

Abstract
Receptor occupancy (RO) assessment by flow cytometry is an important pharmacodynamic (PD) biomarker in the clinical development of large molecules such as monoclonal therapeutic antibodies (mAbs). The total‐drug‐bound RO assay format directly assesses mAb binding to cell surface targets using anti‐drug detection antibodies. Here, we generated a flow cytometry detection antibody specifically binding to mAbs of the IgG1 P329GLALA backbone. Using this reagent, we developed a total‐drug‐bound RO assay format for RG7769, a bi‐specific P329GLALA containing mAb targeting PD‐1 and TIM3 on T cells. In its fit‐for‐purpose validated version, this RO assay has been used in the Phase‐I dose escalation study of RG7769, informing on peripheral T cell RO and RG7769 antibody binding capacity (ABC). We assessed RG7769 RO in checkpoint‐inhibitor (CPI) naïve patients and anti‐PD‐1 CPI experienced patients using our novel assay. Here, we show that in both groups, complete T cell RO can be achieved (~100%). However, we found that the maximum number of T cell binding sites for RG7769 pre‐dosing was roughly twofold lower in patients recently having undergone anti‐PD‐1 treatment. We show that this is due to steric hindrance exerted by competing mAbs masking the available drug binding sites. Our findings highlight the importance of quantitative mAb assessment in addition to relative RO especially in the context of patients who have previously received anti‐PD‐1 treatment.
Keywords: assay validation, checkpoint inhibitors, fit‐for‐purpose, flow cytometry, PD biomarkers, PD‐1, receptor occupancy, steric hindrance
1. INTRODUCTION
Receptor occupancy (RO) assessment in peripheral blood by flow cytometry is a readily accessible biomarker, which can inform on the relative degree of target engagement of a therapeutic monoclonal antibody (mAb) to its cell surface target [1, 2, 3, 4]. RO assays can be used in combination with other parameters to potentially inform on an optimal mAb concentration in vivo in pre‐clinical and clinical settings, for instance in a Phase‐I dose escalation (DE) scenario. Two main types of RO assays exist [1, 2]. Firstly, the entirety of available cell surface target can be assessed in situations where non‐drug competing detection antibodies are available. In the same assay, using drug competing detection mAbs, it can be assessed how much of the target is bound by the therapeutic mAb; this approach is termed “free‐vs‐total” RO assay. Alternatively, if reagent choice is limited and non‐drug competing reagents are not available, a “total‐drug‐bound” format can be chosen. Here, the therapeutic mAb is detected via an anti‐drug antibody approach. However, when therapeutic mAbs of IgG1 or IgG4 isotype are applied, their differentiation from natural antibodies is challenging as typically no specific reagents for the therapeutic mAb exist, apart from anti‐idiotype antibodies. These, however, may interfere with the target binding of the therapeutic mAb and may result in extra challenges in the presence of idiotypic anti‐drug antibodies (ADA) [5]. Determining the degree of RO is particularly important in DE studies with immune‐cell activating mAbs (such as superagonists), given that low doses are usually administered. Here, in order to avoid over‐activation of leukocytes and the induction of cytokine shock, reliable whole‐blood RO data have been found to be of critical value in drug development [6, 7].
Assessing mAb target engagement is also important given the increasingly routine use of cancer immunotherapy (CIT) drugs. In CIT, mAbs are used to treat various cancer types [8]. Prominent examples include checkpoint inhibitors (CPI) pembrolizumab and nivolumab of the IgG4 isotype [9, 10]. These antibodies target programmed cell death protein 1 (PD‐1) on the surface of antigen‐specific dysfunctional (“exhausted”) T cells [11] by binding to defined PD‐1 extracellular epitopes [12]. In addition, several other anti‐PD‐1 therapeutic mAbs are in clinical development or already approved [10, 13, 14, 15], highlighting the importance of this molecule for CIT. In the case of the clinical development of CPIs, some trials included RO by flow cytometry to confirm the degree of PD‐1 occupancy in patients and establish pharmacokinetics–pharmacodynamics (PK–PD) relationships. For instance, in the nivolumab Phase I and III trials, PD‐1 RO was found to be highly stable, resulting in persistently high target engagement (in the range of 60%–80%) even 8 weeks post‐dosing [10, 14].
Studies suggested that in many tumors, upon anti‐PD‐1 therapy, other negative regulators of T cell activation can be co‐expressed, including T cell immunoglobulin and mucin domain‐containing protein 3 (TIM3) [16, 17]. More recently, in order to simultaneously target PD‐1 and TIM3 in cancer patients, Roche developed RG7769 (also termed RO7121661), a bi‐specific mAb with an Fc gamma receptor silent IgG1 P329GLALA backbone [18, 19, 20, 21, 22, 23, 24, 25]. RG7769 was designed so that its anti‐PD‐1 affinity is higher than its anti‐TIM3 affinity in order to avoid the antibody from binding to non‐T cells [24, 26]. It is currently being evaluated in NP40435, a multicenter Phase‐I DE study (NCT03708328, https://clinicaltrials.gov/ct2/show/NCT03708328). Here, we assessed RO on immune cells in the peripheral blood of CPI‐naïve and CPI‐experienced cancer patients.
Importantly, given the increasing fraction of anti‐PD‐1 CPI experienced cancer patients and the relatively small size of cell surface PD‐1 [12], we reasoned that prior exposure to a competitor‐anti‐PD‐1 could impact flow cytometry detection antibody binding to PD‐1 as well as RG7769 target engagement. This phenomenon has been termed “steric hindrance” [27, 28, 29, 30] and could prevent the correct assessment of overall available cell surface PD‐1 on T cells (“total receptor”). The potential under‐estimation of PD‐1 expression in such a situation could entail that RO may report “false‐positive” target engagement for the investigational drug since it would not take into account target masked by competing mAbs (“residual competitor RO”). This, in turn, could result in lower absolute numbers of target receptor engagement by the investigational drug due to residual target competitor RO and could be of relevance with regards to drug MoA and potential PK–PD relationships.
Here, for the RG7769 Phase‐I DE trial, we developed a “total drug bound” RO assay protocol using anti‐PGLALA‐PE specifically binding to the P329G mutation of RG7769 [18, 20] for peripheral T cells in lysed patient whole‐blood. Importantly, this fit‐for‐purpose validated assay not only reports RO as a relative value of target engagement, but also allows for a (quasi‐)quantitative assessment [31] of RG7769 antibody binding capacity (ABC) to patient T cells in CPI experienced and naïve patients. To our knowledge, this is the first time that the effect of a prior anti‐PD‐1 therapeutic antibody on subsequently administered competing biotherapeutics has been monitored by flow cytometry in a clinical setting with regards to RO as a surrogate PD biomarker.
2. MATERIALS AND METHODS
2.1. Ethics statement
Human blood samples from healthy volunteers were collected under the Blood Donation for Research Purposes program at F. Hoffmann‐La Roche, Basel, Switzerland. The clinical study, NP40435, was approved by the Institutional Review Boards or independent ethics committees of the participating centers and followed the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice guidelines. All patients provided informed consent to participate in trial NP40435.
2.2. Preparation of white blood cells and peripheral blood mononuclear cells from healthy donor blood
Healthy donor blood was collected in sodium heparin (NaHep) tubes (BD). For whole‐blood experiments, lysis was performed by mixing 100 μl of whole‐blood with 1 ml of PharmLyse Red Blood Lysis buffer (BD) for 20 min at room temperature (RT) and then washed twice (centrifugation at 300g for 5 min at RT) with PBS (gibco). For peripheral blood mononuclear cell (PBMC) preparation, fresh NaHep blood was diluted with PBS (1:1) and transferred to Leucosep tubes (Greiner Bio‐One) pre‐filled with Ficoll‐Paque Plus (GE Healthcare). Tubes were centrifuged without break (1000 g, 10 min, RT), non‐sedimented leukocytes were transferred to a fresh 50 ml tube (Falcon) and washed twice with PBS.
