

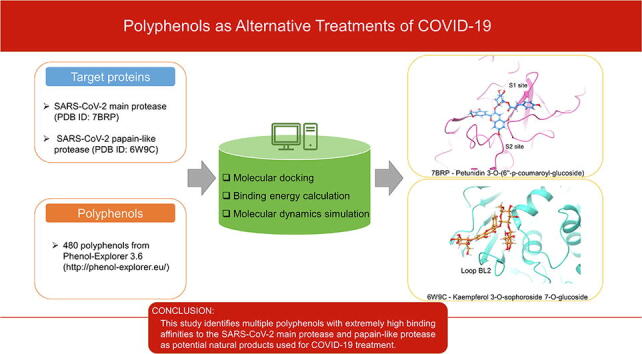
Graphical abstract
Keywords: COVID-19, Virtual screening, Main protease, Papain-like protease, MD simulation, Natural products, Polyphenols
Abstract
Although scientists around the world have put lots of effort into the development of new treatments for COVID-19 since the outbreak, no drugs except Veklury (remdesivir) have been approved by FDA. There is an urgent need to discover some alternative antiviral treatment for COVID-19. Because polyphenols have been shown to possess antiviral activities, here we conducted a large-scale virtual screening for more than 400 polyphenols. Several lead compounds such as Petunidin 3-O-(6″-p-coumaroyl-glucoside) were identified to have promising binding affinities and convincing binding mechanisms. Analyzing the docking results and ADME properties sheds light on the potential efficacy of the top-ranked drug candidates and pinpoints the key residues on the target proteins for the future of drug development.
1. Introduction
In March 2020, the World Health Organization declared a global pandemic of the novel coronavirus disease (COVID-19) [1]. This outbreak continues to wreak havoc around the world, causing over 640,000 deaths in the United States and over 4.5 million deaths worldwide by the end of August 2021 [2]. Thus, rapid discovery of small-molecule antiviral drugs that are therapeutic against COVID-19 continues to be a significant task [3].
SARS-CoV-2, the viral agent responsible for COVID-19, is an enveloped, positive-sense, single-stranded RNA virus [4], [5]. Coronaviruses contain the largest-known RNA virus genomes, being roughly 26–32 kb and made up of at least six open reading frames (ORFs) encoding for proteins [5]. RNA viruses such as SARS-CoV-2 are replicated by releasing these RNA genomes into host cells, which in turn translate that RNA into proteins as if the viral RNA were the host cell’s own RNA. The major ORF of SARS-CoV-2 encodes two overlapping polyproteins, PP1A and PP1B. These proteins are generated in the host cell and then cleaved into 16 non-structural proteins (NSP1-16) by two proteolytic enzymes: the main protease (Mpro) and papain-like protease (PLpro) [6]. These NSPs then initiate replication and transcription of the viral genome by assembling the viral replicase complex on host cells membranes [3], [7], [8]. Thus, Mpro and PLpro play vital roles in the replication of SARS-CoV-2 in the body; without them, the NSPs would not be released from the larger polyproteins, PP1A and PP1B.
The importance of these proteases to the viral life cycle has led researchers to wonder if they could be inactivated, and whether a protease inhibitor would be a useful treatment for COVID-19. Although coronaviruses cannot replicate if the proteolytic activity of PLpro or Mpro is blocked [9], no PLpro and Mpro inhibitors used for COVID-19 treatment have yet been approved. PLpro not only cleaves PP1A and PP1B at three distinct sites between NSP1-4, but also helps coronaviruses to elude the host’s immune response through competitive interaction with ubiquitin and ISG15 [10]. Certain inhibitors of the PLpro from SARS-CoV, the virus responsible for the 2003 global outbreak of severe acute respiratory syndrome (SARS), also target the PLpro from SARS-CoV-2 and exhibit antiviral activity in monkey cells in vitro [10]. Furthermore, PLpro inhibition reduces the cytopathogenic effect and replication of SARS-CoV-2 while maintaining the interferon antiviral response in vitro [11]. On the other hand, Mpro exclusively cleaves PP1A and PP1B at 11 distinct sites immediately following a glutamine residue [6]. Because no human host cell proteases have this substrate specificity, Mpro is an ideal drug target [5], [12], [13]. A drug used to treat feline infectious peritonitis, a lethal coronavirus infection that affects cats, has been found to be capable of inhibiting the SARS-CoV-2 Mpro and blocking viral replication [14]. Two protease inhibitors approved for treating hepatitis C virus have been demonstrated to inhibit the SARS-CoV-2 Mpro and show strong antiviral activity in a mouse model [15]. Thus, further investigation into inhibitors of PLpro and Mpro may very likely lead to the discovery of a safe, effective treatment for COVID-19.
Polyphenols are secondary plant metabolites with a plethora of health benefits, including strong antioxidant properties that defend against oxidative damage by free radicals and prevent chronic disease [16]. In light of the ongoing pandemic, researchers have investigated polyphenols’ antiviral efficacy against COVID-19. Ghosh et al. found that the green tea polyphenols epigallocatechin gallate (EGCG), epicatechingallate, and gallocatechin-3-gallate interact strongly with one or both of the catalytic residues of the SARS-CoV-2 Mpro [17], and later demonstrated that six polyphenols from Broussonetia papyrifera inhibit the catalytic activity of Mpro as well [18]. Khan et al. also found that EGCG interacted strongly with Mpro [19]. Ansari et al. found that luteolin had a higher affinity for PLpro than the FDA-approved antiviral drug, remdesivir [20]. However, these studies were extremely limited in scope. There are around 500 unique polyphenol structures available for download on the Phenol-Explorer 3.6 database created by Neveu et al. [21], and current studies investigating polyphenols as Mpro and PLpro inhibitors are limited to a very small selection of molecules, mainly coming from green tea. Considering that promising results were found using such a small selection of polyphenols, it is possible that there are even better outcomes to be found within a larger sample.
