

Abstract
ELX‐02 is an investigational compound being developed as a therapy for genetic diseases caused by nonsense mutations such as cystic fibrosis. Structurally, ELX‐02 is an aminoglycoside analogue that induces read‐through of nonsense mutations through interaction with the ribosome, resulting in the production of full‐length functional proteins. This phase 1 multiple‐ascending‐dose trial evaluated the safety and pharmacokinetics of ELX‐02 in 62 healthy volunteers. ELX‐02 plasma exposure was dose proportional, with no apparent accumulation, and followed by renal elimination. The most reported adverse event was injection site reactions that were mild to moderate in severity. At the top dose of 5.0 mg/kg, 1 of 6 subjects experienced auditory threshold changes in which ototoxicity could not be clearly ruled out, and 2 of 6 had hearing threshold changes consistent with possible ototoxicity. Two of 3 subjects receiving placebo in the 5.0 mg/kg group also had significant hearing threshold changes. All observed hearing threshold changes resolved or were trending toward resolution after withdrawal of the study drug. No severe or serious adverse events were reported.The results of this study support the evaluation of ELX‐02 in phase 2 clinical trials with patients that have genetic diseases caused by nonsense mutations.
Keywords: cystic fibrosis, ELX‐02, nonsense mutations, pharmacokinetics, phase 1, safety
Globally, nonsense mutations account for ∼11% of all described gene lesions causing inherited monogenetic diseases, two of which are cystic fibrosis (CF) and nephropathic cystinosis.1, 2 CF is a fatal disorder caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. Nearly 10% of the CF patient population bears a nonsense allele mutation that yields no functional CFTR.3 Individuals with these alleles are typically among those with the most severe form of disease. CFTR‐directed small molecule research has provided therapeutic options for many patients with CF; however, to date, these therapies are not effective against CFTR nonsense alleles.
Similarly, cystinosis is an ultra‐rare autosomal recessive lysosomal storage disorder caused by mutations in the cystinosin lysosomal transporter (CTNS) gene that encodes the cystinosin protein.4 Approximately 12% of cystinosis cases are caused by nonsense mutations, resulting in cystine accumulation and formation of intralysosomal crystals associated with widespread tissue and organ damage.5, 6 Early treatment with cysteamine bitartrate may ameliorate cystinosis complications; however, it does not halt disease progression. Therapeutics targeting nonsense mutations are needed to address the unmet medical need for patients with these diseases.
ELX‐02, (6'‐[R]‐Methyl‐5‐O‐[5‐amino‐5,6‐dideoxy‐α‐L‐talofuranosyl]‐paromamine sulfate) is an investigational compound being developed as a therapy for genetic diseases caused by nonsense mutations, including CF and cystinosis. Structurally, it is an aminoglycoside analogue designed as a eukaryotic ribosomal selective glycoside that induces read‐through of nonsense mutations through interaction with the ribosome, resulting in the production of full‐length functional proteins. ELX‐02 was previously studied in a single‐ascending‐dose study over the dose range of 0.3 to 7.5 mg/kg administered subcutaneously (SC) and 0.3 mg/kg administered intravenously and showed an acceptable safety profile with rapid absorption and elimination. In this study, ELX‐02 was shown nearly 100% bioavailable when given SC with a calculated absolute bioavailability of 0.98 as the geometric mean ratio of area under the plasma concentration–time curve (AUC) from time zero to infinity/dose of 0.3 mg/kg SC vs 0.3 mg/kg intravenously. ELX‐02 is not metabolized and is excreted in the urine essentially unchanged as the parent compound.6
This Phase 1 multiple‐ascending‐dose study was conducted to investigate the safety, tolerability, and pharmacokinetics (PK) of ELX‐02 administered SC biweekly, for 29 days, in healthy volunteers.
Methods
This study was conducted between November 22, 2017, and July 18, 2019, at SGS Life Sciences (Antwerpen, Belgium) and PAREXEL International, Medstar Harbor Hospital (Baltimore, Maryland), in accordance with the principles embodied in the Declaration of Helsinki and of the International Council on Harmonisation Good Clinical Practice. The study protocol and other related documents were reviewed and approved by the relevant ethics committee/institutional review board of participating sites. Written informed consent was obtained from all subjects before the study start.
Study Design and Population
This was a phase 1, multiple‐ascending‐dose, randomized, double‐blind, placebo‐controlled study in which 7 sequential cohorts of healthy volunteers received ELX‐02 or placebo, administered SC twice weekly for 9 doses over 29 days to evaluate the safety, tolerability, and PK of ELX‐02. In each of 7 cohorts, 6 subjects were to receive ELX‐02 and 3 subjects were to receive placebo. Dose escalation to the next cohort was made at the recommendation of the general and auditory and vestibular data safety monitoring boards after a treatment‐blinded review of the PK and safety data following a 14‐day observation period to allow for detection of unanticipated or delayed adverse events (AEs). A total of 5 ELX‐02 dose levels and 2 diluted injection concentrations administered in 7 cohorts are summarized in Table 1.
Table 1.
