

Abstract
Cardiac safety and plasma concentration‐QTc interval analyses were completed using data from 2 phase 1 studies of the selective mouse double minute chromosome 2 antagonist, KRT‐232, in patients with solid tumors or multiple myeloma and acute myeloid leukemia (AML) who received KRT‐232 doses of 15 to 480 mg once daily (QD; N = 130). A linear mixed‐effects model related change from baseline Fridericia‐corrected QT interval (ΔQTcF) to KRT‐232 plasma concentrations. The final model included parameters for the intercept (with between‐subject variability), KRT‐232 concentration–ΔQTcF slope, and baseline QTcF effect on the intercept. Diagnostic plots indicated an adequate model fit. Mean (90% confidence interval) predicted ΔQTcF values at the maximum clinical dose (480 mg QD) were 2.04 (0.49‐3.60) milliseconds for patients with solid tumors and 4.52 (2.35‐6.69) milliseconds for patients with AML. Because the 90% confidence interval upper bound of the mean ΔQTcF was predicted to be below 10 milliseconds at doses up to 480 mg QD in patients with solid tumors, multiple myeloma, or AML, KRT‐232 does not result in clinically meaningful QT prolongation at the doses currently under investigation in clinical trials. No significant cardiac safety concerns were identified at these doses.
Keywords: AMG 232, concentration‐QTc, KRT‐232, MDM2 antagonist, QT interval
KRT‐232 (formerly AMG 232; Figure 1) is a selective oral small molecule currently under development for treatment of myeloproliferative neoplasms,1, 2 acute myeloid leukemia (AML),3 and Merkel cell carcinoma.4 It binds to mouse double minute chromosome 2 (MDM2) and inhibits its interactions with tumor suppressor protein 53 (p53; encoded by TP53 gene), leading to p53 activation and increased p53‐mediated transcriptional and cell cycle control.5, 6 KRT‐232 has activity in several tumor xenograft models harboring wild‐type p537, 8 and has been tested clinically as monotherapy in malignancies including solid tumors and multiple myeloma (MM) in Study 20120106, denoted here as Study A9 and AML in Study 20120234, denoted here as Study B.3
Figure 1.

Structure of KRT‐232 and acyl glucuronide metabolite M1.
KRT‐232 is a carboxylic acid (pKa 4.35). Solubility is low at acidic pH and increases with increasing pH above the pKa. KRT‐232 is generally well absorbed at intestinal pH, consistent with high passive permeability of KRT‐232 in Lilly Laboratory Cell Porcine Kidney 1 (LLC‐PK1) and Madin‐Darby Canine Kidney (MDCK) cell monolayers. KRT‐232 is highly protein bound (97.5%), widely distributed, and metabolized primarily to a major circulating acyl glucuronide metabolite (M1) by uridine glucuronosyltransferase isoenzymes UGT1A1, UGT1A3, and UGT1A4.10 M1 is stable in vitro and had 5‐fold less pharmacologic activity than the parent drug in a biochemical homogeneous time‐resolved fluorescence (HTRF) in vitro pharmacologic potency assay in the presence of 15% serum and was equipotent in the absence of serum. In vitro experiments indicated KRT‐232 has the potential to cause a minor drug‐drug interaction with drugs cleared predominantly by cytochrome P450 3A4 (CYP3A4) (inhibition and induction); however, a CYP3A4 physiologically based pharmacokinetic (PBPK) simulation of KRT‐232 effect on the sensitive CYP3A substrate midazolam indicated a clinically significant drug‐drug interaction (ie, >1.25‐fold change in area under the concentration‐time curve [AUC] or maximum concentration [Cmax]) with substrates of CYP3A4 was not predicted.11 Based on generally comparable pharmacokinetics (PK), KRT‐232 can be administered with or without food.12
A KRT‐232 population PK model was developed based on Study A9 and Study B3 plasma.13 The PK profile of KRT‐232 was described by a 2‐compartment model with first‐order absorption. Median apparent oral clearance in a typical solid tumor subject was 29.4 L/hr (61.5% coefficient of variation) with an apparent oral central volume of distribution of 62.9 L and terminal half‐life of 17.1 hours. Apparent oral clearance did not change over the dose range of 15 to 480 mg, indicating that AUC was linear over the therapeutic range. Tumor type and albumin were identified as model covariates affecting clearance. Exposures relative to a 240‐mg daily dose to a solid tumor subject with median albumin of 39 g/dL indicated that patients with relapsed or refractory AML had 61.6% greater steady‐state AUC. Decreased baseline albumin (30 g/L) correlated with a 47.7% increase in steady‐state AUC. KRT‐232 treatment elicited dose‐ and plasma‐concentration–dependent elevation of the serum pharmacodynamic (PD) marker, macrophage inhibitory cytokine‐1 (MIC‐1).14 Overall, human PK were supportive of once daily (QD) dosing. In study A9, mean time to maximum concentration (Tmax) of the metabolite M1 was 2 to 4 hours, and the mean (SD) terminal half‐life (t1/2) was 14.0 (6.07) hours. The mean accumulation for metabolite M1 over a 7‐day QD dosing period was <2‐fold, and the mean metabolite‐to‐parent 24‐hour AUC ratio was 0.461.