2.3. Assessing RG7769 target expression and PD‐1 levels by flow cytometry
For PD‐1 and TIM3 target expression experiments, 1 × 105 freshly prepared PBMCs were seeded in 100 μl of RPMI 1640 with GlutaMAX‐I supplemented with 10% (vol/vol) fetal bovine serum (FBS; life technologies, subsequently termed “R‐10 medium”) in a round‐bottom 96‐well plate and challenged with 1 μg/ml of Staphylococcus aureus enterotoxin B (SEB, Sigma S4881‐1 mg) for 3 days. After washing in PBS, cells were then stained on ice with CD45‐PerCPCy5.5, HI30, BioLegend, 304028 (1:100); CD3‐APC, OKT3, BioLegend, 317318 (1:100); CD56‐PE, HCD56, BioLegend, 318306 (1:100), anti‐PD‐1‐PECy7, EH12.1, BD, 561272 (1:50) and anti‐TIM3‐BV421, BioLegend, F38‐2E2, 345008 (1:50) and analyzed after washing and resuspension in PBS on a BD FACS‐Symphony cytometer (five lasers: 355, 405, 488, 561, 637 nm). In order to assess PD‐1 steric hindrance, freshly prepared PBMCs (1 × 105/condition) were activated with anti‐CD3:anti‐CD28 magnetic beads (DynaBeads, ThermoFisher), for 48 h in R‐10 medium. They were then washed with PBS and incubated with RG7769 or IgG1 PGLALA isotype control (“DP47”) in 96‐well round‐bottom plates (GreinerBioOne) in a total of 100 μl of PBS containing logarithmically increasing drug concentrations (100 pg/ml➔100 μg/ml). After 30 min at 4°C, cells were washed twice with PBS (150 μl/well), then stained with 50 μl of flow cytometry staining master mix containing CD3 APC‐Cy7 (1:100, UCHT1, BD, 300426), CD19 BV421 (1:100, HIB19, BioLegend, 302220) and anti‐PD‐1 PerCP‐Cy5.5 (1:50, EH12.1, BD, 561273). Alternatively, cells were stained with CD3 BV421 (1:100, UCHT1, BD, 562426) and anti‐PD‐1 APC (1:50, MIH4, BD, 558694) or anti‐PD‐1 APC (1:50, NAT105, BioLegend, 367406) or anti‐PD‐1 PerCP‐EF710 (1:50, J105, eBiosciences, 46‐2799‐42). Each experiment contained a fluorescence minus one (FMO) control matching the PD‐1 channel and analyzed on a BD FACS Canto‐II instrument (three lasers: 405, 488, 640 nm). Compensation was performed using VersaComp antibody capture beads (Beckman, B22804). In order to compare the steric hindrance effect, the median fluorescence intensity (MFI) of each PD‐1 flow cytometry detection antibody was normalized to its maximum intensity after subtracting the anti‐PD‐1 channel FMO signal.
2.4. Assessing anti‐PGLALA‐PE IgG Fc specificity
Freshly prepared PBMCs (1 × 105/condition) were incubated with 10 μg of rituximab (Roche), pembrolizumab IgG4 (InvivoGen, 375939001), pembrolizumab‐PGLALA (for comparison reasons, Roche), or isotype controls IgG1 LEAF (BioLegend, 400166), IgG4 LEAF (BioLegend, 403702) or DP47 (Roche) for 30 min on ice, then washed twice with PBS. Anti‐PGLALA binding was assessed by flow cytometry after incubation with a PBS based staining master mix (50 μl/condition) containing CD45‐PerCPCy5.5 (1:100, HI30, BioLegend, 304028), CD3‐APC‐Cy7 (1:100, UCHT1, BD, 300426), CD4‐AF647 (1:100, RPA‐T4, BioLegend, 300520), CD8‐AF488 (1:100, BD, RPA‐T8, 557696), CD19‐BV421 (1:100, HIB19, BioLegend, 302220 and anti‐PGLALA‐PE (Roche, 1:160), for 30 min on ice. Cells were then acquired on a BD FACS‐Symphony cytometer (five lasers: 355, 405, 488, 561, 637 nm). Compensation was performed using VersaComp antibody capture beads (Beckman, B22804). PGLALA‐PE expression was assessed on CD3+ T cells and CD19+ B cells. Refer to Supporting Information for details on anti‐PGLALA‐PE generation.
2.5. Development of RG7769 RO assay in vitro with fresh PBMCs
For RO assay development in vitro, 1 × 105 freshly prepared PBMCs were seeded in 90 μl of R‐10 medium in a round‐bottom 96‐well plate and mixed with 10 μl of 10‐fold concentrated (log10) concentrations of RG7769 or DP47 (in PBS) resulting in final concentrations of 10 pg/ml➔10 μg/ml. Cells were incubated at 37°C for 20 min, washed twice with PBS after centrifugation (300 g, 5 min, RT) and incubated with 50 μl of PBS based flow cytometry staining master mix. Initial experiments contained the following detection antibodies: CD3‐AF488, UCHT1, BioLegend, 300415 (1:100); CD56‐PE, HCD56, BioLegend, 318306 (1:100), and anti‐PGLALA (M1.7.24) custom‐labeled with Alexa Fluor™ 647 antibody labeling kit (Invitrogen, A20186) (10 μg/ml). Occasionally, in vitro RO was determined by staining with a master mix containing CD45‐PerCPCy5.5 (1:100, HI30, BioLegend, 304028), CD4‐V500 (1:100, RPA‐T4, BD Horizon, 560768), CD56‐APC (1:100, HCD56, BioLegend, 318310), CD16‐APC (1:100, 3G8, BD PharMingen, 561248), CD8‐BV421 (1:100, RPA‐T8, BioLegend, 301036), CD3‐AF488 (1:100, SP34‐2, BD PharMingen, 557705) and anti‐PGLALA‐PE (1:160). Cells were analyzed on a BD FACS‐Symphony cytometer (five lasers: 355, 405, 488, 561, 637 nm). Compensation was performed using VersaComp antibody capture beads (Beckman, B22804). RG7769 staining was assessed as the fraction of PE+ CD3+, or CD3+CD4+ or CD3+CD8+ T cells within the CD45+ cell compartment. RO was calculated using three tubes (tube 1—no PGLALA‐PE; tube 2—saturation; tube 3—concentration of interest) where the percentage of anti‐PGLALA+ cells at a given concentration was normalized: .
2.6. Assessing RG7769 competition for PD‐1 in the presence of anti‐PD‐1
Fresh PBMCs (1x105/condition) were incubated with nivolumab (InvivoGen, 375938995), pembrolizumab (InvivoGen, 375939001) or IgG4 LEAF isotype (BioLegend, 403702) in 96‐well round‐bottom plates (GreinerBioOne) in a total of 100 μl of PBS containing 10 pg/ml to 100 μg/ml. After 30 min at 4°C, cells were washed three times with PBS (150 μl/well), then incubated with RG7769 (10 μg/well in PBS). After 30 min at 4°C, cells were again washed three times with PBS (150 μl/well), then stained with 50 μl of master mix containing CD45‐PerCPCy5.5 (1:100, HI30, BioLegend, 304028), CD4‐V500 (1:100, RPA‐T4, BD Horizon, 560768), CD56‐APC (1:100, HCD56, BioLegend, 318310), CD16‐APC (1:100, 3G8, BD PharMingen, 561248), CD8‐BV421 (1:100, RPA‐T8, BioLegend, 301036) and CD3‐AF488 (1:100, SP34‐2, BD PharMingen, 557705) and anti‐PGLALA‐PE (M.1.7.24, 1:160). Cells were analyzed on a BD FACS‐Symphony cytometer (five lasers: 355, 405, 488, 561, 637 nm). Compensation was performed using VersaComp antibody capture beads (Beckman, B22804).
2.7. Assessing RG7769 RO in NP40435 patient peripheral blood
We developed a modified whole‐blood based protocol for RG7769 RO assessment to apply in a multicenter clinical trial. Briefly, three assay tubes were prepared using 100 μl of whole blood (NaHep) each. Tube 2, the positive control, was incubated with RG7769 (100 μg/ml, 37°C, 30 min). All blood was lysed with BD FACS Lyse according to the manufacturer's recommendations. After washing with PBS, tube 1 (negative control) was stained with CD3 FITC (SK7, BioLegend), mIgG2b PE (MPC‐11), BD Biosciences, CD8‐APC (SK1, BioLegend), CD45‐APC‐H7 (2D1, BD Bioscience), CD56‐BV421 (HCD56, BioLegend), CD4‐BV510 (SK3, BioLegend), CD16‐BV605 (3G8), BD Bioscience). Tubes 2 and 3 were stained with CD3‐FITC, anti‐PGLALA‐PE (Roche, 1:160), CD8‐APC, CD45‐APC‐H7, CD56‐BV421, CD4‐BV510, CD16‐BV605. All antibodies were diluted according to the supplier's recommendations. After washing, stained cells were analyzed on a BD FACS Canto cytometer (10 color, 3 laser). The fit‐for‐purpose assay validation was carried out at the service provider. RO was calculated according to the following formula: as illustrated in Figure S1. During the fit‐for‐purpose validation procedure, RO calculation was also compared using the percent of anti‐PGLALA‐PE+ cells. RO was assessed on total (CD3+) T cells and CD4+ and CD8+ T cell subsets as well as total NK cells (CD56+CD16+ and CD56+CD16− cells). The inter‐assay precision of the RO assay was determined in blood from two donors on day 0 post‐pulsing: Donor 1 at 0, 5 and 20 ng/ml and donor 2 at 0, 20 and 100 ng/ml. A single replicate of each sample was analyzed over three independent analytical runs (18 samples total). Sample stability was determined in a similar fashion to the precision assessment in NaHep blood from two donors treated with at 0, 5, 20 or 100 ng/ml. Each sample was analyzed on the day of blood draw (0 days) and 1, 2, 3 and 4 days from blood draw. A 20% CV was considered acceptable. RG7769 ABC was calculated for CD3+, CD4+ and CD8+ T cells using BD QuantiBRITE® beads for tubes 1, 2 and 3. ABC background binding (tube 1) was subtracted from maximum ex vivo binding (tube 2) or RG7769 in vivo binding (tube 3). For patient sample analysis in this research article, FCS raw data files were analyzed using FlowJo v10.5 (BD). For flow cytometry data clustering analyses, the in‐built UMAP (nearest neighbors: 15; minimum distance: 0.5; 5000 cells/condition) and FlowSOM clustering R plugins were used.
