

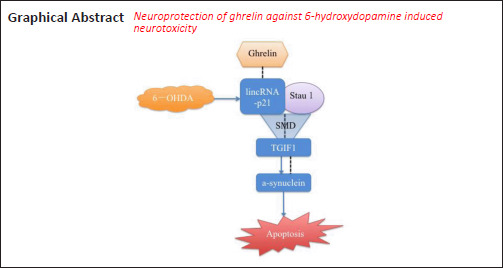
Key Words: 6-hydroxydopamine, apoptosis, ghrelin, lincRNA-p21, neuropeptide, neurotoxicity, Parkinson's disease, STAU1-mediated mRNA decay, TGIF1, α-synuclein
Abstract
Ghrelin is a neuropeptide that has various physiological functions and has been demonstrated to be neuroprotective in a number of neurological disease models. However, the underlying mechanisms of ghrelin in Parkinson’s disease remain largely unexplored. The current study aimed to study the effects of ghrelin in a 6-hydroxydopamine (6-OHDA)-induced Parkinson’s disease model and evaluate the potential underlying mechanisms. In the present study, we treated an SH-SY5Y cell model with 6-OHDA, and observed that pretreatment with different concentrations of ghrelin (1, 10, and 100 nM) for 30 minutes relieved the neurotoxic effects of 6-OHDA, as revealed by Cell Counting Kit-8 and Annexin V/propidium iodide (PI) apoptosis assays. Reverse transcription quantitative polymerase chain reaction and western blot assay results demonstrated that 6-OHDA treatment upregulated α-synuclein and lincRNA-p21 and downregulated TG-interacting factor 1 (TGIF1), which was predicted as a potential transcription regulator of the gene encoding α-synuclein (SNCA). Ghrelin pretreatment was able to reverse the trends caused by 6-OHDA. The Annexin V/PI apoptosis assay results revealed that inhibiting either α-synuclein or lincRNA-p21 expression with small interfering RNA (siRNA) relieved 6-OHDA-induced cell apoptosis. Furthermore, inhibiting lincRNA-p21 also partially upregulated TGIF1. By retrieving information from a bioinformatics database and performing both double luciferase and RNA immunoprecipitation assays, we found that lincRNA-p21 and TGIF1 were able to form a double-stranded RNA-binding protein Staufen homolog 1 (STAU1) binding site and further activate the STAU1-mediated mRNA decay pathway. In addition, TGIF1 was able to transcriptionally regulate α-synuclein expression by binding to the promoter of SNCA. The Annexin V/PI apoptosis assay results showed that either knockdown of TGIF1 or overexpression of lincRNA-p21 notably abolished the neuroprotective effects of ghrelin against 6-OHDA-induced neurotoxicity. Collectively, these findings suggest that ghrelin exerts neuroprotective effects against 6-OHDA-induced neurotoxicity via the lincRNA-p21/TGIF1/α-synuclein pathway.
Introduction
Parkinson’s disease (PD) is categorized as a synucleinopathy because α-synuclein (encoded by the SNCA gene) is the main constituent of Lewy bodies and Lewy neurites, which are the pathological hallmarks of the disease (Spillantini et al., 1998). Abnormal expression of α-synuclein is observed in both familial and sporadic forms of PD (Lashuel et al., 2013), and elevated expression of SNCA mRNA is observed in the affected regions of sporadic PD brains (Chiba-Falek et al., 2006). Despite constant research over the past decades, the underlying mechanisms of aberrant α-synuclein expression have yet to be revealed, and PD remains an incurable neurological disorder.
Ghrelin is a 28-amino-acid peptide that is mainly secreted by X/A-like cells of the gastric mucosa. There are two forms of ghrelin in circulation: the acylated form and the desacylated form. Acylated ghrelin is processed by ghrelin O-acyltransferase, which attaches octanoate to proghrelin (Gutierrez et al., 2008). Despite desacylated ghrelin accounting for 80–90% of total circulating ghrelin, acylated ghrelin is believed to be the active form that binds to the growth hormone secretagogue receptor (GHSR) and exerts physiological functions, such as stimulating growth hormone release, promoting appetite, and regulating energy homeostasis (van der Lely et al., 2004). The present study mainly focused on the biological function of acylated ghrelin, which has been demonstrated to be neuroprotective in a number of neurological disease models (Jiang et al., 2008; Eslami et al., 2018; Dong et al., 2019). The identified underlying mechanisms mainly involve anti-apoptotic, anti-oxidant, and anti-inflammatory actions (Beynon et al., 2013; Morgan et al., 2018). However, it remains unknown whether ghrelin regulates α-synuclein expression in PD remains.
Long non-coding RNAs (lncRNAs) belong to a class of noncoding RNAs with more than 200 nucleotides and a broad range of biological functions (Shi et al., 2013). A recent study suggested that lncRNAs can affect the stability of mRNAs by activating double-stranded RNA-binding protein Staufen homolog 1 (STAU1)-mediated mRNA decay (SMD), which degrades translationally active mRNAs whose 3′-untranslated regions bind to STAU1, which binds to double-stranded RNAs (Gong and Maquat, 2011). An Alu element within an lncRNA and an Alu element within the 3′-untranslated region of a target mRNA can form the STAU1 binding site by imperfect base-pairing. Thus, the lncRNA can downregulate a number of SMD targets, including translationally active mRNAs (Gong and Maquat, 2011). Many lncRNAs are reportedly aberrantly expressed in PD brain specimens, including an upregulation of lincRNA-p21, which precedes the course of PD onset (Kraus et al., 2017). LincRNA-p21 was initially identified as a direct transcriptional target of p53 (Huarte et al., 2010). A recent study by Ye et al. (2018) suggested that lincRNA-p21 can regulate microglial activation and worsen neurodegeneration in lipopolysaccharide- and MPTP-induced PD models, providing evidence that lincRNA-p21 might actively participate in PD pathogenesis.
The current study aimed to study the effects of ghrelin in a 6-hydroxydopamine (6-OHDA)-induced PD model, to assess whether ghrelin regulates the expression of α-synuclein and to explore its underlying mechanisms.
Materials and Methods
Cell culture
Because the cell lines applied in the current study were common commercial cell lines, no special ethical permission needed to be obtained. A human neuroblastoma cell line (SH-SY5Y) and a human embryonic kidney cell line (HEK293T) were obtained from the Cell Bank of the Institute of Biochemistry and Cell Biology (Shanghai, China), and were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium supplemented with 10% fetal bovine serum and 100 U/mL penicillin/streptomycin (Biological Industries, Beit Haemek, Israel) at 37°C in 5% CO2 in a humidified chamber (Thermo Fisher Scientific, Waltham, MA, USA).
Cell viability assays
Cell viability was assessed 24 hours after 6-OHDA treatment using a Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Kumamoto, Japan) according to the manufacturer’s instructions. SH-SY5Y cells were seeded onto 96-well plates and cultured overnight, followed by treatment with different concentrations of ghrelin (0.1 nM, 1 nM, 10 nM, 100 nM, 200 nM, 400 nM) (Enzo Life Sciences, Farmingdale, NY, USA) and 75 μM 6-OHDA (Sigma-Aldrich, St. Louis, MO, USA) in 100 μL DMEM/F12 medium supplemented with 10% fetal bovine serum for 24 hours. At the end of the treatment, 10 μL CCK-8 solution was added to each well and incubated for 1 hour at 37°C. The absorbance of each well was then detected at 450 nm under a microplate reader (Thermo Fisher Scientific). Cell viability was calculated as the ratio of the absorbance value to that of the control group (%).
Cell apoptosis assay with flow cytometry
Cell apoptosis was detected using an Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis assay kit (Bimake, Houston, TX, USA). SH-SY5Y cells were cultured in six-well plates and harvested after 24 hours of different treatments. The cells were then stained with Annexin V-FITC and PI according to the manufacturer’s instructions and analyzed using flow cytometry. The apoptosis rate (%) was calculated as [Annexin V(+)PI(−) cells + Annexin V (+)PI(+) cells]/total number of cells × 100.
