

Abstract
Introduction
Blood‐based Alzheimer's disease (AD) biomarkers provide opportunities for community studies and across ethnic groups. We investigated blood biomarker concentrations in the Washington Heights‐Inwood Columbia Aging Project (WHICAP), a multi‐ethnic community study of aging and dementia.
Methods
We measured plasma amyloid beta (Aβ)40, Aβ42, total tau (t‐tau), phosphorylated tau (p‐tau)181, and p‐tau217, and neurofilament light chain (NfL) in 113 autopsied participants (29% with high AD neuropathological changes) and in 300 clinically evaluated individuals (42% with clinical AD). Receiver operating characteristics were used to evaluate each biomarker. We also investigated biomarkers as predictors of incident clinical AD.
Results
P‐tau181, p‐tau217, and NfL concentrations were elevated in pathologically and clinically diagnosed AD. Decreased Aβ42/Aβ40 ratio and increased p‐tau217 and p‐tau181 were associated with subsequent AD diagnosis.
Discussion
Blood‐based AD biomarker concentrations are associated with pathological and clinical diagnoses and can predict future development of clinical AD, providing evidence that they can be incorporated into multi‐ethnic, community‐based studies.
Keywords: Alzheimer's disease, amyloid, blood‐based biomarkers, neurofilament light chain, tau
1. INTRODUCTION
Alzheimer's disease (AD) is the leading cause of dementia.1 In 1984,2 the National Institute of Neurological and Communicative Diseases and Stroke–Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) diagnostic criteria for AD, categorized three diagnostic levels: definite AD (neuropathological diagnosis), probable AD (clinical diagnosis), and possible AD (clinical diagnosis with comorbidities). The sensitivity and specificity of the clinical criteria for probable AD compared to the post mortem diagnosis was of 81% and 70%.3 In 2011,3 updated criteria recognized that the pathological process begins before the onset of clinical symptoms. The use of magnetic resonance imaging (MRI), positron emission tomography (PET) imaging, and cerebrospinal fluid (CSF) assays were included more systematically into diagnosis. Additional sets of criteria, one for preclinical AD4 and for mild cognitive impairment (MCI) due to AD,4 were introduced that explicitly incorporated biomarkers (MRI, PET, and CSF) into diagnoses. By 2018, a diagnostic scheme5 was recommended based on biomarker evidence of amyloid ("A"), tau ("T"), and neurodegeneration ("N").6
In the clinical setting, incorporating biomarkers into the diagnosis of AD is somewhat easier compared to incorporating them into observational studies. The widespread use of PET and CSF biomarkers is difficult because of limitations in access to radiopharmaceuticals and performing lumbar punctures. Recent developments of AD blood‐based biomarkers7, 8, 9, 10 may overcome these issues, providing an opportunity to improve diagnostic accuracy in observational research.
Here we used stored plasma to measure blood‐based biomarkers in the Washington Heights‐Inwood Columbia Aging Project (WHICAP) cohort, a multi‐ethnic community‐based study, in which diagnosis of AD was defined either clinically or neuropathologically. Our focus was on the current state‐of‐the‐art AD‐related plasma biomarkers, including amyloid beta (Aβ)40 and Aβ42 as markers of amyloid pathology, total tau (t‐tau) and neurofilament light (NfL) chain as markers of neurodegeneration, and phosphorylated tau (p‐tau) 181 and 217 as markers of tau pathology. We compared plasma biomarker concentrations between clinically and pathologically defined diagnostic groups and examined differences by race/ethnicity groups. A subset had undergone florbetaben PET to assess cortical Aβ plaque burden.
2. METHODS
2.1. Participants
We selected all individuals (n = 113) from WHICAP who had brain autopsy with pathological examination and stored plasma. We also selected 300 individuals from the clinical cohort for analysis; the goal was to include equal numbers of participants from each of the three major race/ethnicity groups represented in WHICAP with similar numbers of individuals characterized as having clinical AD at their last available diagnostic visit. Race/ethnicity was self‐reported11 and included non‐Hispanic White (White), Hispanic, and non‐Hispanic Black/African American. Participants were considered for inclusion if they had stored plasma and had been assessed clinically more than once. All participants received neuropsychological testing, structured medical and neurological examinations, and blood sampling at study entry and at 18‐ to 24‐month intervals. An independent consensus committee derived diagnoses of clinical AD,3 control, or other forms of dementia. The diagnosis of clinical AD included individuals with frank dementia and those with a Clinical Dementia Rating (CDR12) of 0.5 deemed by the consensus committee as having a syndrome consistent with very early or mild AD. For primary analyses in the clinical sample, we compared individuals with and without clinical AD at the time of the blood draw used to derive biomarker concentrations. For those without AD, we compared individuals who subsequently developed clinical AD to those who remained unimpaired. A subset of the clinical cohort (n = 40) had received florbetaben PET scanning. Informed consent was obtained from all participants.
2.2. Plasma Aβ42 and Aβ40, t‐tau, and NfL
Centrifuged plasma aliquoted in polypropylene tubes and stored at −80°C was used to measure Aβ42, Aβ40, and t‐tau using SIMOA technology (Quanterix). The multiplex Neuro 3‐plex A kit (#101995), and NfL kit (#103400) were used on 96‐well plates. Rapid‐thawed plasma (25 μL diluted 4‐fold in buffer) was added to kit beads (100 μL) by pipette in each well, the plate was incubated for 15 minutes at 30°C, centrifuged at 10,000xg for 10 min, magnetic‐washed 3x for 5 minutes total, subjected to addition of SBG reagent (100 μL), followed by another incubation for 10 minutes at 30°C, centrifugation at 10,000xg for 10 min, wash again x 5 for 7 minutes total, and reading on the SIMOA SR‐X machine. Each plate assays (in duplicate) 34 samples, 8 calibrators, and 2 controls. We considered the ratio of Aβ42 to Aβ40 (Aβ42/Aβ40) as the primary amyloid biomarker.
RESEARCH IN CONTEXT
Systematic review. The literature on blood‐based biomarkers for Alzheimer's disease (AD) has included White, non‐Hispanic individuals and has been primarily clinic‐based. Here we used the state‐of‐the‐art AD biomarkers in a multi‐ethnic community in northern Manhattan.
