

Abstract
Klebsiella oxytoca is a gram-negative bacterium. It is opportunistic in nature and causes hospital acquired infections. Subtractive proteomics and reverse vaccinology approaches were employed to screen out the best proteins for vaccine designing. Whole proteome of K. oxytoca strain ATCC 8724, consisting of 5483 proteins, was used for designing the vaccine. Total 1670 cytotoxic T lymphocyte (CTL) epitope were predicted through NetCTL while 1270 helper T lymphocyte (HTL) epitopes were predicted through IEDB server. The epitopes were screened for non-toxicity, allergenicity, antigenicity and water solubility. After epitope screening 300 CTL and 250 HTL epitopes were submitted to IFN-γ epitope server to predict their Interferon-γ induction response. The selected IFN-γ positive epitopes were tested for their binding affinity with MHCI-DRB1 by MHCPred. The 15 CTL and 13 HTL epitopes were joined by linkers AAY and GPGPG respectively in vaccine construct. Chain C of Pam3CSK4 (PDB ID; 2Z7X) was linked to the vaccine construct as an adjuvant. A 450aa long vaccine construct was submitted to I-TASSER server for 3D structure prediction. Thirteen Linear B cells were predicted by ABCPred server and 10 sets of discontinues epitopes for 3D vaccine structure were predicted by DiscoTope server. The modeled 3D vaccine construct was docked with human Toll-like receptor 2 (PDB ID: 6NIG) by PatchDock. The docked complexes were refined by FireDock. The selected docked complex showed five hydrogen bonds and one salt bridge. The vaccine sequence was reverse transcribed to get nucleotide sequence for In silico cloning. The reverse transcribed sequence strand was cloned in pET28a(+) expression vector. A clone containing 6586 bp was constructed including the 450 bp of query gene sequence.
Keywords: Linear B cells epitopes, Disulfide engineering, Thermostability curve, Human TLR2, pET28a (+)
Introduction
Klebsiella oxytoca is a non-motile, rod shaped gram-negative bacterium of the family Enterobacteriaceae (Gorkiewicz 2009). It is unscrupulous in nature and causes hospital acquired infections in hospitalized patients, especially newborn infants (Podschun and Ullmann 1998). A large number of infants have been reported to die worldwide due to this infection (Reiss et al. 2000). After Escherichia sp. this genus includes second most infectious pathogens (Yinnon et al. 1996) while K. oxytoca has been reported to be the second most frequent source for bacterial infection next to Klebsiella pneumonia species (Singh et al. 2016). It is also found to be the causal organism of AAHC (antibiotic-associated hemorrhagic colitis) disease (Hoffmann et al. 2010). It uses many virulence factors to overcome the host innate immune system. The virulence factors are adhesions, siderophores, lipopolysaccharides (LPSs) on cell surface and capsular polysaccharides (CPLs). Although LPS have potential to activate infection pathway but K antigen of the capsule have been reported to be the main source of pathogenicity (Simoons-Smit et al. 1986; Podschun and Ullmann 1998). The bacterium attaches to host cell by adhesins as primary step for host cell infections. It attaches to intestinal, urogenital tracts and respiratory epithelial cells by Type 1 pili. It binds to the mannose-containing trisaccharides of the host glycoproteins via Type 4 pili (Podschun and Ullmann 1998).
Being unscrupulous in nature it causes infections in multiple fashions so that multiple drugs are used to treat the disease accordingly. This indiscriminate and heavy use of drugs leads to multiple drug resistance to commonly used antibiotics (Vali et al. 2015; Singh et al. 2016). Resistance development in different species of Klebsiella to different drugs like the oxyimino (carbapenemases), β-lactams (ceftazidime and cefotaxime), penicillins (ampicillin, cephalosporinases, carbenicillin), monobactam and aztreonam have been reported from last so many decades (Decré et al. 2004; Wu et al. 2009). As this bacterium is not causal organism of a particular disease so, in spite of its multidrug resistance, very little attention have been given to the development of effective vaccine against this. Although it causes rare infectious disease but these can lead to life threatening complications in the patients especially in children. The development of an effective vaccine against this pathogen is need of this era.
Development of vaccine is a long tedious and time taking procedure which involves high cost of production even at initial screening steps at laboratory scale (André 2003). Some pathogens are very difficult or even impossible to handle in lab conditions so their vaccine development is very difficult even impossible (Tannock et al. 2020). The development of advanced technologies and improved bioinformatics techniques during recent past have given a breakthrough in many industries including medical technologies. The advanced approaches of bioinformatics can be used in vaccinology, an important branch of healthcare sciences. Reverse vaccinology (RV) is an innovative and smart in silico prediction technique for the identification of new protein-derived vaccine candidates to minimize the in vitro vaccinology limitation (Rappuoli 2000). A classic RV pipeline consists of screening the whole proteome- and choosing proteins that will become vaccine construct through computational methods. RV has been used against different kind of infectious diseases (Delany et al. 2013; De la Fuente et al. 2016; Michalik et al. 2016; Mehla and Ramana 2017; Muruato et al. 2017) and the first successful application of this approach was achieved in Neisseria meningitidis serogroup B (Kelly & Rappuoli 2005). Conventional approaches, most of the time, failed in providing a universal and efficient vaccine for the most affected groups of patients. In contrast, reverse vaccinology has shown to be an efficient tool for the identification of protective vaccine candidates. Here, we have used subtractive proteomics and cheminformatics approaches for the screening the surface exposed proteins of K. oxytoca to design the effective epitopes for vaccine. These surface epitopes from surface exposed proteins can be used to design an effective and human safe vaccine against this bacterium.
Methods
Bacterial Proteome Acquisition and Subtraction
The proteome of K. oxytoca (strain ATCC8724) was retrieved from UniProt (UniProt ID: UP000007843). The whole proteome was passed through the pipeline of subtractive proteomics. The proteins, suitable for designing a potential vaccine against this pathogen infections, were shortlisted.
The proteome dataset may contain some similar or duplicate protein sequences which are unnecessary to carry forward. The proteome was screened through CD-HIT suite. The Paralogous or duplicates proteins arisen during evolution process were identified and then redundant protein entries were discarded at 0.8 cutoff values (Li & Godzik 2006).
The CD-HIT screened files of proteins were submitted to PSORTb (Yu et al. 2010) for the subcellular localization prediction. The proteins localized in four cell regions i.e., cytoplasmic membrane, outer membrane extracellular and periplasmic proteins, were selected. These membrane proteins were selected due to their important role in infection, attachment, and survival of pathogens (Nagpal et al. 2018; Ul-Ain et al. 2018).
The next requirement was to screen out virulent proteins. VFDB is a user-friendly database to find out the virulence factors in a variety of pathogenic bacteria (Chen et al. 2005). Protein BLAST (BLASTp), for similarity search with other infectious proteins, was used for screening in this database. Proteins showing score more than 100 and identity above 30% were selected. These virulent proteins screened by VFDB were kept in dataset to be analyzed further (Chen et al. 2005) and rest were discarded. The virulent proteins were submitted to an online antigenicity prediction server VaxiJen (Doytchinova and Flower 2007) and antigenicity was predicted at 0.4 threshold value (Barh et al. 2013; Hassan et al. 2016a,b). Antigenic virulent proteins were subjected to further analysis. For the selection of small sized proteins we used Expasy:ProtParam (Gasteiger et al. 2005). Proteins with molecular weight < 110 kDa were carried forwarded as potential candidates for vaccine construction. The shortlisted proteins for vaccine designs must be least identical with human protein or may be unique for bacteria. For making sure this condition similarity search of the proteins, with human (Human taxid: 9606), was done by NCBI BLASTp. The resultant proteins showing percent identity less than 35% or not found in human were selected for prediction of different type of epitopes.
MHCI Cytotoxic T Lymphocytes (CTL) Epitopes Prediction
Epitope is the portion of an antigen which is detected by immune system with the help of the B-lymphocytes (Chen et al. 2005). Many of the proteins of a pathogen can act as antigen and the portions of an antigen that attach with antigen binding site on either an antibody or a T-lymphocyte receptor sites are called epitopes which then help in producing cellular or humoral immune response.
NetCTL 1.2 server used to predict CTL epitopes in protein sequences at 0.75 thresholds level (Larsen et al. 2007). These predictions were done on the basis of (i) binding to MHC-I, (ii) proteasomal C-terminus cleavage score, (iii) transport efficiency of transporter associated with antigen processing (TAP).
Weight matrix was used for TAP score calculation while artificial neural network approach was deployed to calculate proteasomal C-terminal cleavage scores and peptide binding to MHC-I.
