

Abstract
Apoptosis, the most extensively studied form of programmed cell death, is essential for organismal homeostasis. Apoptotic cell death has widely been reported as a tumor suppressor mechanism. However, recent studies have shown that apoptosis exerts noncanonical functions and may paradoxically promote tumor growth and metastasis. The hijacking of apoptosis by cancer cells may arise at different levels, either via the interaction of apoptotic cells with their local or distant microenvironment, or through the abnormal pro‐oncogenic roles of the main apoptosis effectors, namely caspases and mitochondria, particularly upon failed apoptosis. In this review, we highlight some of the recently described mechanisms by which apoptosis and these effectors may promote cancer aggressiveness. We believe that a better understanding of the noncanonical roles of apoptosis may be crucial for developing more efficient cancer therapies.
Keywords: apoptosis, cancer, caspases, mitochondria, oncogenesis
Apoptosis exerts noncanonical pro‐oncogenic effects. Firstly, apoptotic cells release various paracrine factors that instruct cancer cells to proliferate, develop drug resistance, or avoid immune surveillance. Secondly, minority MOMP, which is characterized by permeabilization of few mitochondria and nonlethal caspase activation, triggers DNA damage and mutagenesis, and it increases cancer aggressiveness. Thirdly, mitochondria have pro‐oncogenic functions through either their cell‐to‐cell transfer or mitochondrial dynamics.
Abbreviations
- AiP
Apoptosis‐induced proliferation
- Apo‐EVs
Apoptotic‐derived EVs
- ATM
Ataxia telangiectasia mutated
- BAK
BCL2 antagonist killer 1
- BAX
BCL2‐associated X
- BCL2
B‐cell lymphoma 2
- BIK
BCL2‐interacting killer
- CAD
Caspase‐activated DNAse
- cGAS
cyclic GMP‐AMP synthase
- CICD
Caspase‐independent cell death
- COX
Cyclooxygenase
- CSCs
Cancer stem cells
- DAMP
Damage‐associated molecular pattern
- DRP1
Dynamin‐related protein 1
- EGF
Epidermal growth factor
- EMT
Epithelial–mesenchymal transition
- EndoG
Endonuclease G
- EVs
Extracellular vesicles
- FTK
Fractalkine
- HSCs
hematopoietic stem cells
- IAPs
inhibitor of apoptosis proteins
- ICM
Irradiation‐induced cell migration
- iPLA2
Calcium‐independent phospholipase A2
- JNK
c‐Jun N‐Terminal Kinase
- MFN1
Mitofusin 1
- MFN2
Mitofusin 2
- MLL
Mixed lineage leukemia
- MOMP
Mitochondrial outer membrane permeabilization
- PS
Phosphatidylserine
- ROS
Reactive oxygen species
- SMAC
Second mitochondria‐derived activator of caspases
- STAT3
Signal transducer and activator of transcription 3
- STING
stimulation of IFN genes protein
- TAM
Tumor‐associated macrophage
- TGFβ
Transforming growth factor beta
- TNTs
tunneling nanotubes
- Upd
Unpaired
- VEGF
Vascular endothelial growth factor
- Wg
Wingless
- YAP
Yes‐associated protein
Introduction
Programmed cell death plays a crucial role in many biological processes including tissue structuring during embryogenesis, development of the immune system and destruction of damaged cells. Among the different forms of programmed cell death, apoptosis is the most common and best‐studied, relying on the activation of caspase proteases to induce extensive cleavage of hundreds of substrates and rapid cell death [1]. Apoptosis is necessary to remove pathogen‐invaded cells, but is also needed to eliminate malignant as well as auto‐aggressive immune cells. Caspases, the key molecular mediators of this process, can be activated through two distinct but interconnected pathways: the extrinsic and the intrinsic pathway.
On one hand, the extrinsic pathway relies on the activation of death receptors through the binding of their cognate ligand(s) [1]. On the other hand, the intrinsic apoptotic pathway (also known as mitochondrial pathway) relies on widespread mitochondrial outer membrane permeabilization (MOMP) after deadly apoptotic stresses such as cytokine deprivation, DNA damage, or endoplasmic reticulum stress. MOMP is tightly controlled by pro‐apoptotic and anti‐apoptotic members of the BCL‐2 protein family [2].
Upon induction of apoptosis, the pro‐apoptotic effector proteins BCL2‐associated X (BAX) and BCL2 antagonist killer 1 (BAK) undergo direct or indirect activation by pro‐apoptotic BH3‐only proteins in order to form pores across the outer mitochondrial membrane. This process is counteracted by anti‐apoptotic BCL‐2 family proteins. Upon MOMP, mitochondrial intermembrane space proteins such as second mitochondria‐derived activator of caspases (SMAC) and cytochrome c are released into the cytosol. Cytochrome c can then trigger apoptosome assembly and the activation of initiator caspase‐9 and subsequently of effector caspase‐3 and ‐7, leading to apoptosis [2].
The dysregulation of apoptosis characterizes a wide variety of diseases. In cancer, apoptosis is a well‐established tumor suppressor mechanism, and loss of apoptotic control allows cancer cells to survive longer thus acquiring additional oncogenic hits [3]. However, apoptosis may not only serve as a tumor suppressor mechanism but may also have pro‐oncogenic functions by promoting the emergence and maintenance of aggressive tumors. Aggressiveness is commonly used in the literature to describe cancers with a poor prognosis, increased proliferation, and metastatic capacities [4, 5].
Interestingly when defining what drives tumor aggressiveness, a concept is gaining popularity, namely that apoptosis may exert oncogenic effects and sublethal apoptotic stresses may lead to the development of aggressive tumors.
In this review, we discuss the hidden link between apoptotic cell death, its effectors, mitochondria and caspases, and cancer aggressiveness. Our discussion begins by presenting the main oncogenic outcomes of complete apoptosis. Consequently, we highlight how failure to efficiently undergo apoptosis, nonlethal caspase signaling, and mitochondria could mediate a variety of pro‐tumorigenic events. We suggest that understanding the noncanonical roles of apoptosis may improve therapeutic strategies aimed at treating highly aggressive tumors.
Noncanonical oncogenic roles of complete apoptosis
As the most relevant form of programmed cell death, apoptosis has long been regarded as a barrier to carcinogenesis. However, apoptotic cells may also exert noncanonical functions by driving tumor cell proliferation, leading to a reevaluation of the benefits of triggering apoptosis in cancer therapy [6]. Moreover, a high intratumoral apoptotic index is correlated with an increased mitotic index and shorter overall survival in several cancers, while potentiating tumor progression [7, 8, 9, 10]. To address these apparently paradoxical findings, in this section we will discuss some of the main mechanisms underlying the pro‐tumorigenic effects of apoptotic cells.
Pro‐oncogenic effects of apoptotic cell death on tumor proliferation
Apoptosis‐induced proliferation (AiP) is an evolutionarily conserved form of compensatory proliferation in multicellular organisms, by which apoptotic cells actively stimulate surviving cells to divide [11]. This process has been the most extensively studied in Drosophila melanogaster, where apoptotic cells in the imaginal disk were shown to activate mitogenic signaling, thus promoting the proliferation of neighboring cells [12, 13, 14]. In this model, apoptotic cells can stimulate the proliferation of neighboring nonapoptotic cells in a caspase‐dependent manner, through the initiator caspase Dronc (ortholog of human caspase‐9) [15]. AiP is also supported through the recruitment of immune cells, and in particular macrophage‐like hemocytes. Mechanistically, undead imaginal disk cells, sequester activated Dronc at the basal side of the plasma membrane where it activates the NAPDH‐oxidase Duox inducing extracellular reactive oxygen species (eROS) production. This triggers hemocyte recruitment and Eiger (Tumor necrosis factor alpha analogue) production that will sustain c‐Jun N‐terminal kinase (JNK) and caspase activation in undead cells. This cascade ultimately sustains the proliferation of neighboring cells [16, 17, 18]. These caspase‐dependent signaling pathways equally play a critical role in wound healing and regeneration via the activation of p53, the JNK and the secretion of mitogens like Wnt homolog Wingless (Wg), the Epidermal growth factor (EGF) homolog Spitz, and the interleukin‐6 homolog Unpaired (Upd) [19, 20, 21, 22, 23, 24].
Furthermore, in mammals, apoptotic caspases may also contribute to tumor initiation through caspase‐dependent inflammation and sustained AiP [25]. A key mediator of AiP in mammals is prostaglandin E2 (PGE2), which is secreted by apoptotic cells and has been found to mediate their ability to stimulate tumor proliferation (Fig. 1A). Accordingly, Li and collaborators demonstrated that dying cells stimulate proliferation of neighboring cancer cells through a caspase‐3‐dependent mechanism. In these settings, caspases cleave and activate calcium‐independent phospholipase A2 (iPLA2), resulting in an increased production of arachidonic acid, which is converted via cyclooxygenase 1 (COX1) and COX2 to PGE2, which then promotes stem and progenitor cell proliferation [26, 27]. This caspase‐3/PGE2 signaling cascade was also described for dying tumor cells, thus promoting the growth of neighboring tumor cells, but also extends to dying vascular endothelial cells fostering tumor cell growth [28]. Of note, active caspase‐3 was also recently reported to be a key regulator of cell proliferation and organ size without instructing cell elimination, through the activation and nuclear translocation of the yes‐associated protein (YAP), a vital regulator of organ size [29]. Whether this caspase‐3/YAP axis is relevant for the proliferation of cancer cells remains a matter of debate.
Fig. 1.
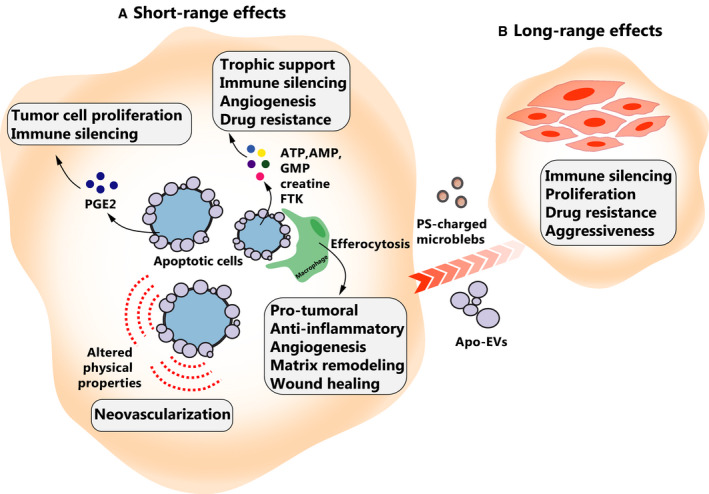
Pro‐oncogenic effects of apoptotic cells. (A) Short‐range effects of apoptotic cell death. Apoptotic cells secrete prostaglandin E2 (PGE2) that can have pleiotropic effects on surrounding cells, including mitogenic and immune‐silencing effects. Apoptotic cells release various ‘eat‐me’ and ‘find‐me’ molecules to signal their removal by phagocytes. These molecules include FKN that promotes angiogenesis or drug resistance and ATP that can be taken up by surrounding tumor cells to provide energy for tumor growth. These signals can also polarize TAMs toward a pro‐oncogenic state (M2 phenotype) that can impair immune surveillance. Apoptotic cancer cells can also exert a physical pull on neighboring cells. (B) Long‐range effects of apoptotic cell death. Apoptotic cells can impact their distant environment by releasing apoptotic‐derived extracellular vesicles (Apo‐EVs) or PS‐charged microblebs that promote tumor survival, proliferation, and migration by transporting oncogenes, splicing factors, and PS.