Interpretation. Plasma phosphorylated tau (p‐tau)217, was strongly associated with post mortem diagnosis of AD, and cerebral amyloidosis on positron emission tomography (PET). P‐tau217 was also the most accurate biomarker to identify clinically diagnosed AD. Reduced amyloid beta (Aβ)42/Aβ40 ratio and increased p‐tau217 were associated with incident AD.
Future directions. These results underscore the precision gained by including blood‐based biomarkers in observational studies of AD in which autopsy is infrequent and where amyloid PET and the acquisition of spinal fluid are limited. The need for increased sample sizes and more diversity is critical. Further development of the risk assessment of these biomarkers will be a priority.
2.3. Plasma p‐tau181 and p‐tau217
The p‐tau assays were optimized to measure disease‐related differences through the selection of monoclonal antibodies. Selection of the monoclonal antibody pair provided a unique combination of sensitivity and selectivity for the tau forms in plasma that differ between AD and healthy control participants. The p‐tau181 assay was modified from that published previously8 to improve the assay and more directly compare between phosphorylation sites. The assays were performed on a streptavidin small spot plate using the Meso Scale Discovery (MSD) platform. For the p‐tau181 assay, Biotinylated‐AT270 was used as a capture antibody (anti‐p‐tau181 antibody) and SULFO‐TAG‐Ru‐4G10‐E2 (anti‐tau monoclonal antibody) for the detector. The assay was calibrated using a synthetic p‐tau181 peptide. For the p‐tau217 assay, Biotinylated‐IBA493 was used as a capture antibody (anti‐p‐tau217 antibody) and SULFO‐TAG‐Ru‐4G10‐E2 (anti‐tau monoclonal antibody) for the detector. The assay was calibrated using a synthetic P‐tau217 peptide. Additional detailed methods for the p‐tau217 and p‐tau181 assays are provided in the supporting information.
2.4. Autopsy
Cases were classified according to National Institute on Aging—Alzheimer's Association guidelines for the neuropathological assessment of AD,13 which characterizes likelihood of AD according to an "ABC" staging. In this scheme, amyloid plaques ("A") are rated according to the method of Thal et al.;14 neurofibrillary tangles are rated according to Braak et al. ("B");15, 16 and neuritic plaques are rated according to Consortium to Establish a Registry for Alzheimer's Disease (CERAD; "C") criteria.17 ABC staging yields a rating for each case, corresponding to one of four categories: not AD, low AD, intermediate AD, or high AD neuropathological change (ADNC). In this study, similar to others that examined plasma biomarkers,8, 18 our primary pathological grouping compared those classified as “high ADNC” with all other groups. Systematic Thal ratings for amyloid plaques were added later in the study and available for n = 38 cases in the current sample, so our primary ADNC classification reflected a CERAD neuritic plaque rating of 2 (moderate) or 3 (severe), and a Braak rating of V or VI without incorporation of Thal ratings. In secondary analyses, we considered biomarker levels across the four degrees of AD neuropathological change among the n = 38 participants with complete ADNC ratings (Thal, Braak, and CERAD).
2.5. Amyloid PET
A subset of participants from the clinical sample had undergone amyloid PET scanning with [18F]Florbetaben (8.1 mCi target dose). The standard uptake value ratio (SUVR) was calculated with data 50 to 70 minutes post‐injection and an inferior cerebellar gray matter reference region in native space. FreeSurfer19 was used to parcellate the brain and derive regions of interest (ROIs) from T1‐weighted MRI. A global composite SUVR was calculated as an average of frontal, temporal, and parietal cortex SUVRs, based on the FreeSurfer‐defined ROIs. An SUVR cut‐score of 1.25 was additionally used as an amyloid positivity threshold.
2.6. Analyses
We examined differences in each individual biomarker between pathological cases characterized as high ADNC and all other groups, between those diagnosed clinically with AD patients and controls, and between PET amyloid positive and negative participants by t‐tests. For autopsy data, we compared biomarker concentrations between those defined as high ADNC and all other groups with t‐tests and examined receiver operating characteristics (ROC) with associated areas under the curve (AUCs) and Aikake and Bayesian information criteria (AIC/BIC) for diagnostic classification. Differences in AUCs were determined with the method of Delong et al.20, 21, 22 We used logistic regression analyses to test how well the biomarkers classified those with high ADNC with and without age as a covariate. For these analyses we chose the p‐tau217 concentrations as the p‐tau biomarker. We used general linear models to examine differences in biomarker concentration across pathological ABC staging (not AD, low ADNC, intermediate ADNC, and high ADNC). In the clinical group, we examined associations between biomarker concentrations and demographic features (age, sex, apolipoprotein E [APOE] ε4 status) and amyloid PET SUVR with Pearson correlations and t‐tests. We used logistic regression analysis with biomarker concentrations entered simultaneously to calculate the model AUCs with respect to diagnostic and amyloid status, and we assessed the additional contribution of body mass index (BMI)23 and age as covariates to the base biomarker model by likelihood ratio test (LRT) and by testing for a change in AUC. Prior to statistical analysis, frequency distributions were inspected for outliers, which were removed from subsequent analyses. Three outliers were identified in the clinical group only: one participant with t‐tau concentration value of 472.3, one with a p‐tau181 concentration of 29.5, and one with NfL concentration of 628.5; these values are greater than 10 times the 90% quantile level of all other values and thus physiologically unlikely. The logistic regression analyses in the clinical sample were repeated with the outlier values included to examine whether they notably affected results.
Finally, we used Cox regression analyses to examine the relationship between biomarker concentrations and clinical diagnosis of AD at the last available clinical follow‐up visit among individuals classified as controls at time of the blood draw, using individuals who remained controls at last visit as reference. These analyses included biomarker concentrations entered simultaneously and the period between the initial blood draw and the last clinical diagnosis as the time‐to‐event adjusted for age at blood draw, sex, APOE ε4, and race/ethnicity. We ran separate models for p‐tau181 and p‐tau217. We repeated these analyses after inclusion of outlier values and with BMI as an additional covariate.
