

Abstract
Background
Augmentation of natriuretic peptide receptor and cyclic GMP (cGMP) signaling has emerged as a therapeutic strategy in heart failure (HF). Cyclic GMP-specific phosphodiesterase 9 (PDE9) inhibition increases cGMP signaling and attenuates stress-induced hypertrophic heart disease in preclinical studies. A novel cGMP-specific PDE9 inhibitor, CRD-733, is currently being advanced in human clinical studies. Here we explore the effects of chronic PDE9 inhibition with CRD-733 in the mouse transverse aortic constriction (TAC) pressure overload HF model.
Methods
Adult male C57BL/6J mice were subjected to TAC and developed significant left ventricular (LV) hypertrophy after 7 days (P<0.001). Mice then received daily treatment with CRD-733 (600mg/kg/day; n=10) or vehicle (n=17), alongside sham-operated controls (n=10).
Results
CRD-733 treatment reversed existing LV hypertrophy compared to vehicle (P<0.001), significantly improved LV ejection fraction (P=0.009) and attenuated left atrial dilation (P<0.001), as assessed by serial echocardiography. CRD-733 prevented elevations in LV end diastolic pressures (P=0.037) compared to vehicle, whilst lung weights, a surrogate for pulmonary edema, were reduced to sham levels. Chronic CRD-733 treatment increased plasma cGMP levels compared to vehicle (P<0.001), alongside increased phosphorylation of Ser273 of cardiac myosin binding protein-C, a cGMP-dependent protein kinase I phosphorylation site.
Conclusions
The PDE9 inhibitor, CRD-733, improves key hallmarks of HF including LV hypertrophy, LV dysfunction, left atrial dilation and pulmonary edema after pressure overload in the mouse TAC HF model. Additionally, elevated plasma cGMP may be used as a biomarker of target engagement. These findings support future investigation into the therapeutic potential of CRD-733 in human HF.
Subject Terms: hypertrophy, remodeling, contractile function, echocardiography, heart failure, hemodynamics
Introduction
Heart failure (HF) remains a leading cause of death and morbidity worldwide. For HF with reduced left ventricular (LV) ejection fraction (HFrEF), multiple medical therapies exist, but the prognosis continues to be poor, and the residual risk on current therapies remains substantial, highlighting the need for novel drug targets and therapies.
With the approval of angiotensin receptor blocker-neprilysin inhibitor therapy for HFrEF, augmentation of intracellular cGMP has emerged as a first line therapeutic strategy for this condition. The neprilysin inhibitor component of this approach enhances natriuretic peptide receptor (NPR) pathway signaling by inhibiting natriuretic peptide (NP) degradation, augmenting downstream signaling by the NPR pathway. There are two cyclic GMP (cGMP)-generating enzymes in cardiovascular tissues, the membrane bound NPR guanylate cyclases activated by NPs, and the cytoplasmic soluble guanylate cyclase activated by nitric oxide. Cyclic GMP-augmenting agents with proven efficacy in clinical studies of chronic HFrEF include the nitric oxide donor isosorbide dinitrate (combined with hydralazine), the soluble guanylate cyclase stimulator vericiguat,1 and the neprilysin inhibitor sacubitril (combined with valsartan), which blocks neprilysin cleavage of NP. Cyclic GMP directly activates the cGMP-dependent protein kinase (PKG), and multiple genetic and pharmacological preclinical studies support that the cGMP-PKG signaling pathway opposes pathological cardiac hypertrophy, remodeling, and dysfunction.2-4 Additional strategies to augment NPR and cGMP signaling that might be of benefit in HFrEF remain of interest.
Inhibition of phosphodiesterases (PDE), which catabolize cyclic nucleotides, represents an additional strategy to promote cyclic nucleotide signaling. Sildenafil, a PDE5 selective inhibitor, proved highly promising in preclinical studies of HF,5 but failed to provide a clinical benefit in HF with preserved ejection fraction.6 In HFrEF, early phase studies support that PDE5 inhibition improves hemodynamic and echocardiographic indices,7-9 but to date, no large scale clinical trials of PDE5 inhibition in HFrEF have been performed. Cardiac expression and activity of PDE9 increases in myocardial tissue from patients with HF and in preclinical models of myocardial hypertrophy, and has been identified recently as the predominant cGMP-phosphodiesterase that regulates cGMP generated by the NPR pathway.10-12 Genetic deletion of PDE9 in mice attenuated cardiac hypertrophy, dysfunction and fibrosis,10 identifying inhibition of PDE9 as a potential therapeutically viable approach. Indeed, recent studies have demonstrated therapeutic effects on LV hypertrophy and fractional shortening for small molecule inhibition of PDE9 in pressure overload and isoproterenol-induced HF models.10-12
PDE9 inhibitors have been administered safely to patients with non-HF conditions, including benign prostatic hypertrophy, dementias,13 and sickle cell disease,14 and to date appear well tolerated. The PDE9 inhibitor, CRD-733 (Cardurion Pharmaceuticals), has been investigated previously in clinical studies of benign prostatic hypertrophy (formerly ASP4901; NCT02038868). However, the effects of CRD-733 on the LV response to pressure overload and heart failure in preclinical models have not been studied, and the effects of PDE9 inhibition on comprehensive HF indices in vivo such as LV filling pressures and atrial remodeling remain incompletely studied in HFrEF. In the current study, we tested the hypothesis that PDE9 inhibition with CRD-733 can reverse established LV hypertrophy and dysfunction, and improve hemodynamic indices of HF in a mouse transverse aortic constriction (TAC) heart failure model.
