

Abstract
Aims
A mutation in the GBA1 gene is the most common genetic risk factor for developing Parkinson's disease. GBA1 encodes the lysosomal enzyme glucosylceramidase beta (glucocerebrosidase, GCase) and mutations decrease enzyme activity. LTI‐291 is an allosteric modulator of GCase, enhancing its activity. These first‐in‐human studies evaluated the safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple ascending doses of LTI‐291 in healthy volunteers.
Methods
In the single ascending dose (SAD) study, 40 healthy volunteers were randomly assigned to LTI‐291 (n = 8 per dose level) or placebo (n = 2 per dose level). Single doses of 3, 10, 30 and 90 mg LTI‐291 were investigated. In the multiple ascending dose (MAD) study, 40 healthy middle‐aged or elderly volunteers were randomly assigned to LTI‐291 (n = 8 per dose level) or placebo (n = 2 per dose level). Fourteen consecutive daily doses of 3, 10, 30 and 60 mg LTI‐291 or placebo were administered. In both the SAD and MAD studies, glycosphingolipid levels were measured and a test battery of neurocognitive tasks was performed.
Results
LTI‐291 was generally well tolerated and no deaths or treatment‐related SAEs occurred and no subject withdrew from a study due to AEs. C max, AUC0–24 and AUC0‐inf increased in a dose proportional manner. The median half‐life was 28.0 hours after multiple dosing. No dose‐dependent glycosphingolipid changes occurred. No neurocognitive adverse effects were detected.
Conclusions
These first‐in‐human studies demonstrated that LTI‐291 was well tolerated when given orally once daily for 14 consecutive days. This supports the continued clinical development and the exploration of LTI‐291 effects in a GBA1‐mutated Parkinson population.
Keywords: clinical trial, disease modification, GBA, Parkinson's disease, phase 1
What is already known about this subject
Parkinson's disease is the second most common neurodegenerative disorder.
Current therapies only alleviate motor symptoms, without addressing non‐motor symptoms or influencing disease progression.
A mutation in the GBA1 gene, typically as a heterozygous mutation in one allele, affecting 4–20% of patients, is the most common genetic risk factor for developing Parkinson's disease known to date and therefore a potential target for a disease‐modifying therapy.
What this study adds
LTI‐291, a glucocerebrosidase enhancer, was shown to be central nervous system penetrant and is intended for use in patients with Parkinson's disease carrying a GBA1 mutation.
LTI‐291 was generally well tolerated in healthy volunteers.
LTI‐291 has favourable pharmacokinetic characteristics for daily single dosing.
1. INTRODUCTION
Parkinson's disease (PD; MIM: 168600) is the second most common neurodegenerative disorder, with a multifactorial disease aetiology, consisting of both environmental and genetic risk factors and their interaction.1 PD is clinically characterized by both motor and non‐motor symptoms, such as mood, sleep, autonomic, cognitive and olfactory disturbances. Currently available pharmacotherapy is primarily directed at the dopaminergic system and primarily alleviates motor symptomatology, without addressing non‐motor symptoms or otherwise influencing disease progression. This leaves a large unmet need for disease‐modifying therapy.
A potential target in PD is the lysosomal enzyme glucosylceramidase beta (glucocerebrosidase, GCase; EC 3.2.1.45), encoded by the GBA1 gene (MIM: 606463). In the mutated state this is the largest genetic risk factor for developing PD known to date, referred to as GBA‐PD.2 In most populations, 4–12% of PD patients carry a heterozygous GBA1 mutation and in Ashkenazi Jewish PD patients, this is approximately 20%.3, 4 For PD patients, odds ratios (OR) of having a GBA1 mutation compared to people without PD usually vary between 2 and 7.3, 4, 5, 6 GBA‐PD on average presents at a younger age with a higher prevalence of non‐motor symptoms, however with a high variability between individual patients.7, 8
GCase is involved in intralysosomal metabolism of glycosphingolipids (GSLs), a class of lipids, essential for membrane and other cellular functions.9 It hydrolyses the glycosphingolipid glucosylcerebroside (GluCer) into glucose and ceramide. In the presence of a GCase mutation, this reaction rate is decreased and this may lead to a lower flux of glycolipid metabolism within the lysosome. In cases of GCase deficiency, the enzyme acid ceramidase can convert GluCer into glucosylsphingosine (GluSph).10 GluSph is postulated to have toxic effects and can also be metabolized by GCase;10 however, the turnover rate is much lower than for GluCer.11 The immediate precursor to GluCer in the lysosomal degradation pathway is lactosylceramide (LacCer). It is still not fully understood how these molecules are affected in GBA‐PD, with conflicting results on whether these accumulate or not.12, 13, 14, 15
LTI‐291 is a central nervous system (CNS) penetrable small‐molecule GCase allosteric modulator, enhancing GCase activity, as a potential treatment of GBA‐PD. Preclinically, activation of GCase using LTI‐291 was shown in recombinant wild type and mutant (the two common variants p.Asn409Ser and p.Glu365Lys, commonly referred to as N370S and E326K) human GCase, using an in vitro micellar environment and the artificial fluorescent 4‐MUG substrate. LTI‐291 at concentrations of 0.1 and 1.0 μM increased wild type GCase activity by 25% and 130%, respectively. GCase resides on the inner membrane surface of the lysosome, and the substrate GluCer resides within the lysosomal membrane. LTI‐291 requires a lipidic surface in which to form an active complex with the GCase enzyme. In vitro, this surface was supplied by micelles, while in vivo activation of GCase probably depends on complex formation within the lysosomal lipid membrane. In transgenic mouse models, in cultured human‐induced pluripotent stem cell‐derived neurons and in ex vivo peripheral blood mononuclear cells (PBMCs) from idiopathic PD and GBA‐PD patients, GluCer levels decreased by 10–30% after treatment with LTI‐291 (observed in GluCer isoforms: C22:0, C22:1, C24:0, C24:1; not observed in GluCer C18:0 and C20:0). Based on the transgenic mouse data, the minimally efficacious plasma concentration lies between 400 and 2500 nM. Higher doses showed comparable reduction of GluCer (~−20%) in transgenic mice, suggesting that once normalization of the pathway flux is achieved, there is a flattening of the dose response. Modelling was used to predict that a dose of 6 mg LTI‐291 was needed to achieve a C max of 1000 nM (~360 ng/mL) in humans. The no‐observed‐adverse‐effect level (NOAEL) was determined in rat as the most sensitive species, with first observed adverse effects of frequent salivation and vomiting at a human equivalent dose of 580 mg per day. Starting dose was determined at 3 mg per day, expected to be pharmacologically inactive and 19‐fold lower than the maximum recommended starting dose (MRSD) of approximately 58 mg. Ascending doses of 10 mg, 30 mg, 60 mg and 90 mg were expected to be pharmacologically active.
GSLs constitute a vast and dynamic network, in which local disturbances can cause multiple changes within the network.9 GluCer, GluSph and LacCer, as proximal molecules to GCase, were investigated as potential biomarkers in various matrices. Plasma is most conveniently obtained, whereas peripheral blood mononuclear cells (PBMCs), which are more labour intensive to obtain, may better reflect the site of action, namely the lysosome. Cerebrospinal fluid (CSF) was included as matrix most proximal to the brain, despite the limited abundance of lipid substances in this hydrophilic fluid. Healthy volunteers are expected to have normal GCase activity and normal GSL levels. It is hypothesized that LTI‐291 may increase the flux through the GSL metabolism by activating GCase and thereby potentially transiently change normal glycosphingolipid levels in healthy volunteers. GSLs were measured to assess potential transient changes and as safety indicator to prevent unexpected unwanted changes. This paper describes the first‐in‐human doses of LTI‐291, assessing safety, tolerability, pharmacokinetics (PK) and pharmacodynamics, in healthy volunteers in a single ascending dose (SAD) study and in healthy middle‐aged volunteers in a multiple ascending dose (MAD) study.
2. METHODS
Both the SAD and the MAD study were randomized, double‐blind and placebo‐controlled. The studies were approved by the Independent Ethics Committee of the Foundation “Evaluation of Ethics in Biomedical Research” (Stichting Beoordeling Ethiek Biomedisch Onderzoek), Assen, The Netherlands. Both studies are registered in the Dutch Trial Registry (Nederlands Trial Register, NTR) under study number NTR6598 (SAD) and NTR6705 (MAD). The SAD study took place between August and September 2017 and the MAD study took place between September and December 2017. All subjects signed an informed consent form prior to any study‐related activity.