3. RESULTS
3.1. Development of an RG7769 specific total‐drug bound RO assay
We developed an RO assay to monitor RG7769 target engagement prior to dosing and on‐treatment in the peripheral blood of cancer patients during DE in the clinical trial NP40435. Since RG7769 was designed to primarily engage T cells by binding to cell surface PD‐1 [22, 24, 32], we evaluated the possibility of assessing total PD‐1 on CD3+ T cells in the presence of RG7769. In freshly isolated PBMCs incubated with RG7769 (100 pg/ml➔100 μg/ml), we found that three typical commercial flow cytometry PD‐1 detection antibody clones (EH12.1, NAT105 and J105) showed a strong signal reduction with increasing RG7769 concentrations (Figure 1A,B). The overall reduction depended on the clone and ranged from 36% of the maximum signal (J105) to 48% (NAT105) (Figure 1B). We also assessed clone MIH4 but confirmed that its PD‐1 detection performance was generally inferior [33] (data not shown). We thus concluded that the development of an RO assay reporting total PD‐1 would not be possible without underestimating the amount of PD‐1 expressed on RG7769+ T cells. Consequently, we developed a “total‐drug‐bound” anti‐drug RO approach similarly to previously used protocols where available cell‐surface target is indirectly assessed by staining for mAbs bound to it [34, 35].
FIGURE 1.
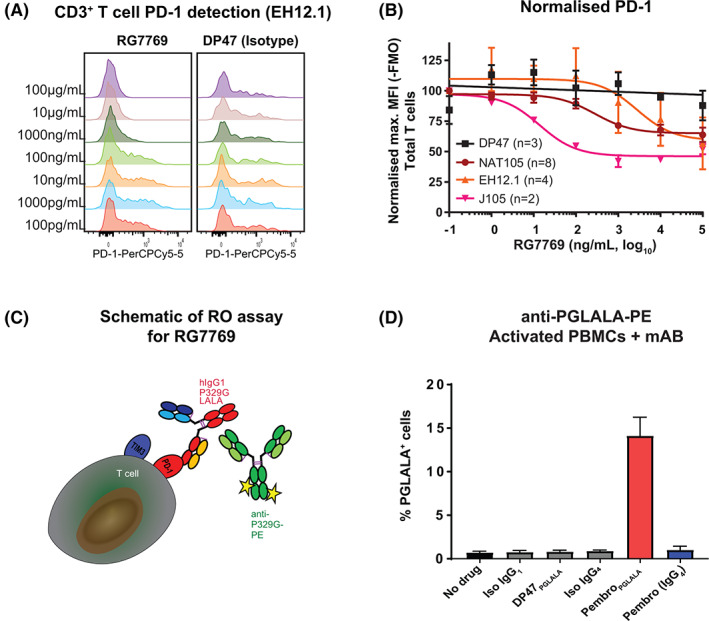
Development of total‐drug‐bound receptor occupancy (RO) assay to detect RG7769 on peripheral blood derived immune cells. (A) CD3‐CD28 activated peripheral blood mononuclear cells (PBMCs) were incubated with increasing concentrations of RG7769 (100 pg/ml➔100 μg/ml). PD‐1 expression on CD3+ T cells was analyzed using anti‐PD‐1, clone EH12.1, by flow cytometry in a representative donor. (B) Pooled normalized relative CD3+ T cell PD‐1 expression from two independent experiments assessing anti‐PD‐1 clones EH12.1, NAT105 and J105. (C) Schematic depicting RG7769 engaging T cells via high‐affinity anti‐PD‐1 and low‐affinity anti‐TIM3 binding on target cells. RG7769 Fc harbors the P329 IgG1 mutation (“PGLALA”) which can be specifically detected via anti‐PGLALA‐PE (M‐1.7.24). (D) PBMCs from n = 3 different donors incubated with pembrolizumab (IgG4 or PGLALA), rituximab (IgG1) or respective isotype controls (Iso IgG1, Iso IgG4, DP47‐PGLALA), and detected via anti‐PGLALA‐PE. Pembrolizumab staining is shown for CD3+ T cells [Color figure can be viewed at wileyonlinelibrary.com]
RG7769 contains the P329G‐LALA mutation [19] (Figure 1C). Consequently, we developed a phycoerythrin (PE) coupled anti‐P329GLALA detection antibody (clone M‐1.7.24; “anti‐PGLALA‐PE”). This clone was previously shown to be highly specific for the IgG1‐P329GLALA antibody format in a soluble assay system [18], but had not been tested in a cell‐based assay by flow cytometry. Using anti‐PGLALA, we showed that RG7769 bound to activated T cells expressing PD‐1 and TIM3, but not activated natural killer (NK) cells which exclusively expressed TIM3 (Figure S1A,B). This confirmed that RG7769 primarily engages T cells via PD‐1 [24]. After determining the optimal staining dilution for a PGLALA‐PE (10 μg/ml; Figure S1C), we incubated activated PBMCs with native pembrolizumab (IgG4); a modified pembrolizumab (IgG1‐PGLALA); and rituximab (wildtype IgG1); or the respective isotype controls (Figure 1D). We stained all conditions with anti‐PGLALA‐PE and analyzed the PE staining pattern on T cells (for pembrolizumab) or B cells (CD19+, for rituximab, data not shown). As expected, anti‐PGLALA‐PE only detected pembrolizumab‐PGLALA on T cells, but not native pembrolizumab or isotype control (DP47) (Figure 1D). It also failed to detect rituximab (wildtype IgG1) on B cells confirming high specificity (not shown). Taken together, these results suggest that anti‐PGLALA‐PE will specifically detect P329GLALA on a cell but no other idiotypes.
Next, in order to determine saturating conditions for RG7769, we incubated activated PBMCs from healthy donors with increasing concentrations (1 pg/ml➔100 μg/ml) of RG7769. This resulted in a greater degree of RG7769+ CD8+ T cells than CD4+ T cells (Figure S1D). As complete binding by RG7769 was achieved between 1 and 10 μg/ml, 100 μg/ml was chosen for saturation in the total‐drug‐bound RO assay for RG7769. For the final fit‐for‐purpose validation of the RO assay in lysed whole‐blood, we developed a three‐tube system, where tube 1, the negative control (=0% RO), would be stained with a mIgG2B‐PE isotype control. Tube 2, the positive control (=100% RO), would consist of whole‐blood saturated ex vivo with RG7769, and tube 3 consisted of the blood containing in vivo bound RG7769 only. Post‐lysis, tubes 2 and 3 would be stained with anti‐PGLALA‐PE to detect RG7769 as illustrated in Figure S1E.
3.2. Superiority of MFI‐based RO calculation method for RG7769
Before use in NP40435, we validated the RO assay at the bioanalytical laboratory. The flow cytometry gating strategy (Figure 2A) focused on total CD3+ T, CD4+ and CD8+ T cells, and NK cells. In healthy volunteer steady‐state blood, dose‐dependent binding of RG7769 to immune cells was assessed (Figures 2A,B and S2A). Importantly, only T cells showed a clear increase in anti‐PGLALA‐PE+, with CD8+ T cells showing a stronger anti‐PGLALA‐PE signal than CD4+ T cells (Figure 2A), while there was no specific RG7769 binding to NK cells and only inconsistent binding to non‐T non‐NK cells (Figure 2B). The preferential binding to CD8+ T cells confirms the purported binding pattern of RG7769, which was designed to preferentially engage cytotoxic T cells in cancer patients [24].
FIGURE 2.