The effect of KRT‐232 plasma concentration on heart rate–corrected QT (QTc) interval was modeled using paired PK and electrocardiogram (ECG) data from Study A9 and Study B3 to identify any QTc prolongation risk at clinical doses. This general approach is consistent with relevant guidance15 and with precedent in the MDM2 antagonist class.16 Cardiac safety data from the 2 studies were also summarized.
Materials and Methods
Studies Used for the Concentration‐QT Analysis
Study A (NCT01723020) was a phase 1, open‐label, first‐in‐human dose‐exploration study that evaluated KRT‐232 in patients with advanced TP53 wild‐type solid tumors (n = 97) or MM (n = 10).9 Patients received 15 to 480 mg of KRT‐232 QD via oral administration (Table 1) in up to thirty‐one 21‐day cycles (7 days on/14 days off). A dose exploration part of the study evaluated the safety, tolerability, PK, and PD of KRT‐232 and determined the maximum tolerated dose using a practical continual reassessment method. The dose expansion part of the study tested the 240‐mg QD dose in additional patients with tumors harboring MDM2 amplification (liposarcoma, glioblastoma, and all other solid tumors), potentially harboring MDM2 overexpression (estrogen receptor–positive metastatic breast cancer), or with MM. Time points with time‐matched ECG and concentration measurements were cycle (C) 1 day (D) 1 (C1D1): 1, 3, 5, 7 and 24 hours post‐dose; C1D7: pre‐dose and 1, 3, 5, 7, 24, and 72 hours post‐dose; C1D15 (unscheduled); and end of study.
Table 1.
Number (%) of Patients in ECG Outlier Categories
| 15, 30 mg | 60 mg | 120 mg | 240 mg | 300, 360 mg | 480 mg | All | |
|---|---|---|---|---|---|---|---|
| Study A | (n = 6) | (n = 4) | (n = 7) | (n = 75) | (n = 8) | (n = 6) | (n = 106) |
| QTcF >450 to ≤480 ms | 0 (0) | 1 (25) | 1 (14.3) | 7 (9.3) | 0 (0) | 0 (0) | 9 (8.5) |
| QTcF >480 to ≤500 msa | 0 (0) | 0 (0) | 0 (0) | 1 (1.3) | 0 (0) | 1 (16.7) | 2 (1.9) |
| ΔQTcF >30 to ≤60 msa | 0 (0) | 0 (0) | 0 (0) | 2 (2.7) | 1 (12.5) | 0 (0) | 3 (2.8) |
| PR >200 ms and ≥25% increase from baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0)b | 0 (0) |
| QRS >120 ms and ≥25% increase from baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (12.5) | 0 (0) | 1 (0.9) |
| HR <50 bpm and ≤25% decrease from baseline | 0 (0) | 0 (0) | 0 (0) | 2 (2.7) | 0 (0) | 0 (0) | 2 (1.9) |
| HR >100 bpm and ≥25% increase from baseline | 1 (16.7) | 0 (0) | 0 (0) | 6 (8.0) | 0 (0) | 0 (0) | 7 (6.6) |
| 60 mg | 90 mg | 180 mg | 240 mg | 360 mg | All | |
|---|---|---|---|---|---|---|
| Study B | (n = 4) | (n = 4) | (n = 5) | (n = 3) | (n = 10) | (n = 26) |
| QTcF >450 to ≤480 ms | 0 (0) | 2 (50.0) | 1 (20.0) | 1 (33.3) | 5 (50.0) | 9 (34.6) |
| QTcF >480 to ≤500 msa | 0 (0) | 1 (25.0) | 0 (0) | 0 (0) | 0 (0) | 1 (3.8) |
| ΔQTcF >30 to ≤60 msa | 0 (0) | 2 (50.0) | 1 (20.0) | 2 (66.7) | 2 (20.0) | 7 (26.9) |
| PR >200 ms and ≥25% increase from baseline | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) | 0 (0) | 1 (3.8) |
| QRS >120 ms and ≥25% increase from baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| HR <50 bpm and ≤25% decrease from baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| HR >100 bpm and ≥25% increase from baseline | 0 (0) | 0 (0) | 1 (20.0) | 1 (33.3) | 1 (10.0) | 3 (11.5) |
ΔQTcF, change from baseline QT corrected using the Fridericia formula; HR, heart rate; n, number of patients in each treatment group.