2.1. Subjects
For the SAD study, healthy men and women of non‐childbearing potential were enrolled for a single oral dose of LTI‐291. Prior concomitant medication was only allowed at the discretion of the investigator and the sponsor. The following dose levels were investigated in ascending order: 3 mg, 10 mg, 30 mg and 90 mg LTI‐291. Treatment was administered as powder in a capsule with 240 mL water. In each cohort, 10 subjects were randomized to receive LTI‐291 or placebo in an 8:2 ratio. Cohort 1 was dosed using a sentinel approach: the first two subjects (1 placebo and 1 active) were randomized and dosed with a 24 hour observation period prior to dosing the remainder of the cohort. The effect of food on PK was investigated in the 10 mg cohort, using a high‐fat breakfast according to Food and Drug Administration (FDA) standards. In that cohort, subjects returned for a second visit for dosing (in the same 8:2 randomization) in the fed state after a wash‐out of at least 1 week following the initial dose.
For the MAD study, healthy middle‐aged and elderly (50–75 years of age) men and women of non‐childbearing potential were enrolled for 14 consecutive daily oral doses of LTI‐291. Prior concomitant medication was only allowed at the discretion of the investigator and the sponsor. The following dose levels were investigated: 3 mg, 10 mg, 30 mg and 60 mg LTI‐291. Treatment was administered as powder in a capsule with 240 mL water. In each dose group, 10 subjects were randomized to receive LTI‐291 or placebo in an 8:2 ratio. Cohort 1 was dosed following a sentinel approach: the first two subjects (1 placebo and 1 active) were randomized and dosed with an observation period equivalent to at least five half‐lives prior to dosing the remainder of the cohort.
For the SAD study, dose escalation took place following thorough review of the safety and tolerability data of at least 24 hours post dose, as well as PK assessments of the preceding dose group. In addition, all available pharmacodynamic data were used in this evaluation. For the MAD study, two dose escalation reviews per cohort were performed: the first review after 7 days of dosing, to initiate the next dose level, and a second review was performed after 14 days of dosing, to confirm continuation of the subsequent cohort.
2.2. Safety
For both studies, a medical screening (medical history, record of prior concomitant medication, subject demographics, height and weight, 12‐lead electrocardiography [ECG], vital signs, routine haematology, biochemistry/electrolytes and urinalysis, urine pregnancy test [for females], virology, urine drug screen, ethanol breath test and physical examination) was performed to assess a subject's eligibility. During study periods, safety was assessed using monitoring of adverse events (AEs), concomitant medication, vital signs, ECG, physical examination and safety chemistry and haematology blood sampling.
2.3. Pharmacokinetics
LTI‐291 levels were measured in K2EDTA plasma and in cerebrospinal fluid (CSF) (MAD only). LTI‐291 levels were measured using a validated high‐performance liquid chromatography with tandem mass spectrometric (LC–MS/MS) detection, by PRA Health Sciences (Assen, the Netherlands). The calibration range was 10.0–20 000 ng/mL with a coefficient of variation of 5.2% in plasma and 0.025–50.0 ng/mL with a coefficient of variation of 4.4% in CSF.
LTI‐291 plasma levels were used for non‐compartmental PK analysis (NCA) in Phoenix 64 build 7.0.0.2535 using WinNonlin 7.0, by Certara QSP (Princeton, NJ, USA). If concentrations were below the assay limit of quantification they were treated as having a concentration of 0 ng/mL prior to the first sample with a measurable concentration or as missing at all other time points. PK NCA was performed on the data from each subject as data permitted. The apparent terminal half‐life was calculated by log‐linear extrapolation of those points determined to be on the apparent terminal phase. The area under the LTI‐291 plasma concentration–time curve (AUC) was calculated from 0 to the last measurement point (AUC0–last). When the terminal phase was sufficiently well characterized and the terminal half‐life was estimated, the AUC from 0 to infinity (AUC0–inf) was derived. The apparent clearance (CL/F) and apparent volume of distribution (Vz/F) were calculated following the first dose, where the AUC0–inf was calculated and following the seventh and fourteenth doses, where the AUC0–24 was calculated. The C max, AUC0–24 and AUC0–inf were plotted against dose level to visually assess dose proportionality. CSF was only collected in the MAD pre‐dose and approximately 4 hours after the fourteenth dose, paired with a plasma sample. Due to this limited sampling, only CSF:plasma ratios were calculated.
2.4. Pharmacodynamics
2.4.1. Glycosphingolipid levels
Biochemical pharmacodynamic markers were measured in K2EDTA plasma, PBMCs and CSF. PBMCs were isolated from venous blood using cell preparation tubes (CPT) containing sodium heparin (Becton Dickinson, NJ, USA) according to manufacturer's instructions. In short, CPTs were centrifuged at 1800g for 30 minutes at room temperature. PBMCs were collected and washed twice with phosphate‐buffered saline (PBS) containing 10% heat‐inactivated human serum (both Gibco, Thermo Scientific, Waltham, MJ, USA). Cells were snap‐frozen at 1 × 107 cells mL−1 in PBS with 0.1% bovine serum albumin (BSA) buffer, and stored at −80°C until analysis. Cells for GluSph analysis were frozen in glass tubes instead of a cryopreservation tube. For CSF collection, a Pencan® 25G atraumatic needle was used. The first mL CSF was discarded to prevent contamination with blood, after which 4 mL was collected in a 15 mL Falcon® tube. CSF was transferred to a glass tube and 0.2% BSA with ascorbic acid added. This was centrifuged at 2000g for 3 minutes at room temperature. Supernatant was transferred to glass tubes, snap frozen and stored at −80°C until shipment for analysis. Time from collection to freezing did not exceed 60 minutes.
GluCer, LacCer and GluSph were measured in K2EDTA plasma and in PBMCs, using a validated LC–MS/MS method, by Ardena Bioanalytical Laboratory (Assen, the Netherlands). The carbon chain of a ceramide group like in GluCer can be of varying length and saturation. For the SAD, both in plasma and PBMCs, concentrations were measured of GluCer C16:0, C18:0, C22:0, C24:0 and C24:1. In plasma, the assay range was 1.00–2500 pmol. In PBMCs, the assay range was 0.0500–12.5 pmol. Biomarker samples were taken pre‐dose and 1, 4, 8 and 24 hours post‐dose.
For the MAD, GluCer and LacCer were measured in plasma, PBMCs and CSF. GluSph was measured in plasma and PBMCs. GluSph is present at too low levels in CSF to be accurately measured. Both in plasma and PBMCs, the same GluCer isomers as in the SAD were measured in the MAD. Additionally, concentrations were measured of LacCer C16:0, C18:0, C20:0, C22:0, C22:1, C24:0 and C24:1. In plasma, the assay range was 1.00–4000 pmol. In PBMCs, the assay range was 0.0500–50.0 pmol. GluSph was measured in plasma with an assay range of 0.0500–10.0 pmol and in PBMCs with an assay range of 0.00500–1.00 pmol. In CSF, GluCer C16:0, C18:0, C22:0, C24:0 and C24:1 were measured and LacCer C16:0, C18:0, C20:0, C22:0, C22:1, C24:0 and C24:1 were measured. The lower limit of quantification (LLOQ) was 0.100 pmol for GluCer C20:0, C22:1, C24:0 and C24:1 and LacCer C20:0, C22:1 and C24:1; 0.300 pmol for GluCer C16:0, C18:0 and C22:0 and LacCer C18:0, C22:0, C24:0 and C24:1; and 4.00 pmol for LacCer C16:0. The upper limit of quantification (ULOQ) was 100 pmol for all analytes. Biomarker samples were taken twice pre‐dose, 2 hours after the seventh dose and 2 hours after the fourteenth (last) dose.
2.4.2. Neurocognitive biomarkers (for safety)
Both in SAD and MAD, the NeuroCart®, a CNS test battery, was used to exclude any adverse effects of LTI‐291 on CNS functioning. The test battery consists of neurophysiological, psychomotor and cognitive tests and has been extensively used previously in clinical drug development.16, 17, 18, 19, 20 In short, measurements consist of saccadic and smooth pursuit eye movements, the adaptive tracking test (a visuo‐motor task sensitive to disturbances in vigilance and attention), the body sway (a test of postural stability), the Bond and Lader test (visual analogue scale [VAS] of alertness, calmness and mood), the Visual Verbal Learning Test (VVLT) (a test of immediate and delayed memory), and pharmaco‐EEG. Tests were performed in a quiet room with ambient illumination with only one subject in the same room (and a research assistant) per session. For the SAD, all measurements were performed at baseline and 1, 3 and 6 hours post‐dose. For the MAD, all measurements were performed at baseline, 1, 2 and 4 hours after the first dose, 2 hours prior to, and 1, 2 and 4 hours after the fourteenth (last) dose. The VVLT, part of the test battery, was only performed at 1 hour post‐dose after first and last dose, to minimize the learning effect for this memory‐based task. All subjects underwent a training session of all CNS tests within 21 days preceding study start to minimize learning effects and to make sure subjects were familiar with the test procedure.
