

Abstract
Busulfan (Bu) is a common component of conditioning regimens before hematopoietic stem cell transplantation (HSCT) and is known for high interpatient pharmacokinetic (PK) variability. This study aimed to develop and externally validate a multicentric, population PK (PopPK) model for intravenous Bu in pediatric patients before HSCT to first study the influence of glutathione‐s‐transferase A1 (GSTA1) polymorphisms on Bu's PK in a large multicentric pediatric population while accounting for fludarabine (Flu) coadministration and, second, to establish an individualized, model‐based, first‐dose recommendation for intravenous Bu that can be widely used in pediatric patients. The model was built using data from 302 patients from five transplantation centers who received a Bu‐based conditioning regimen. External model validation used data from 100 patients. The relationship between body weight and Bu clearance (CL) was best described by an age‐dependent allometric scaling of a body weight model. A stepwise covariate analysis identified Day 1 of Bu conditioning, GSTA1 metabolic groups based on GSTA1 polymorphisms, and Flu coadministration as significant covariates influencing Bu CL. The final model adequately predicted Bu first‐dose CL in the external cohort, with 81% of predicted area under the curves within the therapeutic window. The final model showed minimal bias (mean prediction error, −0.5%; 95% confidence interval [CI], −3.1% to 2.0%) and acceptable precision (mean absolute prediction error percentage, 18.7%; 95% CI, 17.0%–20.5%) in Bu CL prediction for dosing. This multicentric PopPK study confirmed the influence of GSTA1 polymorphisms and Flu coadministration on Bu CL. The developed model accurately predicted Bu CL and first doses in an external cohort of pediatric patients.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Glutathione‐s‐transferase A1 (GSTA1) polymorphisms have been shown to influence busulfan clearance in children receiving myeloablative conditioning regimens. Although fludarabine's impact on intravenous busulfan clearance is not well established, busulfan and fludarabine are increasingly used together in reduced intensity regimens. Whether GSTA1 polymorphisms influence busulfan clearance when coadministered with fludarabine is also unknown.
WHAT QUESTION DID THIS STUDY ADDRESS?
The study aimed to examine the influence of GSTA1 polymorphisms and fludarabine coadministration on busulfan's pharmacokinetic parameters and whether these factors should be considered in busulfan first‐dose recommendations.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This multicenter population pharmacokinetic analysis in a large patient data set confirmed the significant effect of GSTA1 polymorphisms and fludarabine on busulfan clearance. The developed and externally validated model enables to accurately recommend first doses of intravenous busulfan.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The present study suggests that personalized first doses of busulfan should consider both patients’ GSTA1‐related busulfan metabolic capacity and fludarabine coadministration for optimal exposure.
INTRODUCTION
The bifunctional alkylating agent busulfan (Bu) is commonly used as part of chemotherapeutic conditioning regimens for hematopoietic stem cell transplantation (HSCT). Bu is known for its narrow therapeutic window. Pediatric patients’ exposure to Bu has usually been described using the area under the curve (AUC) of its concentration‐time data, with a historical cumulative target AUC range of 59.1–98.5 mg·h/L (14,400–24,000 µM·min) over 4 days of administration as part of a multiagent myeloablative conditioning regimen,1, 2, 3, 4, 5, 6, 7 although a different target could be chosen for reduced conditioning regimens based on the treated disease.8 Achieving this narrow therapeutic window is crucial as small variations in the AUC have substantial effects on the clinical outcome of HSCT. Underexposure to Bu is associated with poor HSCT outcomes, such as disease relapse or graft rejection,1, 4 whereas overexposure leads to higher rates of treatment‐related toxicities, such as sinusoidal obstructive syndrome and treatment‐related mortality.3, 7, 9 Considerable interindividual variability in pharmacokinetic (PK) parameters, especially in pediatric patients, may also contribute to variable clinical outcomes.6, 10, 11 Dose adjustments guided by therapeutic drug monitoring (TDM) are thus still recommended in pediatric HSCT to normalize the patients’ exposure to Bu.12, 13
The current European Medicines Agency6 and U.S. Food and Drug Administration14 first‐dose recommendations, only based on body weight (BW), often result in patients falling outside the target therapeutic window.15 Prediction models based on population PK (PopPK) studies have also been developed, with the aim of reaching the target window immediately after the first dose, optimizing the exposure to Bu. Those studies considered body‐size metrics (actual BW, body surface area, normal fat mass) and/or age‐dependent maturation functions to predict Bu's PK parameters and first doses in HSCT patients.16, 17, 18, 19, 20, 21 However, these models still resulted in 25%–55% of patients with AUCs outside the therapeutic window after the first dose of Bu.15, 22 This is even more valid for patients with extreme glutathione‐s‐transferase A1 (GSTA1) metabolic capacity.15 As the expression of the GSTA1 enzyme is involved in Bu's main metabolic pathway,23, 24 GSTA1 proximal promoter polymorphisms have been associated with differing GSTA1 metabolic capacity.25, 26 Poor GSTA1 metabolic capacity is typically associated with lower Bu clearance (CL) and thus supratherapeutic AUC. These patients are more likely to develop treatment‐related acute toxicities.26 Our group recently developed a PopPK model using data from 112 patients from a single center and incorporating GSTA1 genetically determined metabolic capacity, resulting in a better Bu CL prediction than most other available models.27 However, the model's development was based on data from a single center and did not include patients receiving fludarabine (Flu).