Cohorts by ELX‐02 Dose and Strength
| Cohort | Dose | Injection Concentration |
|---|---|---|
| 1 | 0.1 mg/kg | 50 mg/mL |
| 2 | 0.3 mg/kg | 50 mg/mL |
| 3 | 1.0 mg/kg | 100 mg/mL |
| 4 | 2.5 mg/kg | 100 mg/mL |
| 5 | 1.0 mg/kg | 50 mg/mL |
| 6 | 2.5 mg/kg | 50 mg/mL |
| 7 | 5.0 mg/kg | 50 mg/mL |
Healthy female and male volunteers between the ages of 18 and 55 years, inclusive, with normal renal function (glomerular filtration rate >60 mL/min/1.73 m2), a body mass index of 19.0 to 30.0 kg/m2 (inclusive), and a total body weight >50.0 to <100.0 kg were eligible for study participation. Volunteers were excluded at screening if they had clinically relevant laboratory abnormalities, any history of ear disease or surgeries, persistent dizziness or persistent tinnitus, or any abnormality that indicated the presence of a vestibular pathology, conductive hearing loss, or balance problem. Use of prescription or nonprescription drugs or dietary and herbal supplements within 30 days or 5 half‐lives (whichever was longer) before the administration of the study drug was prohibited, except for acetaminophen/paracetamol, which could be used at doses of ≤2 g/d, and contraception.
Safety Assessments
AEs including the determination of relatedness, severity, and seriousness; AEs leading to discontinuation; the incidence of AEs of special interest (AESIs) (hypersensitivity, nephrotoxicity, and ototoxicity), and local injection site reactions; safety laboratory tests (hematology, clinical chemistry, urinalysis, and coagulation); vital signs; 12‐lead electrocardiogram; and physical examination were included in the safety assessment. AEs were collected from the time informed consent was given up to and including 28 days after the last dose of the study drug was administered.
Laboratory tests (hematology, clinical chemistry, urinalysis, and coagulation) were performed at screening, day –1, and days 1, 2, 3, 7, 14, 21, 28, 29, 30, and 31 and end of study. Auditory and vestibular testing were performed at screening; baseline; days 3, 10, 17, and 24; and end of study.
Pharmacokinetic Assessments and Blood/Urine Sample Collection
The analytic methods for quantification of ELX‐02 in human plasma and urine have been described previously.5 Plasma PK assessments included AUC, maximal concentration (Cmax) and time to maximal concentration (tmax). Additionally, dose proportionality, accumulation over time, apparent volume of distribution (Vd/F), elimination half‐life, and apparent clearance were assessed. Urine PK assessments included the cumulative amount of unchanged drug excreted into the urine, the fraction of the SC administered drug excreted in the urine, maximum accumulation ratio, the time of maximum accumulation ratio, and renal clearance. Blood and urine samples were collected for all enrolled subjects. For PK analyses, serial blood and urine samples were collected for 72 hours after dosing on days 1 and 29. PK blood samples were collected before dosing and 1 hour after dosing for trough and peak concentrations on days 4, 8, 11, 15, 18, and 22.
Statistical Methods and Data Analyses
No formal sample size was calculated. The number of 9 subjects included (6 active and 3 placebo) in each cohort for each dose was deemed sufficient for the examination of exploratory safety/tolerability and PK.
Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, North Carolina). PK parameters were derived based on noncompartmental analysis methods using Phoenix WinNonlin, version 8.0 or higher (Certara, Princeton, New Jersey).
Dose proportionality in AUC and Cmax was assessed using a power model, including the log‐transformed PK parameters as dependent variables and the log‐transformed dose as a fixed effect separately on days 1 and 29. The slope for log‐transformed dose was estimated with its 90% confidence interval (CI) to examine dose proportionality on days 1 and 29. The tmax was compared between dose groups using the nonparametric Kruskal–Wallis test separately on days 1 and 29. Steady‐state achievement was visually assessed using the 1‐hour postdose concentration vs time figure and predose concentration data.
The differences in injection concentrations were evaluated using the ratios of geometric mean, AUC, and Cmax with their 90% CIs and elimination half‐life, with the Kruskal–Wallis test, to compare cohort 3 vs 5 and cohort 4 versus 6 (ie, 100 mg/mL vs 50 mg/mL injection strengths at the 1.0 and 2.5 mg/kg dose levels, respectively).
All AEs were coded using Medical Dictionary of Regulatory Activities (MedDRA) version 20.1 and displayed using system organ class and preferred term. Injection site reactions were graded according to Division of AIDS, National Institutes of Health criteria. Severity of abnormal clinical laboratory measurements was determined using Common Terminology Criteria for Adverse Events, version 4.03 when available. Standardized MedDRA Queries for AESIs included acute renal failure, hearing and vestibular disorders, and hypersensitivity.
Descriptive statistics were used to summarize demographics, baseline characteristics, and safety data for each cohort. Placebo was pooled across cohorts and presented in 1 group.
Results
Summary Demographics of Study Population
A total of 62 healthy volunteers were enrolled, and 55 completed the study. Of the 41 subjects assigned to ELX‐02 treatment, 5 discontinued the study; among the 21 subjects assigned to placebo, 2 discontinued the study (Figure S1). Subjects were exposed to ELX‐02 for a mean (± standard deviation) of 30 (±7) days with a median (min–max) of 9 (6–9) injections. Demographic characteristics of study participants are summarized in Table 2.