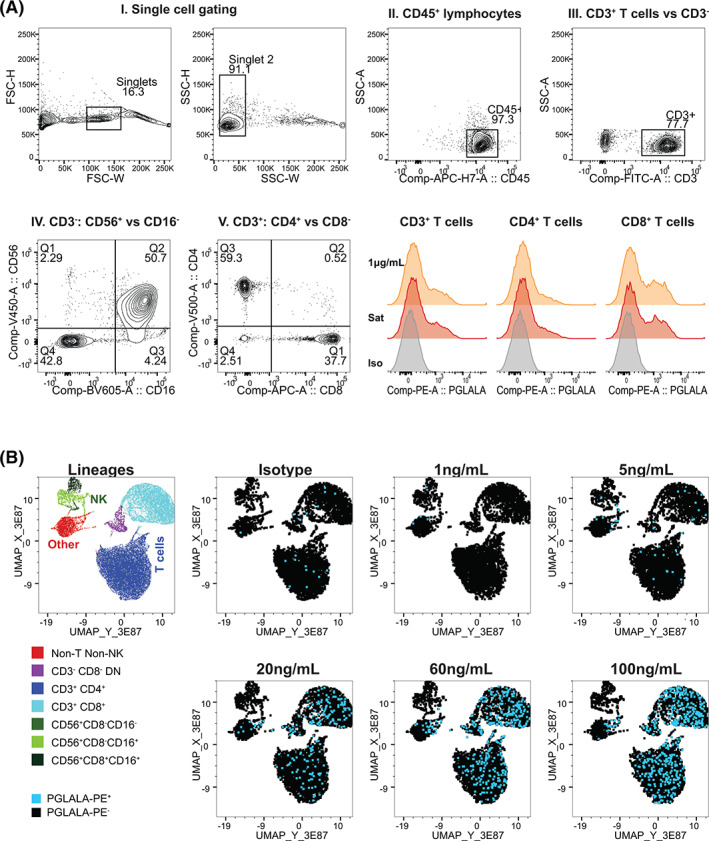
Validation of RG7769 receptor occupancy (RO) assay for whole‐blood T cell RO analysis. (A) Gating strategy for RO calculation. I. In all conditions, single lymphocytes were identified by excluding cell doublets via FSC‐H versus FSC‐W followed by SSC‐H versus SSC‐W. II. Total lymphocytes were gated on via CD45 staining versus SSC‐A. III. Within the CD45+ compartment, T cells were identified via CD3 expression. A non‐T cell gate (NOT‐CD3+) was set. IV. Within total CD3+ T cells, CD4+ (Q3) and CD8+ single‐positive (Q1) T cells were identified. V. In the non‐T cell population, NK cells were identified via CD56 versus CD16 (QI‐III). Total NK cells were defined as CD56+CD16+/−. Lower right: Comparing the anti‐PGLALA‐PE staining intensity of CD3+ T cells, CD4+ and CD8+ T cells pulsed with RG7769. Orange: 1 μg/ml, red: Saturation (100 μg/ml); gray: Negative control (1000 ng/ml + mIgG2b‐isotype‐PE). (B) Dose–response of RG7769 binding to healthy donor lymphocytes. Using the gating strategy in A, CD45+ lymphocyte single cells from blood pulsed ex vivo with RG7769 (0 ng/ml➔100 ng/ml), were identified. The control group (0 ng/ml) was stained with mIgG2b‐PE isotype, cells with RG7769 with anti‐PGLALA‐PE. Five thousand concatenated CD45+ events were clustered according to CD3, CD4, CD8, CD56 and CD16 by UMAP. Left: Cell lineages identified using FlowSOM include CD4+ T cells, CD8+ T cells, CD4−CD8− unconventional T cells as well as NK cell subsets. Right: PE+ (RG7769+) T cells (blue) [Color figure can be viewed at wileyonlinelibrary.com]
Next, the cell subsets of interest and subset RO were validated with regards to sample precision and stability over time. For this purpose, a total of five different runs were carried out in two donors at three different concentrations of RG7769. The assessment of the major cell populations (total CD3+ and CD4+, CD8+ T cells a well as NK cells [CD56+CD16+ and CD56+CD16− combined, Figure 2A.V]) met the acceptance criteria (<20% CV) with overall‐precision ranging from 3.5% CV (CD8+ T cells) to 7.8% CV (total NK cells) in freshly lysed blood. RO calculation precision was assessed based on the MFI of anti‐PGLALA‐PE, which we found to produce more robust results than RO calculated based on the percentage of anti‐PGLALA‐PE+ T cells (Figure S2B and Table S1). Importantly, RG7769 RO met the acceptance criteria for CD3+ cells (9.6% CV) and CD4+ T cells (15% CV). CD8+ T cell RO also passed the acceptance criteria (9.8% CV), while NK cell RO did not (68.2% CV). Details of the precision assessment are summarized in Table S1.
Subsequently, RO stability over time (0–4 days post‐phlebotomy) was assessed in samples from two healthy donors at 3 different RG7769 concentrations: Donor 1: 0, 5 and 20 ng/ml; donor 2: 0, 20 and 100 ng/ml (a total of n = 6 samples over 5 days; Tables S2–S4 and Figure S2B). Mean ROs were determined both for the %‐positive and the MFI‐based calculation method. In all T cell subsets, RO changed least from d1 post‐phlebotomy onwards and was found to be most stable in using the MFI‐based method. For CD3+ T cells, for instance, the relative change of the mean RO ranged from 0.1% (d2 to d1) over 6.7% (d3 to d1) to 11.4% (d4 to d1), while in the %‐positive calculation, changes were greater ranging from 10.9% (d2–d1) over 37.2% (d3–d1) to 3.7% (d4–d1). Using the MFI based approach, T cell RO was consistently, acceptably accurate from d1‐d4 post‐phlebotomy in all subsets. However, when using the %‐positive based method, RO results were only stable up to d2 post‐phlebotomy with relative RO changes from d3 to d1 ranging from 28.5% (CD8+) to 45.9% (CD4+). Details on T cell subset RO stability are summarized in Tables S2–S4. The assay was successfully transferred to the bioanalytical lab for routine sample analysis (Figure S5). In addition to RO, T cell RG7669 ABC was determined in NP40435 patients using the PE‐QuantiBRITE® method [36].
In summary, the MFI‐based RO calculation method proved to be more precise and robust, resulting in more consistent RO reporting in T cells. This highlights the importance of assessing both readouts and calculation methods in a fit‐for‐purpose RO assay validation especially with regards to multicenter clinical trial implementation [3].
3.3. RG7769 ABC analysis is critical to reveal long‐lasting competitor steric hindrance effects on T cell PD‐1
RO, as assessed in a typical total‐drug bound assay format, generally provides information on the relative degree of available cell surface target occupancy at a given time. If several mAbs bind to the same target, drug binding sites may be masked and RO assessment may be misleading in such situations, resulting in high RO but lower overall ABC. Since RG7769 targets PD‐1, the target of several other CPIs, we reasoned that this might impact RG7769 RO in affected patients. Indeed, in NP40435, 26% of the patients were previously treated with anti‐PD‐1 therapeutic antibodies at any time prior to RG7769 administration (Figure 3A). Therefore, they were considered CPI‐experienced. Most anti‐PD‐1 CPI experienced patients (60%) had received the last dose of CPI >14 weeks prior to the first dose of RG7769. Since long wash‐out periods of several weeks have been observed for some anti‐PD‐1 CPIs [10], we reasoned that in anti‐PD‐1 experienced patients, RG7769 target engagement via PD‐1 binding might be affected due to pre‐engagement of PD‐1.
FIGURE 3.