Flu is a purine analog increasingly used along with Bu in conditioning regimens for HSCT in various malignant and nonmalignant indications. A few reports have suggested that the coadministration of Flu significantly influenced Bu CL. De Castro et al.28 reported that patients treated with Flu before oral Bu had 30% lower CL than patients receiving oral Bu followed by cyclophosphamide (Cy). Yeh et al.29 reported higher interdose CL variability between conditioning days among patients receiving Flu before intravenous (i.v.) Bu than among patients receiving Cy before Bu. According to this report, the lower end of the Bu target window should be aimed for when coadministered with Flu.29 However, this interaction of Flu and Bu has not been studied in a larger cohort of pediatric patients while accounting for the effect of GSTA1 polymorphisms.
The present study aimed to confirm the effects of GSTA1 polymorphisms on Bu CL in a large, multicentric, PopPK study including pediatric patients who had received a variety of Bu‐based conditioning regimens. We also investigated the influence of Flu on Bu CL, and we now propose a widely applicable algorithm for individualized Bu first doses in pediatric patients undergoing HSCT.
METHODS
Patient population
This study included pediatric patients from the NCT01257854 prospective observational cohort study registered in clinicaltrial.gov and the ACTRN12612000544875 prospective observational cohort study registered in the Australian New Zealand Clinical Trial Registry. The patients received i.v. Bu in combination with other chemotherapeutic agents as part of a conditioning chemotherapy regimen before autologous or allogeneic HSCT in one of five pediatric transplantation centers. Patients were included in the analysis if their complete data were available in terms of demographic and clinical data (e.g., age, weight, height, sex, diagnosis, conditioning chemotherapy), GSTA1 genetic information (or available DNA samples for genotyping), and measured plasma concentrations of Bu.
Treatment regimen, PK analysis, and genotyping
Bu (Busulfex®, Otsuka Pharmaceuticals or Busilvex®, Pierre Fabre Laboratory) was administered to patients every 6 h (q6h), every 12 h (q12h), or every 24 h (q24h) via i.v. infusions from 2 to 4 h. Bu first‐dose calculations, dosing schedules, sampling schedules, and the analytical TDM methods used in each transplantation center are presented in Table S1. The GSTA1 genotyping methods25, 30 are detailed in Supplemental Material S2, and GSTA1 promoter‐haplotype determination is detailed in Supplemental Material S3. Based on their GSTA1 promoter diplotypes, patients were classified into three GSTA1 metabolic groups as rapid (G1), intermediate (G2), and poor metabolizers (G3) according to their GSTA1 gene expression potential, as previously reported (Supplemental Material S4).15, 26, 27 An external validation data set of 25% of the patients (not used in the model training) was sampled from the full data set using stratified random sampling in R software (version 4.0.0; R Foundation for Statistical Computing; Supplemental Material S5). The stratification criteria were the GSTA1 metabolic groups and the presence of Flu in the conditioning regimen.