MHCII Helper T Lymphocyte (HTL) Epitopes Prediction
The prediction of MHCII HTL epitopes was done taking 27 human HLAs as a reference set, by Immune Epitope Database server (IEDB; Fleri et al. 2017). The server predicts the HTL epitopes based on their percentile rank or MHCII binding affinity which is expressed in terms of IC50 values which are inversely related to the binding affinity towards the MHC-II (Patronov & Doytchinova 2013). Ten epitopes from each protein were selected based on, the low percentile rank because the binding affinity to MHC-II, of predicted epitopes, is inversely proportional to the percentile rank (Paul et al. 2016).
Epitope Screening
Allergenicity Antigenicity, Toxicity and Water Solubility Prediction for Epitopes
Allergenicity prediction with better precision was achieved by the AlgPred server (Christensen et al. 2003). The multiple algorithms approach was employed by the server. The accuracy of the server is nearly 85% for the hybrid algorithm at threshold (− 0.4). Allows to predict allergens based on SVM modules using amino acid composition and similarity with known allergenic epitopes. It does prediction of allergens based on similarity of known epitope with any region of protein.
VaxiJen server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html/) was employed to predict antigenicity of the vaccine sequence. This server predicts the antigenic score wholly based on the query amino acid sequence and the target strain was selected as bacteria. 0.4 was the threshold value selected for it. VaxiJen precision for antigenicity is quite better, 70–89%. The antigenic scores for the vaccine sequence at 0.4 thresholds for designating the vaccine. Prediction of toxicity of peptides by using computational methods not only saves resources, but also helps in designing better least toxic therapeutic peptides by retaining the functionalities undisturbed (Gupta et al. 2013).
The list of the non-allergen and antigenic epitopes was imported in ToxinPred online server as batch submission for their toxicity prediction. The non-toxic epitopes were selected and rest were discarded.
INNOVAGEN peptide property calculator tool PepCalc was used to calculate the water solubility of peptides. Water soluble peptides were shortlisted.
Interferon-γ Epitope Prediction
The IFN-γ cytokine is a very important factor induced in response of pathogen attack. It activates macrophages as an immune response in the cell. It enhances the response of MHCII to antigens. A potential candidate of vaccine is a peptide not only be epitope for B and T cell but also be capable of inducing the appropriate immune cells to generate desired immunity response (Shtrichman and Samuel 2001). The vaccine candidate required to induce IFN-γ to destroy the infection agents in the cell (Flynn et al. 1993; Reljic 2007). The IFN-γ epitope server (Dhanda et al. 2013) was used to identify IFN-γ epitopes. The IFN-γ epitopes in CTL and HTL epitopes was predicted by IFN-γ epitope server by using support vector machine (SVM) hybrid approach. The epitopes with value IC50 < 100 with positive results were selected.
Prediction of CTL Epitope-HLA-DRB1complex Binding Affinity
The peptides which do not bind to any of an individual’s allelic MHC variants cannot be antigenic within a cellular context (Hattotuwagama et al. 2004). The human version of MHC complex is human leukocytes antigen complex (HLA). The 9-mer CTL epitopes were docked with HLA-DRB1*101 allele through MHCPred (Guan et al. 2003). MHCPred is an online available (http://www.jenner.ac.uk/MHCPred) used to predict binding affinity of CTL epitope-HLA-DRB1complex in terms of IC50 values. The 15-mer HTL epitopes were predicted through IEDB on the basis of seven HLA-DRB alleles (Wang et al. 2010) that’s why they were not docked by MHCPred server.
Vaccine Construct
MHC class I and II binding epitopes were screened based on their high binding affinity and non-allergenic nature for vaccine construction. AAY linkers are used to join the CTL epitopes and GPGPG linkers for HTL epitopes (Hasan et al. 2019). These linkers are helpful for differentiation and improvement of epitope presentation (Saadi et al. 2017; Dorosti et al. 2019). The adjuvant protein sequence SKKKK, of TLR2 agonist Pam3CSK4 Chain C, was attached in the start of vaccine construct sequence. This adjuvant enhances the immunogenic property of the designed vaccine. The adjuvant was joined to CTL epitope by another linker sequence EAAAK at N-terminus of vaccine construct (Arai et al. 2001).
Antigenicity, Allergenicity and Thermostability Test of the Vaccine Construct
The antigenicity and non-allergenicity of the vaccine construct was predicted by VaxiJen v 2.0 and AllerTOP v 2.0 respectively. Thermal stability of the vaccine construct was measured by an online available server SCooP (prediction of the stability curve of proteins). It measures the thermal stability in terms of Tm (the protein melting point) which shows the temperature at which half of the folds can become unfolded and denatured. It also measures the values of change in free energy (ΔG), standard enthalpy of folding (ΔHm) and standard heat capacity of folding (ΔCp) of protein.
Tertiary Structure Prediction, Refinement and Validation
The vaccine construct sequence was subjected to NCBI BLASTp but no suitable template was found for homology based modeling so it was submitted to an online available server I-TASSER for ab initio based structure prediction. The quality of the predicted structure was improved by using ModRefiner.
B-Cell Epitope Prediction
Receptors present in the B-lymphocytes surface recognize and binds B cell epitopes. ABCpred server was used for linear B cell epitopes prediction. The accuracy of the ABCpred server is 75% (0.75 specificities and 0.49 sensitivity) (Saha and Raghava 2006). The amino acid sequence of vaccine construct was submitted for 16-mer B cell linear epitope prediction. Discontinuous B-cell epitopes were predicted by an online server Discotope2.0 (Kringelum et al. 2012) by submitting refined 3D structure of vaccine construct. Discotope2.0 conformational B cell epitopes prediction is based on amino acids composition ratio between residues of epitope and non-epitope. The server specificity is 0.75, whereas the sensitivity is 0.47, at 3.7 threshold value.
Cysteine Mutation to Improve Disulfide Bonding
Cysteine is a sulfur containing amino acid. Two cysteine molecules combine to form cysteine by forming disulfide bond which improves protein stability (Liu et al. 2016). An online freely available server Disulfide by Design 2 v 2.13 (DbD2) was used to enhance disulfide bonding in vaccine construct. The server can predicts the suitable pairs of amino acids to be mutated with cysteine for disulfide bond formation. A list of potential amino acid pairs was generated and suitable pairs were selected keeping in view the resultant bond energy and B-factor (Craig & Dombkowski 2013).
Molecular Docking with Human Toll Like Receptor 2 (TLR-2)
The three dimensional structure of human TLR2 (PDB ID: 6NIG) was downloaded from RCSB Protein Data Bank (PDB). The structure was refined by using Chimera and chain A was selected for docking purpose. Vaccine construct was docked with Human TLR2 by online available server PatchDock. The resulting solutions were submitted to FireDock for refinement. The best refined complex model with lowest binding energy was selected. The interactions in docked complex were visualized by LIGPLOT: DIMPLOT (Laskowski & Swindells 2011).
Reverse Translation and Expression Vector Cloning of Vaccine Construct
The vaccine construct was prepared and optimized to be incorporated in expression vector for expression. The amino acid sequence of vaccine was reverse translated and optimized by JCAT server (JAVA Codon Adaptation Tool) server. GC content and codon adaptation (CAI) index was calculated for optimized sequence to ensure its suitability to be carried forward in cloning vector. The DNA sequence obtained from revere translation was modified by conjugation with XhoI and NdeI restriction site at the N-terminal and C-terminal sites respectively through SnapGene software. The modified DNA sequence was inserted in pET28a (+) vector at multiple cloning site (MCS) between the XhoI (158) and NdeI (1450) by using SnapGene restriction cloning module.
Results
Proteome Subtraction
The whole proteome of K. oxytoca strain ATCC 8724 (UniProt ID: UUP000007843), containing 5483 proteins was retrieved from the UniProt database. The whole proteome was passed through the subtractive proteomics pipeline.
CD-HIT refined the proteome and 5470 non-paralogous proteins at the 80% threshold were carried on for further analysis. Prediction of subcellular localization was the next step executed by submitting proteins to PSORTb. The proteins from four cellular location i.e., periplasm (188), extracellular (54), outer membrane (108) and cytoplasmic membrane (1398) were selected and carried forward. These total 1748 screened proteins were submitted to Expasy ProtParam to get small sized proteins. The proteins with less than 110 kDa were selected for vaccine construction (Hassan et al. 2016a,b) and number of proteins reduced to 197. NCBI BLASTp was used for taking off the non-human homologous proteins keeping “Human taxid: 9606” as the reference organism. The proteins showing more than 35% identity with human proteins were discarded. The number of qualified proteins reduced to 172.
Virulent proteins interact with different host pathways and play important role in pathogenicity that’s why are considered best for vaccine construction (Baseer et al. 2017). BLASTp search of VFDB (virulence factor database) was performed for virulent proteins identification (Ul Ain et al. 2018). Proteins showing identity and bit score greater than 30% and 100 respectively, were considered virulent and were further evaluated for antigenicity. Total 156 virulent proteins were submitted to VaxiJen v 2.0 server (Doytchinova & Flower 2007) for antigenicity score prediction at 0.4 threshold value (Naz et al. 2015; Hassan et al. 2016a,b). Total 127 proteins with VaxiJen score greater than 0.4 were selected and all other were discarded. Antigenic virulent proteins were subjected to further analysis. The number of protein reduced from 5483 to only 127 and these virulent and antigenic proteins were selected for CTLs and HTL epitope prediction and further analysis (Fig. 1).