Methods
The investigation conforms to the guide for the care and use of laboratory animals published by the US national institutes of health (NIH Publication No. 85-23, revised 1985), as approved by the Institutional Animal Care and Use Committee of Tufts University School of Medicine and Tufts Medical Center.
Data Requests
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Transverse Aortic Constriction
TAC was performed as previously described.15 Briefly, male C57BL/6J mice (8 weeks old; 24g±3g; Jackson Labs, ME) were anesthetized with 2.5% isoflurane and body temperature maintained at 37°C on a heat pad. Mice were administered 1 mg/kg Buprenorphine SR-LAB (ZooPharm, USA) by subcutaneous injection for prophylactic pain relief. The transverse aorta was then constricted to the diameter of a 27G needle between the brachiocephalic and left common carotid arteries using 7-0 nylon suture, as previously described.15 Sham-operated mice underwent a similar procedure, without constriction.
Echocardiography
Comprehensive echocardiography was performed as previously described using a Vevo 2100 (Fujifilm VisualSonics).15 Mice underwent echocardiography prior to TAC (baseline), seven days after TAC (day 7) and prior to sacrifice (day 21). All data was analyzed using VevoLab 3.1 (Fujifilm VisualSonics).
In vivo drug administration
CRD-733 was dissolved in 50% saline/50% PEG-400 for a stock concentration of 60 mg/mL. At day 7, mice were randomized to daily administration of CRD-733 for 14 days (day 7 to day 21) at 10μL/g body weight by oral gavage (p.o.) for a final dose of 600mg/kg/day, or vehicle control (50% saline/50% PEG-400). Sham mice were administered vehicle as a control.
Statistical Analysis
All data is presented as mean ± standard error. Statistical tests used in analyses are stated in each figure legend alongside replicate (n) numbers. All statistical tests were performed using GraphPad Prism v8.3.0 (La Jolla, CA). A P-value of <0.05 was taken as statistically significant.
Detailed Materials and Methods are available in the Data Supplement.
Results
TAC-induced hypertrophy
Treatment of naïve mice with CRD-733 at 600 mg/kg/day for 14 days had no significant effects on baseline LV mass or cardiac function (Figure I in the Data Supplement). To test the effects of CRD-733 on established cardiac hypertrophy, mice were subjected to TAC, followed by daily treatment with CRD-733 starting 7 days after TAC (Figure 1A). Significant LV pressure overload at day 7 post-TAC was confirmed by measurement of echocardiography-derived trans-TAC pressure gradients, showing peak gradients of 71.0±4.49 mmHg and 65.2±4.05 mmHg for prospective vehicle and CRD-733 TAC treatment groups, compared to 2.18±0.20 mmHg in sham controls (P<0.001; Figure 1B). TAC surgery also induced significant cardiac hypertrophy after 7 days, with echocardiography-derived LV mass/body weight ratios of 4.99±0.34 and 4.79±0.30 for prospective TAC vehicle (P<0.001) and TAC CRD-733 (P=0.007) treatment groups respectively, compared to 3.20±0.10 for sham controls (Figure 1C). At day 7, prior to treatment, there were no statistically significant differences in LV hypertrophy (P=0.90) or LV pressure gradients (P=0.57) between treatment groups.
Figure 1. Study timeline, equal pressure gradients and left ventricular hypertrophy in TAC groups at day 7, prior to drug treatment.
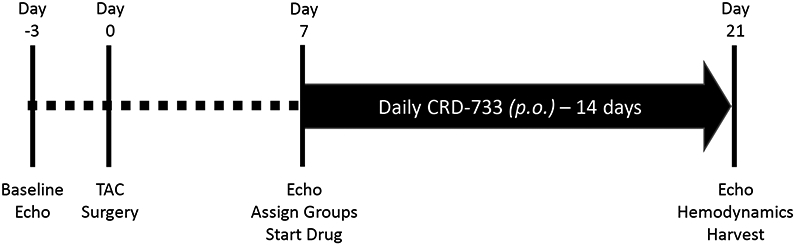


A, Illustration of experimental study design. B, Pulsed-wave Doppler-derived peak TAC gradients calculated using the Bernouilli equation are elevated vs. sham to a similar extent in treatment groups. C, Echocardiography-derived left ventricular mass is increased to a similar extent in TAC groups prior to drug treatment. Results are expressed as mean ± SEM. Comparisons were performed using 1-way ANOVA with Tukey’s post-test; n=10-17.
CRD-733 reverses cardiac hypertrophy
At day 21 post-TAC, terminal normalized LV weights were increased in vehicle treated TAC mice after 21 days compared to sham (mean difference, 2.67 mg/mm [95% CI, 1.77-3.56]; P<0.001). In TAC mice treated with CRD-733, LV weight normalized to tibia length was not statistically different to sham (P=0.23; Figure 2A), and was significantly reduced compared with TAC vehicle treated mice (LV mass/tibia length 7.71±0.29 mg/mm in TAC vehicle, 5.71±0.18 mg/mm in TAC CRD-733, P<0.001). Serial echocardiography demonstrated that in vehicle-treated TAC mice, average LV mass increased by 10.1±3.77 mg from the start of treatment at day 7 to day 21 (Figure 2B). In contrast, whereas LV mass increased in 12 out of 17 TAC mice receiving vehicle, LV mass decreased in 9 of 10 of mice treated with CRD-733 during the same period (mean decrease from day 7 to day 21 in TAC CRD-733 of 20.6±5.64 mg; P<0.001). This reduction in LV mass may have been due in part to reductions in interventricular septal and LV posterior wall thicknesses in CRD-733 treated mice, although these specific parameters failed to reach statistical significance vs. TAC vehicle (Figure 2C; P=0.05 and 2D; P=0.07). Normalized kidney weights did not differ significantly between sham, TAC vehicle, or TAC CRD-733 groups (Figure II in the Data Supplement).