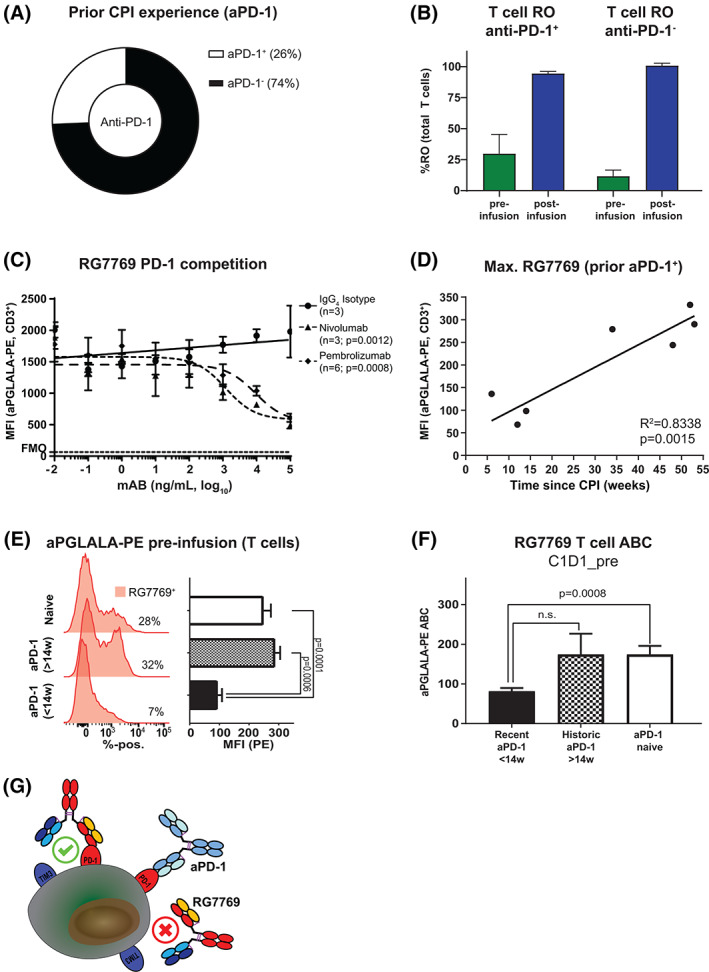
Assessing RG7769 binding to T cells in anti‐PD‐1 experienced patients. (A) Comparison of the frequency of NP40435 patients with prior aPD‐1 experience with aPD‐1 naïve patients. (B) Comparison of RG7769 CD3+ T cell receptor occupancy (RO) pre‐ and post‐infusion in NP40435 patients. (C) Control in vitro experiment assessing the effect of PD‐1 competition on RG7769 binding to CD3+ T cells. Activated peripheral blood mononuclear cells (PBMCs) were incubated with IgG4 isotype, nivolumab or pembrolizumab (100 pg/ml➔100 μg/ml), then pulsed with RG7769 (10 μg/ml), and analyzed for T cell anti‐PGLALA‐PE binding. The pooled MFI for PE is shown (data from two independent experiments). (D) Linear regression of max. RG7769 (MFI) to CD3+ T cells in NP40435 anti‐PD‐1+ patients prior to RG7769 infusion (C1D1_pre) as a function of the last reported prior anti‐PD‐1 administration. R 2 = .8338, p = 0.0015. (E) anti‐PGLALA‐PE staining on CD3+ T cells of representative NP40435 patients. Maximum anti‐PGLALA‐PE assessed post ex vivo blood saturation with RG7769, in recently (up to 14 weeks) or historically experienced (>14 weeks) patients versus naïve patients prior to RG7769 (C1D1_pre). Anti‐PGLALA–PE intensities are shown for ex‐vivo saturation and neg. control. (F) CD3+ T cell RG7769 mean ABC in recently (<14 weeks) or historically (>14 weeks) aPD‐1+ patients versus naïve. Data are pooled from (E). ABCs were compared to aPD‐1 naïve (Welch ANOVA test). (G) Schematic of RG7769 binding to T cells in aPD‐1+ patients. Left: RG7769 engages T cells via high‐affinity anti‐PD‐1 engagement if PD‐1 is freely available. Right: In situations where prior aPD‐1 is engaging PD‐1, RG7769 has to displace prior bound aPD‐1 antibodies. Steric hindrance may occur if competing mAbs target an epitope in the PD‐L1 interaction side of PD‐1 [Color figure can be viewed at wileyonlinelibrary.com]
Initially, when we compared CD3+ T cell RO in anti‐PD‐1 experienced versus anti‐PD‐1 naïve patients, we found no apparent difference between these groups. Both CPI experienced as well as naïve patients had ~100% CD3+ T cell RO after the first infusion at C1D1 (Figure 3B). This was independent of RG7769 dosing (not shown). However, this finding did not exclude the possibility of potentially reduced cell‐surface PD‐1 available for RG7769 binding, caused by previously administered anti‐PD‐1. Consequently, in order to better understand if and how prior anti‐PD‐1 experience affected the amount of drug binding sites, we analyzed if RG7769 competed with anti‐PD‐1 CPIs in a control experiment in vitro (Figure 3C). We incubated activated PBMCs with nivolumab, pembrolizumab or IgG4 isotype (10 pg/ml➔10 μg/ml) followed by subsequent RG7769. Compared to the isotype control, nivolumab resulted in a 75% signal reduction (p = 0.001), while pembrolizumab resulted in a 69% reduction (p = 0.0008) at the highest concentration. This suggested that in such a situation, RO alone would not capture the fact that a large percentage of total cell‐surface PD‐1 would not be available for immediate RG7769 binding.
With regards to patient RO, we reasoned that the greatest PD‐1 steric hindrance would be detectable prior to any RG7769 administration. We also expected competitor PD‐1 residual occupancy to fade over time. When re‐analyzing the anti‐PGLALA‐PE signal on T cells (Figures 3D and S3A), we found indeed that anti‐PGLALA‐PE‐MFI negatively correlated with the time elapsed since the last reported administration of competitor anti‐PD‐1. Concretely, for CD3+ T cells, our analysis suggested that recently experienced patients (<14 weeks since anti‐PD‐1) bound less RG7769 than historically (>14 weeks) experienced patients. Further analysis of RG7769 binding to CD3+ T cells suggested that historically anti‐PD‐1 experienced patients had a highly comparable anti‐PGLALA‐PE MFI signal compared to CPI naïve patients, while recently experienced patients showed a significant 2.7‐fold signal reduction (p < 0.0001, Figure 3D,E). This effect was stronger in CD4+ T cells than in CD8+ T cells (Figure S3B).
We decided to quasi‐quantitatively assess the number of RG7769 binding sites per T cell (ABC) using the PE quantiBRITE® method in CPI naïve and historically or recently CPI experienced patients (Figures 3F and S3C). This comparison suggested that recent anti‐PD‐1 experience resulted in a twofold reduction in the number of binding sites per CD3+ T cell available to RG7769 pre‐infusion (mean ABC = 81.25 in recently CPI+ patients versus174 in historically CPI+ or CPI naïve patients, p = 0.0008) (Figure 3F). Similar results were found for CD4+ and CD8+ T cells (Figure S3C). In summary, the detailed analysis of RO in combination with (semi)quantitative ABC assessment suggests that, while RG7769 effectively binds to virtually all available binding sites already at C1D1, the actual number of RG7769 binding sites initially available may be roughly twofold reduced in recently, but not historically, anti‐PD‐1 experienced patients. This finding may likely be explained by the restricted PD‐L1 interacting epitope on PD‐1 targeted by anti‐PD‐1 mAbs (Figure 3G) [12]. This highlights the potential functional implications of long‐lasting PD effects of anti‐PD‐1 therapies, and the importance of rational PD‐1 biomarker panel design in affected patients.
4. DISCUSSION
RO by flow cytometry is a PD biomarker approach used in large molecule development to confirm target binding and inform on dose selection [1, 7, 37]. RO is a measure of the degree of target occupied at a given drug dose and can be assessed on immune cells using flow cytometry [1]. Different RO formats exist: The “free‐vs‐total” assay format requires non‐competing detection antibodies for the mAb cell surface target, while the “total‐drug‐bound” format requires highly specific anti‐drug detection antibodies [3, 37]. A conceptual drawback of the latter format is that the anti‐drug detection method only provides information on the relative degree of cell surface target available for binding at a given time. In situations where different mAbs bind to the same target [12, 27], RO values may report “false positive” target engagement, since they would report complete RO in situations where a competitor drug still maintains residual target occupancy.
CIT aiming to re‐invigorate “exhausted” T cells includes CPIs of the IgG4 isotype targeting PD‐1 on the surface of antigen‐specific T cells [11, 12]. Here, during the clinical development, RO by flow cytometry was used as a PD biomarker using an anti‐IgG4 total‐drug‐bound approach [14, 35]. Importantly, conventional therapeutic antibodies [38, 39] can interact with Fc gamma receptors on non‐effector cells present for instance in the tumor microenvironment. This in turn can lead to internalization and removal of the therapeutic antibody, impacting the activation of antigen‐presenting cells [40] as well as the local anti‐tumor responses [39], which has led to the development of Fc gamma receptor inert therapeutic mAbs, including the fully Fc effector silenced IgG1 format, IgG1‐P329GLALA [18, 19, 20, 25]. In order to specifically detect mAbs with this backbone on target cells, in the present study, we adapted an anti‐P329G antibody [20] previously used in soluble assay formats [18] for flow cytometric analysis by generating anti‐PGLALA‐PE. This is an advantage since idiotype‐specific detection mAbs do not allow to distinguish between different drugs of the same idiotype. Further restrictions of this approach include anti‐drug antibodies (ADAs) which may bind to idiotypic therapeutic mAbs; this phenomenon can interfere with the readout [5]. We show that anti‐PGLALA‐PE specifically detects IgG1‐PGLALA bound to immune cells. Consequently, we propose that this would also be of advantage in situations where ADAs against therapeutic mAbs are formed [5, 41, 42, 43]. Currently, a number of IgG1‐PGLALA based bispecific antibodies, immunocytokines and antibody fusion proteins are in pre‐clinical or clinical development [24, 44, 45, 46, 47, 48, 49] requiring custom tailored PD biomarker assays. We believe that anti‐PGLALA‐PE represents a powerful detection antibody for the development of immunophenotyping and other flow cytometry based PD assay protocols as it allows the specific detection of the drug that otherwise would not be possible in a matrix with human immunoglobulins.