QTcF, QT corrected using the Fridericia formula.
HR in bpm was calculated as 60,000/RR (ms).
No patients had QTcF >500 ms or ΔQTcF >60 ms.
n = 5 (1 patient did not have any PR data).
Study B (NCT02016729) was a Phase 1b, open‐label, nonrandomized study that evaluated KRT‐232 alone (n = 26) and in combination with trametinib (n = 10; excluded from this analysis) in patients with relapsed or refractory AML.3 Patients received up to forty‐six 14‐day cycles (7 days on/7 days off) of treatment at 60, 90, 180, 240, and 360 mg QD via oral administration. The monotherapy arm determined the maximum tolerated dose and evaluated the safety, tolerability, PK, and PD of KRT‐232. Time points with time‐matched ECG and concentration measurements were C1D1: 1, 2, 4, 6, and 24 hours post‐dose; C1D7: pre‐dose and 1, 2, 4, 6, 24, and 72 hours post‐dose; and pre‐dose on C2D1, C2D3, C2D8, C3D1, C7D1, C11D1, C15D1, C27D1, and C31D1.
Studies A and B were conducted in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Study A was conducted between December 2012 and September 2017 at 8 centers in the United States: the Greenville Health System Institute for Translational Oncology (Greenville, South Carolina), Norwalk Hospital (Norwalk, Connecticut), the Dana Farber Cancer Institute (Boston, Massachusetts), the Sarcoma Oncology Research Center LLC (Santa Monica, California), Massachusetts General Hospital (Boston, Massachusetts), Beth Israel Comprehensive Cancer Center (Boston, Massachusetts), Hackensack University Medical Center (Hackensack, New Jersey), and the Memorial Sloan Kettering Cancer Center (New York, New York). The protocol and the informed consent were approved by the Institutional Review Board (IRB‐C) for Oncology Research Office for Research Compliance and Administration (Greenville, South Carolina), Norwalk Hospital IRB (Norwalk, Connecticut), the Dana‐Farber Cancer Institute Institutional Review Board (Boston, Massachusetts), the Wester Institutional Review Board (Puyallup, Washington), and the Memorial Sloan Kettering Cancer IRB (New York, New York). Written informed consent was obtained from all individual participants included in the study.9
Study B was conducted between April 2014 and July 2017 at 5 centers in the United States: Seattle Cancer Care Alliance (Seattle, Washington), University of Alabama at Birmingham Comprehensive Cancer Center (Birmingham, Alabama), University of Utah Huntsman Cancer Institute (Salt Lake City, Utah), Roswell Park Cancer Institute (Buffalo, New York), and the Levine Cancer Institute (Columbia, Maryland). The protocol and the informed consent were approved by the Western Institutional Review Board (Puyallup, Washington), the University of Utah IRB (Salt Lake City, Utah), the Roswell Park Cancer Institute IRB (Buffalo, New York), and the Chesapeake IRB (Columbia, Maryland). Written informed consent was obtained from all individual participants included in the study.3
Bioanalytical Methods and Pharmacokinetics in Study A and Study B
Plasma concentrations of KRT‐232 and KRT‐232 glucuronide were measured using a validated liquid chromatography–tandem mass spectrometry method described by Erba et al.3 Briefly, internal standards were D6‐AMG 232 and D6‐AMG 232 glucuronide. Plasma was processed by protein precipitation with acetonitrile. Samples were chromatographed on a Phenomenex Kinetex C18 analytical column (2.6 µm, 50 × 3.00 mm; Phenomenex, Torrance, California) with gradient elution at a flow rate of 600 µL/min. Mass spectrometry was performed using electrospray ionization with negative ion multiple reaction monitoring of parent to product ion pairs m/z 566.1→64.1 for AMG 232, m/z 574.3→64.1 for D6‐AMG 232, m/z 742.5→566.0 for AMG 232 glucuronide, and m/z 750.4→574.3 for D6‐AMG 232 glucuronide. The lower limit of quantitation was 0.5 ng/mL in Study A and 1 ng/mL in Study B. The precision (% coefficient of variation) and accuracy (% bias) observed ranged from 4.4 to 8.6 and –6.2 to 1.2 for AMG 232; and 3.9 to 9.7 and –5.4 to 0.5 for AMG 232 glucuronide, respectively. Noncompartmental analysis was performed using Phoenix WinNonlin version 8.0 (Certara USA, Inc., Princeton, New Jersey).17
Electrocardiographic Measurements
All 12‐lead ECG data were captured in triplicate with tracings approximately 30 seconds apart. ECGs were taken with patients in the supine position for approximately 5 minutes before PK samples were drawn. The ECGs were read centrally. ECG measurements included QT interval, PR interval, RR interval, and duration of QRS complex. For each replicate, the measured QT interval was corrected using the Fridericia correction to remove the dependence on heart rate (HR; inverse of RR): QTcF = QT/RR1/3 where RR is measured in seconds.