Reverse transcription quantitative polymerase chain reaction
The everse transcription quantitative polymerase chain reaction (qRT-PCR) experiments were carried out 24 hours after 6-OHDA treatment or 24 hours after transfection. Total RNA was isolated using RNAiso Plus (Takara, Dalian, China). RNA quality and concentration were assessed using the 260/280 nm absorbance ratio with a NanoDrop device (Thermo Fisher Scientific). The primer sequences are listed in Table 1. Primer efficacy was assessed using the log2 values for the dilution ratio (two- to eight-fold dilution of the cDNA stock) against Ct values, as described by Uslu et al. (2014). The qRT-PCR was performed using a One-Step SYBR PrimeScript RT-PCR Kit (Takara) with an ABI 7500 Fast RT-PCR System (Thermo Fisher Scientific). The results were analyzed using the 2–ΔΔCt method, normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an endogenous control (Bonet-Ponce et al., 2016).
Table 1.
Primer information
| Name | Sequence (5′–3′) | Product size (bp) |
|---|---|---|
| LincRNA-p21 | Forward: CTG CCT ACA CCA ATC AGA AAC A | 131 |
| Reverse: GCG ATA GAC CCG CAC ATA CA | ||
| SNCA | Forward: AAG AGG GTG TTC TCT ATG TAG GC | 106 |
| Reverse: GCT CCT CCA ACA TTT GTC ACT T | ||
| TGIF1 | Forward: GGG ATT GGC TGT ATG AGC ACC | 186 |
| Reverse: GGC GGG AAA TTG TGA ACT GA |
TGIF1: TG-interacting factor 1; SNCA: the gene encoding α-synuclein.
Western blot assay
SH-SY5Y cells were harvested 24 hours after 6-OHDA treatment and extracted using radioimmunoprecipitation assay lysis buffer containing proteases and phosphatase inhibitors. Protein concentration was determined using a bicinchoninic acid kit (Beyotime, Shanghai, China). Equal amounts of proteins from each group were separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA, USA). The membranes were then blocked with 5% skim milk in Tris-buffered saline with 0.1% Tween-20 for 1 hour at room temperature before being incubated with primary antibodies overnight at 4°C. Primary antibodies included mouse monoclonal anti-α-synuclein antibody (1:2000; #610787; BD Biosciences, Franklin Lakes, NJ, USA), mouse monoclonal anti-TGFI1 antibody (1:1000; #MAB7555; R&D Systems, Minneapolis, MN, USA), and anti-GAPDH antibody (1:10 000; #60004-1-Ig; Proteintech Group, Chicago, IL, USA). After three washes with Tris-buffered saline with 0.1% Tween-20, the membranes were incubated with peroxidase-conjugated secondary antibodies for 2 hours at room temperature. The horseradish peroxidase-conjugated anti-mouse (1:5000, #AB-2305) and anti-rabbit (1:5000, #ZB2301) antibodies were obtained from ZSGB-BIO (Beijing, China), and the horseradish peroxidase-conjugated anti-sheep antibody (1:5000; abs20008) was obtained from Absin Bioscience (Shanghai, China). Immunoreactivity was detected using an enhanced chemiluminescence detection kit (Millipore). Band intensities were quantified by densitometric analysis using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and GAPDH was used as a loading control. The intensity of each band was normalized to the GAPDH band.
Cell transfection
SH-SY5Y cells were treated as follows: Control, 6-OHDA, ghrelin (1 nM), 6-OHDA + ghrelin (0.1 nM), 6-OHDA + ghrelin (1 nM), 6-OHDA + ghrelin (10 nM), 6-OHDA + ghrelin (100 nM), 6-OHDA + ghrelin (200 nM), 6-OHDA + ghrelin (400 nM), negative control (NC), α-synuclein small interfering RNA (siRNA), α-synuclein siRNA + 6-OHDA, lincRNA-p21 siRNA, lincRNA-p21 siRNA + 6-OHDA, STAU1 siRNA, STAU1 siRNA + 6-OHDA, pcDNA-p21, 6-OHDA + 1 nM ghrelin + TG-interacting factor 1 (TGIF1) siRNA, and 6-OHDA + 1 nM ghrelin + pcDNA-p21. The siRNA targeting lincRNA-p21 was synthesized by RiboBio (Guangzhou, China). For the α-synuclein, STAU1, and TGIF1 knockdowns, siRNAs were synthesized by GenePharma (Shanghai, China). Luciferase reporter vectors were constructed with either the SNCA sequence with the assumed TGIF1 binding site or the relative deletion of the binding sites amplified by PCR and cloned into the pGL4.10 dual-luciferase vector. Full-length lincRNA-p21 and TGIF1 were cloned into pcDNA3.1(+) vectors (Invitrogen, Carlsbad, CA, USA). SH-SY5Y cells were plated onto 24-well plates or six-well plates, depending on the experimental purpose, at a density of 1 × 105 cells/mL. Cells were cultured for 24 hours, and jetPRIME reagent (Polyplus Transfection, Strasbourg, France) was applied for the siRNA and plasmid transfections as instructed by the manufacturer. Briefly, when the seeded cells reached 70% confluency, 6 μL jetPRIME was mixed with 2 μL siRNA or pcDNA and added to the cells. The medium was replaced with fresh medium either 24 hours or 4 hours after transfection (He et al., 2018). The silencing and overexpression efficiency was assessed using qRT-PCR.
Immunofluorescence staining
SH-SY5Y cells were cultured on coverslips. After receiving the corresponding treatments for 24 hours, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 15 minutes, followed by permeabilization and blocking with 0.3% Triton X-100 and 5% goat serum in phosphate-buffered saline for 1 hour at room temperature. The cells were then incubated with anti-α-synuclein antibody at 4°C overnight followed by incubation with Alexa Fluor 594-conjugated goat anti-rabbit for 2 hours at room temperature. Nuclei were visualized by incubating the cells with 4′,6-diamidino-2-phenylindole for 5 minutes at room temperature. Images were acquired using a Nikon 300 microscope (Nikon, Tokyo, Japan).
RNA immunoprecipitation
RNA immunoprecipitation was performed using an RNA-ChIP-IT® Magnetic Chromatin Immunoprecipitation Kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer’s protocol. The cells were collected 24 hours after transfection and the isolated RNA samples were measured using a spectrophotometer and further analyzed by qRT-PCR.
Dual-luciferase reporter assay
HEK293T cells were seeded onto 96-well plates and co-transfected with the appropriate plasmids (pGL4.10-SNCA promoter (WT), (pGL4.10-SNCA promoter (MUT), pcDNA3.1-3FLAG, pcDNA3.1-TGIF1-3FLAG). Relative luciferase activities were assessed 48 hours after transfection using the Dual-Luciferase Reporter Assay System (Promega Madison, Fitchburg, WI, USA). The relative luciferase activity was normalized to the Renilla luciferase activity.
Statistical analysis
The data are expressed as the mean ± standard deviation (SD). GraphPad Prism 6.0 software (GraphPad, San Diego, CA, USA) was used for the statistical analyses. Significant differences between two groups were analyzed using the Student’s t-test, and the statistical analysis of multiple groups was performed using one-way analysis of variance followed by Bonferroni post hoc tests. A value of P < 0.05 was considered statistically significant.