Figure 2. CRD-733 reverses TAC-induced left ventricular (LV) hypertrophy.


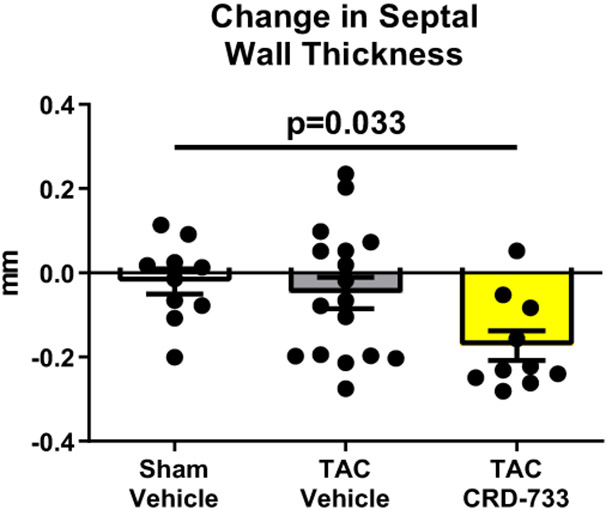




A, Left ventricular wet weight normalized to tibia length after 21 days. B, Change in echo-derived LV mass from start of treatment (day 7) to end of study (day 21), C, septal wall thickness and D, LV posterior wall thickness after 14 days of drug treatment. Apical LV tissue gene expression for E, Nppa, F, Nppb and G, Myh7. Results are expressed as mean ± SEM. Comparisons were performed using 1-way ANOVA with Tukey’s post-test; n=10-17 (A-D) or n=7-8 (E-G).
Gene expression of Nppa, a marker of pathological hypertrophy, was significantly upregulated in TAC vehicle mice compared to sham (P=0.006), but this change was significantly attenuated in TAC mice treated with CRD-733 (P=0.002 TAC vehicle vs. TAC CRD-733; Figure 2E). Similarly, expression of Nppb and Myh7 both increased in TAC vehicle mice compared to sham (P=0.009 and P=0.004 respectively), but not in mice treated with CRD-733 (P=0.65 and P=0.62 for Nppb and Myh7 respectively, in sham compared with TAC CRD-733; Figure 2F and 2G).
CRD-733 treatment improves cardiac function
Systolic LV function was assessed following 21 days of TAC (14 days of treatment, Figure 3). At day 21, vehicle-treated mice had significantly reduced ejection fractions compared to sham mice (58.8%±1.66% in sham versus 46.7%±2.12% in TAC vehicle; P=0.001). In contrast, TAC mice treated with CRD-733 had significantly improved ejection fraction compared to vehicle-treated TAC mice (46.7±2.12% in TAC vehicle-treated vs. 56.5±2.56% in TAC CRD-733; P=0.009). At day 21, ejection fraction in CRD-733 treated TAC mice did not differ significantly from that of sham-operated mice (Figure 3A, CRD-733-treated vs sham mice, P=0.79). Fractional shortening also improved in CRD-733 treated TAC mice compared with vehicle-treated TAC mice (P=0.011; Figure 3B). Representative LV M-Mode echocardiography traces show reduced contractility for TAC mice treated with vehicle compared to sham controls and TAC mice treated with CRD-733 (Figure 3C). Speckle-tracking echocardiography was used to assess global longitudinal strain rate and revealed a significant reduction for vehicle-treated TAC mice vs. sham (P=0.013), which was not observed for TAC mice treated with CRD-733 (Figure 3D). Although treatment with CRD-733 did not lead to a significant improvement in global longitudinal strain rate vs. TAC vehicle (P=0.07), there was no significant difference between TAC CRD-733 treated mice and sham controls (P=0.87).
Figure 3. CRD-733 reverses TAC-induced left ventricular dysfunction.




A, Ejection fraction and B, Fractional shortening at 21 days post-TAC. C, Representative parasternal long axis M-Mode images obtained during echocardiography. D, Global longitudinal strain rate. Results are expressed as mean ± SEM. Comparisons were performed using 1-way ANOVA with Tukey’s post-test; n=10-17.
CRD-733 treatment reverses TAC-induced left atrial dilation
Echocardiographic assessment of left atrial area, a marker of diastolic dysfunction and increased LV filling pressures in mice,15, 16 demonstrated significant atrial dilation in both prospective TAC groups compared to sham control after 7 days of TAC (mean difference, 3.77 mm2 [95% CI, 2.95 to 4.59]; P<0.001). Representative images of the apical 4-chamber view are shown at baseline, 7 days after TAC and 21 days after TAC, (Figure 4A). Left atrial dilation did not differ significantly at day 7 (pre-treatment) between TAC groups (P=0.98; Figure 4A and 4B). While TAC mice treated with vehicle had no significant change in left atrial area during treatment (mean difference day 7-21, −0.30 mm2 [95% CI, −0.95 to 0.35]; P=0.51), left atrial dilation was reversed in TAC mice treated with CRD-733 (mean difference day 7-21, −1.95 mm2 [95% CI, −2.79 to −1.10]; P<0.001). At day 21 there was a significant reduction in left atrial area in TAC CRD-733 treated compared to TAC vehicle-treated mice (6.93±0.40 mm2 in TAC vehicle vs. 5.28±0.38 mm2 in TAC CRD-733; P<0.001; Figure 4A and 4B).
