

Abstract
Cerebrovascular‐related amyloidogenesis is found in over 80% of Alzheimer's disease (AD) cases, and amyloid β (Aβ) generation is increased in the peripheral macrophages during infection of Porphyromonas gingivalis (P. gingivalis), a causal bacterium for periodontitis. In this study, we focused on receptor for advanced glycation end products (RAGE), the key molecule involves in Aβ influx after P. gingivalis infection to test our hypothesis that Aβ transportation from periphery into the brain, known as “Aβ influx,” is enhanced by P. gingivalis infection. Using cultured hCMEC/D3 cell line, in comparison to uninfected cells, directly infection with P. gingivalis (multiplicity of infection, MOI = 5) significantly increased a time‐dependent RAGE expression resulting in a dramatic increase in Aβ influx in the hCMEC/D3 cells; the P. gingivalis‐up‐regulated RAGE expression was significantly decreased by NF‐κB and Cathepsin B (CatB)‐specific inhibitors, and the P.gingivalis‐increased IκBα degradation was significantly decreased by CatB‐specific inhibitor. Furthermore, the P. gingivalis‐increased Aβ influx was significantly reduced by RAGE‐specific inhibitor. Using 15‐month‐old mice (C57BL/6JJmsSlc, female), in comparison to non‐infection mice, systemic P. gingivalis infection for three consecutive weeks (1 × 108 CFU/mouse, every 3 days, intraperitoneally) significantly increased the RAGE expression in the CD31‐positive endothelial cells and the Aβ loads around the CD31‐positive cells in the mice's brains. The RAGE expression in the CD31‐positive cells was positively correlated with the Aβ loads. These observations demonstrate that the up‐regulated RAGE expression in cerebral endothelial cells mediates the Aβ influx after P. gingivalis infection, and CatB plays a critical role in regulating the NF‐κB/RAGE expression.
Cover Image for this issue: https://doi.org/10.1111/jnc.15073
Keywords: amyloid β, cathepsin B, cerebral endothelial cells, NF‐κB, Porphyromonas gingivalis, receptor for advanced Glycation end products
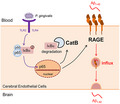
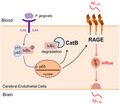
We demonstrated that CatB /NF‐κB‐dependent up‐regulated RAGE expression in cerebral endothelial cells mediates cerebrovascular‐related Aβ influx into brain during P. gingivalis infection. Together with our previous findings of increased Aβ production peripherally by P. gingivalis infection, thus provide a new mechanism for the involvement of periodontitis in the AD pathological process.
Cover Image for this issue: https://doi.org/10.1111/jnc.15073
Abbreviations
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- Aβ
amyloid β protein
- BBB
blood–brain barrier
- BMDM
bone marrow‐derived macrophages
- BSA
bovine serum albumin
- C3
complement component 3
- C5
complement component 5
- CatB
cathepsin B
- CD31
cluster of differentiation 31
- CFU
colony‐forming unit
- CLMS
confocal laser microscope system
- hCMEC/D3
human cerebral microvascular endothelial cell line
- HRP
horseradish peroxidase
- ICLAC
International Cell Line Authentication Committee
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B‐cell inhibitor, alpha
- LPS
lipopolysaccharide
- LRP1
low‐density lipoprotein receptor‐related protein1
- MOI
multiplicity of infection
- Na‐F
sodium fluorescein
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- PBS
phosphate buffer saline
- PCR
polymerase chain reaction
- PFA
paraformaldehyde
- P. gingivalis
porphyromonas gingivalis
- P.gLPS
lipopolysaccharide from P. gingivalis
- pIκBα
phosphorylated IκBα
- RAGE
advanced glycation end products
- RRID
Research Resource Identifier (see scicrunch.org)
- SDS‐PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TBS
tris‐buffered saline
- TLR2
toll‐like receptor 2
- TLR4
toll‐like receptor 4
1. INTRODUCTION
Alzheimer's disease (AD), the most common cause of dementia, affects more than 50 million people worldwide. The prevalence of AD continues to grow because of the increase in the aging population (Hodson, 2018). As the major pathological marker of AD brain, amyloid β protein (Aβ), a polypeptide consisting of 39–43 acid residues, is proteolytically generated by cleavage of amyloid precursor protein (Chen et al., 2019). Aβ accumulation in and around the cerebral vasculature, known as cerebral amyloid angiopathy (CAA), has been found in more than 80% of AD patients (Kovari, Herrmann, Hof, & Bouras, 2013).
Brain Aβ in AD has long been thought to be generated in the brain itself by neurons; however, Aβ was recently found to be generated peripherally by platelets, skeletal muscle cells, skin fibroblasts, and monocyte/macrophages (Evin, Zhu, Holsinger, Masters, & Li, 2003; Kuo et al., 2000; Nie et al., 2019; Roher & Kokjohn, 2009). Recent studies have also shown that peripheral Aβ can be transferred into the brain using a model of parabiosis between APPswe/PS1dE9 transgenic AD mice and their wild‐type littermates, which thus determines the human Aβ originating from transgenic AD model mice in the circulation and CAA in the brains of wild‐type mice after a 12‐month period of parabiosis (Bu et al., 2018).
The physical and functional integrity and health of the mammalian brain are basically maintained by the blood–brain barrier (BBB), which is specialized to prevent pathogens and circulating cells from entering the brain and causing damage. Cerebral endothelial cells are part of the BBB, connected by tight junctions that function as a “physical barrier” and prevent molecular traffic between the blood and brain (Iadecola, 2013). Cerebral endothelial cells also act as a “transport barrier” because of the presence of specific transport systems regulating the transcellular traffic of small molecules (Abbott, Ronnback, & Hansson, 2006). Regarding Aβ transportation in the cerebral endothelial cells, low‐density lipoprotein receptor‐related protein 1 (LRP1) is the receptor for Aβ transport from the brain to the peripheral blood (Aβ efflux), while receptor for advanced glycation end products (RAGE) is accepted as the receptor for Aβ transport from the peripheral blood to the brain (Aβ influx) (Deane et al., 2003; Sagare, Deane, & Zlokovic, 2012). RAGE up‐regulation has been found in the brains of both AD patients and AD mouse models, suggesting that RAGE is involved in the pathological Aβ accumulation in the AD brain (Miller et al., 2008; Du Yan et al., 1996).
Numerous clinical studies have shown that periodontitis, a common oral infection disease, is positively linked to cognitive decline in elderly people (Noble et al., 2009; Sparks Stein et al., 2012) as well as AD patients (Ide et al., 2016). Furthermore, the components of Porphyromonas gingivalis, a key pathogen involved in periodontitis, including lipopolysaccharide (LPS) and gingipain, have been found in human AD brains (Dominy et al., 2019; Poole, Singhrao, Kesavalu, Curtis, & Crean, 2013). These findings suggest the influence of periodontitis on the AD onset as well as its pathological processing.
We previously showed that chronic exposure to LPS from P. gingivalis induced CatB‐dependent AD‐like phenotypes that included memory impairment, microglia‐mediated neuroinflammation and intracellular Aβ accumulation in neurons of middle‐aged mice (Wu et al., 2017). Furthermore, other researchers have found that oral infection with P. gingivalis induced Aβ formation in the brain of adult mice (Ilievski et al., 2018). However, the role of Aβ accumulation in the brain during systemic P. gingivalis infection remains unclear.
P. gingivalis has been recognized as a keystone pathogen for periodontitis because it is a master evader of the host's immune system (Bielecka et al., 2014; Reife et al., 2006). P. gingivalis is resistant to destruction by complements that are dependent on gingipains degrading complement 3 (C3) and C5, thereby preventing the deposition of C3b and C5a on the surface of P. gingivalis (Slaney, Gallagher, Aduse‐Opoku, Pell, & Curtis, 2006). The immune evasion strategies of P. gingivalis prolong its stay in the systemic circulation, providing more opportunities for this bacterium to make direct contact with cerebral endothelial cells. We recently demonstrated the generation of Aβ in the inflammatory monocytes/macrophages in the gingival tissues of periodontitis patients and in the liver of middle‐aged mice after chronic systemic P. gingivalis infection, indicating that the peripheral pools of Aβ expand during chronic systemic P. gingivalis infection (Nie et al., 2019).
In this study, we tested our hypothesis that RAGE in cerebral endothelial cells contributes to the peripheral Aβ influx during chronic P. gingivalis infection, adding new insight into the influence of systemic periodontal bacterial infection on AD.
2. MATERIALS AND METHODS
All reagents in this study were purchased within 2 years and stored properly according to the manufactures. All experiments were performed and repeated according to the same time schedule. The study was not pre‐registered. The study was exploratory. The experimenters were not blinded to experimental condition.