Fig. 1.

Subtractive proteomic schema for vaccine construction against Klebsiella oxytoca
Cytotoxic T Lymphocytes (CTL) and Helper T Lymphocyte (HTL) Epitopes Prediction
CTLs (MHC-I binding) 1670 (9-mer) epitopes were predicted by NetCTL 1.2 server. The Immune Epitope Database (IEDB) server predicted 1270 HTL (MHC-II binding), 15-mer, epitopes against a reference set of seven human leukocyte antigen (HLAs) alleles.
Epitope Screening
Allergenicity Antigenicity, Toxicity and Water Solubility Prediction for Epitopes
Allergenicity scores were predicted through AlgPred 2.0 server. The results showed 1322 CTL and 1107 HTL epitopes which can activate the immune response in the cell (Sharma et al. 2020). VaxiJen filtered out 1109 CTL and 968 HTL epitopes. Total 675 CTL and 548 HTL nontoxic epitopes were predicted by ToxinPred server. PepCalc tool was used to predict water soluble peptides. Total 300 CTL and 250 HTL epitopes were screened out from water solubility filter.
Epitope Interferon-γ Induction Prediction
The vaccine candidate 12 HTL epitopes were selected based on IFN-γ induction (positive score) and MHCII binding affinity (Table 1). Fifty six IFN-γ positive CTL epitopes were further analyzed to qualify to be part of vaccine construct.
Table 1.
HTL epitopes selected for vaccine construct against Klebsiella oxytoca
| Protein ID | Alleles | Start | End | Method | Peptide sequence | Percentile rank | Adjusted rank | Allergenicity score | Antigenicity | IFN epitope score |
|---|---|---|---|---|---|---|---|---|---|---|
| A0A0H3HBY6 | HLA-DRB5*01:01 | 3 | 17 | Consensus (smm/nn/sturniolo) | EKKVVMRIRALHKRF | 0.21 | 0.21 | − 0.4281 | 0.57 | 0.030451 |
| HLA-DRB5*01:01 | 6 | 20 | Consensus (smm/nn/sturniolo) | VVMRIRALHKRFGAQ | 0.58 | 0.58 | − 0.598 | 1.24 | 0.013655 | |
| HLA-DRB5*01:01 | 2 | 16 | Consensus (smm/nn/sturniolo) | SEKKVVMRIRALHKR | 0.21 | 0.21 | − 0.5404 | 0.46 | 0.188259 | |
| A0A0H3HCH0 | HLA-DRB3*02:02 | 142 | 156 | NetMHCIIpan | QQRVAIARALAMKPS | 3.6 | 3.6 | − 4.5988 | 0.73 | 0.336966 |
| A0A0H3HAB9 | HLA-DRB3*01:01 | 117 | 131 | Consensus (comb.lib./smm/nn) | HVKGMKRDEAKAIAM | 0.71 | 0.71 | − 0.5438 | 2.1 | 0.206067 |
| HLA-DRB3*01:01 | 115 | 129 | Consensus (comb.lib./smm/nn) | LLHVKGMKRDEAKAI | 0.72 | 0.72 | − 0.4656 | 0.75 | 0.055908 | |
| HLA-DRB3*01:01 | 118 | 132 | Consensus (comb.lib./smm/nn) | VKGMKRDEAKAIAMQ | 0.72 | 0.72 | − 0.5340 | 0.49 | 0.170946 | |
| HLA-DRB3*01:01 | 116 | 130 | Consensus (comb.lib./smm/nn) | LHVKGMKRDEAKAIA | 0.74 | 0.74 | − 0.5529 | 0.63 | 0.105924 | |
| A0A0H3HGA0 | HLA-DRB5*01:01 | 85 | 99 | Consensus (smm/nn/sturniolo) | TVSRFMLLRPGTHKR | 0.88 | 0.88 | − 0.7711 | 0.42 | 0.602909 |
| HLA-DRB1*03:01 | 15 | 29 | Consensus (smm/nn/sturniolo) | GQRRVLSDISLTLKP | 1.1 | 1.1 | − 9.901 | 0.53 | 0.3684091 | |
| A0A0H3HDE1 | HLA-DRB1*03:01 | 151 | 165 | Consensus (smm/nn/sturniolo) | HGWSAVNVDDRWLFR | 0.24 | 0.24 | − 0.5072 | 0.58 | 0.937815 |
| A0A0H3H729 | HLA-DRB5*01:01 | 214 | 228 | Consensus (smm/nn/sturniolo) | RSVTRAFNHMAAGVK | 0.16 | 0.16 | − 0.697 | 1.43 | 0.509832 |
Prediction of CTL Epitope-HLA-DRB1 Complex Binding Affinity
The IC50 values are the most widely quoted binding affinity measures (Ruppert et al. 1994) and it is inversely proportional to the nominal binding affinity (Guan et al. 2003). If the IC50 value is above 5000 then the peptide cannot bind to the MHC molecules so, the peptides with lower IC50 values (< 100) were selected (Table 2).
Table 2.
CTL epitopes selected for vaccine construct against Klebsiella oxytoca
| Cellular location | Protein ID | Peptide sequence | VaxiJen score | IFN epitope score | Water solubility | MHCPred | AllerTOP v. 2.0 | ToxinPred |
|---|---|---|---|---|---|---|---|---|
| Periplasmic | A0A0H3HK70 | TYDKLAKKY | 0.5864 | 0.045 | Good | 1.53 | ||
| A0A0H3H8B0 | MQEQRASAY | 0.5668 | 0.213 | Good | 6.32 | Non-allergenic | Non toxic | |
| A0A0H3HFM6 | LAEGLRADY | 0.4985 | 0.378 | Good | 13.40 | Non-allergenic | Non toxic | |
| A0A0H3HAJ2 | QSDAEKSGV | 1.6633 | 0.486 | Good | 18.49 | Non-allergenic | Non toxic | |
| Outer membrane | A0A0H3H611 | DSSSSDSVY | 1.4043 | 0.521 | Good | 19.19 | Non-allergenic | Non toxic |
| SSNHSADIY | 0.7153 | 0.085 | Good | 23.99 | Non-allergenic | Non toxic | ||
| FSDIDLSDV | 0.5651 | 0.089 | Good | 3.33 | Non-allergenic | Non toxic | ||
| A0A0H3H6X7 | NSDDTSYAR | 0.9284 | 0.211 | Good | 5.30 | Non-allergenic | Non toxic | |
| A0A0H3H9M6 | RTDSRQLDS | 2.1329 | 0.006 | Good | 6.17 | Non-allergenic | Non toxic | |
| Extracellular | A0A0H3H7X1 | SEEPITFEY | 0.6935 | 0.416 | Good | 15.63 | Non-allergenic | Non toxic |
| A0A0H3HDA8 | HLEQIELRY | 1.8453 | 0.191 | Good | 6.95 | Non-allergenic | Non toxic | |
| Cytoplasmic membrane | A0A0H3HF34 | QSDEVAAAA | 0.9697 | 0.348 | Good | 5.40 | Non-allergenic | Non toxic |
| A0A0H3HFF1 | HIELRALSY | 1.5293 | 0.667 | Good | 0.89 | Non-allergenic | Non toxic | |
| SADSASALM | 0.5090 | 0.977 | Good | 7.80 | Non-allergenic | Non toxic | ||
| A0A0H3HCS4 | ATSRARQPY | 0.4767 | 0.059 | Good | 41.78 | Non-allergenic | Non toxic |
Vaccine Construct
Fifteen CTL (MHCI class) and 13 HTL (MHCII class) epitopes were selected, based on their high binding affinity and non-allergenic nature, for vaccine construction (Fig. 2).
Fig. 2.

Amino acid sequence of vaccine construct for Klebsiella oxytoca
Antigenicity, Allergenicity and Thermostability Test of the Vaccine Construct
The vaccine construct was predicted as non-allergenic and antigenic in nature. SCooP (prediction of the stability curve of proteins) showed Tm = 61.5 °C, ΔGr = − 7.9 kcal/mol, ΔCp = − 5.19 kcal/(mol K) and ΔHm = − 170.5 kcal/mol (Fig. 3). Which reflect the stability of protein.
Fig. 3.