Figure 4. CRD-733 reverses TAC-induced left atrial dilation.


A, Representative apical 4-chamber images obtained from echocardiography. Left atrial areas are indicated by the dotted yellow line; RV: right ventricle, LV: left ventricle, RA: right atrium, LA: left atrium. B, Quantification of left atrial area over time. Results are expressed as mean ± SEM. Comparisons were performed using repeated measures 2-way ANOVA with Tukey’s post-test; n=10-17.
CRD-733 treatment improves pulmonary-related hemodynamic indices of heart failure
Invasive hemodynamic analyses were used in a subset of mice after 21 days of TAC. Full hemodynamic data are presented in Table I in the Data Supplement. Pressure-volume loops showed elevated LV end diastolic pressure in TAC mice treated with vehicle (Figure 5A), compared to sham mice and mice treated with CRD-733. The average LV end diastolic pressures were elevated in TAC mice treated with vehicle compared to sham mice (6.40±1.94 mmHg in sham vs. 21.0±3.09 mmHg in TAC vehicle; P=0.001). Compared with TAC vehicle-treated mice, TAC mice treated with CRD-733 had significantly reduced LV end diastolic pressures (21.0±3.09 mmHg TAC vehicle vs. 13.0±1.4 mmHg in TAC CRD-733; P=0.037; Figure 5B). LV end diastolic pressures were not significantly elevated in CRD-733-treated mice compared to sham controls (P=0.11). Furthermore, the ventricular arterial coupling ratio of Arterial Elastance (Ea) normalized to end systolic pressure volume relation (ESPVR), an index of cardiovascular performance,17 was significantly elevated in vehicle-treated TAC mice (P=0.017 vs. sham), indicating LV dysfunction, whereas treatment with CRD-733 significantly attenuated this increase (P=0.013) and led to values similar to that of sham mice (Figure 5C). The improvement in LV end diastolic pressure due to CRD-733 was observed in the presence of sustained, elevated systolic LV pressures vs. sham for TAC vehicle (mean difference, 64.4 mmHg [95% CI, 30.3 to 98.5]; P<0.001) and TAC CRD-733 (mean difference, 46.0 mmHg [95% CI, 13.9 to 78.1]; P=0.005). Systolic LV pressures were not statistically different between TAC groups (P=0.29; Figure 5D). The trans-TAC pressure gradient was also not statistically different between TAC groups after 21 days (P=0.62; Figure 5E), suggesting that differences in LV end diastolic pressure and other LV indices were not attributable to differences in TAC severity. Normalized terminal lung weights, a surrogate for the extent of pulmonary edema, were significantly lower in TAC mice treated with CRD-733 compared to vehicle treatment (P=0.041; Figure 5F). Normalized lung weight did not differ significantly between TAC CRD-733 mice and sham-operated controls (P=0.81), supporting that CRD-733 protected against the development of pulmonary edema in this model of heart failure (Figure 5F). We did not observe any significant differences in kidney weights or morphology following treatment with CRD-733 (Figure II in the Data Supplement).
Figure 5. CRD-733 reverses TAC-induced elevations in left ventricular (LV) end diastolic pressure and lung weight, despite sustained maximal LV pressure and TAC gradients.






A, Representative pressure-volume loops showing end diastolic volumes denoted by blue arrows. B, LV end diastolic pressure C, Ventricular-arterial coupling ratio and D, Maximal LV pressure obtained by invasive hemodynamics. E, Echocardiography-derived peak trans-TAC gradient after treatment. F, Normalized terminal lung weights after 21 days of TAC and 14 days of treatment. Results are expressed as mean ± SEM. Comparisons were performed using 1-way ANOVA with Tukey’s post-test; n=5-8 (B-D) and n=10-17 (E-F).
CRD-733 treatment attenuates fibrotic gene expression, but does not reverse collagen deposition
We next quantified collagen deposition in LV tissue sections using picrosirius red staining. There were no statistically significant differences between the percentage of interstitial fibrosis (Figure 6A and 6B) or perivascular fibrosis observed (Figure 6A and 6C) for TAC mice treated with either vehicle or CRD-733. In contrast, LV tissue gene expression of Col1α1, Fn1 and Ctgf were all significantly reduced in mice treated with CRD-733 compared to vehicle-treated TAC mice (P=0.022, P=0.018 and P=0.013 respectively; Figure 6D-F). CRD-733 restored expression of these fibrotic genes to basal levels, similar to that of sham-operated mice (P≥0.85).
Figure 6. CRD-733 does not reverse TAC-induced left ventricular (LV) cardiac fibrosis, but attenuates fibrotic gene expression.






A, Representative images for LV interstitial fibrosis (upper panels) and perivascular fibrosis (lower panels); scale bar: 200μm. B, Quantification of interstitial fibrosis and C, Quantification of perivascular fibrosis. Apical LV gene expression is shown for D, Col1a1, E, Ctgf and F, Fn1. Results are expressed as mean ± SEM. Comparisons were performed using 1-way ANOVA with Tukey’s post-test; n=10-17 (B-C), n=7-8 (D-F).