The role of PGE2 is particularly relevant in the context of standard antitumor treatments. Highly aggressive cancers, like melanoma, undergo tumor repopulation after cytotoxic therapy, suggesting that current therapies are often ineffective in eliminating all of the melanoma tumor cells. This tumor repopulation is orchestrated by PGE2 in a caspase 3‐mediated manner, where dying melanoma cells stimulate the growth of living cells after cytotoxic therapy [30, 31]. Thus, targeting PGE2 either by a PGE2‐neutralizing antibody, by COX2 inhibitor celecoxib or by broader acting nonsteroidal anti‐inflammatory drugs has been shown to effectively abrogate cancer stem cell (CSC) repopulation, enhance chemotherapeutic response in bladder urothelial carcinomas, and reduce incidence and mortality of colorectal cancer [32, 33, 34]. Moreover, selective inhibition of the PGE2 receptor EP4 by the small molecule inhibitor, L‐161,982 enhances oxaliplatin efficacy in resistant colon cancer cells, supporting the role for the COX‐2/PGE2/EP4 signaling axis in resistance to therapy [35]. Interestingly, PGE2 can also have immune‐silencing effects and promote angiogenesis independently of Vascular endothelial growth factor (VEGF) signaling [36, 37] (Fig. 1A). Aside from iPLA2, activated effector caspases in dying cancer cells are involved in the activation of protein kinase C delta, which in turn mediates phosphorylation of Akt, p38, and JNK1/2, leading to mitogen production and tumor repopulation following radiation therapy [38].
Paradoxically, these studies suggest that cancer cell apoptosis can instruct the proliferation of neighboring cancer cells, an insidious interaction that is currently not taken into consideration when designing cancer therapies.
The oncogenic impact of apoptotic cells on the tumor microenvironment
Apoptotic cells are part of a dynamic microenvironment where they release the so‐called ‘find‐me’ and ‘eat‐me’ signals responsible for efficient phagocytosis. These signals include fractalkine (FTK, also known as C‐X3‐C motif chemokine ligand 1 or CX3CL1), phosphatidylserine (PS), lysophosphatidylcholine, sphingosine‐1‐phosphate, and nucleotides ATP and UTP [39]. These molecules are often thought to act locally with a high affinity for immune cell activation. Nevertheless, growing evidence indicates that these signals can be used by cancer cells in their own interest to replicate and survive (Fig. 1A).
This has been reported for FTK, the role of which as a chemokine extends beyond the recruitment of phagocytes. Several recent studies investigating the noncanonical roles of FTK in oncogenesis have been carried out in hepatocellular carcinoma, prostate, or breast cancer where FTK was linked with angiogenesis, hypoxia‐induced proliferation, or enhanced ERBB2 receptor signaling [40, 41, 42, 43]. FTK also protects glial cells from Fas‐mediated cell death by affecting the expression and inhibitory phosphorylation of BCL2 family proteins (Fig. 1A) [44]. Additionally, FTK stimulates metastasis formation through the activation of the Src/FAK signaling pathway and M2 macrophage recruitment [45, 46, 47]. The pro‐oncogenic effects of FTK, mainly those related to cancer cell migration, metastasis, and angiogenesis, were extensively reviewed recently [48].
It has been reported that ‘find‐me’ signals produced by apoptotic cells could induce inflammation and tissue damage. However, in most cases of physiological apoptotic cell death, inflammation is avoided through efferocytosis, the mechanism at play in the removal of apoptotic cells by macrophages. Interestingly, in the context of cancer, efferocytosis creates an immunosuppressive phenotype within tumors through a coordinated series of signaling events [49]. Efferocytosis limits the presence of pro‐inflammatory damage‐associated molecular patterns (DAMPs) released by uncleared cell corpses and shifts cytokine production toward an immunosuppressive profile (e.g., IL‐4, IL‐10, IL‐13, and TGF‐β). During efferocytosis, tumor‐associated macrophages (TAMs) are polarized toward an M2‐like wound‐healing phenotype and FOXP3+ regulatory T cells are recruited, which potently suppress the effector functions of CD4+ and CD8+ effector T cells within the tumor microenvironment (Fig. 1A). Of note, M2 macrophage polarization is also promoted by PGE2 signaling, by inducing the expression of the KLF4 transcription factor through the activation of the CREB/BDNF/TrkB signaling pathway [50, 51]. Accumulation of TAMs was reported to be correlated with poor prognosis in various human malignancies and the contribution of TAMs to cancer progression has been extensively explored [49]. In 2015, Ford and collaborators reported that apoptotic tumor cells promote coordinated tumor growth, angiogenesis, and accumulation of TAMs in aggressive B‐cell lymphomas and malignant melanoma. According to their model, in wild‐type lymphoma, spontaneous apoptotic tumor cells are engulfed by macrophages, thus polarizing them into TAMs. Lymphoma TAMs exhibit an efferocytic, pro‐angiogenic, matrix‐remodeling, and wound‐healing phenotype, which contributes to accelerated tumor progression [7].
A recent study showed that a specific subset of metabolites, composed of spermidine, AMP, GMP, creatine, G3P, and ATP, is actively released by various apoptotic cell types such as T cells or macrophages after caspase‐dependent pannexin‐1 cleavage and activation into plasma membrane channels. In a paracrine manner, these metabolites then promote neighboring cell proliferation, wound‐healing and inflammation suppression (Fig. 1A) [52]. Before the clearance of dying cells, ‘find‐me’ signals such as nucleotides can also have a trophic role on tumor cells [53]. For instance, extracellular ATP can be taken up by tumors via macropinocytosis to supplement their additional energetic needs for cancer growth, survival, and drug resistance (Fig. 1A) [54, 55].
Of note, other types of programmed cell death could influence the tumor microenvironment. For instance, RIPK3 activation during necroptosis was shown to induce cytokine production (notably CXCL1, CXCL2 and CCL2) even after loss of membrane integrity and thus to promote inflammation [56].
Apoptotic cells can also affect their microenvironment through their altered biophysical properties. For example, the negative surface charge caused by PS exposure was shown to induce endothelial cell sprouting and contribute to tumor neovascularization [57] (Fig. 1A). Interestingly, Fernández‐Sánchez et al. showed that the mechanical pressure exerted by hyper‐proliferative tumor cells results in the activation of the β‐catenin pathway and mediates the formation of early precancerous intestinal crypt foci, highlighting the impact of mechanical forces on tumor progression [58]. In Drosophila, it was elegantly shown that apoptotic cells display an apical‐basal acto‐myosin pulling force responsible for tissue folding [59, 60]. This concept is supported by the work of Yamaguchi et al. [61] that highlights the role of apoptotic cells in facilitating neural tube closure. These studies suggest that apoptotic cells may play an active role in signaling to environmental cells through their distinctive biophysical properties. How this crosstalk can actually impact cell migration, metastasis, or access to nutrients and oxygen is still under investigation.
Long‐range oncogenic effects of apoptotic cells
The concept of the ‘onco‐regenerative niche’ proposed by Gregory and Paterson suggests that cell death in tumors provides a central driving force in shaping the tumor microenvironment in short and distal settings, by orchestrating a series of interconnected cellular responses and extracellular modifications that promote tumor growth, progression, and relapse [62]. In this concept, TAMs (activated by apoptotic cells) and extracellular vesicles (EVs, produced by apoptotic cells) are proposed as two important components of the onco‐regenerative niche [62].
Since apoptotic cells are active players in a dynamic microenvironment, it is important to highlight that they are able to communicate with their neighboring cells even over a long distance, through the release of bioactive molecules into the extracellular environment and through production of membrane‐bound EVs, including apoptotic bodies (Fig. 1B). EVs are classified on the basis of their morphology, biogenesis, or contents into three main categories: apoptotic bodies, microvesicles, and exosomes. Among them, apoptotic bodies produced by cells undergoing programmed cell death are the largest, with sizes ranging from 50 to 5000 nm in diameter [63]. Cargoes associated with apoptotic‐derived EVs (Apo‐EVs) and apoptotic bodies include integral plasma membrane proteins, enzymes, and RNAs, with apoptotic bodies also harboring organelles and nuclear components including DNA and histones. With direct relevance for cancer, apoptotic bodies are known to be able to take up oncogenes, possibly driving tumor growth and progression [62] (Fig. 1B). In addition, Apo‐EVs from endothelial cells were shown to have immunomodulatory properties [64, 65]. The pro‐oncogenic role of Apo‐EVs is supported by a recent study in which Pavlyukov and collaborators reported that apoptotic cell‐derived EVs promote malignancy of glioblastoma through the intercellular transfer of splicing factors. In this study, the authors demonstrated that apoptotic glioblastoma cells paradoxically promoted proliferation and resistance to therapy of surviving tumor cells by secreting Apo‐EVs enriched in various components of spliceosomes. These Apo‐EVs alter RNA splicing in recipient cells, thereby promoting their resistance to therapy and aggressive migratory phenotype [66]. Apo‐EVs could also mediate drug resistance. For instance, in pancreatic ductal adenocarcinoma, EVs produced by cancer‐associated fibroblasts as a consequence of chemotherapy transfer Snail and miR‐146a to cancer cells, enhancing both their survival and proliferation during treatment [65].
Phosphatidylserine is another well‐established ‘eat‐me’ signal exposed by early apoptotic cells on the plasma membrane, in a process mediated by active caspases [67, 68]. One compelling finding is that apoptotic cells release PS‐charged microblebs (small EVs) that can alter the behavior of normal cells, both in proximity of and at a distance from the site of apoptosis. Specifically, microblebs derived from apoptotic endothelial cells can be taken up by target surviving endothelial cells via the PS receptor and mediate the protection of endothelial cells from camptothecin‐induced cell death by inhibiting p38 activity (Fig. 1B) [69].