2.5. GBA1 genotyping
The full GBA1 gene was sequenced in all subjects, with a validated method accounting for the GBA1 pseudogene, described in detail elsewhere.21 Considering the low predictive clinical value of having a GBA1 mutation, results of the genotyping were not shared with subjects, unless the subject specifically requested this.
2.6. Statistical analysis
Repetitively measured pharmacodynamic data were analysed with a mixed model analysis of variance (ANOVA) with fixed factors treatment, time and treatment by time, random factor subject and the average pre‐value as covariate. Single measured VVLT data were compared with a one‐way ANOVA with factor treatment (for SAD). Repetitively measured VVLT data without pre‐value were analysed with a mixed model ANOVA with fixed factors treatment, time and treatment by time and random factor subject (for MAD). Single measured CSF data with pre‐value were compared with a one‐way ANOVA with factor treatment and pre‐value as covariate (MAD only).
Biomarkers are measured in an exploratory hypothesis‐generating setting and are therefore not corrected for multiple testing.
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.22
3. RESULTS
3.1. Demographics and baseline characteristics
In the SAD study, a total of 40 healthy subjects received a single dose of LTI‐291 or placebo. The cohorts were comparable regarding mean body mass index (BMI). Age varied both within and between cohorts. In the 90 mg group, three subjects with a heterozygous GBA1 mutation were identified, while in all other subjects no GBA1 mutation could be detected (referred to as wild type). Of the three subjects with a GBA1 mutation, two carried the p.Thr408Met variant and one carried the p.Glu365Lys variant. For many GBA1 mutations it is known that in homozygous state they can potentially cause the lysosomal storage disorder Gaucher's disease (GD). The two currently found variants are both relatively mild and do not cause GD in homozygous state, but are a risk factor for developing Parkinson's disease (in GD literature these mutations are historically referred to as T369M and E326K, respectively).23, 24 Demographic data of the SAD study are summarized in Table 1.
TABLE 1.
SAD study summary of baseline subject characteristics
| SAD demographics | |||||
|---|---|---|---|---|---|
| LTI‐291 or placebo | 3 mg LTI‐291 | 10 mg LTI‐291 | 30 mg LTI‐291 | 90 mg LTI‐291 | Placebo |
| n | 8 | 8 | 8 | 8 | 8 |
| Male/female | 6/2 | 4/4 | 7/1 | 8/0 | 6/2 |
| Age (years), mean (range) | 29.5 (18–56) | 49.5 (22–63) | 27.5 (18–56) | 38.1 (23–64) | 46.0 (30–63) |
| BMI (kg/m2), mean (range) | 23.8 (19.7–27.3) | 23.9 (18.9–28.3) | 23.7 (19–28.6) | 24.9 (19.7–29.8) | 24.2 (22.3–27.6) |
| Race | |||||
| Caucasian | 8 | 6 | 7 | 8 | 7 |
| Black | 0 | 1 | 1 | 0 | 1 |
| Asian | 0 | 1 | 0 | 0 | 0 |
| GBA1 wild type/mutation | 8/0 | 8/0 | 8/0 | 5/3 | 8/0 |
In the MAD study, a total of 39 healthy middle‐aged subjects received 14 consecutive daily doses of LTI‐291 or placebo. One subject randomized to 3 mg LTI‐291 withdrew pre‐dose due to personal reasons and was not replaced. The cohorts were comparable regarding mean age and BMI. Two subjects (one randomized to 10 mg LTI‐291 and one to 60 mg LTI‐291) had a heterozygous GBA1 mutation, both carrying the relatively mild p.Thr408Met variant. Demographic data of the MAD study are summarized in Table 2.
TABLE 2.
MAD study summary of baseline subject characteristics.
| MAD demographics | |||||
|---|---|---|---|---|---|
| LTI‐291 or placebo | 3 mg LTI‐291 | 10 mg LTI‐291 | 30 mg LTI‐291 | 60 mg LTI‐291 | Placebo |
| n | 7 | 8 | 8 | 8 | 8 |
| Male/female | 7/0 | 4/4 | 4/4 | 5/3 | 6/2 |
| Age (years), mean (range) | 65.0 (57–74) | 69.4 (60–75) | 67.3 (57–74) | 65.9 (56–75) | 67.4 (52–73) |
| BMI (kg/m2), mean (range) | 24.9 (22.2–28.1) | 24.9 (21.7–29.5) | 25.4 (19.7–28.7) | 26.7 (23.1–31.9) | 26.1 (22.8–30.8) |
| Race | |||||
| Caucasian | 6 | 7 | 7 | 8 | 8 |
| Asian | 0 | 1 | 0 | 0 | 0 |
| Latino | 1 | 0 | 0 | 0 | 0 |
| Mixed | 0 | 0 | 1a | 0 | 0 |
| GBA1 wild type/mutation | 7/0 | 7/1 | 8/0 | 7/1 | 8/0 |
The subject of mixed race was of Dutch‐Indonesian and German‐Indonesian descent
3.2. Safety and tolerability
Single administrations of LTI‐291 up to the highest dose of 90 mg and 14 consecutive daily administrations of LTI‐291 up to the highest dose of 60 mg were generally well tolerated in healthy volunteers. No serious AEs (SAEs) were reported and no AEs led to discontinuation. No clinically relevant changes in blood chemistry, haematology, urinalysis, vital signs, ECG or CNS tests were identified. In the SAD study, all AEs were mild, except for a single moderate AE, which was considered unlikely related to treatment with LTI‐291 (vasovagal collapse after venipuncture). In the MAD study, dose‐dependent increases in AE frequency could be seen in the nervous, gastrointestinal and musculoskeletal system organ classes. Headache, somnolence and myalgia were relatively highly reported in LTI‐291 dose groups in general and in the 60 mg LTI‐291 dose group specifically compared to placebo. Headache was reported by 39% of all LTI‐291 treated subjects vs 0% of placebo subjects; however, no dose‐dependent increase was observed. None of the reported headaches ameliorated in supine position or otherwise seemed related to a lumbar puncture. Somnolence was reported by 50% of 60 mg LTI‐291 subjects vs 12.5% of placebo subjects; however, for most subjects this was of short duration and no abnormalities were found in CNS measurements that are known to be associated with a decline in arousal. Myalgia was reported by 37.5% of 60 mg LTI‐291 subjects vs 12.5% of placebo subjects. No clinically significant creatine kinase abnormalities were found in the 60 mg group, thereby excluding rhabdomyolysis. These AEs were assessed as possibly related to administration of LTI‐291. Three moderate AEs were reported, all three assessed as unlikely related to LTI‐291 treatment. One subject (3 mg LTI‐291) had a transient increase in liver enzymes after dose 13, preceded by abdominal pain in the upper right quadrant, sweating, increased blood pressure and an urge to move around, assessed as a passing gallstone. In this subject, transaminases were highest pre‐dose on day 14 of dosing (AST 702 U/L [reference range: 0–35 U/L], ALT 994 U/L [reference range: 0–45 U/L], GammaGT 286 U/L [reference range: 0–55 U/L] and Alkaline Phosphatase 143 U/L [reference range: 0–115 U/L]). Total bilirubin was not significantly elevated: 20 umol/L (reference range: 0–17 umol/L), decreased on day 15 (AST 85 U/L, ALT 376 U/L, GammaGT 212 U/L and Alkaline Phosphatase 126 U/L) and normalized at follow‐up. A second subject (10 mg LTI‐291) had hypertension on day 1 of dosing, most likely due to suboptimal use of anti‐hypertensives, which normalized after increasing the prior concomitant medication. A third subject (60 mg LTI‐291) had an asymptomatic urinary tract infection on day 1, treated with antibiotics. An overview of all AEs by treatment is given in Table 3.
TABLE 3.