A categorical analysis for each study identified the number and proportion of patients in each dose group with QTcF, ΔQTcF, PR, QRS, and HR measurements in high or low ranges of interest.
QT Interval Correction for RR and Exploratory Analysis
Because the QT interval tends to increase with RR (ie, decrease with HR), for each replicate, the QTcF was used to remove this dependence. If this was insufficient, a secondary analysis with an alternative population– or study‐specific QT correction could be considered. Exploration of the analysis data set also assessed independence of change from baseline RR vs concentration, trend in ΔQTc over time (if any), lack of hysteresis (delay between changes in drug concentration and ΔQTc), and linearity of the concentration‐ΔQTc relationship.
Concentration‐QTc Methodology
The concentration‐QTc modeling started with a modified version of the prespecified linear mixed‐effects model described in Garnett et al,15 retaining only those parameters for which the 95% confidence interval (CI) did not include zero (other than concentration slope, retained regardless of significance). If the exploratory analysis suggested nonlinearity, maximum response (Emax) and power‐of‐concentration models could be tested.
The initial model structure included KRT‐232 plasma concentration and baseline QTcF as continuous covariates. Between‐subject variability (BSV) was included as an additive random effect on intercept and slope. The initial model was
where ΔQTcij is the ΔQTc of subject i at time j; θ0 is the population mean intercept; η0,i is the BSV random effect on the intercept; θ1 is the population mean slope of the assumed linear concentration‐ΔQTcij relationship; η1,i is the random BSV around this slope; Cij is the plasma concentration of KRT‐232 for subject i at time j; θ2 is the fixed effect associated with baseline QTci, j=0; QTc0,mean is the overall mean baseline QTc; and εij is the residual error for subject i at time j. The random effects were assumed to be normally distributed with mean zero; covariance between the 2 random effects was allowed. Residuals were assumed to be normally distributed with mean zero.
Because no placebo treatment was included in either study, the effects of nominal time point and study visit were not initially included in the model but could be included later, if warranted from inspection of standardized residuals vs nominal time point plots. Study effects (ie, effects of data coming from Study B, not Study A) were investigated on the intercept, slope, and residual error. Tumor type (AML, solid tumor, or MM) and gender were also tested on the intercept and slope. Covariate effects were retained in the model only if significant at the α = 0.05 level based on the likelihood ratio test, and if the 95%CI for the effect excluded zero.
Based on the final model, mean ΔQTc and the associated 2‐sided 90%CI were predicted at the steady‐state mean maximum concentration (Cmax) for the therapeutic dose and other doses of clinical interest. Due to the absence of placebo treatment data, placebo‐corrected ΔQTc was not predicted. The upper bound of the 90%CIs for the mean ΔQTc predicted at the Cmax values were compared to 10‐ and 20‐ms thresholds of regulatory concern for investigational oncology drugs. Data assembly and analysis were performed using R version 3.5.2 (R Foundation, Vienna, Austria).18, 19
Results
Cardiac Safety Analyses
No deaths or serious adverse events (AEs) occurred that were deemed related to KRT‐232 during Study A9 and Study B.3 Categorical analysis of ECG outliers (which did not require time‐matched concentration records) used a total of 1604 post‐baseline ECG records from 132 patients in the 2 studies (Table 1). There were no QTcF measurements >500 milliseconds or ΔQTcF >60 milliseconds. In Study A, 2.8% of patients had a ΔQTcF >30 milliseconds; no dose‐response relationship was identified. In Study B, 26.9% of patients had a ΔQTcF >30 milliseconds, but again with no conclusive dose‐response relationship. Of the 10 patients in the combined study data who had a ΔQTcF >30 milliseconds, 6 had concurrent hypokalemia, and 9 received concomitant medication associated with risk of torsades de pointes.
No patients in Study A had a PR outlier (PR >200 milliseconds and ≥25% increase from baseline), and <2% had either QRS >120 milliseconds with an increase of ≥25% from baseline or HR <50 beats per minute (bpm) with a decrease of ≤25% from baseline. Similarly, in Study B, only 1 patient (3.8%) had a PR outlier, and none had a QRS outlier or HR <50 bpm with an increase of ≥25% from baseline. Seven patients (6.6%) in Study A and 3 patients (11.5%) in Study B had HR >100 bpm with an increase of ≥25% from baseline; no conclusive dose‐response relationship was found.