Thus, to find potential therapeutic agents against COVID-19, this study screens a large number of polyphenols to bind to SARS-CoV-2 Mpro or PLpro. The structures of 480 polyphenols were obtained from the aforementioned Phenol-Explorer 3.6 database, and molecular docking was conducted using Maestro. MM-GBSA scores were collected to quantify the affinity of the molecules for the proteins, and then ADME (Absorption, distribution, metabolism, and excretion) and drug-likeness properties were analyzed for further screening. Finally, several polyphenols with high affinities are identified for both proteases: Petunidin 3-O-(6″-p-coumaroyl-glucoside), Malvidin 3,5-O-diglucoside, and Cyanidin 3-O-(6″-p-coumaroyl-glucoside) bind to Mpro (−101.21 kcal/mol, −95.07 kcal/mol, and −90.17 kcal/mol, respectively), while Kaempferol 3-O-sophoroside 7-O-glucoside, Cyanidin 3-O-sambubioside 5-O-glucoside, and Malvidin 3-O-(6″-p-coumaroyl-glucoside) are the top polyphenols bound to PLpro (−87.97 kcal/mol, −87.33 kcal/mol, and −85.70 kcal/mol, respectively). This study identifies multiple polyphenols with extremely high binding affinities to the SARS-CoV-2 Mpro and PLpro as potential natural products used for COVID-19 treatment.
2. Materials and methods
2.1. Ligand preparation
The 3D structures of 480 tested polyphenols were retrieved from Phenol-Explorer 3.6 (http://phenol-explorer.eu/). All the polyphenolic compounds were prepared using Ligprep in Maestro 12.4 (Schrödinger). The force field was OPLS3e by default [22]. The process of preparation includes adding hydrogens, computing correct partial charges, and generating possible conformations.
2.2. Protein preparation
The protein structures of Mpro (PDB ID: 7BRP) and PLpro (PDB ID: 6W9C) from RCSB’s Protein Data Bank (https://www.rcsb.org/) [23] were prepared for use by Maestro in three steps: preprocessing, optimization, and minimization. The preprocessing included assigning bond orders, adding hydrogens, creating zero-order bonds to metals, creating disulfide, filling in missing side chains using Prime, deleting water molecules beyond 5.00 Å from het groups and generating het states using Epik [24]. PROPKA’s default setting (pH = 7.0) and the OPLS3e force field were applied in optimization and minimization.
2.3. Ligand-protein docking
To estimate the interactions between target proteins and polyphenols, we conducted ligand–protein docking by using the Ligand Docking panel in Maestro. Before running docking jobs, a receptor grid box was generated based on existing ligands in protein structures. For the structure of Mpro (PDB ID: 7BRP), the existing ligand boceprevir was used to generate a receptor grid. In the structure of PLpro (PDB ID: 6W9C), the receptor grid was generated according to the same site on SARS-CoV PLpro (PDB ID: 3E9S). The size of the receptor grid box was set as default (20 Å). Ligand-protein docking was performed in extra-precision (XP) mode.
2.4. MM-GBSA calculation
To predict the binding energies of polyphenols bound to Mpro or PLpro, we performed Prime MM-GBSA (molecular mechanics generalized Born surface area) in Maestro. In the MM-GBSA panel, the pose viewer files of the docked complex were uploaded into the MM-GBSA panel. The force field was OPLS3e.
2.5. ADME and drug-likeness properties prediction
After binding energies calculation, we applied Qikprop module in Maestro to predict ADME and drug-likeness properties for further screening [25]. For Qikprop, the top-ranked polyphenols were prepared by using Ligprep. Finally, the descriptors such as RuleOfFive (RO5) and RuleOfThree (RO3) were applied to analyze the candidates.
2.6. AutoQSAR analysis
AutoQSAR module of Schrodinger is a machine learning tool to build and apply quantitative structure–activity relationship (QSAR) models [26]. To build machine learning models, 61 Mpro inhibitors and 20 PLpro inhibitors were retrieved from BindingDB [27]. The best model of each protease was used to predict half-maximal inhibitory concentration (IC50) of the top-ranked polyphenols. The details of the polyphenols along with their predicted IC50 values are presented in Table 1, Table 3.
Table 1.
The results of the top 3 polyphenols bound to Mpro.
| Compound | Estimated binding energy (kcal/mol) | QPlogSa | RO5b | RO3c | Predicted IC50 (nM) |
|---|---|---|---|---|---|
| Petunidin 3-O-(6″-p-coumaroyl-glucoside) | −101.21 | −4.479 | 3 | 2 | 44.47 |
| Malvidin 3,5-O-diglucoside | −95.07 | −2.335 | 3 | 2 | 45.73 |
| Cyanidin 3-O-(6″-p-coumaroyl-glucoside) | −90.17 | −2.303 | 3 | 2 | 43.00 |
| *Saquinavir | −93.58 | −2.164 | 3 | 2 | 52.48 |
| **Epigallocatechin gallate (EGCG) | −65.04 | −3.554 | 2 | 2 | 60.73 |
| ***Papyriflavonol A | −58.38 | −6.287 | 0 | 2 | 76.96 |
| ****Boceprevir | −72.56 | −4.372 | 1 | 0 | 46.72 |
*The best-scored potential drug identified by our previous study [36]
**The best-scored potential drug identified by a previous study [17]
***The best-scored potential drug identified by a previous study [18]
**** The original ligand of 7BRP [37]
a. QPlogS is predicted aqueous solubility. The recommended range is −6.5 ∼ 0.5.
b. RO5: number of violations of Lipinski’s rule of five [38]. The recommended range: maximum is 4.
c. RO3: Number of violations of Jorgensen’s rule of three [39]. The recommended range: maximum is 3.
Table 3.
The results of the top 3 polyphenols bound to PLpro.
| Compound | Estimated binding energy (kcal/mol) | QPlogSa | RO5b | RO3c | Predicted IC50 (nM) |
|---|---|---|---|---|---|
| Kaempferol 3-O-sophoroside 7-O-glucoside | −87.97 | −1.440 | 3 | 2 | 253.25 |
| Cyanidin 3-O-sambubioside 5-O-glucoside | −87.33 | −1.618 | 3 | 2 | 253.25 |
| Malvidin 3-O-(6″-p-coumaroyl-glucoside) | −85.70 | −4.664 | 3 | 2 | 243.37 |
| *GRL-0617 | −60.75 | −4.952 | 0 | 0 | 483.16 |
| **Luteolin | −43.53 | −3.067 | 0 | 0 | 253.25 |
| ***VIR251 | −64.40 | −0.066 | 2 | 2 | 261.62 |
*a known prodrug of PLpro identified in our previous study [10].
**the best-scored potential drug identified by the previous study [20].
*** a peptide inhibitor in the structure of 6WX4 [75].
a. QPlogS is predicted aqueous solubility. The recommended range is −6.5 ∼ 0.5.
b. RO5: number of violations of Lipinski’s rule of five [38]. The recommended range: maximum is 4.
c. RO3: Number of violations of Jorgensen’s rule of three [39]. The recommended range: maximum is 3.
2.7. Molecular dynamics simulation
To further investigate the dynamic interactions between target proteins (Mpro and PLpro) and the top two polyphenols, we conducted Molecular dynamics (MD) simulations by using GROMACS version 2018.1 and CHARMM36 force field [28]. The starting coordinates of the protein–ligand complex were obtained from a ligand–protein docking study. Then, we used CHARMM-GUI to build the MD simulation solution boxes which were cubic boxes with length of 109 Å for 6W9C and 104 Å for 7BRP, and were filled with water [29], [30], [31]. Next, the minimized structures were equilibrated using an NVT ensemble (constant Number of particles, Volume, and Temperature) and NPT ensemble (the Number of particles, Pressure, and Temperature). The target equilibration temperature was 300 K. Finally, MD simulations were performed for 100 ns. After the MD simulations, we calculated the root-mean-square deviation (RMSD) and the energies.
3. Results
3.1. Docking analysis of polyphenols against SARS-CoV-2 Mpro
To seek effective inhibitors from polyphenols against SARS-CoV-2 Mpro, 480 polyphenols (ligand) were docked onto SARS-CoV-2 Mpro (protein: PDB ID: 7BRP). Based on the docking poses, binding energies between the protein and ligand were calculated by MM-GBSA. Compared to three top drug candidates proposed by previous studies [17], [18], in Table 1, the best three protein–ligand complexes, 7BRP- Petunidin 3-O-(6″-p-coumaroyl-glucoside) (−101.21 kcal/mol), 7BRP-Malvidin 3,5-O-diglucoside (−95.07 kcal/mol), and 7BRP- Cyanidin 3-O-(6″-p-coumaroyl-glucoside) (−90.17 kcal/mol) all have better estimated binding affinities. These top three polyphenols are members of anthocyanins, which can be found in black raspberry [32]. Meanwhile, we selected four ligands: saquinavir (−93.58 kcal/mol), EGCG (−65.04 kcal/mol), papyriflavonol A (−58.38 kcal/mol), and boceprevir (−72.56 kcal/mol) as a control group. From Table 1, we find that the binding energy of Saquinavir is slightly better than that of Cyanidin 3-O-(6″-p-coumaroyl-glucoside) (−90.17 kcal/mol), however, the binding energies of Petunidin 3-O-(6″-p-coumaroyl-glucoside) and Malvidin 3,5-O-diglucosideare are better than those of the control group, which suggests the potential inhibitory effects of these polyphenols against SARS-CoV-2 Mpro.
By comparing the 2D ligand–protein interactions of the top three polyphenols with Mpro (Fig. 1), we find that they all interact with Glu166 by forming hydrogen bonds: two hydrogen bonds for Petunidin 3-O-(6″-p-coumaroyl-glucoside), three for Malvidin 3,5-O-diglucoside, and one for cyanidin 3-O-(6″-p-coumaroyl-glucoside) (Table 2). This result suggests that Glu166 is an essential residue in the binding pocket. According to the previous studies, Glu166 plays an important role in connecting the substrate binding site with the dimer interface [33], and it also forms critical interactions with the residues of N-terminal finger on the heterologous monomer [33], [34]. In addition, the equivalent Glu169 on the Mpro of MERS-CoV is also a key residue, which is crucial in both dimerization and catalysis [35].
Fig. 1.

The docking poses and 2-D ligand–protein interaction diagrams of 7BRP and the top three ligands: A, Petunidin 3-O-(6″-p-coumaroyl-glucoside); B, Malvidin 3,5-O-diglucoside; C, Cyanidin 3-O-(6″-p-coumaroyl-glucoside). For the docking poses, S1/S2 subsites are being shown. The purple arrow indicates the hydrogen bond; the green line represents π-π stacking; the red line represents π -cation interaction. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
The number of hydrogen bonds formed between the top three polyphenols and essential residues of SARS-CoV-2 Mpro.
| Petunidin 3-O-(6″-p-coumaroyl-glucoside) | Malvidin 3,5-O-diglucoside | Cyanidin 3-O-(6″-p-coumaroyl-glucoside) | |
|---|---|---|---|
| Thr26 | 2 | 1 | 1 |
| Phe140 | 1 | 1 | |
| Leu141 | 1 | 1 | 1 |
| Asn142 | 1 | ||
| Gly143 | 1 | 1 | |
| Glu166 | 2 | 3 | 1 |
| Asp187 | 1 | 1 | |
| Gln189 | 1 | ||
| Gln192 | 1 | 1 | 1 |
Furthermore, these top three polyphenols all interact with Thr26, Leu141, and Gln192 by forming hydrogen bonds. Petunidin 3-O-(6″-p-coumaroyl-glucoside) and Cyanidin 3-O-(6″-p-coumaroyl-glucoside) interact with Phe140 and Gly143 by forming hydrogen bonds. Petunidin 3-O-(6″-p-coumaroyl-glucoside) and Malvidin 3,5-O-diglucoside both interact with Asp187 by forming one hydrogen bond each. Additionally, His41 is also a critical residue which can interact with the top three polyphenols. Petunidin 3-O-(6″-p-coumaroyl-glucoside) interacts with His41 by forming two π-π stackings and one π-cation interaction, and Cyanidin 3-O-(6″-p-coumaroyl-glucoside) interacts with His41 via one π-cation interaction. The number of hydrogen bonds between the top three polyphenols and essential residues are listed in Table 1. Accordingly, we propose that Petunidin 3-O-(6″-p-coumaroyl-glucoside), Malvidin 3,5-O-diglucoside, and Cyanidin 3-O-(6″-p-coumaroyl-glucoside) are the three best drug candidates among all 480 polyphenols tested against SARS-CoV-2 Mpro.
To further screen the drug candidates for Mpro, we calculated drug-likeness properties and predicted IC50 on the top three polyphenols by using Qikprop and AutoQSAR, respectively. The results are shown in Table 1. The polyphenols whose Qikprop descriptors (QPlogS, RuleOfFive, and RuleOfThree) fell out of the recommended range were excluded. For the results of AutoQSAR, it is interesting to note that the predicted IC50 values of the top three polyphenols are all lower than 50 nM and Cyanidin 3-O-(6″-p-coumaroyl-glucoside) shows the best predicted IC50, which is generally consistent with the docking results.
3.2. Docking analysis of polyphenols against SARS-CoV-2 PLpro
To identify the best inhibitors of SARS-CoV-2 PLpro, we also docked 480 polyphenols on SARS-CoV-2 PLpro (PDB ID: 6W9C) by performing ligand–protein docking and MM-GBSA calculations (see Table 3). Consequently, the three best compounds with the top MM-GBSA binding energies were Kaempferol 3-O-sophoroside 7-O-glucoside (−87.97 kcal/mol), Cyanidin 3-O-sambubioside 5-O-glucoside (−87.33 kcal/mol), and Malvidin 3-O-(6″-p-coumaroyl-glucoside) (−85.70 kcal/mol). Kaempferol 3-O-sophoroside 7-O-glucoside belongs to the group of flavonols. Cyanidin 3-O-sambubioside 5-O-glucoside and Malvidin 3-O-(6″-p-coumaroyl-glucoside) belong to the group of anthocyanins. Meanwhile, a known PLpro inhibitor GRL-0617 [10], the best-scored potential drug luteolin [20], and a peptide inhibitor in the structure 6WX4 were selected as control. From Table 3, we find that the top three polyphenols show better binding energies than the control compounds, which indicates that these polyphenols might have stronger inhibitory effects.
From the 2D ligand–protein interactions in Fig. 2, we find that the top three compounds all can interact with Asp164 and Arg166 by forming hydrogen bonds. Kaempferol 3-O-sophoroside 7-O-glucoside and Malvidin 3-O-(6″-p-coumaroyl-glucoside) interact with Asp164 by forming one hydrogen bond, respectively. Cyanidin 3-O-sambubioside 5-O-glucoside interacts with Asp164 by forming two hydrogen bonds. These three compounds form one hydrogen bond with Arg166, respectively. Moreover, Kaempferol 3-O-sophoroside 7-O-glucoside can interact with Gly266 and Asn267 by forming one hydrogen bond, respectively. Cyanidin 3-O-sambubioside 5-O-glucoside interacts with Gly266 and Tyr268 by forming one hydrogen bond, respectively. Notably, Gly266, Asn267, and Tyr268 are residues on blocking loop 2 (BL2), and BL2 plays an important role in inhibitor binding [40]. Hence, the interactions between the top two polyphenols and BL2 suggest strong inhibitory effects. Accordingly, we conclude that these two polyphenols can tightly bind onto the binding pocket. The number of hydrogen bonds between these three compounds and the essential residues are listed in Table 4.
Fig. 2.

The docking poses and 2-D ligand–protein interaction diagrams of 6W9C and the top three ligands: A, Kaempferol 3-O-sophoroside 7-O-glucoside; B, Cyanidin 3-O-sambubioside 5-O-glucoside; C, Malvidin 3-O-(6″-p-coumaroyl-glucoside). The pink arrow indicates the hydrogen bond; the green line represents π-π stacking; the red line represents π-cation interaction. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 4.
The number of hydrogen bonds formed between the top three polyphenols and essential residues of SARS-CoV-2 PLpro.
| Kaempferol 3-O-sophoroside 7-O-glucoside | Cyanidin 3-O-sambubioside 5-O-glucoside | Malvidin 3-O-(6″-p-coumaroyl-glucoside) | |
|---|---|---|---|
| Gly163 | 1 | ||
| Asp164 | 1 | 2 | 1 |
| Arg166 | 1 | 1 | 1 |
| Glu167 | 1 | 1 | |
| Ser170 | 1 | ||
| Pro248 | 1 | ||
| Tyr264 | 1 | ||
| Gly266 | 1 | 1 | |
| Asn267 | 1 | ||
| Tyr268 | 1 | ||
| Tyr273 | 1 | 1 | |
| Thr301 | 1 | 1 | |
| Asp302 | 1 |
3.3. Molecular dynamics (MD) simulation
To further analyze the stability of complexes, we conducted MD simulation to calculate RMSD and energy for the top two candidates. First, RMSD can be used to assess the stability of a protein–ligand complex. As shown in Fig. 3A, the RMSD for the complex of 7BRP- Petunidin 3-O-(6″-p-coumaroyl-glucoside) stabilized around 0.25 nm between 5 ns and 100 ns. In Fig. 3B, the RMSD of 7BRP-Malvidin 3,5-O-diglucoside stabilized at 0.25 nm from 10 ns to 40 ns, and then stabilized around 0.3 nm after 40 ns. The RMSD of 6W9C with Kaempferol 3-O-sophoroside 7-O-glucoside stabilized at 0.25 nm from 4 to 100 ns (Fig. 3C). From Fig. 3D, we find that the RMSD of 6W9C with Cyanidin 3-O-sambubioside 5-O-glucoside stabilized at 0.25 nm before 30 ns, and then stabilized around 0.35 nm from 30 to 90 ns. Moreover, the total energies of these four complexes are shown in Fig. 4. The energies of 7BRP complexes stabilized at around −1.13 × 106 KJ/mol (Fig. 4A and B). Interestingly, the energy of 6W9C complexes stabilized at around −1.365 × 106 KJ/mol (Fig. 4C and D). The RMSD and energy analysis show that the complexes of Mpro or PLpro with their respective top two polyphenols stay stable during the simulation process.
Fig. 3.

The RMSD of protein–ligand complexes. A, 7BRP- Petunidin 3-O-(6″-p-coumaroyl-glucoside); B, 7BRP- Malvidin 3,5-O-diglucoside; C, 6W9C- Kaempferol 3-O-sophoroside 7-O-glucoside; D, 6W9C- Cyanidin 3-O-sambubioside 5-O-glucoside.
Fig. 4.

The energy of protein–ligand complexes. A, 7BRP- Petunidin 3-O-(6″-p-coumaroyl-glucoside); B, 7BRP- Malvidin 3,5-O-diglucoside; C, 6W9C- Kaempferol 3-O-sophoroside 7-O-glucoside; D, 6W9C- Cyanidin 3-O-sambubioside 5-O-glucoside.
4. Discussion
Many studies have shown natural products possessing antiviral properties against the Epstein-Barr virus [41], [42], herpes simplex virus [43], [44], influenza virus [45], and other viruses targeting the respiratory tract [43], [46], [47], [48], [74]. Since the 2003 outbreak of SARS, a number of natural products have been reported to inhibit the coronavirus which causes SARS (SARS-CoV) or its target proteins [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [63], [64]. In addition, previous studies demonstrate that many polyphenols, including quercetin and its glycosylated derivatives, inhibit cell proliferation of tumor cells or microorganisms [65], [66], [67], [68]. Glycosylation of polyphenols could enhance water solubility, bioavailability, and their binding affinities to significant enzymes and improve the drug efficacy [69], [70], [71]. This result not only suggests that polyphenols are potential drug candidates, but also indicates that this study discovered more polyphenols for COVID-19 treatment.
Based on the docking pose of each target protein, we find that the key residue(s) can interact with the best inhibitor candidate via multiple interactions. First, in the structure of Mpro, Glu166 is a key residue for Mpro dimerization and substrate binding pocket creation [72]. The best inhibitor candidate Petunidin 3-O-(6″-p-coumaroyl-glucoside) interacts with Glu166 side chain by forming two hydrogen bonds (Fig. 5A), which makes an impact on increasing the binding energy. This result is consistent with the finding in our previous research which showed that saquinvir interacts with Glu166 [36]. Accordingly, Glu166 is a key residue for Mpro inhibitor discovery. In the structure of PLpro, the residues (Gly266-Gly271) in the BL2 loop are critical for inhibitor binding [7], [73]. From Fig. 5B, we find that the best candidate of PLpro Kaempferol 3-O-sophoroside 7-O-glucoside forms hydrogen bonds with Gly266 and Asn267 which are the residues in the BL2 loop. Additionally, the second-best candidate Cyanidin 3-O-sambubioside 5-O-glucoside form one hydrogen bond with Tyr268 which is also an important residue in the BL2 loop. Therefore, more interactions between the inhibitor and the BL2 loop may increase the binding affinity.
Fig. 5.

3D interaction diagrams showing the interactions between the best ligand and the key residue(s). A, Petunidin 3-O-(6″-p-coumaroyl-glucoside) interacts with the side chain of Glu166 via hydrogen bonds (yellow dash lines) on Mpro (PDB ID: 7BRP); B, Kaempferol 3-O-sophoroside 7-O-glucoside interacts with Gly266 and Asn267 via hydrogen bonds (yellow dash lines) on PLpro (PDB ID: 6W9C). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In summary, this study demonstrates the potential of polyphenols being an alternative treatment of COVID-19. The docking results agree with previous studies identifying the key residues interacting with the binding inhibitors or prodrugs, but the proposed inhibitors in this study possess even better estimated binding affinities. However, most of the top-ranked polyphenols cannot be ordered for validation experiments currently. We are attempting to obtain some of them from other labs or produce them ourselves. The much better estimated binding affinities than previously identified compounds and rational binding mechanisms support their potential efficacy and provide the clues for the future drug development.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
In this research, YW, LL, and ZRX were supported by a faculty seed grant from the office of research, University of Georgia and KYC was supported by Ministry of Science and Technology, R.O.C. under MOST-108-2221-E-019-052. We would like to acknowledgment Georgia Advanced Computing Resource Center (GACRC) and the College of Engineering’s IT department of the UGA for technical support. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) Bridges GPU at the Pittsburgh Supercomputing Center through allocation TGDPP180005.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.09.022.
Contributor Information
Kuan Y. Chang, Email: kchang@email.ntou.edu.tw.
Zhong-Ru Xie, Email: paulxie@uga.edu.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Cucinotta D., Vanelli M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020;91:157–160. doi: 10.23750/abm.v91i1.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klemm T., Ebert G., Calleja D.J., Allison C.C., Richardson L.W., Bernardini J.P. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39:1–17. doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.V’kovski P., Kratzel A., Steiner S., Stalder H., Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19:155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorganic Med Chem Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu J., Yuan X., Wang B., Gu R., Li W., Xiang X. Severe acute respiratory syndrome coronavirus 2: from gene structure to pathogenic mechanisms and potential therapy. Front Microbiol. 2020;11:1576. doi: 10.3389/fmicb.2020.01576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Báez-Santos Y.M., St. John S.E., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J.C., Spence R.A., Currier P.F., Lu X., Denison M.R. Coronavirus protein processing and RNA synthesis is inhibited by the cysteine proteinase inhibitor E64d. Virology. 1995;208:1–8. doi: 10.1006/viro.1995.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freitas B.T., Durie I.A., Murray J., Longo J.E., Miller H.C., Crich D. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect Dis. 2020;6:2099–2109. doi: 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- 11.Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baby K., Maity S., Mehta C.H., Suresh A., Nayak U.Y., Nayak Y. Targeting SARS-CoV-2 main protease: a computational drug repurposing study. Arch Med Res. 2021;52:38–47. doi: 10.1016/j.arcmed.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science (80-) 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2020;11 doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiao J., Li Y.-S., Zeng R., Liu F.-L., Luo R.-H., Huang C. SARS-CoV-2 M pro inhibitors with antiviral activity in a transgenic mouse model. Science (80-) 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandey KB, Rizvi SI. Plant polyphenols as dietary antioxidants in human health and disease. 2009. 10.1016/B978-0-12-849873-6.00001-7%250Ahttp://saber.ucv.ve/ojs/index.php/rev_venes/article/view/1112%250Ahttps://www.bps.go.id/dynamictable/2018/05/18/1337/persentase-panjang-jalan-tol-yang-beroperasi-menurut-operatornya-2014.html. [DOI] [PMC free article] [PubMed]
- 17.Ghosh R., Chakraborty A., Biswas A., Chowdhuri S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. J Biomol Struct Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1779818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh R., Chakraborty A., Biswas A., Chowdhuri S. Identification of polyphenols from Broussonetia papyrifera as SARS CoV-2 main protease inhibitors using in silico docking and molecular dynamics simulation approaches. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1802347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan M.F., Khan M.A., Khan Z.A., Ahamad T., Ansari W.A. Identification of dietary molecules as therapeutic agents to combat COVID-19 using molecular docking studies. Res Sq. 2020:1–17. [Google Scholar]
- 20.Ansari W.A., Ahamad T., Khan M.A., Khan Z.A., Luteolin Khan MF. A dietary molecule as potential anti-COVID-19 agent. Res Sq. 2020:1–10. [Google Scholar]
- 21.Neveu V., Perez-Jiménez J., Vos F., Crespy V., du Chaffaut L., Mennen L. Phenol-explorer: an online comprehensive database on polyphenol contents in foods. Database (Oxford) 2010;2010 doi: 10.1093/database/bap024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J.Y. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 23.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. http://www.ncbi.nlm.nih.gov/pubmed/10592235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shelley J.C., Anuradha A.E., Ae C., Ae L.L.F., Greenwood J.R., Mathew A.E. Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 25.Schrödinger Release 2021-3: QikProp, Schrödinger, LLC, New York, NY, 2021.
- 26.Dixon S.L., Duan J., Smith E., Von Bargen C.D., Sherman W., Repasky M.P. AutoQSAR: an automated machine learning tool for best-practice quantitative structure-activity relationship modeling. Future Med Chem. 2016;8:1825–1839. doi: 10.4155/fmc-2016-0093. [DOI] [PubMed] [Google Scholar]
- 27.Liu T., Lin Y., Wen X., Jorissen R.N., Gilson M.K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35 doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vanommeslaeghe K., Hatcher E., Acharya C. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J., Cheng X., Swails J.M., Yeom M.S., Eastman P.K., Lemkul J.A. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput. 2016;12:405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jo S., Kim T., Iyer V.G., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 31.Brooks B.R., Brooks C.L., Mackerell A.D., Nilsson L., Petrella R.J., Roux B. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Artemio Z., Tulio J., Reese R.N., Wyzgoski F.J., Rinaldi P.L., Fu R. Cyanidin 3-rutinoside and cyanidin 3-xylosylrutinoside as primary phenolic antioxidants in black raspberry. J Agric Food Chem. 2008;56:1880–1888. doi: 10.1021/jf072313k. [DOI] [PubMed] [Google Scholar]
- 33.Cheng S.C., Chang G.G., Chou C.Y. Mutation of glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys J. 2010;98:1327–1336. doi: 10.1016/j.bpj.2009.12.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu M.F., Kuo C.J., Chang K.T., Chang H.C., Chou C.C., Ko T.P. Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho B.L., Cheng S.C., Shi L., Wang T.Y., Ho K.I., Chou C.Y. Critical assessment of the important residues involved in the dimerization and catalysis of MERS Coronavirus Main Protease. PLoS ONE. 2015;10:1–18. doi: 10.1371/journal.pone.0144865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Y., Chang K.Y., Lou L., Edwards L.G., Doma B.K., Xie Z.R. In silico identification of drug candidates against COVID-19. Informatics Med Unlocked. 2020;21 doi: 10.1016/j.imu.2020.100461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun. 2020;11 doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 39.Jorgensen W.L., Duffy E.M. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54:355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 40.Lee H., Lei H., Santarsiero B.D., Gatuz J.L., Cao S., Rice A.J. Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem Biol. 2015;10:1456–1465. doi: 10.1021/cb500917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Leo A., Arena G., Lacanna E., Oliviero G., Colavita F., Mattia E. Resveratrol inhibits Epstein Barr Virus lytic cycle in Burkitt’s lymphoma cells by affecting multiple molecular targets. Antiviral Res. 2012;96:196–202. doi: 10.1016/j.antiviral.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Yiu C.-Y., Chen S.-Y., Chang L.-K., Chiu Y.-F., Lin T.-P. Inhibitory effects of resveratrol on the Epstein-Barr virus lytic cycle. Molecules. 2010;15:7115–7124. doi: 10.3390/molecules15107115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Annunziata G., Maisto M., Schisano C., Ciampaglia R., Narciso V., Tenore G.C. Resveratrol as a novel anti-herpes simplex virus nutraceutical agent: an overview. Viruses. 2018;10 doi: 10.3390/v10090473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faith S.A., Sweet T.J., Bailey E., Booth T., Docherty J.J. Resveratrol suppresses nuclear factor-B in herpes simplex virus infected cells. Antiviral Res. 2006;72:242–251. doi: 10.1016/j.antiviral.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 45.Lin C.J., Lin H.J., Chen T.H., Hsu Y.A., Liu C.S., Hwang G.Y. Polygonum cuspidatum and its active components inhibit replication of the influenza virus through Toll-like receptor 9-induced interferon beta expression. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0117602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zang N., Xie X., Deng Y., Wu S., Wang L., Peng C. Resveratrol-mediated gamma interferon reduction prevents airway inflammation and airway hyperresponsiveness in respiratory syncytial virus-infected immunocompromised mice. J Virol. 2011;85:13061–13068. doi: 10.1128/JVI.05869-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu T., Zang N., Zhou N., Li W., Xie X., Deng Y. Resveratrol Inhibits the TRIF-dependent pathway by upregulating sterile alpha and armadillo motif protein, contributing to anti-inflammatory effects after respiratory syncytial virus infection. J Virol. 2014;88:4229–4236. doi: 10.1128/JVI.03637-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mastromarino P., Capobianco D., Cannata F., Nardis C., Mattia E., De Leo A. Resveratrol inhibits rhinovirus replication and expression of inflammatory mediators in nasal epithelia. Antiviral Res. 2015;123:15–21. doi: 10.1016/j.antiviral.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 49.Cho J.K., Curtis-Long M.J., Lee K.H., Kim D.W., Ryu H.W., Yuk H.J. Geranylated flavonoids displaying SARS-CoV papain-like protease inhibition from the fruits of Paulownia tomentosa. Bioorg Med Chem. 2013;21:3051–3057. doi: 10.1016/j.bmc.2013.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim H.Y., Eo E.Y., Park H., Kim Y.C., Park S., Shin H.J. Medicinal herbal extracts of Sophorae radix, Acanthopanacis cortex, Sanguisorbae radix and Torilis fructus inhibit coronavirus replication in vitro. Antivir Ther. 2010;15:697–709. doi: 10.3851/IMP1615. [DOI] [PubMed] [Google Scholar]
- 51.Chen C.N., Lin C.P.C., Huang K.K., Chen W.C., Hsieh H.P., Liang P.H. Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3,3′- digallate (TF3) Evidence-based Complement Altern Med. 2005;2:209–215. doi: 10.1093/ecam/neh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin L.T., Hsu W.C., Lin C.C. Antiviral natural products and herbal medicines. J Tradit Complement Med. 2014;4:24–35. doi: 10.4103/2225-4110.124335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryu Y.B., Jeong H.J., Kim J.H., Kim Y.M., Park J.Y., Kim D. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CLpro inhibition. Bioorg Med Chem. 2010;18:7940–7947. doi: 10.1016/j.bmc.2010.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng P.W., Ng L.T., Chiang L.C., Lin C.C. Antiviral effects of saikosaponins on human coronavirus 229E in vitro. Clin Exp Pharmacol Physiol. 2006;33:612–616. doi: 10.1111/j.1440-1681.2006.04415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin C.W., Tsai F.J., Tsai C.H., Lai C.C., Wan L., Ho T.Y. Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antiviral Res. 2005;68:36–42. doi: 10.1016/j.antiviral.2005.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lau K.M., Lee K.M., Koon C.M., Cheung C.S.F., Lau C.P., Ho H.M. Immunomodulatory and anti-SARS activities of Houttuynia cordata. J Ethnopharmacol. 2008;118:79–85. doi: 10.1016/j.jep.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li S.Y., Chen C., Zhang H.Q., Guo H.Y., Wang H., Wang L. Identification of natural compounds with antiviral activities against SARS-associated coronavirus. Antiviral Res. 2005;67:18–23. doi: 10.1016/j.antiviral.2005.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sonja A. Rasmussen, MD, MS JCS. Dieckol, a SARS-CoV 3CLpro inhibitor, isolated from the edible brown algae Ecklonia cava. Bioorganic Med Chem J. 2020; January:19–21. [DOI] [PMC free article] [PubMed]
- 59.Park H.R., Yoon H., Kim M.K., Lee S.D., Chong Y. Synthesis and antiviral evaluation of 7-O-arylmethylquercetin derivatives against SARS-associated coronavirus (SCV) and hepatitis C virus (HCV) Arch Pharm Res. 2012;35:77–85. doi: 10.1007/s12272-012-0108-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee C., Lee J.M., Lee N.R., Kim D.E., Jeong Y.J., Chong Y. Investigation of the pharmacophore space of Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) NTPase/helicase by dihydroxychromone derivatives. Bioorg Med Chem Lett. 2009;19:4538–4541. doi: 10.1016/j.bmcl.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee C., Lee J.M., Lee N.R., Jin B.S., Jang K.J., Kim D.E. Aryl diketoacids (ADK) selectively inhibit duplex DNA-unwinding activity of SARS coronavirus NTPase/helicase. Bioorg Med Chem Lett. 2009;19:1636–1638. doi: 10.1016/j.bmcl.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park J.Y., Yuk H.J., Ryu H.W., Lim S.H., Kim K.S., Park K.H. Evaluation of polyphenols from Broussonetia papyrifera as coronavirus protease inhibitors. J Enzyme Inhib Med Chem. 2017;32:504–512. doi: 10.1080/14756366.2016.1265519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ho T.Y., Wu S.L., Chen J.C., Li C.C., Hsiang C.Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antiviral Res. 2007;74:92–101. doi: 10.1016/j.antiviral.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yi L., Li Z., Yuan K., Qu X., Chen J., Wang G. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. J Virol. 2004;78:11334–11339. doi: 10.1128/JVI.78.20.11334-11339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahbub A.A., Le Maitre CL, Haywood-small S., Cross N.A., Jordan-mahy N. Polyphenols enhance the activity of alkylating agents in leukaemia cell lines. Oncotarget. 2019;10:4570–4586. doi: 10.18632/oncotarget.27068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren K.W., Li Y.H., Wu G., Ren J.Z., Bin Lu.H., Li Z.M. Quercetin nanoparticles display antitumor activity via proliferation inhibition and apoptosis induction in liver cancer cells. Int J Oncol. 2017;50:1299–1311. doi: 10.3892/ijo.2017.3886. [DOI] [PubMed] [Google Scholar]
- 67.Turumtay H., Midilli A., Turumtay E.A., Demir A., Selvi E.K., Budak E.E. Gram (−) microorganisms DNA polymerase inhibition, antibacterial and chemical properties of fruit and leaf extracts of Sorbus acuparia and Sorbus caucasica var. yaltirikii. Biomed Chromatogr. 2017;31 doi: 10.1002/bmc.3901. e3901. [DOI] [PubMed] [Google Scholar]
- 68.Nosrati M., Shakeran Z., Shakeran Z. Frangulosid as a novel hepatitis B virus DNA polymerase inhibitor: a virtual screening study. In Silico Pharmacol. 2018;6:10. doi: 10.1007/s40203-018-0047-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu Y., Hsieh T., Wu J.M., Wang X., Christopher J.S., Pham A.H. Elucidating the inhibitory effect of resveratrol and its structural analogs on selected nucleotide- related enzymes. Biomolecules. 2020;10:1223. doi: 10.3390/biom10091223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lepak A., Gutmann A., Kulmer S.T., Nidetzky B. Creating a water-soluble resveratrol-based antioxidant by site-selective enzymatic glucosylation. ChemBioChem. 2015;16:1870–1874. doi: 10.1002/cbic.201500284. [DOI] [PubMed] [Google Scholar]
- 71.Hollman P.C.H., Bijsman M.N.C.P., Van Gameren Y., Cnossen E.P.J., De Vries J.H.M., Katan M.B. The sugar moiety is a major determinant of the absorption of dietary flavonoid glycosides in man. Free Radic Res. 1999;31:569–573. doi: 10.1080/10715769900301141. [DOI] [PubMed] [Google Scholar]
- 72.Gupta S., Singh A.K., Kushwaha P.P., Prajapati K.S., Shuaib M., Senapati S. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1776157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci USA. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mhatre Susmit, Gurav Nitisha, Shah Mansi, Patravale Vandana. Entry-inhibitory role of catechins against SARS-CoV-2 and its UK variant. Comput Biol Med. 2021;135:104560. doi: 10.1016/j.compbiomed.2021.104560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rut Wioletta, Lv Zongyang, Zmudzinski Mikolaj, Patchett Stephanie, Nayak Digant. Activity profiling and crystal structures of inhibitorbound SARS-CoV-2 papain-like protease: A frameworkfor anti–COVID-19 drug design. Sci Adv. 2020;6 doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.