MAD study adverse events by treatment. All AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 20.0. Light blue rows depict system organ classes and numbers are a summation of all preferred terms in that class. Multiple AEs could be reported by the same subject
| MAD adverse events | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 3 mg LTI‐291 | 10 mg LTI‐291 | 30 mg LTI‐291 | 60 mg LTI‐291 | Placebo | ||||||
| n = 7 | n = 8 | n = 8 | n = 8 | n = 8 | ||||||
| System organ class/preferred term | Events (n) | Subjects (n (%)) | Events (n) | Subjects (n (%)) | Events (n) | Subjects (n (%)) | Events (n) | Subjects (n (%)) | Events (n) | Subjects (n (%)) |
| Any events | 17 | 5 (71.4) | 15 | 6 (75.0) | 19 | 7 (87.5) | 27 | 8 (100.0) | 11 | 5 (62.5) |
| Cardiac disorders | 1 | 1 (14.3) | – | – | – | – | – | – | – | – |
| Bundle branch block right | 1 | 1 (14.3) | – | – | – | – | – | – | – | – |
| Eye disorders | – | – | 1 | 1 (12.5) | – | – | 1 | 1 (12.5) | 1 | 1 (12.5) |
| Ocular hyperaemia | – | – | 1 | 1 (12.5) | – | – | – | – | – | – |
| Vision blurred | – | – | – | – | – | – | 1 | 1 (12.5) | 1 | 1 (12.5) |
| Gastrointestinal disorders | 3 | 3 (42.9) | 1 | 1 (12.5) | 3 | 2 (25.0) | 4 | 2 (25.0) | – | – |
| Abdominal pain upper | 1 | 1 (14.3) | 1 | 1 (12.5) | – | – | 1 | 1 (12.5) | – | – |
| Constipation | 1 | 1 (14.3) | – | – | – | – | – | – | – | – |
| Diarrhoea | 1 | 1 (14.3) | – | – | – | – | – | – | – | – |
| Faeces soft | – | – | – | – | – | – | 1 | 1 (12.5) | – | – |
| Flatulence | – | – | – | – | – | – | 1 | 1 (12.5) | – | – |
| Frequent bowel movements | – | – | – | – | 1 | 1 (12.5) | – | – | – | – |
| Nausea | – | – | – | – | 2 | 1 (12.5) | 1 | 1 (12.5) | – | – |
| General disorders and administration site conditions | 4 | 3 (42.9) | 2 | 2 (25.0) | 2 | 2 (25.0) | 3 | 3 (37.5) | 2 | 2 (25.0) |
| Fatigue | 1 | 1 (14.3) | – | – | – | – | – | – | 1 | 1 (12.5) |
| Feeling cold | – | – | – | – | – | – | 1 | 1 (12.5) | – | – |
| Feeling hot | – | – | 1 | 1 (12.5) | – | – | – | – | – | – |
| Medical device site eczema | – | – | – | – | 1 | 1 (12.5) | – | – | 1 | 1 (12.5) |
| Medical device site erythema | – | – | – | – | – | – | 1 | 1 (12.5) | – | – |
| Medical device site reaction | 2 | 2 (28.6) | – | – | 1 | 1 (12.5) | – | – | – | – |
| Pain | – | – | – | – | – | – | 1 | 1 (12.5) | – | – |
| Puncture site pain | 1 | 1 (14.3) | – | – | – | – | – | – | – | – |
| Vessel puncture site vesicles | – | – | 1 | 1 (12.5) | – | – | – | – | – | – |
| Infections and infestations | 1 | 1 (14.3) | – | – | – | – | 1 | 1 (12.5) | – | – |
| Nasopharyngitis | 1 | 1 (14.3) | – | – | – | – | – | – | – | – |
| Urinary tract infection | – | – | – | – | – | – | 1 | 1 (12.5) | – | – |
| Investigations | 1 | 1 (14.3) | 1 | 1 (12.5) | – | – | 1 | 1 (12.5) | – | – |
| Gamma‐glutamyltransferase increased | – | – | – | – | – | – | 1 | 1 (12.5) | – | – |
| Hepatic enzyme increased | 1 | 1 (14.3) | 1 | 1 (12.5) | – | – | – | – | – | – |
| Musculoskeletal and connective tissue disorders | 1 | 1 (14.3) | 4 | 4 (50.0) | 4 | 2 (25.0) | 9 | 6 (75.0) | 5 | 4 (50.0) |
| Arthralgia | – | – | – | – | 2 | 2 (25.0) | – | – | – | – |
| Back pain | – | – | 2 | 2 (25.0) | – | – | 2 | 2 (25.0) | 2 | 2 (25.0) |
| Muscle fatigue | – | – | 1 | 1 (12.5) | – | – | 1 | 1 (12.5) | – | – |
| Muscle spasms | – | – | – | – | 1 | 1 (12.5) | 2 | 2 (25.0) | – | – |
| Musculoskeletal stiffness | – | – | – | – | – | – | 1 | 1 (12.5) | 1 | 1 (12.5) |
| Myalgia | 1 | 1 (14.3) | – | – | 1 | 1 (12.5) | 3 | 3 (37.5) | 1 | 1 (12.5) |
| Neck pain | – | – | 1 | 1 (12.5) | – | – | – | – | – | – |
| Pain in extremity | – | – | – | – | – | – | – | – | 1 | 1 (12.5) |
| Nervous system disorders | 4 | 3 (42.9) | 3 | 2 (25.0) | 6 | 5 (62.5) | 8 | 5 (62.5) | 3 | 3 (37.5) |
| Dizziness | 1 | 1 (14.3) | – | – | 1 | 1 (12.5) | – | – | 2 | 2 (25.0) |
| Head discomfort | – | – | 1 | 1 (12.5) | – | – | 1 | 1 (12.5) | – | – |
| Headache | 2 | 2 (28.6) | 2 | 2 (25.0) | 5 | 5 (62.5) | 3 | 3 (37.5) | – | – |
| Somnolence | 1 | 1 (14.3) | – | – | – | – | 4 | 4 (50.0) | 1 | 1 (12.5) |
| Renal and urinary disorders | 1 | 1 (14.3) | – | – | – | – | – | – | – | – |
| Polyuria | 1 | 1 (14.3) | – | – | – | – | – | – | – | – |
| Respiratory, thoracic and mediastinal disorders | 1 | 1 (14.3) | 1 | 1 (12.5) | 2 | 1 (12.5) | – | – | – | – |
| Nasal congestion | 1 | 1 (14.3) | 1 | 1 (12.5) | 1 | 1 (12.5) | – | – | – | – |
| Oropharyngeal pain | – | – | – | – | 1 | 1 (12.5) | – | – | – | – |
| Skin and subcutaneous tissue disorders | – | – | 1 | 1 (12.5) | 2 | 1 (12.5) | – | – | – | – |
| Hyperhidrosis | – | – | 1 | 1 (12.5) | 2 | 1 (12.5) | – | – | – | – |
| Vascular disorders | – | – | 1 | 1 (12.5) | – | – | – | – | – | – |
| Hypertension | – | – | 1 | 1 (12.5) | – | – | – | – | – | – |
3.3. Pharmacokinetics
Following oral dosing of LTI‐291 to fasted subjects' plasma concentrations increased immediately with no apparent lag time. The average T max was 2.2 hours (range 1.0–8.2 h) following single oral administration and was similar following multiple administrations at 2.3 hours (range 1.0–8.8 h) (Figure 1 for SAD and Figure 2 for MAD). In the SAD study group, geometric mean half‐life varied between 21.2 and 23.3 hours and plasma concentrations declined in a mono‐ or biphasic manner following C max. Other PK parameters estimated for both SAD and MAD studies are summarized in Tables 4 and 5. In the fed state there was a lag in absorption of LTI‐291 resulting in an average T max of 8.0 hours (range 4.0–12.0 h) vs 1 hour (range 1.0–4.0 h) in the fasted state, but there appeared to be no difference in AUC0–inf, AUC0–last, elimination rate constant (λz) or half‐life between fed and fasted dosing, as assessed in the 10 mg SAD cohort (Figure 1, Table 4). Plasma exposure was dose‐proportional both in single and in multiple doses (Tables 4 and 5). For example, in the SAD study over a 30‐fold dose range (3–90 mg) the C max, AUC0–last and AUC0–inf all increased in a dose‐proportional manner. Steady state plasma concentrations were reached after 7 days of daily dosing, resulting in an approximate 1.8‐fold increase in C max and 2.5‐fold increase in AUC0–24. The mean CSF:plasma concentration ratios ranged from 0.0124 to 0.0134 at 4–5 hours after the 14th dose and were similar at all dose levels (Table 6). Over the 20‐fold dose range (3–60 mg) the CSF concentrations increased in a dose‐proportional manner relative to the plasma concentrations.
FIGURE 1.
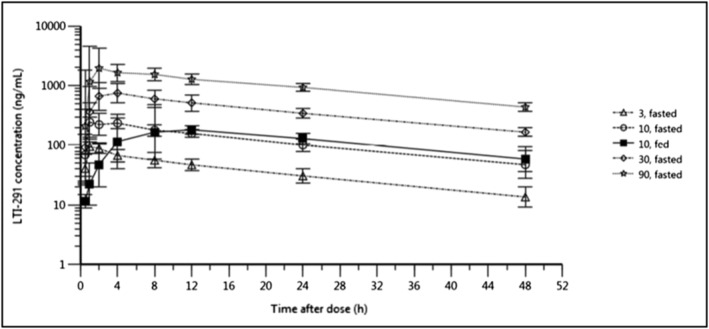
SAD study: Geometric mean (± geometric SD) plasma concentration of LTI‐291 against time (h) after dose following a single oral dose of 3, 10, 30 and 90 mg LTI‐291 in the fasted state and 10 mg LTI‐291 in the fed state
FIGURE 2.