3. RESULTS
3.1. Participant characteristics
3.1.1. Autopsy group
Average age at death and at last clinical follow‐up were 88.63 (standard deviation [SD] = 6.80) and 85.64 (SD = 7.13) years, respectively (time interval between last evaluation/blood draw and autopsy, 2.99 [SD = 2.77] years). Thirty‐four (30%) had a clinical diagnosis of AD at their last WHICAP follow‐up visit. Thirty‐three (29%) had an ABC score indicating high ADNC. Table 1 displays demographic characteristics of the autopsy group. Cases with high ADNC were similar in age and race/ethnicity distribution but had a higher proportion of women and APOE ε4 carriers than those with lower ADNC.
TABLE 1.
Demographic and biomarker concentration characteristics in participants classified pathologically, clinically, and according to amyloid PET status
| Pathological status | Clinical status | PET amyloid status | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Controls | high AD | Total | Statistic | Controls | AD | Total | Statistic | – | + | Total | Statistic | |
| N | 80 | 33 | 113 | – | 169 | 131 | 300 | – | 32 | 8 | 40 | – |
| Age, mean (SD) years | 84.93 (7.45) | 87.38 (6.04) | 85.64 (7.13) | t = 1.67, P = 0.09 | 81.01 (6.31) | 82.99 (6.49) | 81.87(6.46) | t = 2.66 P = 0.008 | 82.16 (5.19) | 84.25 (4.55) | 82.58 (5.08) | t = 1.04, P = 0.30 |
| Sex, n (%) women | 41 (51%) | 28 (85%) | 69 (61%) | Χ2 = 11.09, P = 0.001 | 109 (64%) | 91 (69%) | 200 (67%) | Χ2 = 0.82, P = 0.36 | 19 (59%) | 5 (63%) | 24 (60%) | Χ2 = 0.03, P = 0.87 |
| APOE, n (%) ε4 | 16 (20%) | 13 (41%) | 32 (29%) | Χ2 = 4.89, P = 0.02 | 46 (27%) | 38 (29%) | 84 (28%) | Χ2 = 0.11, P = 0.73 | 11 (34%) | 6 (75%) | 17 (43%) | Χ2 = 4.32, P = 0.04 |
| Race/ethnicity, n (%) | ||||||||||||
| White | 37 (46%) | 15 (45%) | 52 (46%) | Χ2 = 1,90, P = 0.38 | 63 (37%) | 37 (28%) | 100 (33%) | Χ2 = 2.71, P = 0.25 | 11 (34%) | 4 (50%) | 15 (38%) | Χ2 = 2.15, P = 0.34 |
| Black | 25 (31%) | 7 (21%) | 32 (28%) | 53 (31%) | 47 (36%) | 100 (33%) | 17 (53%) | 2 (25%) | 19 (48%) | |||
| Hispanic | 18 (25%) | 11 (33%) | 29 (26%) | 53 (31%) | 47 (36%) | 100 (33%) | 4 (13%) | 2 (25%) | 6 (15%) | |||
| Plasma biomarker concentration, mean (SD) pg/mL | ||||||||||||
| Aβ42/Aβ40 | 0.06 (0.04) | 0.05 (0.01) | 0.06 (0.03) | t = 1.24, P = 0.21 | 0.05 (0.01) | 0.06 (0.01) | 0.05 (0.01) | t = 0.57, P = 0.56 | 0.06 (0.01) | 0.05 (0.005) | 0.06 (0.009) | t = 0.83, P = 0.40 |
| T‐tau | 4.30 (3.22) | 4.12 (2.37) | 4.25 (2.98) | t = 0.29, P = 0.76 | 4.94 (2.13) | 4.90 (2.06) | 4.92 (2.09) | t = 0.15, P = 0.88 | 4.55 (2.13) | 5.64 (1.43) | 4.77 (2.04) | t = 1.36, P = 0.18 |
| P‐tau181 | 1.06 (0.81) | 1.93 (1.14) | 1.32 (1.00) | t = 4.54, P < 0.001 | 0.86 (0.73) | 1.24 (1.09) | 1.02 (0.92) | t = 3.55, P < 0.001 | 0.73 (0.61) | 1.35 (0.54) | 0.86 (0.64) | t = 2.59, P = 0.01 |
| P‐tau217 | 0.19 (0.16) | 0.51 (0.40) | 0.28 (0.29) | t = 5.99, P < 0.001 | 0.18 (0.17) | 0.32 (0.32) | 0.25 (0.26) | t = 4.80, P < 0.001 | 0.15 (0.14) | 0.39 (0.18) | 0.20 (0.18) | t = 3.86, P < 0.001 |
| NfL | 51.45 (51.11) | 34.41 (25.35) | 46.43 (45.63) | t = 1.82, P = 0.07 | 31.10 (28.96) | 36.55 (24.63) | 33.48 (27.25) | t = 1.76, P = 0.08 | 30.29 (20.73) | 30.31 (14.15) | 30.30 (19.43) | t < 0.01, P = 0.99 |
Notes: For the autopsy group, "high AD" refers to ADNC classification of high degrees of AD neuropathological change and controls include all other groups. For the clinical group, AD diagnosis is based on standardized clinical classification without consideration of biomarker status. For the PET subgroup, amyloid positivity was determined according to an SUVR threshold of 1.25.
Abbreviations: AD, Alzheimer's disease; ADNC, AD neuropathological change; PET, positron emission tomography; SD, standard deviation; SUVR, standardized uptake value ratio.
3.1.2. Clinical group
Demographic characteristics at the time of the blood draw used to derive biomarker concentrations of participants included in the clinical cohort are in Table 1. Compared to controls, those with clinical AD were similar for sex distribution, race/ethnicity distribution, and APOE ε4 allele frequency, but were slightly older.
Of those characterized as controls at time of blood draw, 71 subsequently had a clinical diagnosis of AD by the last assessment. They were similar in age (t = 0.52, P = .46), sex (χ2 < 0.01, P = .99), APOE ε4 allele frequency (χ2 = 0.52, P = .46), and race/ethnicity distribution (χ2 = 1.72, P = .42) as those who remained controls. The last available diagnosis took place an average of 4.29 (SD = 3.04) years after the blood draw used to derive biomarker concentrations.