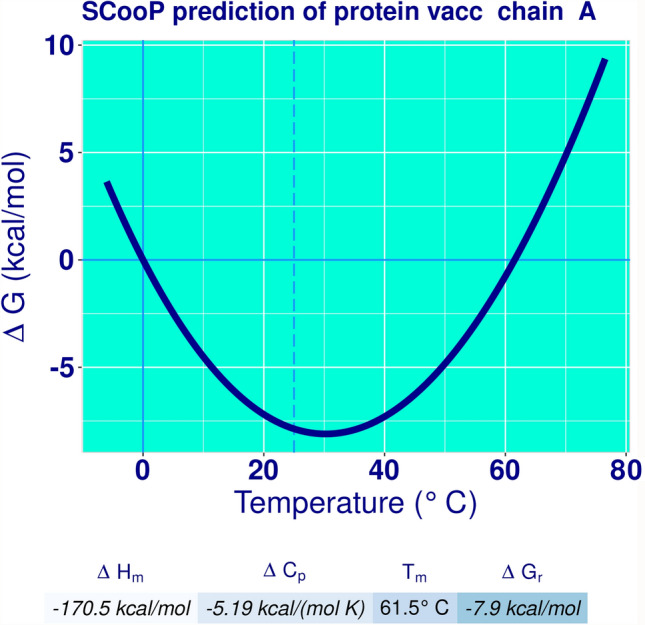
Thermostability curve of Klebsiella oxytoca vaccine protein
Tertiary Structure Prediction, Refinement and Validation of Vaccine Construct
The vaccine construct sequence was subjected to NCBI BLASTp but no suitable template was found for homology based modeling so it was submitted to an online available server I-TASSER for ab initio based structure prediction. Refinement of the predicted model was done by using ModRefiner (Fig. 4).
Fig. 4.
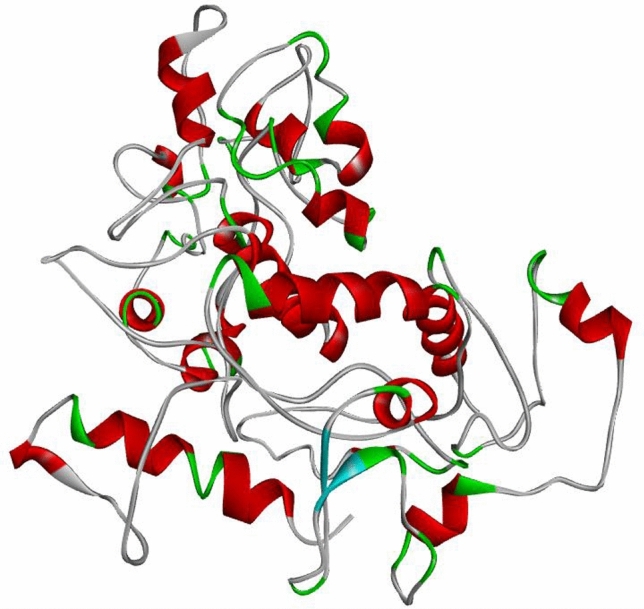
Predicted 3D structure of Klebsiella oxytoca vaccine construct
Prediction of B (Lymphocytes) Cell Epitopes
The B-cell (lymphocytes) epitopes identification is very important for the vaccine development research (Shirai et al. 2014). Eight (16-mer) linear B-cell epitopes were predicted by ABCprederver at 0.85 threshold level (Table 3). B-cell epitopes are mostly present at discontinuous locations within the sequence far apart from each other but brought together during folding in 3D structure (Kringelum et al. 2013). Thirty four discontinuous epitopes were predicted by DiscoTope server at threshold of − 3.7 (Table 4).
Table 3.
Predicted B cell linear epitopes in vaccine construct for Klebsiella oxytoca
| Rank | Sequence | Start position | Score |
|---|---|---|---|
| 1 | GPGGQRRVLSDISLTL | 393 | 0.93 |
| 2 | RALAMKPSGPGPGLSW | 243 | 0.92 |
| 3 | SRQGPGPGTHDLQLAK | 348 | 0.91 |
| 3 | SDSRYAAYTVDALNSE | 162 | 0.91 |
| 3 | YGMAHKADVYAAYETT | 145 | 0.91 |
| 4 | YHLETLELRYAAYKTN | 73 | 0.89 |
| 5 | PGPGMHNSVMRLTISN | 292 | 0.88 |
| 5 | PGPGEKKVVMRIRALH | 192 | 0.88 |
| 6 | PGPGPGHGWSAVNVDD | 410 | 0.87 |
| 7 | QGVETDLPQLRALDGP | 277 | 0.86 |
| 7 | QAVSYGPGPGEKKVVM | 186 | 0.86 |
| 8 | GTHKRGPGPGGQRRVL | 386 | 0.85 |
| 8 | VVMRIRALHKRFGAGP | 217 | 0.85 |
Table 4.
Predicted discontinuous epitopes in 3D structure of vaccine construct for Klebsiella oxytoca
| Nos | Residues | Contact numbers | DiscoTope score |
|---|---|---|---|
| 1 | GLU123, GLU130, GLY238 | 0 | − 1.906, − 3.442, − 0.980 |
| 2 | SER66, HIS67, THR329, SER66, | 1 | 1.392, 0.663, − 2.111, 0.667 |
| 3 | GLY65, ARG70, ASN124, ASN176, VAL330, ARG70, ASN124 | 3 | 1.621, − 1.187, − 2.030, − 3.616, − 3.040, − 1.044, − 1.73 |
| 4 | LEU69, GLU173 VAL330 | 4 | − 2.100, − 2.387, − 3.220 |
| 5 | GLU71, TYR121, PRO236 | 5 | − 1.514, − 3.301, 0.379 |
| 6 | GLY65, GLU173 | 6 | 1.094, − 3.147 |
| 7 | LYS72, GLU180, THR219, LYS285 | 7 | − 3.456, − 3.119, − 0.831, − 3.436 |
| 8 | MET68, | 8 | − 0.689 |
| 9 | SER73, GLU183, MET68, LYS285 | 9 | − 2.647, − 2.797, − 1.266, − 3.673 |
| 10 | ALA127, ASN217 | 10 | − 2.900, − 1.729 |
Cysteine Mutation to Improve Disulfide Bonding
The analysis provides option of 61 pairs of amino acids which can be potentially mutated to cysteine along with estimated bond energy χ3 and β factors as a result of mutation. Residues pairs with lower bond energy < 1.5 were mutated to cysteine 184Val-218Val, 156Ala-163Asp, 420Ala-424Asp, 43Asp-264Leu and 179Ala-221Ile (Table 5). The χ3 of selected pairs ranged from − 93.27 to 101.17 and β factor 10.42 to 23.92. The disulfide bonds appeared as yellow cylinders in the structure (Fig. 5). The mutated residues are encircled in the sequence (Fig. 6). The thermal stability of the vaccine can be enhanced by increasing disulfide bindings without affecting its efficacy (Bouvier et al. 2003). Our current study proves this finding. The mutated sequence 3D vaccine structure submitted to SCooP resulted in 1 point rise in Tm (Fig. 7). This shows that the thermostability of vaccine can be enhanced by disulfide engineering.
Table 5.
Mutated amino acids pairs in vaccine construct of Klebsiella oxytoca
| Amino acid | Bond | |||||
|---|---|---|---|---|---|---|
| Seq# | AA | Seq# | AA | χ3 | Energy (kcal/mol) | Sum B-factors |
| 184 | Val | 218 | Val | − 86.21 | 0.07 | 10.42 |
| 156 | Ala | 163 | Asp | − 83.27 | 0.72 | 12.78 |
| 420 | Ala | 424 | Asp | 94.41 | 0.99 | 14.29 |
| 43 | Asp | 264 | Leu | 96.71 | 1.07 | 11.12 |
| 179 | Ala | 221 | Ile | − 76.81 | 1.07 | 11.90 |
Fig. 5.
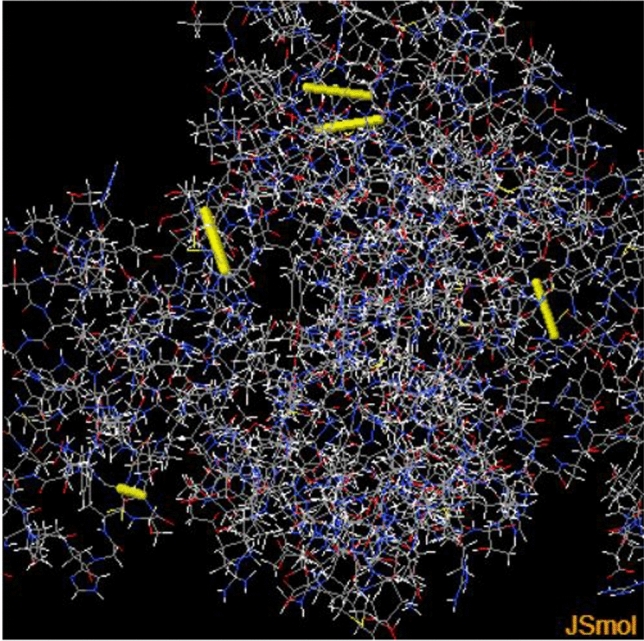
Disulfide engineered structure of Klebsiella oxytoca vaccine construct
Fig. 6.