PopPK analysis
PopPK analysis was performed using nonlinear mixed effects modeling in Phoenix NLME software (version 8.2; Certara USA Inc.). A first‐order conditional estimation algorithm was used for this analysis. The reduction in the objective function value (OFV), based on the log‐likelihood ratio test, was the main criterium for model comparison, assuming a χ2 distribution. A decrease in the OFV of more than 3.84 (p < 0.05) for one additional model parameter was considered statistically significant. The minimization of the Akaike information criterion, the adequacy of goodness‐of‐fit plots, the minimization of random effect estimates, and the precision of fixed and random effects parameters were also considered in the selection and evaluation of models.
Structural and statistical model
Bu i.v. infusion was assumed to be zero order. One‐compartment and two‐compartment models were tested, and nonlinear elimination models were also examined. The random variability of PK parameters was estimated using an exponential‐error model, as shown in Equation (1):
| (1) |
where Pij is the parameter value for the i th individual on the j th occasion, θP is the typical value for the PK parameter in the study population, ηi is the random interindividual variability (IIV), and κj the random interoccasion variability (IOV). Additive, proportional, and mixed error models describing the residual variability were evaluated.
Base model selection
Because of their crucial role in pediatric PK, body size, age, or age‐dependent metabolism maturation descriptors were included in the base model. Body‐size metrics such as actual BW, fat‐free mass,31 normal‐fat mass,32 and body surface area were investigated to assess which factor best predicted Bu's PK parameters. To best describe the relationship between individual PK parameters, the allometric scaling of body size was tested using Equation (2):
| (2) |
where BSi is the individual value of the body‐size descriptor, standard BS is either the median value or a standard value (e.g., BW =70 kg) for the size descriptor in the model population, and pwr is the allometric exponent. Several allometric scaling methods based on previous pediatric PopPK studies were evaluated.19, 33, 34 The inclusion of an age‐dependent maturation function for Bu metabolism was evaluated last.16, 35 The details of the calculation of body‐size metrics and the maturation function are shown in Table S6, and the different base models tested are shown in Supplemental Material S7.
Covariate modeling
Covariate analysis was performed using a stepwise covariate addition/elimination approach. In the forward addition steps, a covariate was deemed significant if its addition resulted in a decrease in the OFV of more than 3.84 (p < 0.05) for one additional parameter; it was then retained in the model. If more than one covariate was significant, the covariate retained was the one resulting in the greatest decrease in the OFV. The remaining covariates were tested again in subsequent forward steps until no additional covariate significantly improved the model. During backward elimination, covariates were removed from the full covariate model one at a time. The covariate's significance was confirmed if its removal resulted in an increase in the OFV by a significant value corresponding to the χ2 distribution for p < 0.01 (e.g., 6.63 for one less parameter). The clinical and genetic covariates were selected based on their previously reported or potential influence on Bu PK. The tested covariates were the sex, the diagnosis, GSTA1 polymorphisms, Flu coadministration, the day of conditioning, and the treatment center. More details on the different tested settings of these covariates are in Supplemental Material S8.
Model validation
The bootstrap method was performed using 1000 replicates resampled from the modeling data set to evaluate the model's robustness. The model was considered stable if the bootstrap's estimates corresponded to the final model's estimates and if the final parameter estimates were within the 95% confidence interval (CI) of the bootstrap estimates.
To externally validate the model, we used the prediction‐corrected visual predictive check (pcVPC) simulation method to evaluate the model's predictive performance based on 1000 replicates from the external data set. The model was also refitted to the external data set, and the final parameter estimates obtained with the validation data set were compared with those obtained with the model‐building data set.
Model's predictive performance
We evaluated the new model's ability to predict first‐dose Bu CL—the PK parameter used for the first‐dose calculation. Using the external data set, simulated CL was assessed in terms of mean prediction error (MPE) for accuracy, mean absolute prediction error (MAPE), and root mean square relative prediction error (RMSE) for precision using the following equations36:
| (3) |
| (4) |
| (5) |
with CLobs representing the observed first‐dose CL determined by Non‐compartmental analysis in Phoenix WinNonLin software (version 8.2; Certara Inc.), CLpred being the model‐predicted CL, and n being the number of observations. Other i.v. Bu dosing algorithms enabling CL calculations were included in the analysis for comparison.16, 17, 18, 19, 27 These were the best performing algorithms according to two previous studies.15, 22 The acceptability interval was set at a ±25% deviation from the CLpred, which corresponds to the difference between the center value of the target therapeutic window for q24h AUC (19.7 mg·h/L), with the lower and upper ranges of 14.8 mg·h/L and 24.6 mg·h/L, respectively. We used the Wilcoxon signed‐rank test in SPSS software (version 26; IBM Corp.) and the McNemar test for related samples for the pairwise comparison of the new model with each of the other tested models. The significance level was set as p = 0.01 in consideration of the Bonferroni correction for five pairwise comparisons.