3.1.3. Amyloid PET subgroup
We compared demographic features between amyloid positive and negative participants (see Table 1). Amyloid positive individuals were more likely to be APOE ε4 allele carriers, but were similar to amyloid negative participants in age, sex, and race/ethnicity distribution. Fifty percent of amyloid positive individuals (n = 4) were considered to have clinical AD at the diagnostic visit closest to the blood draw whereas 25% (n = 8) amyloid negative individuals met clinical criteria for AD (χ2 = 1.90, P = .16). PET scans were completed 3.79 (SD = 20.8) months on average after the blood draw used to derive biomarker concentrations.
3.2. Biomarker concentration and neuropathological diagnosis
Individuals with high ADNC had higher concentrations of p‐tau181 and p‐tau217, but similar Aβ42/Aβ40, t‐tau, and NfL concentrations compared to those with lower ADNC (see Table 1). P‐tau217 and p‐tau181 showed good diagnostic classification, but concentrations for the other biomarkers did not (see Table 2 and Figure 1A‐D). Although sample sizes were small and confidence intervals relatively wide, classification accuracy for p‐tau181 and particularly p‐tau217 concentrations was numerically better in non‐Hispanic Black and Hispanic cases relative to Whites (see Table 2 and Figure 1). When we considered classification accuracy of all biomarkers together in a logistic regression analysis in the overall sample, the resulting ROC area was 0.925. Adding age at death to the model increased the ROC area to 0.932. Although age improved model fit by LRT (P = .019), the increase in ROC area was not statistically significant (P = .597). There was a monotonic increase in p‐tau181 (F = 5.04, P = .005) and p‐tau217 (F = 6.27, P = .002) but not the other biomarkers (F values range = 0.12–0.25, P‐values range = 0.70–0.94) across neuropathological classification groups (see Figure 2).
TABLE 2.
AUC statistics for each biomarker in the total sample and stratified by race/ethnicity, together with 95% confidence intervals, and P‐values
| Table 2 A (autopsy) | N | AUC | 95% CI | P | AIC/BIC | Aβ42/40 | Tau | P‐tau181 | P‐tau217 | NfL |
|---|---|---|---|---|---|---|---|---|---|---|
| Whole sample | 113 | Statistical comparison between ROC curves, P‐value | ||||||||
| Aβ42/40 | 112 | 0.58 | 0.47 to 0.68 | 0.1769 | 138/143 | |||||
| Tau | 112 | 0.48 | 0.37 to 0.60 | 0.7614 | 140/145 | 0.3183 | ||||
| P‐tau181 | 112 | 0.77 | 0.67 to 0.87 | <0.0001 | 120/126 | 0.0039 | 0.0017 | |||
| P‐tau217 | 112 | 0.84 | 0.75 to 0.92 | <0.0001 | 108/113 | 0.0001 | <0.0001 | 0.0116 | ||
| NfL | 112 | 0.59 | 0.48 to 0.70 | 0.0396 | 136/141 | 0.8387 | 0.1185 | 0.0345 | 0.0017 | |
| Non‐Hispanic White | ||||||||||
| Aβ42/40 | 52 | 0.60 | 0.44 to 0.77 | 0.1769 | 65/69 | |||||
| Tau | 52 | 0.56 | 0.39 to 0.73 | 0.4007 | 66/70 | 0.7328 | ||||
| P‐tau181 | 52 | 0.65 | 0.48 to 0.83 | 0.2058 | 65/69 | 0.6377 | 0.5378 | |||
| P‐tau217 | 52 | 0.75 | 0.61 to 0.89 | 0.0156 | 61/65 | 0.1444 | 0.1805 | 0.0480 | ||
| NfL | 52 | 0.64 | 0.49 to 0.79 | 0.0103 | 60/64 | 0.7167 | 0.4227 | 0.9216 | 0.3471 | |
| Non‐Hispanic Black | ||||||||||
| Aβ42/40 | 31 | 0.53 | 0.31 to 0.75 | 0.9436 | 38/40 | |||||
| Tau | 31 | 0.58 | 0.37 to 0.80 | 0.4466 | 37/39 | 0.5973 | ||||
| P‐tau181 | 31 | 0.94 | 0.86 to 1.00 | 0.0001 | 28/31 | 0.0020 | 0.0061 | |||
| P‐tau217 | 31 | 0.96 | 0.90 to 1.00 | <0.0001 | 21/23 | 0.0005 | 0.0020 | 0.4304 | ||
| NfL | 31 | 0.54 | 0.27 to 0.82 | 0.5863 | 37/40 | 0.9340 | 0.7570 | 0.0138 | 0.0078 | |
| Hispanic | ||||||||||
| Aβ42/40 | 29 | 0.62 | 0.42 to 0.82 | 0.4671 | 42/45 | |||||
| Tau | 29 | 0.67 | 0.45 to 0.90 | 0.2676 | 42/44 | 0.6928 | ||||
| P‐tau181 | 29 | 0.82 | 0.66 to 0.98 | 0.0029 | 34/36 | 0.1008 | 0.1998 | |||
| P‐tau217 | 29 | 0.85 | 0.69 to 1.00 | 0.0001 | 28/31 | 0.0754 | 0.1671 | 0.5223 | ||
| NfL | 29 | 0.44 | 0.20 to 0.67 | 0.7539 | 43/45 | 0.2516 | 0.0579 | 0.0026 | 0.0024 | |
| Table 2 B (clinical) | N | AUC | 95% CI | P | AIC/BIC | Aβ42/40 | Tau | P‐tau181 | P‐tau217 | NfL |