Table 2.
Summary of Demographic Characteristics
| ELX‐02 Dose | |||||||
|---|---|---|---|---|---|---|---|
| Demographic Parameters | All Placebo (N = 21) | Cohort 1 0.1 mg/kg (N = 6) | Cohort 2 0.3 mg/kg (N = 5) | Cohort 3 and 5 1.0 mg/kg (N = 12) | Cohort 4 and 6 2.5 mg/kg (N = 12) | Cohort 7 5.0 mg/kg (N = 6) | All ELX‐02 (N = 41) |
| Malea | 7 (33) | 5 (83) | 0 | 4 (33) | 2 (17) | 3 (50) | 14 (34) |
| Femalea | 14 (67) | 1 (17) | 5 (100) | 8 (67) | 10 (83) | 3 (50) | 27 (66) |
| Racea | |||||||
| White | 19 (91) | 6 (100) | 5 (100) | 12 (100) | 12 (100) | 0 | 35 (85) |
| Black | 2 (9) | 0 | 0 | 0 | 0 | 6 (100) | 6 (15) |
| Age,b y | 39 (11) | 41 (12) | 36 (9) | 38 (12) | 41 (11) | 34 (8) | 38 (11) |
| Height,c cm | 172 (10) | 180 (13) | 164 (6) | 171 (6) | 170 (4) | 170 (80) | 171 (8) |
| Weight,c kg | 73 (11) | 81 (12) | 62 (7) | 73 (10) | 70 (8) | 78 (12) | 73 (11) |
| BMI,d kg/m2 | 25 (3) | 25 (2) | 23 (3) | 25 (3) | 24 (3) | 27 (3) | 25 (3) |
Data are shown as number (percent).
Age calculated based on date of informed consent.
Data are shown as mean (standard deviation).
BMI indicates body mass index = weight (kg) / height (m)2.
No protocol deviations reported during the study were considered to have an impact on the interpretation of the study results. All subjects were included in the safety and PK analyses.
Pharmacokinetics
When comparing the 100 mg/mL vs 50 mg/mL concentrations, no significant differences in plasma PK parameters were observed between cohorts 3 and 5 (1 mg/kg) on days 1 and 29 or between cohorts 4 and 6 (2.5 mg/kg) on day 1. The 90% CIs for the geometric mean ratios of Cmax, AUC12h, and AUC72h for cohort 3 vs 5 (1 mg/kg) on days 1 (Table S1) and 29 (Table S2) and for cohorts 4 vs 6 (2.5 mg/kg) on day 1 all covered 1, indicating no formulation‐related differences in PK. The findings were similar for ELX‐02 urinary parameters with no suggestion of a formulation‐related impact on urinary excretion. Therefore, the results from the 100 and 50 mg/kg concentrations in cohorts 3 and 5 and in cohorts 4 and 6 were combined for the 1 and 2.5 mg/kg groups, respectively.
On days 1 and 29, ELX‐02 administered SC was rapidly absorbed across a dose range of 0.1 to 5.0 mg/kg (Figure 1) with peak plasma concentrations achieved at a median tmax ranging from 45 minutes to 1 hour after dosing. No differences (P > .05) were found for tmax among the 5 dose groups (Figure S2). The slope of the log‐transformed power model was close to 1 and the 90% CI of the slope for Cmax and AUC from time zero to infinity covers 1 on days 1 and 29; hence, these parameters increase proportionally with dose (Table 3). A summary of major plasma PK parameters on days 1 and 29 are presented in Table 4.
Figure 1.
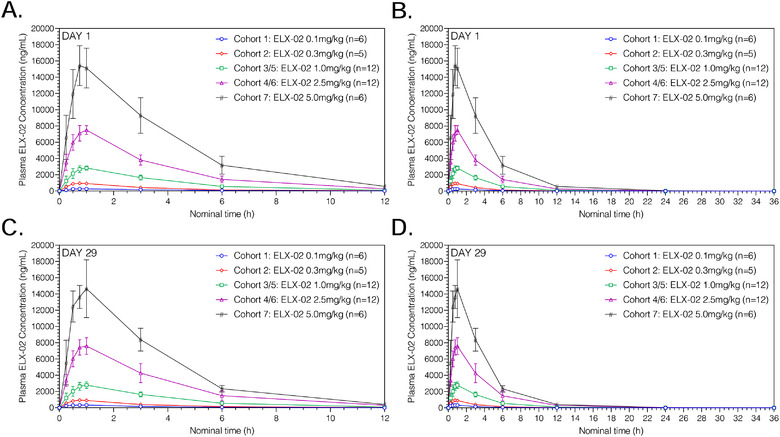
Mean (standard deviation) plasma ELX‐02 concentration vs time by cohort on day 1 from 0 to 12 hours (A), from 0 to 36 hours (B), day 29 from 0 to 12 hours (C), and from 0 to 36 hours (D).
Table 3.