Results
Ghrelin protects SH-SY5Y cells against 6-OHDA-induced neurotoxicity
Based on the results of our previous study (He et al., 2018), 6-OHDA was applied at a concentration of 75 μM in the current study because this concentration induced moderate neurotoxicity. To assess the effects of ghrelin on a 6-OHDA-induced cell model, SH-SY5Y cells were pretreated with different concentrations of ghrelin for 30 minutes before the addition of 6-OHDA. Phase contrast microscopy revealed that 6-OHDA induced a notable decrease in the number of adherent cells and led to more round cells that had lost their processes. The 1 nM, 10 nM, and 100 nM ghrelin pretreatments partially preserved the morphology of SH-SY5Y cells upon 6-OHDA treatment (Figure 1A). The CCK-8 assay revealed that 6-OHDA caused a notable decrease in cell viability, which was reversed by 1 nM, 10 nM, and 100 nM ghrelin pretreatment (P < 0.01). Unexpectedly, however, the neuroprotective effects of ghrelin were weakened with its increasing concentration, and no protective effects were detected with ghrelin pretreatment at concentrations of over 200 nM (Figure 1B). To assess whether this biological effect was related to GHSR-1a activation, we applied [D-Lys3]-GHRP-6, a GHSR antagonist, at different concentrations. Treatment with 10 μM [D-Lys3]-GHRP-6 significantly abolished the neuroprotective effects of ghrelin (P < 0.01; Additional Figure 1 (443.9KB, tif) ).
Figure 1.
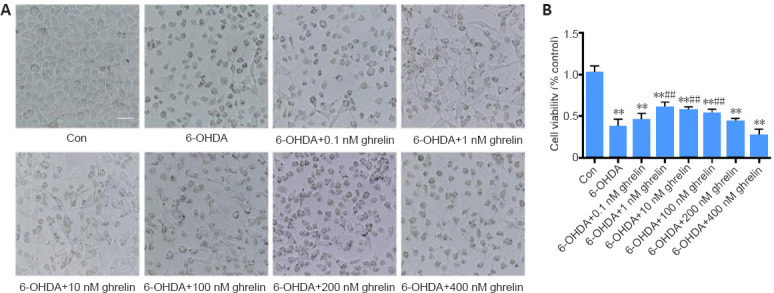
Ghrelin is neuroprotective against 6-OHDA neurotoxicity.
(A) Phase contrast images of SH-SY5Y cells treated with different concentrations of ghrelin (0.1, 1, 10, 100, 200, and 400 nM) for 30 minutes followed by 6-OHDA treatment for 24 hours. Images revealed a notable decrease of adherent cells and more round cells that had lost processes after 6-OHDA treatment. These changes were partially reversed by 1 nM, 10 nM, and 100 nM ghrelin treatment; Scale bar: 50 μm. (B) Cell viability was assessed using Cell Counting Kit-8. The data are shown as the mean ± SD (n = 5; one-way analysis of variance followed by Bonferroni post hoc test). **P < 0.01, vs. control group; ##P < 0.01, vs. 6-OHDA treatment group. 6-OHDA: 6-Hydroxydopamine; con: control; TGIF1: TG-interacting factor 1.
α-Synuclein is upregulated and involved in 6-OHDA-induced injury
SH-SY5Y cells were stimulated with 75 μM 6-OHDA for 24 hours, and were then harvested for qRT-PCR and western blot analyses to evaluate the α-synuclein expression. As shown in Figure 2A and B, 6-OHDA significantly upregulated α-synuclein expression (P < 0.01). To assess whether α-synuclein upregulation was responsible for 6-OHDA-induced neurotoxicity, α-synuclein was downregulated using siRNA. The transfection rate was assessed by qRT-PCR (Figure 2C), and cell toxicity was evaluated using the Annexin/PI apoptosis assay. As shown in Figure 2D, α-synuclein siRNA significantly lowered the percentage of apoptotic cells induced by 6-OHDA (to approximately 15%), suggesting that α-synuclein upregulation is at least partially responsible for 6-OHDA-induced neurotoxicity.
Figure 2.
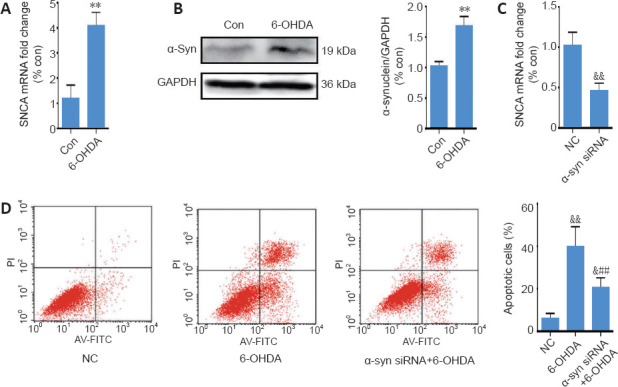
6-OHDA exerts neurotoxicity by upregulating α-synuclein expression.
(A) Reverse transcription quantitative polymerase chain reaction analysis of SNCA mRNA levels in SH-SY5Y cells after treatment with 6-OHDA for 24 hours; GAPDH served as the internal control. (B) Representative western blot and quantification data of α-synuclein in SH-SY5Y cells treated with 6-OHDA for 24 hours. (C) Reverse transcription quantitative polymerase chain reaction analysis of SNCA mRNA levels in SH-SY5Y cells transfected with SNCA siRNA for 24 hours. (D) Cell apoptosis was assessed using Annexin V-FITC/propidium iodide. The data are shown as the mean ± SD (n = 5; A–C: Student’s t-test; D: one-way analysis of variance followed by Bonferroni post hoc test); **P < 0.01, vs. control group; ##P < 0.01, vs. 6-OHDA treatment group; &P < 0.05; &&P < 0.01, vs. NC group. 6-OHDA: 6-Hydroxydopamine; Con: control; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; NC: negative control; siRNA: small interfering RNA; SNCA: the gene encoding α-synuclein; TGIF1: TG-interacting factor 1; α-syn: α-synuclein.
LincRNA-p21 is upregulated in a 6-OHDA-induced PD model and mediates α-synuclein expression and 6-OHDA-induced neurotoxicity
Treatment for 24 hours with 6-OHDA induced a nearly four-fold upregulation of lincRNA-p21 (Figure 3A). To further explore the role of lincRNA-p21 upregulation in 6-OHDA-induced neurotoxicity, we downregulated lincRNA-p21 using siRNA; the transfection rate was confirmed by qRT-PCR 48 hours after transfection (Figure 3B). Transfected cells were then treated with 6-OHDA for 24 hours, and western blot assay revealed that lincRNA-p21 siRNA suppressed the 6-OHDA-induced upregulation of α-synuclein (P < 0.01; (Figure 3C). Furthermore, the Annexin/PI apoptosis assay demonstrated that 6-OHDA treatment resulted in a marked increase in the number of apoptotic cells, and that inhibiting lincRNA-p21 alleviated the 6-OHDA-induced apoptosis (P < 0.01; Figure 3D).
Figure 3.
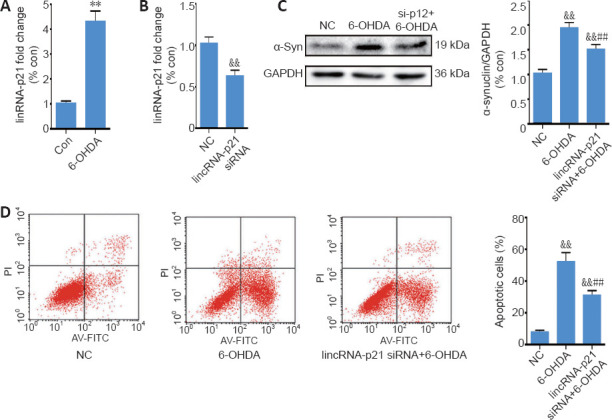
6-OHDA-induced lincRNA-p21 upregulation modulates α-synuclein expression and neurotoxicity.