2.1. Animals
Fifteen‐month‐old female mice (C57BL/6JJmsSlcJapan SLC, Incorporation, MGI Cat# 5658738, RRID:MGI:5658738) were kept in a pathogen‐free condition at the Kyushu University Faculty of Dental Sciences. All animal experiments were conducted in according to the protocols approved by the Animal Care and Use Committee of Kyushu University (A27‐232‐0). The mice (n = 6) were intraperitoneally injected with P. gingivalis (1 × 108 CFU/mouse) in 100 μL phosphate‐buffered saline (PBS) every 3 days and last for three consecutive weeks. The age‐matched control mice (n = 6) were injected with 100 μL PBS in the same time course. Then the mice were anesthetized via an intraperitoneal injection of widely used somnopentyl (50 mg/kg Kyoritsu Seiyaku Cat# 7212101) and sacrificed by cardiac perfusion with PBS followed by 4% paraformaldehyde, and then the brain samples were collected immediately. The active ingredient of somnopentyl is pentobarbital sodium, which binds to the gamma‐aminobutyric acid‐A receptor and causes central nervous system depression (Luo et al., 2015). We fixed three mice per group for immunostaining in this study. The sample size determination was based on the behavior test data of our previously publication (Wu et al., 2017). G *Power software (G *Power 3.1.9.7, means: difference between two independent means, two tails) was used to verify the Power of the behavior test (Charan & Kantharia, 2013). The α err prob was set at 0.05, and the sample size of each group was 6. The effect size d was 5.6537 which was determined by mean and SD of the two groups (group 1: mean 300, SD 1; group 2: mean 180, SD 30). As the Power was 1 after calculation, six mice in each group were used for the behavior test in this study.
2.2. Step‐through passive avoidance test
The step‐through passive avoidance test was used to test memory decline in this paper. The device included a bright compartment and a dark compartment with an electrifiable grid floor, which were separated by a sliding door. In acquisition phase, each mouse was placed in the bright compartment and was able to freely explore for 30 s. After the door was opened, mice entered into the dark compartment. Then the door was closed and an electric shock (0.2 mA, 2 s) was delivered through the grid. The endpoint of the acquisition phase was that mice stay in the bright compartment for 300 s without entering the dark compartment. From the first day of P. gingivalis injection, the step‐through passive avoidance test was carried out every 7 days until 3 weeks after P. gingivalis injection. The latency was set up to 300 s and no electric shock was given. To minimize animal pain in procedures, the mice were gently picked out from or put back to their cages, and were returned to their familiar environment as soon as possible after behavior test or injection. No other procedures involved animal pain, suffering or distress. Mice failed to enter the dark compartment would be excluded, but no mouse was excluded or died during experiments.
2.3. Cell culture
The hCMEC/D3 cell line (Millipore Cat# SCC066, RRID:CVCL_U985) was maintained in EBM‐2 medium with supplements (Lonza Cat# CC‐3202) and grown to confluence in rat‐tail collagen type I coated cell culture dishes at 37°C and 5% CO2 in humid atmosphere. The hCMEC/D3 was infected by P. gingivalis (ATCC33277, MOI (multiplicity of infection) =5) for indicated time points. For the immunofluorescent staining, the number of hCMEC/D3 was 5 × 104 and the corresponding number of P. gingivalis was 2.5 × 105. For the real‐time quantitative polymerase chain reaction (PCR) analysis and immunoblotting analysis, the number of hCMEC/D3 was 1 × 106 and the corresponding number of P. gingivalis was 5 × 106. For the Na‐F permeability and Aβ1‐42 transportation, the number of hCMEC/D3 was 7 × 105 and the corresponding number of P. gingivalis was 3.5 × 106. All the inhibitors were added 1 hr before P. gingivalis infection. The maximum number of passages for the hCMEC/D3 cell line was 30 passages. The cell line was last authenticated in 2018 by the supplier and this cell line is not listed as a commonly misidentified cell line by the ICLAC.
2.4. Bacterial strain and culture conditions
P. gingivalis ATCC33277 was maintained on blood agar plate containing 40 mg/mL Trypto‐Soya Ager (Nissui Cat# 05516), 5 mg/mL Brain Heart Infusion (Becton Dickinson Cat# 211059), 1 mg/mL L‐Cysteine Hydrochloride (Wako Cat#039‐05274), 1 μg/mL Menadione (Sigma‐AldrichCat# M9429‐25G), 5 μg/mL Hemin (Sigma‐Aldrich Cat# 51280‐1G) and 5% defibrinated sheep blood (ThermoFisher Cat# R54020). When P. gingivalis was ready to use, it was grown in liquid medium containing 37 mg/mL Brain Heart Infusion, 2.5 mg/mL Yeast Extract (Becton Dickinson Cat# 212750), 1 mg/mL Cysteine, 1 μg/mL Menadione and 5 μg/mL Hemin. We used an anaerobic chamber with 10% CO2, 10% H2, and 80% N2 maintaining at 37°C for P. gingivalis culture. Before P. gingivalis infection, the liquid medium was centrifuged (6,000 g, 10 min) and replaced by EBM‐2 medium without antibiotics.
2.5. Immunofluorescent staining
For the hCMEC/D3 cells staining, cells were fixed with 4% Formaldehyde solution (Sigma‐Aldrich Cat# 11‐0720‐5) for 1 hr at 4°C, and permeated with 0.1% Triton X‐100 (Sigma‐Aldrich Cat# 30‐5140‐5) for 5 min at 25ºC. After blocking with 3% Albumin from Bovine Serum (BSA) (Wako Cat# 017‐21273) in PBS, the cells were incubated with goat anti‐RAGE (1:1,000; R&D Systems Cat# AF1179, RRID:AB_354649) or rabbit anti‐p65 (1:1,000; Abcam Cat# ab16502, RRID:AB_443394) at 4°C overnight. After washing with PBS for three times, the cells were then incubated with donkey anti‐goat Alexa Fluor 488 (1:1,000; Jackson ImmunoResearch Labs Cat# 705‐545‐147, RRID:AB_2336933) or donkey anti‐rabbit Alexa Fluor 488 (1:1,000; Jackson ImmunoResearch Labs Cat# 711‐545‐152, RRID:AB_2313584) at 4°C for 2 hr. After washing with PBS for three times, cell nuclear were stained with Hoechst (1:1,000; Sigma‐Aldrich Cat#14533) for 5 min and mounted in Vectashield anti‐fading medium (Vector Laboratories Cat# H‐1000, RRID:AB_2336789). In the case of F‐actin staining, Texas Red™‐XPhalloidins (Invitrogen Cat# T7471) were used at the recommended concentration for 2 hr at 4°C.
The brain samples of mice were cut into serial coronal frozen sections (10 μm) (n = 3 in each group). Then the sections were incubated with 3% BSA in PBS containing 0.1% Triton X‐100 for 1 hr at 25ºC. After that, 0.05% Sudan Black B (Sigma‐Aldrich Cat# 199664‐25G) was used to block non‐specific staining. Then the sections were incubated with goat anti‐CatB (1:1,000; Santa Cruz Biotechnology Cat# sc‐6493, RRID:AB_2086939), rabbit anti‐p65, goat anti‐RAGE, rabbit anti‐Aβ1‐42 (1:1,000; Thermo Fisher Scientific Cat# 44‐344, RRID:AB_2533636), and rat anti‐CD31 (1:1,000; BD Biosciences Cat# 553370, RRID:AB_394816) at 4°C overnight. After washing with PBS for three times, the sections were then incubated with donkey anti‐goat Alexa Fluor 488 (1:1,000; Jackson ImmunoResearch Labs Cat# 705‐545‐147, RRID:AB_2336933) and donkey anti‐rat Cy3 (1:1,000; Jackson ImmunoResearch Labs Cat# 712–165–153, RRID:AB_2340667) at 4°C for 2 hr. After washing with PBS for three times, the sections were stained with Hoechst for 5 min and mounted in Vectashield anti‐fading medium. Fluorescence images were taken using a CLMS (2si Confocal Laser Microscope, Nikon).