Amino acid sequence of Klebsiella oxytoca vaccine construct with cysteine mutations
Fig. 7.
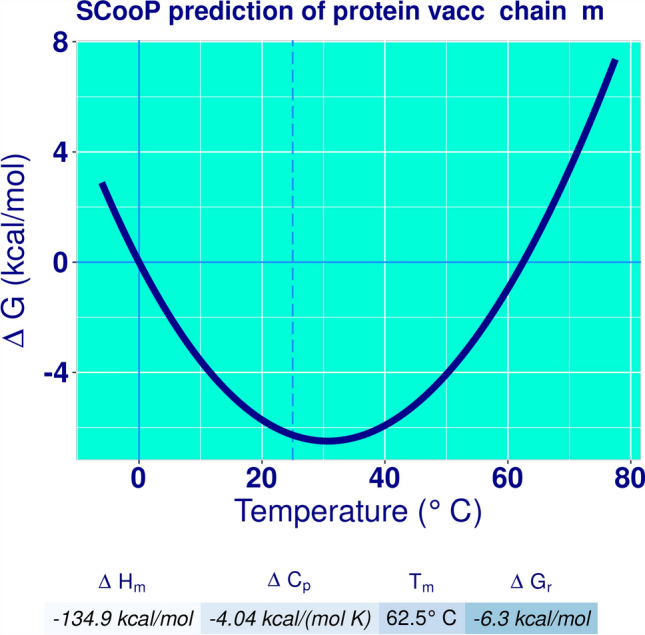
Thermostability curve of Klebsiella oxytoca vaccine protein after disulfide engineering
Molecular Docking of Vaccine Construct with Human TLR-2
Human toll like receptor 2 (TLR-2) is widely used as vaccine target receptor (Basto and Leitão 2014) it is a protein that in humans is encoded by the TLR2 gene (Rock et al. 1998). It is a membrane protein, a receptor, which is produced on the surface of certain cells and detects foreign invaders and transmits the appropriate signals to immune cells (Borrello et al. 2011). The 3D model of human TLR2 (PDBID: 6NIG) was downloaded from RCSB Protein Data Bank (PDB). The refund chain A is shown in Fig. 8. Vaccine construct was docked with Human TLR2 by online available server PatchDock. The vaccine-HTLR2 docked models (solutions) were received via email as top 20 models with their respective docking score. The top 10 solutions (vaccine-HTLR2 complexes) were refined using FireDock. The solution/complex showing the highest refinement rank (solution 6) with lowest binding affinity of − 85.71 kcal/mol, was selected. Five hydrogen bonds with bond distance less than 3 Å were formed between vaccine and HTLR2. A salt bridge was found between Arg375 (HTL2) and Glu141 (Vaccine) (Fig. 9). Three dimensional interaction complex was visualized by BIOVADiscovery Studio (Fig. 10).
Fig. 8.
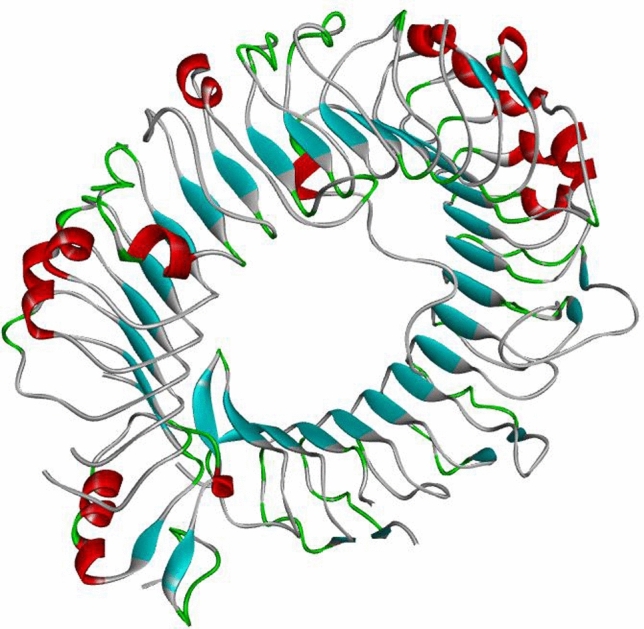
Chain A of human toll like receptor 2 (PDB ID: 6NIG)
Fig. 9.

Two dimensional interactions in docked complex of human TLR2 and Klebsiella oxytoca vaccine construct
Fig. 10.
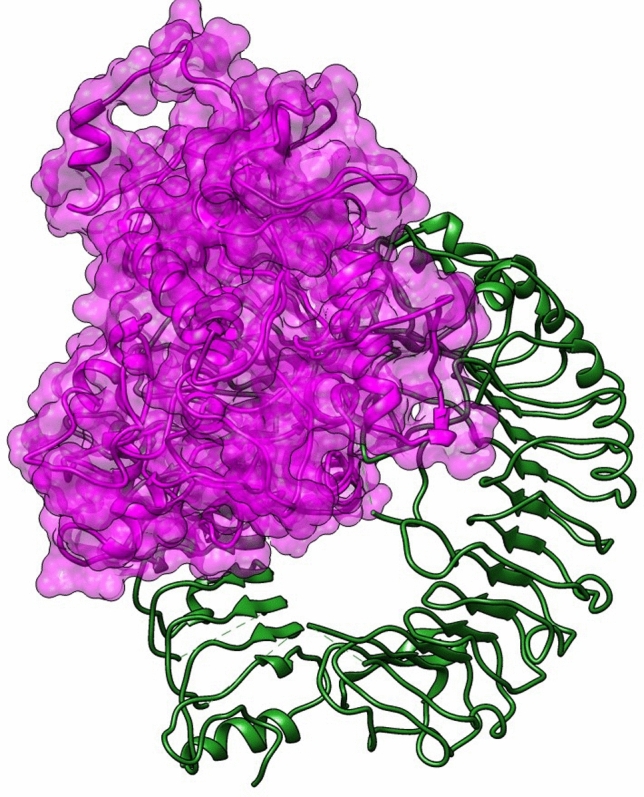
Three dimensional structure of docked complex of human TLR2 and Klebsiella oxytoca vaccine construct (vaccine construct: purple and solid, HTLR2: green)
Reverse Translation and Expression Vector Cloning of Vaccine Construct
Reverse translation of the primary sequence of vaccine construct was done through JCAT (Grote et al. 2005) by selection Escherichia coli as reference organism. The human expression system is different from that of E. coli that’s why codon adaptation index (CAI) was calculated to ensure high accuracy of translated sequence (Khatoon et al. 2017). CAI is a measure of the synonymous DNA/RNA codon usage bias and reflects the resemblance in the synonymous codon usage of query gene set and that of a reference set (Sharp and Li 1987; Carbone et al. 2003). Vaccine protein was reverse transcribed and codon adaptation index of the adapted codons was found to be 0.991 indicating the higher proportion of most abundant similar codons. The frequency of G and C base pair is called GC content (Carbone et al. 2003). The GC content calculated for the optimized codons was 55.45%. Computational cloning, by using the SnapGene software, was done to check the expression of inserted gene, The optimized codon sequence, of vaccine, obtained by reverse translation, was inserted into pET28a (+) vector plasmid at multiple cloning site (MCS) between XhoI and NdeI restriction sites (Sikarwar et al. 2013; Hasan et al. 2019; Khan et al. 2020). Multiple cloning sites are a feature that allows for the insertion of foreign DNA without disrupting the rest of the plasmid which makes it extremely useful in biotechnology, bioengineering, and molecular genetics (Clark 2005). The pET vector system is a powerful and widely used system for expressing recombinant proteins in E. coli (Studier et al. 1990). The gene of interest is initially cloned into the pET vector in a bacteria host that lacks the T7 RNA polymerase gene. This eliminates plasmid instability due to expression of proteins of interest that may be harmful to host cells (Dubendorf and Studier 1991). The construct codons lacked restriction sites for XhoI and NdeI which were added manually, ensuring its compatibility with vector restriction site, for the cloning purpose (Fig. 11). A clone containing 6586 bp was constructed including the 450 bp of query gene sequence. The query sequence was shown as red region in pET28a (+) vector sequence (Fig. 12).
Fig. 11.

The engineered Klebsiella oxytoca vaccine construct map for cloning
Fig. 12.