(A) Reverse transcription quantitative polymerase chain reaction analysis of lincRNA-p21 expression in SH-SY5Y cells treated with 6-OHDA for 24 hours; GAPDH served as the internal control. (B) Reverse transcription quantitative polymerase chain reaction analysis of lincRNA-p21 levels in SH-SY5Y cells transfected with lincRNA-p21 siRNA for 24 hours. (C) Representative western blot and quantification data of α-synuclein in SH-SY5Y cells transfected with lincRNA-p21 siRNA and then treated with 6-OHDA for 24 hours. (D) Cell apoptosis was assessed using Annexin V-FITC/propidium iodide. The data are shown as the mean ± SD (n = 5; A, B: Student’s t-test; C, D: one-way analysis of variance followed by Bonferroni post hoc test); **P < 0.01, vs. control group; ##P < 0.01, vs. 6-OHDA treatment group; &&P < 0.01, vs. NC group. 6-OHDA: 6-Hydroxydopamine; Con: control; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; NC: negative control; siRNA: small interfering RNA; TGIF1: TG-interacting factor 1; α-syn: α-synuclein.
Ghrelin reverses the 6-OHDA-induced upregulation of lincRNA-p21 and α-synuclein
Based on the CCK-8 results, SH-SY5Y cells were pretreated with 1 nM ghrelin for 30 minutes followed by co-treatment with 75 μM 6-OHDA for 24 hours. The qRT-PCR results revealed that ghrelin inhibited the 6-OHDA-induced upregulation of lincRNA-p21 (P < 0.01; Figure 4A). Western blot assay further revealed that ghrelin pretreatment downregulated the 6-OHDA-induced expression of α-synuclein in a dose-dependent manner (Figure 4B). α-Synuclein immunostaining also revealed lower levels of α-synuclein with ghrelin pretreatment (Figure 4C).
Figure 4.

Ghrelin can reverse the 6-OHDA-induced upregulation of lincRNA-p21 and α-synuclein.
(A) Reverse transcription quantitative polymerase chain reaction analysis of lincRNA-p21 levels in SH-SY5Y cells treated with 1 nM ghrelin with or without 6-OHDA for 24 hours; GAPDH served as the internal control. (B) Representative western blot and quantification data of α-synuclein in SH-SY5Y cells pretreated with different concentrations of ghrelin for 30 minutes followed by incubation with 6-OHDA for 24 hours. (C) Immunostaining of α-synuclein in SH-SY5Y cells treated with 1 nM ghrelin with or without 6-OHDA for 24 hours (α-synuclein in red, and nuclei in blue). Scale bar: 50 μm. The data are shown as the mean ± SD (n = 5; A, B: one-way analysis of variance followed by Bonferroni post hoc test); **P < 0.01, vs. control group; ##P < 0.01, vs. 6-OHDA treatment group. 6-OHDA: 6-Hydroxydopamine; Con: control; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; TGIF1: TG-interacting factor 1; α-syn: α-synuclein.
6-OHDA-induced upregulation of lincRNA-p21 targets TGIF1 by SMD
To elucidate the pathological role of lincRNA-p21 in the 6-OHDA-induced PD model, a bioinformatics analysis was performed using the bioinformatics database IntaRNA. Both lincRNA-p21 and TGIF1 mRNA have Alu elements within 3′-untranslated regions, and TGIF1 mRNA might be a putative target of lincRNA-p21 in a sequence-specific manner. It has been hypothesized that they may form a STAU1 binding site and bind to STAU1 to activate SMD. The potential binding sequence between lincRNA-p21 and TGIF1 mRNA is shown in Figure 5A. To assess this hypothesis, qRT-PCR was first conducted to examine the expression of TGIF1 in the 6-OHDA-induced PD model. As shown in Figure 5B, in contrast to the upregulation of lincRNA-p21 and α-synuclein, 6-OHDA led to a notable decrease in TGIF1 (P < 0.01). Moreover, inhibiting lincRNA-p21 markedly reversed the downregulation of TGIF1 by 6-OHDA (P < 0.01). The RNA immunoprecipitation assay using anti-STAU1 antibody revealed that lincRNA-p21 siRNA significantly reduced the enrichment of TGIF1 mRNA by the STAU1 antibody (P < 0.01; Figure 5C). In addition, the inhibition of STAU1 by approximately 50% (as confirmed using qRT-PCR, Figure 5D) significantly reversed the 6-OHDA-induced downregulation of TGIF1 (P < 0.01; Figure 5E). Moreover, western blot assay revealed that inhibiting either lincRNA-p21 or STAU1 expression effectively reversed the reduced expression of TGIF1 after 6-OHDA treatment (P < 0.05; Figure 5F). Collectively, these findings suggest that TGIF1 is a target for SMD degradation, which may be promoted by the 6-OHDA-induced upregulation of lincRNA-p21.
Figure 5.
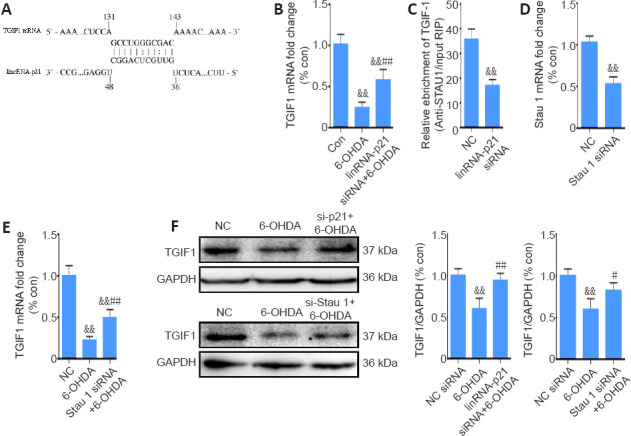
6-OHDA-induced lincRNA-p21 upregulation targets TGIF1 by STAU1-mediated mRNA decay.
(A) The predicted lincRNA-p21 binding site within TGIF13′-untranslated regions. (B) Reverse transcription quantitative polymerase chain reaction analysis of TGIF1 mRNA levels in SH-SY5Y cells transfected with lincRNA-p21 siRNA and treated with 6-OHDA for 24 hours; GAPDH served as the internal control. (C) Interaction of TGIF1 mRNA with STAU1 was detected using the RNA immunoprecipitation assay in SH-SY5Y cells transfected with lincRNA-p21 siRNA. (D) Reverse transcription quantitative polymerase chain reaction analysis of STAU1 mRNA levels in SH-SY5Y cells transfected with STAU1 siRNA for 24 hours. The data are shown as the mean ± SD (n = 5); &&P < 0.01, vs. NC group (Student’s t-test). (E) Reverse transcription quantitative polymerase chain reaction analysis of TGIF1 mRNA levels in SH-SY5Y cells transfected with STAU1 siRNA and treated with 6-OHDA for 24 hours. (F) Representative western blot and quantification data of TGIF1 in SH-SY5Y cells transfected with lincRNA-p21 siRNA or STAU1 siRNA, and treated with 6-OHDA for 24 hours. The data are shown as the mean ± SD (n = 5; one-way analysis of variance followed by Bonferroni post hoc test). &&P < 0.01, vs. NC siRNA group; #P < 0.05, ##P < 0.01, vs. 6-OHDA treatment group. 6-OHDA: 6-Hydroxydopamine; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; siRNA: small interfering RNA; STAU1: double-stranded RNA-binding protein Staufen homolog 1; TGIF1: TG-interacting factor 1. Con: Control; NC; negative control.