Statistical Assessment of ELX‐02 Dose Proportionality Based on the Slope of the Log‐Transformed Power Model on Days 1 and 29
| Parameter | N | Intercept | Slope | 90%CI for the Slope |
|---|---|---|---|---|
| Day 1 | ||||
| Cmax, ng/mL | 41 | 8.017 | 1.015 | 0.987‐1.042 |
| AUCt, ng • h/mL | 41 | 9.311 | 1.037 | 1.004‐1.070 |
| AUCinf, ng • h/mL | 40 | 9.326 | 1.028 | 0.995‐1.062 |
| Day 29 | ||||
| Cmax, ng/mL | 35 | 8.029 | 0.975 | 0.941‐1.009 |
| AUCt, ng • h/mL | 35 | 9.313 | 1.016 | 0.972‐1.060 |
| AUCinf, ng • h/mL | 35 | 9.333 | 1.001 | 0.958‐1.044 |
AUCinf, area under the plasma concentration–time curve from time zero to infinity; AUCt, area under the plasma concentration–time curve over the dosing interval; Cmax, maximum plasma concentration; CI, confidence interval.
Table 4.
Summary of Plasma PK Parameters of ELX‐02 on Days 1 and 29
| ELX‐02 Dose | ||||||
|---|---|---|---|---|---|---|
| Parameter | Statistics | 0.1 mg/kg (N = 6) | 0.3 mg/kg (N = 5) | 1.0 mg/kg (N = 12) | 2.5 mg/kg (N = 12) | 5.0 mg/kg (N = 6) |
| Day 1 | ||||||
| Cmax, ng/mL | Mean | 290 | 1005 | 2898 | 7736 | 16099 |
| SD | 60 | 58 | 338 | 512 | 2566 | |
| tmax, h | Median | 1.17 | 0.75 | 0.92 | 0.90 | 0.86 |
| Min, Max | 0.50, 3.00 | 0.50, 1.00 | 0.75, 1.00 | 0.75, 1.00 | 0.73, 1.00 | |
| AUCt, ng • h/mL | Mean | 1074 | 3083 | 10942 | 28545 | 63147 |
| SD | 184 | 409 | 1477 | 4611 | 13356 | |
| AUC24h, ng • h/mL | Mean | 1115 | 3150 | 11097 | 28569 | 62994 |
| SD | 170 | 452 | 1412 | 4489 | 13127 | |
| AUC48h, ng • h/mL | Mean | 1118 | 3152 | 11117 | 28676 | 63254 |
| SD | 171 | 454 | 1438 | 4564 | 13343 | |
| AUC72h, ng • h/mL | Mean | 1118 | 3152 | 11118 | 28677 | 63260 |
| SD | 171 | 454 | 1439 | 4566 | 13349 | |
| AUCinf, ng • h/mL | Mean | 1118 | 3153 | 11118 | 28679 | 63262 |
| SD | 171 | 455 | 1439 | 4566 | 13351 | |
| C1h, ng/mL | Mean | 264 | 918 | 2822 | 7511 | 15132 |
| SD | 52 | 61 | 252 | 539 | 2454 | |
| t1/2, h | Mean | 2.31 | 2.06 | 2.32 | 2.87 | 2.96 |
| SD | 0.61 | 0.23 | 0.51 | 0.52 | 0.48 | |
| Vd/F, L | Mean | 23.38 | 17.57 | 22.27 | 25.04 | 26.42 |
| SD | 4.72 | 1.74 | 3.38 | 3.01 | 4.66 | |
| CL/F, L/h | Mean | 7.14 | 5.93 | 6.79 | 6.24 | 6.25 |
| SD | 1.10 | 0.53 | 1.02 | 1.30 | 1.06 | |
| Day 29 | ||||||
| Cmax, ng/mL | Mean | 345 | 964 | 2840 | 7908 | 15533 |
| SD | 42 | 73 | 436 | 1010 | 2091 | |
| tmax, h | Median | 0.80 | 0.80 | 0.93 | 0.86 | 0.90 |
| Min, Max | 0.50, 1.00 | 0.75, 1.00 | 0.75, 1.00 | 0.75, 1.00 | 0.73, 0.98 | |
| AUCt, ng • h/mL | Mean | 1163 | 3046 | 10748 | 30388 | 55206 |
| SD | 201 | 483 | 1607 | 7277 | 7947 | |
| AUC24h, ng • h/mL | Mean | 1226 | 3145 | 10947 | 30381 | 55100 |
| SD | 182 | 542 | 1578 | 7148 | 7770 | |
| AUC72h, ng • h/mL | Mean | 1228 | 3150 | 10964 | 30524 | 55295 |
| SD | 183 | 546 | 1597 | 7252 | 7919 | |
| AUCinf, ng • h/mL | Mean | 1228 | 3150 | 10964 | 30526 | 55296 |
| SD | 183 | 546 | 1597 | 7253 | 7920 | |
| C1h, ng/mL | Mean | 333 | 900 | 2780 | 7594 | 14652 |
| SD | 31 | 50 | 407 | 1020 | 3560 | |
| t1/2, h | Mean | 2.09 | 2.39 | 2.29 | 2.99 | 3.00 |
| SD | 0.51 | 0.25 | 0.48 | 0.67 | 0.59 | |
| Vd/F, L | Mean | 19.98 | 20.38 | 22.09 | 25.22 | 33.66 |
| SD | 5.07 | 1.27 | 4.01 | 4.67 | 5.69 | |
| CL/F, L/h | Mean | 6.67 | 5.95 | 6.80 | 6.07 | 7.51 |
| SD | 1.34 | 0.58 | 1.17 | 1.47 | 0.89 | |
| Rac(AUC24h) | Mean | 1.09 | 0.99 | 0.99 | 1.07 | 0.92 |
| SD | 0.08 | 0.06 | 0.08 | 0.15 | 0.13 | |
| Rac(Cmax) | Mean | 1.23 | 0.96 | 0.98 | 1.03 | 0.94 |
| SD | 0.18 | 0.07 | 0.2 | 0.16 | 0.01 | |
AUC24h, area under the plasma concentration–time curve from time zero to 24 hours; AUC48h, area under the plasma concentration–time curve from time zero to 48 hours; AUC72h, area under the plasma concentration–time curve from time zero to 72 hours; AUCinf, area under the plasma concentration–time curve from time zero to infinity; AUCt, area under the plasma concentration–time curve over the dosing interval; C1h, 1‐hour postdose concentration; Cmax, maximum plasma concentration; CL/F, apparent clearance; PK, pharmacokinetic; Rac, accumulation ratio; t1/2, elimination half‐life; tmax, time to maximum concentration; Vd/F, apparent volume of distribution.