The cloned pET28a (+) vector showing Klebsiella oxytoca vaccine construct as red region
Discussion
The modern developments in Bioinformatics techniques had opened diversified ways to develop protection against human pathogens (Khan et al. 2019). The use of a vaccine against a particular pathogen is an efficient way to protect against infections. The classical vaccine development process against infectious microorganisms is successfully working in medical science worldwide (Serruto & Rappuoli 2006; Heldens, et al. 2008). However vaccine development by these traditional approaches have becoming more tedious laborious and expensive, which is not suitable in current era of advancement in science (Khan et al. 2019). Computational based methods are employed efficiently to overcome these drawbacks of conventional vaccine development. These computational approaches are rapid, inexpensive, and reliable with particular results (Saha and Raghava 2006). With the passage of time computational strategy for vaccines development of has become lucrative for scientist because of its safety, simplicity and specificity (Sette & Rappuoli 2010). So far, no approved vaccine for the K. oxytoca has been produced; an effective vaccine is required to efficiently manage the clinical borne infections by this pathogen. In silico multiepitope vaccine designing have many benefits compared to conventional vaccines (Li et al. 2014). The vaccine designed by using this method allows the immune system of the host to target specifically appropriate antigenic epitopes, thus avoiding non-protective reactions, the autoimmune response from the host and pathogen evasion of the immune system (Wiederstein and Sippl 2007; Li et al. 2014). We used a subtractive proteomics pipeline to identify antigenic proteins for designing vaccine against K. oxytoca. CD-HIT tools were used to refine the proteome refined the proteome and resulted in 5470 non-paralogous proteins at the 80% threshold which were carried on for further analysis (Hassan et al. 2016a,b). The proteins of four location i.e., periplasm, extracellular, outer membrane and cytoplasmic membrane were selected by PSORTb. The proteins from these four locations were selected on the basis of their importance in vaccine designing (Nagpal et al. 2018) and due to their important role in infection, attachment, and survival of pathogens (Ul Ain et al. 2018). Smaller sized proteins are good candidates for vaccine design (Hassan et al. 2016a,b) so we selected protein with weight less than 110 kDa.
Virulence is one of the vaccine’s vital characteristics. Virulent proteins can potentially evolve the infection pathways as compared to non-virulent proteins (Naz et al. 2015). We selected virulent protein from VFDB generated results. Human homologs proteins were removed, to avoid autoimmune response, followed by the removal of paralogs and proteins that are not essential for the survival of the pathogen (Solank et al. 2021). We removed all the protein having more than 30% identity with human protein. The final set of screened proteins subjected to immunoinformatics tools for designing an effective vaccine. MHCs, also called human leukocyte alleles (HLA) are among the most polymorphic proteins in higher vertebrates, with more than 6000 class I and class II MHC molecules listed in IMGT/HLA. The selections of these alleles are the prerequisite for vaccine development. MHC class I and MHC class II binding epitopes were predicted at the first step of the immunoinformatics pipeline. The immune system has T cells that readily identify MHC class I and MHC class II epitopes. MHC molecules are present on antigenic cell surface recognized by the T cell receptors (Chen et al. 2005). Class I MHCs protein are present on almost all the nucleated body cells (Sanchez-Trincado et al. 2017). MHC class I represents the CTL, peptides that have been through the cytosolic pathway are represented by MHC-I, these peptides are epitopes, either antigenic portion of pathogen or endogenous proteins (Wieczorek et al. 2017). MHC class II represents HTLs, peptides that have been through the endocytic pathways, the peptides are antigenic portions of the pathogen surface (Wosen et al. 2018). The oligopeptides from the pathogen protein bind to MHC II protein and these bond pathogenic proteins are recognized by T-cells (Jones et al. 2006). Appropriate number of the HTL epitopes results in the induced pathogen specific immune response triggered by T cell. Class II MHCs are expressed on antigen presenting cells (APCs) such as B cells, macrophages and dendritic cells (Nielsen et al. 2003). In the current study we predicted 1670 CTL and 1270 HTL epitopes.
The final vaccine construct consisted of a good number of B and T cell epitopes. Thermostability of a vaccine is one of the most important property of a successful vaccine (Yadav, et al 2021a) and thermostability was checked by SCooP. High thermal stability of a protein can be judged by values of standard enthalpy of folding measured at the melting temperature intervals (ΔHm), in combination with Tm values (Pucci & Rooman 2016). To attain thermal stability, proteins need large negative values of ΔH that make the value of ΔG more negative, moreover a less negative value of ΔCp increases the Tm values and in turn the thermal stability. The large Tm values with large negative ΔH and ΔG values give evidence for the observed thermal stability of the enzymes standard heat capacity of folding (ΔCp), and free energy change (ΔG) correlated with the protein folding with respect to temperature (Yadav et al. 2021a). In silico studies have proved high melting temperature as an indicator of thermostability (Khairudin and Mazlan 2013). The values of Tm = 61.5 °C, ΔGr = − 7.9 kcal/mol, ΔCp = − 5.19 kcal/(mol K) and ΔHm = − 170.5 kcal/mol shows stability of vaccine protein. The vaccine was thermostable, non-allergen, nontoxic and antigenic in natures which reflects the standards of a good vaccine (Liu and Irvine 2015). The tertiary structure provides information such as a normal function of the protein, dynamics, interaction with other proteins and ligands (Mittag et al. 2010). The suitable templates homology modeling of vaccine construct were not found so, online available serer I TASEER was used for tertiary structure prediction by using multiple templates (Zhang 2008). Five models were generated for query sequence and good quality structure with high C score was selected. C-score is a confidence score for quality estimation of I-TASSER generated models. It is calculated on the basis of significance of threading template alignments and the convergence parameters of the structure assembly simulations. C-score is generally in the range between − 5 and 2 and high value reflects the high confidence level on predicted structure (Yang et al. 2015). The PDB files of vaccine construct and HTLR2 were submitted to PatchDock server for protein–protein docking. PatchDock is a geometry-based molecular docking algorithm (Duhovny et al. 2002). The results received via email in the form of 20 complex solution PDB files. Top 10 complex solution were selected for refinement. This algorithm is aimed at finding docking transformations that yield good molecular shape complementarity (Schneidman-Duhovny et al. 2005) a transformation file was also generated along with the complex PDB files. The PDB file of vaccine and HTLR2 and transformation file for top ten complexes were submitted to FireDock for refinement. The methods used by this server include optimization of side-chain conformations and rigid-body orientation and allows a high-throughput refinement (Mashiach et al. 2008). The protocol of FireDock thoroughly tested (Andrusier et al. 2007) on protein–protein docking benchmarks (Mintseris et al. 2005) which makes the results more reliable. JCat server was used for the optimization of codons, in order to increase the expression of vaccine protein in the E. coli system. The vaccine was reverse translated and CAI and GC content were calculated by this server. CAI is a measure of the synonymous DNA/RNA codon usage bias and reflects the resemblance in the synonymous codon usage of query gene set and that of a reference set (Sharp & Li 1987; Carbone et al. 2003). The CAI was 0.991 and GC content 55.4% have been reported for our designed vaccine. These values (CAI and GC content) support the fact that vaccine will be highly expressed in E. coli expression system (Khan et al. 2019). Vaccines are generally tested on model organisms before getting approval for human use. However, most of the time, the vaccine may show promising results in model organisms but can’t show same successful results in human being. This might be because of the complicated human immune system. The in silico designed vaccines are computationally designed strictly by using human proteins and immune system which make it specific and effective.
Conclusion
We presented a study about the prediction of an effective, non-toxic and non-allergenic vaccine against of K. oxytoca strain ATCC 8724. A smart and comprehensive approach of subtractive proteomics was employed to screen out the potential 28 peptides (CTL and HTL epitopes) sequences out of 5483 protein. Epitopes were joined with appropriate linkers to design 450 aa long vaccine construct. The predicted 3D structure of vaccine construct was further tested for stability and B cell epitopes. It showed good binding affinity with human Toll like receptor protein. The vaccine construct was reverse transcribed and confirmed its expression by inserting in pET28a (+) vector. This predicted vaccine construct showed stability up to 61.5 °C temperature. This computational investigation can be used as baseline study for vaccine designating in wet labs. The preliminary study result can save time and resources in vaccine production experiments.
Author Contributions
QY: Conceived idea, designed methodology, helped in analysis and Manuscript writing. HA: Helped in data interpretation and write-up. SB: Helped in result interpretation drafting. RR: Execution of methodology (partly). MSK: Execution of methodology (partly). HA: drafting and analysis (partly). AM: Helped in manuscript writing and analysis.
Funding
No funding was obtained for this research work.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declarations
Conflict of interest
The authors declared no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Qudsia Yousafi, Email: qudsia@cuisahiwal.edu.pk.
Humaira Amin, Email: humaira.amin96@gmail.com.
Shabana Bibi, Email: dazzling.grin@gmail.com.
Rafea Rafi, Email: rafia.rafi000@gmail.com.
Muhammad S. Khan, Email: saad.khan@cuisahiwal.edu.pk
Hamza Ali, Email: maikeehamza@gmail.com.
Ashir Masroor, Email: masroorakb@gmail.com.