The analysis was also extended to dosing nomograms not enabling to calculate CL.6, 14, 21 Each model's ability to predict first doses resulting in AUCs within the therapeutic window was assessed. The model‐predicted dose (Dosemodel) calculations using each model are shown in Supplemental Material S9, with the AUCs predicted by each model (AUCmodel) calculated using Equation (6):
| (6) |
Bu exposure was expressed as AUC in mg·h/L, in keeping with international attempts to harmonize the expression of Bu plasma exposure.37 The first‐dose target AUC was set to 19.7 mg·h/L (4800 µM·min), and the therapeutic window was set to 14.8–24.6 mg·h/L (3600–6000 µM·min) for q24h dosing. Pairwise comparisons between the new model and each of the other models tested used the McNemar test for related samples, with significance set at p = 0.00625 according to a Bonferroni correction for eight pairwise comparisons.
RESULTS
Patient population
Our population included 402 pediatric HSCT recipients (0–20 years old), and a subset of 112 patients was analyzed in a previous PopPK model.27 Patients’ clinical and demographic characteristics are presented in Table 1.
TABLE 1.
Patient clinical characteristics
| Characteristics | Model cohort | Validation cohort | ||
|---|---|---|---|---|
| n (%) | Median (range) | n (%) | Median (range) | |
| Total | 302 | 100 | ||
| Sex | ||||
| Male | 176 (58.3) | 56 (56.0) | ||
| Female | 126 (41.7) | 44 (44.0) | ||
| Age, years | 5.2 (0.1–20.1) | 6.0 (0.2–20.0) | ||
| Weight, kg | 19.5 (2.9–101.1) | 19.1 (4.5–86.0) | ||
| 0–9 kg | 36 (11.9) | 15 (15.0) | ||
| 9–16 kg | 85 (28.1) | 27 (27.0) | ||
| 16–23 kg | 51 (16.9) | 16 (16.0) | ||
| 23–34 kg | 45 (14.9) | 13 (13.0) | ||
| > 34 kg | 85 (28.1) | 29 (29.0) | ||
| Body mass index (kg/m2) | 17.4 (12.6–40.9) | 16.6 (10.4–29.6) | ||
| Diagnosis | ||||
| Malignancies | 191 (63.2) | 67 (67.0) | ||
| AML | 79 (26.2) | 27 (27.0) | ||
| ALL | 31 (10.3) | 13 (13.0) | ||
| MDS | 32 (10.6) | 9 (9.0) | ||
| MPS | 6 (2.0) | 2 (2.0) | ||
| Neuroblastoma | 21 (7.0) | 11 (11.0) | ||
| Other malignanciesa | 22 (7.3) | 4 (4.0) | ||
| Nonmalignancies | 111 (36.8) | 33 (33.0) | ||
| Immune deficiencies | 38 (12.6) | 9 (9.0) | ||
| BMFS | 3 (1.0) | 1 (1.0) | ||
| Metabolic diseases | 20 (6.6) | 11 (11.0) | ||
| Hemoglobinopathies | 22 (7.3) | 7 (7.0) | ||
| CGD | 14 (4.6) | 4 (4.0) | ||
| HLH | 13 (4.3) | 2 (2.0) | ||
| Chronic relapsing polychondritis | 1 (0.3) | 0 (0.0) | ||
| Number of HSCTs | ||||
| 1 | 287 (95.0) | 95 (95.0) | ||
| 2 | 15 (5.0) | 3 (3.0) | ||
| 3 or more | 0 (0.0) | 2 (2.0) | ||
| Bu dosing schedule | ||||
| q6h | 114 (37.7) | 35 (35.0) | ||
| q12h | 1 (0.3) | 0 (0.0) | ||
| q24h | 187 (61.9) | 65 (65.0) | ||
| Conditioning regimen | ||||
| Non‐flu regimens | 169 (56.0) | 56 (56.0) | ||
| BuCy | 110 (36.4) | 37 (37.0) | ||
| BuCyVP16 | 11 (3.6) | 0 (0.0) | ||
| BuMel | 27 (8.9) | 15 (15.0) | ||
| BuCyMel | 19 (6.3) | 4 (4.0) | ||
| BuMelAraC | 1 (0.3) | 0 (0.0) | ||
| BuMelGe | 1 (0.3) | 0 (0.0) | ||
| Flu regimens | 133 (44.0) | 44 (44.0) | ||
| FluBu | 96 (31.8) | 24 (24.0) | ||
| FluBuCy | 9 (3.0) | 1 (1.0) | ||
| FluBuTTP | 8 (2.6) | 7 (7.0) | ||
| FluBuMel | 18 (5.9) | 12 (12.0) | ||
| FluBuTTPCy | 2 (0.7) | 0 (0.0) | ||
| GSTA1 group | ||||
| 1 | 54 (17.9) | 18 (18.0) | ||
| 2 | 195 (64.6) | 64 (64.0) | ||