|---|---|---|---|---|---|---|---|---|---|---|
| Whole sample | Statistical comparison between AUC curves, P‐value | |||||||||
| Aβ42/40 | 297 | 0.49 | 0.43 to 0.56 | 0.4951 | 411/418 | |||||
| Tau | 297 | 0.49 | 0.43 to 0.56 | 0.8586 | 411/418 | 0.9763 | ||||
| P‐tau181 | 297 | 0.61 | 0.54 to 0.67 | 0.0003 | 398/405 | 0.0245 | 0.0348 | |||
| P‐tau217 | 297 | 0.63 | 0.57 to 0.70 | <0.0001 | 384/391 | 0.0076 | 0.0079 | 0.1208 | ||
| NfL | 297 | 0.59 | 0.52 to 0.66 | 0.0800 | 408/415 | 0.0517 | 0.0943 | 0.5684 | 0.2638 | |
| Non‐Hispanic White | ||||||||||
| Aβ42/40 | 99 | 0.50 | 0.39 to 0.63 | 0.9423 | 135/140 | |||||
| Tau | 99 | 0.53 | 0.42 to 0.65 | 0.8990 | 135/140 | 0.7510 | ||||
| P‐tau181 | 100 | 0.69 | 0.59 to 0.80 | 0.0011 | 125/130 | 0.0261 | 0.0392 | |||
| P‐tau217 | 100 | 0.71 | 0.61 to 0.82 | <0.0001 | 119/124 | 0.0114 | 0.0298 | 0.4363 | ||
| NfL | 99 | 0.65 | 0.54 to 0.77 | 0.1383 | 133/138 | 0.1074 | 0.0853 | 0.5661 | 0.4325 | |
| Non‐Hispanic Black | ||||||||||
| Aβ42/40 | 98 | 0.50 | 0.39 to 0.61 | 0.3616 | 139/144 | |||||
| Tau | 98 | 0.56 | 0.44 to 0.67 | 0.3041 | 138/144 | 0.5543 | ||||
| P‐tau181 | 98 | 0.63 | 0.51 to 0.74 | 0.1093 | 137/142 | 0.1758 | 0.2455 | |||
| P‐tau217 | 98 | 0.68 | 0.57 to 0.78 | 0.0030 | 131/136 | 0.0539 | 0.0690 | 0.0455 | ||
| NfL | 98 | 0.64 | 0.53 to 0.76 | 0.0040 | 131/136 | 0.0797 | 0.1453 | 0.7547 | 0.5945 | |
| Hispanic | ||||||||||
| Aβ42/40 | 100 | 0.45 | 0.35 to 0.57 | 0.9405 | 142/147 | |||||
| Tau | 100 | 0.58 | 0.47 to 0.70 | 0.1528 | 140/145 | 0.1264 | ||||
| P‐tau181 | 100 | 0.51 | 0.40 to 0.64 | 0.0672 | 139/144 | 0.5267 | 0.4876 | |||
| P‐tau217 | 100 | 0.52 | 0.40 to 0.64 | 0.0344 | 138/143 | 0.5163 | 0.4787 | 0.9224 | ||
| NfL | 100 | 0.48 | 0.37 to 0.6 | 0.4999 | 142/147 | 0.6974 | 0.1470 | 0.7688 | 0.7443 | |
Notes: Fit statistics are presented as AIC and BIC. P‐values for statistical comparisons among AUCs are presented. Table 2A displays results from the autopsy sample. Table 2B presents results from the clinical sample. Bolded values are statistically significant.
Abbreviations: Aβ, amyloid beta; AIC, Aikake information criterion; AUC, area under the receiver operating characteristic curve; BIC, Bayesian information criterion; CI, confidence interval; NfL, neurofilament light.
FIGURE 1.
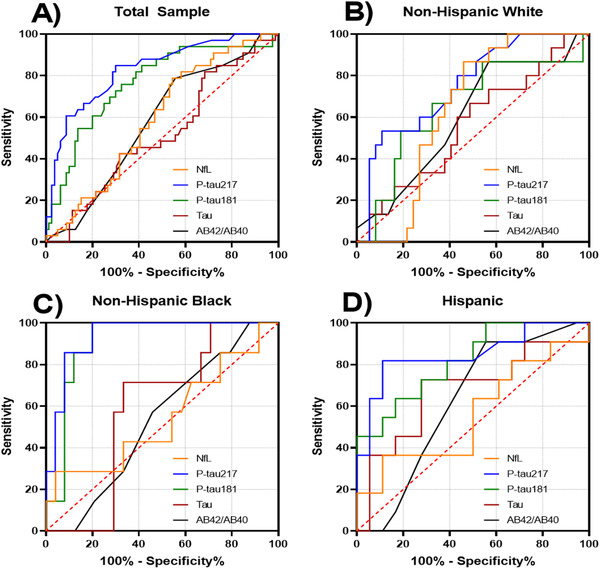
Receiver operating curves for classification of post mortem diagnosis of Alzheimer's disease. A, Total sample. B, Non‐Hispanic Whites. C, Non‐Hispanic Blacks. D, Hispanics
FIGURE 2.
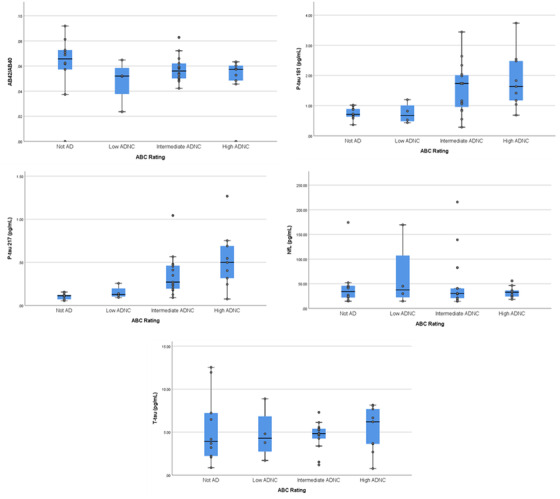
Biomarker concentrations across pathological "ABC" ratings. Midline represents median, box represents 25th and 75th percentile, and T‐bars represent 95% confidence interval. Individual subject data points are superimposed. AD, Alzheimer's disease; ADNC, AD neuropathological change;
3.3. Biomarker concentration and clinical characteristics
3.3.1. Demographic features
Increased age was associated with t‐tau (r = 0.149, P = .01), p‐tau181 (r = 0.214, P < .001), p‐tau217 (r = 0.192, P = .001), and NfL (r = 0.286, P < .001), but not Aβ42/Aβ40 (r = –0.099, P = .08) concentrations. Biomarker concentrations did not differ between men and women (t value range = 0.14–1.12, P value range = 0.25–0.88). Compared to non‐carriers, APOE ε4 allele carriers had higher concentrations of p‐tau181 (mean ± SD = 1.20 ± 0.95 vs. 0.96 ± 0.91, t = 2.03, P = .04) and p‐tau217 (mean ± SD = 0.31 ± 0.23 vs. 0.22 ± 0.26, t = 2.78, P = .006), but were similar for all other biomarker concentration levels (t value range 0.16–1.30, P value range 0.86–0.19). Increased BMI was associated with lower p‐tau181 (r = –0.20, P = .001), p‐tau217 (r = –0.20, P = .001), and NfL (r = –0.15, P = .02) concentrations but was unrelated to Aβ42/Aβ40 (r = –0.06, P = .32) and t‐tau concentrations (r = –0.07, P = .28) concentrations. Biomarker concentrations were similar across the three race/ethnicity groups (F value range 0.61–2.15, P value range 0.54–0.11).