2.6. Real‐time quantitative PCR analysis
The mRNA was isolated from hCMEC/D3 cell cultures after stimulation with P. gingivalis, or pre‐treatment with Bay11‐7082 (10 μM; Sigma‐Aldrich Cat#B5556‐10MG), SN50 (20 μM; Sigma‐Aldrich Cat#SML1471‐1MG) or CA‐074Me (10 μM; Peptide Cat# 4323‐v), respectively, with RNAiso Plus (Takara Cat# 9109) under the manufacturer's instructions. And then up to 1 μg of extracted mRNA was reverse transcribed to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen Cat# 205314). The cDNA was amplified in duplicate using a Rotor‐Gene SYBR Green RT‐PCR Kit (Qiagen Cat#204174) with a Corbett Rotor‐Gene RG‐3000A Real‐Time PCR System. The data were evaluated using the RG‐3000A software program (version Rotor‐Gene 6.1.93 Corbett). The sequences of primer pairs were as follows: human RAGE, forward 5’CCAGGAGGAAGAGGAGGAG‐3’ and reverse 5’‐GCTGATGGATGGGATCTGTC‐3’; human CatB, forward 5’‐AACACGT
CACCGGAGAGATGA‐3’ and reverse 5’‐CCCAGTCAGTGTTCCAGGAGTT‐3’; human TLR2, forward 5’GCCAAAGTCTTGATTGATTGG‐3’ and reverse 5’‐TTGA
AGTTCTCCAGCTCCTG‐3’; human TLR4, forward 5’‐TACAAAATCCCCGACAA
CCTCC‐3’ and reverse 5’‐GCTGCCTAAATGCCTCAGGG‐3’; human Actin, forward 5’‐CATCTCTTGCTCGAAGTCCA‐3’ and reverse 5’‐ATCATGTTTGAGACCTTCA ACA‐3’
2.7. Immunoblotting analysis
The hCMEC/D3 cells were grown to confluence and were harvested after P. gingivalis infection for indicated time points, and protein was extracted after harvesting samples. For the nuclear lysate, cells were harvested and nuclear were isolated using the Nuclear Extraction Kit (AbcamCat#ab113474). Each sample was homogenized and separated using 12% SDS‐PAGE gel. After electrophoresis, the proteins were transferred to nitrocellulose membranes. After blocking with 5% milk in TBS, the membranes were incubated with primary antibodies at 4°C overnight. The primary antibodies were as follows: rabbit anti‐RAGE (1:1,000; Abcam Cat# ab3611, RRID:AB_303947); rabbit anti‐p65; goat anti‐CatB (1:1,000;Santa Cruz Biotechnology Cat# sc‐6493, RRID:AB_2086939); rabbit anti‐IκBα (1:1,000; Cell Signaling Technology Cat# 8993, RRID:AB_2797687); mouse anti‐pIκBα (1:1000; Cell Signaling Technology Cat# 9246, RRID:AB_2267145); and rabbit anti‐Lamin B1 (1:1,000, Cell Signaling Technology Cat# 13435, RRID:AB_2737428). After washing, the membranes were incubated with HRP‐labeled second antibodies and mouse anti‐actin (1:1,000; Abcam Cat# ab20272, RRID:AB_445482) at 25ºC for 2 hr. The second antibodies were as follows: anti‐rabbit (1:1,000; GE Healthcare Cat#NA934V), anti‐goat (1:1,000; R&D Systems Cat# HAF109, RRID:AB_357236), and anti‐mouse (1:1,000; GE Healthcare Cat#NA931V). Finally, the membrane‐bound, HRP‐labeled antibodies were detected by an Immobilon ECL Ultra Western HRP Substrate (Millipore Cat#WBULS0100) with an image analyzer (LAS‐1000 Fuji Photo Film).
For the secreted CatB, culture medium was collected after P. gingivalis infection for indicated time points. The medium was centrifuged to pellet cell debris at 750 g for 5 min. Then supernatant was collected and protein concentration was determined. The loading quantity of each sample was 10 μg and the following procedures were the same as above.
2.8. Na‐F permeability
The permeability of endothelial monolayer was determined using Sodium Fluorescein (Na‐F) (100 ng/mL; Sigma‐Aldrich Cat# F6377). The transcytosis system was constructed by confluence hCMEC/D3 cells on inserts with collagen‐coated polycarbonate transwell membrane filters (cell culture inserts; Millicell Cell Culture Inserts, RRID:SCR_015799). The hCMEC/D3 monolayer was cultured in the apical side of a 24‐well tissue culture insert. The EBM‐2 medium was added into both the apical side and the basolateral side. After P. gingivalis infection at the apical side for 12 hr, all EBM‐2 medium was replaced by warmed Assay Buffer. Fluorescence labeled Na‐F was added into the apical Assay Buffer, after 5 min, inserts were removed and the fluorescence of the basolateral Assay Buffer was determined by a reader (infiniteM200 TECAN, excitation wavelength, 485 nm; measurement wavelength, 535 nm). These experiments were carried out at 37°C.
2.9. Aβ1‐42 transportation
The transcytosis system was the same with Na‐F permeability experiment. After P. gingivalis infection at the apical side for 12 hr, all EBM‐2 medium was replaced by warmed Assay Buffer. Thirty minutes after adding HiLyte Fluor 488‐labeled Aβ1‐42 (50 nM; AnaSpec Cat#AS‐60479‐01) into the apical side, inserts were removed and the fluorescence of the basolateral Assay Buffer was determined by the reader (infiniteM200 TECAN, excitation wavelength, 485 nm; measurement wavelength, 535 nm). The RAGE‐specific inhibitor FPS‐ZM1 (10 μM; Abcam Cat#ab235552) was added 1 hr before P. gingivalis infection. These experiments were carried out at 37°C. For the Aβ1‐42 transportation and Na‐F permeability, the following calculation was used: P [cm/s]=VD/(A × MD)×(ΔMR/Δt). Where VD is the apical side volume, A is the membrane surface area, MD is the apical amount, and Δt is the time and ΔMR is deduced from a standard curve (Di Marco et al., 2019).
2.10. Statistical analyses
The data are represented as the means ± SEM. All data presented represent the mean of at least three replicates of independent cell culture preparations. The statistical analyses were performed by one‐way ANOVA, two‐way ANOVA, Student's t test, or multiple t test using the GraphPad Prism software package (GraphPad Software 7.00). A value of p < .05 was considered to indicate statistical significance. Normality was determined using the Shapiro–Wilk normality test. There is no test for outliers and no data point was excluded.
3. RESULTS
3.1. RAGE expression was up‐regulated in hCMEC/D3 cells after P. gingivalis infection
We used the hCMEC/D3 cell line because it has been extensively characterized for the brain endothelial phenotype and is a model of the human BBB function. The time course of in vitro experiments was set up to 12 hr after P. gingivalis infection, as P. gingivalis can be kept alive for up to 12 hr in our culture system (Liu et al., 2017; Nie et al., 2019). Compared with the uninfected cells (None group), the mRNA expression of RAGE was significantly increased in the hCMEC/D3 cells from 3 hr (two‐fold) to 12 hr (2.8‐fold) after P. gingivalis infection (Figure 1a), the protein expression of RAGE in hCMEC/D3 cells was significantly increased from 6 to 12 hr after P. gingivalis infection (Figure 1b and c), and the immunofluorescent density of RAGE in hCMEC/D3 cells was significantly increased at 12 hr after P. gingivalis infection (Figure 1d and e).These observations showed that the RAGE expression in hCMEC/D3 cells was up‐regulated after P. gingivalis infection.
FIGURE 1.
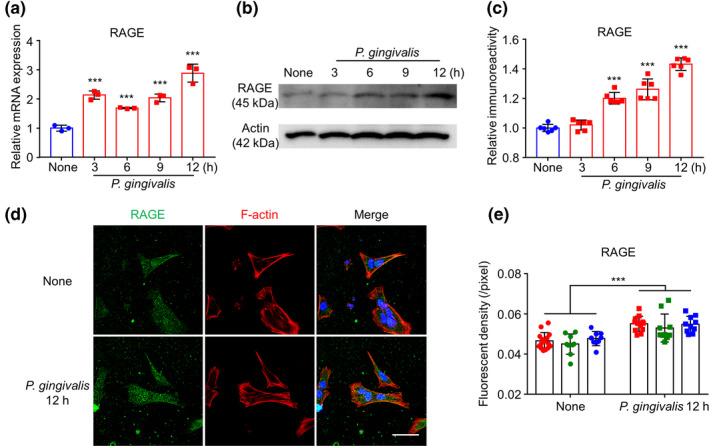
RAGE expression increased in hCMEC/D3 cells after P. gingivalis infection. (a) The mean mRNA expression level of RAGE increased in cultured hCMEC/D3 cells after P. gingivalis infection (n = 3 independent cell culture preparations, ***p < .001; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “None” group. (b) The immunoblots show RAGE increased in hCMEC/D3 cells after P. gingivalis infection. (c) The quantitative analysis of RAGE in the immunoblots in (b) (n = 6 independent cell culture preparations, ***p < .001; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “None” group. (d) The immunofluorescent CLMS images of RAGE (green) in hCMEC/D3 cells with Hoechst‐stained nuclei (blue) and F‐actin (red) 12 hr after P. gingivalis infection. Scale bar, 50 μm. (e) The quantitative analysis of RAGE fluorescence density in the images in (d) (each color represents one independent cell culture preparations, ***p < .01; multiple t‐test). Asterisks indicate a statistically significant difference versus “None” group
3.2. RAGE up‐regulation in hCMEC/D3 cells was dependent on NF‐κB activation after P. gingivalis infection
We next examined the involvement of the NF‐κB signaling pathway in the RAGE expression in hCMEC/D3 cells after P. gingivalis infection. We focused on TLR2 and TLR4 as well as NF‐κB activation in hCMEC/D3 cells after P. gingivalis infection because P. gingivalis activated NF‐κB via TLR2 and TLR4 in bone marrow‐derived macrophages (BMDMs) and RAW264.7 cells (Nie et al., 2019; Papadopoulos et al., 2017). Compared with uninfected cells, the mRNA expression of TLR2 in hCMEC/D3 cells was significantly increased at 3 hr (1.9‐fold) and then returned to the baseline level from 6 to 12 hr after P. gingivalis infection (Figure 2a). In contrast, the mRNA expression of TLR4 in hCMEC/D3 cells was significantly increased from 6 hr (1.7‐fold) to 12 hr (3.7‐fold) after P. gingivalis infection (Figure 2a).
FIGURE 2.