How failed apoptosis shapes cancer aggressiveness
The widely accepted dogma is that apoptosis is an obstacle to cancer development. This assumption however is not compatible with an increasing body of evidence implying that proteins involved in apoptosis execution can in fact drive oncogenesis. Indeed, in several cancers, there is a strong correlation between high anti‐apoptotic BCL‐2 levels and favorable prognosis [70, 71, 72, 73], whereas high expression of pro‐apoptotic BAX is correlated with a poor outcome in certain cancers [74, 75]. Far from being mutated or lost through genetic alterations, caspase‐3 overexpression was reported in a variety of cancers, arguing in favor of noncanonical roles for caspases in oncogenesis [76]. In addition, caspase‐3 is associated with poor survival in several cancers such as colorectal, gastric, ovarian, cervical, or oral carcinoma [77, 78, 79].
Recent data show that apoptosis execution in cancer cells can be faulty, with cancer cells undergoing failed apoptosis. This is characterized by limited MOMP (minority MOMP) and subsequent low‐level caspase activation, which can induce DNA damage and genomic instability and ultimately favors tumorigenesis. In this section, we will therefore discuss recent studies on the role of nonlethal caspase activation in cancer.
Caspases and genomic instability
Most studies on caspases have focused on their roles as cell killers. However, increasing evidence highlights a variety of roles for these cysteine proteases within the cell, being either normal or cancerous. These functions are as diverse as signal transduction and cytoskeletal remodeling, and caspases are now known to play an essential role in cell proliferation, migration, and differentiation, which is particularly relevant in the context of cancer [80, 81].
Recently, it was demonstrated that radiotherapy, chemotherapeutic drugs or BH3‐only proteins such as BCL2‐interacting killer (BIK) can activate caspases at nonlethal levels in surviving cells following minority MOMP [82, 83, 84]. Despite the fact that these studies proposed different DNA damaging mechanisms (Caspase‐Activated DNAse or CAD and endonuclease G, EndoG, respectively), they all reported that surviving cells are heavily impacted by unresolved DNA damage leading to genomic instability and tumorigenesis (Fig. 2). In addition, several reports shed light on a dark side of caspase activity impacting cell survival and tumorigenesis. Remarkably, sublethal doses of TRAIL or Fas ligand were shown to induce DNA damage and genetic mutations in a caspase‐8‐ and CAD‐dependent manner [85, 86]. Similarly, a recent study associated nonlethal caspase activation with the transformation of Barrett’s esophageal cells to cancer cells following bile acid‐induced repetitive minority MOMP [87]. This conflicting role for caspases has drawn attention to their use in cancer therapy, since the BH3‐mimetic ABT‐737, the prototype of anticancer drugs targeting the pro‐survival BCL2 family proteins, induces low‐level caspase activation accompanied by DNA damage [88]. In addition, cells undergoing anastasis, which is a process of reversing late‐stage apoptosis, are also heavily impacted by DNA damage, which in some cells leads to oncogenic transformation [89].
Fig. 2.
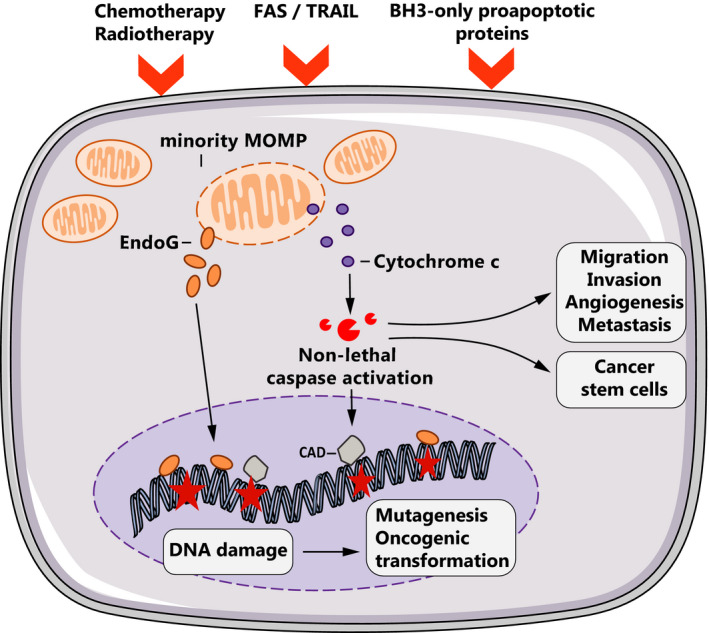
Oncogenic effects of nonlethal caspase activation. Suboptimal apoptotic stimuli can engage minority MOMP, which releases several apoptogenic factors such as cytochrome c or EndoG. Cytochrome c leads to nonlethal effector caspase activation and derepression of caspase‐activated DNAse (CAD). Together with EndoG, CAD cleaves genomic DNA causing oncogenic mutations that can lead to genomic instability and tumorigenesis. Nonlethal caspase activation can also promote cancer cell migration, invasion, and stemness (induction of CSCs).
Recently, Cartwright et al. further explored the role of sublethal caspase‐3 activation in facilitating Myc‐induced genetic instability and carcinogenesis. They proposed that EndoG, an apoptotic nuclease downstream of caspase‐3, was directly responsible for Myc‐induced genetic instability and malignant transformation in mammalian cells [90].
In recent years, there has been an increasing amount of literature supporting a causal role for caspase‐dependent DNA damage in genomic instability and increased predisposition to certain cancers. Translocations of the Mixed Lineage Leukemia (MLL) gene are very common in infant or secondary (therapy‐related) acute leukemia [91]. Interestingly, etoposide‐induced MLL translocation was shown to require caspase activity and CAD [92]. The mitochondrial‐resident EndoG was also proposed to cleave MLL locus under aphidicolin‐induced replicative stress [91].
Sustained DNA damage can also lead to a process known as chromothripsis, a mechanism of chromosome fragmentation that can promote the acquisition of numerous cancer‐driving mutations [93]. Since DNA cleavage is a key aspect of programmed cell death, it has been proposed that chromothripsis may occur when apoptosis is transiently initiated and then aborted [89, 94]. Cells that survive such a drastic event and hence evade apoptosis may sustain several pro‐oncogenic alterations, such as gene fusions, loss of tumor suppressor functions, and/or the amplification of certain oncogenes [95].
These findings suggest that minority MOMP and the subsequent nonlethal caspase activation could favor oncogenic transformation and promote the emergence of more aggressive tumor cells. Hence, understanding these caspase‐mediated oncogenic effects may also give rise to potentially important therapeutic possibilities.
Caspases and metastasis
Metastasis involves the spreading of cancer cells from the primary tumor to other sites in the body and is one of the deadliest consequences of cancer, resulting in the majority of cancer‐related deaths [96]. Thus, understanding the mechanisms that lead to invasion and metastasis are critical for the identification of novel therapeutic targets. To achieve enhanced motility, cells continuously deform and rearrange their cytoskeleton. Caspases are major modifiers of cytoskeletal structure during apoptosis; therefore, it is conceivable that caspases may also play a role in controlling cell motility (Fig. 2). Several studies demonstrated a role for caspases in cell invasion and cellular transformation. However, an activating or inhibiting role for caspases on metastasis depends on the model studied [97].
In mammalian models, a role for caspases has been uncovered in cell migration. Zhou et al. demonstrated that CASP3 knockout colon cancer cells are markedly less invasive and more sensitive to radiotherapy in vitro and in vivo. More interestingly, cells deficient in caspase‐3 were less prone to generate pulmonary metastasis when inoculated subcutaneously or intravenously [98].
Caspase‐8 is also involved in cell migration and metastasis. Caspase‐8 mutations in head and neck squamous cell carcinoma ES confer resistance to death receptor‐mediated apoptosis and enhance migration, invasion, and tumor growth [99]. Mechanistically, caspase‐8 interacts with multicomplex proteins to enhance cleavage of focal adhesion substrates and cell migration. Senft et al. [100] reported that caspase‐8 contributes to cell migration via its interaction with p85, a subunit of phosphatidylinositol 3‐kinase. Furthermore, caspase‐8 knockdown disrupts metastasis in neuroblastoma in vivo [101]. Similarly, it was also reported that inhibition of caspase‐3 and caspase‐8 decreased glioblastoma cell migration and invasion and that this caspase‐dependent motility was mediated by the cleavage of the motility‐associated gelsolin protein [102]. These studies support a role for caspases in promoting cell migration and metastasis (Fig. 2). To further investigate the link between nonlethal caspase activation and cancer aggressiveness, Berthenet et al. recently isolated melanoma cells undergoing failed apoptosis following BH3‐only protein expression or chemotherapy [103]. Interestingly, these cells displayed enhanced migration and invasion through a JNK‐dependent mechanism, supporting recent publications [104, 105]. In addition, they defined a failed apoptotic gene signature which can discriminate primary from metastatic melanoma tumors. Cancer cells undergoing anastasis also present a phenotypic switch from proliferation to migration, with activation of Transforming growth factor beta signaling and Snail expression that are known to promote epithelial–mesenchymal transition (EMT) [106].
Aside from these migration‐associated features, caspases are also involved in angiogenesis, a process allowing tumor neovascularization. Angiogenesis plays a key role in cancer aggressiveness by providing tumor cells with oxygen and nutrients, and by opening new paths for cancer dissemination. Interestingly, Bernard et al. [107] recently showed that activated caspase‐3 can act as a transcription factor to induce VEGFA expression and promote angiogenesis. Accordingly, anastatic cells also have a higher expression of angiogenesis‐related genes (PGF, EPHA2, and SPRY2), suggesting an increased potential to trigger tumor neovascularization [106, 108].
In contrast, several studies in flies have proposed an antagonistic role for caspase in cell migration. In a model of larval wing epithelium migration, Rudrapatna et al. generated undead epithelial cells via the expression of Hid (SMAC/Diablo orthologue) while preventing lethal effector caspase activation through the expression of the baculovirus caspase inhibitor p35. Intriguingly, these cells gained in migratory and invasive properties, partly due to JNK‐driven metalloproteinase 1 expression [109, 110]. In contrast, a recent study from Arama et al. showed that caspases act as inhibitors of cell motility, invasion and EMT in a model of epithelial cell migration in irradiated developing Drosophila wings [97]. Using elegant genetic and imaging approaches, they showed that nonlethal levels of caspase activation inhibit irradiation‐induced cell migration (ICM) in Drosophila. They further showed that ICM requires the Rho family of GTPases and the ATR DNA repair pathway. An inhibitory nonapoptotic role for caspase in cell migration is also supported by another recent study, showing that Dronc activation coupled with ROS production delays thorax closure, which is a model of wound healing and that requires collective cell migration [111]. An interesting fact, which may help to reconcile this apparent conflicting role of caspases in ICM, is that enhanced cell motility is only observed when the Drosophila tissue is entirely mutated for caspases, suggesting that exogenous factors cleaved by caspases are present in the invaded tissue to limit ICM [97]. These studies in Drosophila contradict the study using CASP3 KO cancer cells injected in otherwise wild‐type mice showing that cell invasion could be antagonized by caspase inhibition [98]. These paradoxical results in Drosophila and in mammalian cells may be due to the fact that caspases positively regulate cell invasion in a cell‐autonomous manner and negatively in a nonautonomous fashion in the invaded tissue. These different findings could also arise from the choice of experimental setup. Firstly, for caspase activation Rudrapatna et al. opted for Dronc activation, while effector caspases were inhibited, which is a cellular model known to elicit a strong secretory phenotype, and the Arama group triggered caspase activation further upstream of the apoptotic cascade, by using irradiation. Secondly, different types of cellular migration (ICM triggers an EMT‐like invasion centered on single cell invasion while thorax closure implies collective cell migration) may involve different caspases and levels of activation.