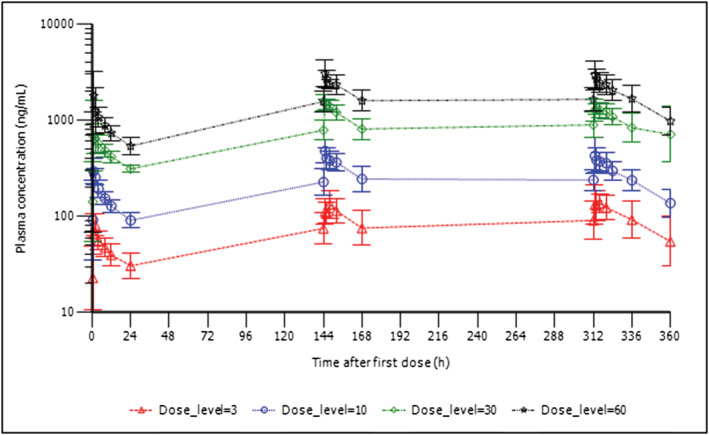
MAD study: Geometric mean (± geometric SD) plasma concentration of LTI‐291 against time (h) after dose following the first (top), seventh (middle) and fourteenth (bottom) oral dose of 3, 10, 30 or 60 mg LTI‐291
TABLE 4.
Arithmetic means ± standard deviation of different pharmacokinetic parameters of the SAD study, measured in plasma. SAD, single ascending dose
| SAD pharmacokinetics in plasma | |||||
|---|---|---|---|---|---|
| LTI‐291 dose | 3 mg (n = 8) | 10 mg (n = 8) | 10 mg (n = 8) | 30 mg (n = 8) | 90 mg (n = 8) |
| Nutritional state | Fasted | Fasted | Fed | Fasted | Fasted |
| Cmax (ng/mL) | 110 ± 13 | 350 ± 131 | 243 ± 75 | 997 ± 283 | 2750 ± 786 |
| t½ (h) | 23.6 ± 11.1 | 23.6 ± 8.3 | 25.3 ± 10.1 | 24.3 ± 7.7 | 23.9 ± 6.1 |
| λz (h−1) | 0.0366 ± 0.0185 | 0.0334 ± 0.0137 | 0.0324 ± 0.0156 | 0.0314 ± 0.0109 | 0.0304 ± 0.0064 |
| AUC0‐last (ng*h/mL) | 1600 ± 383 | 5760 ± 965 | 5950 ± 1130 | 18 700 ± 3920 | 48 400 ± 8790 |
| AUC0‐inf (ng*h/mL) | 2350 ± 703 | 7820 ± 2300 | 8860 ± 2670 | 24 800 ± 3050 | 64 000 ± 10 200 |
| CL/F (L) | 1.38 ± 0.40 | 1.37 ± 0.35 | 1.22 ± 0.36 | 1.23 ± 0.15 | 1.44 ± 0.21 |
| Vz/F (L) | 42.7 ± 15.8 | 43.5 ± 9.9 | 40.7 ± 9.9 | 43.4 ± 15.8 | 49.1 ± 12.4 |
| Cmax/dose (ng/mL/mg) | 36.7 ± 4.4 | 35 ± 13.1 | 24.3 ± 7.5 | 33.2 ± 9.4 | 30.5 ± 8.7 |
| AUC0‐inf/dose (ng*h/mL/mg) | 783 ± 234 | 782 ± 230 | 886 ± 267 | 827 ± 102 | 711 ± 113 |
| Median T max (h) (min, max) | 1.48 (1.00, 2.00) | 1.01 (1.00, 4.02) | 8.00 (4.00, 12.00) | 3.02 (1.00, 4.00) | 2.00 (1.00, 8.17) |
TABLE 5.
Arithmetic means ± standard deviation of different pharmacokinetic parameters of day 1, day 7 and day 14 of the MAD study, measured in plasma. All PK doses were in fasted state. MAD, multiple ascending dose
| MAD pharmacokinetics in plasma | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 7 | Day 14 | ||||||||||
| LTI‐291 dose | 3 mg (n = 7) | 10 mg (n = 8) | 30 mg (n = 8) | 60 mg (n = 8) | 3 mg (n = 7) | 10 mg (n = 8) | 30 mg (n = 8) | 60 mg (n = 8) | 3 mg (n = 7) | 10 mg (n = 8) | 30 mg (n = 8) | 60 mg (n = 8) |
| Cmax (ng/mL) | 91 ± 18 | 316 ± 80 | 927 ± 340 | 2070 ± 702 | 145 ± 33 | 516 ± 109 | 1610 ± 186 | 3290 ± 891 | 159 ± 47 | 509 ± 141 | 1510 ± 204 | 3200 ± 1070 |
| Cmin (ng/mL) | 32 ± 9 | 92 ± 18 | 312 ± 22 | 547 ± 109 | 77 ± 30 | 237 ± 70 | 797 ± 183 | 1580 ± 368 | 95 ± 38 | 239 ± 56 | 847 ± 220 | 1650 ± 493 |
| t½ (h) | 35.2 ± 30.2 | 20.9 ± 3.0 | 27.4 ± 9.9 | 26.0 ± 7.5 | 32.6 ± 13.1 | 29.9 ± 9.9 | 26.2 ± 5.0 | 38.6 ± 17.7 | 40.1 ± 19.7 | 33.0 ± 8.3 | 96.9 ± 72.5 | 33.5 ± 6.9 |
| λz (h−1) | 0.0296 ± 0.017 | 0.0338 ± 0.0051 | 0.0280 ± 0.0090 | 0.0287 ± 0.0088 | 0.0231 ± 0.0093 | 0.0251 ± 0.0072 | 0.0276 ± 0.0071 | 0.0206 ± 0.0069 | 0.0207 ± 0.0089 | 0.0222 ± 0.0053 | 0.0128 ± 0.013 | 0.0214 ± 0.0042 |
| AUC0–24 (ng*h/mL) | 1060 ± 182 | 3450 ± 625 | 10 700 ± 1470 | 19 900 ± 4080 | 2010 ± 805 | 7900 ± 1750 | 26 900 ± 4930 | 53 000 ± 10 200 | 2900 ± 896 | 7650 ± 1580 | 26 500 ± 4410 | 52 300 ± 13 800 |
| CL/F (L) | 1.41 ± 0.77 | 1.66 ± 0.29 | 1.35 ± 0.29 | 1.59 ± 0.50 | 1.63 ± 0.65 | 1.33 ± 0.32 | 1.15 ± 0.22 | 1.17 ± 0.25 | 1.14 ± 0.41 | 1.36 ± 0.28 | 1.17 ± 0.24 | 1.21 ± 0.27 |
| Vz/F (L) | 49.9 ± 9.1 | 49.4 ± 8.0 | 50.4 ± 9.4 | 56.2 ± 11.9 | 70.5 ± 0.0322 | 54.7 ± 12.5 | 42.2 ± 5.2 | 61.3 ± 17.3 | 60.3 ± 16.2 | 63.6 ± 15.8 | 149 ± 95.6 | 57.2 ± 12.7 |
| Cmax/dose (ng/mL/mg) | 30.2 ± 5.9 | 31.6 ± 8.0 | 30.9 ± 11.3 | 34.5 ± 11.7 | 48.2 ± 11.1 | 51.6 ± 10.9 | 53.6 ± 6.19 | 54.9 ± 14.9 | 53.0 ± 15.5 | 50.9 ± 14.1 | 50.4 ± 6.8 | 53.3 ± 17.9 |
| AUC0–24/dose (ng*h/mL/mg) | 353.3 ± 60.7 | 345.0 ± 62.5 | 356.7 ± 49.0 | 331.7 ± 68.0 | 670.0 ± 268.3 | 790.0 ± 175.0 | 896.7 ± 164.3 | 883.3 ± 170.0 | 966.7 ± 298.7 | 765.0 ± 158.0 | 883.3 ± 147.0 | 871.7 ± 230.0 |
| Median T max (h) (min, max) | 2.0 (1.0, 4.0) | 1.0 (1.0, 2.0) | 1.0 (1.0, 8.0) | 1.0 (1.0, 8.1) | 4.0 (1.0, 4.0) | 1.0 (1.0, 8.0) | 1.5 (1.0, 4.0) | 1.0 (1.0, 4.0) | 1.0 (1.0, 8.8) | 1.1 (1.0, 8.0) | 1.5 (1.0, 8.0) | 1.0 (1.0, 2.0) |
| Accumulation index | ‐ | ‐ | ‐ | ‐ | 2.51 ± 0.77 | 2.35 ± 0.58 | 2.13 ± 0.28 | 2.86 ± 1.05 | 2.95 ± 1.17 | 2.53 ± 0.49 | 6.35 ± 4.34 | 2.56 ± 0.41 |
TABLE 6.