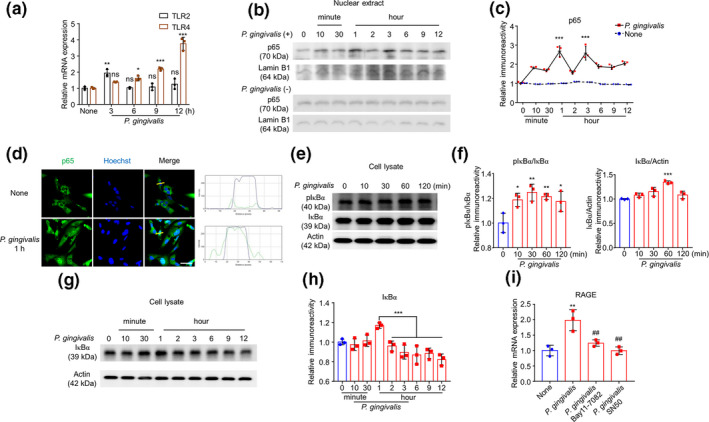
RAGE elevation was dependent on NF‐κB activation in hCMEC/D3 cells after P. gingivalis infection. (a) The mean mRNA expression level of TLR2/4 in cultured hCMEC/D3 cells after P. gingivalis infection (n = 3 independent cell culture preparations, ns: no significant difference, *p < .05, **p < .01, ***p < .001; one‐way ANOVA). Asterisksindicate a statistically significant difference versus “None” group. (b) The immunoblot images show p65 in nuclear extract with or without P. gingivalis infection. (c) The quantitative analysis of p65 in the immunoblots shown in (b) (n = 3 independent cell culture preparations, ***p < .001; two‐way ANOVA). Asterisks indicate a statistically significant difference versus “None” group. (d) The immunofluorescent CLMS images indicating the nuclear translocation of p65 (green) in hCMEC/D3 cells with Hoechst‐stained nuclei (blue) after P. gingivalis infection for 1 hr. Scale bar, 50 μm. (e) The immunoblot images show pIκBα and IκBα in cell lysate in hCMEC/D3 cells after P. gingivalis infection. (f) The quantitative analysis of immunoblots shown in (e), pIκBα/IκBα (left) and IκBα/Actin (right) (n = 3 independent cell culture preparations, *p < .05, **p < .01, ***p < .001; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “0” group. (g) The immunoblots show IκBα degradation in cell lysate after P. gingivalis infection. (h) The quantitative analysis of IκBα in the immunoblots shown in (g) (n = 3 independent cell culture preparations; one‐way ANOVA) (i) The effects of Bay11‐7082 and SN50, two specific NF‐κB inhibitors, on the mRNA expression of RAGE in hCMEC/D3 cells after P. gingivalis infection for 12 hr (n = 3 independent cell culture preparations, **p < .01, ## p < .01; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “None” group and pounds indicate a statistically significant difference versus “P. gingivalis” group
Nuclear extracts and cell lysates were prepared, and western blotting was conducted. The immunoreactivity of p65 in both with or without P. gingivalis infection groups were compared with corresponding Lamin B1, and then the relative immunoreactivity of the two groups was analyzed with two‐way ANOVA. After analysis, we found that the translocation of p65 into the nucleus of hCMEC/D3 cells was significantly increased at 1 and 3 hr and lasting until 12 hr after P. gingivalis infection (Figure 2b and c). The P. gingivalis infection‐induced increase in nuclear p65 localization in hCMEC/D3 cells was further confirmed by immunofluorescent staining (Figure 2d).
We further examined the phosphorylation and degradation of IκBα in hCMEC/D3 cells after P. gingivalis infection, as the ubiquitin proteasome‐ and autophagy lysosome‐dependent degradation of IκBα has been reported to be involved in the early and late phases of NF‐κB activation, respectively (Ni, 2015). Western blotting revealed a significant increase in pIκBα in hCMEC/D3 cells from 10 to 120 min after P. gingivalis infection (Figure 2e and f). Significant decrease in IκBα band was found between 2 and 12 hr after P. gingivalis infection (Figure 2g and h), indicating the obvious degradation of IκBα at the late phase of P. gingivalis infection.
To further confirm the regulating functions of NF‐κB for the RAGE expression in hCMEC/D3 cells after P. gingivalis infection, we pre‐treated the P. gingivalis infected hCMEC/D3 cells with Bay11‐7082 (inhibit IκBα phosphorylation) and SN50 (inhibit p65 combining with DNA). Compared to the P. gingivalis‐infected hCMEC/D3 cells, pretreatment with both Bay11‐7082 and SN50 significantly inhibited the P. gingivalis‐induced increased RAGE expression in hCMEC/D3 cells (Figure 2i). These observations showed that the up‐regulated RAGE expression in hCMEC/D3 cells was dependent on the ubiquitin–proteasome degradation of IκBα in the early phase and on the IκBα degradation in the late phase after P. gingivalis infection.
3.3. Cathepsin B (CatB) involvement in RAGE up‐regulation via NF‐κB activation after P. gingivalis infection
We next examined the involvement of CatB in the RAGE expression in hCMEC/D3 cells, as we previously found that CatB was involved in the NF‐κB activation in phagocytic cells (Ni et al., 2015; Nie et al., 2019). Compared with uninfected cells, the mRNA expression of CatB in hCMEC/D3 cells significantly increased from 3 to 12 hr after P. gingivalis infection (Figure 3a), while also the intracellular CatB protein level significantly increased from 3 to 9 hr after P. gingivalis infection (Figure 3b and c). The extracellular CatB protein level was also detected from 3 to 12 hr after P. gingivalis infection (Figure 3b).
FIGURE 3.
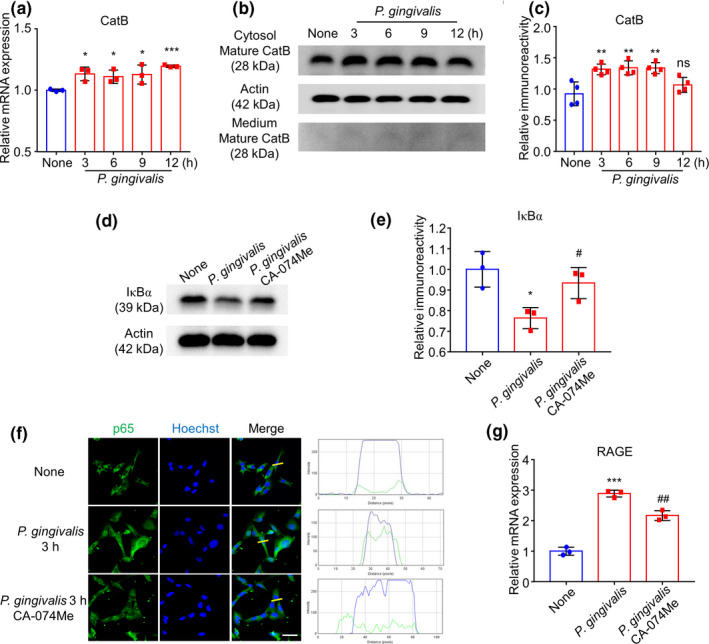
CatB/NF‐κB pathway regulated late‐phase expression of RAGE after P. gingivalis infection. (a) The mean mRNA expression level of CatB of cultured hCMEC/D3 cells increased after P. gingivalis infection (n = 3 independent cell culture preparations, *p < .05, ***p < .001; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “None” group. (b) The immunoblots show mature CatB in cytosol and medium in hCMEC/D3 after P. gingivalis infection. (c) The quantitative analysis of cytosol CatB in (b) (n = 4 independent cell culture preparations, ns: no significant difference, **p < .01; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “None” group. (d) The effect of CA‐074Me, a specific CatB inhibitor, on IκBα degradation after P. gingivalis infection for 12 hr. (e) The quantitative analysis of (d) (n = 3 independent cell culture preparations, *p < .05, # p < .05; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “None” group and pounds indicate a statistically significant difference versus “P. gingivalis” group. (f) The effect of CA‐074Me on the nuclear translocation of p65 (green) in hCMEC/D3 cells with Hoechst‐stained nuclei (blue) at 3 hr after P. gingivalis infection. Scale bar, 50 μm. (g) The effect of CA‐074Me on the mRNA expression of RAGE in hCMEC/D3 cells after P. gingivalis infection (n = 3 independent cell culture preparations, ***p < .001, ## p < .01; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “None” group and pounds indicate a statistically significant difference versus “P. gingivalis” group
The fact that the CatB increase paralleled the IκBα degradation prompted us to explore the involvement of CatB in IκBα degradation in hCMEC/D3 cells in the late phase after P. gingivalis infection. Treatment with the CatB‐specific inhibitor CA‐074Me partially reversed the P. gingivalis infection‐induced IκBα degradation at 12 hr after P. gingivalis infection (Figure 3d and e). The inhibition of the P. gingivalis infection‐increased nuclear p65 localization in hCMEC/D3 cells by CA‐074Me was further confirmed by immunofluorescent staining (Figure 3f). Furthermore, CA‐074Me pretreatment partially inhibited the P. gingivalis infection‐increased RAGE expression in hCMEC/D3 cells at 12 hr after P. gingivalis infection (Figure 3g). These observations indicate that CatB is one of the protease digesting IκBα and partially mediates the RAGE expression in hCMEC/D3 cells after P. gingivalis infection.