Any values below the limit of quantitation were set to zero.
Measurable plasma ELX‐02 concentrations were observed in most subjects until 12 hours after dosing at ELX‐02 doses of 0.1 and 0.3 mg/kg and up to 24 hours after dosing at ELX‐02 doses ranging from 1.0 to 5.0 mg/kg (Figure 1). Concentrations appeared to decline in a monophasic manner following attainment of maximum concentration.
The mean 1‐hour postdose concentration values over 29 days remained similar over time in every dose group (Figure 2). All predose values were below the limit of quantitation. Accumulation ratios, Rac(Cmax) and Rac(AUC24h), based on days 1 and 29 Cmax and AUC24h, were close to 1 in all dose groups, indicating that there was no accumulation of plasma ELX‐02 associated with the twice‐weekly regimen for up to 9 doses.
Figure 2.
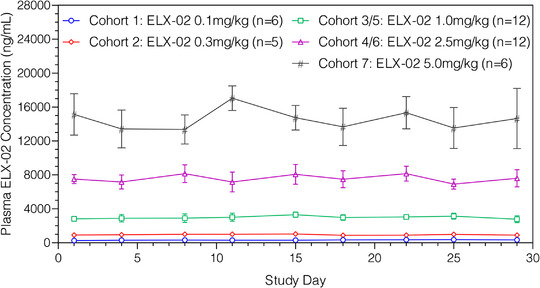
Mean (standard deviation) plasma ELX‐02 1‐hour postdose concentration vs study day by cohort.
The Vd/F on both days 1 and 29, ranged from 18 to 34 L, suggesting distribution beyond the blood compartment. The mean Vd/F values were generally dose independent, although those values in the 2.5‐ and 5.0‐mg/kg groups were slightly higher but within the observed variation. Total apparent clearance, ranging from 6 to 8 L/h, was high on days 1 and 29, and independent of dose level.
Key urinary PK parameters for days 1 and 29 are presented in Figure 3 and Table 5. Urinary excretion is rapid, with the fraction of ELX‐02 dose beginning to plateau after 12 hours on both days 1 and 29 (Table 5). The majority (77%–94%) of the ELX‐02 dose was excreted unchanged in urine within 12 hours after dosing across all dose groups on both days 1 and 29. Excretion was rapid following absorption of the SC dose, with median peak excretion occurring at 0.5 to 1.5 hours after dosing on both days 1 and 29. Twenty‐four‐hour renal clearance was high, ranging from 4.8 to 7.3 L/h on days 1 and 29, and dose independent.
Figure 3.
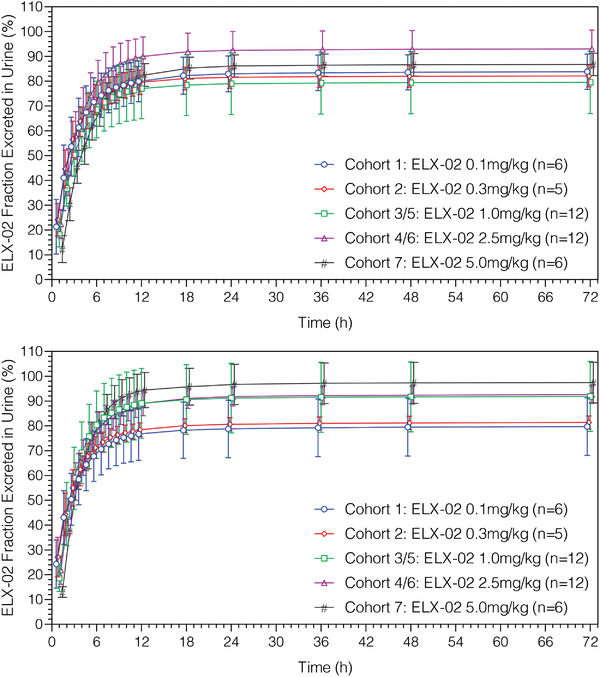
Mean (standard deviation) ELX‐02 fraction excreted vs midpoint of urinary collection time interval by cohort on days 1 (upper panel) and 29 (lower panel).