Caspases and stemness
Cancer stem cells are involved in tumorigenesis, metastasis, drug resistance, and relapse [112]. Within the tumor bulk, CSCs have the capacity to self‐renew, differentiate, and give rise to a new tumor. This process is precisely regulated by various modulators, including WNT/β‐catenin, NOTCH and Hedgehog signaling, chromatin remodeling complexes, and noncoding RNAs [112].
Caspases were described to regulate stem cell properties in a wide variety of embryonic and adult tissues, and they are thus prevalent in many cellular processes during development and adulthood. More specifically, caspases play an important role in stem cell differentiation. Fujita et al. showed that caspase activity increases during retinoic acid‐driven differentiation, a scenario requiring cleavage and inactivation of the stemness transcriptional factor NANOG by active caspase‐9. Of note, chemical inhibition of caspase activation using pan‐caspase inhibitors or caspase‐3 KO hinders the differentiation process [113]. At a more physiological level, caspases can also influence muscle regeneration from resident stem cells called satellite cells. The stemness of satellite cells is maintained by the transcription factor Pax7. Importantly, caspase‐3 can cleave Pax‐7, initiating the myogenic differentiation program [114]. Fernando et al. [115] reached a similar conclusion in an in vitro model of skeletal muscle differentiation. The authors identified Mst1 as being activated by caspase‐mediated proteolysis and initiating myogenesis. The same group extended their findings to neurosphere differentiation [116]. Cardiomyocyte regeneration equally requires nonlethal caspase activation: Low‐level stresses trigger caspase activation that further drives cardiomyocyte differentiation [117].
Intense interest surrounds induced pluripotent stem cells that can be obtained from virtually all differentiated cells through the expression of a particular mix of transcription factors [118]. One study links the re‐induction of pluripotency with OCT4‐driven expression of caspase‐3 and caspase‐8. Mechanistically, the authors demonstrated that caspase‐8 cleaves and inactivates RB, a differentiation state enforcer [119]. Caspases can also be involved in maintaining stem cells quiescence. For hematopoietic stem cells (HSCs), cellular quiescence is particularly important to ensure durable life‐long repopulation of all blood and immune cells. In caspase‐3 KO mice, HSCs have an increased sensitivity to exogenous proliferation signals leading to accelerated differentiation and proliferation [120]. The authors therefore concluded that caspases limit the proliferating signals and enforce the quiescence of HSCs [120].
Since caspases play an important role in orchestrating the fate of normal stem cells, several studies tested this interplay in CSCs (Fig. 2). The group of Chuan‐Yuan Li recently described that a large panel of cancer cells sustained spontaneous DNA double‐strand breaks through limited mitochondrial permeabilization and nonlethal activation of caspases and nucleases [121]. With relevance for CSCs, these DNA lesions chronically activate an Ataxia telangiectasia mutated (ATM)‐signal transducer and activator of transcription 3 (STAT3)‐NF‐κB axis that sustains tumorigenicity and promotes cancer stemness in a patient‐derived glioma model [121]. The role of ATM in CSCs is also supported by studies in glioblastoma and breast cancer where ATM activation was correlated with increased survival and radioresistance of CSCs [122, 123, 124]. The population of CSCs can also increase after failed apoptosis in human mammary carcinoma. Xu et al. referred to this as ‘reverse apoptosis’, reporting that mammary carcinoma cells initiating apoptosis under staurosporine or paclitaxel treatment could recover with increased capacity to induce lymph node metastasis. This is associated with an elevated percentage of cells with CSC characteristics (CD44+/CD24− surface markers) in the surviving cells [125]. Along this line, BIK‐triggered failed apoptosis in breast cancer cells was also reported to increase their stem‐like characteristics [84].
These results suggest that nonlethal caspase activation can promote cancer aggressiveness through the induction and maintenance of CSCs (Fig. 2). However, the mechanisms underlying this noncanonical regulation need to be further explored, in order to understand the full impact of nonlethal caspase activation in tumor cells.
Mitochondria, key apoptotic mediators of tumor aggressiveness
Mitochondria are vital organelles involved in critical processes such as energy production, calcium homeostasis, or redox balance. There are several major pro‐oncogenic mechanisms involving mitochondria such as oncogenic mtDNA mutations, oxidative stress, regulation of cell death, or metabolic reprogramming. Since these canonical functions were extensively described in recent reviews, [126, 127] we will briefly outline here how mitochondria can initiate and foster the oncogenic process through less known mechanisms involving partial mitochondrial permeabilization, mitochondrial transfer, and mitochondrial dynamics (Fig. 3).
Fig. 3.
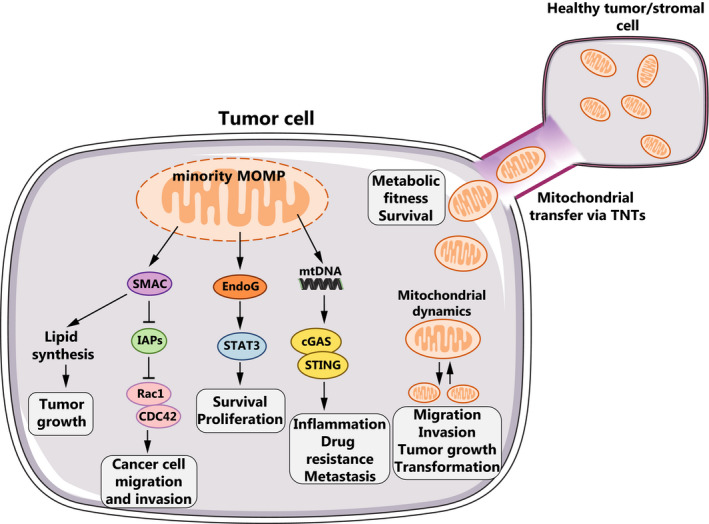
Apoptosis‐related oncogenic roles of mitochondria. Minority MOMP can release several pro‐apoptotic molecules such as SMAC, EndoG and mtDNA. Specifically, SMAC promotes growth and metastasis in surviving tumor cells by increasing lipid synthesis and lifting IAP‐mediated inhibition on the Rho GTPases Rac1 or Cdc42. EndoG promotes survival and proliferation through the activation of the STAT3 signaling pathway. MtDNA released from permeabilized mitochondria is sensed by the cGAS‐STING pathway and stimulates inflammation and metastasis. Mitochondrial dynamics (fission and fusion) can also sustain cancer cell migration, tumor growth and oncogenic transformation. Mitochondrial transfer via TNTs from neighboring healthy cells promotes tumor cell survival by enhancing the metabolic fitness of damaged cancer cells.
Impact of limited mitochondrial permeabilization in cancer progression
It was commonly believed that MOMP irreversibly induces apoptosis when triggered in all mitochondria. However, as discussed above, some cells can resist apoptosis due to the engagement of MOMP in a limited number of mitochondria without killing the cell [12, 82, 103]. Several cell signaling modulators can thus be released upon minority MOMP that may have a significant impact on tumor progression. For example, the SMAC/Diablo protein, which usually resides in the mitochondrial intermembrane space, is released following MOMP and promotes apoptosis by negatively regulating inhibitor of apoptosis proteins (IAPs). Interestingly, IAPs can function as direct E3 ubiquitin ligases for the Rho GTPase Rac1, inducing its proteasomal degradation (Fig. 3). As a consequence, SMAC‐mediated stabilization of Rac1 promotes a mesenchymal mode of migration in tumor cells [128, 129]. Similarly, the same group reported that melanoma‐IAP regulates C‐RAF stability and cell migration, while XIAP can also promote cancer cell migration through its contribution to Cdc42 homeostasis [130, 131, 132]. However, it remains unclear whether SMAC/Diablo released from the mitochondria upon minority MOMP can have an impact on cancer cell migration by negatively regulating IAP expression. Recently, another study strongly supported the role of the SMAC/Diablo protein in tumorigenesis. Indeed, SMAC/Diablo was reported to be over‐expressed in cancer cells despite its role in promoting cell death, suggesting it might possess nonapoptotic functions. In this research, the authors demonstrated that SMAC/Diablo displays additional roles related to the regulation of lipid synthesis essential for cancer growth and development, which may partially explain the overexpression of this protein in various cancers, including lung, B‐cell lymphoma, testis, colon, stomach, breast, prostate, and skin cancer [133, 134]. EndoG is another factor released from the mitochondria and as mentioned before it promotes the activation of NF‐κB and STAT3 signaling pathways, leading to the survival and proliferation of tumor cells (Fig. 3) [121].
Additionally, mtDNA could be involved in tumorigenesis, metastasis, and chemoresistance. For instance, Neuzil et al. reported that in the absence of mtDNA, tumor cells display a remarkably delayed tumor growth. MtDNA‐depleted tumor cells may actively acquire mtDNA from surrounding cells, thereby restoring respiratory function and even tumorigenic potential [135, 136]. This finding indicates that tumor cells may seek for and obtain functional mtDNA from the surrounding microenvironment via horizontal transfer. Therefore, it could be hypothesized that mtDNA released upon minority MOMP could also have an impact on cancer. Acting as a DAMP, mtDNA can be a potent innate immunity activator. Indeed, mtDNA released from mitochondria as a result of the permeabilizing activity of pro‐apoptotic BAX and BAK proteins can trigger the activation of the interferon pathway via the cyclic GMP‐AMP synthase (cGAS) and the stimulator of IFN genes protein (STING) pathway, which stimulates interferon production [137, 138, 139]. Although crucial for innate immunity and fight against viral pathogens, sustained and uncontrolled activation of cGAS‐STING and the downstream signaling cascade is responsible for chronic inflammation, resistance to chemotherapy and metastasis [140, 141, 142, 143] (Fig. 3). While cGAS‐STING activation is undeniably involved in cancer progression, the exact contribution of mtDNA to this effect remains unclear, as well as the relevance of minority MOMP to release sufficient amounts of mtDNA to efficiently activate this inflammatory pathway. A more plausible way through which cancer cells could maintain chronic cGAS‐STING‐driven inflammation would be through the acquisition of extra‐tumoral mtDNA [143].