Ghrelin modulates α-synuclein by inhibiting lincRNA-p21 expression and preserving TGIF1 expression in a 6-OHDA-induced PD model
To assess whether ghrelin can inhibit α-synuclein expression via TGIF1 modulation, we first assessed the expression of TGIF1 after ghrelin treatment. As shown in Figure 6A, in contrast to the expression of α-synuclein and lincRNA-p21, ghrelin reversed the 6-OHDA-induced downregulation of TGIF1, suggesting that TGIF1 might inhibit α-synuclein expression. By querying the bioinformatics database, a number of binding sites were identified at the promoter region of SNCA with two binding motifs (5′-AAGGAA-3′ and 5′-CAGCTG-3′) of TGIF1, suggesting that α-synuclein may be a direct target of TGIF1 (Additional Figure 2 (1.2MB, tif) ). A dual-luciferase gene reporter assay was then performed to evaluate the predicted binding. As shown in Figure 6B, TGIF1 overexpression markedly increased the luciferase activity in HEK293T cells transfected with pGL4.10-SNCA promoter (WT) compared with cells transfected with the luciferase reporter vector. In contrast, luciferase activity was decreased in cells co-transfected with the pGL4.10-SNCA promoter (MUT) and pcDNA3.1-TGIF1-3FLAG (P < 0.01), suggesting a transcription regulatory role of TGIF1 in α-synuclein expression; unexpectedly, this role was as a transcriptional activator. To further evaluate whether TGIF1 upregulation by ghrelin led to α-synuclein downregulation, SH-SY5Y cells were transfected with TGIF1 siRNA, and transfection was confirmed by qRT-PCR (Figure 6C). Both qRT-PCR and western blot assay revealed that α-synuclein levels were higher in the TGIF1 siRNA + ghrelin + 6-OHDA group compared with the NC siRNA + ghrelin + 6-OHDA group (P < 0.01; Figure 6D and H), suggesting that the inhibitory effects of ghrelin on α-synuclein expression are caused by TGIF1 upregulation. With respect to the luciferase results, an α-synuclein-overexpressing SH-SY5Y cell model was used to further evaluate the transcription regulatory role of TGIF1 on α-synuclein. An opposite effect of TGIF1 siRNA was detected in this model; TGIF1 siRNA induced a further increase in the α-synuclein levels (Additional Figure 3 (661.3KB, tif) ). Together, these findings indicate that TGIF1 might act as a transcriptional activator or repressor depending on the cellular context. To further confirm that ghrelin regulates the expression of TGIF1 and α-synuclein via lincRNA-p21, SH-SY5Y cells were transfected with lincRNA-p21-overexpressing vector, and the overexpression was confirmed using qRT-PCR (Figure 6E). After being transfected for 24 hours with the lincRNA-p21-overexpressing vector, cells were then treated with ghrelin for 30 minutes before the addition of 6-OHDA for 24 hours. lincRNA-p21 overexpression abolished the ghrelin-induced TGIF1 upregulation and led to a notable increase in α-synuclein levels compared with the 6-OHDA + 1 nM ghrelin group (P < 0.01; Figure 6F–H).
Figure 6.

Ghrelin modulates α-synuclein expression by inhibiting lincRNA-p21 expression and preserving TGIF1 levels.
(A) Representative western blot assay and quantification of TGIF1 in SH-SY5Y cells pretreated with different concentrations of ghrelin for 30 minutes followed by incubation with 6-OHDA for 24 hours. (B) Results of the dual-luciferase reporter assay. (C) qRT-PCR analysis of TGIF1 mRNA levels in SH-SY5Y cells transfected with TGIF1 siRNA for 24 hours. (D) qRT-PCR analysis of SNCA mRNA levels in SH-SY5Y cells transfected with TGIF1 siRNA and treated with 6-OHDA with or without the presence of 1 nM ghrelin for 24 hours. (E) qRT-PCR analysis of lincRNA-p21 levels in SH-SY5Y cells transfected with pcDNA-p21 for 24 hours. (F) qRT-PCR analysis of TGIF1 mRNA levels in SH-SY5Y cells transfected with pcDNA-p21 and treated with 6-OHDA with or without the presence of 1 nM ghrelin for 24 hours. (G) qRT-PCR analysis of SNCA mRNA levels in SH-SY5Y cells transfected with pcDNA-p21 and treated with 6-OHDA with or without the presence of 1 nM ghrelin for 24 hours. (H) Representative western blot and quantification data of α-synuclein and TGIF1 in SH-SY5Y cells transfected with TGIF1 siRNA or pcDNA-p21 and treated with 6-OHDA with or without the presence of 1 nM ghrelin for 24 hours. The data are shown as the mean ± SD (n = 5; one-way analysis of variance followed by Bonferroni post hoc test); *P < 0.05, vs. control group; #P < 0.05, ##P < 0.01, vs. 6-OHDA treatment group; &P < 0.05, &&P < 0.01, vs. NC group; ††P < 0.01, vs. 6-OHDA + 1 nM ghrelin group; ^^P < 0.01, vs. pGL4.10-SNCA promoter (WT) + pcDNA3.1-3FLAG group; $$P < 0.01, vs. pGL4.10-SNCA promoter (MUT) + pcDNA3.1-3FLAG group. 6-OHDA: 6-Hydroxydopamine; Con: control; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; Luc: luciferase; NC: negative control; qRT-PCR: reverse transcription quantitative polymerase chain reaction; R-Luc: Renilla luciferase; siRNA: small interfering RNA; SNCA: the gene encoding α-synuclein; TGIF1: TG-interacting factor 1; α-syn: α-synuclein.
Ghrelin exerts its neuroprotective effects via the lincRNA-p21/TGIF1/α-synuclein pathway in a 6-OHDA-induced PD model
To assess whether ghrelin exerts its neuroprotective effects against 6-OHDA via the lincRNA-p21/TGIF1/α-synuclein pathway, SH-SY5Y cells were transfected with the lincRNA-p21-overexpressing vector or TGIF1 siRNA, followed by 1 nM ghrelin pretreatment for 30 minutes and 6-OHDA treatment for 24 hours. Next, the Annexin/PI apoptosis assay was performed to examine cell toxicity. Either the overexpression of lincRNA-p21 or the knockdown of TGIF1 abolished the anti-apoptotic effects of ghrelin in 6-OHDA-treated SH-SY5Y cells (Figure 7). These results suggest that ghrelin protects SH-SY5Y cells against 6-OHDA neurotoxicity by regulating the lincRNA-p21/TGIF1/α-synuclein pathway.
Figure 7.

Ghrelin exerts its neuroprotective effects via the lincRNA-p21/TGIF1/α-synuclein pathway.
SH-SY5Y cells transfected with pcDNA-p21 or TGIF1 siRNA and treated with 6-OHDA with or without the presence of 1 nM ghrelin for 24 hours. Cell apoptosis was assessed using Annexin V-FITC/propidium iodide. The data are shown as the mean ± SD (n = 5; one-way analysis of variance followed by Bonferroni post hoc test); &&P < 0.01, vs. NC group; #P < 0.05, vs. 6-ODHA group; †P < 0.05, ††P < 0.01, vs. 6-OHDA + 1 nM ghrelin group. 6-OHDA: 6-Hydroxydopamine; NC: negative control; siRNA: small interfering RNA; TGIF1: TG-interacting factor 1.
Discussion
PD is one of the most common neurological disorders, affecting 1–2 individuals per 1000 of the population at any time (Tysnes and Storstein, 2017; Arena et al., 2021; Xie et al., 2021; Zhao et al., 2021). Despite increasing research in the field of PD, there are no disease-modifying treatments. Ghrelin is a neuropeptide that is secreted by gastric cells under the stimulation of hunger. It has been identified as a promising neuroprotective agent in a number of neurological disorders (Jiang et al., 2008; Eslami et al., 2018; Dong et al., 2019). Furthermore, several studies have suggested a possible link between ghrelin and PD. Song et al. (2017) reported that PD patients have lower plasma ghrelin levels than healthy individuals, and their ghrelin response to meals is also impaired. In addition, downregulating GHSR in dopaminergic neurons in the substantia nigra can evoke PD-like motor dysfunction (Suda et al., 2018). Moreover, ghrelin enhances the firing rate of dopaminergic neurons in the substantia nigra pars compacta and promotes dopamine availability during the course of neurodegeneration (Andrews et al., 2009). In the current study, we propose a new mechanism by which ghrelin exerts neuroprotective effects in the 6-OHDA-induced SH-SY5Y cell model. Ghrelin was able to reverse the 6-OHDA-induced upregulation of lincRNA-p21, suppress lincRNA-p21-mediated TGIF1 degradation via SMD, and inhibit α-synuclein expression.