Plasma cGMP concentration is elevated with CRD-733
We next examined the effects of PDE9 inhibition with CRD-733 on terminal blood and LV tissue cGMP concentrations at day 21. Plasma cGMP increased significantly in TAC mice treated with CRD-733 compared to vehicle-treated TAC mice (mean difference, 3.17 pmol/mL [95% CI, 1.76-4.58]; P<0.001; Figure 7A). In contrast, cGMP in LV tissue trended higher in TAC groups, but did not reach statistical significance (P=0.06; Figure 7B). In CRD-733 treated TAC LVs, phosphorylation of Ser273 on cardiac myosin binding protein-C (cMyBP-C), a PKGIα phosphorylation site,18 increased compared with vehicle-treated TAC LVs (P=0.048, Figure 7C), indicating activation of LV PKGIα signaling by CRD-733. The protein expression of calcineurin, a phosphatase involved in the hypertrophic response, trended lower in mice treated with CRD-733, but failed to reach statistical significance (P=0.11; Figure 7D).
Figure 7. CRD-733 increases plasma cGMP levels.




A, Plasma cGMP levels from blood taken immediately prior to sacrifice (n=10-17). B, Left ventricular (LV) tissue levels of cGMP (n=5-9). C, Protein expression for phosphorylated myosin binding protein-C at Ser273, normalized to GAPDH (n=5-7) and D, Protein expression for calcineurin from cardiac tissue, normalized to GAPDH (n=5-7). Results are expressed as mean ± SEM. Comparisons were performed using 1-way ANOVA with Tukey’s post-test (A-B) or Mann-Whitney U test (C-D).
Discussion
In the present study we show that inhibition of phosphodiesterase 9 by CRD-733, a novel PDE9 inhibitor, reverses cardiac hypertrophy and left atrial dilation, improves LV function and LV end diastolic pressure, and increases plasma cGMP in a 3-week mouse model of LV pressure overload. In this study, we administered CRD-733 beginning one week after initiation of pressure overload, at which time cardiac hypertrophy had already been established.
Lee and colleagues first identified the potential pathological role of PDE9 activity in HF by demonstrating increased myocardial PDE9 expression and activity in cardiac tissue from mice subjected to TAC-induced HF and from HF patients.10 In the same study, genetic deletion of PDE9 in mice prevented the development of pressure overload-induced cardiac hypertrophy, dysfunction and fibrosis. Chemical inhibition of PDE9 reverses cardiac hypertrophy and dysfunction in preclinical pressure overload and pharmacological models of HF.10-12, 19 In the present TAC study, we observed significant reversal of TAC-induced LV hypertrophy with CRD-733 treatment, as well as reversal of left atrial dilation, supporting that CRD-733 treatment reduces left-sided filling pressures in this HF model. The reduced LV end diastolic pressure observed at day 21 in CRD-733 treated mice also directly supports a beneficial effect of PDE9 inhibition on LV filling pressures. These hemodynamic benefits of CRD-733 were seen in addition to the observed improvement in LV ejection fraction.
In patients, LV hypertrophy predicts mortality20 and its regression correlates with improved outcomes in hypertensive patients with LV hypertrophy.21 Further, elevation of left atrial and LV end diastolic pressures drive many of the symptoms seen in HF patients and contribute to pulmonary edema and other morbidities of this condition. The observations reported here in the TAC model identify hemodynamic effects of PDE9 inhibition that may prove clinically relevant in humans with HF. Our data also show that CRD-733 did not significantly affect LV systolic pressure in these studies, suggesting that PDE9 inhibition might represent a strategy to promote NPR pathway signaling and PKG-mediated anti-remodeling effects that avoids the hypotension observed with other pharmacologic approaches to activate these myocardial pathways. While we detected no effects of CRD-733 on blood pressure measured by invasive hemodynamics, we did not measure this in conscious mice and therefore future studies will be required to confirm lack of blood pressure effects of this drug. CRD-733 has been studied previously in human trials for a non-cardiac indication (NCT02038868). Therefore, the beneficial effects of PDE9 inhibition with CRD-733 in this preclinical HF model provide support for exploring the effects of CRD-733 in human HF.
In the current study, the functional effects of CRD-733 on LV systolic function argue for at least some intrinsic myocardial effects. The improvements observed in fetal gene expression patterns with CRD-733 provide further evidence for direct myocardial effects of PDE9 inhibition in this model, which is consistent with previous studies in which genetic and pharmacological inhibition of PDE9 exerts direct antihypertrophic effects on isolated cardiac myocytes.10, 11 In a recent large animal HF model, administration of a single PDE9 inhibitor dose acutely reduced LV filling pressures and improved urine output,12 raising the possibility that effects of PDE9 inhibition on LV end diastolic pressure and lung mass may also arise in part due to extracardiac effects on preload. While our study did not test these effects directly, our prior work demonstrates no significant effects of 4 week TAC on renal function.15 Further, we detected no gross effects of CRD-733 on kidney morphology in the present study. Thus, while primary renal effects do not appear to explain our chronic TAC findings, future studies may elucidate acute beneficial effects of CRD-733 on renal hemodynamics. Additionally, the effects of TAC and of CRD-733 on other upstream cGMP generating systems, such as the circulating NPs, were not studied and thus remain unknown.
Here, we determined that PDE9 inhibition with CRD-733 decreased the expression of fibrosis-related genes in LV tissue, consistent with previous reports.10 In recent studies of other cGMP or dual cAMP/cGMP selective PDE isoforms, PDE1,22 PDE2,23 PDE3,24 PDE55 or PDE1025 inhibition or genetic ablation reversed or prevented cardiac fibrosis in mouse models of chronic pressure overload, which supports a role of cGMP in opposing pathological fibrosis. Though CRD-733 reduced fibrotic gene expression, we still observed similar degrees of collagen deposition between vehicle and CRD-733 treated groups, suggesting the possibility that the relatively short duration of TAC in our study was insufficient to detect more chronic effects on collagen deposition.
Finally, we observed that CRD-733 increased plasma cGMP in the chronic TAC model, supporting plasma cGMP as a biomarker of target engagement and inhibition of PDE9 activity. Other cGMP-augmenting drugs such as sacubitril/valsartan also increase plasma cGMP,26-28 and a recent study in a sheep HF model demonstrated acute effects of another PDE9 inhibitor on plasma cGMP.12 Our findings therefore support that circulating cGMP may serve as a biomarker of PDE9 inhibitory or therapeutic effect. We speculate that measuring cGMP might serve as a strategy both to detect response to cGMP-augmenting drugs, and to identify patients who could potentially benefit from these drugs.
Notably, in our model we did not observe decreased cGMP in either plasma or LV tissue from TAC-vehicle mice, compared with sham, at the end of the study. This raises the question of whether the LV hypertrophy and dysfunction observed in TAC arise from any derangements of cGMP signaling. Our findings do agree with prior observations in which plasma cGMP29 or net LV cGMP5, 10, 29 do not decrease and even increase in the setting of LV dysfunction and remodeling. Further, the clinical and physiological significance of plasma cGMP remains unclear. Some studies in humans suggest that circulating cGMP correlates with NP activity and thus predicts, rather than protects from, incident HF.30, 31 Based on the observed increased PKG substrate phosphorylation with CRD-733 in LV tissue, we interpret our findings to support that PDE9 inhibition acts in the LV through cGMP-PKG signaling to improve LV structure and function. We further interpret the CRD-733 augmentation of plasma cGMP in TAC as a readout of effective PDE9 inhibition. However, future work will be important to determine whether plasma cGMP represents only a marker of drug effect, or whether it also mediates any of the therapeutic effects of this drug class.
We also note that though CRD-733 augmented plasma cGMP as discussed above, we did not detect gross increases in LV cGMP in the CRD-733 TAC group. We interpret the lack of statistically increased cGMP in the LV to represent a limitation of assay sensitivity. Specifically, since PDE9 inhibition augments only a membrane-associated pool of myocardial cGMP, but not the NO-sensitive pool,10 it is likely that resultant changes in the PDE9-specific pool of cGMP may not raise net myocardial cGMP substantially despite having an important physiological effect. The CRD-733 induced increase in phosphorylation of cMyBP-C, which is phosphorylated by PKGIα in LV tissue and in the cardiomyocyte in a cGMP-dependent manner,18 supports that PDE9 inhibition with CRD-733 sufficiently activated anti-remodeling PKGIα signaling. Since phosphorylation of the cMyBP-C M-domain promotes LV systolic and diastolic function32, 33 and opposes pathological LV remodeling,34 we speculate that CRD-733 improved LV function and hypertrophy through PKGIα-mediated phosphorylation of cMyBP-C and other PKGIα mediated substrates in the cardiomyocyte and in LV tissue.2
Despite the positive findings in this study, we do note a few limitations. First, this study focused solely on the pressure overload model of cardiac remodeling. We therefore cannot conclude whether CRD-733 affects LV remodeling and function in the setting of other pathological stress, such as ischemia or neurohormonal activation. Additionally, whether the short-term effects on LV structure and function we observe in this study would be sustained after discontinuation of drug remains untested. Further, while the 21 day severe TAC model has been demonstrated to induce increased myocardial PDE9 expression,10 our studies did not measure this directly. Finally, though CRD-733 has been administered to humans for other conditions, the current study focused on an animal preclinical model, and thus further investigation will be required to test whether PDE9 inhibition with CRD-733 or other agents can improve LV structure and function in humans.
In summary, we have demonstrated that the PDE9 inhibitor CRD-733 improves pre-existing LV hypertrophy and dysfunction after pressure overload, as well as reducing hemodynamic markers of HF such as left atrial dilation, increased LV end diastolic pressure, and pulmonary edema. These findings identify novel physiological mechanisms improved by PDE9 inhibition and support future investigation of CRD-733 as a therapeutic agent in human HF.
Supplementary Material
Clinical Perspective.
What is new?
We demonstrate that CRD-733, a phosphodiesterase 9 inhibitor, reverses pre-existing LV hypertrophy when administered one week after induction of pressure overload by transverse aortic constriction.
In this experimental model, CRD-733 also improved: left atrial area, LV end diastolic pressure and other hemodynamic parameters; LV ejection fraction; plasma cGMP; as well as molecular markers of pathologic cardiac remodeling.
These findings identify physiological mechanisms through which inhibition of cGMP breakdown improves LV structure and function after pressure overload.
What are the clinical implications?
Augmentation of cGMP signaling represents an emerging therapeutic strategy in heart failure with reduced LV ejection fraction, but mortality for this condition remains high. The enzyme phosphodiesterase 9 (PDE9) breaks down cGMP, and thus PDE9 inhibition promotes cGMP signaling.