Thus, it is tempting to speculate that the release of mitochondrial factors upon minority MOMP may promote tumor progression, supporting a noncanonical role for mitochondria beyond the induction of apoptosis.
Mitochondrial transfer
The mechanisms underlying cancer cell recovery from apoptosis are under investigation; however, it seems that mitochondrial transfer from healthy cells may rescue apoptotic cells from death (Fig. 3). Therefore, recent work suggests that this may also be relevant for tumor cell survival following an early apoptotic signaling. Wang et al. found that the delivery of functional mitochondria from healthy cells to early apoptotic cells via tunneling nanotubes (TNTs) mediates the recovery of UV‐treated PC12 cells in the early stages of apoptosis [144]. Mitochondrial transfer has also been reported between cardiomyocytes and cardiac myofibroblasts, along membrane nanotubes to rescue distressed cardiomyocytes from apoptosis [145].
Remarkably, this process has also been described in cancer cells and particularly in the leukemic microenvironment, where mitochondrial transfer between bone marrow mesenchymal stromal cells and leukemic cells protects cancer cells from cytarabine treatment [146]. Moreover, the uptake of mitochondria by leukemic cells increases oxidative phosphorylation and favors survival (Fig. 3) [146, 147]. Pasquier et al. also investigated the formation of heterocellular TNTs between stromal, mesenchymal or endothelial cells, and cancer cells. These TNTs preferentially mediated mitochondrial exchange from stromal to cancer cells, resulting in the emergence of chemoresistance to doxorubicin. The preferential mitochondrial shuttling from stromal cells toward cancer cells adds another layer of complexity to the metastatic niche [148]. Given that TNT‐mediated mitochondrial transfer is almost exclusively unidirectional, from healthy to damaged cells, one might wonder whether molecular cues expressed by apoptotic cells can also act as ‘rescue me’ signals, attracting mitochondrial‐loaded TNTs. This issue was addressed by Liu et al. by shielding surface‐exposed PS on apoptotic endothelial cells with Annexin V. Unexpectedly, this blocked the formation of TNTs between donor and apoptotic cells, indicating that PS selectively guides TNTs toward damaged cells [147, 149]. These findings are important for cancer therapy, where mitochondrial transfer to cancer cells damaged by chemo‐ or radiotherapy could protect certain cancer cells from therapy. One might envision combining chemotherapy with inhibitors of TNT formation and guidance such as actin polymerization inhibitors or masking PS on apoptotic cancer cells [147].
Understanding the role of mitochondrial transfer in defined biological contexts is limited by the experimental tools available. This was improved by the MitoCeption protocol developed by the Vignais team that permitted direct and quantitative mitochondrial transfer from one cell to another. They showed that the transfer of mesenchymal stem cell‐derived mitochondria boosts the energetic metabolism in recipient cancer cells while fostering cancer cell proliferation and invasion [150]. More recently, they also demonstrated the role of mitochondrial transfer as a novel mechanism in the regulation of T cell function, whereas uptake of mitochondria promotes an anti‐inflammatory phenotype in Th17 cells [151]. Surprisingly, recent studies found that circulating blood contains cell‐free respiratory competent mitochondria [152]. This implies that cancer cell‐derived mitochondria might transmit their pro‐oncogenic signals at the level of the whole organism. In summary, mitochondrial transfer seems to arise as a relevant survival strategy for cancer cells in order to recover from apoptosis and enhance tumor progression. Thus, this pro‐survival mechanism should seriously be taken into account and specifically targeted when developing cancer therapies.
Mitochondrial dynamics
The mitochondrial network is highly dynamic since it undergoes perpetual cycles of fusion and fission. Mechanistically, mitochondrial fusion and fission are executed by evolutionarily conserved GTPases that either constrict and fragment the mitochondria [dynamin‐related protein 1 (DRP1)] or tether and fuse together shorter mitochondria to form elongated organelles [Mitofusin 1 (MFN1), MFN2, and Optic atrophy protein 1] [153, 154]. Interestingly, during early apoptotic stages, the mitochondrial network is dramatically altered, leaning toward complete fission [155]. Similarly, abnormal mitochondrial fusion and fission are observed in cancer cells where they could impact tumor migration and invasion. This notion is supported by Zhao et al., who found an upregulation of mitochondrial fission protein DRP1 expression in human invasive breast carcinoma and metastases to lymph nodes. This was associated with a higher mitochondrial fission and preferential distribution of fragmented mitochondria in lamellipodia, which are wave‐like cellular extensions at the leading edge of migrating cells. Their results suggest that DRP1‐dependent mitochondrial fission is critical for breast cancer cell migration and invasion (Fig. 3) [156]. These findings reflect those of Desai et al. who also found that preferential anterior distribution of mitochondria is correlated with faster and more persistent cancer cell migration. This correlation is particularly relevant during confined cell migration conditions, similar to those encountered by metastatic cells in vivo [157]. With a direct impact on cancer therapy, the distribution of elongated and active mitochondria within the lamellipodia was also observed as an adaptive response of cancer cells to treatment with PI3K inhibitors that are currently used in clinical applications. As a consequence, mitochondrial repositioning in the cortical cytoskeleton sustains the turnover of focal adhesion complexes and therefore facilitates cell migration and invasion [158, 159, 160]. Mechanistically, Caino et al. established that cancer cells reprogram a network of proteins involved in mitochondrial intracellular trafficking centered on syntaphilin to shuttle mitochondria to the cortical cytoskeleton and sustain metastasis [161, 162].
Well‐characterized oncogenic signaling pathways can also hijack mitochondrial dynamics and favor cancer aggressiveness (Fig. 3). Specifically, oncogenic Myc transcriptionally controls the expression of several proteins involved either in mitochondrial trafficking (RHOT1, RHOT2, TRAK2, and Kif5B) or fission (DRP1). This facilitates the redistribution of mitochondria in lamellipodia to boost tumor cell invasion [163]. DRP1‐mediated mitochondrial fission was also shown to be a crucial regulator of MAPK‐driven tumor growth, while also mediating Ras‐induced transformation [164, 165]. These studies suggest that mitochondrial dynamics are linked with tumor progression (Fig. 3) raising the question whether one can use mitochondrial shape and intracellular localization to specifically identify highly metastatic cancer cells. Moreover, inhibition of either fusion or fission might be considered when targeting cancer metastasis.
Concluding remarks
Triggered by radiotherapy and most chemotherapeutic drugs, apoptosis blunts tumor growth in a wide variety of cancers. However, in the context of cancer therapy, inducing apoptosis might not always be pertinent. As we have discussed in this review, apoptosis per se or failed apoptosis exert oncogenic functions in certain circumstances, with caspases and mitochondria mediating most of the pro‐oncogenic effects. These noncanonical tumorigenic functions range from tumor cell proliferation, survival, genetic instability, stemness, angiogenesis, and metastasis, all of which may promote tumor growth and emergence of more aggressive cancers. Therefore, since apoptosis might be deleterious toward cancer treatment in certain contexts, we could consider inducing other forms of cell death, such as MOMP‐induced caspase‐independent cell death (or CICD). Indeed, CICD is highly immunogenic and it lacks the capacity to induce the proliferation of surviving cancer cells, making it a promising alternative to apoptosis when envisioning novel therapeutic approaches [166, 167, 168, 169]. Other strategies could involve the use of caspase inhibitors to prevent unwanted effects linked with nonlethal caspase activation or caspase activators to raise this nonlethal caspase activation to lethal levels and effectively kill cancer cells. PGE2 inhibitors could also be used to prevent AiP.
Taken together, we suggest that understanding the mechanisms regulating the balance between the tumor suppressor and oncogenic functions of apoptotic signaling may lead to more effective therapeutic strategies in the treatment of highly aggressive tumors.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
All authors drafted, edited, and revised the manuscripts.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.15624
[Correction added on 04 February 2021, after first online publication: URL for peer review history has been corrected.]
Acknowledgements
This work was supported by funding from LabEx DEVweCAN (University of Lyon), Agence Nationale de la Recherche (ANR) Young Researchers Project (ANR‐18‐CE13‐0005‐01), Rhône‐Alpes and Saône‐et‐Loire Ligue Contre le Cancer grant, ANR PLAsCAN Institute and a fellowship from Fondation de France (00108257). We would like to thank Brigitte Manship for editorial suggestions.