3.3.2. Clinical diagnosis at time of blood draw
We examined the differences in biomarker concentrations between participants with clinically diagnosed AD and controls (Table 1 and Figure 3). Participants with AD had higher concentrations of p‐tau181, p‐tau217, and NfL, and similar Aβ42/Aβ40 and t‐tau levels compared to controls. We constructed ROCs for each biomarker as a function of diagnostic status (Figure 4), which revealed the best diagnostic classification for p‐tau concentrations (Table 2). A logistic regression model with all biomarkers resulted in an ROC area of 0.663. Inclusion of three outliers did not reveal any striking changes to this model comparing coefficients or ROC area (0.668). Comparing models after adding age and BMI covariates did not significantly improve the model fit or prediction (LRT P = .251, ROC comparison P = .235). When stratified by race/ethnicity, classifications were comparable among non‐Hispanic White and Black participants but AUCs among Hispanic participants were relatively low for all biomarkers measured (Table 2 and Figure 4).
FIGURE 3.
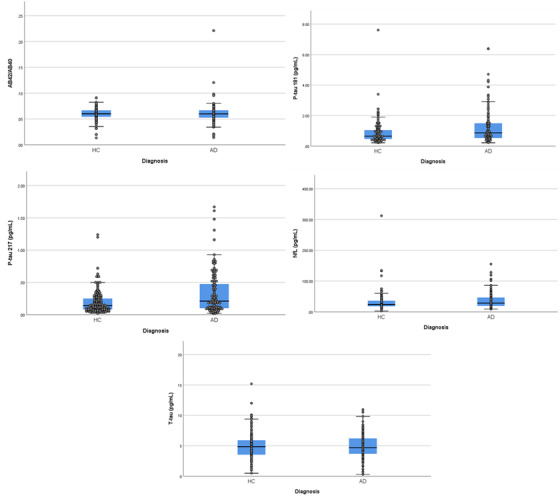
Differences between clinically diagnosed patients with Alzheimer's disease (AD) and healthy controls (HC) in absolute concentrations of each plasma biomarker. Midline represents median, box represents 25th and 75th percentile, and T‐bars represent 95% confidence interval. Individual subject data points are superimposed
FIGURE 4.
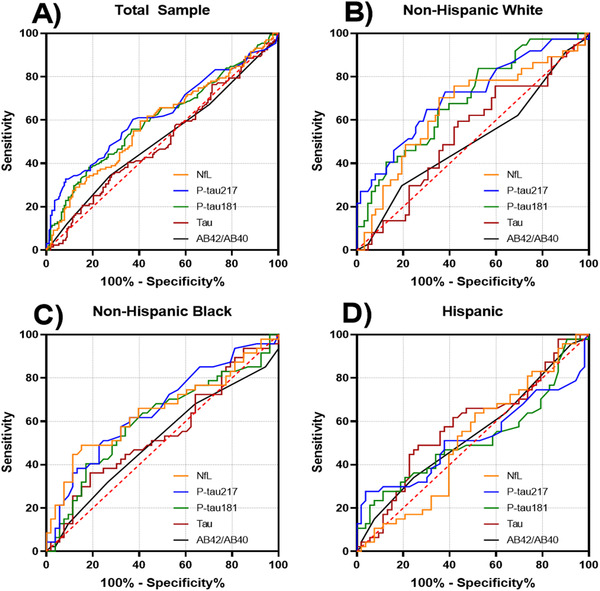
Receiver operating curves for classification of clinical diagnosis of Alzheimer's disease. A, Total sample. B, Non‐Hispanic Whites. C, Non‐Hispanic Blacks. D, Hispanics
3.3.3. Amyloid PET
Compared to amyloid negative individuals the amyloid positive individuals had higher levels of p‐tau217 and p‐tau181 (Table 2). Total tau concentrations trended higher in amyloid positive individuals, but Aβ42/Aβ40 and NfL levels were quite similar between the groups. There were strong correlations of amyloid SUVR with p‐tau217 (r = 0.48, P = .002) and p‐tau181 (r = 0.36, P = .02), more modest associations with t‐tau (r = 0.23, P = .15) and Aβ42/Aβ40 (r = –0.16, P = .30), and trivial correlations with the other biomarkers (r value range = 0.01–0.07, P value range = 0.93–0.65). When we examined classification of amyloid positivity, the findings paralleled the other results: AUCs were greatest for p‐tau217 (AUC: 0.84, 95% confidence interval [CI]: 0.68–0.99) and p‐tau181 (AUC: 0.82, 95%CI: 0.65–0.99); AUCs for the other biomarkers ranged from 0.40 (95%CI: 0.22–0.59, Aβ42/Aβ40) to 0.56 (95%CI: 0.34–0.79, NfL). A multiple logistic regression model with all biomarkers resulted in an ROC area of 0.840. Comparing models after adding age and BMI covariates did not improve the model fit or prediction (LRT P = .868, ROC comparison P = .732). We did not examine differences across race/ethnicity groups because of the reduced sample size and low number of amyloid positive participants.