The aberrant expression and aggregation of α-synuclein is a key feature of PD. α-Synuclein is a small, cytosolic protein consisting of 140 amino acid residues. It is located in presynaptic nerve terminals and is widely expressed throughout the brain (Hasegawa et al., 2016). Multiplications and various mutations in SNCA, the gene encoding α-synuclein, have been proposed to be related to PD progression and severity (Devine et al., 2011). In addition, at both the mRNA and protein levels, α-synuclein expression is increased in sporadic PD brains (Murphy et al., 2014). So far, however, the role of α-synuclein in PD remains controversial. Overexpressing α-synuclein can increase reactive oxygen species levels, decrease dopaminergic neurotransmission, and cause signs of motor deficits that are associated with PD (Hansen et al., 2013; Butler et al., 2017; Perfeito et al., 2017). In the present study, we consistently observed a marked upregulation of α-synuclein in our 6-OHDA-induced PD model. Furthermore, inhibiting α-synuclein expression by around 50% greatly alleviated the neurotoxic effects of 6-OHDA, thus supporting a pathological role of α-synuclein upregulation in PD. However, in a previous study, silencing α-synuclein in mature nigral neurons led to rapid neuroinflammation and toxicity (Benskey et al., 2018), while knockdown of α-synuclein by 35% did not affect motor function or lead to nigral dopaminergic neuron degeneration. Long-term inhibition of endogenous α-synuclein expression in the substantia nigra for 12 months does not cause neurodegeneration and may even confer neuroprotection in rotenone-exposed rats (Zharikov et al., 2015, 2019). Because α-synuclein exerts physiological functions, such as mediating neurotransmitter release, enhancing ATP synthesis efficiency, and modulating DNA repair (Ludtmann et al., 2016; Logan et al., 2017; Schaser et al., 2019), the degree of α-synuclein knockdown, the kinetics of knockdown, and the targeted cell type may affect outcomes.
LncRNAs can regulate gene expression at transcriptional, post-transcriptional, and epigenetic levels (Mercer and Mattick, 2013). In PD, a number of dysregulated lncRNAs have been identified to mediate disease progression. In the current study, 6-OHDA induced a marked increase in lincRNA-p21, which has also been reported in lipopolysaccharide- and MPP+-induced PD models (Ye et al., 2018; Ding et al., 2019). Inhibiting lincRNA-p21 not only confers neuroprotection against 6-OHDA, but also decreases α-synuclein levels. In an MPP+-induced PD model, Xu et al. (2018) reported an indirect regulatory role of lincRNA-p21 on α-synuclein expression via sponging miR-1277-5p. Herein, we identified a novel mechanism of lincRNA-p21 that involved activating SMD and affecting the stability of TGIF1, which may further inhibit α-synuclein expression. Moreover, overexpressing lincRNA-p21 led to α-synuclein upregulation and abolished the neuroprotective effects of ghrelin. Given that lincRNA-p21 is a transcriptional target of p53 and can feed back to enhance p53 transcriptional activity (Wu et al., 2014), and α-synuclein is a novel transcription target of p53 (Duplan et al., 2016), there may be more than one mechanism that mediates the regulatory effects of lincRNA-p21 on α-synuclein expression. Collectively, these findings point to the crucial role of lincRNA-p21 in mediating α-synuclein expression and PD pathogenesis, and indicate that ghrelin confers at least partial neuroprotection against 6-OHDA via the inhibition of lincRNA-p21 expression.
One notable finding in the current study was that we identified TGIF1 as a novel regulator of α-synuclein expression. TGIF1 is a homeodomain transcription factor that belongs to the three-amino-acid-loop extension superfamily. It was first identified by its ability to bind the retinol-binding protein 2 (RBP2) gene and reduce activation by retinoid X receptors (Bertolino et al., 1995). It can also inhibit the transforming growth factor β pathway by binding mothers against decapentaplegic homolog 2 (SMAD2) and recruiting corepressors such as mSin3 (Seo et al., 2004). Studies have revealed that TGIF1 exerts crucial functions in the central nervous system (Sha et al., 2012; Fu et al., 2019), and mutations of TGIF1 are closely associated with holoprosencephaly (Gripp et al., 2000). In the present study, TGIF1 was identified as a transcriptional regulator of α-synuclein in PD; the luciferase assay revealed several binding sites of TGIF1 on the promoter region of SNCA. However, in contrast to our luciferase assay results that suggested TGIF1 as a transcriptional activator, we found that inhibiting TGIF1 yielded higher levels of 6-OHDA-induced α-synuclein. Moreover, in our α-synuclein-overexpressing SH-SY5Y cell model, TGIF1 inhibition led to lower levels of α-synuclein, suggesting a dual transcriptional regulation. In light of the literature, a number of transcription factors may exert dual roles on transcriptional regulation, depending on the cellular context and their interaction with other regulatory elements (Kuhn and Grummt, 1992; Weill et al., 2003; Palavecino-Ruiz et al., 2017); this might explain our contrasting findings. It is possible that TGIF1 exerts two opposite transcriptional regulatory functions through a mechanism that is yet to be defined. Because TGIF1 siRNA abolished the neuroprotective effects of ghrelin in our 6-OHDA-induced PD model, it is likely that TGIF1 plays a protective role in this situation. Similarly, Yeger-Lotem et al. (2009) reported that TGIF1 overexpression can promote cellular survival in a yeast model of α-synuclein toxicity. To date, there have been very few studies on the role of TGIF1 in PD; thus, further research is necessary.
There are some limitations in the current study. First, we only collected cells 24 hours after 6-OHDA treatment, when nearly half of the cells were impaired. Considering that α-synuclein has a number of different physiological functions, its upregulation during the early stage (when fewer cells are injured) might be a compensatory protective mechanism; this remains to be revealed. Second, we mainly focused on the effects of ghrelin on α-synuclein synthesis. However, α-synuclein monomers can further form oligomers or even fibrils to exert toxic effects, and further study is required to evaluate whether ghrelin affects the formation of these aggregates. Because the pathogenesis of PD remains unclear, future in vivo studies using different PD models will be necessary to fully elucidate the functions of ghrelin in PD.
In conclusion, ghrelin was able to attenuate 6-OHDA-induced neurotoxicity by regulating the lincRNA-p21/TGIF1/α-synuclein pathway. The present study highlighted a pathological role of lincRNA-p21 in PD development, and elucidated a regulatory relationship between TGIF1 and α-synuclein. Because the pathogenesis of PD remains elusive, the effects of ghrelin and the regulatory mechanisms of the effects of TGIF1 on α-synuclein require further investigation using different PD models. Overall, the findings herein provide novel targets for developing PD therapies and a theoretical basis of ghrelin for future clinical applications.
Additional files:
Additional Figure 1 (443.9KB, tif) : The neuroprotective effect of ghrelin is dependent on its receptor activation.
The neuroprotective effect of ghrelin is dependent on its receptor activation.
SH-SY5Y cells were pretreated with 1 nM ghrelin with or without co-treatment with different concentrations of growth hormone secretagogue receptor (GHSR) inhibitor, [D-Lys3]-GHRP-6. Cell viability was assessed with CCK-8. The data were shown as the mean±SD (n=5, one-way analysis of variance followed by Bonferroni post hoc test); **P < 0.01, vs. control group; ##P < 0.01, vs. 6-OHDA treatment group;$$P < 0.01, vs. 6-OHDA+ghrelin (1 nM) group;$$P < 0.01, vs. 6-OHDA+ghrelin (1 nM) group.
Additional Figure 2 (1.2MB, tif) : The predicted binding sites of TG-interacting factor 1 (TGIF1) with the promoter region of SNCA.
The predicted binding sites of TG-interacting factor 1 (TGIF1) with the promoter region of SNCA.