References
- 1.Fuchs Y & Steller H (2015) Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol 16, 329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DWet al. (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfeffer CM & Singh ATK (2018) Apoptosis: a target for anticancer therapy. Int J Mol Sci 19, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thoma C (2017) Prostate cancer: potential biomarkers of aggressive disease. Nat Rev Urol 14, 639. [DOI] [PubMed] [Google Scholar]
- 5.La Porta CAM & Zapperi S (2018) Explaining the dynamics of tumor aggressiveness: at the crossroads between biology, artificial intelligence and complex systems. Semin Cancer Biol 53, 42–47. [DOI] [PubMed] [Google Scholar]
- 6.Labi V & Erlacher M (2015) How cell death shapes cancer. Cell Death Dis 6, e1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ford CA, Petrova S, Pound JD, Voss JJ, Melville L, Paterson M, Farnworth SL, Gallimore AM, Cuff S, Wheadon Het al. (2015) Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr Biol 25, 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alcaide J, Funez R, Rueda A, Perez‐Ruiz E, Pereda T, Rodrigo I, Coveñas R, Muñoz M & Redondo M (2013) The role and prognostic value of apoptosis in colorectal carcinoma. BMC Clin Pathol 13, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dworakowska D, Jassem E, Jassem J, Karmoliński A, Lapiński M, Tomaszewski D, Rzyman W, Jaśkiewicz K, Sworczak K & Grossman AB (2009) Prognostic value of the apoptotic index analysed jointly with selected cell cycle regulators and proliferation markers in non‐small cell lung cancer. Lung Cancer 66, 127–133. [DOI] [PubMed] [Google Scholar]
- 10.Langendijk H,Thunnissen E, Arends JW, de Jong J , ten Velde G , Lamers R, Guinee D, Holden J & Wouters M (2000) Cell proliferation and apoptosis in stage III inoperable non‐small cell lung carcinoma treated by radiotherapy. Radiother Oncol 56, 197–207. [DOI] [PubMed] [Google Scholar]
- 11.Mollereau B, Perez‐Garijo A, Bergmann A, Miura M, Gerlitz O, Ryoo HD, Steller H & Morata G (2013) Compensatory proliferation and apoptosis‐induced proliferation: a need for clarification. Cell Death Differ 20, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ichim G & Tait SW (2016) A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer 16, 539–548. [DOI] [PubMed] [Google Scholar]
- 13.Martin FA, Perez‐Garijo A & Morata G (2009) Apoptosis in Drosophila: compensatory proliferation and undead cells. Int J Dev Biol 53, 1341–1347. [DOI] [PubMed] [Google Scholar]
- 14.Mollereau B & Ma D (2014) The p53 control of apoptosis and proliferation: lessons from Drosophila. Apoptosis 19, 1421–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fogarty CE & Bergmann A (2015) The sound of silence: signaling by apoptotic cells. Curr Top Dev Biol 114, 241–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez E, Lindblad JL & Bergmann A (2017) Tumor‐promoting function of apoptotic caspases by an amplification loop involving ROS, macrophages and JNK in Drosophila. Elife 6, e26747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fogarty CE, Diwanji N, Lindblad JL, Tare M, Amcheslavsky A, Makhijani K, Brückner K, Fan Y & Bergmann A (2016) Extracellular reactive oxygen species drive apoptosis‐induced proliferation via drosophila macrophages. Curr Biol 26, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amcheslavsky A, Wang S, Fogarty CE, Lindblad JL, Fan Y & Bergmann A (2018) Plasma membrane localization of apoptotic caspases for non‐apoptotic functions. Dev Cell 45, 450–464 e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryoo HD, Gorenc T & Steller H (2004) Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell 7, 491–501. [DOI] [PubMed] [Google Scholar]
- 20.Fan Y, Wang S, Hernandez J, Yenigun VB, Hertlein G, Fogarty CE, Lindblad JL & Bergmann A (2014) Genetic models of apoptosis‐induced proliferation decipher activation of JNK and identify a requirement of EGFR signaling for tissue regenerative responses in Drosophila. PLoS Genet 10, e1004131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shlevkov E & Morata G (2012) A dp53/JNK‐dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ 19, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wells BS, Yoshida E & Johnston LA (2006) Compensatory proliferation in Drosophila imaginal discs requires Dronc‐dependent p53 activity. Curr Biol 16, 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez‐Garijo A, Martin FA & Morata G (2004) Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 131, 5591–5598. [DOI] [PubMed] [Google Scholar]
- 24.Santabarbara‐Ruiz P, López‐Santillán M, Martínez‐Rodríguez I, Binagui‐Casas A, Pérez L, Milán M, Corominas M & Serras F (2015) ROS‐induced JNK and p38 signaling is required for unpaired cytokine activation during drosophila regeneration. PLoS Genet 11, e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fogarty CE & Bergmann A (2017) Killers creating new life: caspases drive apoptosis‐induced proliferation in tissue repair and disease. Cell Death Differ 24, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagedorn EJ, Durand EM, Fast EM & Zon LI (2014) Getting more for your marrow: boosting hematopoietic stem cell numbers with PGE2. Exp Cell Res 329, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS & Li CY (2010) Apoptotic cells activate the "phoenix rising" pathway to promote wound healing and tissue regeneration. Sci Signal 3, ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao P, Smith L, Xie W & Wang M (2013) Dying endothelial cells stimulate proliferation of malignant glioma cells via a caspase 3‐mediated pathway. Oncol Lett 5, 1615–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yosefzon Y, Soteriou D, Feldman A, Kostic L, Koren E, Brown S, Ankawa R, Sedov E, Glaser F & Fuchs Y (2018) Caspase‐3 regulates YAP‐dependent cell proliferation and organ size. Mol Cell 70, 573–587 e574. [DOI] [PubMed] [Google Scholar]
- 30.Donato AL, Huang Q, Liu X, Li F, Zimmerman MA & Li CY (2014) Caspase 3 promotes surviving melanoma tumor cell growth after cytotoxic therapy. J Investig Dermatol 134, 1686–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O'Sullivan B, He Z, Peng Y, Tan ACet al. (2011) Caspase 3‐mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 17, 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PLet al. (2015) Blocking PGE2‐induced tumor repopulation abrogates bladder cancer chemoresistance. Nature 517, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP & Meade TW (2010) Long‐term effect of aspirin on colorectal cancer incidence and mortality: 20‐year follow‐up of five randomised trials. Lancet 376, 1741–1750. [DOI] [PubMed] [Google Scholar]
- 34.Ulrich CM, Bigler J & Potter JD (2006) Non‐steroidal anti‐inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer 6, 130–140. [DOI] [PubMed] [Google Scholar]
- 35.Huang H, Aladelokun O, Ideta T, Giardina C, Ellis LM & Rosenberg DW (2019) Inhibition of PGE2/EP4 receptor signaling enhances oxaliplatin efficacy in resistant colon cancer cells through modulation of oxidative stress. Sci Rep 9, 4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SAet al. (2015) Cyclooxygenase‐dependent tumor growth through evasion of immunity. Cell 162, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garber K (2018) Cancer stem cell pipeline flounders. Nat Rev Drug Discov 17, 771–773. [DOI] [PubMed] [Google Scholar]
- 38.Cheng J, Tian L, Ma J, Gong Y, Zhang Z, Chen Z, Xu B, Xiong H, Li C & Huang Q (2015) Dying tumor cells stimulate proliferation of living tumor cells via caspase‐dependent protein kinase Cdelta activation in pancreatic ductal adenocarcinoma. Mol Oncol 9, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park SY & Kim IS (2017) Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Exp Mol Med 49, e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li F, Wang Z, Liu Y & Li J (2010) Down‐regulation of fractalkine inhibits the in vitro and in vivo angiogenesis of the hepatocellular carcinoma HepG2 cells. Oncol Rep 24, 669–675. [PubMed] [Google Scholar]
- 41.Tang J, Chen Y, Cui R, Li D, Xiao L, Lin P, Du Y, Sun H, Yu X & Zheng X (2015) Upregulation of fractalkine contributes to the proliferative response of prostate cancer cells to hypoxia via promoting the G1/S phase transition. Mol Med Rep 12, 7907–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tardaguila M & Manes S (2013) CX3CL1 at the crossroad of EGF signals: relevance for the progression of ERBB2 breast carcinoma. Oncoimmunology 2, e25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tardaguila M, Mira E, García‐Cabezas MA, Feijoo AM, Quintela‐Fandino M, Azcoitia I, Lira SA & Mañes S (2013) CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res 73, 4461–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boehme SA, Lio FM, Maciejewski‐Lenoir D, Bacon KB & Conlon PJ (2000) The chemokine fractalkine inhibits Fas‐mediated cell death of brain microglia. J Immunol 165, 397–403. [DOI] [PubMed] [Google Scholar]
- 45.Sun C, Hu A, Wang S, Tian B, Jiang L, Liang Y, Wang H & Dong J (2020) ADAM17‐regulated CX3CL1 expression produced by bone marrow endothelial cells promotes spinal metastasis from hepatocellular carcinoma. Int J Oncol 57, 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu P, Liang Y, Jiang L, Wang H, Wang S & Dong J (2018) CX3CL1/fractalkine enhances prostate cancer spinal metastasis by activating the Src/FAK pathway. Int J Oncol 53, 1544–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishida Y, Kuninaka Y, Yamamoto Y, Nosaka M, Kimura A, Furukawa F, Mukaida N & Kondo T (2020) Pivotal involvement of the CX3CL1‐CX3CR1 axis for the recruitment of M2 tumor‐associated macrophages in skin carcinogenesis. J Investig Dermatol 140, 1951–1961. [DOI] [PubMed] [Google Scholar]
- 48.Korbecki J, Simińska D, Kojder K, Grochans S, Gutowska I, Chlubek D & Baranowska‐Bosiacka I (2020) Fractalkine/CX3CL1 in neoplastic processes. Int J Mol Sci 21, 3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Werfel TA & Cook RS (2018) Efferocytosis in the tumor microenvironment. Semin Immunopathol 40, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bi C, Fu Y, Zhang Z & Li B (2020) Prostaglandin E2 confers protection against diabetic coronary atherosclerosis by stimulating M2 macrophage polarization via the activation of the CREB/BDNF/TrkB signaling pathway. FASEB J 34, 7360–7371. [DOI] [PubMed] [Google Scholar]
- 51.Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S & Montminy M (2015) CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci USA 112, 15642–15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Medina CB, Mehrotra P, Arandjelovic S, Perry JSA, Guo Y, Morioka S, Barron B, Walk SF, Ghesquière B, Krupnick ASet al. (2020) Metabolites released from apoptotic cells act as tissue messengers. Nature 580, 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma Pet al. (2009) Nucleotides released by apoptotic cells act as a find‐me signal to promote phagocytic clearance. Nature 461, 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang H, Geng YH, Wang P, Yang H, Zhou YT, Zhang HQ, He HY, Fang WG & Tian XX (2020) Extracellular ATP promotes breast cancer invasion and chemoresistance via SOX9 signaling. Oncogene 39, 5794–5810. [DOI] [PubMed] [Google Scholar]
- 55.Qian Y, Wang X, Li Y, Cao Y & Chen X (2016) Extracellular ATP a new player in cancer metabolism: NSCLC cells internalize ATP in vitro and in vivo using multiple endocytic mechanisms. Mol Cancer Res 14, 1087–1096. [DOI] [PubMed] [Google Scholar]