No cardiac or QT prolongation treatment‐emergent AEs were deemed related to KRT‐232 in Study B. In Study A, 3 such events occurred, all Grade 1 in severity, and resolved: QT prolonged in a 66‐year‐old man on a 240‐mg dose, palpitations in a 52‐year‐old woman on a 240‐mg dose, and sinus tachycardia in a 47‐year‐old woman on a 120‐mg dose. No additional related AEs that were suggestive of proarrhythmic potential, such as torsades de pointes, ventricular tachycardia, ventricular fibrillation and flutter, syncope, or seizures were reported in either study.
Concentration‐QTc Analyses
A total of 1427 post‐baseline time‐matched concentration‐QTc pairs from 130 patients (104 in Study A9 and 26 in Study B3) were included in the concentration‐QTc analysis, after exclusions for QT and PK measurements that were missing or taken more than 30 minutes apart. Among these patients, 50% were female in Study A, and 35% were female in Study B. Mean (standard deviation [SD]) baseline QTcF was 411.5 (20.9) milliseconds in Study A and 420.2 (13.5) milliseconds in Study B.
The Fridericia correction (QTcF) adequately corrected for RR effects on QT (Figure 2); similar plots by study did not show any consistent trends. Data exploration did not show an influence of KRT‐232 exposure on RR. No clear evidence of hysteresis was observed. Plots of ΔQTcF vs KRT‐232 concentration showed a positive linear trend, suggesting adequacy of the linear mixed‐effects model. A largely flat trend was found vs metabolite (KRT‐232 acyl glucuronide M1) concentration.
Figure 2.
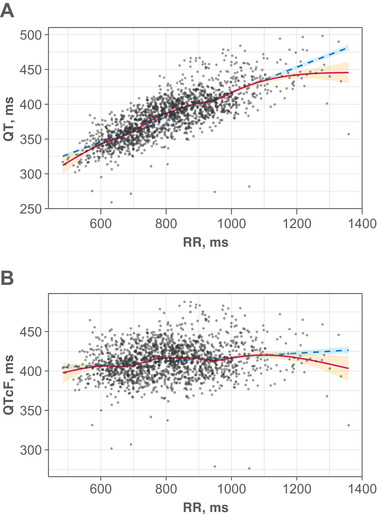
Relationship between time‐matched (A) QT vs RR and (B) QTcF vs RR. The filled circles indicate individual observed data from Studies A and B. The red line is the smooth regression. The dashed blue line is the linear regression (R2 = 0.605 for QT vs RR; R2 = 0.0255 for QTcF vs RR). The orange and blue shaded areas represent 95% CI of smooth and linear regressions, respectively. CI, confidence interval; QTcF, QT corrected using the Fridericia formula.
The final model for ΔQTcF included parameters for intercept, KRT‐232 concentration–ΔQTcF slope, baseline QTcF effect on the intercept, additive BSV on the intercept (but not on the slope, as this BSV was not significant), and residual error SD with a study effect (Table 2). All parameters were statistically significant. The final model equation for a subject's typical value of ΔQTcF (omitting BSV and residual error) was
where Conc is KRT‐232 concentration in ng/mL, and baseline QTcF and mean baseline QTcF (the overall mean in the data set) are in milliseconds. Baseline QTcF had a negative effect on the intercept: every millisecond that a subject's baseline QTcF exceeded the overall mean reduced ΔQTcF by 0.140 milliseconds. Residual error SD for Study B was estimated to be 54% higher than that for Study A, indicating more unexplained variability for the AML study. No significant effects of study, gender, or tumor type were identified on the intercept or slope.
Table 2.
Final Model Parameter Estimatesa
| Parameter | Estimate (SE) | RSE | P value | 95%CI |
|---|---|---|---|---|
| Intercept (ms) | −2.14 (0.811) | 37.9% | 0.0084 | (−3.73 to −0.552) |
| Concentration slope (ms/[ng/mL]) | 0.00214 (0.000445) | 20.8% | <0.0001 | (0.00127 to 0.00302) |
| Baseline QTcF coefficient | −0.140 (0.0378) | 27.0% | 0.0003 | (−0.214 to −0.0648) |
| Intercept BSV SD (ms) | 7.76 (0.593) | 7.64% | … | (6.69 to 9.01) |
| Residual error SD, Study A (ms) | 10.7 (0.237) | 2.21% | … | (10.3 to 11.2) |
| Ratio of residual error SD, Study A:B | 1.54 (0.0744) | 4.83% | … | (1.40 to 1.69) |
BSV, between‐subject variability; CI, confidence interval; QTcF, QT corrected using the Fridericia formula; SD, standard deviation; SE, standard error; RSE, relative standard error.
The marginal coefficient of determination for the model (variance explained by the fixed effects only; ie, intercept, slope, and baseline) is 0.0634; the conditional coefficient of determination (variance explained by fixed plus random effects; ie, intercept variability—a substantial component of total variability) is 0.385.