References
- Andrusier N, Nussinov R, Wolfson HJ. FireDock: fast interaction refinement in molecular docking. Proteins. 2007;69:139–159. doi: 10.1002/prot.21495. [DOI] [PubMed] [Google Scholar]
- André FE. Vaccinology: past achievements, present roadblocks and future promises. Vaccine. 2003;21(7–8):593–595. doi: 10.1016/S0264-410X(02)00702-8. [DOI] [PubMed] [Google Scholar]
- Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001;14(8):529–532. doi: 10.1093/protein/14.8.529. [DOI] [PubMed] [Google Scholar]
- Barh D, Barve N, Gupta K, Chandra S, Jain N, Tiwari S, Azevedo V. Exoproteome and secretome derived broad spectrum novel drug and vaccine candidates in Vibrio cholerae targeted by Piper betel derived compounds. PLoS ONE. 2013;8(1):e52773. doi: 10.1371/journal.pone.0052773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baseer S, Ahmad S, Ranaghan KE, Azam SS. Towards a peptide-based vaccine against Shigella sonnei: a subtractive reverse vaccinology based approach. Biologicals. 2017;50:87–99. doi: 10.1016/j.biologicals.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Basto AP, Leitão A. Targeting TLR2 for vaccine development. J Immunol Res. 2014 doi: 10.1155/2014/619410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrello S, Nicolo C, Delogu G, Pandolfi F, Ria F. TLR2: a crossroads between infections and autoimmunity? Int J Immunopathol Pharmacol. 2011;24(3):549–556. doi: 10.1177/039463201102400301. [DOI] [PubMed] [Google Scholar]
- Bouvier A, Chapline J, Boerner R, Jeyarajah S, Cook S, Acharya PS, Shepard SR. Identifying and modulating disulfide formation in the biopharmaceutical production of a recombinant protein vaccine candidate. J Biotechnol. 2003;103(3):257–271. doi: 10.1016/S0168-1656(03)00106-8. [DOI] [PubMed] [Google Scholar]
- Carbone A, Zinovyev A, Képes F. Codon adaptation index as a measure of dominating codon bias. Bioinformatics. 2003;19(16):2005–2015. doi: 10.1093/bioinformatics/btg272. [DOI] [PubMed] [Google Scholar]
- Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, Jin Q. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33(Suppl_1):D325–D328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JK, Lamberth K, Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lund O. Selecting informative data for developing peptide-MHC binding predictors using a query by committee approach. Neural Comput. 2003;15(12):2931–2942. doi: 10.1162/089976603322518803. [DOI] [PubMed] [Google Scholar]
- Clark DP (2005) Molecular biology. Academic, p 611. ISBN 0-12-175551-7
- Craig DB, Dombkowski AA. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013;14(1):1–7. doi: 10.1186/1471-2105-14-S19-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decré D, Burghoffer B, Gautier V, Petit JC, Arlet G. Outbreak of multi-resistant Klebsiella oxytoca involving strains with extended-spectrum β-lactamases and strains with extended-spectrum activity of the chromosomal β-lactamase. J Antimicrob Chemother. 2004;54(5):881–888. doi: 10.1093/jac/dkh440. [DOI] [PubMed] [Google Scholar]
- Delany I, Rappuoli R, Seib KL. Vaccines, reverse vaccinology, and bacterial pathogenesis. Cold Spring Harb Perspect Med. 2013;3:a012476. doi: 10.1101/cshperspect.a012476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorosti H, Eslami M, Negahdaripour M, Ghoshoon MB, Gholami A, Heidari R, Ghasemi Y. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J Biomol Struct Dyn. 2019;37(13):3524–3535. doi: 10.1080/07391102.2018.1519460. [DOI] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumor antigens and subunit vaccines. BMC Bioinform. 2007;8(1):1–7. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubendorf JW, Studier FW. Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J Mol Biol. 1991;219(1):45–59. doi: 10.1016/0022-2836(91)90856-2. [DOI] [PubMed] [Google Scholar]
- Duhovny D, Nussinov R, Wolfson HJ (2002) Efficient unbound docking of rigid molecules. In: Guigo R, Gusfield D (eds) Proceedings of the fourth international workshop on algorithms in bioinformatics, 17–21 September 2002, Rome, Italy. Springer, pp 185–200
- Fleri W, Paul S, Dhanda SK, Mahajan S, Xu X, Peters B, Sette A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front Immunol. 2017;8:278. doi: 10.3389/fimmu.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Fuente J, Kopacek P, Lew-Tabor A, Maritz-Olivier C. Strategies for new and improved vaccines against ticks and tick-borne diseases. Parasite Immunol. 2016;38:754–769. doi: 10.1111/pim.12339. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: The proteomics protocols handbook. Humana Press, Totowa, pp 571–607
- Gorkiewicz G. Nosocomial and antibiotic-associated diarrhoea caused by organisms other than Clostridium difficile. Int J Antimicrob Agents. 2009;33:S37–S41. doi: 10.1016/S0924-8579(09)70015-9. [DOI] [PubMed] [Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33(Suppl_2):W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan P, Doytchinova IA, Zygouri C, Flower DR. MHCPred: a server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res. 2003;31:3621–3624. doi: 10.1093/nar/gkg510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP, Open Source Drug Discovery Consortium In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8(9):e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M, Ghosh PP, Azim KF, Mukta S, Abir RA, Nahar J, Khan MMH. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb Pathog. 2019;130:19–37. doi: 10.1016/j.micpath.2019.02.023. [DOI] [PubMed] [Google Scholar]
- Hassan A, Naz A, Obaid A, Paracha RZ, Naz K, Awan FM, Ali A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genomics. 2016;17(1):1–25. doi: 10.1186/s12864-016-2951-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A, Naz A, Obaid A, Paracha RZ, Naz K, Awan FM, Muhmmad SA, Janjua HA, Ahmad J, Ali A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genomics. 2016;17(1):732. doi: 10.1186/s12864-016-2951-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattotuwagama CK, Guan P, Doytchinova IA, Zygouri C, Flower DR. Quantitative online prediction of peptide binding to the major histocompatibility complex. J Mol Graph Model. 2004;22:195–207. doi: 10.1016/S1093-3263(03)00160-8. [DOI] [PubMed] [Google Scholar]
- Heldens JGM, Patel JR, Chanter N, Ten Thij GJ, Gravendijck M, Schijns VEJC, Schetters TP. Veterinary vaccine development from an industrial perspective. Vet J. 2008;178(1):7–20. doi: 10.1016/j.tvjl.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann KM, Deutschmann A, Weitzer C, Joainig M, Zechner E, Högenauer C, Hauer AC. Antibiotic-associated hemorrhagic colitis caused by cytotoxin-producing Klebsiella oxytoca. Pediatrics. 2010;125(4):e960–e963. doi: 10.1542/peds.2009-1751. [DOI] [PubMed] [Google Scholar]
- Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6(4):271–282. doi: 10.1038/nri1805. [DOI] [PubMed] [Google Scholar]
- Kelly DF, Rappuoli R (2005) Reverse vaccinology and vaccines for serogroup B Neisseria meningitides. In: Hot topics in infection and immunity in children II. Springer, Boston, pp 217–223 [DOI] [PubMed]
- Khairudin NBA, Mazlan NSF. Molecular docking study of beta-glucosidase with cellobiose, cellotetraose and cellotetriose. Bioinformation. 2013;9:813–815. doi: 10.6026/97320630009813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Khan A, Rehman AU, Ahmad I, Ullah S, Khan AA, Ali SS, Gul S, Wei DQ. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in silico expression. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2019;73:390–400. doi: 10.1016/j.meegid.2019.06.006. [DOI] [PubMed] [Google Scholar]
- Khan S, Ali SS, Zaheer I, Saleem S, et al. Proteome-wide mapping and reverse vaccinology-based B and T cell multi-epitope subunit vaccine designing for immune response reinforcement against Porphyromonas gingivalis. J Biomol Struct Dyn. 2020;1:15. doi: 10.1080/07391102.2020.1819423. [DOI] [PubMed] [Google Scholar]
- Khatoon N, Pandey RK, Prajapati VK. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep. 2017;7(1):1–12. doi: 10.1038/s41598-017-08842-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol. 2012;8(12):e1002829. doi: 10.1371/journal.pcbi.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kringelum JV, Nielsen M, Padkjaer SB, Lund O. Structural analysis of B-cell epitopes in antibody: protein complexes. Mol Immunol. 2013;53:24–34. doi: 10.1016/j.molimm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007;8(1):1–12. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. CD-Hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Li W, Joshi M, Singhania S, Ramsey K, Murthy A. Peptide vaccine: progress and challenges. Vaccines. 2014;2(3):515–536. doi: 10.3390/vaccines2030515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Irvine DJ. Guiding principles in the design of molecular bioconjugates for vaccine applications. Bioconjug Chem. 2015;26(5):791–801. doi: 10.1021/acs.bioconjchem.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Wang Y, Luo X, Li J, Reed SA, Xiao H, Young S, Schultz PG. Enhancing protein stability with extended disulfide bonds. Proc Natl Acad Sci USA. 2016;113(21):5910–5915. doi: 10.1073/pnas.1605363113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ. FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008;36(Suppl_2):W229–W232. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehla K, Ramana J. Surface proteome mining for identification of potential vaccine candidates against Campylobacter jejuni: an in silico approach. Funct Integr Genomics. 2017;17(1):27–37. doi: 10.1007/s10142-016-0530-z. [DOI] [PubMed] [Google Scholar]
