nos. 31971035 (to BML), 31771182 (to BML), 81471116 (to BML); and the Natural Science Foundation of Jiangxi Province of China, Nos. 20171BAB204019 (to CS), 20192ACB20022 (to CS).
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was supported by the National Natural Science Foundation of China, Nos. 31971035 (to BML), 31771182 (to BML), 81471116 (to BML); and the Natural Science Foundation of Jiangxi Province of China, Nos. 20171BAB204019 (to CS), 20192ACB20022 (to CS). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement:The study was approved by the Institutional Animal Care Committee of Nanchang University (approval No. 2017-0006) on December 29, 2017.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open peer reviewers:Diana Amantea, University of Calabria, Italy; Ubaldo Armato, University of Verona, Italy.
References
- 1.Berdichevsky E, Riveros N, Sánchez-Armáss S, Orrego F. Kainate, N-methylaspartate and other excitatory amino acids increase calcium influx into rat brain cortex cells in vitro. Neurosci Lett. 1983;36:75–80. doi: 10.1016/0304-3940(83)90489-5. [DOI] [PubMed] [Google Scholar]
- 2.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 3.Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, Xu L, Duan WH, Xiong ZQ. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke. 2008;39:3042–3048. doi: 10.1161/STROKEAHA.108.521898. [DOI] [PubMed] [Google Scholar]
- 4.Chinese Pharmacopoeia Commission. Beijing: China Medical Science and Technology Publishing House; 2015. Chinese Pharmacopoeia. [Google Scholar]
- 5.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 7.Chu J, Liu CX, Song R, Li QL. Ferrostatin-1 protects HT-22 cells from oxidative toxicity. Neural Regen Res. 2020;15:528–536. doi: 10.4103/1673-5374.266060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorsett CR, McGuire JL, DePasquale EA, Gardner AE, Floyd CL, McCullumsmith RE. Glutamate neurotransmission in rodent models of traumatic brain injury. J Neurotrauma. 2017;34:263–272. doi: 10.1089/neu.2015.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Endo H, Nito C, Kamada H, Nishi T, Chan PH. Activation of the Akt/GSK3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1479–1489. doi: 10.1038/sj.jcbfm.9600303. [DOI] [PubMed] [Google Scholar]
- 10.Fei F, Su N, Li X, Fei Z. Neuroprotection mediated by natural products and their chemical derivatives. Neural Regen Res. 2020;15:2008–2015. doi: 10.4103/1673-5374.282240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 12.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- 14.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 15.Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 16.Huberfeld G, Vecht CJ. Seizures and gliomas--towards a single therapeutic approach. Nat Rev Neurol. 2016;12:204–216. doi: 10.1038/nrneurol.2016.26. [DOI] [PubMed] [Google Scholar]
- 17.Huo TG, Li WK, Zhang YH, Yuan J, Gao LY, Yuan Y, Yang HL, Jiang H, Sun GF. Excitotoxicity induced by realgar in the rat hippocampus: the involvement of learning memory injury, dysfunction of glutamate metabolism and NMDA receptors. Mol Neurobiol. 2015;51:980–994. doi: 10.1007/s12035-014-8753-2. [DOI] [PubMed] [Google Scholar]
- 18.Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury. Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 19.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 20.Ishii T, Moriyoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M, Nakanishi S. Molecular characterization of the family of the N-methyl-D-aspartate receptor subunits. J Biol Chem. 1993;268:2836–2843. [PubMed] [Google Scholar]
- 21.Jiang LL, Zhu B, Zhao Y, Li X, Liu T, Pina-Crespo J, Zhou L, Xu W, Rodriguez MJ, Yu H, Cleveland DW, Ravits J, Da Cruz S, Long T, Zhang D, Huang TY, Xu H. Membralin deficiency dysregulates astrocytic glutamate homeostasis leading to ALS-like impairment. J Clin Invest. 2019;129:3103–3120. doi: 10.1172/JCI127695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lafon-Cazal M, Perez V, Bockaert J, Marin P. Akt mediates the anti-apoptotic effect of NMDA but not that induced by potassium depolarization in cultured cerebellar granule cells. Eur J Neurosci. 2002;16:575–583. doi: 10.1046/j.1460-9568.2002.02124.x. [DOI] [PubMed] [Google Scholar]
- 23.Lai TW, Shyu WC, Wang YT. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med. 2011;17:266–275. doi: 10.1016/j.molmed.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- 26.Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu W, Xu Z, Yang T, Xu B, Deng Y, Feng S. Memantine, a low-affinity NMDA receptor antagonist, protects against methylmercury-induced cytotoxicity of rat primary cultured cortical neurons, involvement of Ca(2+) dyshomeostasis antagonism, and indirect antioxidation effects. Mol Neurobiol. 2017;54:5034–5050. doi: 10.1007/s12035-016-0020-2. [DOI] [PubMed] [Google Scholar]