| 3 | 53 (17.5) | 18 (18.0) | ||
Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; AraC, cytarabine; BMFS, bone marrow failure syndrome; Bu, busulfan; CGD, chronic granulomatous disease; Cy, cyclophosphamide; Flu, fludarabine; Ge, gemcitabine; HLH, hemophagocytic lymphohistiocytosis; HSCT, hematopoietic stem cell transplantation; MDS, myelodysplastic syndrome; Mel, melphalan; MPS, myeloproliferative disease; q12h, twice‐daily Bu dosing; q24h, once‐daily Bu dosing; q6h, four times daily Bu dosing; TTP, thiotepa; VP16, etoposide.
Lymphomas, Ewing sarcoma, chronic myeloid leukemia, and juvenile myelomonocytic leukemia.
A total of 5293 Bu plasma concentrations and 994 Bu PK profiles were available for analysis, with 1286 plasma concentrations (mean of 8.6 per patient; range, 2–22) and 294 PK profiles (mean of 1.7 per patient) available for q6h patients, 29 plasma concentrations and five PK profiles from a sole patient receiving q12h doses, and 3978 concentrations (mean of 15.8 per patient; range, 2–30) and 740 PK profiles (mean of 2.9 per patient) available for q24h patients. Concentrations below the lower limits of quantification accounted for 5.2% of the data (277 concentrations). Their impact is therefore negligible, and they were not included in the analysis.38 Only 19.6% of the q6h patients had their Bu concentrations measured beyond Day 1 of administration compared with 79.0% in the q24h patients. The infusion durations were missing for 13.4% of patients and were in these cases set to 2 h for q6h doses and to 3 h for q24h doses.
Base model
A two‐compartment model with first‐order elimination and proportional residual error was the best structural model to fit the Bu concentration‐time data. The estimated PK parameters were the CL, volume of distribution of central compartment (V1), the intercompartmental CL (Q), and the peripheral volume of distribution (V2). The model incorporating the postmenstrual age in years (PMA) dependent allometric scaling of BW was superior to the other base models tested in terms of minimizing the OFV and IIV of CL. The allometric scaling factor varied from 1.17 in neonates to 0.65 in 20‐year‐old patients. Compared with the theoretical allometric scaling factor of 0.75, this model resulted in a significant decrease in the OFV (ΔOFV = −368) and a decrease in the IIV of CL from 29% to 24%. Using the body size descriptors of fat‐free mass, normal‐fat mass, or body surface area did not improve the model. We assumed linear relationships between V1, Q, V2, and BW. Including IOV in CL and V1 significantly improved model predictions (ΔOFV = −287).
Covariate search
The results of the stepwise covariate search are illustrated in Table S10. The covariate analysis identified the day of Bu infusion (Day 1 vs. others), GSTA1 metabolic groups, and Flu presence/absence as significant covariates of Bu CL. No significant covariate of V1 was observed. CL on subsequent days of Bu administration was 9% lower than on Day 1. Compared with G2 patients, G1 patients exhibited 10% higher CL, whereas G3 patients exhibited 12% lower CL. Haplotypes based on a single polymorphism (−69C/T; *A*A, *A*B, and *B*B) resulted in less decrease in the OFV than GSTA1 metabolic groups (ΔOFV = −57.0 vs. ΔOFV = −67.9, respectively). Patients receiving Flu exhibited significantly lower CL (−7%).