3.4. RAGE expression mediated Aβ1‐42 transportation after P. gingivalis infection in a transcytosis system
The hCMEC/D3 cell line is typically used for studies of molecule transport from blood‐to‐brain, including Aβ. To determine the involvement of RAGE in Aβ transportation after P. gingivalis infection, we developed a transcytosis system of hCMEC/D3 cells. We cultured hCMEC/D3 cells on transwell inserts, which were then inserted in 24‐well plates for 7 days once the endothelial monolayer had formed. To confirm the integrity of the hCMEC/D3 monolayer, we first applied 488‐Na‐F to the apical side of the transcytosis system. The density of fluorescence in the medium from the basolateral side was determined after 5‐min incubation. We detected an obvious fluorescent signal in the no‐cell group, which was set as a negative control. Compared with the no‐cell group, an 81% reduction in the fluorescent signal was detected in the groups with an hCMEC/D3 monolayer, thus demonstrating the feasibility of the hCMEC/D3 monolayer in the transcytosis system (Figure 4a and b). The transportation of Aβ1‐42 by the hCMEC/D3 monolayer was then analyzed (Figure 4c). Compared to the uninfected cells, the Aβ1‐42 fluorescent signal was significantly increased in the medium at the basolateral side of hCMEC/D3 cells after P. gingivalis infection. Pre‐treatment with the RAGE‐specific inhibitor FPS‐ZM1 significantly inhibited the fluorescent Aβ1‐42 (51%) transportation from the apical side to the basolateral side in the P. gingivalis‐infected hCMEC/D3 cells at 12 hr after P. gingivalis infection (Figure 4d). It was noted that neither P. gingivalis nor FPS‐ZM1 affected the integrity of the hCMEC/D3 monolayer, as no significant increase in 488‐Na‐F was detected in the basolateral medium (Figure 4b). These observations suggest that Aβ1‐42 transportation via hCMEC/D3 cells was induced by P. gingivalis infection, and the increased expression of RAGE was responsible for Aβ1‐42 transportation during P. gingivalis infection.
FIGURE 4.
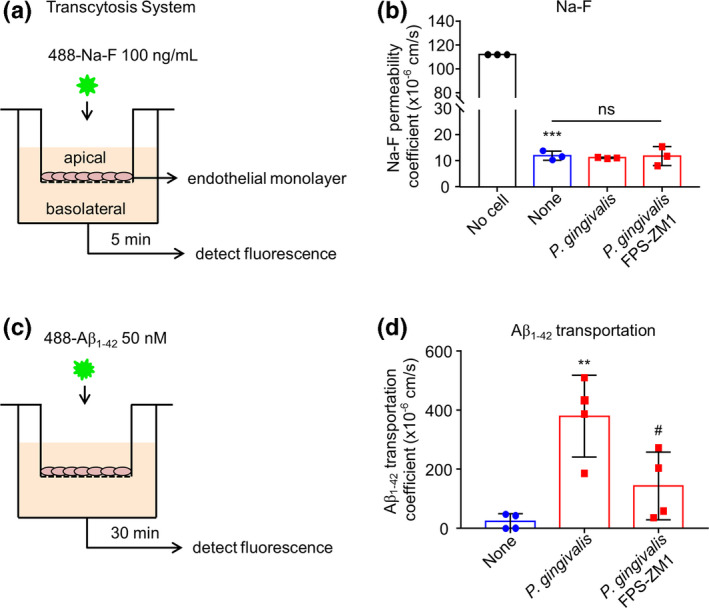
RAGE expression mediated Aβ1‐42 transportation after P. gingivalis infection in a transcytosis system. (a) The schematic of the in vitro transcytosis system for Na‐F transportation. (b) The quantitative analysis of Na‐F (100 ng/mL) permeability after P. gingivalis infection for 12 hr in the presence or absence of FPS‐ZM1 pre‐treatment. The “No cell” means no cell in the transcytosis system (n = 3 independent cell culture preparations, ns: no significant difference, ***p < .001; one‐way ANOVA). Asterisks indicate a statistically significant difference versus “No cell” group and “ns” indicates the result compared with “None” group. (c) The schematic of the in vitro transcytosis system for Aβ1‐42 transportation. (d) The transportation of Aβ1‐42 from apical to basolateral compartment after P. gingivalis infection for 12 hr. (n = 4 independent cell culture preparations, **p < .01, # p < .05, one‐way ANOVA). Asterisks indicate a statistically significant difference compared with “None” group and pounds indicate a statistically significant difference compared with “P. gingivalis” group
3.5. Increased RAGE expression in cerebral endothelial cells and the induction of Aβ1‐42 around cerebral endothelial cells in mice after systemic P. gingivalis infection
To determine the RAGE expression is increased in the cerebral endothelial cell which is related to Aβ influx into brain in vivo, we finally examined the RAGE expression on cerebral endothelial cells in mouse brains after systemic P. gingivalis infection. The timing of systemic P. gingivalis infection is shown as an illustration (Figure 5a). The expression level of CatB and RAGE, as well as the localization of p65 and Aβ1‐42 in cerebral endothelial cells were determined by immunofluorescent staining. Compared to the saline‐treated mice, the CatB expression in CD31‐positive cerebral endothelial cells was significantly increased (Figure 5b and d). The co‐localization between p65 and Hoechst was significantly increased, which means more NF‐κB activation in cerebral endothelial cells after systemic P. gingivalis infection (Figure 5c and e). Additionally, RAGE expression in CD31‐positive cerebral endothelial cells was significantly increased (1.8‐fold), and the co‐localization between RAGE and CD31 was significantly increased (2.7‐fold) in the mouse brain after 3 weeks of systemic P. gingivalis infection (Figure 6a, c and d). Furthermore, compared to the scarce Aβ1‐42 signal in the brain of saline‐treated mice, the Aβ1‐42 signal in the brain parenchyma localized around the CD31‐positive cerebral endothelial cells was significantly increased (6.8‐fold) in mice after systemic P. gingivalis infection (Figure 6b and e). In addition, the expression of RAGE on cerebral endothelial cells was positively correlated with that of Aβ1‐42 in the systemic P. gingivalis infected mice (r = 0.6205, Figure 6f). Furthermore, significant memory decline was found at week 3 of the mice with P. gingivalis infection in comparison to that in saline‐treated mice (Figure 6g). These observations showed that the inducted Aβ1‐42 in the brain parenchyma was close to the significantly increased level of RAGE‐brain endothelial cells that were expressed in middle‐aged mice after chronic systemic P. gingivalis infection.
FIGURE 5.
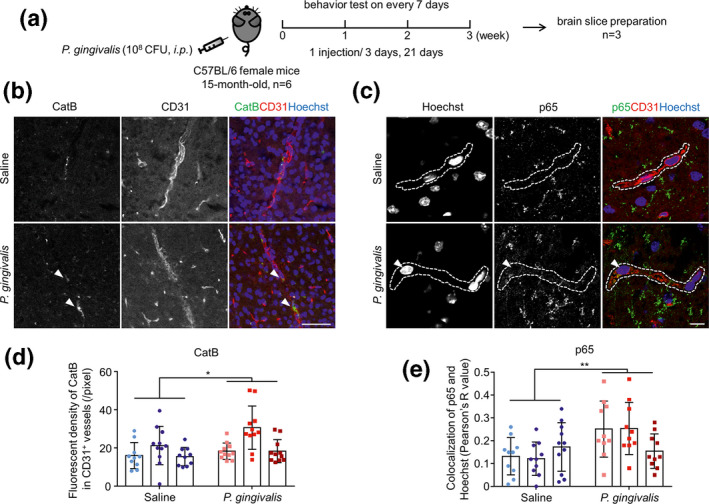
Increased CatB and translocation of p65 in cerebral endothelial cells of mice after systemic P. gingivalis infection. (a) Time schedule of systemic P. gingivalis infection and behavior test. (b) The immunofluorescent CLMS images of brain slice stained with CatB (green), CD31 (red), and Hoechst (blue) after systemic P. gingivalis infection. (c) The immunofluorescent CLMS images of brain slice stained with p65 (green), CD31 (red), and Hoechst (blue) after systemic P. gingivalis infection. Dash lines marked the location of blood vessels. (d) The quantitative analysis of CatB fluorescence density in CD31+ vessels in the images in (b). (e) Co‐localization analysis of p65 and Hoechst in the images in (c) using ImageJ (COLOC 2 plugin of Fiji is just ImageJ). Three mice per group were used and each mouse has its own color. Asterisks indicate a statistically significant difference versus “Saline” group using multiple t‐test. *p < .05, **p < .01. Scale bars in b: 50 μm; c: 10 μm
FIGURE 6.