Table 5.
Key Summary of Urinary PK Parameters of ELX‐02 on Days 1 and 29
| ELX‐02 Dose | ||||||
|---|---|---|---|---|---|---|
| Parameter | Statistics | 0.1 mg/kga (N = 6) | 0.3 mg/kg (N = 5) | 1.0 mg/kg (N = 12) | 2.5 mg/kg (N = 12) | 5.0 mg/kg (N = 6) |
| Day 1 | ||||||
| Ae12h, mg | Mean | 6.39 | 14.62 | 56.97 | 155.16 | 319.94 |
| SD | 1.04 | 1.26 | 11.29 | 18.43 | 53.52 | |
| fe12h, % | Mean | 79.91 | 78.92 | 76.38 | 89.98 | 82.47 |
| SD | 8.22 | 4.95 | 12.17 | 7.84 | 4.59 | |
| Rmax, mg | Mean | 1.92 | 4.77 | 16.31 | 42.40 | 69.83 |
| SD | 0.58 | 0.32 | 2.45 | 8.45 | 9.59 | |
| TRmax, h | Median | 1.00 | 0.50 | 1.50 | 1.50 | 1.50 |
| Minimum | 0.50 | 0.50 | 0.50 | 0.50 | 1.50 | |
| Maximum | 1.05 | 1.50 | 1.50 | 1.50 | 1.50 | |
| CLR24h, L/h | Mean | 5.99 | 4.84 | 5.34 | 5.79 | 5.39 |
| SD | 1.27 | 0.47 | 1.02 | 1.27 | 0.85 | |
| Day 29 | ||||||
| Ae12h, mg | Mean | 6.28 | 14.52 | 64.99 | 157.50 | 389.93 |
| SD | 1.60 | 1.22 | 10.17 | 20.71 | 55.45 | |
| fe12h, % | Mean | 76.74 | 78.44 | 89.08 | 89.04 | 94.47 |
| SD | 10.67 | 2.83 | 14.07 | 4.98 | 7.41 | |
| Rmax, mg | Mean | 2.04 | 5.08 | 18.84 | 41.25 | 90.43 |
| SD | 0.61 | 0.76 | 5.09 | 7.94 | 12.89 | |
| TRmax, h | Median | 0.50 | 0.50 | 1.50 | 1.50 | 1.50 |
| Minimum | 0.50 | 0.50 | 0.50 | 0.50 | 1.50 | |
| Maximum | 1.50 | 1.50 | 1.50 | 1.50 | 2.05 | |
| CLR24h, L/h | Mean | 5.32 | 4.81 | 6.14 | 5.63 | 7.25 |
| SD | 1.39 | 0.52 | 0.98 | 1.55 | 0.57 | |
Ae12h, amount of unchanged drug excreted into the urine at 12 hours; CLR24h, renal clearance at 24 hours; fe12h, fraction of the drug excreted in the urine at 12 hours; PK, pharmacokinetic; Rmax, maximum accumulation ratio; TRmax, median peak excretion.
n = 5 for CLR24h in the 0.1 mg/kg group.
Safety and Tolerability
A total of 37 (90%) subjects in the ELX‐02 treatment groups and 14 (67%) subjects in the placebo group experienced at least 1 treatment‐emergent adverse event (TEAE). There were no deaths and no severe or serious TEAEs. A total of 4 subjects (1 in the placebo group and 3 in the ELX‐02 group) had TEAEs leading to study drug discontinuation: 1 receiving placebo (depression), 1 receiving ELX‐02 at 2.5 mg/kg (100 mg/mL; ototoxicity, hyperacusis, and tinnitus), and 2 receiving ELX‐02 at 5.0 mg/kg (50 mg/mL; abnormal audiogram). Additionally, 2 subjects withdrew consent during the trial (1 receiving placebo at the 1.0‐mg/kg dose, 1 receiving ELX‐02 at the 1.0‐mg/kg dose). Additionally, 1 subject receiving ELX‐02 at the 5.0‐mg/kg dose was withdrawn at the discretion of the investigator (classified as “other”).
Thirty‐four (83%) subjects in the all ELX‐02 treatment group and 9 (43%) subjects in the placebo group experienced treatment‐related TEAEs. The most common (≥10%) treatment‐related TEAE were injection site reactions in both the ELX‐02 treatment group (34 subjects) and the placebo group (6 subjects). All injection site reactions were mild to moderate in severity. Other treatment‐related TEAEs are summarized in Table 6. When the final diluted injection concentration was reduced from 100 to 50 mg/mL, the duration of all TEAEs under the MedDRA high‐level term of “injection site reactions” were reduced from 28 to 12 days for the 1.0 mg/kg dose and from 45 to 19 days for the 2.5 mg/kg dose.
Table 6.