Model adequacy was assessed with goodness‐of‐fit (diagnostic) plots (Figure 3). Plots of observations vs population or individual predictions (top left and top middle) showed a reasonably balanced distribution of the observations around the identity line. Plots of standardized residuals vs population prediction and KRT‐232 concentration (bottom left and middle) show the residuals randomly and evenly scattered around zero as desired. Finally, residuals appeared largely normally distributed (rightmost plots). Predicted ΔQTcF vs KRT‐232 concentration with 90%CI was overlaid with the observed data in Figure 3. A reasonable fit of the linear predictions to the data was confirmed by a similar plot with the ΔQTcF data summarized by 90%CI bars plotted at the midpoint of each concentration decile.20
Figure 3.
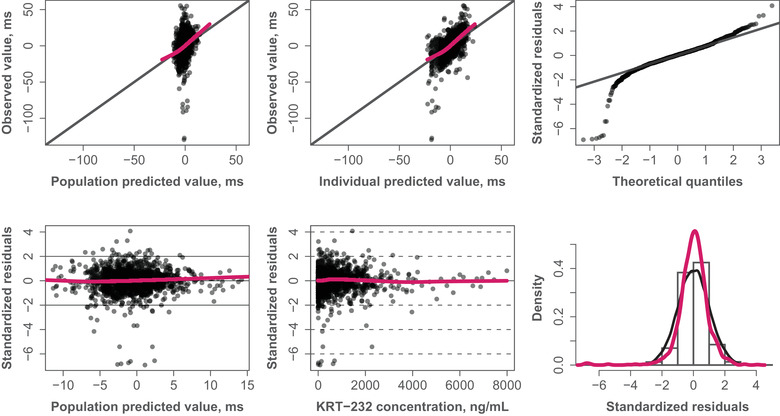
Goodness‐of‐fit plots. Red curves represent LOESS (locally weighted scatterplot smoothing). The filled circles represent individual data points. The black solid lines represent the identity line or line through first and third quartiles (quantile‐quantile plot).
Concentration‐QTc Predictions
The model evaluations show that the final model fit the observed data adequately. The model was then used to predict the mean and 90%CIs of mean ΔQTcF at mean steady‐state Cmax for doses up to the maximum clinical dose of 480 mg QD, in patients with solid tumors or AML. Patients with AML typically have higher KRT‐232 exposure than patients with solid tumors have.13 A population PK model simulation predicted that for the 480‐mg QD regimen, day 7 mean Cmax was 1951 ng/mL for patients with solid tumors, increasing to 3108 ng/mL for patients with AML (data on file). Corresponding mean (90%CI) ΔQTcF predictions, with baseline QTcF at its overall mean value, were 2.04 (0.49‐3.60) milliseconds for patients with solid tumors and 4.52 (2.35‐6.69) milliseconds for patients with AML, well under the 10‐millisecond threshold.
KRT‐232 concentrations at which the 90%CI upper bounds of mean ΔQTcF were predicted to reach 10 and 20 milliseconds were 4298 and 7821 ng/mL, respectively (Figure 4). These concentrations are 2.2‐ and 4.0‐fold higher, respectively, than the mean steady‐state Cmax for 480‐mg QD KRT‐232 predicted for patients with solid tumors, and 1.4‐ and 2.5‐fold higher, respectively, than the corresponding mean steady‐state Cmax in patients with AML.
Figure 4.
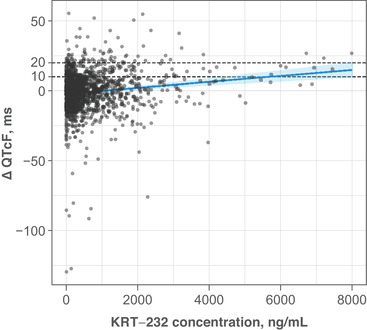
Predicted relationship of ΔQTcF with KRT‐232 concentration. The filled circles indicate individual observed data from studies A and B. The solid blue line represents model‐predicted ΔQTcF over observed range of concentrations. The shaded blue area is the 90% confidence interval of model‐predicted mean ΔQTcF. The horizontal dashed black lines represent the ΔQTcF 10 and 20 ms thresholds. ΔQTcF change from baseline QT corrected using the Fridericia formula. The marginal coefficient of determination for the model (variance explained by the fixed effects only; ie, intercept, slope, and baseline) is 0.0634; the conditional coefficient of determination (variance explained by fixed plus random effects; ie, intercept variability—a substantial component of total variability) is 0.385. ΔQTcF, change from baseline Fridericia‐corrected QT interval.