Final model
The final model's equation for CL prediction is:
where CLi is the individual CL, CLpop is the population CL for a patient weighting 20 kg, BW is the actual BW, l is the allometric scaling exponent, PMA is the postmenstrual age in years, and M is the exponent for the variation of L with the patient's age. F day1 is the correction factor for CL on Day 1 compared with subsequent days. F GSTA1 is the factor according to the GSTA1 metabolic group (in G1 and G3 patients), and F Flu is the correction factor for patients receiving Flu in their conditioning regimen. According to the initial covariate search results, considering both F GSTA1 and F Flu would predict that G3 patients receiving Flu would have a 18% lower CL (data not shown). To avoid underestimating CL in this particular group of patients, Flu's effect on CL was assessed separately with each GSTA1 metabolic group. This showed that the population estimate of Flu's effect on Bu CL was similar among G1 and G2 patients (8% lower CL), whereas among G3 patients, after considering FGSTA1, Flu's effect on CL was a decrease of only 4%. Flu's effect on Bu CL among G3 patients was therefore estimated separately. The final model code is provided in Supplemental Material S11, and the final model estimates and bootstrap simulation results are shown in Table 2.
TABLE 2.
Final model table
| Parameters | Final model estimates | Refit on external data set | Bootstrap estimates | ||||
|---|---|---|---|---|---|---|---|
| Estimate | RSE% | Estimate | RSE% | Estimate | 95% CI | ||
| Fixed effects | |||||||
| CL (L/h) | 5.01 | 1 | 4.87 | 2 | 4.92 | 4.65–5.23 | |
| V1 (L) | 12.98 | 1 | 13.02 | 2 | 12.95 | 12.90–13.02 | |
| Q (L/h) | 1.85 | 6 | 1.79 | 11 | 1.77 | 0.91–1.94 | |
| V2 (L) | 2.14 | 4 | 2.07 | 9 | 2.09 | 1.60–2.24 | |
| l in | 1.17 | 1 | 1.15 | 4 | 1.14 | 1.05–1.22 | |
| M in | −0.19 | 5 | −0.20 | 13 | −0.20 | −0.21 to −0.13 | |
| Covariates | |||||||
| FGSTA1 G1 | 1.10 | 3 | 1.09 | 35 | 1.10 | 1.09–1.11 | |
| FGSTA1 G3 | 0.88 | 3 | 0.89 | 26 | 0.88 | 0.87–0.89 | |
| FFlu in G1 and G2 patients | 0.92 | 10 | 0.92 | 31 | 0.92 | 0.92–0.95 | |
| FFlu in G3 patients | 0.97 | 4 | 0.96 | 20 | 0.96 | 0.96–0.97 | |
| Fday 1 | 1.09 | 4 | 1.08 | 22 | 1.09 | 1.08–1.10 | |
| Random effects | |||||||
| Interindividual variability | |||||||
| IIV CL (%) | 17 | 4 | 17 | 5 | 17 | 16–18 | |
| IIV V1 (%) | 11 | 7 | 10 | 4 | 11 | 10–12 | |
| IIV Q (%) | 68 | 9 | 65 | 20 | 66 | 54–77 | |
| IIV V2 (%) | 67 | 11 | 68 | 32 | 64 | 58–69 | |
| Interoccasion variability | |||||||
| IOV CL (%) | 13 | 6 | 12 | 13 | 13 | 12–13 | |
| IOV V (%) | 15 | 7 | 15 | 10 | 14 | 14–15 | |
| Proportional residual error (%) | 6 | 5 | 6 | 1 | 6 | 5–8 | |
| Eps shrinkage (%) | 19 | 17 | |||||
Abbreviations: BW, body weight; CI, confidence interval; CL, clearance; CLi, individual clearance; CLpop, population estimate for clearance; Eps shrinkage, shrinkage attributed to residual variability; Fday 1, correction factor to be used on day 1 of Bu infusion; FFlu, correction factor for patient receiving Flu; FGSTA1 G1, correction factor for G1 patients (rapid metabolizers); FGSTA1 G3, correction factor for G3 patients (slow metabolizers); IIV, random interindividual variability; IOV, random interoccasion variability; l, allometric scaling exponent; M, exponent for the variation of l with PMA; PMA, postmenstrual age in years; Q, intercompartment clearance; RSE%, percentage of relative standard error; V1, volume of distribution of central compartment; V2, volume of distribution of peripheral compartment.