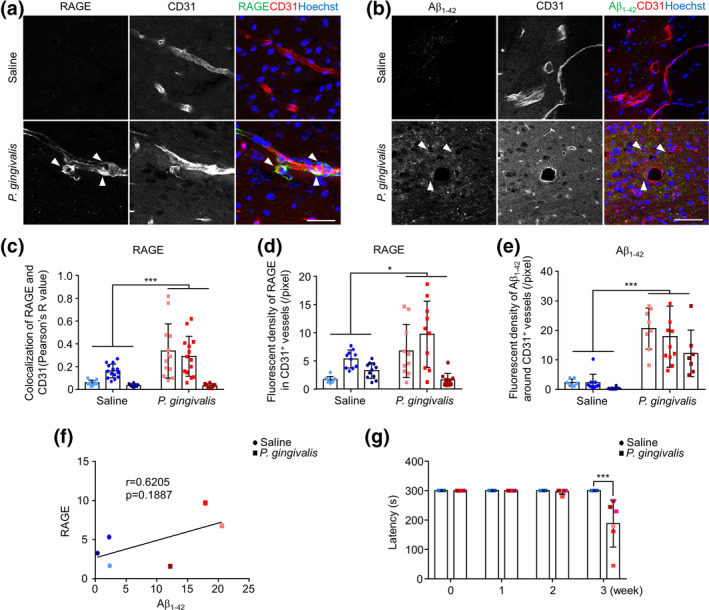
Increased RAGE expression in cerebral endothelial cells and the induction of Aβ1‐42 around cerebral endothelial cells of mice after systemic P. gingivalis infection. (a) The immunofluorescent CLMS images of brain slice stained with RAGE (green), CD31 (red), and Hoechst (blue) after systemic P. gingivalis infection. (b) The immunofluorescent CLMS images of brain slice stained with Aβ1‐42 (green), CD31 (red), and Hoechst (blue) after systemic P. gingivalis infection. (c) Co‐localization analysis of RAGE and CD31 in the images in (a) using COLOC 2 plugin. (d)The quantitative analysis of RAGE fluorescence density in CD31+ vessels in the images in (a). (e) The quantitative analysis of Aβ1‐42 fluorescence density around CD31+ vessels in the images in (b). (f) Pearson's correlation between the fluorescence density of RAGE and Aβ1‐42. (a–f) Three mice per group were used and each mouse has its own color. (g) Memory decline in mice after 3 weeks of systemic P. gingivalis infection (n = 6 mice/group). Asterisks indicate a statistically significant difference versus “Saline” group using multiple t‐test. *p < .05, **p < .01, ***p < .001. Scale bar, 50 μm
4. DISCUSSION
The major finding of this study was our determination that the CatB/NF‐κB‐dependent RAGE expression mediates the Aβ influx after P. gingivalis infection using an in vitro model of the BBB and middle‐aged mice (Summarized in Figure 7). We thus demonstrated a new mechanism underlying the influence of systemic periodontal bacterial infection on the initiation and pathological progression of AD.
FIGURE 7.
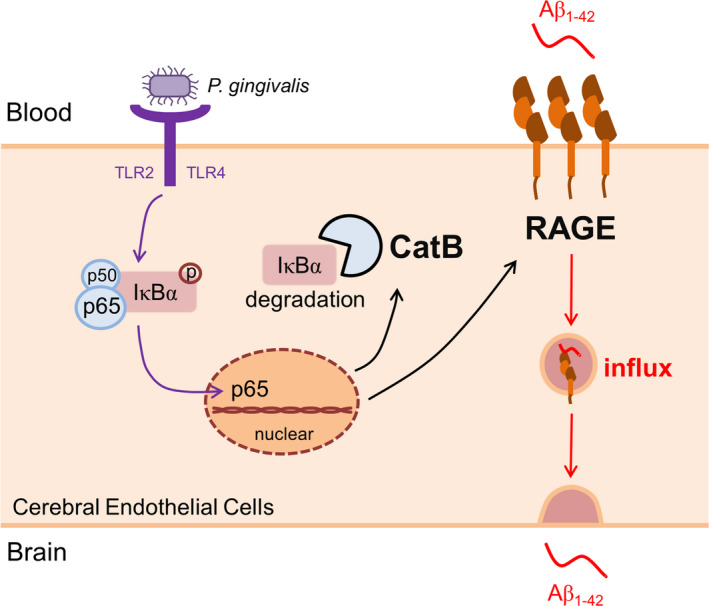
The schematic of the critical role of RAGE in cerebral endothelial cell after P. gingivalis infection. P. gingivalis activates NF‐κB pathway through binding TLR2/4, triggers transcription of RAGE and CatB. The increased CatB further involves in RAGE up‐regulation via regulating NF‐κB activation. At last, elevated RAGE mediates the influx of Aβ from blood to the brain
Structurally as a transmembrane receptor, RAGE is expressed in multiple types of cells, including cerebral endothelial cells (Derk, MacLean, Juranek, & Schmidt, 2018). P. gingivalis increased the RAGE expression in hCMEC/D3 cells, which is consistent with the previous study that brain endothelial RAGE expression was increased in AD mouse models and AD patients (Miller et al., 2008). P. gingivalis has been reported to activate TLR2/4/MyD88/NF‐κB pathway in macrophage (Papadopoulos et al., 2013). In this study, we found that the mRNA level of both TLR2 and TLR4 were increased at early and late phase, respectively, suggesting that TLR2 and TLR4 cooperatively responded to the P. gingivalis in hCMEC/D3 cells. It is commonly accepted that TLR2 and TLR4 could lead to a signaling cascade that ultimately resulted in the activation of NF‐κB. In this study, we also detected the activation of NF‐κB in hCMEC/D3 cells after P. gingivalis treatment. However, the activation of NF‐κB showed two peaks revealing by the nuclear translocation of p65 at 1 and 3 hr after P. gingivalis treatment. To further study the activation status of NF‐κB, we separately examined the IκBα phosphorylation/proteasome degradation system and IκBα/CatB degradation system. We found P. gingivalis induced IκBα phosphorylation/proteasome‐dependent activation of NF‐κB at early phase, while induced CatB‐dependent activation of NF‐κB at late phase in hCMEC/D3 cells. RAGE promoter possesses three NF‐κB‐binding sites, the increased RAGE mRNA may be because of the two times activation of NF‐κB (Li, 1997). It was noted that P. gingivalis induced NF‐κB activation and CatB production may be synergized because CatB promoter also possessed NF‐κB‐binding sites (Bien et al., 2004).
The RAGE expression in cerebral endothelial cells plays an important role in facilitating the influx of circulating Aβ into the brain across the BBB (Deane et al., 2003; Schmidt, Yan, Yan, & Stern, 2001). In this study, an increase in the Aβ1‐42 transportation across the P. gingivalis‐infected hCMEC/D3 cells compared to the uninfected cells was noted, strongly suggesting that the Aβ1‐42 influx was dramatically up‐regulated after P. gingivalis infection (Figure 4). In our in vitro transcytosis system, the Na‐F permeability across the monolayer of hCMEC/D3 cells were not reduced by P. gingivalis infection at MOI = 5 for 12 hr, confirming that the P. gingivalis infection‐induced Aβ1‐42 influx via hCMEC/D3 cells was mediated by the “transport barrier” without disrupting the “physical barrier” (cell monolayer) (Poggesi, Pasi, Pescini, Pantoni, & Inzitari, 2016). The P. gingivalis infection‐induced Aβ1‐42 influx was markedly reduced by pre‐treatment with FPS‐ZM1, a specific inhibitor of RAGE, providing the first evidence that the RAGE expression in cerebral endothelial cells mediates the Aβ influx after P. gingivalis infection. Of note, the P. gingivalis infection‐induced mild increase in the RAGE expression in the cerebral endothelial cells was related to a dramatic Aβ influx in the present in vitro Aβ transportation system, strongly indicating that the increased RAGE expression in the cerebral endothelial cells efficiently affected the Aβ influx during P. gingivalis infection.
The present in vitro observations were further supported by the parallel increase in the RAGE expression in cerebral endothelial cells and the Aβ loads in the brain parenchyma around the cerebral vasculature in middle‐aged mice with a significant memory decline after 3 weeks of chronic systemic P. gingivalis infection. The increased expression level of CatB in cerebral endothelial cells was also found in P. gingivalis infected mice. To our knowledge, this is the first evidence of cerebrovascular‐related amyloid genesis induced by systemic P. gingivalis infection, highlighting a new mechanism underlying the effect of periodontitis on AD pathologies, as cerebrovascular‐related amyloidogenesis has been found in over 80% of AD patients (Attems & Jellinger, 2014; Jellinger & Attems, 2005) and this condition positively correlates with a cognitive decline (Thal, Griffin, de Vos, & Ghebremedhin, 2008; Weller, Boche, & Nicoll, 2009). The finding that systemic P. gingivalis infection induced the cerebral RAGE expression/cerebrovascular amyloid genesis in middle‐aged mice is significant because around 70% of periodontitis morbidity has been found in middle‐aged individuals (Eke et al., 2012, 2015) and the RAGE up‐regulation with continues up until middle age (Osgood, Miller, Messier, Gonzalez, & Silverberg, 2017). In combination with our recent findings that the peripheral pools of Aβ are increased during P. gingivalis infection (Nie et al., 2019), the synergic interplay between the cerebral RAGE expression and peripheral Aβ generation contributes to the cerebral amyloidogenesis during chronic P. gingivalis infection, as the circulating Aβ can be transferred into the brain (Deane & Zlokovic, 2007), thereby increasing the Aβ levels in the brains of AD patients (Donahue et al., 2006). Given the contribution of RAGE up‐regulation and Aβ/RAGE interaction to AD pathogenesis (Donahue et al., 2006; Jeynes & Provias, 2008; Kook et al., 2012), an elevated cerebral RAGE expression may be a new therapeutic strategy for AD (Zlokovic, 2008). We are currently investigating the influence of P. gingivalis/LPS on other outcomes of the cerebral microenvironment in a separate study.