3.4. Risk of subsequent clinical AD among those without dementia at the first blood draw
We used the median value of Aβ42/Aβ40, p‐tau181, and p‐tau217 to estimate the risk of developing AD. The overall effect of biomarker concentration on subsequent development of clinical AD was similar for the model that included p‐tau181 (χ2 = 26.27, P = .002), and p‐tau217 (χ2 = 21.98, P = .009) in independent analyses. Including outlier values did not notably affect the model that included P‐tau181 (χ2 = 29.08, P < .001) and had no impact on the model that examined p‐tau217. Similarly, including BMI as an additional covariate did not markedly effect either the model that examined p‐tau181 (χ2 = 25.04, P = .005) or p‐tau217 (χ2 = 26.26, P = .004); BMI was not predictive of outcome in either model (B = 0.002, P = .95 and B = 0.006, P = .83). We found reduced Aβ42/Aβ40 ratio and an increase in either p‐tau217 or p‐tau181 to be associated with increased risk of developing a clinical diagnosis of AD (Table 3). None of the other biomarker concentrations were associated with increased risk, but APOE ε4 allele was associated with a slightly higher risk.
TABLE 3.
The association between plasma biomarker concentrations derived at blood draw and the subsequent clinical AD diagnosis ≈4 years later
| Table 3 A | B | P | OR (95% CI) |
|---|---|---|---|
| P‐tau181 (pg/mL) | 1.01 | 0.001 | 2.74 (1.54–4.86) |
| Aβ42/Aβ40 | 0.60 | 0.02 | 1.82 (1.09–3.03) |
| T‐tau (pg/mL) | ‐0.095 | 0.06 | 0.90 (0.82–1.00) |
| NfL (pg/mL) | 0.005 | 0.18 | 1.00 (0.99–1.01) |
| Age | 0.06 | 0.03 | 1.06 (1.00–1.12) |
| APOE e4 allele | ‐0.25 | 0.38 | 0.77 (0.43–1.37) |
| Race/ethnicity | |||
| Non‐Hispanic White | 1.0 | reference | |
| Black | ‐0.19 | 0.57 | 0.82 (0.43–1.59) |
| Hispanic | 0.09 | 0.77 | 1.09 (0.58–2.04) |
| Sex | 0.40 | 0.14 | 1.49 (0.87–2.58) |
| Table 3 B | B | P | OR (95% CI) |
|---|---|---|---|
| P‐tau217 (pg/mL) | 0.82 | 0.003 | 2.27 (1.31–3.93) |
| Aβ42/Aβ40 | 0.57 | 0.03 | 1.78 (1.05–3.00) |
| T‐tau (pg/mL) | ‐0.04 | 0.41 | 0.96 (0.87–1.05) |
| NfL (pg/mL) | 0.007 | 0.05 | 1.00 (1.00–1.01) |
| Age | ‐0.027 | 0.31 | 0.97 (0.92–1.02) |
| APOE e4 allele | ‐0.38 | 0.19 | 0.67 (0.37–1.21) |
| Race/ethnicity | |||
| Non‐Hispanic White | 1.0 | – | Reference |
| Black | –0.21 | 0.52 | 0.80 (0.42–1.54) |
| Hispanic | –0.41 | 0.22 | 0.66 (0.34–1.29) |
| Sex | 0.27 | 0.32 | 1.31 (0.76–2.25) |
Notes: We used Cox regression in which the time to event was calculated as the period between the blood draw and last diagnostic assessment. Biomarker predictors were dichotomized according to their median (Ab42/Ab40 the higher value was the reference; P‐tau181 and P‐tau217 the lower values were the references); outcome was coded as 1 = healthy control, 2 = incident AD and we adjusted for age at last visit, sex (reference group is male sex), ethnic group, and APOE‐ε4; Table 3A shows the adjusted results using p‐tau181, while Table 3B displays the adjusted results using p‐tau217.
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; APOE, apolipoprotein E; CI, confidence interval; NfL, neurofilament light; OR, odds ratio.
4. DISCUSSION
We found that the plasma biomarker concentrations of phosphorylated tau, particularly p‐tau217, were strongly associated with autopsy‐confirmed AD. As expected, this observation did not completely translate to clinically diagnosed participants due to known limitations of clinical diagnosis for AD. Nonetheless, at the time of blood draw those diagnosed clinically with AD had similar plasma biomarker profiles to the participants who were autopsy‐confirmed AD, including higher p‐tau217 and NfL concentrations and classification accuracy was generally quite good. P‐tau biomarkers were also associated with amyloid pathology on PET, more so than other plasma biomarkers, including Aβ42/Aβ40. Among individuals classified as controls at time of blood draw, lower Aβ42/Aβ40 ratio, and higher p‐tau217 or p‐tau181 concentrations, were associated with increased risk of subsequent AD diagnosis.
Plasma levels of p‐tau217 outperformed all other biomarkers across analyses, achieving AUCs of 0.84 overall, and 0.85 and 0.96 in Hispanic and Black participants with pathological diagnosis. However, in the analysis of those who developed subsequent AD dementia, both p‐tau181 and p‐tau217 as well as the Aβ42/Aβ40 ratio were reliable predictors, consistent with previous observations.18 It is clear that among the currently available plasma biomarkers, p‐tau217 concentrations reflect underlying tau pathology with the greatest fidelity and are useful to aid the clinical diagnosis of AD. As expected, biomarker concentrations were somewhat age‐dependent and varied with respect to APOE ε4 status. Concentrations did not differ across race/ethnicity or sex, although larger studies will be necessary to understand fully the potentially moderating effects of these demographic variables.
The results we observed for p‐tau181 are similar with those of Janelidze et al.7 In addition, p‐tau217 concentrations were numerically more strongly associated with post mortem and amyloid PET outcomes, consistent with Palmqvist et al., who showed p‐tau217 was superior to p‐tau181 for determination of pathology by PET or neuropathology.10, 18 Our p‐tau217 results are similar to Palmqvist et al.,18 although we did not observe large differences between p‐tau217 and p‐tau181, possibly attributable to differences in assay design that incorporated the 4G10E2 antibody in place of the LRL antibody for the p‐tau181 assay. The slight differences may partially be attributed to the older ages in this study and higher likelihood of comorbidities.24 Additionally, Janelidze et al.7 separated AD from other forms of dementia, aiding in the specificity of p‐tau. For this study, we used strict criteria to select individuals with clinical AD and did not include other dementias.