One CIT candidate with the IgG1‐PGLALA configuration currently undergoing clinical development is RG7769, a bispecific mAb targeting PD‐1 and TIM3 [22]. RG7769 is designed to primarily engage T cells via sell‐surface PD‐1 but would only engage TIM3 via an avidity‐driven mechanism [24]. We confirm that this antibody binds to PD‐1+ T cells with no binding to steady‐state NK or other cells. In vitro, RG7769 is unable to engage activated NK cells exclusively expressing TIM3.
In CIT, therapeutic mAbs are often administered at saturating doses [10]. Despite this, precise knowledge of RO remains an important PD parameter [37]. It helps assess the degree and duration of mAb‐target interaction in patients that are no longer dosed with a particular mAb, or treated in short succession with different mAbs to the same target or cell populations. Here, in order to develop an RO assay for bioanalytical use in early clinical trials of RG7769, we assessed the feasibility of a free‐vs‐total format requiring PD‐1 detection antibodies not competing with therapeutic anti‐PD‐1 mAbs. We screened detection antibodies for PD‐1 assessment in the presence of RG7769 and pembrolizumab and nivolumab. We confirm previous reports [33] claiming that there are no commercial antibodies allowing for the simultaneous assessment of therapeutic anti‐PD‐1 and total cell surface PD‐1. Consequently, we developed a “total‐drug bound” RO assay relying on mAb detection via anti‐PGLALA‐PE, to support dose finding in the clinical development of RG7769. In its fit‐for‐purpose validated format, the RO assay provides more reliable results using the anti‐PGLALA‐PE MFI calculation method as opposed to using the percentage of anti‐PGLALA‐PE+ cells. This highlights the importance to assess both calculation methods in RO assay fit‐for‐purpose validations and selecting the most appropriate readout [3]. One conceptual drawback of this method is that RO here only assesses available cell surface target. In NP40435, we expected that some patients might be pre‐exposed to competitor anti‐PD‐1 mAbs potentially resulting in residual PD‐1 occupancy and confounding true RG7769 target engagement. Hence, we aimed to simultaneously measure target engagement in relative terms binding and RG7769 binding capacity using the (quasi)quantitative ABC method [31, 36].
In our current study, we found that 26% of patients presented with previous anti‐PD‐1 therapy. With regards to overall RO using our specific anti‐drug detection approach, we observed no difference in anti‐PD‐1 experienced versus naïve patients, with RG7769 RO reaching ~100% in both groups. Importantly, though, since nivolumab and pembrolizumab were shown to bind to an epitope in the PD‐L1 interaction side of PD‐1 [12], and given the relatively confined extracellular domain responsible for PD‐1 interaction with PD‐L1, we assessed these mAbs with regards to RG7769 binding competition. We found that residual competitor RO reduced the binding of RG7769 to activated T cells in vitro. In line with this, we observed a reduction in the maximum anti‐PGLALA‐PE signal on peripheral blood T cells of NP40435 patients dosed with RG7769. This strongly suggests that therapeutic PD‐1 inhibitors may lead to very long‐lasting PD effects on the assumed target cells. This effect diminished over time but was still prevalent up to 14 weeks post‐competitor anti‐PD‐1 dosing. Indeed, for PD‐1 targeting CPIs, stable PD effects have previously been suggested. High nivolumab RO prevailed over at least 8 weeks post‐dosing [10, 50]. Our results suggest that this residual RO may entail steric hindrance for other PD‐1 targeting mAbs for at least up to 14 weeks. If unaccounted for, residual competitor RO could, for instance, result in reduced numbers of novel PD‐1 specific mAbs (including bispecifics) to T cells. Such a scenario might entail suboptimal target engagement and T cell activation especially in the case of low‐affinity anti‐PD‐1 binders. In recently (<14 weeks) CPI experienced patients, residual competitor PD‐1 RO resulted roughly in a twofold reduction of RG7769 ABC.
We assume that RG7769 will displace pre‐existing competitor anti‐PD‐1 even in situations of residual competitor PD‐1 RO due to its high‐affinity anti‐PD‐1 binding [24]. Consequently, we do not expect this to be a limiting factor in NP40435. Detailed analyses of the RG7769 PK–PD relationship and RO over time are not shown in this manuscript and are currently being performed.
In summary, our results have strong implications for flow cytometry immunophenotyping, RO assay development and data interpretation in samples with previous exposure to anti‐PD‐1 antibodies [51]. They suggest that steric hindrance by prior anti‐PD‐1 treatment may lead to the false‐negative estimation of T cell PD‐1 using routine flow cytometry reagents. With regards to CPI PD biomarker assay development, this could systematically entail incorrect assessment of target engagement since therapeutic mAbs can then only bind to remaining, non‐pre‐engaged PD‐1 [29]. In the case of RG7769, this effect is masked by the relative nature of the RO value, which does not contain information on the number of drug molecules bound to T cells with “complete” target engagement. Consequently, we propose to routinely quantitatively assess mAb binding to target cells in addition to RO in patients with previous administration of therapeutic anti‐PD‐1 in order to provide more mechanistic granularity to RO. More generally, our findings could also be of relevance for the development of other large molecules such as agonistic mAbs where dosing is critical for patient safety and ROs below 100% need to be achieved during DE [6, 7].
AUTHOR CONTRIBUTIONS
Fabian Junker: Conceptualization; data curation; formal analysis; investigation; methodology; software; supervision; validation; visualization; writing‐original draft; writing‐review & editing. Pratiksha Gulati: Conceptualization; formal analysis; methodology; software; visualization; writing‐review & editing. Uwe Wessels: Conceptualization; methodology; resources; writing‐original draft. Stefan Seeber: Conceptualization; methodology. Kay‐Gunnar Stubenrauch: Conceptualization; methodology. Laura Codarri Deak: Conceptualization. Christoph Markert: Investigation; project administration; writing‐review & editing. Christian Klein: Investigation; methodology; writing‐review & editing. Priscila Camillo Teixeira: Conceptualization; project administration; resources; supervision; writing‐original draft; writing‐review & editing. Henry Kao: Conceptualization; project administration; resources; supervision; writing‐review & editing.
CONFLICT OF INTEREST
All authors are current or former employees of F. Hoffmann‐La Roche Ltd and may own company stock. CK in addition discloses ownership of F. Hoffmann‐La Roche Ltd patents and stock.
Supporting information
Appendix S1. Checklist: MIFlowCyt‐Compliant Items
Appendix S2. Supporting Information
FIGURE S1 A. Analysis of PD‐1 and TIM3 cell surface expression in PBMCs from n = 3 healthy donors. PBMCs were analyzed after preparation (d0) and after 3d in culture in the presence of SEB (1000 ng/mL). CD3+ T cells and CD56+ NK cells were assessed; shown is the percentage of target‐positive cells minus unstained (FM) control. B. Activated PBMCs of n = 3 donors were incubated with RG7769 or DP47 isotype (10 pg/mL➔10 μg/mL) and subsequently stained with anti‐PGLALA‐A647. Shown is the MFI for CD3+ CD56+ cells. C. Titration of anti‐PGLALA‐PE (M‐1.7.24) used in the fit‐for‐purpose validated RO assay for RG7769. Freshly isolated CD3‐CD28 activated PBMCs were incubated with 10 μg/mL of RG7769 followed by incubation with anti‐PGLALA‐PE (100 ng/mL➔100 μg/mL). Shown is the total single‐cell lymphoid PBMC cell fraction. D. Comparison of RG7769 binding to PBMCs (via anti‐PGLALA‐PE) on CD3+, CD4+ and CD8+ T cells of a representative healthy donor. E. Schematic illustrating sample preparation for RO analysis. Peripheral blood was collected into three NaHep coated tubes and pulsed with 1000 ng/mL of RG7769. Tube 1 serves as negative control. Tube 2 serves as a positive control ‐ following initial incubation, tube 2 was re‐challenged with excess RG7769 (100 μg/mL). All three samples underwent red blood cell lysis followed by incubation with flow cytometry antibodies. Tube 1 was stained with a lineage mix including mIgG2b‐PE isotype, while tubes 2 and 3 were incubated with the same lineage mix including anti‐PGLALA‐PE.
FIGURE S2 A. Dose‐dependent RO increase in whole‐blood incubated with 0 ng/mL, 5 ng/mL, 20 ng/mL, 60 ng/mL or 100 ng/mL of RG7769 and analyzed 24 hrs post‐incubation. RG7769 RO was calculated for CD3+, CD3+CD4+ and CD3+CD8+ T cells based on MFI (PE). B. RO stability assessment. Blood of 2 healthy donors was incubated with 0 ng/mL or 20 ng/mL of RG7769 and analyzed immediately (0d) or 1–4 days post challenge. RO was calculated using anti‐PE (MFI) and assessed over time (top) or the percentage of anti‐PGLALA‐PE positive cells (bottom). Only time points from d1 (but not d0) were considered relevant (dashed line).