Arithmetic means ± standard deviation of CSF concentrations of LTI‐291 and CSF:plasma ratios of the MAD study. Three CSF samples could not be obtained (due to a repeated dry tap, a refusal of a subject and an error in lab handling). MAD, multiple ascending dose; CSF, cerebrospinal fluid; SD, standard deviation
| MAD (day 14) CSF pharmacokinetics | ||||
|---|---|---|---|---|
| LTI‐291 dose | 3 mg (n = 6) | 10 mg (n = 8) | 30 mg (n = 7) | 60 mg (n = 7) |
| LTI‐291 CSF concentration (ng/mL) | ||||
| Mean ± SD | 1.82 ± 0.69 | 4.77 ± 1.40 | 18.2 ± 6.73 | 30.7 ± 7.56 |
| Minimum, maximum | 1.01, 3.08 | 2.51, 6.40 | 7.95, 26.5 | 19.8, 43.3 |
| CSF:plasma concentration ratio ([ng/mL]/[ng/mL]) | ||||
| Mean ± SD | 0.0130 ± 0.00262 | 0.0127 ± 0.00317 | 0.0134 ± 0.00414 | 0.0124 ± 0.00267 |
| Minimum, maximum | 0.0105, 0.0171 | 0.00904, 0.0175 | 0.00642, 0.0194 | 0.00947, 0.0176 |
3.4. Pharmacodynamics
3.4.1. Glycosphingolipid levels
In the SAD study, different GluCer isomers were measured in plasma and PBMCs. No significant dose‐dependent effects of LTI‐291 were detected. See Table S1 in the Supporting Information for details. The three subjects with a GBA1 mutation showed a comparable pattern of GluCer variables in plasma and PBMCs to the subjects in the same cohort without a GBA1 mutation before and after a single dose of LTI‐291 (data not shown).
In the MAD study, different GluCer and LacCer isomers were measured in plasma, PBMCs and CSF. GluSph was measured in plasma and PBMCs. No significant dose‐dependent effects of LTI‐291 were detected. Some isolated decreases and increases in GSLs were detected at different dose levels in different matrices, but no consistent pattern was observed. See Tables S2 and S3 in the Supporting Information for details. Similar as in the SAD study, the two subjects with a GBA1 mutation (randomized to 10 mg and 60 mg) did not show a distinct pattern of glycosphingolipid variables compared to controls (data not shown).
For both studies, no correction for multiple testing was performed, considering the hypothesis‐generating nature of these measurements. Isolated glycosphingolipid changes are therefore at risk of being chance findings.
3.4.2. Neurocognitive biomarkers
There were no significant, dose‐dependent effects of LTI‐291 on any of the CNS tests, either after a single dose, or after prolonged daily dosing. Some isolated differences from placebo were seen in single, mostly submaximal, dose levels, but, due to the lack of dose dependency, these were considered chance findings due to multiple testing. Details can be found in Table S4 for the SAD study and Table S5 for the MAD study in the Supporting Information.
4. DISCUSSION
Here we report the results of the first‐in‐human administration of a first‐in‐class drug aimed at enhancing glucocerebrosidase activity with the goal of slowing down disease progression of Parkinson's disease. In these studies, safety, tolerability, PK and pharmacodynamics of LTI‐291 were evaluated in healthy volunteers. LTI‐291 was administered as a single dose at 3, 10, 30 and 90 mg and as 14 consecutive daily doses at 3, 10, 30 and 60 mg. LTI‐291 was generally well tolerated and no treatment‐related SAEs or deaths occurred and no subject withdrew from a study due to AEs.
Drug levels as measured by C max, AUC0–24 and AUC0–inf increased in a dose‐proportional manner (Figures 1 and 2). In the SAD study at the 3 mg dose the mean C max and AUC0–last were 110 ± 13.2 ng/mL and 1600 ± 383 ng*h/mL, respectively, whereas at the 90 mg dose the mean C max and AUC0–last were 2750 ± 786 ng/mL and 48 400 ± 8790 ng*h/mL, respectively, demonstrating a 25‐fold increase in C max and 30‐fold increase in AUC0–last (Table 4). Similarly, in the MAD study on day 14 at the 3 mg dose, the mean C max and AUC0–24 were 159 ± 46.6 ng/mL and 3200 ± 1070 ng*h/mL, respectively, whereas at the 60 mg dose the mean C max and AUC0–24 were 2900 ± 896 ng/mL and 52 300 ± 13 800 ng*h/mL, respectively, demonstrating an 18‐fold increase in C max and 16‐fold increase in AUC0–last (Table 5). With a median half‐life of 28.0 hours after multiple dosing, LTI‐291 is a suitable candidate for daily single dosing. In a single subject in the 30 mg MAD cohort, an estimated half‐life of 250 hours at day 14 sampling was observed. This subject had a relatively low C max this day with an apparently slowed absorption, possibly contributing to the long half‐life based on the 48 hours post‐dose sampling, combined with a variable elimination. This was seen to a lesser extent in other subjects. Within the 30 mg dose group on day 14, the half‐lives recorded were 15.5, 52.9, 56.1, 70.8, 85.2, 99.4, 145 and 250 hours. Nothing unusual in the higher range subjects or in the cohort happened that could explain this wide range, so it is likely an expression of a variable elimination, or possibly of flip‐flop kinetics, despite the powder being in a capsule formulation. The 24‐hour concentrations following all the multiple doses were consistent, as were the AUC0–24 values following the 7th and 14th doses. Pharmacokinetic steady state is reached by the 7th daily dose (Table 5). The median ratios of the AUC0–24 following multiple doses were 2.52 and 2.59 after the 7th and 14th doses, respectively, whereas the median ratios of the C max were 1.62 and 1.55 after the 7th and 14th doses. The median accumulation index was 2.28 and 2.70 after the 7th and 14th doses (Table 5).
Since LTI‐291 is a highly permeable molecule and, based on the preclinical rat neuro PK data where the unbound partition coefficient (Kpuu) between brain and plasma was determined to be 1.02, the compound has excellent brain penetration and is at distribution equilibrium between the plasma, brain and CSF compartments. On this basis one can anticipate that in humans at steady state the CSF concentration will be similar to the unbound drug in brain (Cu,b) and plasma (Cu,p). In the MAD study, the mean CSF:plasma concentration ratios ranged from 0.0124 to 0.0134 at 4–5 hours after the 14th dose and were similar at all dose levels (Table 6). Over the 20‐fold dose range, the CSF concentrations increased in a dose‐proportional manner relative to the plasma concentrations. For example, at the 3 mg dose the LTI‐291 concentration was 1.82 ± 0.687 ng/mL, whereas at the 60 mg dose the LTI‐291 concentration was 30.7 ± 7.56 ng/mL, demonstrating a 17‐fold increase. These results show that LTI‐291 has good CNS/brain distribution.
The LTI‐291 drug levels achieved in the MAD study suggest that all doses could potentially result in GCase activation. Based on preclinical in vitro studies (unpublished), 18% activation of GCase occurs at drug levels of 36 ng/mL (0.1 μM) and 100% activation of GCase occurs at drug levels of 360 ng/mL (1 μM). The 10 mg daily dose would achieve 100% GCase activation, since at day 14 the average plasma C max was 509 ± 141 ng/mL and based on the long half‐life the C min was 239 ± 56 ng/mL (Table 5).
LTI‐291 is expected to increase GCase activity, which might lead to increased cleavage of the substrates GluCer and GluSph of GCase, thus leading to lower levels of these substrates in the lysosome. This could indirectly affect the upstream glycosphingolipid LacCer upon stimulation of this sphingolipid pathway. It is still unclear how this would translate to levels measured in whole cells, since, for example, GluCer is primarily located outside of the lysosome.25 Lack of a clear treatment effect in this study can be explained by LTI‐291 having been given to healthy subjects, who can be expected to have a normally functioning GCase enzyme. Additionally, it is unclear how a change in intralysosomal levels is reflected in total cellular levels. No clear transient effects on GSLs were detected. It is expected that the drug would have a stronger effect in patients with compromised GCase activity. In the MAD study, two healthy subjects with a mild GBA1 mutation (T369M) were identified, of which one randomized to the highest dose. Even though these subjects might have had an altered GCase activity, the variability in biomarker effects between subjects was too large to be able to conclude that these two GBA1 mutated subjects respond differently on GluCer, LacCer and GluSph variables compared to the non‐mutated subjects.