The WHICAP cohort is racially/ethnically diverse and community‐based. Most previous studies that incorporated plasma biomarkers included clinic‐based samples with minimal racial and ethnic diversity. Here, we provide evidence that plasma biomarker data can be incorporated successfully into community‐based research, correspond with neuropathological changes seen in AD, and perform equally well or better in racial/ethnic groups typically under‐represented in aging research.
While the observations shown in this study highlight the promise and potential of plasma‐based biomarkers in identification of AD pathology and risk of developing AD, they fall short as stand‐alone diagnostics. Based on the findings here, p‐tau181 or p‐tau217 would augment the clinical diagnostic accuracy of AD as well as the presence of pathology preceding detectable symptoms.
Although the plasma biomarkers showed modest associations with clinical dementia, the p‐tau associations improved when confirmed with PET or post mortem evidence of high ADNC. Pathological and PET diagnoses are more definitive than clinical diagnoses. Clinical AD is a heterogeneous condition, determined by multiple pathologies that may vary across individuals.25 While our study was not designed to test the specificity of the plasma biomarkers in distinguishing AD from other dementias, future research will need to consider other forms of dementia. Admittedly, the small number of amyloid positive individuals may increase the likelihood of a Type 1 statistical error, although the p‐tau181 and p‐tau217 observations were consistent, validating observations with autopsy and clinical data.
Prior research on AD biomarkers across racial/ethnic groups has been challenged by small sample sizes and selection biases and has mostly focused on comparing non‐Hispanic Whites to Blacks, while inclusion of Hispanics is less frequent.26 ‐30 Large population studies are required to provide key information as to the role race/ethnicity has in disease prevalence or the socioeconomic factors that contribute to dementia. This study provides evidence that p‐tau217 may be useful as an indicator of pathology that will aid in the evaluation of other biomarkers as well as understanding race/ethnicity and socioeconomic factors in the context of large community‐based studies.
Taken together the results here provide encouraging data for the use of blood‐based biomarkers in diverse cohorts across clinical settings and in observational, epidemiological studies. Establishing a universal cut‐point for p‐tau181 or p‐tau217 should be a priority in future studies of these biomarkers.
CONFLICTS OF INTEREST
The following authors have no conflicts of interest to report: Adam M. Brickman, Jennifer J. Manly, Lawrence S. Honig, Danurys Sanchez, Dolly Reyes‐Dumeyer, Rafael A. Lantigua, Patrick J. Lao, Yaakov Stern, Jean Paul Vonsattel, Andrew F. Teich and Richard Mayeux. David C. Airey, Nicholas Kyle Proctor, and Jeffrey L. Dage are employees and stock holders of Eli Lilly and Company.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
Data collection and sharing for this project was supported by the Washington Heights‐Inwood Community Aging Project (WHICAP; P01AG07232, RF1AG054023, R01AG037212, R56AG034189, R01AG034189, R01AG054520) funded by the National Institute on Aging (NIA). This manuscript has been reviewed by WHICAP investigators for scientific content and consistency of data interpretation with previous WHICAP Study publications. We acknowledge the WHICAP study participants and the WHICAP research and support staff for their contributions to this study.
Florbetaben was provided to the study by Life Molecular Imaging (formally Piramal Imaging) under an Investigator Initiated Study.
Brickman AM, Manly JJ, Honig LS, et al. Plasma p‐tau181, p‐tau217, and other blood‐based Alzheimer's disease biomarkers in a multi‐ethnic, community study. Alzheimer's Dement. 2021;17:1353–1364. 10.1002/alz.12301
REFERENCES
- 1.Carmona S, Hardy J, Guerreiro R. The genetic landscape of Alzheimer disease. Handb Clin Neurol. 2018;148:395‐408. [DOI] [PubMed] [Google Scholar]
- 2.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA work group under the auspices of department of health and human services task force on Alzheimer's Disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 3.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jack CR Jr, Bennett DA, Blennow K, Research Framework NIA‐AA . Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jack CR Jr, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer's disease biomarkers. Neurology. 2016;87:539‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020;26:379‐386. [DOI] [PubMed] [Google Scholar]
- 8.Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mielke MM, Hagen CE, Xu J, et al. Plasma phospho‐tau181 increases with Alzheimer's disease clinical severity and is associated with tau‐ and amyloid‐positron emission tomography. Alzheimers Dement. 2018;14:989‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med. 2019;11:e11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bureau USC . Census of Population and Housing. Summary Population and Housing Characteristics. Washington, DC: Bureau of the Census; 2001. [Google Scholar]
- 12.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412‐2414. [DOI] [PubMed] [Google Scholar]
- 13.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791‐1800. [DOI] [PubMed] [Google Scholar]
- 15.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer's disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 17.Mirra SS, Heyman A, McKeel D, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479‐486. [DOI] [PubMed] [Google Scholar]
- 18.Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of Plasma Phospho‐tau217 for Alzheimer's Disease vs other neurodegenerative disorders. JAMA, J Am Med Assoc. 2020;324:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dale AM, Fischl B, Sereno MI. Cortical surface‐based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9:179‐194. [DOI] [PubMed] [Google Scholar]
- 20.Cleves MA. From the help desk: comparing areas under receiver operating characteristic curves from two or more probit or logit models. Stata J. 2002;2:301‐313. [Google Scholar]
- 21.Robin X, Turck N, Hainard A, et al. pROC: an open‐source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeLong ER, DeLong DM, Clarke‐Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837‐845. [PubMed] [Google Scholar]
- 23.Manouchehrinia A, Piehl F, Hillert J, et al. Confounding effect of blood volume and body mass index on blood neurofilament light chain levels. Ann Clin Transl Neurol. 2020;7:139‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134:171‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris JC, Schindler SE, McCue LM, et al. Assessment of racial disparities in biomarkers for Alzheimer Disease. JAMA Neurol. 2019;76:264‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information