The novel PDE9 inhibitor CRD-733 has been administered to patients in non-cardiac clinical studies.
Our findings in an LV pressure overload heart failure model that CRD-733 reverses LV hypertrophy and normalizes LV function thus support further investigation of CRD-733 in humans with heart failure.
Acknowledgements
The authors thank Sakthivel Sadayappan for generously sharing the antibody to the phosphoserine 273 of cardiac myosin binding protein-C.
Sources of Funding
These studies were funded by a research grant from Cardurion Pharmaceuticals. R.M. Blanton also funded by the NIH R01HL131831.
Non-standard Abbreviations and Acronyms
- cGMP
Cyclic guanosine monophosphate
- cMyBP-C
Cardiac myosin binding protein-C
- ESPVR
End systolic pressure volume relationship
- HF
Heart Failure
- HFrEF
Heart failure with reduced ejection fraction
- LV
Left ventricle
- NP
Natriuretic peptide
- NPR
Natriuretic peptide receptor
- PDE1
Phosphodiesterase 1
- PDE2
Phosphodiesterase 2
- PDE3
Phosphodiesterase 3
- PDE5
Phosphodiesterase 5
- PDE9
Phosphodiesterase 9
- PDE10
Phosphodiesterase 10
- PKG
Protein kinase G
- PKG1α
Protein kinase G 1-alpha
- TAC
Transverse aortic constriction
Footnotes
Disclosures
M.E. Mendelsohn and D.M. Bloomfield are employees of Cardurion Pharmaceuticals.
References
- 1.Armstrong PW, Pieske B, Anstrom KJ, Ezekowitz J, Hernandez AF, Butler J, Lam CSP, Ponikowski P, Voors AA, Jia G, et al. Vericiguat in patients with heart failure and reduced ejection fraction. The New England journal of medicine. 2020;382:1883–1893 [DOI] [PubMed] [Google Scholar]
- 2.Kong Q, Blanton RM. Protein kinase g i and heart failure: Shifting focus from vascular unloading to direct myocardial antiremodeling effects. Circ Heart Fail. 2013;6:1268–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura T, Zhu G, Ranek MJ, Kokkonen-Simon K, Zhang M, Kim GE, Tsujita K, Kass DA. Prevention of pkg-1alpha oxidation suppresses antihypertrophic/antifibrotic effects from pde5 inhibition but not sgc stimulation. Circ Heart Fail. 2018;11:e004740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park M, Sandner P, Krieg T. Cgmp at the centre of attention: Emerging strategies for activating the cardioprotective pkg pathway. Basic Res Cardiol. 2018;113:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic gmp phosphodiesterase 5a prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222 [DOI] [PubMed] [Google Scholar]
- 6.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. Jama. 2013;309:1268–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pde5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: Results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2011;4:8–17 [DOI] [PubMed] [Google Scholar]
- 8.Behling A, Rohde LE, Colombo FC, Goldraich LA, Stein R, Clausell N. Effects of 5'-phosphodiesterase four-week long inhibition with sildenafil in patients with chronic heart failure: A double-blind, placebo-controlled clinical trial. Journal of cardiac failure. 2008;14:189–197 [DOI] [PubMed] [Google Scholar]
- 9.Guazzi M, Samaja M, Arena R, Vicenzi M, Guazzi MD. Long-term use of sildenafil in the therapeutic management of heart failure. J Am Coll Cardiol. 2007;50:2136–2144 [DOI] [PubMed] [Google Scholar]
- 10.Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, et al. Phosphodiesterase 9a controls nitric-oxide-independent cgmp and hypertrophic heart disease. Nature. 2015;519:472–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang PX, Li ZM, Cai SD, Li JY, He P, Huang Y, Feng GS, Luo HB, Chen SR, Liu PQ. C33(s), a novel pde9a inhibitor, protects against rat cardiac hypertrophy through upregulating cgmp signaling. Acta Pharmacol Sin. 2017;38:1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott NJA, Rademaker MT, Charles CJ, Espiner EA, Richards AM. Hemodynamic, hormonal, and renal actions of phosphodiesterase-9 inhibition in experimental heart failure. J Am Coll Cardiol. 2019;74:889–901 [DOI] [PubMed] [Google Scholar]
- 13.Frolich L, Wunderlich G, Thamer C, Roehrle M, Garcia M Jr., Dubois B. Evaluation of the efficacy, safety and tolerability of orally administered bi 409306, a novel phosphodiesterase type 9 inhibitor, in two randomised controlled phase ii studies in patients with prodromal and mild alzheimer's disease. Alzheimer's research & therapy. 2019;11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charnigo RJ, Beidler D, Rybin D, Pittman DD, Tan B, Howard J, Michelson AD, Frelinger Al III, Clarke N. Pf-04447943, a phosphodiesterase 9a inhibitor, in stable sickle cell disease patients: A phase ib randomized, placebo-controlled study. Clinical and translational science. 2019;12:180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards DA, Aronovitz MJ, Calamaras TD, Tam K, Martin GL, Liu P, Bowditch HK, Zhang P, Huggins GS, Blanton RM. Distinct phenotypes induced by three degrees of transverse aortic constriction in mice. Scientific reports. 2019;9:5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schnelle M, Catibog N, Zhang M, Nabeebaccus AA, Anderson G, Richards DA, Sawyer G, Zhang X, Toischer K, Hasenfuss G, et al. Echocardiographic evaluation of diastolic function in mouse models of heart disease. J Mol Cell Cardiol. 