Periodontal disease positive association with AD was thought to be induced by systemic inflammation (Gaur & Agnihotri, 2015). Chronic periodontitis induced increased pro‐inflammatory cytokines in periphery tissues and the circulating system decreased BBB integrity, and activated microglia. Therefore, chronic periodontitis and infection with P. gingivalis have been identified as a significant risk factor for developing Aβ plaque, dementia, and AD (Dominy et al., 2019). Beside of this indirect relationship between periodontal disease and AD, we found P. gingivalis induced increased Aβ influx directly through RAGE in brain endothelial cells. Therefore, it will be interesting to study the positive association between periodontitis, RAGE expression in brain endothelial cell, and brain Aβ accumulation in patients with AD.
In this study, the P. gingivalis‐induced IκBα degradation was partially reduced by the CatB‐specific inhibitor in hCMEC/D3 cells, indicating that CatB participates in chronic NF‐κB activation in cerebral endothelial cells after P. gingivalis infection. The present finding was supported by the previous report that CatB is involved in NF‐κB activation by lysosome degradation in microglia, the primary immune cells in the brain (Ni et al., 2015). This, along with our previous findings of the involvements of CatB in Aβ generation in neurons of the brain as well as in peripheral inflammatory monocytes/macrophages after P. gingivalis infection/exposure to P. gingivalis LPS (Nie et al., 2019; Wu et al., 2017), suggests that CatB may involve in the pathogenesis of AD for prolonging NF‐κB activation with its enzymatic functions during P. gingivalis infection. Of note, increased CatB in the hCMEC/D3 cells (human cerebral endothelial cells) was induced by P. gingivalis infection at MOI = 5, while the expression of CatB in human umbilical vein endothelial cells (endothelial cells of peripheral blood vessels) had no change after P. gingivalis infection at MOI = 200 (Huck, Elkaim, Davideau, & Tenenbaum, 2012), suggesting different endothelial cells have different responses to P. gingivalis infection. The sensitive response to microbial infection of cerebral endothelial cells may contribute to the CatB/NF‐κB/RAGE‐dependent Aβ influx.
In conclusion, this study demonstrated that the CatB/NF‐κB activation‐dependent RAGE up‐regulation in cerebral endothelial cells mediates cerebrovascular‐related amyloidogenesis during P. gingivalis infection, highlighting a new mechanism for the involvement of periodontitis in AD and pathological process.
CONFLICT OF INTEREST
The authors F. Zeng, J. Ni, Y. Liu, W. Huang, H. Qing, T. Kadowaki, H. Kashiwazaki, and Z. Wu declare that they have no conflict of interest.
OPEN RESEARCH BADGES
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. More information about the Open Science badges can be found at https://cos.io/our‐services/open‐science‐badges/.
Supporting information
ACKNOWLEDGMENTS
This work is supported by the Japanese KAKENHI Grant Number 16K11478 (Grants‐in‐Aid for Scientific Research to Z.W.), Grant Number JP17K17093 (a Grant‐in‐Aid for Young Scientists to J.N.), and the research grant for OBT research center from the Kyushu University (to Z.W.).
All experiments were conducted in compliance with the ARRIVE guidelines.
Zeng F, Liu Y, Huang W, et al. Receptor for advanced glycation end products up‐regulation in cerebral endothelial cells mediates cerebrovascular‐related amyloid β accumulation after Porphyromonas gingivalis infection. J. Neurochem..2021;158:724–736. 10.1111/jnc.15096
Cover Image for this issue: https://doi.org/10.1111/jnc.15073
REFERENCES
- Abbott, N. J., Ronnback, L., & Hansson, E. (2006). Astrocyte‐endothelial interactions at the blood‐brain barrier. Nature Reviews Neuroscience, 7, 41–53. 10.1038/nrn1824 [DOI] [PubMed] [Google Scholar]
- Attems, J., & Jellinger, K. A. (2014). The overlap between vascular disease and Alzheimer’s disease‐lessons from pathology. BMC Medicine, 12, 206. 10.1186/s12916-014-0206-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielecka, E., Scavenius, C., Kantyka, T., Jusko, M., Mizgalska, D., Szmigielski, B., … Potempa, J. (2014). Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. Journal of Biological Chemistry, 289, 32481–32487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien, S., Ritter, C. A., Gratz, M., Sperker, B., Sonnemann, J., Beck, J. F., & Kroemer, H. K. (2004). Nuclear factor‐kappaB mediates up‐regulation of cathepsin B by doxorubicin in tumor cells. Molecular Pharmacology, 65, 1092–1102. [DOI] [PubMed] [Google Scholar]
- Bu, X. L., Xiang, Y., Jin, W. S., Wang, J., Shen, L.‐L., Huang, Z.‐L., … Wang, Y.‐J. (2018). Blood‐derived amyloid‐beta protein induces Alzheimer's disease pathologies. Molecular Psychiatry, 23, 1948–1956. [DOI] [PubMed] [Google Scholar]
- Charan, J., & Kantharia, N. D. (2013). How to calculate sample size in animal studies? Journal of Pharmacology and Pharmacotherapeutics, 4, 303–306. 10.4103/0976-500X.119726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. D., Zeldich, E., Khodr, C., Camara, K., Tung, T. Y., Lauder, E. C., … Abraham, C. R. (2019). Small molecule amyloid‐beta protein precursor processing modulators lower amyloid‐beta peptide levels via cKit signaling. Journal of Alzheimer's Disease, 67, 1089–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane, R., Du Yan, S., Submamaryan, R. K., LaRue, B., Jovanovic, S., Hogg, E., … Zlokovic, B. (2003). RAGE mediates amyloid β peptide transport across the blood‐brain barrier and accumulation in brain. Nature Medicine, 9, 907. 10.1038/nm890 [DOI] [PubMed] [Google Scholar]
- Deane, R., & Zlokovic, B. V. (2007). Role of the blood‐brain barrier in the pathogenesis of Alzheimer's disease. Current Alzheimer Research, 4, 191–197. [DOI] [PubMed] [Google Scholar]
- Derk, J., MacLean, M., Juranek, J., & Schmidt, A. M. (2018). The receptor for advanced glycation endproducts (RAGE) and mediation of inflammatory neurodegeneration. Journal of Alzheimer’s Disease & Parkinsonism, 8(1), 421. 10.4172/2161-0460.1000421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco, A., Gonzalez Paz, O., Fini, I., Vignone, D., Cellucci, A., Battista, M. R., … Muñoz‐Sanjuán, I. (2019). Application of an in vitro blood‐brain barrier model in the selection of experimental drug candidates for the treatment of huntington's disease. Molecular Pharmaceutics, 16, 2069–2082. 10.1021/acs.molpharmaceut.9b00042 [DOI] [PubMed] [Google Scholar]
- Dominy, S. S., Lynch, C., Ermini, F., Malgorzata, B., Marczyk, A., Konradi, A., … Potempa, J. (2019). Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small‐molecule inhibitors. Science Advances, 5, eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue, J. E., Flaherty, S. L., Johanson, C. E., Duncan, J. A., Silverberg, G. D., Miller, M. C., … Stopa, E. G. (2006). RAGE, LRP‐1, and amyloid‐beta protein in Alzheimer's disease. Acta Neuropathologica, 112, 405–415. 10.1007/s00401-006-0115-3 [DOI] [PubMed] [Google Scholar]
- Eke, P. I., Dye, B. A., Wei, L., Slade, G. D., Thornton‐Evans, G. O., Borgnakke, W. S., … Genco, R. J. (2015). Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. Journal of Periodontology, 86, 611–622. 10.1902/jop.2015.140520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eke, P. I., Dye, B. A., Wei, L., Thornton‐Evans, G. O., Genco, R. J., Surveillance, C. P. D., & workgroup: James Beck, G. D. R. P. , (2012). Prevalence of periodontitis in adults in the United States: 2009 and 2010. Journal of Dental Research, 91, 914–920. 10.1177/0022034512457373 [DOI] [PubMed] [Google Scholar]
- Evin, G., Zhu, A., Holsinger, R. M. D., Masters, C. L., & Li, Q.‐X. (2003). Proteolytic processing of the Alzheimer's disease amyloid precursor protein in brain and platelets. Journal of Neuroscience Research, 74, 386–392. 10.1002/jnr.10745 [DOI] [PubMed] [Google Scholar]
- Gaur, S., & Agnihotri, R. (2015). Alzheimer's disease and chronic periodontitis: Is there an association? Geriatr Gerontol Int, 15, 391–404. 10.1111/ggi.12425 [DOI] [PubMed] [Google Scholar]
- Hodson, R. (2018). Alzheimer's disease. Nature, 559, S1. 10.1038/d41586-018-05717-6 [DOI] [PubMed] [Google Scholar]
- Huck, O., Elkaim, R., Davideau, J. L., & Tenenbaum, H. (2012). Porphyromonas gingivalis and its lipopolysaccharide differentially regulate the expression of cathepsin B in endothelial cells. Mol. Oral Microbiol., 27, 137–148. 10.1111/j.2041-1014.2012.00638.x [DOI] [PubMed] [Google Scholar]
- Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron, 80, 844–866. 10.1016/j.neuron.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide, M., Harris, M., Stevens, A., Sussams, R., Hopkins, V., Culliford, D., … Holmes, C. (2016). Periodontitis and cognitive decline in Alzheimer's disease. PLoS One, 11, e0151081. 10.1371/journal.pone.0151081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilievski, V., Zuchowska, P. K., Green, S. J., Toth, P. T., Ragozzino, M. E., Le, K., … Watanabe, K. (2018). Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One, 13, e0204941. 10.1371/journal.pone.0204941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger, K. A., & Attems, J. (2005). Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease. Journal of the Neurological Sciences, 229–230, 37–41. 10.1016/j.jns.2004.11.018 [DOI] [PubMed] [Google Scholar]
- Jeynes, B., & Provias, J. (2008). Evidence for altered LRP/RAGE expression in Alzheimer lesion pathogenesis. Current Alzheimer Research, 5, 432–437. [DOI] [PubMed] [Google Scholar]
- Kook, S. Y., Hong, H. S., Moon, M., Ha, C. M., Chang, S., & Mook‐Jung, I. (2012). Abeta(1)(‐)(4)(2)‐RAGE interaction disrupts tight junctions of the blood‐brain barrier via Ca(2)(+)‐calcineurin signaling. Journal of Neuroscience, 32, 8845–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovari, E., Herrmann, F. R., Hof, P. R., & Bouras, C. (2013). The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer's disease. Neuropathology and Applied Neurobiology, 39, 498–509. 10.1111/nan.12003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, Y. M., Kokjohn, T. A., Watson, M. D., Woods, A. S., Cotter, R. J., Sue, L. I., … Roher, A. E. (2000). Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. American Journal of Pathology, 156, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J., & Schmidt, A. M. (1997). Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. Journal of Biological Chemistry, 272, 16498–16506. 10.1074/jbc.272.26.16498 [DOI] [PubMed] [Google Scholar]
- Liu, Y., Wu, Z., Nakanishi, Y., Ni, J., Hayashi, Y., Takayama, F., … Nakanishi, H. (2017). Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain‐mediated activation of protease‐activated receptor‐2 in mice. Scientific Reports, 7, 11759. 10.1038/s41598-017-12173-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, C., Zhang, Y.‐L., Luo, W., Zhou, F. H., Li, C.‐Q., Xu, J.‐M., & Dai, R.‐P. (2015). Differential effects of general anesthetics on anxiety‐like behavior in formalin‐induced pain: Involvement of ERK activation in the anterior cingulate cortex. Psychopharmacology (Berl), 232, 4433–4444. 10.1007/s00213-015-4071-2 [DOI] [PubMed] [Google Scholar]
- Miller, M. C., Tavares, R., Johanson, C. E., Hovanesian, V., Donahue, J. E., Gonzalez, L., … Stopa, E. G. (2008). Hippocampal RAGE immunoreactivity in early and advanced Alzheimer's disease. Brain Research, 1230, 273–280. 10.1016/j.brainres.2008.06.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, J., Wu, Z., Peterts, C., Yamamoto, K., Qing, H., & Nakanishi, H. (2015). The critical role of Proteolytic relay through cathepsins B and E in the phenotypic change of microglia/macrophage. Journal of Neuroscience, 35, 12488–12501. 10.1523/JNEUROSCI.1599-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie, R., Wu, Z., Ni, J., Zeng, F., Yu, W., Zhang, Y., & Zhou, Y. (2019). Porphyromonas gingivalis infection induces amyloid‐beta accumulation in monocytes/macrophages. Journal of Alzheimer's Disease, 72, 479–494. [DOI] [PubMed] [Google Scholar]
- Noble, J. M., Borrell, L. N., Papapanou, P. N., Elkind, M. S. V., Scarmeas, N., & Wright, C. B. (2009). Periodontitis is associated with cognitive impairment among older adults: Analysis of NHANES‐III. Journal of Neurology, Neurosurgery and Psychiatry, 80, 1206–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osgood, D., Miller, M. C., Messier, A. A., Gonzalez, L., & Silverberg, G. D. (2017). Aging alters mRNA expression of amyloid transporter genes at the blood‐brain barrier. Neurobiology of Aging, 57, 178–185. 10.1016/j.neurobiolaging.2017.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, G., Shaik‐Dasthagirisaheb, Y. B., Huang, N., Viglianti, G. A., Henderson, A. J., Kantarci, A., & Gibson, F. C.III (2017). Immunologic environment influences macrophage response to Porphyromonas gingivalis. Molecular Oral Microbiology, 32, 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, G., Weinberg, E. O., Massari, P., Gibson, F. C., Wetzler, L. M., Morgan, E. F., & Genco, C. A. (2013). Macrophage‐specific TLR2 signaling mediates pathogen‐induced TNF‐dependent inflammatory oral bone loss. The Journal of Immunology, 190, 1148–1157. 10.4049/jimmunol.1202511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggesi, A., Pasi, M., Pescini, F., Pantoni, L., & Inzitari, D. (2016). Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: A review. Journal of Cerebral Blood Flow and Metabolism, 36, 72–94. 10.1038/jcbfm.2015.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole, S., Singhrao, S. K., Kesavalu, L., Curtis, M. A., & Crean, S. (2013). Determining the presence of periodontopathic virulence factors in short‐term postmortem Alzheimer's disease brain tissue. Journal of Alzheimer's Disease, 36, 665–677. 10.3233/JAD-121918 [DOI] [PubMed] [Google Scholar]
- Reife, R. A., Coats, S. R., Al‐Qutub, M., Dixon, D. M., Braham, P. A., Billharz, R. J., … Darveau, R. P. (2006). Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: Differential activities of tetra‐ and penta‐acylated lipid A structures on E‐selectin expression and TLR4 recognition. Cellular Microbiology, 8, 857–868. 10.1111/j.1462-5822.2005.00672.x [DOI] [PubMed] [Google Scholar]
- Roher, A. E., & Kokjohn, T. A. (2009). Commentary on "Alzheimer's disease drug development and the problem of the blood‐brain barrier." Alzheimer's disease drugs: More than one barrier to breach. Alzheimer's & Dementia: the Journal of the Alzheimer's Association, 5, 437–438. [DOI] [PubMed] [Google Scholar]
- Sagare, A. P., Deane, R., & Zlokovic, B. V. (2012). Low‐density lipoprotein receptor‐related protein 1: A physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharmacology & Therapeutics 136, 94–105. 10.1016/j.pharmthera.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, A. M., Yan, S. D., Yan, S. F., & Stern, D. M. (2001). The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. Journal of Clinical Investigation, 108, 949–955. 10.1172/JCI200114002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaney, J. M., Gallagher, A., Aduse‐Opoku, J., Pell, K., & Curtis, M. A. (2006). Mechanisms of resistance of Porphyromonas gingivalis to killing by serum complement. Infection and Immunity, 74, 5352–5361. 10.1128/IAI.00304-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks Stein, P., Steffen, M. J., Smith, C., Jicha, G., Ebersole, J. L., Abner, E., & Dawson, D. (2012). Serum antibodies to periodontal pathogens are a risk factor for Alzheimer's disease. Alzheimer's & Dementia: the Journal of the Alzheimer's Association, 8, 196–203. 10.1016/j.jalz.2011.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal, D. R., Griffin, W. S., de Vos, R. A., & Ghebremedhin, E. (2008). Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathologica, 115, 599–609. 10.1007/s00401-008-0366-2 [DOI] [PubMed] [Google Scholar]
- Weller, R. O., Boche, D., & Nicoll, J. A. (2009). Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathologica, 118, 87–102. 10.1007/s00401-009-0498-z [DOI] [PubMed] [Google Scholar]
- Wu, Z., Ni, J., Liu, Y., Teeling, J. L., Takayama, F., Collcutt, A., … Nakanishi, H. (2017). Cathepsin B plays a critical role in inducing Alzheimer's disease‐like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain, Behavior, and Immunity, 65, 350–361. 10.1016/j.bbi.2017.06.002 [DOI] [PubMed] [Google Scholar]
- Yan, S. D., Chen, X. I., Fu, J., Chen, M., Zhu, H., Roher, A., … Schmidt, A. M. (1996). RAGE and amyloid β peptide neurotoxicity in Alzheimer's disease. Nature, 382, 685. 10.1038/382685a0 [DOI] [PubMed] [Google Scholar]
- Zlokovic, B. V. (2008). New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics: the Journal of the American Society for Experimental NeuroTherapeutics, 5, 409–414. 10.1016/j.nurt.2008.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials