

ABSTRACT
Introduction: Systemic lupus erythematosus [SLE] is a chronic, autoimmune condition characterized by the formation of autoantibodies directed against nuclear components and by oxidative stress. Recently, a number of studies have demonstrated the essential role of iron in the immune response and there is growing evidence that abnormal iron homeostasis can occur in the chronic inflammatory state seen in SLE. Not only is iron vital for hematopoiesis, it is also important for a number of other key physiological processes, in particular in maintaining healthy mitochondrial function.
Areas covered: In this review, we highlight the latest understanding with regards to how patients with SLE may be at risk of cellular iron depletion as a result of both absolute and functional iron deficiency. Furthermore, we aim to explain the latest evidence of mitochondrial dysfunction in the pathogenesis of the disease.
Expert opinion: Growing evidence suggests that both abnormal iron homeostasis and subsequent mitochondrial dysfunction can impair effector immune cell function. Through a greater understanding of these abnormalities, therapeutic options that directly target iron and mitochondria may ultimately represent novel treatment targets that may translate into clinical care of patients with SLE in the near future.
KEYWORDS: Ferroptosis, functional iron deficiency, immunometabolism, Iron, mitochondria, systemic lupus erythematosus
1. Introduction
1.1. Systemic lupus erythematosus
Systemic lupus erythematosus [SLE] is a chronic, multisystem autoimmune disease characterized by the formation of autoantibodies directed against nuclear components. The prevalence is reported to be in the region of 1:1000 with a significant female predominance [~90% of cases] [1]. It is highly variable disease in terms of both clinical symptoms and immunological profile [2]. The wide-ranging clinical manifestations of SLE include rash [often associated with photosensitivity], arthralgia and arthritis [3], alopecia, mucosal ulceration, and fevers [4]. The most severe manifestations of the disease typically result from end organ involvement such as renal disease [lupus nephritis] [5], neurological involvement [6], cardiac [7] and pulmonary disease [8].
Serologically, SLE is associated with a wide array of autoantibodies with anti-nuclear antibodies [ANA] present in approximately 95% of cases [9]. Associated extractable nuclear antigens [ENA] include anti-Ro, anti-La, anti-Sm, anti-RNP, and anti-double stranded DNA [dsDNA] antibodies. Other common serological abnormalities include an elevated erythrocyte sedimentation rate [ESR] and low levels of complement C3 and C4. In clinical practice, anti-dsDNA antibody titers and C3 levels can be useful in evaluating disease activity.
1.2. SLE pathogenesis
The principal underlying pathways involved in the pathogenesis are not fully understood but are believed to involve both the innate and adaptive immune response [10]. A loss of self-tolerance is widely believed to result in the formation of pathogenic autoreactive B and T-cells that in turn result in tissue damage [11,12]. The precise mechanism through which this occurs is not known, however, it is felt to involve a combination of genetic factors [13], environmental triggers [such as ultraviolet radiation and possible viral exposure] [14] and, given the marked female predominance of the disease, the influences of hormones [15].
Observed pathogenic innate immune responses include dysfunction of macrophages that appear to be defective in removing apoptotic material [16,17], a feature of the so-called ‘waste disposal’ theory that has been suggested as a key mechanism in the pathogenesis of SLE in which prolonged failure to clear debris induces production of autoantibodies directed against self-antigens [18]. Macrophages also display abnormal polarization in both patients with SLE [19] and murine models of the disease [20]. Plasmacytoid dendritic cells [pDCs] are another important innate immune driver that have been observed to play a key role in the production of interferon-alpha [INFα] and subsequent generation of reactive oxygen species [ROS] [21], which is an important mediator in the pathogenesis of SLE [22]. Furthermore, pDCs have been shown to interact with B-cells and therefore have an influence on the adaptive immune response seen in SLE [23].
The role of the adaptive immune system in the pathogenesis of SLE is better understood. Autoreactive B-cells are a hallmark of the disease and display abnormal activation as well as aberrant expression [12]. Furthermore, B-cells play a vital role in the development of immune complexes that contain self-antigen [24]. These can be deposited within a variety of tissues and the resultant engagement of the Fc receptor and the complement cascade can in turn promote activation of a number of proinflammatory processes including release of Interleukin [IL]-1β and IL-6 [25–27]. In addition, autoreactive T-cells have been shown to play an essential role in the adaptive responses observed in the pathogenesis of SLE [28]. CD4+ helper T cells play a key role in orchestrating the immunological response through the release of a variety of cytokines, including IL-2 that has a vital regulatory effect. Previous studies have demonstrated that CD4+ T cells derived from patients with SLE show defective production of IL-2 [29]. In addition, regulatory T cells [Tregs] have been noted to have reduced number and function in SLE, suggesting that unregulated immune cell activation can occur [30]. Furthermore, CD8+ T cells have been shown to have impaired cytotoxicity in SLE when compared with healthy controls [31].
More recently there has been growing interest in the role of abnormal immune cell metabolism in the pathogenic pathways of SLE. In particular, a number of metabolites that are essential for establishing and maintaining the immune response are being identified as playing a role in pathogenesis. In this review, we highlight the role of one such metabolite, iron, and its potential role in SLE, as well as the downstream implications for mitochondrial function in the disease.
1.3. Iron metabolism and the mitochondria
Aside from its role in hemopoiesis, iron has an essential role in mitochondrial function [32]. Mitochondria are specialized organelles that conduct a wide array of vital cellular process, most notably known for their key role in energy metabolism via the production of adenosine triphosphate [ATP]. The generation of ATP is dependent upon oxidative phosphorylation [OXPHOS] that results from a series of metabolic processes that are broadly referred to as mitochondrial respiration. The primary site of this reaction is across the electron transport chain [ETC] on the inner mitochondrial membrane [as summarized in Figure 1]. The ETC is comprised of five individual complexes [I–V], which drive protons across the membrane in order to generate an electrochemical gradient that is required for the conversion of adenosine diphosphate [ADP] to ATP via the fifth complex [also known as ATP synthase]. Iron plays an important role in complexes I, II, and II, where it is contained within iron-sulfur [IS] clusters in the ferrous [Fe2+] state. Clinically, abnormalities relating to IS clusters have been shown to play a pathogenic role in both Parkinson’s disease [33,34] and Friedreich ataxia [35]. However, before considering the role of abnormal iron metabolism in SLE, it is important to understand the way in which iron homeostasis is maintained in health.
Figure 1.
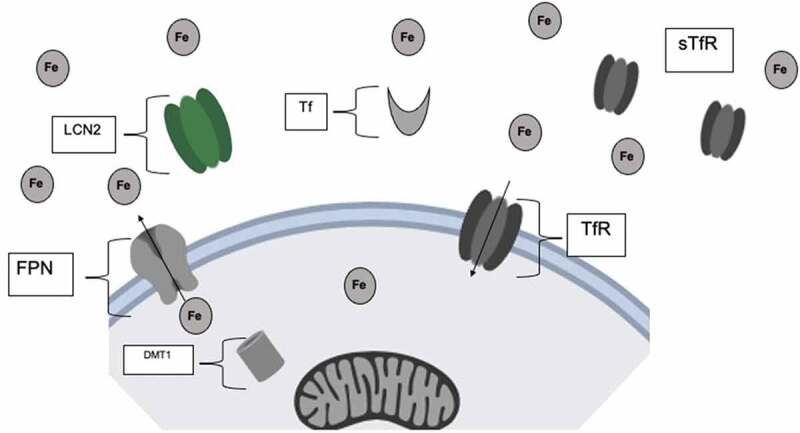
Free ferric (Fe3+) iron is bound to the transporter protein, transferrin, in the circulation. Transferrin bound iron can then enter the cell through the membrane transferrin receptor (TfR). Iron is exported from the cell by Ferroportin (FPN). Excessive free iron may be sequestered by lipocalin, which is released on innate immune system activation. Iron may also be bound to soluble transferrin receptors (sTfR) in the circulation. Fe, Free Ferric (Fe3+) iron; FPN, Ferroportin; Tf, Transferrin; TfR, Transferrin receptor; sTfR, soluble transferrin receptor; LCN-2, Lipocalin-2
2. Iron homeostasis in health
Iron is biologically available in numerous oxidative states although is most abundant in ferrous [Fe2+] and ferric [Fe3+] forms within the body. Oxidation and reduction of these two ionic forms are essential for a number of essential cellular processes. There are however some important differences between both states, with ferrous [Fe2+] iron more soluble and bioavailable than the ferric [Fe3+] state of iron. In excessive levels, iron is cytotoxic and is therefore tightly controlled through several vital metabolic processes [36]. Furthermore, unbound iron can result in free radical production and oxidative stress [37]. It is therefore important to remember that in order to prevent this, iron is commonly bound to a variety of transporter or storage proteins.
2.1. Iron absorption
Under physiological conditions, 20–25 mg of dietary iron is required each day, the majority of which is used for erythropoiesis [38]. Iron is absorbed across the duodenum at a rate that ensures sufficient quantities are absorbed to maintain iron stores without leading to excessive amounts of iron accumulation. Divalent metal transporter 1 [DMT-1] is a specialist transporter of ferrous [Fe2+] iron found on the apical luminal membrane of enterocytes that assists in the passage of iron from the gut to the bloodstream. Ferric [Fe3+] iron cannot be directly absorbed and therefore must be reduced to ferrous [Fe2+] iron by the transmembrane enzyme, duodenal cytochrome B-like ferrireductase [Dcytb], located on the luminal side of the enterocyte before entering the cell [39]. Iron may then either be stored within the enterocyte as ferritin or exported into the circulation by Ferroportin-1 [FPN-1], which is the only known mammalian cellular iron exporter [40].
2.2. Iron transport in the circulation and cellular uptake
In the circulation, ferrous [Fe2+] iron is then oxidized to the ferric state [Fe3+] by the ferroxidase enzyme Ceruloplasmin [and the specialist enterocyte equivalent, Hephaestin], before it can then bind to transferrin, a soluble transporter protein predominantly produced by the liver that can bind two molecules of iron. When iron is bound to transferrin, it is known as holo-transferrin and when transferrin is not binding iron it is referred to as apo-transferrin. Once transported to a cell that requires iron to be taken up, transferrin binds to the cell surface transferrin receptor, which has a high affinity for holo-transferrin [41].
In turn, the cellular transferrin receptor bound to transferrin is internalized and fused with endosome intracellularly. Protons are then taken up by the endosome, which in turn acidifies the endosomal pH and promotes the release of iron molecules [Fe3+] that are bound to the internalized transferrin receptor. However, the pH is maintained at a level that does not degrade either transferrin or its receptor, thus allowing for recycling of the molecules after the endosome binds with the cell surface following the release of iron from the endosome into the cytoplasm. Importantly, ferric iron [Fe3+] cannot be exported from the endosome and instead must be reduced to the ferrous form by the metalloreductase six-transmembrane epithelial antigen of prostate 3 [Steap3] [42,43]. The ferrous iron is then able to leave the endosome via DMT-1, which transports it into the cytoplasm. Ferrous iron that remains in the cytoplasm is termed the labile iron pool. Alternatively, iron may be transported to mitochondria [where it is incorporated into heme in erythroid cells] [32] or stored as intracellular ferritin [that may be degraded into haemosiderin].
In addition to the cell membrane-bound transferrin receptor, a soluble form has also been identified in human serum. This soluble transferrin receptor [sTfR] is a truncated monomer of the tissue receptor that can form complexes with transferrin and its receptor. Whereas reticulocytes are the main source of the membrane-bound receptor, the soluble form is predominantly found on erythroblasts and as such sTfR is felt to be representative of erythropoietic activity [44]. In the context of reduced erythropoiesis, sTfR levels are decreased; however, if erythropoietic activity is stimulated by hemolysis levels of serum sTfR can be elevated.
When holo-transferrin reaches the liver via the hepatic circulation, hepatocytes detect circulating iron concentrations. When high amounts of iron are present, hepatocytes will release the key iron regulatory peptide Hepcidin that in turn degrades FPN-1 and prevents iron transport from both enterocytes and other cellular stores [such as those abundant in the liver and macrophages] into the blood.
The majority of circulating iron is taken up by the bone marrow, which has a high demand given that iron is required by erythroid precursors that incorporate it into hemoglobin. A key driver of erythropoiesis is the glycoprotein cytokine, erythropoietin [EPO] that is released by the interstitial fibroblasts within the kidney in the context of cellular hypoxia [45]. Red blood cells will survive for approximately 120 days, after which splenic macrophages are responsible for their clearance and recycle the iron, which is then released back into circulation where it is again bound to transferrin. Free hemoglobin released by erythrocytes is in turn recycled after forming a complex with the protein Haptoglobin [Hp] that is subsequently removed by the reticuloendothelial system. Clinically, reduced serum Hp levels can be a useful measure of intravascular hemolysis [as is the case in hemolytic anemia] [46].
When there is an excessive amount of iron within the circulation it may be stored in the liver where it is bound to ferritin. This is essential as excessive iron can be cytotoxic and impair a number of physiological processes. Furthermore, free iron may also increase the risk of infection through the stimulation of bacterial pathogen growth. In order to reduce the risk of bacterial iron uptake, Lipocalin-2 [LCN-2] is released on activation of the innate immune system and sequesters iron by binding to bacterial iron-containing siderophores [47].
2.3. Iron excretion
Iron excretion can occur as a result of gastric loss, for example, in the context of high hepcidin expression. Iron is still taken into enterocytes by DMT1 although is not released into the circulation due to FPN-1 degradation. Ultimately, enterocytes containing high concentration of iron molecules desquamate from the lining of the gut and are replaced every 72 hours resulting in iron containing enterocytes being excreted in the feces. To a lesser degree, iron contained within the skin is lost in a similar fashion. In females it is also important to consider that iron is lost during menstruation [48].
An overview of the key regulators of cellular iron transport is summarized in Figure 2.
Figure 2.
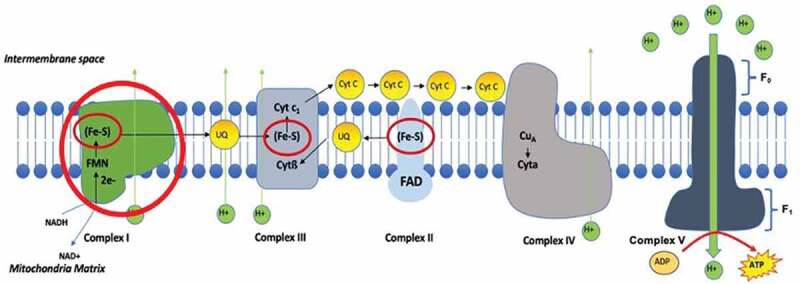
The electron transport chain on the inner mitochondrial membrane is the primary site of ATP production as a result of oxidative phosphorylation. The chain is comprised of five separate complexes that conduct a number of processes that generates an electrochemical proton gradient across the membrane, which in turn catalyses the generation of ATP from ADP. Complex I, II and III contain iron molecules in the form of iron sulfur cluster. FMN, Flavin mononucleotide; NAD+, Nicotinamide adenine dinucleotide; NADH, Nicotinamide adenine dinucleotide hydrogen; H+, Hydrogen; Fe-S, Iron-sulfide cluster; Cyt, Cytochrome; UQ, Ubiquinone; FAD, Flavin adenine dinucleotide; Cu, copper; ATP, adenosine triphosphate; ADP, adenosine diphosphate
3. The interface between iron metabolism and immune cell function
Iron is an essential metal that maintains normal cellular processes in all living organisms. It is able to be used in many forms as it can switch between multiple oxidation states which makes it an important co-factor, in particular in the regulation of the mitochondrial electron transport chain [49]. The regulation of iron metabolism is important for the maintenance of many immunometabolism pathways, as iron metabolism regulators act as co-regulators for many different pathways and are mediated by pro-inflammatory cytokines such as IL-6 and interferon alpha [IFN- α].
The role of iron in the immune response to infection has previously been well documented. Iron is an essential nutrient for bacterial growth. A study investigating the effects of iron therapy in the prophylaxis of malaria in children in Zanzibar reported significant adverse effects including life-threatening infection following treatment with iron [50]. It was suggested that supplementary iron treatment was associated with substantially higher rates of infection, thus showing how a state of iron deficiency may actually be protective against infection. In addition, elevated levels of hepcidin have been noted in patients suffering from malaria and it is believed that limiting serological iron availability may be protective against the disease [51]. Furthermore, patients suffering from hemochromatosis [a disease characterized by excessive circulatory iron], have been reported to have a significantly higher risk of bacterial infection [52,53].
A number of studies have evaluated the impacts of iron deficiency on the adaptive immune response. Frost et al recently demonstrated that mice treated with a hepcidin mimic developed a hypoferremic state, before being treated with a viral vaccine vector. It was found that the iron deficient mice demonstrated impaired effector and memory T-cell responses to the vaccine [54]. Howden et al have also demonstrated that activated CD4+ and CD8+ T-cells significantly upregulate their cell surface transferrin receptor, thus suggesting that iron plays an important role in T-cell activation [55]. Iron deprivation has also been shown to induce metabolic reprogramming in macrophage energy metabolism, with a reduction in mitochondrial oxidative phosphorylation and conversely an increase in glycolysis noted in those treated in iron deficient conditions [56]. The authors also assessed for the impact of iron deficiency on rat models of glomerulonephritis and interestingly noted that this resulted in an anti-inflammatory macrophage phenotype, thus suggesting that by changing iron availability it may be possible to influence effector immune cell function directly.
4. Abnormal iron metabolism in SLE
4.1. Absolute iron deficiency
Given the complex interaction of cellular processes required for physiological regulation of iron availability, the state of iron deficiency may occur as a result of disruption in any of these mechanisms. The most common causes of iron deficiency relate to an absolute deficit of iron both in the circulation and within stores. This may be due to insufficient dietary iron intake or as a result of excessive iron loss with the leading cause of iron deficiency resulting from heavy menstrual bleeding [menorrhagia] in women. We have previously demonstrated that symptoms of menorrhagia are present in almost half of pre-menopausal females with SLE [this is almost twice the rate reported in the general healthy population] [57,58]. In addition, patients who experienced more symptoms of menorrhagia reported higher incidence of anemia and history of iron supplementation when compared with those with fewer symptoms. This is an important, and often underappreciated symptom, that also represents an easily reversible cause of iron deficiency in women with SLE. Furthermore, conditions that impair gastric iron absorption may also result in deficiency [such as Celiac disease and inflammatory bowel disease].
4.2. Functional iron deficiency
Interleukin-6 [IL-6] is a pro-inflammatory cytokine that is commonly produced in an inflammatory state in SLE, through endocrine, autocrine, and paracrine actions on a large number of target cells. IL-6 drives terminal B cell differentiation and increases the secretion of immunoglobulins [Ig] from plasma cells. IL-6 is important in immune regulation, one of the most important roles that IL-6 contributes to in SLE is its ability to stimulate the final stages of B cell maturation therefore playing a key role in B cell hyperactivity. B cell hyperactivity causes the over-production of autoantibodies in SLE which then leads to deposition of self-antigen-antibody complexes [immune complexes] in multiple organ types. The kidneys are vulnerable to immune complex deposition due to the kidneys filtering macromolecules in proportion to the large endothelial cell surface relative to its weight which causes an accumulation of immune complexes in the glomerulus causing further inflammation and increased hepcidin expression [59].
The key central regulator of iron metabolism is hepcidin, which when present in high levels prevents the release of iron from stores and can result in a lack of biological availability of iron for effector cells. Hepcidin has been strongly linked to anemia in SLE patients as it is responsible for regulating iron availability and increased IL-6 expression causes a chronic inflammatory response [60]. Furthermore, studies in SLE prone mice found that those treated with hepcidin had a reduction in intra-renal iron accumulation and less tubular injury. It was therefore suggested that hepcidin analogues may be a potential novel treatment for SLE, however, human studies are yet to take place [61].
Hepcidin, ferritin, and haptoglobin are all produced in the liver and act as acute-phase reactants and IL-6 expression plays a large role on the production of these iron regulators. The increased expression of IL-6 and consequently increased hepcidin has been reported several times to have an impact on the pathogenesis of anemia [62]. Due to the pro-inflammatory response caused by IL-6, this induces the retention of iron in macrophages as IL-6 downregulates ferroportin, which is regulated by hepcidin and is the only known mammalian exporter that is responsible for the release of iron into the bloodstream [63].
Targeting IL-6 signaling may be a possible therapeutic option for SLE patients not only due to IL-6 having a large involvement on the production of B cell hyperactivity in SLE but also because IL-6 signaling plays a large role on the expression of hepcidin through STAT3 [64]. Tocilizumab is humanized monoclonal antibody that binds the Il-6 receptor and inhibits IL-6 activity and has been suggested as an alternative therapeutic option for SLE treatment which could also regulate iron homeostasis. However, a phase II clinical trial of Tocilizumab in SLE failed to show superiority when compared with placebo [65].
In addition to impaired iron release from stores through elevated hepcidin expression, the state of functional iron deficiency may also be brought about through reduced availability of key iron transporter molecules. Previous studies have found that serum transferrin levels are lower in patients with SLE when compared with healthy controls [66] and that serum transferrin concentrations inversely correlate with disease activity [67]. Furthermore, LCN-2 [which scavenges free iron in the event of innate immune system activation] has previously been found to correlate with proteinuria and renal flares in SLE when measured in the urine [68]. In addition, it has been suggested that LCN-2 may directly exacerbate lupus nephritis through promoting Th1 differentiation [69]. This also suggests that this mechanism of enhanced iron sequestration may ultimately result in a reduction of physiologically available iron within the circulation.
Red cell distribution width [RDW], a measure of variability in erythrocyte size and volume, has been suggested as a useful surrogate marker of functional iron deficiency. Interestingly, previous studies have demonstrated that patients with SLE have an elevated RDW when compared with healthy controls irrespective of anemia status [70]. In addition, Hu et al reported in 2013 that RDW shows correlation with erythrocyte sedimentation rate [ESR], C-reactive protein [CRP] and SLE Disease Activity Index 2000 [SLEDAI-2 K] scores in a retrospective study of 131 patients with SLE [71]. Furthermore, Zou et al noted that an elevated RDW at diagnosis correlates with disease activity and also predicts worse therapeutic outcomes in SLE [72]. We have also observed that RDW correlates with fatigue scores in three diverse groups of patients with the disease [73]. In comparison, there was no correlation between fatigue scores and the commonly used serological markers of disease or disease activity scores, thus suggesting that abnormal iron metabolism rather than disease activity per se may play a role in the pathogenesis of this symptom.
5. The links between iron deficiency and abnormal mitochondrial dysfunction in SLE
Abnormal iron metabolism can alter cellular metabolism through highly potent iron-free radicals that are nonselective and highly toxic. Reactive oxygen species [ROS] that are produced as by-products by the electron transport chain can then liberate ferrous iron [Fe2+] and ferric iron [Fe3+] therefore increasing mitochondrial stress and rendering the conditions pathophysiological [49] thus representing a link between altered metabolism and abnormalities in iron homeostasis.
As has already been highlighted, iron is a key component within Complexes I, II and III of the mitochondrial electron transport chain [the site of oxidative phosphorylation], where it is contained within iron-sulfur clusters. This is demonstrated in Figure 1. In addition, an in vitro study of T-cell Complex I activity revealed this complex to be the primary source of mitochondrial-derived reactive oxygen species [ROS] [74]. Furthermore, a recent study in mice found that those who were iron deficient showed reduced mitochondrial ATP production than those with normal iron levels, thus demonstrating that a lack of cellular iron availability can directly impair OXPHOS [54].
It is therefore possible that cellular iron deficiency in SLE may ultimately result in impaired mitochondrial physiology. Recently there has been growing evidence for a number of abnormalities in mitochondrial phenotype and function that have been linked to the pathogenesis of SLE. Furthermore, a number of authors have suggested that by restoring mitochondrial homeostasis it may be possible to improve SLE symptoms clinically, thus suggesting that the mitochondria represent a novel therapeutic target for the disease.
One particular example in which abnormal iron metabolism and mitochondrial dysfunction may occur is within the kidney. Lupus nephritis is a serious manifestation of the disease, which if not promptly identified and adequately treated can result in irreversible damage and in some cases lead to end-stage renal failure [requiring renal replacement therapy]. The most common cause of renal injury is glomerulonephritis, in which there is inflammation within the glomerulus. This is an important site of nutrient reabsorption and as such has the highest concentration of mitochondria within the kidney [75]. At present, very little is known with regards to the precise way in which the glomerulus handles circulating iron but previous studies have noted that podocytes [a specialist glomerular epithelial cell type found within Bowman’s capsule] are capable of taking up iron bound to transferrin and store it as ferritin [76]. As a result, some authors have proposed elevated urinary transferrin levels to be a useful biomarker of potentially active lupus nephritis [77].
5.1. Abnormal mitochondrial energy metabolism in SLE
All cells require energy in order to maintain cellular functions, for example, upon immune cell activation effector cells demand a rapid shift in energy metabolism in order to proliferate and maintain the immunological response. Adenosine triphosphate [ATP], the universal energy currency, can be generated through either glycolysis or oxidative phosphorylation [which takes place within the mitochondria]. A number of recent studies have implicated abnormalities relating to cellular bioenergetics in both animal models and human studies of SLE. In a study of peripheral blood mononuclear cells [PBMCs] from lupus patients, Complex I was found to have markedly reduced activity when compared with healthy controls [78].
T-cell metabolism has been a particular area of interest as these cells demonstrate significant changes in energy state upon immunological activation. In health, activation of the naïve T-cell occurs via the T-cell receptor, which in turn induces increased ATP production primarily through glycolytic pathways [79]. These activated effector cells will then transition to memory T-cell phenotypes and revert back to OXPHOS as their predominant source of energy generation [80]. However, chronic T-cell activation, as can be the case in SLE, results in increased mitochondrial dependent ATP production [81]. Murine lupus models have shown that CD4+ T-cells from lupus mice display significantly increased energy production via both OXPHOS and glycolysis when compared with healthy mice [82]. This suggests that cellular energy metabolism is enhanced in hyperactivated T-cells in SLE. The authors also observed similar findings in T-cells derived from patients with lupus. In addition to having significantly higher energy demands, T-cells from patients with SLE have been identified to have an increased mitochondrial transmembrane potential [ΔΨm]. This suggests that these mitochondria appear to be in a state of persistent hyperpolarisation and appear to be primed for a rapid upregulation in relation to pro-inflammatory effector functions [83]. In addition, increased ΔΨm may have implications for cell death via apoptosis, a pathway that is well established to be defective in the pathogenesis of SLE. It has been described that disruption in ΔΨm is a vital and irreversible process in the induction of apoptosis [84] and it may therefore suggest that abnormal mitochondrial membrane potential could have a role in SLE pathogenesis with implications for apoptotic pathways in the disease.
Monocytes are also known to play a central role in the storage and transport of iron within the body. They are also implicated in the failure to clear apoptotic debris that has been suggested as a hallmark in the pathogenesis of SLE. Monocyte mitochondrial function has previously been investigated by Gkirtzimanaki et al. who reported that INFα directly lead to dysregulated mitochondrial function, in particular in relation to impaired autophagic degradation, which in turn resulted in an accumulation of mtDNA within the cytosol [85].
5.2. Mitochondria-derived reactive oxygen species and oxidative stress in SLE
In addition to demonstrating abnormal energy metabolism, mitochondrial dysfunction in SLE has been linked with the generation of reactive oxygen species [ROS], another hallmark of the disease. Excessive ROS generation is often a result of an imbalance between ROS production [though incomplete reduction of oxygen molecules generated from OXPHOS] and reduction in the physiological antioxidant pathways that in turn results in decreased neutralization. The increased presence of ROS brings about a pro-inflammatory state known as oxidative stress.
Mitochondrial-derived ROS have previously been identified in in higher numbers in T-cells derived from patients with SLE and have been found in higher numbers in those with more active disease [86]. This suggests that impaired T-cell OXPHOS within the mitochondria could be producing excessive numbers of ROS due to impaired bioenergetics at the electron transport chain, which is in turn generating these toxic by-products. It is also important to consider that ROS play a key role as an induction signal for apoptosis, and therefore excessive ROS may upregulate the rates of apoptosis, which may ultimately result in increased cellular debris that could be a target for auto-antibody formation [as suggested by the ‘waste disposal’ theory of lupus pathogenesis [87]. ROS have also been implicated in the pathogenesis of lupus nephritis, where abnormal lipid peroxidation at the glomerular basement membrane has been implicated in the generation of toxic ROS, which can in turn result in tubular damage. It has been reported that process is dependent upon the breakdown of a number of intracellular metals, in particular iron [88].
ROS have also been demonstrated to damage DNA directly and this is a potential mechanism through which autoantibodies directed against DNA antigens may occur [89]. In addition, to inducing damage to genomic DNA, ROS can also damage mitochondrial DNA [mtDNA]. Previous studies have reported that the mtDNA segment particularly at risk of damage is the region encoding for the electron transport chain, which may also be an explanation for impaired mitochondrial energy metabolism [90]. Direct ROS damage to mitochondria may in turn result in the release of mtDNA into the circulation. A previous study has found that patients with lupus nephritis have elevated levels of cell-free mtDNA and the authors suggest that could potentially represent a novel biomarker for the disease [91]. A prompt clearance of this cell-free DNA is vital as failure to remove mtDNA can in turn induce pro-inflammatory cytokine release [92]. Furthermore, cell-free mtDNA may also in turn become an antigenic target for autoantibodies with previous anti-mtDNA antibodies recently being identified at higher levels in the blood of patients with SLE than healthy controls [93].
5.3. Therapeutic targeting of mitochondrial function in SLE
There is now growing focus on targeting mitochondrial function as a novel therapeutic option in the management of SLE. A number of pre-clinical studies are providing early evidence that correction of mitochondrial function may in turn normalize the abnormal immune responses seen in the disease. A recent study evaluated the use of Idebenone [2,3-dimethoxy-5methyl-6- [10]-hydroxydecyl]-1,4-benzoquinononenoben] which is a drug that has been previously tested for other diseases involving mitochondrial dysfunction [94]. This drug is a compound of coenzyme Q10, an essential electron carrier in the mitochondrial respiratory chain, that improves the function of the electron transfer chain by bypassing deficient complex I activity of the mitochondria. This improves mitochondrial physiology by the amount of ATP that is able to be newly synthesized. Q10 is found in all tissues and cells and has been suggested to protect cells against enhanced ROS toxicity [95]. It has been shown that treatment with idebenone led to a decreased number of effector memory CD4+ T cells in mice; however, the mechanism of this effect is unclear as this could be a direct result of altering the cellular metabolism or possibly decreasing the number of circulating pro-inflammatory cytokines.
Recent evidence suggests that it may be possible to repurpose drugs used in other conditions to improve mitochondrial metabolism in lupus. One drug that is showing promise for normalizing mitochondrial function in vitro is N-acetylcysteine [NAC]. A study by Doherty et al. noted that NAC selectivity inhibited Complex I within the mitochondrial electron transport chain in peripheral lymphocytes isolated from patients with SLE [74]. Another drug that is already available and has been trialed in SLE is Sirolimus, which acts on the mammalian target of Rapamycin [mTOR] that plays an essential role in regulating mitochondrial function [96]. In murine models of SLE, mice treated with the drug were found to have significantly lower levels of anti-dsDNA, proteinuria, and reduced mortality rates compared with those that were not treated with the drug [97]. A small study in patients with SLE found that administration of the drug to those suffering from refractory disease resulted in significant improvements in disease activity scores [98].
5.4. Ferroptosis: a potential link between abnormal iron homeostasis, mitochondrial dysfunction, and the pathogenesis of SLE
Until recently a central, unifying pathway of abnormal immune function, altered iron homeostasis and mitochondrial dysfunction in SLE had not been found. However, a novel potential link that is emerging is the role of ferroptosis in the pathogenesis of the disease. Ferroptosis is a recently discovered mechanism of programmed cell death that is iron dependent. The process is associated with an accumulation of cellular iron and also characteristic changes in mitochondrial phenotype, which makes it distinct from other forms of cell death [99,100].
6. Conclusions
In conclusion, iron homeostasis is maintained by a variety of tightly regulated physiological processes. However, patients with SLE may ultimately be at risk of cellular iron deficiency due to either increased rates of absolute iron deficiency [a fundamental lack of iron within the body], or via a state of functional iron deficiency [whereby elevated levels of proinflammatory cytokines impair iron transport and prevent the release of iron from stores]. This has a number of wide-ranging consequences as iron is an essential nutrient for numerous essential cellular processes. This includes DNA synthesis, enzyme activity and, importantly, energy metabolism. Within the immune system, iron deficiency has been demonstrated to impair priming to vaccine response, whilst iron supplementation has been shown to increase the risk of infection through enabling bacterial proliferation.
Iron is a key component within the mitochondrial electron transport chain and there is growing evidence that changes in the homeostatic mechanisms that maintain physiological iron regulation may also in turn result in abnormal mitochondrial function. Furthermore, abnormal mitochondrial energy metabolism has become a recent focus of a number of mechanistic studies investigating the pathogenesis of SLE. Whilst there is a paucity of data relating to the direct impact of abnormal iron metabolism on mitochondrial function in lupus, the proinflammatory state has been shown to induce a state of functional iron deficiency through impaired iron release from stores under the influence of hepcidin. In addition, we have demonstrated that a reduction in circulating transferrin means iron that is liberated from stores is not effectively transported to effector cells at an adequate rate to meet physiological demand. In addition, both monocytes and CD4+ T cells cultured in iron deficient conditions have been shown to impair normal mitochondrial function [68]. A proposed mechanism through which this could occur is summarized in Figure 3.
Figure 3.
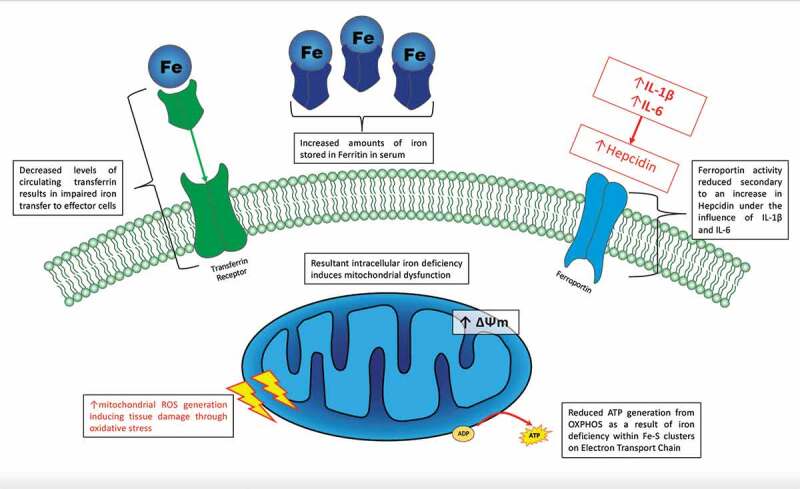
Abnormalities of iron metabolism in SLE
Future research may seek to assess whether immune regulation could be restored through correcting abnormalities within pathways responsible for iron regulation and altering mitochondrial function. Potential future therapeutic options could include the use of intravenous iron therapy as a means of correcting both absolute and functional iron deficiency, which may in turn restore mitochondrial function. At present, studies investigating the impact of correction of cellular iron concentrations in relation to mitochondrial physiology are lacking. Furthermore, newer agents that target abnormal pathways in iron metabolism are currently being investigated in a variety of other conditions [aside from SLE] that may potentially be trialed in lupus in the future.
Targeting abnormal iron metabolism and correction of abnormal mitochondrial function both represent exciting and novel possible therapeutic strategies for drug development that could translate into new treatment options for patients with SLE in the future.
7. Expert opinion
Whilst there have been significant advances in our understanding of the pathogenesis of SLE over recent decades, this is only now beginning to translate into new therapeutic options that are now translating into clinical care. However, in spite of this the precise mechanisms that lead to the development of disease are still largely unknown. In turn, there are a number of significant unmet needs that remain. This includes a fundamental lack of understanding as to why patients suffer from a number of debilitating symptoms such as fatigue, an increased atherosclerotic risk and difficulty in stratifying treatment to individual patients.
There is now growing evidence that both absolute and functional iron deficiency occur in SLE and this represents a potential new avenue for treatment options in the future. Intravenous iron therapy has already been demonstrated to improve fatigue, quality of life and exercise capacity in patients with cardiac failure, malignancy and Parkinson’s disease [101–103]. Studies assessing the benefits of iron therapy in patients with SLE are currently lacking though. Another possible therapeutic strategy that could potentially target iron metabolism in clinical practice is through hepcidin analogues and antagonists, although these are predominantly in the pre-clinical stage of development.
Upstream targets for hepcidin inhibition include the blockade of IL-6, with Tocilizumab an already available drug [used in the management of both rheumatoid arthritis and giant cell arteritis]. Although previous small studies have found IL-6 inhibition to be safe and effective in the management of SLE [104,105], this has not translated into clinical care as yet.
The field of immunometabolism is an area of immunology that has undergone rapid expansion in the last decade. Changes in cellular energetics have been identified in a number of pathological immunological scenarios including cancer immunity, infection, and more recently in autoimmune conditions. Several recent studies have found abnormal mitochondrial function in leukocytes derived from patients with SLE. The possibility of targeting metabolism with the aim of normalizing mitochondrial function thus represents an exciting future therapeutic option. Furthermore, therapies targeting the mitochondria may help to bring about immune regulation without inducing immunosuppression. Pre-clinical studies are showing promising data from a number of drug compounds that restore mitochondrial function and it remains an area of potential development for the future.
Finally, it is important to consider that SLE is a highly heterogenous disease with regards to both clinical symptoms and immunological manifestations. This poses clinicians significant challenges, not only with regards to making a diagnosis but also when planning treatment. Although a number of new treatments will soon be licensed for the disease, not every patient responds to treatment in the same way. Stratification of the disease through immunometabolism may help to inform future treatment decisions.
Funding Statement
CW is supported by a grant from Versus Arthritis [ref 21992].
Article highlights
Systemic lupus erythematosus [SLE] is a chronic autoimmune disorder characterised by abnormalities within both the innate and adaptive immune response. In order to maintain normal immune functions in health, leukocytes require a number of essential nutrients, such as iron, to initiate and establish the effector or regulatory response.
Patients with SLE are at risk of cellular iron deficiency, which can result from insufficient physiological supplies [absolute iron deficiency], which may be as a result of either reduced dietary intake or heavy menstrual loss. A state of functional iron deficiency, in which there are sufficient stores of iron within the body but it is not possible to either mobilise from stores or transport iron at a sufficient rate to meet physiological demands, may also occur in SLE.
Iron is well known to play a central role in haematopoiesis but also has a number of other vital physiological functions within the body. It is a key component of the mitochondrial electron transport chain, where it is found within iron-sulphur clusters. Recent evidence shows that cellular iron deficiency may ultimately impair mitochondrial energy metabolism through oxidative phosphorylation.
Mitochondrial dysfunction has recently been identified in the aberrant immune responses typically seen in SLE. Abnormal mitochondrial energy metabolism and subsequent oxidative stress have been identified in leukocytes derived from patients with SLE and this represents a possible novel therapeutic target for future drug development.
Declaration of interest
The author(s) have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewers disclosure
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
References
- 1.Rees F, Doherty M, Grainge M, et al. The incidence and prevalence of systemic lupus erythematosus in the UK, 1999-2012. Ann Rheum Dis. 2016;75(1):136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manson JJ, Rahman A.. Systemic lupus erythematosus. Orphanet J Rare Dis. 2006;1(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ceccarelli F, Perricone C, Cipriano E, et al. Joint involvement in systemic lupus erythematosus: from pathogenesis to clinical assessment. Semin Arthritis Rheum. 2017;47(1):53–64. [DOI] [PubMed] [Google Scholar]
- 4.Segura BT, Bernstein BS, McDonnell T, et al. Damage accrual and mortality over long-term follow-up in 300 patients with systemic lupus erythematosus in a multi-ethnic British cohort. Rheumatol [Oxford, England]. 2019. DOI: 10.1093/rheumatology/kez292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanly JG, Su L, Urowitz MB. et al. A longitudinal analysis of outcomes of lupus nephritis in an international inception cohort using a multistate model approach. Arthritis Rheumatol [Hoboken, NJ]. 2016;68(8):1932–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanly JG, Li Q, Su L, et al. Peripheral nervous system disease in systemic lupus erythematosus: results from an international inception cohort study. Arthritis Rheumatol [Hoboken, NJ]. 2020;72(1):67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doria A, Iaccarino L, Sarzi-Puttini P, et al. Cardiac involvement in systemic lupus erythematosus. Lupus. 2005;14(9):683–686. [DOI] [PubMed] [Google Scholar]
- 8.Tselios K, Urowitz MB. Cardiovascular and pulmonary manifestations of systemic lupus erythematosus. Curr Rheumatol Rev. 2017;13(3):206–218. [DOI] [PubMed] [Google Scholar]
- 9.Pisetsky DS. Evolving story of autoantibodies in systemic lupus erythematosus. J Autoimmun. 2019;110:102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katsiari CG, Liossis SN, Sfikakis PP. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheum. 2010;39(6):491–503. [DOI] [PubMed] [Google Scholar]
- 11.Zhou H, Li B, Li J, et al. Dysregulated T Cell activation and aberrant cytokine expression profile in systemic lupus erythematosus. Mediators Inflamm. 2019;2019:8450947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Y, Yang M, Wu H, et al. The pathological role of B cells in systemic lupus erythematosus: from basic research to clinical. Autoimmunity. 20. 20;53(2): 56-64. [DOI] [PubMed] [Google Scholar]
- 13.Song K, Liu L, Zhang X, et al. An update on genetic susceptibility in lupus nephritis. Clin Immunol. 2019;210:108272. [DOI] [PubMed] [Google Scholar]
- 14.Stannard JN, Kahlenberg JM. Cutaneous lupus erythematosus: updates on pathogenesis and associations with systemic lupus. Curr Opin Rheumatol. 2016;28(5):453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones BG, Penkert RR, Surman SL, et al. Matters of life and death: how estrogen and estrogen receptor binding to the immunoglobulin heavy chain locus may influence outcomes of infection, allergy, and autoimmune disease. Cell Immunol. 2019;346:103996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bijl M, Reefman E, Horst G, et al. Reduced uptake of apoptotic cells by macrophages in systemic lupus erythematosus: correlates with decreased serum levels of complement. Ann Rheum Dis. 2006;65(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tas SW, Quartier P, Botto M, et al. Macrophages from patients with SLE and rheumatoid arthritis have defective adhesion in vitro, while only SLE macrophages have impaired uptake of apoptotic cells. Ann Rheum Dis. 2006;65(2):216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22(1):431–456. [DOI] [PubMed] [Google Scholar]
- 19.Mohammadi S, Saghaeian-Jazi M, Sedighi S, et al. Immunomodulation in systemic lupus erythematosus: induction of M2 population in monocyte-derived macrophages by pioglitazone. Lupus. 2017;26(12):1318–1327. [DOI] [PubMed] [Google Scholar]
- 20.Li F, Yang Y, Zhu X, et al. Macrophage polarization modulates development of systemic lupus erythematosus. Cell Physiol Biochem. 2015;37(4):1279–1288. [DOI] [PubMed] [Google Scholar]
- 21.Eloranta ML, Lovgren T, Finke D, et al. Regulation of the interferon-alpha production induced by RNA-containing immune complexes in plasmacytoid dendritic cells. Arthritis Rheumatism. 2009;60(8):2418–2427. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3(73):73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menon M, Blair PA, Isenberg DA, et al. A regulatory feedback between plasmacytoid dendritic cells and regulatory B cells is aberrant in systemic lupus erythematosus. Immunity. 2016;44(3):683–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pickering MC, Botto M, Taylor PR, et al. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. 2000;76:227–324. [DOI] [PubMed] [Google Scholar]
- 25.Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. 2012;18(6):871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidson A. What is damaging the kidney in lupus nephritis? Nat Rev Rheumatol. 2016;12(3):143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thanadetsuntorn C, Ngamjanyaporn P, Setthaudom C, et al. The model of circulating immune complexes and interleukin-6 improves the prediction of disease activity in systemic lupus erythematosus. Sci Rep. 2018;8(1):2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stratigou V, Doyle AF, Carlucci F, et al. Altered expression of signalling lymphocyte activation molecule receptors in T-cells from lupus nephritis patients-a potential biomarker of disease activity. Rheumatol [Oxford, England]. 2017;56(7):1206–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moulton VR, Suarez-Fueyo A, Meidan E, et al. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol Med. 2017;23(7):615–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. 2015;45(2):344–355. [DOI] [PubMed] [Google Scholar]
- 31.Gravano DM, Hoyer KK. Promotion and prevention of autoimmune disease by CD8+ T cells. J Autoimmun. 2013;45:68–79. [DOI] [PubMed] [Google Scholar]
- 32.Shaw GC, Cope JJ, Li L, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440(7080):96–100. [DOI] [PubMed] [Google Scholar]
- 33.Betarbet R, Sherer TB, MacKenzie G, et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3(12):1301–1306. [DOI] [PubMed] [Google Scholar]
- 34.Greenamyre JT, Hastings TG. Biomedicine. Parkinson’s–divergent causes, convergent mechanisms. Science. 2004;304(5674):1120–1122. [DOI] [PubMed] [Google Scholar]
- 35.Huang ML, Becker EM, Whitnall M, et al. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc Natl Acad Sci U S A. 2009;106(38):16381–16386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Umemura M, Kim JH, Aoyama H, et al. The iron chelating agent, deferoxamine detoxifies Fe[Salen]-induced cytotoxicity. J Pharmacol Sci. 2017;134(4):203–210. [DOI] [PubMed] [Google Scholar]
- 37.Jomova K, Valko M. Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr Pharm Des. 2011;17(31):3460–3473. [DOI] [PubMed] [Google Scholar]
- 38.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117(3):285–297. [DOI] [PubMed] [Google Scholar]
- 39.Frazer DM, Wilkins SJ, Becker EM, et al. A rapid decrease in the expression of DMT1 and Dcytb but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut. 2003;52(3):340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wallace DF. The regulation of iron absorption and homeostasis. Clin Biochem Rev. 2016;37(2):51–62. [PMC free article] [PubMed] [Google Scholar]
- 41.Kleven MD, Jue S, Enns CA. Transferrin receptors TfR1 and TfR2 bind transferrin through differing mechanisms. Biochemistry. 2018;57(9):1552–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lambe T, Simpson RJ, Dawson S, et al. Identification of a Steap3 endosomal targeting motif essential for normal iron metabolism. Blood. 2009;113(8):1805–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang F, Tao Y, Zhang Z, et al. Metalloreductase Steap3 coordinates the regulation of iron homeostasis and inflammatory responses. Haematologica. 2012;97(12):1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beguin Y. Soluble transferrin receptor for the evaluation of erythropoiesis and iron status. Clin Chim Acta. 2003;329(1–2):9–22. [DOI] [PubMed] [Google Scholar]
- 45.Souma T, Nezu M, Nakano D, et al. Erythropoietin synthesis in renal myofibroblasts is restored by activation of hypoxia signaling. J Am Soc Nephrol. 2016;27(2):428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shih AW, McFarlane A, Verhovsek M. Haptoglobin testing in hemolysis: measurement and interpretation. Am J Hematol. 2014;89(4):443–447. [DOI] [PubMed] [Google Scholar]
- 47.Nasioudis D, Witkin SS. Neutrophil gelatinase-associated lipocalin and innate immune responses to bacterial infections. Med Microbiol Immunol. 2015;204(4):471–479. [DOI] [PubMed] [Google Scholar]
- 48.Bruinvels G, Burden R, Brown N, et al. The prevalence and impact of heavy menstrual bleeding [Menorrhagia] in elite and non-elite athletes. PloS One. 2016;11(2):e0149881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cronin SJF, Woolf CJ, Weiss G, et al. The role of iron regulation in immunometabolism and immune-related disease. Front Mol Biosci. 2019;6(116). DOI: 10.3389/fmolb.2019.00116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sazawal S, Black RE, Ramsan M, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet [London, England]. 2006;367(9505):133–143. [DOI] [PubMed] [Google Scholar]
- 51.Portugal S, Carret C, Recker M, et al. Host-mediated regulation of superinfection in malaria. Nat Med. 2011;17(6):732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Höpfner M, Nitsche R, Rohr A, et al. Yersinia enterocolitica infection with multiple liver abscesses uncovering a primary hemochromatosis. Scand J Gastroenterol. 2001;36(2):220–224. [DOI] [PubMed] [Google Scholar]
- 53.Barton JC, Acton RT. Hemochromatosis and Vibrio vulnificus wound infections. J Clin Gastroenterol. 2009;43(9):890–893. [DOI] [PubMed] [Google Scholar]
- 54.Frost JNTT, Abbas M, Wideman SK, et al. Hepcidin-mediated hypoferremia disrupts immune responses to vaccination and infection. Med [Cell Press]. 2020;2(2):164–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howden AJM, Hukelmann JL, Brenes A, et al. Quantitative analysis of T cell proteomes and environmental sensors during T cell differentiation. Nat Immunol. 2019;20(11):1542–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pereira M, Chen TD, Buang N, et al. Acute iron deprivation reprograms human macrophage metabolism and reduces inflammation in vivo. Cell Rep. 2019;28(2):498–511.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wincup C, McDonnell TCR, Rahman A. Menorrhagia: an underappreciated problem in pre-menopausal women with systemic lupus erythematosus. Lupus. 2019;28(7):916–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fraser IS, Mansour D, Breymann C, et al. Prevalence of heavy menstrual bleeding and experiences of affected women in a European patient survey. Int J Gynaecol Obstet. 2015;128(3):196–200. [DOI] [PubMed] [Google Scholar]
- 59.Davidson A, Berthier C, Kretzler M. Chapter 18 - Pathogenetic mechanisms in lupus nephritis. In: Wallace DJ, Hahn BH, editors. Dubois’ lupus erythematosus and related syndromes [eighth edition]. Philadelphia: W.B. Saunders; 2013. p. 237–255. [Google Scholar]
- 60.Indrakanti DL, Alvarado A, Zhang X, et al. The interleukin-6-hepcidin-hemoglobin circuit in systemic lupus erythematosus flares. Lupus. 2017;26(2):200–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scindia Y, Wlazlo E, Ghias E, et al. Therapeutic benefit of regulating iron metabolism in spontaneous lupus nephritis. J Immunol. 2020;204(1 Supplement):236.7. [Google Scholar]
- 62.Vadhan-Raj S, Zhou X, Bueso-Ramos CE, et al. Interleukin-6, hepcidin, and other biomarkers in Anemia of Chronic Disease [ACD] and Chemotherapy-Induced Anemia [CIA]: potential therapeutic targets. Blood. 2012;120(21):2086. [Google Scholar]
- 63.Wang C-Y, Babitt JL. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol. 2016;23(3):189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108(9):3204–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wallace DJ, Strand V, Merrill JT, et al. Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: a phase II dose-ranging randomised controlled trial. Ann Rheum Dis. 2017;76(3):534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taysi S, Gul M, Sari RA, et al. Serum oxidant/antioxidant status of patients with systemic lupus erythematosus. Clin Chem Lab Med. 2002;40(7):684–688. [DOI] [PubMed] [Google Scholar]
- 67.Wincup C, McDonnell CMT, Robinson G, et al. Disease activity and dysregulated iron metabolism: a potentially overlooked mechanism for anaemia in patients with systemic lupus erythematosus? [Abstract]. Arthritis Rheumatol. 2019;71(suppl 10). [Google Scholar]
- 68.Yang CC, Hsieh SC, Li KJ, et al. Urinary neutrophil gelatinase-associated lipocalin is a potential biomarker for renal damage in patients with systemic lupus erythematosus. J Biomed Biotechnol. 2012;2012:759313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen W, Li W, Zhang Z, et al. Lipocalin-2 exacerbates lupus nephritis by promoting Th1 cell differentiation. J Am Soc Nephrol. 2020;31(10):2263–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vaya A, Alis R, Hernandez JL, et al. RDW in patients with systemic lupus erythematosus. Influence of anaemia and inflammatory markers. Clin Hemorheol Microcirc. 2013;54(3):333–339. [DOI] [PubMed] [Google Scholar]
- 71.Hu ZD, Chen Y, Zhang L, et al. Red blood cell distribution width is a potential index to assess the disease activity of systemic lupus erythematosus. Clin Chim Acta. 2013;425:202–205. [DOI] [PubMed] [Google Scholar]
- 72.Zou XL, Lin XJ, Ni X, et al. Baseline red blood cell distribution width correlates with disease activity and therapeutic outcomes in patients with systemic lupus erythematosus, irrespective of anemia status. Clin Lab. 2016;62(10):1841–1850. [DOI] [PubMed] [Google Scholar]
- 73.Wincup C, Parnell C, Cleanthous S, et al. Red cell distribution width correlates with fatigue levels in a diverse group of patients with systemic lupus erythematosus irrespective of anaemia status. Clin Exp Rheumatol. 2019;37(5):852–854. [PubMed] [Google Scholar]
- 74.Doherty E, Oaks Z, Perl A. Increased mitochondrial electron transport chain activity at complex I is regulated by N-acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid Redox Signal. 2014;21(1):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13(10):629–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bányai E, Balogh E, Fagyas M, et al. Novel functional changes during podocyte differentiation: increase of oxidative resistance and H-ferritin expression. Oxid Med Cell Longev. 2014;2014:976394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Urrego T, Ortiz-Reyes B, Vanegas-García AL, et al. Utility of urinary transferrin and ceruloplasmin in patients with systemic lupus erythematosus for differentiating patients with lupus nephritis. Reumatol Clin [Engl Ed]. 2020;16(1):17–23. [DOI] [PubMed] [Google Scholar]
- 78.Leishangthem BD, Sharma A, Bhatnagar A. Role of altered mitochondria functions in the pathogenesis of systemic lupus erythematosus. Lupus. 2016;25(3):272–281. [DOI] [PubMed] [Google Scholar]
- 79.Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16(6):769–777. [DOI] [PubMed] [Google Scholar]
- 80.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Byersdorfer CA, Tkachev V, Opipari AW, et al. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood. 2013;122(18):3230–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yin Y, Choi SC, Xu Z, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perl A, Hanczko R, Doherty E. Assessment of mitochondrial dysfunction in lymphocytes of patients with systemic lupus erythematosus. Methods Mol Biol [Clifton, NJ]. 2012;900:61–89. [DOI] [PubMed] [Google Scholar]
- 84.Susin SA, Zamzami N, Castedo M, et al. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med. 1997;186(1):25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gkirtzimanaki K, Kabrani E, Nikoleri D, et al. IFNα impairs autophagic degradation of mtDNA promoting autoreactivity of SLE monocytes in a STING-dependent fashion. Cell Rep. 2018;25(4):921–33.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oates JC, Gilkeson GS. The biology of nitric oxide and other reactive intermediates in systemic lupus erythematosus. Clin Immunol. 2006;121(3):243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jabs T. Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem Pharmacol. 1999;57(3):231–245. [DOI] [PubMed] [Google Scholar]
- 88.Wlazlo E, Mehrad B, Morel L, et al. Iron metabolism: an under investigated driver of renal pathology in lupus nephritis. Front Med [Lausanne]. 2021;8:643686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cooke MS, Mistry N, Wood C, et al. Immunogenicity of DNA damaged by reactive oxygen species–implications for anti-DNA antibodies in lupus. Free Radic Biol Med. 1997;22(1–2):151–159. [DOI] [PubMed] [Google Scholar]
- 90.Lemarie A, Grimm S. Mitochondrial respiratory chain complexes: apoptosis sensors mutated in cancer? Oncogene. 2011;30(38):3985–4003. [DOI] [PubMed] [Google Scholar]
- 91.Truszewska A, Wirkowska A, Gala K, et al. Cell-free DNA profiling in patients with lupus nephritis. Lupus. 2020;29(13):1759–1772. [DOI] [PubMed] [Google Scholar]
- 92.West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17(6):363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Becker Y, Loignon RC, Julien AS, et al. Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci Rep. 2019;9(1): 4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Blanco LP, Pedersen HL, Wang X, et al. Improved mitochondrial metabolism and reduced inflammation following attenuation of murine lupus with Coenzyme Q10 analog Idebenone. Arthritis Rheumatol. 2020;72(3):454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Dev Disabilities Res Rev. 2010;16(2):183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.de la Cruz López KG, Toledo Guzmán ME, Sánchez EO, et al. mTORC1 as a regulator of mitochondrial functions and a therapeutic target in cancer. Front Oncol. 2019;9:1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Warner LM, Adams LM, Sehgal SN. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheumatism. 1994;37(2):289–297. [DOI] [PubMed] [Google Scholar]
- 98.Fernandez D, Bonilla E, Mirza N, et al. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheumatism. 2006;54(9):2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li J, Cao F, Yin HL, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grellier N, Deray G, Yousfi A, et al. Functional iron deficiency, inflammation and fatigue after radiotherapy. Bull Cancer. 2015;102(9):780–785. [DOI] [PubMed] [Google Scholar]
- 102.Comin-Colet J, Enjuanes C, Gonzalez G, et al. Iron deficiency is a key determinant of health-related quality of life in patients with chronic heart failure regardless of anaemia status. Eur J Heart Fail. 2013;15(10):1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zuo LJ, Yu SY, Hu Y, et al. Serotonergic dysfunctions and abnormal iron metabolism: relevant to mental fatigue of Parkinson disease. Sci Rep. 2016;6(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Illei GG, Shirota Y, Yarboro CH, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheumatism. 2010;62(2):542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ocampo V, Haaland D, Legault K, et al. Successful treatment of recurrent pleural and pericardial effusions with tocilizumab in a patient with systemic lupus erythematous. BMJ Case Rep. 2016 Aug 8;2016:bcr2016215423. [DOI] [PMC free article] [PubMed] [Google Scholar]