Because the concentration‐QTc analysis of KRT‐232 did not predict clinically significant QTc prolongation at the maximum intended clinical dose, the concentration‐QTc relationship for the KRT‐232 acyl glucuronide metabolite M1 was not evaluated.
Discussion
The potential of KRT‐232 to delay cardiac repolarization, as measured by QTc prolongation, was analyzed using concentration‐QTc linear mixed‐effects modeling with ΔQTcF as the dependent variable. Two phase 1 studies with doses of 15 to 480 mg supplied time‐matched PK and ECG data (Study A9 and Study B3). Significant effects included a small positive slope in KRT‐232 concentration, a negative effect of baseline QTcF, and higher residual error SD for patients with AML in Study B. The analysis found no clinically meaningful QT prolongation at doses up to 480 mg QD for the analysis population, including patients with AML, who typically have higher concentrations for a given dose than patients with solid tumors.
No patients had QTcF exceeding 500 milliseconds or ΔQTcF exceeding 60 milliseconds. The frequency of QTcF or ΔQTcF outliers (QTcF >450 milliseconds or ΔQTcF >30 milliseconds) was higher in Study B (34.6% and 26.9%, respectively) than in Study A (≤8.5%), but no dose‐response relationship was identified in either study (albeit with very small‐dose groups). These outliers were not always associated with high KRT‐232 concentrations (concentrations ranged from 19.3 to 2370 ng/mL for ΔQTcF outliers), suggesting that some could be due to KRT‐232–independent variability related, for example, to electrolyte imbalance or concomitant medications that prolong QT. The proportions of patients with PR, QRS, or HR outliers in each study was <7%, except that 3 patients (11.5%) in Study B had an HR >100 bpm with ≥25% increase from baseline.
These results were consistent with in vitro pharmacology experiments, in which KRT‐232 demonstrated a low potential to inhibit the human ether‐à‐go‐go‐related gene (hERG) channel, with a half maximal inhibitory concentration (IC50) >300 μM (>171 000 ng/mL). Based on population PK‐derived parameters for patients with AML6, the no‐effect level on hERG function of 10 μM is approximately 14.6‐fold greater than the anticipated 60‐mg QD steady‐state Cmax of 683 nM total KRT‐232 concentration. This corresponds to a plasma concentration at a 480‐mg dose that is 1.8‐fold (14.6/8‐fold) greater than the steady‐state Cmax at 480 mg. Additionally, no abnormal electrocardiographic findings were attributable to the administration of KRT‐232 in a Good Laboratory Practice 28‐day oral toxicology study in cynomolgus monkeys (unpublished data).
Analysis limitations included a modest sample size for the AML subpopulation and the lack of placebo or control arms, like other oncology studies. However, the analysis followed International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use recommendations, using data pooled across studies that covered a wide range of doses, using common methods of ECG measurement with central laboratory evaluation, and using a robust concentration‐QTc modeling approach that included baseline QTc as a covariate.
In conclusion, because the mean ΔQTcF and its 90%CI upper bound were predicted to be below 10 milliseconds at doses of up to 480 mg QD in patients with solid tumors, MM, and AML, KRT‐232 does not result in clinically meaningful QT prolongation at the doses currently under investigation in clinical trials. No significant cardiac safety concerns were identified at these doses.
Conflicts of Interest
A.T., M.A., and B.P. were Certara employees contracted by Kartos Therapeutics to perform and report this analysis. D.L. and J.G.S. are employees of Kartos Therapeutics.
A.T. is employed by Certara and has provided consultancy services as an employee of Certara; she has equity ownership of various for‐profit health care companies through broad market mutual funds. D.L. is employed by Kartos Therapeutics. M.A. is employed by Telios Pharma (formerly employed by Certara and CytomX Therapeutics) and provided consultancy services to Certara. B.P. is employed by Certara and has provided consultancy services as an employee of Certara; he has equity ownership of various for‐profit health care companies through broad market mutual funds. J.G.S. is employed by Kartos Therapeutics, has equity ownership with AstraZeneca, and has divested equity in Amgen.
Acknowledgments and Funding Information
Study 20120106 and Study 20120234 were sponsored by Amgen Inc., Thousand Oaks, California. This analysis was funded by Kartos Therapeutics, Inc., Redwood City, California. Medical writing and editorial assistance were provided by Team 9 Science, LLC, funded by Kartos Therapeutics, Inc.
References
- 1.Garcia‐Delgado R, McLornan DP, Rejtő L, et al. An open‐label, phase 2 study of KRT‐232, a first‐in‐class, oral small molecule inhibitor of MDM2, for the treatment of patients with myelofibrosis (MF) who have previously received treatment with a JAK inhibitor. Blood. 2019;134(suppl 1):2945‐2945. [Google Scholar]
- 2.Gotlib J, Gabrail N, O'Connell CL, et al. A randomized, open‐label, multicenter, phase 2 study to evaluate the efficacy, safety, and pharmacokinetics of KRT‐232 compared with ruxolitinib in patients with phlebotomy‐dependent polycythemia vera. Blood. 2019;134(suppl):4168‐4168. [Google Scholar]
- 3.Erba HP, Becker PS, Shami PJ, et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019;3(13):1939‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong MKK, Kelly CM, Burgess MA, et al. KRT‐232, a first‐in‐class, murine double minute 2 inhibitor (MDM2i), for TP53 wild‐type (p53WT) Merkel cell carcinoma (MCC) after anti–PD‐1/L1 immunotherapy. J Clin Oncol. 2020;38(15 suppl):10072‐10072. [Google Scholar]
- 5.Sun D, Li Z, Rew Y, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2‐p53 inhibitor in clinical development. J Med Chem. 2014;57(4):1454‐1472. [DOI] [PubMed] [Google Scholar]
- 6.Konopleva M, Martinelli G, Daver N, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020;34(11):2858‐2874. [DOI] [PubMed] [Google Scholar]
- 7.Canon J, Osgood T, Olson SH, et al. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53‐inducing cytotoxic agents. Mol Cancer Ther. 2015;14(3):649‐658. [DOI] [PubMed] [Google Scholar]
- 8.Shattuck‐Brandt RL, Chen SC, Murray E, et al. Metastatic melanoma patient‐derived xenografts respond to MDM2 inhibition as a single agent or in combination with BRAF/MEK inhibition. Clin Cancer Res. 2020;26(14):3803‐3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gluck WL, Gounder MM, Frank R, et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild‐type solid tumors or multiple myeloma. Invest New Drugs. 2020;38(3):831‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye Q, Jiang M, Huang WT, et al. Pharmacokinetics and metabolism of AMG 232, a novel orally bioavailable inhibitor of the MDM2‐p53 interaction, in rats, dogs and monkeys: in vitro–in vivo correlation. Xenobiotica. 2015;45(8):681‐692. [DOI] [PubMed] [Google Scholar]
- 11.Templeton IE, Podoll T, Krejsa CM, Slatter JG.A mechanistic physiologically based pharmacokinetic (PBPK) drug interaction model for the mouse double minute 2 (MDM2) inhibitor KRT‐232. Blood. 2020;136(suppl 1):9‐10. [Google Scholar]
- 12.Wong S, Krejsa CM, Lee D, et al. Effect of food on MDM2 inhibitor KRT‐232 pharmacokinetics and macrophage inhibitory cytokine‐1 (MIC‐1) response in healthy volunteers. Blood. 2020;136(suppl 1):7‐8. [Google Scholar]
- 13.Ma SC, Wada R, Allard M, Slatter G.Population pharmacokinetic analysis of the MDM2 inhibitor KRT‐232 (formerly AMG 232) in subjects with advanced solid tumors, multiple myeloma or acute myeloid leukemia. Blood. 2019;134(suppl 1):5766‐5766. [Google Scholar]
- 14.Allard MWD, Krejsa CM, Slatter JG. Exposure–macrophage inhibitory cytokine‑1 (MIC‑1) response analysis of the MDM2 antagonist KRT‑232 in patients with advanced solid tumors, multiple myeloma, or acute myeloid leukemia. HemaSphere. 2020;4:S1, Abstract EP519. [Google Scholar]
- 15.Garnett C, Bonate PL, Dang Q, et al. Scientific white paper on concentration‐QTc modeling. J Pharmacokinet Pharmacodyn. 2018;45(3):383‐397. [DOI] [PubMed] [Google Scholar]
- 16.Blotner S, Chen LC, Ferlini C, Zhi J.Phase 1 summary of plasma concentration‐QTc analysis for idasanutlin, an MDM2 antagonist, in patients with advanced solid tumors and AML. Cancer Chemother Pharmacol. 2018;81(3):597‐607. [DOI] [PubMed] [Google Scholar]
- 17.Phoenix® WinNonlin® version 8.0. Princeton, NJ: Certara USA Inc; 2020.
- 18.R Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. http://www.R-project.org/. Accessed April 2020.
- 19.Ihaka R, Gentleman R.R: A language for data analysis and graphics. J Comput Graph Stat. 1996;5(3):299‐314. [Google Scholar]
- 20.Taylor A, Allard M, Kresja C, Lee D, Slatter G.Concentration‐QTc (C‐QTc) analysis of the MDM2 inhibitor KRT‐232 (formerly AMG 232) in subjects with advanced solid tumors, multiple myeloma, or acute myeloid leukemia. Blood. 2019;134(suppl 1):5768‐5768. [Google Scholar]