Treatment‐Related TEAEs by Preferred Term, Treatment, and Dose (Safety Population)
| ELX‐02 Dose | |||||||
|---|---|---|---|---|---|---|---|
| Preferred Term | All Placebo (N = 21) n (%), E | Cohort 1 0.1 mg/kg (N = 6) n (%), E | Cohort 2 0.3 mg/kg (N = 5) n (%), E | Cohort 3 and 5 1.0 mg/kg (N = 12) n (%), E | Cohort 4 and 6 2.5 mg/kg (N = 12) n (%), E | Cohort 7 5.0 mg/kg (N = 6) n (%), E | All ELX‐02 (N = 41) n (%), E |
| ≥1 TEAE | 9 (43), 12 | 1 (17), 1 | 5 (100), 11 | 10 (83), 36 | 12 (100), 87 | 6 (100), 78 | 34 (83), 213 |
| Injection site reaction | 6 (29), 7 | 1 (17), 1 | 5 (100), 8 | 10 (83), 23 | 12 (100), 59 | 6 (100), 66 | 34 (83), 157 |
| Injection site erythema | 0 | 0 | 0 | 3 (25), 3 | 4 (33), 8 | 0 | 7 (17), 11 |
| Injection site discoloration | 0 | 0 | 0 | 0 | 0 | 5 (83), 5 | 5 (12), 5 |
| Injection site induration | 0 | 0 | 0 | 2 (17), 5 | 3 (25) 5 | 0 | 5 (12), 10 |
| Injection site pain | 0 | 0 | 0 | 1 (8), 1 | 4 (33), 8 | 0 | 5 (12), 9 |
| Injection site pruritus | 0 | 0 | 0 | 1 (8), 1 | 0 | 0 | 1 (2), 1 |
| Injection site scab | 0 | 0 | 0 | 0 | 0 | 1 (17), 1 | 1 (2), 1 |
| Injection site hematoma | 2 (10), 3 | 0 | 0 | 0 | 3 (25), 3 | 0 | 3 (7), 3 |
| Auditory reactions | |||||||
| Audiogram abnormala | 0 | 0 | 0 | 0 | 0 | 2 (33), 4 | 2 (5), 4 |
| Ear discomfort | 0 | 0 | 0 | 0 | 2 (17), 2 | 0 | 2 (5), 2 |
| Erythemaa | 0 | 0 | 0 | 1 (8) | 0 | 0 | 1 (2) |
| Hyperacusisa | 0 | 0 | 0 | 0 | 0 | 1 (17), 1 | 1 (2), 1 |
| Ototoxicitya | 0 | 0 | 0 | 0 | 1 (8), 1 | 0 | 1 (2), 1 |
| Tinnitusa | 0 | 0 | 0 | 0 | 0 | 1 (17), 1 | 1 (2), 1 |
| Hyperhidrosis | 0 | 0 | 1 (20), 1 | 0 | 0 | 0 | 1 (2), 1 |
| Rasha | 0 | 0 | 0 | 1 (8), 1 | 0 | 0 | 1 (2), 1 |
| Eye pruritus | 1 (5), 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Headache | 1 (5), 1 | 0 | 2 (40), 2 | 2 (17), 2 | 1 (8), 1 | 0 | 5 (12), 5 |
E, number of events for that particular TEAE; TEAE, treatment‐emergent adverse event.
If a subject experienced more than 1 event within a given preferred term, that subject is counted only once for that preferred term.
Indicates an adverse event of interest.
A total of 4 subjects (10%) receiving ELX‐02 experienced 9 AESIs. Standardized MedDra Queries used to identify AESI included acute renal failure, hearing and vestibular disorders, and hypersensitivity. By System Organ Class, 3 subjects (7%) experienced 7 AESIs of hearing impairment and 1 subject (2%) experienced 2 AESIs of hypersensitivity (erythema and rash) occurring in the 1.0‐mg/kg dosing group. Of the 3 subjects who experienced 7 AESIs related to transient hearing changes, 1 subject had a hearing‐related AESI (ototoxicity) that occurred in the 2.5‐mg/kg dosing group. Two subjects experienced 6 hearing‐related AESIs that occurred in the 5.0‐mg/kg dosing group (audiogram abnormal [n = 4], hyperacusis [n = 1], and tinnitus [n = 1]). Following detailed analyses of the audiograms, 1 of 12 subjects in the 2.5‐mg/kg group was confirmed with auditory threshold changes in which ototoxicity could not be clearly ruled out. Three of 6 subjects in the 5.0‐mg/kg group had transient frequency changes primarily in the high‐frequency range. Of these, 2 out of 6 subjects showed hearing threshold changes consistent with possible ototoxicity and 1 additional subject of 6 showed results in which ototoxicity could not be clearly ruled out. Some variability in the audiometry test results may have contributed to the findings for the 5.0‐mg/kg group because 2 of 3 subjects receiving placebo in cohort 7 also had threshold changes meeting the American Speech‐Language‐Hearing Association significant change criteria for the high‐frequency range. The observed hearing threshold changes were largely confined to the high‐frequency audiometry range (10–16 kHz), which is above the conventional range (0.25–8 kHz) needed for normal human communication and were asymptomatic to the subjects. The hearing threshold changes observed in the 2.5‐ and 5.0‐mg/kg groups resolved or were trending toward resolution after withdrawal of the study drug. No notable trends were observed for subjects’ tympanometry, Tinnitus Handicap Inventory, or Dizziness Handicap Inventory results. No evidence of ototoxicity based on audiometric tests was observed in the 0.1‐, 0.3‐, or 1.0‐mg/kg groups.