By querying the bioinformatics database, a number of binding sites at the promoter region of SNCA with two binding motifs (5'-3' AAGGAA and CAGCTG) of TGIF1 were identified (highlighted).
Additional Figure 3 (661.3KB, tif) : TG-interacting factor 1 (TGIF1) functions as a transcription regulator of α-synuclein.
TG-interacting factor 1 (TGIF1) functions as a transcription regulator of α-synuclein.
(A) Representative western blot of α-synuclein in lentivirus-mediated α-synuclein overexpressing SH-SY5Y cell model. (B) Representative western blot of TGIF1 and α-synuclein in lentivirus-mediated α-synuclein overexpressing SH-SY5Y cell model transfected with TGIF1 siRNA for 48 h.
Footnotes
C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editors: Gardner B, Haase R, Qiu Y, Song LP; T-Editor: Jia Y
Chinese Library Classification No. R452; R363; R364
Conflicts of interest:The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support:This work was supported by the National Natural Science Foundation of China, No. 81901417 (to XH); the Natural Science Foundation Doctoral Research Initiation Plan of Liaoning Province of China, No. 2019-BS-287 (to XH); the China Postdoctoral Science Foundation, No. 2019M661173 (to XH). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement:The current study is an in vitro study based on commercial cell lines without any involvement of animal or human object. Thus the ethical approval is not needed.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Funding:This work was supported by the National Natural Science Foundation of China, No. 81901417 (to XH); the Natural Science Foundation Doctoral Research Initiation Plan of Liaoning Province of China, No. 2019-BS-287 (to XH); the China Postdoctoral Science Foundation, No. 2019M661173 (to XH).
References
- 1.Arena G, Modjtahedi N, Kruger R. Exploring the contribution of the mitochondrial disulfide relay system to Parkinson’s disease: the PINK1/CHCHD4 interplay. Neural Regen Res. 2021;16:2222–2224. doi: 10.4103/1673-5374.310679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews ZB, Erion D, Beiler R, Liu ZW, Abizaid A, Zigman J, Elsworth JD, Savitt JM, DiMarchi R, Tschoep M, Roth RH, Gao XB, Horvath TL. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci. 2009;29:14057–14065. doi: 10.1523/JNEUROSCI.3890-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benskey MJ, Sellnow RC, Sandoval IM, Sortwell CE, Lipton JW, Manfredsson FP. Silencing alpha synuclein in mature nigral neurons results in rapid neuroinflammation and subsequent toxicity. Front Mol Neurosci. 2018;11:36. doi: 10.3389/fnmol.2018.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertolino E, Reimund B, Wildt-Perinic D, Clerc RG. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J Biol Chem. 1995;270:31178–31188. doi: 10.1074/jbc.270.52.31178. [DOI] [PubMed] [Google Scholar]
- 5.Beynon AL, Brown MR, Wright R, Rees MI, Sheldon IM, Davies JS. Ghrelin inhibits LPS-induced release of IL-6 from mouse dopaminergic neurones. J Neuroinflammation. 2013;10:40. doi: 10.1186/1742-2094-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonet-Ponce L, Saez-Atienzar S, da Casa C, Sancho-Pelluz J, Barcia JM, Martinez-Gil N, Nava E, Jordan J, Romero FJ, Galindo MF. Rotenone induces the formation of 4-hydroxynonenal aggresomes. Role of ROS-mediated tubulin hyperacetylation and autophagic flux disruption. Mol Neurobiol. 2016;53:6194–6208. doi: 10.1007/s12035-015-9509-3. [DOI] [PubMed] [Google Scholar]
- 7.Butler B, Sambo D, Khoshbouei H. Alpha-synuclein modulates dopamine neurotransmission. J Chem Neuroanat. 2017;83-84:41–49. doi: 10.1016/j.jchemneu.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiba-Falek O, Lopez GJ, Nussbaum RL. Levels of alpha-synuclein mRNA in sporadic Parkinson disease patients. Mov Disord. 2006;21:1703–1708. doi: 10.1002/mds.21007. [DOI] [PubMed] [Google Scholar]
- 9.Devine MJ, Gwinn K, Singleton A, Hardy J. Parkinson’s disease and alpha-synuclein expression. Mov Disord. 2011;26:2160–2168. doi: 10.1002/mds.23948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding XM, Zhao LJ, Qiao HY, Wu SL, Wang XH. Long non-coding RNA-p21 regulates MPP(+)-induced neuronal injury by targeting miR-625 and derepressing TRPM2 in SH-SY5Y cells. Chem Biol Interact. 2019;307:73–81. doi: 10.1016/j.cbi.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 11.Dong R, Chen M, Liu J, Kang J, Zhu S. Temporospatial effects of acyl-ghrelin on activation of astrocytes after ischaemic brain injury. J Neuroendocrinol. 2019;31:e12767. doi: 10.1111/jne.12767. [DOI] [PubMed] [Google Scholar]
- 12.Duplan E, Giordano C, Checler F, Alves da Costa C. Direct alpha-synuclein promoter transactivation by the tumor suppressor p53. Mol Neurodegener. 2016;11:13. doi: 10.1186/s13024-016-0079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eslami M, Sadeghi B, Goshadrou F. Chronic ghrelin administration restores hippocampal long-term potentiation and ameliorates memory impairment in rat model of Alzheimer’s disease. Hippocampus. 2018;28:724–734. doi: 10.1002/hipo.23002. [DOI] [PubMed] [Google Scholar]
- 14.Fu Y, Wu Z, Guo Z, Chen L, Ma Y, Wang Z, Xiao W, Wang Y. Systems-level analysis identifies key regulators driving epileptogenesis in temporal lobe epilepsy. Genomics. 2019;112:1768–1780. doi: 10.1016/j.ygeno.2019.09.020. [DOI] [PubMed] [Google Scholar]
- 15.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- 17.Gutierrez JA, Solenberg PJ, Perkins DR, Willency JA, Knierman MD, Jin Z, Witcher DR, Luo S, Onyia JE, Hale JE. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl Acad Sci U S A. 2008;105:6320–6325. doi: 10.1073/pnas.0800708105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen C, Bjorklund T, Petit GH, Lundblad M, Murmu RP, Brundin P, Li JY. A novel alpha-synuclein-GFP mouse model displays progressive motor impairment, olfactory dysfunction and accumulation of alpha-synuclein-GFP. Neurobiol Dis. 2013;56:145–155. doi: 10.1016/j.nbd.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 19.Hasegawa M, Nonaka T, Masuda-Suzukake M. alpha-Synuclein: experimental pathology. Cold Spring Harb Perspect Med. 2016;6:a024273. doi: 10.1101/cshperspect.a024273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He X, Yuan W, Li Z, Hou Y, Liu F, Feng J. 6-Hydroxydopamine induces autophagic flux dysfunction by impairing transcription factor EB activation and lysosomal function in dopaminergic neurons and SH-SY5Y cells. Toxicol Lett. 2018;283:58–68. doi: 10.1016/j.toxlet.2017.11.017. [DOI] [PubMed] [Google Scholar]
- 21.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T, Rinn JL. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang H, Li LJ, Wang J, Xie JX. Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp Neurol. 2008;212:532–537. doi: 10.1016/j.expneurol.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Kraus TFJ, Haider M, Spanner J, Steinmaurer M, Dietinger V, Kretzschmar HA. Altered long noncoding RNA expression precedes the course of Parkinson’s disease-a preliminary report. Mol Neurobiol. 2017;54:2869–2877. doi: 10.1007/s12035-016-9854-x. [DOI] [PubMed] [Google Scholar]