- 28.Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, Bennett DA, Colaiácovo MP, Yankner BA. REST and stress resistance in ageing and Alzheimer’s disease. Nature. 2014;507:448–454. doi: 10.1038/nature13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schütz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- 30.Martel MA, Ryan TJ, Bell KF, Fowler JH, McMahon A, Al-Mubarak B, Komiyama NH, Horsburgh K, Kind PC, Grant SG, Wyllie DJ, Hardingham GE. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron. 2012;74:543–556. doi: 10.1016/j.neuron.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol. 2013;698:6–18. doi: 10.1016/j.ejphar.2012.10.032. [DOI] [PubMed] [Google Scholar]
- 32.Micale V, Kucerova J, Sulcova A. Leading compounds for the validation of animal models of psychopathology. Cell Tissue Res. 2013;354:309–330. doi: 10.1007/s00441-013-1692-9. [DOI] [PubMed] [Google Scholar]
- 33.Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- 34.Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 35.Park E, Lee GJ, Choi S, Choi SK, Chae SJ, Kang SW, Pak YK, Park HK. The role of glutamate release on voltage-dependent anion channels (VDAC)-mediated apoptosis in an eleven vessel occlusion model in rats. PLoS One. 2010;5:e15192. doi: 10.1371/journal.pone.0015192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 37.Rueda CB, Llorente-Folch I, Traba J, Amigo I, Gonzalez-Sanchez P, Contreras L, Juaristi I, Martinez-Valero P, Pardo B, Del Arco A, Satrustegui J. Glutamate excitotoxicity and Ca2+-regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs) Biochim Biophys Acta. 2016;1857:1158–1166. doi: 10.1016/j.bbabio.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 38.Saxton J, Hofbauer RK, Woodward M, Gilchrist NL, Potocnik F, Hsu HA, Miller ML, Pejović V, Graham SM, Perhach JL. Memantine and functional communication in Alzheimer’s disease: results of a 12-week, international, randomized clinical trial. J Alzheimers Dis. 2012;28:109–118. doi: 10.3233/JAD-2011-110947. [DOI] [PubMed] [Google Scholar]
- 39.Shi RL, Hu JF, Kong LL, Niu F, WU MM, He X, Li PF, Chen NH. Inhibitory effect of Polygalasaponin F on the apoptosis of PC12 cells induced by sodium dithionite-caused oxygen-glucose deprivation and reperfusion. Zhongguo Yaolixue Tongbao. 2013;29:333–336. [Google Scholar]
- 40.Shi ZL, Zhang H, Fan ZY, Ma W, Yuan H, Yang B. Nerve conduits of chitosan/polyvinyl alcohol with brain-derived neurotrophic factor microspheres for peripheral nerve defects in rats. Zhongguo Zuzhi Gongcheng Yanjiu. 2021;25:1555–1559. [Google Scholar]
- 41.Song C, Zhang WH, Wang XH, Zhang JY, Tian XL, Yin XP, Pan BX. Acute stress enhances the glutamatergic transmission onto basoamygdala neurons embedded in distinct microcircuits. Mol Brain. 2017;10:3. doi: 10.1186/s13041-016-0283-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that mediate phosphatidylinositol 3, 4, 5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 44.Sun F, Sun JD, Han N, Li CJ, Yuan YH, Zhang DM, Chen NH. Polygalasaponin F induces long-term potentiation in adult rat hippocampus via NMDA receptor activation. Acta Pharmacol Sin. 2012;33:431–437. doi: 10.1038/aps.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F, Liang W, Lei C, Kinden R, Sang H, Xie Y, Huang Y, Qu Y, Xiong L. Combination of HBO and Memantine in Focal Cerebral Ischemia: Is There a Synergistic Effect. Mol Neurobiol. 2015;52:1458–1466. doi: 10.1007/s12035-014-8949-5. [DOI] [PubMed] [Google Scholar]
- 46.Wei W, Yuan YH, Gao YN, Yan WF, Li CJ, Zhang DM, Chen NH. Polygalasaponin F inhibits secretion of inflammatory cytokines via NF-κB pathway regulation. J Asian Nat Prod Res. 2014;16:865–875. doi: 10.1080/10286020.2014.918962. [DOI] [PubMed] [Google Scholar]
- 47.Wu MM, Yuan YH, Chen J, Li CJ, Zhang DM, Chen NH. Polygalasaponin F against rotenone-induced apoptosis in PC12 cells via mitochondria protection pathway. J Asian Nat Prod Res. 2014;16:59–69. doi: 10.1080/10286020.2013.864283. [DOI] [PubMed] [Google Scholar]
- 48.Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie W, Wulin H, Shao G, Wei L, Qi R, Ma B, Chen N, Shi R. Polygalasaponin F inhibits neuronal apoptosis induced by oxygen-glucose deprivation and reoxygenation through the PI3K/Akt pathway. Basic Clin Pharmacol Toxicol. 2020;127:196–204. doi: 10.1111/bcpt.13408. [DOI] [PubMed] [Google Scholar]
- 50.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 51.Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C, Aso Y, Descombes P, Bading H. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009;5:e1000604. doi: 10.1371/journal.pgen.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou WL, Zhang JT, Xu W, Sun JH. Protective effects of polygalasaponin F on oxidative stress and apoptosis-induced ischemic myocardial injury in neonatal rats with hypoxic-ischemic brain damage. Neuroreport. 2019;30:1148–1156. doi: 10.1097/WNR.0000000000001330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.