- 56.Orozco SL, Daniels BP, Yatim N, Messmer MN, Quarato G, Chen‐Harris H, Cullen SP, Snyder AG, Ralli‐Jain P, Frase Set al. (2019) RIPK3 activation leads to cytokine synthesis that continues after loss of cell membrane integrity. Cell Rep 28, 2275–2287. e2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weihua Z, Tsan R, Schroit AJ & Fidler IJ (2005) Apoptotic cells initiate endothelial cell sprouting via electrostatic signaling. Cancer Res 65, 11529–11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernandez‐Sanchez ME, Barbier S, Whitehead J, Béalle G, Michel A, Latorre‐Ossa H, Rey C, Fouassier L, Claperon A, Brullé Let al. (2015) Mechanical induction of the tumorigenic beta‐catenin pathway by tumor growth pressure. Nature 523, 92–95. [DOI] [PubMed] [Google Scholar]
- 59.Monier B, Gettings M, Gay G, Mangeat T, Schott S, Guarner A & Suzanne M (2015) Apico‐basal forces exerted by apoptotic cells drive epithelium folding. Nature 518, 245–248. [DOI] [PubMed] [Google Scholar]
- 60.Ambrosini A, Gracia M, Proag A, Rayer M, Monier B & Suzanne M (2017) Apoptotic forces in tissue morphogenesis. Mech Dev 144, 33–42. [DOI] [PubMed] [Google Scholar]
- 61.Yamaguchi Y,Shinotsuka N, Nonomura K, Takemoto K, Kuida K, Yosida H & Miura M (2011) Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. J Cell Biol 195, 1047–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gregory CD & Paterson M (2018) An apoptosis‐driven 'onco‐regenerative niche': roles of tumor‐associated macrophages and extracellular vesicles. Philos Trans R Soc Lond B Biol Sci 373, 20170003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kakarla R, Hur J, Kim YJ, Kim J & Chwae YJ (2020) Apoptotic cell‐derived exosomes: messages from dying cells. Exp Mol Med 52, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caruso S & Poon IKH (2018) Apoptotic cell‐derived extracellular vesicles: more than just debris. Front Immunol 9, 1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lynch C, Panagopoulou M & Gregory CD (2017) Extracellular vesicles arising from apoptotic cells in tumors: roles in cancer pathogenesis and potential clinical applications. Front Immunol 8, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pavlyukov MS, Yu H, Bastola S, Minata M, Shender VO, Lee Y, Zhang S, Wang J, Komarova S, Wang Jet al. (2018) Apoptotic cell‐derived extracellular vesicles promote malignancy of glioblastoma via intercellular transfer of splicing factors. Cancer Cell 34, 119–135 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suzuki J, Denning DP, Imanishi E, Horvitz HR & Nagata S (2013) Xk‐related protein 8 and CED‐8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406. [DOI] [PubMed] [Google Scholar]
- 68.Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F & Nagata S (2014) Caspase‐mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168. [DOI] [PubMed] [Google Scholar]
- 69.Jansen F, Yang X, Hoyer FF, Paul K, Heiermann N, Becher MU, Abu Hussein N, Kebschull M, Bedorf J, Franklin BSet al. (2012) Endothelial microparticle uptake in target cells is annexin I/phosphatidylserine receptor dependent and prevents apoptosis. Arterioscler Thromb Vasc Biol 32, 1925–1935. [DOI] [PubMed] [Google Scholar]
- 70.Huang Q, Li S, Cheng P, Deng M, He X, Wang Z, Yang C‐H, Zhao X‐Y & Huang J (2017) High expression of anti‐apoptotic protein Bcl‐2 is a good prognostic factor in colorectal cancer: result of a meta‐analysis. World J Gastroenterol 23, 5018–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Watson NF, Madjd Z, Scrimegour D, Spendlove I, Ellis IO, Scholefield JH & Durrant LG (2005) Evidence that the p53 negative / Bcl‐2 positive phenotype is an independent indicator of good prognosis in colorectal cancer: a tissue microarray study of 460 patients. World J Surg Oncol 3, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pillai K, Pourgholami MH, Chua TC & Morris DL (2013) Does the expression of BCL2 have prognostic significance in malignant peritoneal mesothelioma? Am J Cancer Res 3, 312–322. [PMC free article] [PubMed] [Google Scholar]
- 73.Vargas‐Roig LM, Cuello‐Carrión FD, Fernández‐Escobar N, Daguerre P, Leuzzi M, Ibarra J, Gago FE, Nadin SB & Ciocca DR (2008) Prognostic value of Bcl‐2 in breast cancer patients treated with neoadjuvant anthracycline based chemotherapy. Mol Oncol 2, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kohler T, Schill C, Deininger MW, Krahl R, Borchert S, Hasenclever D, Leiblein S, Wagner O & Niederwieser D (2002) High Bad and Bax mRNA expression correlate with negative outcome in acute myeloid leukemia (AML). Leukemia 16, 22–29. [DOI] [PubMed] [Google Scholar]
- 75.Kulsoom B, Shamsi TS, Afsar NA, Memon Z, Ahmed N & Hasnain SN (2018) Bax, Bcl‐2, and Bax/Bcl‐2 as prognostic markers in acute myeloid leukemia: are we ready for Bcl‐2‐directed therapy? Cancer Manag Res 10, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boudreau MW, Peh J & Hergenrother PJ (2019) Procaspase‐3 overexpression in cancer: a paradoxical observation with therapeutic potential. ACS Chem Biol 14, 2335–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flanagan L, Meyer M, Fay J, Curry S, Bacon O, Duessmann H, John K, Boland KC, McNamara DA, Kay EWet al. (2016) Low levels of Caspase‐3 predict favourable response to 5FU‐based chemotherapy in advanced colorectal cancer: caspase‐3 inhibition as a therapeutic approach. Cell Death Dis 7, e2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu Q, Peng J, Liu W, He X, Cui L, Chen X, Yang M, Liu H, Liu S & Wang H (2014) Elevated cleaved caspase‐3 is associated with shortened overall survival in several cancer types. Int J Clin Exp Pathol 7, 5057–5070. [PMC free article] [PubMed] [Google Scholar]
- 79.Liu PF, Hu YC, Kang BH, Tseng YK, Wu PC, Liang CC, Hou YY, Fu TY, Liou HH, Hsieh ICet al. (2017) Expression levels of cleaved caspase‐3 and caspase‐3 in tumorigenesis and prognosis of oral tongue squamous cell carcinoma. PLoS One 12, e0180620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McArthur K & Kile BT (2018) Apoptotic caspases: multiple or mistaken identities? Trends Cell Biol 28, 475–493. [DOI] [PubMed] [Google Scholar]
- 81.Connolly PF, Jager R & Fearnhead HO (2014) New roles for old enzymes: killer caspases as the engine of cell behavior changes. Front Physiol 5, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos Det al. (2015) Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57, 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP & Li CY (2015) Caspase‐3 promotes genetic instability and carcinogenesis. Mol Cell 58, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pandya V, Githaka JM, Patel N, Veldhoen R, Hugh J, Damaraju S, McMullen T, Mackey J & Goping IS (2020) BIK drives an aggressive breast cancer phenotype through sublethal apoptosis and predicts poor prognosis of ER‐positive breast cancer. Cell Death Dis 11, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miles MA & Hawkins CJ (2017) Executioner caspases and CAD are essential for mutagenesis induced by TRAIL or vincristine. Cell Death Dis 8, e3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lovric MM & Hawkins CJ (2010) TRAIL treatment provokes mutations in surviving cells. Oncogene 29, 5048–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu Y, Surman DR, Diggs L, Xi S, Gao S, Gurusamy D, McLoughlin K, Drake J, Feingold P, Brown Ket al. (2020) Bile acid‐induced "Minority MOMP" promotes esophageal carcinogenesis while maintaining apoptotic resistance via Mcl‐1. Oncogene 39, 877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Song JH, Kandasamy K, Zemskova M, Lin YW & Kraft AS (2010) The BH3 mimetic ABT‐737 induces cancer cell senescence. Cancer Res 71, 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, Wong CS, Wu HY, Law HT, Liu Ket al. (2012) Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell 23, 2240–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cartwright IM, Liu X, Zhou M, Li F & Li CY (2017) Essential roles of Caspase‐3 in facilitating Myc‐induced genetic instability and carcinogenesis. Elife 6, e26371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gole B, Baumann C, Mian E, Ireno CI & Wiesmuller L (2015) Endonuclease G initiates DNA rearrangements at the MLL breakpoint cluster upon replication stress. Oncogene 34, 3391–3401. [DOI] [PubMed] [Google Scholar]
- 92.Hars ES, Lyu YL, Lin CP & Liu LF (2006) Role of apoptotic nuclease caspase‐activated DNase in etoposide‐induced treatment‐related acute myelogenous leukemia. Cancer Res 66, 8975–8979. [DOI] [PubMed] [Google Scholar]
- 93.Koltsova AS, Pendina AA, Efimova OA, Chiryaeva OG, Kuznetzova TV & Baranov VS (2019) On the complexity of mechanisms and consequences of chromothripsis: an update. Front Genet 10, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tubio JM & Estivill X (2011) Cancer: when catastrophe strikes a cell. Nature 470, 476–477. [DOI] [PubMed] [Google Scholar]
- 95.Rode A, Maass KK, Willmund KV, Lichter P & Ernst A (2016) Chromothripsis in cancer cells: an update. Int J Cancer 138, 2322–2333. [DOI] [PubMed] [Google Scholar]
- 96.Lambert AW, Pattabiraman DR & Weinberg RA (2017) Emerging biological principles of metastasis. Cell 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gorelick‐Ashkenazi A, Weiss R, Sapozhnikov L, Florentin A, Tarayrah‐Ibraheim L, Dweik D, Yacobi‐Sharon K & Arama E (2018) Caspases maintain tissue integrity by an apoptosis‐independent inhibition of cell migration and invasion. Nat Commun 9, 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou M, Liu X, Li Z, Huang Q, Li F & Li CY (2018) Caspase‐3 regulates the migration, invasion and metastasis of colon cancer cells. Int J Cancer 143, 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li C, Egloff AM, Sen M, Grandis JR & Johnson DE (2014) Caspase‐8 mutations in head and neck cancer confer resistance to death receptor‐mediated apoptosis and enhance migration, invasion, and tumor growth. Mol Oncol 8, 1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Senft J, Helfer B & Frisch SM (2007) Caspase‐8 interacts with the p85 subunit of phosphatidylinositol 3‐kinase to regulate cell adhesion and motility. Cancer Res 67, 11505–11509. [DOI] [PubMed] [Google Scholar]
- 101.Barbero S, Mielgo A, Torres V, Teitz T, Shields DJ, Mikolon D, Bogyo M, Barilà D, Lahti JM, Schlaepfer Det al. (2009) Caspase‐8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res 69, 3755–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gdynia G, Grund K, Eckert A, Böck BC, Funke B, Macher‐Goeppinger S, Sieber S, Herold‐Mende C, Wiestler B, Wiestler ODet al. (2007) Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol Cancer Res 5, 1232–1240. [DOI] [PubMed] [Google Scholar]
- 103.Berthenet K, Castillo Ferrer C, Fanfone D, Popgeorgiev N, Neves D, Bertolino P, Gibert B, Hernandez‐Vargas H & Ichim G (2020) Failed apoptosis enhances melanoma cancer cell aggressiveness. Cell Rep 31, 107731. [DOI] [PubMed] [Google Scholar]
- 104.Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park JSet al. (2016) Tead and AP1 coordinate transcription and motility. Cell Rep 14, 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter Let al. (2015) Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 6, 6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun G, Guzman E, Balasanyan V, Conner CM, Wong K, Zhou HR, Kosik KS & Montell DJ (2017) A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J Cell Biol 216, 3355–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bernard A, Chevrier S, Beltjens F, Dosset M, Viltard E, Lagrange A, Derangère V, Oudot A, Ghiringhelli F, Collin Bet al. (2019) Cleaved caspase‐3 transcriptionally regulates angiogenesis‐promoting chemotherapy resistance. Cancer Res 79, 5958–5970. [DOI] [PubMed] [Google Scholar]