Several GluCer and LacCer isoforms in CSF decreased in 10 mg or 60 mg LTI‐291‐treated subjects compared to placebo. However, this could be a chance finding, because the placebo group had an average increase compared to baseline for these isoforms, whereas LTI‐291‐treated subjects remained unchanged. Glycosphingolipid levels are not expected to change significantly in this timeframe in untreated subjects, so this was likely a result of natural variability.
The desired mode of action for treating GBA‐PD is an ongoing debate.26, 27, 28, 29, 30 More than 400 different mutations in the GBA1 gene are known from GD.31, 32 These different mutations can have different effects, e.g. enzymes that reach the lysosome albeit with decreased activity, or retention in the endoplasmic reticulum (ER), possibly resulting in an ER stress response. Correction of protein misfolding, assisted transport to the lysosome and substrate reduction therapies have also been suggested.30 The multitude of different mutant enzymes makes enzyme activation challenging. Since most GBA‐PD patients have a heterozygous mutation, a common factor is the remainder of the wild type GCase enzyme. Enhancement of the wild type enzyme, as is shown preclinically for LTI‐291, seems crucial when targeting enzyme activity. GCase activation by LTI‐291 could not directly be measured ex vivo in cell lysate, because LTI‐291 requires a lipidic surface to function (in vivo the lysosomal membrane). The live cell assay for lysosomal GCase activity (using 5‐(Pentafluorobenzoylamino) Fluorescein Di‐beta‐D‐Glucopyranoside (PFB‐FDGlu) substrate) is not compatible with LTI‐291 since it blocks the allosteric site. It remains unclear what the best biomarkers would be for future studies in GBA‐PD.
In conclusion, LTI‐291 was observed to be well tolerated generally when given orally once daily for 14 consecutive days at all dose levels tested in this healthy subject population. Results from this study support the continued clinical development of LTI‐291 and the exploration of LTI‐291 effects in a GBA1‐mutated Parkinson population.
COMPETING INTERESTS
K.B., R.S., C.J., L.D., P.L., V.C. and D.H. were employees of Lysosomal Therapeutics, Inc. at time of execution of these studies. There are no other conflicts of interest to declare.
CONTRIBUTORS
1) Research project: A. Conception, B. Organization, C. Execution;
2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique;
3) Manuscript: A. Writing of the first draft, B. Review and Critique.
Jonas M. den Heijer: 1A,1B,1C,2A,2C,3A,3B; Annelieke C. Kruithof: 1A,1B,1C,2A,2C,3B; Guido van Amerongen: 1A,1B,1C,2A,2C,3B; Marieke L. de Kam: 1A,2A,2B,2C,3B; Eva Thijssen: 1C,2C,3B; Hendrika W. Grievink: 1A,1B,1C,2C,3B; Matthijs Moerland: 1A,1B,2C,3B; Mike Walker: 1A,2A,2B,2C,3B; Kees Been: 1A,1B,2C,3B; Renato Skerlj: 1A,1B,2A,2C,3B; Craig Justman: 1A,1B,2A,2C,3B; Lindsay Dudgeon: 1A,1B,2C,3B; Peter Lansbury: 1A,1B,2A,2C,3B; Valerie C. Cullen: 1A,1B,2A,2C,3B; Dana C. Hilt: 1A,1B,2A,2C,3B; Geert Jan Groeneveld: 1A,1B,1C,2A,2C,3B.
Supporting information
TABLE S1 SAD study: Summary of GluCer analysis results in plasma and PBMCs. Overall treatment P‐value is given for all treatment groups combined. Specific analysis per dose group compared to placebo were also done. Results of these comparisons are shown in the table as estimates of the least square mean difference with 95% confidence interval (CI) and P‐value. GluCer, glucosylceramide.
TABLE S2 MAD study: Summary of GluCer, LacCer and GluSph analysis results in plasma, PBMCs and CSF. Specific analysis per dose group compared to placebo are given. Results of these comparisons are shown in the table as estimates of the least square mean differences with 95% confidence interval and P‐value. Log‐transformed parameters were back‐transformed after analysis and therefore these parameters are shown as percentage change. For GluCer and LacCer ‘nn:n’, the first number indicates the number of carbon atoms in the fatty acid arm (part of the ceramide [Cer] moiety), the second number indicates the number of unsaturated bonds in this carbon chain. For technical reasons, GluCer22:0 in plasma and PBMCs could only be compared between specific batches: a Placebo of cohorts 1 and 2; b Placebo of cohorts 3 and 4; c Placebo of cohorts 1, 2 and 3; d Placebo of cohort 4. An overall treatment effect P‐value is given in Table S3. MAD, multiple ascending dose; GluCer, glucosylceramide; LacCer, lactosylceramide; GluSph, glucosylsphingosine; PBMCs, peripheral blood mononuclear cells; CSF, cerebrospinal fluid.
TABLE S3 MAD study: Summary of GluCer, LacCer and GluSph analysis results in plasma, PBMCs and CSF. Overall treatment P‐value is given for all treatment groups combined. Specific analysis per dose group compared to placebo were also done. Results of these comparisons are shown in the table as estimates of the least square mean differences with 95% confidence interval and P‐value. Log‐transformed parameters were back‐transformed after analysis and therefore these parameters are shown as percentage change. For GluCer and LacCer ‘nn:n’ the first number indicates the number of carbon atoms in the fatty acid arm (part of the ceramide [Cer] moiety), the second number indicates the number of unsaturated bonds in this carbon chain. For technical reasons, GluCer22:0 in plasma and PBMCs could only be compared between specific batches: a Placebo of cohorts 1 and 2; b Placebo of cohorts 3 and 4; c Placebo of cohorts 1, 2 and 3; d Placebo of cohort 4. MAD, multiple ascending dose; GluCer, glucosylceramide; LacCer, lactosylceramide; GluSph, glucosylsphingosine; PBMCs, peripheral blood mononuclear cells; CSF, cerebrospinal fluid.
TABLE S4 SAD study: Summary of analysis results per NeuroCart parameter. The table shows overall treatment P‐value (all dose groups compared to placebo) and comparisons of each LTI‐291 dose against placebo. Estimates of the difference with 95% confidence intervals (CI) and P‐values are shown for comparison. Log‐transformed parameters were back‐transformed after analysis and therefore these parameters are shown as percentage change. SAD, single ascending dose; RT, reaction time; VAS, visual analogue scale; EEG, electro‐encephalogram; VVLT, Visual Verbal Learning Test.
TABLE S5 MAD study: Summary of analysis results per NeuroCart parameter. Table shows overall treatment P‐value (all treatment groups combined) and subsequent comparisons of each LTI‐291 dose against placebo. Estimates of the difference with 95% confidence intervals and P‐values are shown for comparison. Log‐transformed parameters were back‐transformed after analysis and therefore these parameters are shown as percentage change. MAD, multiple ascending dose; RT, reaction time; VAS, visual analogue scale; EEG, electro‐encephalogram.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. E. ‘t Hart and Dr. K. Kanhai for their supervision and assistance in clinical execution. These studies were funded by Lysosomal Therapeutics, Inc.