- 24.Kuhn A, Grummt I. Dual role of the nucleolar transcription factor UBF: trans-activator and antirepressor. Proc Natl Acad Sci U S A. 1992;89:7340–7344. doi: 10.1073/pnas.89.16.7340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Logan T, Bendor J, Toupin C, Thorn K, Edwards RH. alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci. 2017;20:681–689. doi: 10.1038/nn.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ludtmann MH, Angelova PR, Ninkina NN, Gandhi S, Buchman VL, Abramov AY. Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J Neurosci. 2016;36:10510–10521. doi: 10.1523/JNEUROSCI.1659-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20:300–307. doi: 10.1038/nsmb.2480. [DOI] [PubMed] [Google Scholar]
- 29.Morgan AH, Rees DJ, Andrews ZB, Davies JS. Ghrelin mediated neuroprotection - A possible therapy for Parkinson’s disease. Neuropharmacology. 2018;136:317–326. doi: 10.1016/j.neuropharm.2017.12.027. [DOI] [PubMed] [Google Scholar]
- 30.Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, Cooper A, Garner B, Halliday GM. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain. 2014;137:834–848. doi: 10.1093/brain/awt367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palavecino-Ruiz M, Bermudez-Moretti M, Correa-Garcia S. Unravelling the transcriptional regulation of Saccharomyces cerevisiae UGA genes: the dual role of transcription factor Leu3. Microbiology. 2017;163:1692–1701. doi: 10.1099/mic.0.000560. [DOI] [PubMed] [Google Scholar]
- 32.Perfeito R, Ribeiro M, Rego AC. Alpha-synuclein-induced oxidative stress correlates with altered superoxide dismutase and glutathione synthesis in human neuroblastoma SH-SY5Y cells. Arch Toxicol. 2017;91:1245–1259. doi: 10.1007/s00204-016-1788-6. [DOI] [PubMed] [Google Scholar]
- 33.Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, Weston LJ, Owen N, Weissman TA, Luna E, Raber J, Luk KC, McCullough AK, Woltjer RL, Unni VK. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep. 2019;9:10919. doi: 10.1038/s41598-019-47227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo SR, Lallemand F, Ferrand N, Pessah M, L’Hoste S, Camonis J, Atfi A. The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J. 2004;23:3780–3792. doi: 10.1038/sj.emboj.7600398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sha L, Kitchen R, Porteous D, Blackwood D, Muir W, Pickard B. SOX11 target genes: implications for neurogenesis and neuropsychiatric illness. Acta Neuropsychiatr. 2012;24:16–25. doi: 10.1111/j.1601-5215.2011.00583.x. [DOI] [PubMed] [Google Scholar]
- 36.Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339:159–166. doi: 10.1016/j.canlet.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 37.Song N, Wang W, Jia F, Du X, Xie A, He Q, Shen X, Zhang J, Rogers JT, Xie J, Jiang H. Assessments of plasma ghrelin levels in the early stages of parkinson’s disease. Mov Disord. 2017;32:1487–1491. doi: 10.1002/mds.27095. [DOI] [PubMed] [Google Scholar]
- 38.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suda Y, Kuzumaki N, Sone T, Narita M, Tanaka K, Hamada Y, Iwasawa C, Shibasaki M, Maekawa A, Matsuo M, Akamatsu W, Hattori N, Okano H, Narita M. Down-regulation of ghrelin receptors on dopaminergic neurons in the substantia nigra contributes to Parkinson’s disease-like motor dysfunction. Mol Brain. 2018;11:6. doi: 10.1186/s13041-018-0349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tysnes OB, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm (Vienna) 2017;124:901–905. doi: 10.1007/s00702-017-1686-y. [DOI] [PubMed] [Google Scholar]
- 41.Uslu V, Petretich M, Ruf S, Langenfeld K, Fonseca N, Marioni J, Spitz F. Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat Genet. 2014;46:753–758. doi: 10.1038/ng.2971. [DOI] [PubMed] [Google Scholar]
- 42.van der Lely AJ, Tschop M, Heiman ML, Ghigo E. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004;25:426–457. doi: 10.1210/er.2002-0029. [DOI] [PubMed] [Google Scholar]
- 43.Weill L, Shestakova E, Bonnefoy E. Transcription factor YY1 binds to the murine beta interferon promoter and regulates its transcriptional capacity with a dual activator/repressor role. J Virol. 2003;77:2903–2914. doi: 10.1128/JVI.77.5.2903-2914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, Cai Y, Huang H, Yang Y, Liu Y, Xu Z, He D, Zhang X, Hu X, Pinello L, Zhong D, He F, Yuan GC, Wang DZ, Zeng C. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation. 2014;130:1452–1465. doi: 10.1161/CIRCULATIONAHA.114.011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie WJ, Xia TJ, Zhou QY, Liu YJ, Gu XP. Role of microglia-mediated neuronal injury in neurodegenerative diseases. Zhongguo Zuzhi Gongcheng Yanjiu. 2021;25:1109–1115. [Google Scholar]
- 46.Xu X, Zhuang C, Wu Z, Qiu H, Feng H, Wu J. LincRNA-p21 inhibits cell viability and promotes cell apoptosis in Parkinson’s disease through activating alpha-synuclein expression. Biomed Res Int. 2018;2018:8181374. doi: 10.1155/2018/8181374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye Y, He X, Lu F, Mao H, Zhu Z, Yao L, Luo W, Sun X, Wang B, Qian C, Zhang Y, Lu G, Zhang S. A lincRNA-p21/miR-181 family feedback loop regulates microglial activation during systemic LPS- and MPTP-induced neuroinflammation. Cell Death Dis. 2018;9:803. doi: 10.1038/s41419-018-0821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeger-Lotem E, Riva L, Su LJ, Gitler AD, Cashikar AG, King OD, Auluck PK, Geddie ML, Valastyan JS, Karger DR, Lindquist S, Fraenkel E. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat Genet. 2009;41:316–323. doi: 10.1038/ng.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao J, Kumar M, Sharma J, Yuan Z. Arbutin effectively ameliorates the symptoms of Parkinson’s disease: the role of adenosine receptors and cyclic adenosine monophosphate. Neural Regen Res. 2021;16:2030–2040. doi: 10.4103/1673-5374.308102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zharikov A, Bai Q, De Miranda BR, Van Laar A, Greenamyre JT, Burton EA. Long-term RNAi knockdown of alpha-synuclein in the adult rat substantia nigra without neurodegeneration. Neurobiol Dis. 2019;125:146–153. doi: 10.1016/j.nbd.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zharikov AD, Cannon JR, Tapias V, Bai Q, Horowitz MP, Shah V, El Ayadi A, Hastings TG, Greenamyre JT, Burton EA. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J Clin Invest. 2015;125:2721–2735. doi: 10.1172/JCI64502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The neuroprotective effect of ghrelin is dependent on its receptor activation.
SH-SY5Y cells were pretreated with 1 nM ghrelin with or without co-treatment with different concentrations of growth hormone secretagogue receptor (GHSR) inhibitor, [D-Lys3]-GHRP-6. Cell viability was assessed with CCK-8. The data were shown as the mean±SD (n=5, one-way analysis of variance followed by Bonferroni post hoc test); **P < 0.01, vs. control group; ##P < 0.01, vs. 6-OHDA treatment group;$$P < 0.01, vs. 6-OHDA+ghrelin (1 nM) group;$$P < 0.01, vs. 6-OHDA+ghrelin (1 nM) group.
The predicted binding sites of TG-interacting factor 1 (TGIF1) with the promoter region of SNCA.
By querying the bioinformatics database, a number of binding sites at the promoter region of SNCA with two binding motifs (5'-3' AAGGAA and CAGCTG) of TGIF1 were identified (highlighted).
TG-interacting factor 1 (TGIF1) functions as a transcription regulator of α-synuclein.
(A) Representative western blot of α-synuclein in lentivirus-mediated α-synuclein overexpressing SH-SY5Y cell model. (B) Representative western blot of TGIF1 and α-synuclein in lentivirus-mediated α-synuclein overexpressing SH-SY5Y cell model transfected with TGIF1 siRNA for 48 h.