- 108.Gong YN, Crawford JC, Heckmann BL & Green DR (2019) To the edge of cell death and back. FEBS J 286, 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rudrapatna VA, Bangi E & Cagan RL (2013) Caspase signalling in the absence of apoptosis drives Jnk‐dependent invasion. EMBO Rep 14, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Portela M & Richardson HE (2013) Death takes a holiday–non‐apoptotic role for caspases in cell migration and invasion. EMBO Rep 14, 107–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fujisawa Y, Kosakamoto H, Chihara T & Miura M (2019) Non‐apoptotic function of Drosophila caspase activation in epithelial thorax closure and wound healing. Development 146, dev169037. [DOI] [PubMed] [Google Scholar]
- 112.Lytle NK, Barber AG & Reya T (2018) Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer 18, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fujita J, Crane AM, Souza MK, Dejosez M, Kyba M, Flavell RA, Thomson JA & Zwaka TP (2008) Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dick SA, Chang NC, Dumont NA, Bell RA, Putinski C, Kawabe Y, Litchfield DW, Rudnicki MA & Megeney LA (2015) Caspase 3 cleavage of Pax7 inhibits self‐renewal of satellite cells. Proc Natl Acad Sci USA 112, E5246–E5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fernando P, Kelly JF, Balazsi K, Slack RS & Megeney LA (2002) Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci USA 99, 11025–11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fernando P, Brunette S & Megeney LA (2005) Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J 19, 1671–1673. [DOI] [PubMed] [Google Scholar]
- 117.Bulatovic I, Ibarra C, Österholm C, Wang H, Beltrán‐Rodríguez A, Varas‐Godoy M, Månsson‐Broberg A, Uhlén P, Simon A & Grinnemo KH (2015) Sublethal caspase activation promotes generation of cardiomyocytes from embryonic stem cells. PLoS One 10, e0120176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Takahashi K & Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- 119.Li F, He Z, Shen J, Huang Q, Li W, Liu X, He Y, Wolf F & Li CY (2010) Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell 7, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Janzen V, Fleming HE, Riedt T, Karlsson G, Riese MJ, Celso CL, Reynolds G, Milne CD, Paige CJ, Karlsson Set al. (2008) Hematopoietic stem cell responsiveness to exogenous signals is limited by caspase‐3. Cell Stem Cell 2, 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu X, Li F, Huang Q, Zhang Z, Zhou L, Deng Y, Zhou M, Fleenor DE, Wang H, Kastan MBet al. (2017) Self‐inflicted DNA double‐strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res 27, 764–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD & Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. [DOI] [PubMed] [Google Scholar]
- 123.Yin H & Glass J (2011) The phenotypic radiation resistance of CD44+/CD24(‐or low) breast cancer cells is mediated through the enhanced activation of ATM signaling. PLoS One 6, e24080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stagni V, Oropallo V, Fianco G, Antonelli M, Cinà I & Barilà D (2014) Tug of war between survival and death: exploring ATM function in cancer. Int J Mol Sci 15, 5388–5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xu Y, So C, Lam HM, Fung MC & Tsang SY (2018) Apoptosis reversal promotes cancer stem cell‐like cell formation. Neoplasia 20, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grasso D, Zampieri LX, Capeloa T, Van de Velde JA & Sonveaux P (2020) Mitochondria in cancer. Cell Stress 4, 114–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Giampazolias E & Tait SW (2016) Mitochondria and the hallmarks of cancer. FEBS J 283, 803–814. [DOI] [PubMed] [Google Scholar]
- 128.Oberoi TK, Dogan T, Hocking JC, Scholz RP, Mooz J, Anderson CL, Karreman C, Meyer zu Heringdorf D, Schmidt G, Ruonala Met al. (2012) IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J 31, 14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Oberoi‐Khanuja TK, Murali A & Rajalingam K (2013) IAPs on the move: role of inhibitors of apoptosis proteins in cell migration. Cell Death Dis 4, e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oberoi‐Khanuja TK, Karreman C, Larisch S, Rapp UR & Rajalingam K (2012) Role of melanoma inhibitor of apoptosis (ML‐IAP) protein, a member of the baculoviral IAP repeat (BIR) domain family, in the regulation of C‐RAF kinase and cell migration. J Biol Chem 287, 28445–28455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Murali A, Shin J, Yurugi H, Krishnan A, Akutsu M, Carpy A, Macek B & Rajalingam K (2017) Ubiquitin‐dependent regulation of Cdc42 by XIAP. Cell Death Dis 8, e2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dubrez L & Rajalingam K (2015) IAPs and cell migration. Semin Cell Dev Biol 39, 124–131. [DOI] [PubMed] [Google Scholar]
- 133.Paul A, Krelin Y, Arif T, Jeger R & Shoshan‐Barmatz V (2018) A new role for the mitochondrial pro‐apoptotic protein SMAC/Diablo in phospholipid synthesis associated with tumorigenesis. Mol Ther 26, 680–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Arbiser JL (2018) Diablo: a double‐edged sword in cancer? Mol Ther 26, 678–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dong LF, Kovarova J, Bajzikova M, Bezawork‐Geleta A, Svec D, Endaya B, Sachaphibulkij K, Coelho AR, Sebkova N, Ruzickova A (2017) Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA‐deficient cancer cells. Elife 6, e22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tan AS, Baty JW, Dong LF, Bezawork‐Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan Bet al. (2015) Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21, 81–94. [DOI] [PubMed] [Google Scholar]
- 137.Riley JS & Tait SW (2020) Mitochondrial DNA in inflammation and immunity. EMBO Rep 21, e49799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CYet al. (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie MEet al. (2014) Apoptotic caspases suppress mtDNA‐induced STING‐mediated type I IFN production. Cell 159, 1549–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl Cet al. (2009) Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ahn J, Xia T, Konno H, Konno K, Ruiz P & Barber GN (2014) Inflammation‐driven carcinogenesis is mediated through STING. Nat Commun 5, 5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NKet al. (2018) Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kwon J & Bakhoum SF (2020) The cytosolic DNA‐sensing cGAS‐STING pathway in cancer. Cancer Discov 10, 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang X & Gerdes HH (2015) Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 22, 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shen J, Zhang JH, Xiao H, Wu JM, He KM, Lv ZZ, Li ZJ, Xu M & Zhang YY (2018) Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cardiomyocytes from apoptosis. Cell Death Dis 9, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, Saland E, Castellano R, Pouyet L, Collette Yet al. (2016) Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 128, 253–264. [DOI] [PubMed] [Google Scholar]
- 147.Griessinger E, Moschoi R, Biondani G & Peyron JF (2017) Mitochondrial transfer in the leukemia microenvironment. Trends Cancer 3, 828–839. [DOI] [PubMed] [Google Scholar]
- 148.Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu‐Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii Set al. (2013) Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liu K, Ji K, Guo L, Wu W, Lu H, Shan P & Yan C (2014) Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia‐reperfusion model via tunneling nanotube like structure‐mediated mitochondrial transfer. Microvasc Res 92, 10–18. [DOI] [PubMed] [Google Scholar]
- 150.Caicedo A, Fritz V, Brondello JM, Ayala M, Dennemont I, Abdellaoui N, de Fraipont F, Moisan A, Prouteau CA, Boukhaddaoui Het al. (2015) MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci Rep 5, 9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Luz‐Crawford P, Hernandez J, Djouad F, Luque‐Campos N, Caicedo A, Carrère‐Kremer S, Brondello JM, Vignais ML, Pène J & Jorgensen C (2019) Mesenchymal stem cell repression of Th17 cells is triggered by mitochondrial transfer. Stem Cell Res Ther 10, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, Arena G, Lasorsa L, Bennett A, Grange Tet al. (2020) Blood contains circulating cell‐free respiratory competent mitochondria. FASEB J 34, 3616–3630. [DOI] [PubMed] [Google Scholar]
- 153.Giacomello M, Pyakurel A, Glytsou C & Scorrano L (2020) The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21, 204–224. [DOI] [PubMed] [Google Scholar]
- 154.Altieri DC (2019) Mitochondrial dynamics and metastasis. Cell Mol Life Sci 76, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Otera H & Mihara K (2012) Mitochondrial dynamics: functional link with apoptosis. Int J Cell Biol 2012, 821676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW & Tu Y (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32, 4814–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Desai SP, Bhatia SN, Toner M & Irimia D (2013) Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J 104, 2077–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, Rampini P, Kossenkov AV, Aird KM, Zhang Ret al. (2015) PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci USA 112, 8638–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Caino MC & Altieri DC (2015) Cancer cells exploit adaptive mitochondrial dynamics to increase tumor cell invasion. Cell Cycle 14, 3242–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Furnish M & Caino MC (2020) Altered mitochondrial trafficking as a novel mechanism of cancer metastasis. Cancer Rep 3, e1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Caino MC, Seo JH, Aguinaldo A, Wait E, Bryant KG, Kossenkov AV, Hayden JE, Vaira V, Morotti A, Ferrero Set al. (2016) A neuronal network of mitochondrial dynamics regulates metastasis. Nat Commun 7, 13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Majumdar R, Steen K, Coulombe PA & Parent CA (2019) Non‐canonical processes that shape the cell migration landscape. Curr Opin Cell Biol 57, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Agarwal E, Altman BJ, Ho Seo J, Bertolini I, Ghosh JC, Kaur A, Kossenkov AV, Languino LR, Gabrilovich DI, Speicher DWet al. (2019) Myc regulation of a mitochondrial trafficking network mediates tumor cell invasion and metastasis. Mol Cell Biol 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM & Kashatus DF (2015) Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK‐driven tumor growth. Mol Cell 57, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki Het al. (2015) Mitochondrial division is requisite to RAS‐induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell 57, 521–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Giampazolias E, Zunino B, Dhayade S, Bock F, Cloix C, Cao K, Roca A, Lopez J, Ichim G, Proïcs Eet al. (2017) Mitochondrial permeabilization engages NF‐kappaB‐dependent anti‐tumor activity under caspase deficiency. Nat Cell Biol 19, 1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Giampazolias E & Tait SWG (2017) Caspase‐independent cell death: an anti‐cancer double‐whammy. Cell Cycle 17, 269–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Roumane A, Berthenet K, El Fassi C & Ichim G (2018) Caspase‐independent cell death does not elicit a proliferative response in melanoma cancer cells. BMC Cell Biol 19, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Messmer MN, Snyder AG & Oberst A (2019) Comparing the effects of different cell death programs in tumor progression and immunotherapy. Cell Death Differ 26, 115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]