There were no reported AESIs of nephrotoxicity, and no notable trends were observed in clinical laboratory parameters, vital sign measurements, or physical examination findings among the different treatment groups. No notable trends in renal injury biomarkers were noted among the different treatment groups.
Discussion
The objectives of this phase 1, multiple‐ascending‐dose, randomized, double‐blind, placebo‐controlled study were to assess the safety, tolerability, and PK of ELX‐02 in healthy volunteers who, in sequential cohorts, received ELX‐02, or placebo, administered SC twice weekly for 9 doses over 29 days.
The PK profile in this multiple‐ascending‐dose ELX‐02 study was highly reproducible and is consistent with that previously reported following single ascending doses of ELX‐02.6 ELX‐02 was rapidly absorbed following SC administration across a dose range of 0.1 to 5.0 mg/kg, with peak plasma concentrations achieved at approximately 45 minutes to 1 hour after dosing. Plasma ELX‐02 exposure was dose proportional across the dose range, and no evidence of accumulation was observed following twice‐weekly administration for up to 9 doses. The volume of distribution was beyond the blood compartment.
Systemic ELX‐02 was rapidly excreted in urine, with approximately 76% to 94% of the administered dose excreted within 12 hours after dosing. Both plasma and renal clearance were high and independent of dose, consistent with the short elimination half‐life of approximately 2 to 3 hours. Plasma ELX‐02 exposure was generally similar for the 50‐ and 100‐mg/mL injection solution strengths, indicating no formulation‐related differences in PK.
There were no severe, serious, or unexpected TEAEs reported in any cohort during this study. The most common treatment‐related TEAEs across all groups were injection site reactions, which were of mild or moderate severity, and occurred less frequently in subjects receiving placebo and lower doses (0.1 and 1.0 mg/kg) of ELX‐02. By reducing the diluted injection concentration from 100 to 50 mg/mL, the mean duration of injection site reactions was reduced from 28 to 12 days at the 1.0‐mg/kg dose level, and from 45 to 19 days at the 2.5‐mg/kg dose level. Other than injection site reactions, the safety profiles were similar between the 2 diluted injection concentrations.
Overall, ELX‐02 was well tolerated across this dosage range, and the safety profile in this multiple‐ascending‐dose study is consistent with that previously reported following single ascending doses of ELX‐02.5 Since ELX‐02 is an aminoglycoside analogue, possible events of hypersensitivity, nephrotoxicity, and ototoxicity, which are common to aminoglycoside antibiotics, were prospectively defined as AESIs. In this study, mild to moderate injection site reactions were common, but there was only 1 event of hypersensitivity at the 1.0‐mg/kg dose, and there were no events of nephrotoxicity. Cumulatively, 4 total subjects had transient high‐frequency hearing changes: 1 of 12 at the 2.5‐mg/kg dose and 3 of 6 at the 5.0‐mg/kg dose. However, these results are somewhat difficult to interpret since 2 out of 3 subjects receiving placebo during the 5.0‐mg/kg cohort also experienced meaningful high‐frequency threshold shifts at isolated time points. Furthermore, no irreversible hearing threshold shift or clinical impact was observed in any subject, nor did any subject demonstrate significant tinnitus or vestibular disorders that are commonly affiliated with ototoxicity. Thus, clinically impactful signals for ototoxicity were absent.
Although further study is required, the emerging ELX‐02 safety profile appears to compare favorably to that of aminoglycoside antibiotics.
Conclusion
The results of this study further support the evaluation of ELX‐02 in phase 2 clinical trials with patients who have genetic diseases caused by nonsense mutations.
Conflicts of Interest
A.L., M.‐Y.H., E.G., K.T., K.B., and T.H. are employees of Eloxx Pharmaceuticals, Inc. F.V. was a principal investigator for the study. K.P. and K.C. are consultants to Eloxx Pharmaceuticals, Inc.
Funding
This study was funded by Eloxx Pharmaceuticals, Inc.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Acknowledgments
The authors thank the volunteers who participated in this study.
References
- 1.Mort M, Ivanov D, Cooper DN, et al. A meta‐analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29(8):1037‐1047. [DOI] [PubMed] [Google Scholar]
- 2.Linde L, Kerem B.Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552‐563. [DOI] [PubMed] [Google Scholar]
- 3.McCague AF, Raraigh KS, Pellicore MJ, et al. Correlating cystic fibrosis transmembrane conductance regulator function with clinical features to inform precision treatment of cystic fibrosis. Am J Respir Crit Care Med. 2019;199(9):1116‐1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gahl WA, Thoene JG, Schneider JA.Cystinosis N Engl J Med. 2002;347:111‐121. [DOI] [PubMed] [Google Scholar]
- 5.Brasell EJ, Chu LL, Akpa MM, et al. The novel aminoglycoside, ELX‐02, permits CTNSW138X translational read‐through and restores lysosomal cystine efflux in cystinosis. PLoS One. 2019;14(12):e0223954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leubitz A, Frydman‐Marom A, Sharpe N, van Duzer J, Campbell KCM, Vanhoutte F.Safety, tolerability, and pharmacokinetics of single ascending doses of ELX‐02, a potential treatment for genetic disorders caused by nonsense mutations, in healthy volunteers. Clin Pharmacol Drug Dev. 2019;8:984‐994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.