FIGURE S3 Linear regression of max. RG7769 binding (MFI) to T cells in NP40435 aPD‐1+ patients prior to RG7769 infusion (C1D1_pre) as a function of the last reported prior aPD‐1 administration on CD4+ (left) or CD8+ T cells (right). B. anti‐PGLALA‐PE staining on T cells of representative NP40435 patients. Maximum anti‐PGLALA‐PE was assessed post ex vivo blood saturation with RG7769, in recently (up to 14 weeks) or historically aPD‐1+ experienced (>14 weeks) vs naïve patients prior to RG7769 infusion (C1D1_pre). Anti‐PGLALA–PE staining intensities are shown for the ex‐vivo saturation RG7769 negative control on CD4+ T cells (left) or CD8+ T cells (right). C. RG7769 ABC to CD3+ T cells in NP40435 patients in recently anti‐PD‐1‐experienced (up to 14 weeks) or historically experienced (>14 weeks) patients vs naïve patients. ABCs were compared to aPD‐1 naïve (Welch ANOVA test).
ACKNOWLEDGMENTS
We would like to thank the following people who helped with certain aspects at various stages of this study: Merlind Muecke, Anne Freimoser‐Grundschober, Andreas Thommen, Juha Lindner, Lori Jukofsky and Lin‐Chi Chen. We would also like to thank Bernhard Reis for providing feedback on the design and validation of the fit‐for‐purpose RO assay.
Junker F, Gulati P, Wessels U, et al. A human receptor occupancy assay to measure anti‐PD‐1 binding in patients with prior anti‐PD‐1. Cytometry. 2021;99:832–843. 10.1002/cyto.a.24334
Funding information F. Hoffmann‐La Roche
REFERENCES
- 1.Green CL, Stewart JJ, Hogerkorp CM, Lackey A, Jones N, Liang M, et al. Recommendations for the development and validation of flow cytometry‐based receptor occupancy assays. Cytometry B Clin Cytom. 2016;90:141–9. [DOI] [PubMed] [Google Scholar]
- 2.Liang M, Schwickart M, Schneider AK, Vainshtein I, Del Nagro C, Standifer N, et al. Receptor occupancy assessment by flow cytometry as a pharmacodynamic biomarker in biopharmaceutical development. Cytometry B Clin Cytom. 2016;90:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stevenson L, Richards S, Pillutla R, Torri A, Kamerud J, Mehta D, et al. 2018 White Paper on Recent Issues in Bioanalysis: focus on flow cytometry, gene therapy, cut points and key clarifications on BAV (part 3—LBA/cell‐based assays: immunogenicity, biomarkers and PK assays). Bioanalysis. 2018;10:1973–2001. [DOI] [PubMed] [Google Scholar]
- 4.Stewart JJ, Green CL, Jones N, Liang M, Xu Y, Wilkins DE, et al. Role of receptor occupancy assays by flow cytometry in drug development. Cytometry B Clin Cytom. 2016;90:110–6. [DOI] [PubMed] [Google Scholar]
- 5.Gunn GR, Evans C, Yang E. Immunogenicity and biomarkers: bioanalytical challenges and considerations. Bioanalysis. 2017;9:1729–32. [DOI] [PubMed] [Google Scholar]
- 6.Waibler Z, Sender LY, Kamp C, Muller‐Berghaus J, Liedert B, Schneider CK, et al. Toward experimental assessment of receptor occupancy: TGN1412 revisited. J Allergy Clin Immunol. 2008;122:890–2. [DOI] [PubMed] [Google Scholar]
- 7.Moulard M, Ozoux ML. How validated receptor occupancy flow cytometry assays can impact decisions and support drug development. Cytometry B Clin Cytom. 2016;90:150–8. [DOI] [PubMed] [Google Scholar]
- 8.Robert C. A decade of immune‐checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11:3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600–9. [DOI] [PubMed] [Google Scholar]
- 10.Agrawal S, Feng Y, Roy A, Kollia G, Lestini B. Nivolumab dose selection: challenges, opportunities, and lessons learned for cancer immunotherapy. J Immunother Cancer. 2016;4:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18:153–67. [DOI] [PubMed] [Google Scholar]
- 12.Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, et al. An unexpected N‐terminal loop in PD‐1 dominates binding by nivolumab. Nat Commun. 2017;8:14369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu J, Wang F, Dong LH, Xing MJ, Cheng X, Wei S, et al. Receptor occupancy measurement of anti‐PD‐1 antibody drugs in support of clinical trials. Bioanalysis. 2019;11:1347–58. [DOI] [PubMed] [Google Scholar]
- 14.Naing A, Infante J, Goel S, Burris H, Black C, Marshall S, et al. Anti‐PD‐1 monoclonal antibody MEDI0680 in a phase I study of patients with advanced solid malignancies. J Immunother Cancer. 2019;7:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naing A, Gainor JF, Gelderblom H, Forde PM, Butler MO, Lin CC, et al. A first‐in‐human phase 1 dose escalation study of spartalizumab (PDR001), an anti‐PD‐1 antibody, in patients with advanced solid tumors. J Immunother Cancer. 2020;8:e000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD‐1(+) CD8(+) T cell pool with predictive potential in non‐small‐cell lung cancer treated with PD‐1 blockade. Nat Med. 2018;24:994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20:173–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wessels U, Schick E, Ritter M, Kowalewsky F, Heinrich J, Stubenrauch K. Novel drug and soluble target tolerant antidrug antibody assay for therapeutic antibodies bearing the P329G mutation. Bioanalysis. 2017;9:849–59. [DOI] [PubMed] [Google Scholar]
- 19.Schlothauer T, Herter S, Koller CF, Grau‐Richards S, Steinhart V, Spick C, et al. Novel human IgG1 and IgG4 Fc‐engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29:457–66. [DOI] [PubMed] [Google Scholar]
- 20.Darowski D, Jost C, Stubenrauch K, Wessels U, Benz J, Ehler A, et al. P329G‐CAR‐J: a novel Jurkat‐NFAT‐based CAR‐T reporter system recognizing the P329G Fc mutation. Protein Eng Des Sel. 2019;32:207–218. [DOI] [PubMed] [Google Scholar]
- 21.Friedlaender A, Addeo A, Banna G. New emerging targets in cancer immunotherapy: the role of TIM3. ESMO Open. 2019;4:e000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herrera‐Camacho I, Anaya‐Ruiz M, Perez‐Santos M, Millan‐Perez Pena L, Bandala C, Landeta G. Cancer immunotherapy using anti‐TIM3/PD‐1 bispecific antibody: a patent evaluation of EP3356411A1. Expert Opin Ther Pat. 2019;29:587–93. [DOI] [PubMed] [Google Scholar]
- 23.Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, et al. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. 2019;154:21–31. [DOI] [PubMed] [Google Scholar]
- 24.Codarri Deak L, Seeber S, Perro M, Weber P, Lauener L, Chen S, et al. Abstract 2270: RG7769 (PD1‐TIM3), a novel heterodimeric avidity‐driven T cell specific PD‐1/TIM‐3 bispecific antibody lacking Fc‐mediated effector functions for dual checkpoint inhibition to reactivate dysfunctional T cells. Proceedings of the Annual Meeting of the American Association for Cancer Research 2020; 2020.
- 25.Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA‐T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell. 2017;31:396–410. [DOI] [PubMed] [Google Scholar]
- 26.Vega‐Carrascal I, Bergin DA, McElvaney OJ, McCarthy C, Banville N, Pohl K, et al. Galectin‐9 signaling through TIM‐3 is involved in neutrophil‐mediated gram‐negative bacterial killing: an effect abrogated within the cystic fibrosis lung. J Immunol. 2014;192:2418–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cowan R, Underwood PA. Steric effects in antibody reactions with polyvalent antigen. J Theor Biol. 1988;132:319–35. [DOI] [PubMed] [Google Scholar]
- 28.De Vita M, Catzola V, Buzzonetti A, Fossati M, Battaglia A, Zamai L, et al. Unexpected interference in cell surface staining by monoclonal antibodies to unrelated antigens. Cytometry B Clin Cytom. 2014;88(5):352–354. [DOI] [PubMed] [Google Scholar]
- 29.Matos DM. Steric hindrance: a practical (and frequently forgotten) problem in flow cytometry. Cytometry B Clin Cytom. 2020. https://onlinelibrary.wiley.com/doi/10.1002/cyto.b.21959. [DOI] [PubMed] [Google Scholar]
- 30.De Vita M, Catzola V, Buzzonetti A, Fossati M, Battaglia A, Zamai L, et al. Unexpected interference in cell surface staining by monoclonal antibodies to unrelated antigens. Cytometry B Clin Cytom. 2015;88:352–4. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Gaigalas AK, Marti G, Abbasi F, Hoffman RA. Toward quantitative fluorescence measurements with multicolor flow cytometry. Cytometry A. 2008;73:279–88. [DOI] [PubMed] [Google Scholar]
- 32.Codarri‐Deak L, Fertig G, Fischer J, Klein C, Levitski V, Lifke V, et al. Bispecific antibodies for PD1 and TIM3. European patent EP3356411A1. EU; 2016.