den Heijer JM, Kruithof AC, van Amerongen G, et al. A randomized single and multiple ascending dose study in healthy volunteers of LTI‐291, a centrally penetrant glucocerebrosidase activator. Brit Jnl Clinical Pharma. 2021;87(9):3561–3573. 10.1111/bcp.14772
The authors confirm that the Principal Investigator for this paper is Geert Jan Groeneveld and that he had direct clinical responsibility for participants.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. The Lancet Neurol. 2006;5(6):525‐535. [DOI] [PubMed] [Google Scholar]
- 2.Schapira AH. Glucocerebrosidase and Parkinson disease: Recent advances. Mol Cell Neurosci. 2015;66(Pt A):37‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gan‐Or Z, Amshalom I, Kilarski LL, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. 2015;84(9):880‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruskey JA, Greenbaum L, Ronciere L, et al. Increased yield of full GBA sequencing in Ashkenazi Jews with Parkinson's disease. Eur J Med Genet. 2019;62(1):65‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesage S, Anheim M, Condroyer C, et al. Large‐scale screening of the Gaucher's disease‐related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet. 2011;20(1):202‐210. [DOI] [PubMed] [Google Scholar]
- 6.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009;361(17):1651‐1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrucci S, Ginevrino M, Trezzi I, et al. GBA‐related Parkinson's disease: dissection of genotype‐phenotype correlates in a large Italian cohort. Mov Disord. 2020;35(11):2106‐2111. [DOI] [PubMed] [Google Scholar]
- 8.Mata IF, Leverenz JB, Weintraub D, et al. GBA variants are associated with a distinct pattern of cognitive deficits in Parkinson's disease. Mov Disord. 2016;31(1):95‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merrill AH Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. 2011;111(10):6387‐6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferraz MJ, Marques AR, Appelman MD, et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016;590(6):716‐725. [DOI] [PubMed] [Google Scholar]
- 11.Dekker N, van Dussen L, Hollak CE, et al. Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood. 2011;118(16):e118‐e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alcalay RN, Levy OA, Waters CC, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. 2015;138(Pt 9):2648‐2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huebecker M, Moloney EB, van der Spoel AC, et al. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson's disease. Mol Neurodegener. 2019;14(1):40–61. 10.1186/s13024-019-0339-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moors TE, Paciotti S, Ingrassia A, et al. Characterization of brain lysosomal activities in GBA‐related and sporadic Parkinson's disease and dementia with Lewy bodies. Mol Neurobiol. 2019;56(2):1344‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papagiannakis N, Xilouri M, Koros C, et al. Lysosomal alterations in peripheral blood mononuclear cells of Parkinson's disease patients. Mov Disord. 2015;30(13):1830‐1834. [DOI] [PubMed] [Google Scholar]
- 16.Muehlan C, Brooks S, Zuiker R, van Gerven J, Dingemanse J. Multiple‐dose clinical pharmacology of ACT‐541468, a novel dual orexin receptor antagonist, following repeated‐dose morning and evening administration. Eur Neuropsychopharmacol. 2019;29(7):847‐857. [DOI] [PubMed] [Google Scholar]
- 17.Baakman AC, Alvarez‐Jimenez R, Loewen G, et al. No synergistic effect of subtherapeutic doses of donepezil and EVP‐6124 in healthy elderly subjects in a scopolamine challenge model. Alzheimers Dement (N Y). 2019;5:89‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groeneveld GJ, Hay JL, Van Gerven JM. Measuring blood–brain barrier penetration using the NeuroCart, a CNS test battery. Drug Discov Today Technol. 2016;20:27‐34. [DOI] [PubMed] [Google Scholar]
- 19.van Steveninck AL, Schoemaker HC, Pieters MS, Kroon R, Breimer DD, Cohen AF. A comparison of the sensitivities of adaptive tracking, eye movement analysis and visual analog lines to the effects of incremental doses of temazepam in healthy volunteers. Clin Pharmacol Ther. 1991;50(2):172‐180. [DOI] [PubMed] [Google Scholar]
- 20.Chen X, de Haas S, de Kam M, van Gerven J. An overview of the CNS‐pharmacodynamic profiles of nonselective and selective GABA agonists. Adv Pharm Sci. 2012;2012:134523–134533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.den Heijer JM, Cullen VC, Quadri M, et al. A large‐scale full GBA1 gene screening in Parkinson's disease in the Netherlands. Mov Disord. 2020;35(9):1667‐1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176(S1):S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y, Deng L, Zhong Y, Yi M. The association between E326K of GBA and the risk of Parkinson's disease. Parkinson's Disease. 2018;2018:1048084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mallett V, Ross JP, Alcalay RN, et al. GBA p.T369M substitution in Parkinson disease: polymorphism or association? A meta‐analysis. Neurol Gen. 2016;2(5):e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuller M, Rozaklis T, Lovejoy M, Zarrinkalam K, Hopwood JJ, Meikle PJ. Glucosylceramide accumulation is not confined to the lysosome in fibroblasts from patients with Gaucher disease. Mol Genet Metab. 2008;93(4):437‐443. [DOI] [PubMed] [Google Scholar]
- 26.Aflaki E, Borger DK, Moaven N, et al. A new glucocerebrosidase chaperone reduces α‐synuclein and glycolipid levels in iPSC‐derived dopaminergic neurons from patients with Gaucher disease and Parkinsonism. J Neurosci. 2016;36(28):7441‐7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullin S, Smith L, Lee K, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized noncontrolled trial. JAMA Neurol. 2020;77(4):427‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sidransky E, Arkadir D, Bauer P, et al. Substrate reduction therapy for GBA1‐associated Parkinsonism: are we betting on the wrong mouse? Mov Disord. 2020;35(2):228‐230. [DOI] [PubMed] [Google Scholar]
- 29.Silveira CRA, MacKinley J, Coleman K, et al. Ambroxol as a novel disease‐modifying treatment for Parkinson's disease dementia: protocol for a single‐centre, randomized, double‐blind, placebo‐controlled trial. BMC Neurol. 2019;19(1):20‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Sam R, Sharma P, Chen L, Do J, Sidransky E. Glucocerebrosidase as a therapeutic target for Parkinson's disease. Expert Opin Ther Targets. 2020;24(4):287‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567‐583. [DOI] [PubMed] [Google Scholar]
- 32.Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Hum Genet. 2017;136(6):665‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 SAD study: Summary of GluCer analysis results in plasma and PBMCs. Overall treatment P‐value is given for all treatment groups combined. Specific analysis per dose group compared to placebo were also done. Results of these comparisons are shown in the table as estimates of the least square mean difference with 95% confidence interval (CI) and P‐value. GluCer, glucosylceramide.
TABLE S2 MAD study: Summary of GluCer, LacCer and GluSph analysis results in plasma, PBMCs and CSF. Specific analysis per dose group compared to placebo are given. Results of these comparisons are shown in the table as estimates of the least square mean differences with 95% confidence interval and P‐value. Log‐transformed parameters were back‐transformed after analysis and therefore these parameters are shown as percentage change. For GluCer and LacCer ‘nn:n’, the first number indicates the number of carbon atoms in the fatty acid arm (part of the ceramide [Cer] moiety), the second number indicates the number of unsaturated bonds in this carbon chain. For technical reasons, GluCer22:0 in plasma and PBMCs could only be compared between specific batches: a Placebo of cohorts 1 and 2; b Placebo of cohorts 3 and 4; c Placebo of cohorts 1, 2 and 3; d Placebo of cohort 4. An overall treatment effect P‐value is given in Table S3. MAD, multiple ascending dose; GluCer, glucosylceramide; LacCer, lactosylceramide; GluSph, glucosylsphingosine; PBMCs, peripheral blood mononuclear cells; CSF, cerebrospinal fluid.
TABLE S3 MAD study: Summary of GluCer, LacCer and GluSph analysis results in plasma, PBMCs and CSF. Overall treatment P‐value is given for all treatment groups combined. Specific analysis per dose group compared to placebo were also done. Results of these comparisons are shown in the table as estimates of the least square mean differences with 95% confidence interval and P‐value. Log‐transformed parameters were back‐transformed after analysis and therefore these parameters are shown as percentage change. For GluCer and LacCer ‘nn:n’ the first number indicates the number of carbon atoms in the fatty acid arm (part of the ceramide [Cer] moiety), the second number indicates the number of unsaturated bonds in this carbon chain. For technical reasons, GluCer22:0 in plasma and PBMCs could only be compared between specific batches: a Placebo of cohorts 1 and 2; b Placebo of cohorts 3 and 4; c Placebo of cohorts 1, 2 and 3; d Placebo of cohort 4. MAD, multiple ascending dose; GluCer, glucosylceramide; LacCer, lactosylceramide; GluSph, glucosylsphingosine; PBMCs, peripheral blood mononuclear cells; CSF, cerebrospinal fluid.
TABLE S4 SAD study: Summary of analysis results per NeuroCart parameter. The table shows overall treatment P‐value (all dose groups compared to placebo) and comparisons of each LTI‐291 dose against placebo. Estimates of the difference with 95% confidence intervals (CI) and P‐values are shown for comparison. Log‐transformed parameters were back‐transformed after analysis and therefore these parameters are shown as percentage change. SAD, single ascending dose; RT, reaction time; VAS, visual analogue scale; EEG, electro‐encephalogram; VVLT, Visual Verbal Learning Test.
TABLE S5 MAD study: Summary of analysis results per NeuroCart parameter. Table shows overall treatment P‐value (all treatment groups combined) and subsequent comparisons of each LTI‐291 dose against placebo. Estimates of the difference with 95% confidence intervals and P‐values are shown for comparison. Log‐transformed parameters were back‐transformed after analysis and therefore these parameters are shown as percentage change. MAD, multiple ascending dose; RT, reaction time; VAS, visual analogue scale; EEG, electro‐encephalogram.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.