2018;114:20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guarracino F, Baldassarri R, Pinsky MR. Ventriculo-arterial decoupling in acutely altered hemodynamic states. Critical care. 2013;17:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thoonen R, Giovanni S, Govindan S, Lee DI, Wang GR, Calamaras TD, Takimoto E, Kass DA, Sadayappan S, Blanton RM. Molecular screen identifies cardiac myosin-binding protein-c as a protein kinase g-ialpha substrate. Circ Heart Fail. 2015;8:1115–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kokkonen-Simon KM, Saberi A, Nakamura T, Ranek MJ, Zhu G, Bedja D, Kuhn M, Halushka MK, Lee DI, Kass DA. Marked disparity of microrna modulation by cgmp-selective pde5 versus pde9 inhibitors in heart disease. JCI insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. American heart journal. 2001;141:334–341 [DOI] [PubMed] [Google Scholar]
- 21.Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, et al. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (life): A randomised trial against atenolol. Lancet. 2002;359:995–1003 [DOI] [PubMed] [Google Scholar]
- 22.Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, et al. Pde1c deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016;113:E7116–E7125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baliga RS, Preedy MEJ, Dukinfield MS, Chu SM, Aubdool AA, Bubb KJ, Moyes AJ, Tones MA, Hobbs AJ. Phosphodiesterase 2 inhibition preferentially promotes no/guanylyl cyclase/cgmp signaling to reverse the development of heart failure. Proc Natl Acad Sci U S A. 2018;115:E7428–E7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polidovitch N, Yang S, Sun H, Lakin R, Ahmad F, Gao X, Turnbull PC, Chiarello C, Perry CGR, Manganiello V, et al. Phosphodiesterase type 3a (pde3a), but not type 3b (pde3b), contributes to the adverse cardiac remodeling induced by pressure overload. J Mol Cell Cardiol. 2019;132:60–70 [DOI] [PubMed] [Google Scholar]
- 25.Chen S, Zhang Y, Lighthouse JK, Mickelsen DM, Wu J, Yao P, Small EM, Yan C. A novel role of cyclic nucleotide phosphodiesterase 10a in pathological cardiac remodeling and dysfunction. Circulation. 2020;141:217–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobalava Z, Kotovskaya Y, Averkov O, Pavlikova E, Moiseev V, Albrecht D, Chandra P, Ayalasomayajula S, Prescott MF, Pal P, et al. Pharmacodynamic and pharmacokinetic profiles of sacubitril/valsartan (lcz696) in patients with heart failure and reduced ejection fraction. Cardiovascular therapeutics. 2016;34:191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu J, Noe A, Chandra P, Al-Fayoumi S, Ligueros-Saylan M, Sarangapani R, Maahs S, Ksander G, Rigel DF, Jeng AY, et al. Pharmacokinetics and pharmacodynamics of lcz696, a novel dual-acting angiotensin receptor-neprilysin inhibitor (arni). Journal of clinical pharmacology. 2010;50:401–414 [DOI] [PubMed] [Google Scholar]
- 28.Morrow DA, Velazquez EJ, DeVore AD, Prescott MF, Duffy CI, Gurmu Y, McCague K, Rocha R, Braunwald E. Cardiovascular biomarkers in patients with acute decompensated heart failure randomized to sacubitril-valsartan or enalapril in the pioneer-hf trial. Eur Heart J. 2019;40:3345–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Methawasin M, Strom J, Borkowski T, Hourani Z, Runyan R, Smith JE 3rd, Granzier H. Phosphodiesterase 9a inhibition in mouse models of diastolic dysfunction. Circ Heart Fail. 2020;13:e006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao D, Guallar E, Vaidya D, Ndumele CE, Ouyang P, Post WS, Lima JA, Ying W, Kass DA, Hoogeveen RC, et al. Cyclic guanosine monophosphate and risk of incident heart failure and other cardiovascular events: The aric study. J Am Heart Assoc. 2020;9:e013966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ying W, Zhao D, Ouyang P, Subramanya V, Vaidya D, Ndumele CE, Guallar E, Sharma K, Shah SJ, Kass DA, et al. Associations between the cyclic guanosine monophosphate pathway and cardiovascular risk factors: Mesa. J Am Heart Assoc. 2019;8:e013149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tong CW, Wu X, Liu Y, Rosas PC, Sadayappan S, Hudmon A, Muthuchamy M, Powers PA, Valdivia HH, Moss RL. Phosphoregulation of cardiac inotropy via myosin binding protein-c during increased pacing frequency or beta1-adrenergic stimulation. Circ Heart Fail. 2015;8:595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosas PC, Liu Y, Abdalla MI, Thomas CM, Kidwell DT, Dusio GF, Mukhopadhyay D, Kumar R, Baker KM, Mitchell BM, et al. Phosphorylation of cardiac myosin-binding protein-c is a critical mediator of diastolic function. Circ Heart Fail. 2015;8:582–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein c phosphorylation is cardioprotective. Proc Natl Acad Sci U S A. 2006;103:16918–16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: An open-source platform for biological-image analysis. Nature methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calamaras TD, Baumgartner RA, Aronovitz MJ, McLaughlin AL, Tam K, Richards DA, Cooper CW, Li N, Baur WE, Qiao X, et al. Mixed lineage kinase-3 prevents cardiac dysfunction and structural remodeling with pressure overload. Am J Physiol Heart Circ Physiol. 2019;316:H145–H159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.