- Michalik MM, Djahanshiri B, Leo JC, Linke D. Reverse vaccinology: the pathway from genomes and epitope predictions to tailored recombinant vaccines. Methods Mol Biol. 2016;1403:87–106. doi: 10.1007/978-1-4939-3387-7_4. [DOI] [PubMed] [Google Scholar]
- Mintseris J, Wiehe K, Pierce B, Anderson R, Chen R, Janin J, Weng Z. Protein–protein docking benchmark 2.0: an update. Proteins. 2005;60:214–216. doi: 10.1002/prot.20560. [DOI] [PubMed] [Google Scholar]
- Mittag T, Kay LE, Forman-Kay JD. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit Interdiscip J. 2010;23(2):105–116. doi: 10.1002/jmr.961. [DOI] [PubMed] [Google Scholar]
- Muruato LA, Tapia D, Hatcher CL, Kalita M, Brett PJ, Gregory AE, et al. The use of reverse vaccinology in the design and construction of nano-glycoconjugate vaccines against Burkholderia pseudomallei. Clin Vaccine Immunol. 2017;24(11):e00206–e217. doi: 10.1128/CVI.00206-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal G, Usmani SS, Raghava GPS. A web resource for designing subunit vaccine against major pathogenic species of bacteria. Front Immunol. 2018;9:2280. doi: 10.3389/fimmu.2018.02280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz A, Awan FM, Obaid A, Muhammad SA, Paracha RZ, Ahmad J, Ali A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: a reverse vaccinology based approach. Infect Genet Evol. 2015;32:280–291. doi: 10.1016/j.meegid.2015.03.027. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patronov A, Doytchinova I. T-cell epitope vaccine design by immunoinformatics. Open Biol. 2013;3(1):120139. doi: 10.1098/rsob.120139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Sidney J, Sette A, Peters B. TepiTool: a pipeline for computational prediction of T cell epitope candidates. Curr Protoc Immunol. 2016;114(1):18–19. doi: 10.1002/cpim.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podschun R, Ullmann U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. 1998;11(4):589–603. doi: 10.1128/CMR.11.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucci F, Rooman M. Towards an accurate prediction of the thermal stability of homologous proteins. J Biomol Struct Dyn. 2016;34(5):1132–1142. doi: 10.1080/07391102.2015.1073631. [DOI] [PubMed] [Google Scholar]
- Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3(5):445–450. doi: 10.1016/S1369-5274(00)00119-3. [DOI] [PubMed] [Google Scholar]
- Reiss I, Borkhardt A, Füssle R, Sziegoleit A, Gortner L. Disinfectant contaminated with Klebsiella oxytoca as a source of sepsis in babies. Lancet. 2000;356(9226):310. doi: 10.1016/S0140-6736(00)02509-5. [DOI] [PubMed] [Google Scholar]
- Reljic R. IFN-gamma therapy of tuberculosis and related infections. Interferon Cytokine Res. 2007;27:353–364. doi: 10.1089/jir.2006.0103. [DOI] [PubMed] [Google Scholar]
- Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci USA. 1998;95(2):588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell. 1994;74:929–934. doi: 10.1016/0092-8674(93)90472-3. [DOI] [PubMed] [Google Scholar]
- Saadi M, Karkhah A, Nouri HR. Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect Genet Evol. 2017;51:227–234. doi: 10.1016/j.meegid.2017.04.009. [DOI] [PubMed] [Google Scholar]
- Saha S, Raghava GPS. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct Funct Bioinform. 2006;65(1):40–48. doi: 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- Sanchez-Trincado JL, Gomez-Perosanz M, Reche PA. Fundamentals and methods for T- and B-cell epitope prediction. J Immunol Res. 2017 doi: 10.1155/2017/2680160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(Suppl_2):W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serruto D, Rappuoli R. Post-genomic vaccine development. FEBS Lett. 2006;580(12):2985–2992. doi: 10.1016/j.febslet.2006.04.084. [DOI] [PubMed] [Google Scholar]
- Sette A, Rappuoli R. Reverse vaccinology: developing vaccines in the era of genomics. Immunity. 2010;33(4):530–541. doi: 10.1016/j.immuni.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Patiyal S, Dhall A, Pande A, Arora C, Raghava GP. AlgPred 2.0: an improved method for predicting allergenic proteins and mapping of IgE epitopes. Brief Bioinform. 2020 doi: 10.1093/bib/bbaa294. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acid Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai H, Prades C, Vita R, Marcatili P, Popovic B, Xu J, Overington JP, Hirayama K, Soga S, Tsunoyama K, Clark D, Lefranc M-P, Ikeda K. Antibody informatics for drug discovery. Biochim Biophys Acta. 2014;1844(11):2002–2015. doi: 10.1016/j.bbapap.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Shtrichman R, Samuel CE. The role of gamma interferon in antimicrobial immunity. Curr Opin Microbiol. 2001;4:251–259. doi: 10.1016/S1369-5274(00)00199-5. [DOI] [PubMed] [Google Scholar]
- Sikarwar J, Kaushik S, Sinha M, Kaur P, Sharma S, Singh TP. Cloning, expression, and purification of nucleoside diphosphate kinase from Acinetobacter baumannii. Enzyme Res. 2013 doi: 10.1155/2013/597028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoons-Smit AM, Verweij-van Vught AMJJ, MacLaren DM. The role of K antigens as virulence factors in Klebsiella. J Med Microbiol. 1986;21(2):133–137. doi: 10.1099/00222615-21-2-133. [DOI] [PubMed] [Google Scholar]
- Singh L, Cariappa MP, Kaur M. Klebsiella oxytoca: an emerging pathogen? Med J Armed Forces India. 2016;72:S59–S61. doi: 10.1016/j.mjafi.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanki V, Sharma S, Tiwari V. Subtractive proteomics and reverse vaccinology strategies for designing a multiepitope vaccine targeting membrane proteins of Klebsiella pneumoniae. Int J Pept Res Ther. 2021;27:1177–1195. doi: 10.1007/s10989-021-10159-2. [DOI] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-C. [DOI] [PubMed] [Google Scholar]
- Tannock GA, Kim H, Xue L. Why are vaccines against many human viral diseases still unavailable; an historic perspective? J Med Virol. 2020;92(2):129–138. doi: 10.1002/jmv.25593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Ain Q, Ahmad S, Azam SS. Subtractive proteomics and immunoinformatics revealed novel B-cell derived T-cell epitopes against Yersinia enterocolitica: an etiological agent of Yersiniosis. Microb Pathog. 2018;125:336–348. doi: 10.1016/j.micpath.2018.09.042. [DOI] [PubMed] [Google Scholar]
- Vali L, Dashti AA, El-Shazly S, Jadaon MM. Klebsiella oxytoca with reduced sensitivity to chlorhexidine isolated from a diabetic foot ulcer. Int J Infect Dis. 2015;34:112–116. doi: 10.1016/j.ijid.2015.03.021. [DOI] [PubMed] [Google Scholar]
- Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010;11:568. doi: 10.1186/1471-2105-11-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, Freund C. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. 2017;8:292. doi: 10.3389/fimmu.2017.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Web Server Issue):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosen JE, Mukhopadhyay D, Macaubas C, Mellins ED. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol. 2018;9:2144. doi: 10.3389/fimmu.2018.02144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KM, Li LH, Yan JJ, Tsao N, Liao TL, Tsai HC, et al. Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J Bacteriol. 2009;191(14):4492–4501. doi: 10.1128/JB.00315-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav PD, Ella R, Kumar S, Patil DR, Mohandas S, Shete AM, Bhargava B. Immunogenicity and protective efficacy of inactivated SARS-CoV-2 vaccine candidate, BBV152 in rhesus macaques. Nat Commun. 2021;12(1):1–11. doi: 10.1038/s41467-021-21639-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Pandey AK, Dubey SK. Molecular modeling, docking and simulation dynamics of β-glucosidase reveals high-efficiency, thermo-stable, glucose tolerant enzyme in Paenibacillus lautus BHU3 strain. Int J Biol Macromol. 2021;168:371–382. doi: 10.1016/j.ijbiomac.2020.12.059. [DOI] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yinnon AM, Butnaru A, Raveh D, Jerassy Z, Rudensky B. Klebsiella bacteraemia: community versus nosocomial infection. QJM Int J Med. 1996;89(12):933–942. doi: 10.1093/qjmed/89.12.933. [DOI] [PubMed] [Google Scholar]
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Brinkman FS. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.