- 33.Zelba H, Bochem J, Pawelec G, Garbe C, Wistuba‐Hamprecht K, Weide B. Accurate quantification of T‐cells expressing PD‐1 in patients on anti‐PD‐1 immunotherapy. Cancer Immunol Immunother. 2018;67:1845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo L, Zhang H, Chen B. Nivolumab as programmed death‐1 (PD‐1) inhibitor for targeted immunotherapy in tumor. J Cancer. 2017;8:410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pluim D, Ros W, Miedema IHC, Beijnen JH, Schellens JHM. Multiparameter flow cytometry assay for quantification of immune cell subsets, PD‐1 expression levels and PD‐1 receptor occupancy by nivolumab and pembrolizumab. Cytometry A. 2019;95:1053–65. [DOI] [PubMed] [Google Scholar]
- 36.Pannu KK, Joe ET, Iyer SB. Performance evaluation of QuantiBRITE phycoerythrin beads. Cytometry. 2001;45:250–8. [DOI] [PubMed] [Google Scholar]
- 37.Spitz S, Zhang Y, Fischer S, McGuire K, Sommer U, Amaravadi L, et al. 2020 White Paper on Recent Issues in Bioanalysis: BAV guidance, CLSI H62, biotherapeutics stability, parallelism testing, CyTOF and regulatory feedback (part 2A—recommendations on biotherapeutics stability, PK LBA regulated bioanalysis, biomarkers assays, cytometry validation & innovation Part 2B—regulatory agencies' inputs on bioanalysis, biomarkers, immunogenicity, gene & cell therapy and vaccine). Bioanalysis. 2021;13(5):295–361. [DOI] [PubMed] [Google Scholar]
- 38.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–25. [DOI] [PubMed] [Google Scholar]
- 39.Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor‐associated macrophage‐mediated resistance pathway in anti‐PD‐1 therapy. Sci Transl Med. 2017;9:eaal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junker F, Gordon J, Qureshi O. Fc gamma receptors and their role in antigen uptake, presentation, and T cell activation. Front Immunol. 2020;11:1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davda J, Declerck P, Hu‐Lieskovan S, Hickling TP, Jacobs IA, Chou J, et al. Immunogenicity of immunomodulatory, antibody‐based, oncology therapeutics. J Immunother Cancer. 2019;7:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Vugt MJH, Stone JA, De Greef R, Snyder ES, Lipka L, Turner DC, et al. Immunogenicity of pembrolizumab in patients with advanced tumors. J Immunother Cancer. 2019;7:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agrawal S, Statkevich P, Bajaj G, Feng Y, Saeger S, Desai DD, et al. Evaluation of immunogenicity of nivolumab monotherapy and its clinical relevance in patients with metastatic solid tumors. J Clin Pharmacol. 2017;57:394–400. [DOI] [PubMed] [Google Scholar]
- 44.Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, et al. A novel carcinoembryonic antigen T‐cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016;22:3286–97. [DOI] [PubMed] [Google Scholar]
- 45.Brunker P, Wartha K, Friess T, Grau‐Richards S, Waldhauer I, Koller CF, et al. RG7386, a novel tetravalent FAP‐DR5 antibody, effectively triggers FAP‐dependent, avidity‐driven DR5 hyperclustering and tumor cell apoptosis. Mol Cancer Ther. 2016;15:946–57. [DOI] [PubMed] [Google Scholar]
- 46.Regula JT, Lundh von Leithner P, Foxton R, Barathi VA, Cheung CM, Bo Tun SB, et al. Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol Med. 2016;8:1265–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klein C, Waldhauer I, Nicolini VG, Freimoser‐Grundschober A, Nayak T, Vugts DJ, et al. Cergutuzumab amunaleukin (CEA‐IL2v), a CEA‐targeted IL‐2 variant‐based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL‐2‐based immunocytokines. Onco Targets Ther. 2017;6:e1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson LB, Bell CJM, Howlett SK, Pekalski ML, Brady K, Hinton H, et al. A long‐lived IL‐2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun. 2018;95:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan M, Aziz AA, Shafi NA, Abbas T, Khanani AM. Targeting angiopoietin in retinal vascular diseases: a literature review and summary of clinical trials involving faricimab. Cell. 2020;9(8):1869.. 10.3390/cells9081869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Centanni M, Moes D, Troconiz IF, Ciccolini J, van Hasselt JGC. Clinical pharmacokinetics and pharmacodynamics of immune checkpoint inhibitors. Clin Pharmacokinet. 2019;58:835–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD‐1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD‐1 blockade therapies. Nat Immunol. 2020;21:1346–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Checklist: MIFlowCyt‐Compliant Items
Appendix S2. Supporting Information
FIGURE S1 A. Analysis of PD‐1 and TIM3 cell surface expression in PBMCs from n = 3 healthy donors. PBMCs were analyzed after preparation (d0) and after 3d in culture in the presence of SEB (1000 ng/mL). CD3+ T cells and CD56+ NK cells were assessed; shown is the percentage of target‐positive cells minus unstained (FM) control. B. Activated PBMCs of n = 3 donors were incubated with RG7769 or DP47 isotype (10 pg/mL➔10 μg/mL) and subsequently stained with anti‐PGLALA‐A647. Shown is the MFI for CD3+ CD56+ cells. C. Titration of anti‐PGLALA‐PE (M‐1.7.24) used in the fit‐for‐purpose validated RO assay for RG7769. Freshly isolated CD3‐CD28 activated PBMCs were incubated with 10 μg/mL of RG7769 followed by incubation with anti‐PGLALA‐PE (100 ng/mL➔100 μg/mL). Shown is the total single‐cell lymphoid PBMC cell fraction. D. Comparison of RG7769 binding to PBMCs (via anti‐PGLALA‐PE) on CD3+, CD4+ and CD8+ T cells of a representative healthy donor. E. Schematic illustrating sample preparation for RO analysis. Peripheral blood was collected into three NaHep coated tubes and pulsed with 1000 ng/mL of RG7769. Tube 1 serves as negative control. Tube 2 serves as a positive control ‐ following initial incubation, tube 2 was re‐challenged with excess RG7769 (100 μg/mL). All three samples underwent red blood cell lysis followed by incubation with flow cytometry antibodies. Tube 1 was stained with a lineage mix including mIgG2b‐PE isotype, while tubes 2 and 3 were incubated with the same lineage mix including anti‐PGLALA‐PE.
FIGURE S2 A. Dose‐dependent RO increase in whole‐blood incubated with 0 ng/mL, 5 ng/mL, 20 ng/mL, 60 ng/mL or 100 ng/mL of RG7769 and analyzed 24 hrs post‐incubation. RG7769 RO was calculated for CD3+, CD3+CD4+ and CD3+CD8+ T cells based on MFI (PE). B. RO stability assessment. Blood of 2 healthy donors was incubated with 0 ng/mL or 20 ng/mL of RG7769 and analyzed immediately (0d) or 1–4 days post challenge. RO was calculated using anti‐PE (MFI) and assessed over time (top) or the percentage of anti‐PGLALA‐PE positive cells (bottom). Only time points from d1 (but not d0) were considered relevant (dashed line).
FIGURE S3 Linear regression of max. RG7769 binding (MFI) to T cells in NP40435 aPD‐1+ patients prior to RG7769 infusion (C1D1_pre) as a function of the last reported prior aPD‐1 administration on CD4+ (left) or CD8+ T cells (right). B. anti‐PGLALA‐PE staining on T cells of representative NP40435 patients. Maximum anti‐PGLALA‐PE was assessed post ex vivo blood saturation with RG7769, in recently (up to 14 weeks) or historically aPD‐1+ experienced (>14 weeks) vs naïve patients prior to RG7769 infusion (C1D1_pre). Anti‐PGLALA–PE staining intensities are shown for the ex‐vivo saturation RG7769 negative control on CD4+ T cells (left) or CD8+ T cells (right). C. RG7769 ABC to CD3+ T cells in NP40435 patients in recently anti‐PD‐1‐experienced (up to 14 weeks) or historically experienced (>14 weeks) patients vs naïve patients. ABCs were compared to aPD‐1 naïve (Welch ANOVA test).