Model validation
The bootstrap simulation estimates did not differ from the final model estimates as final model estimates were within the 95% CI of the bootstrap results.
The goodness‐of‐fit plots illustrated in Figure 1 show good agreement between the model‐predicted concentrations and observed concentrations. Plots of the conditional weighted residuals (CWRES) showed a normal distribution with mean 0. However, in the CWRES versus predicted concentrations plot (Figure 1d), the distribution of the residuals was slightly wider toward the lower concentrations.
FIGURE 1.
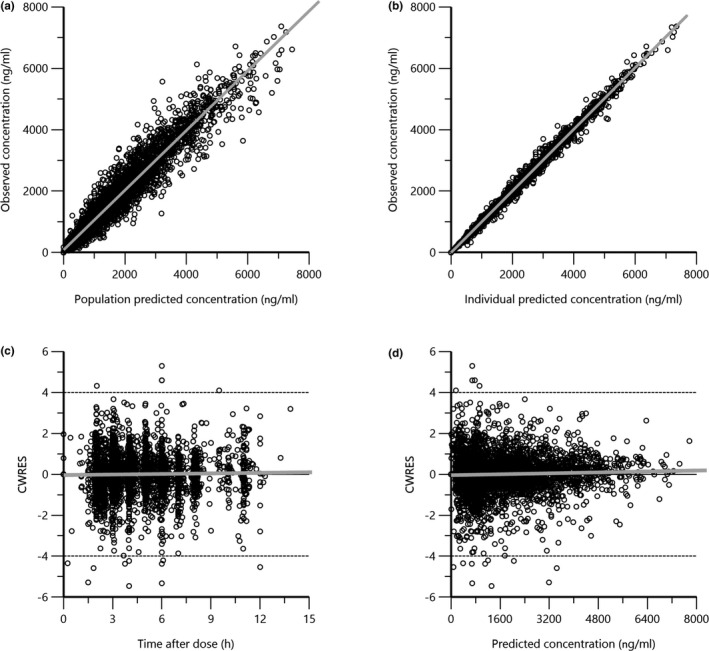
Final model goodness‐of‐fit plots: (a) observed busulfan concentrations versus population predicted concentrations, (b) observed busulfan concentrations versus individual predicted concentrations, (c) CWRES versus time after infusion initiation, and (d) CWRES versus population‐predicted concentrations. CWRES, conditional weighted residuals
The plots of the pcVPC simulation, performed using the external data set and illustrated in Figure 2, showed a satisfactory agreement between the observed and predicted data. The new model adequately described Bu PK in the external validation cohort. Moreover, refitting the model on the external data set resulted in parameters that did not deviate from the final model estimates.
FIGURE 2.
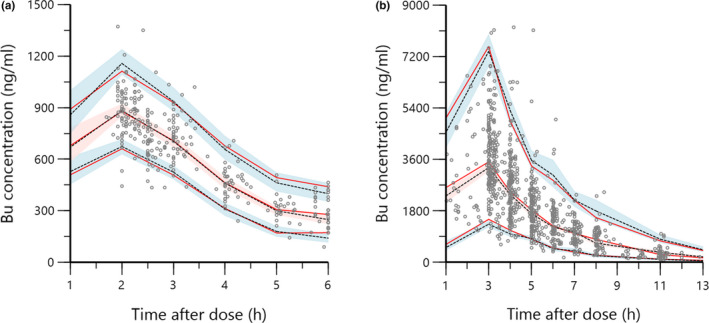
Prediction‐corrected visual predictive check of the final model using the external validation data set: (a) doses every 6 h and (b) doses every 24 h. The marks represent the observed concentrations. Red solid lines represent the 5th, 50th, and 95th percentiles of observed concentrations; dashed black lines represent the 5th, 50th, and 95th percentiles of predicted concentrations; and shaded areas represent 95% confidence intervals of the predicted 5th, 50th, and 95th percentiles. The observed data's percentiles were in concordance with the predicted data's percentiles. Bu, busulfan
Models’ predictive performance
The tested models’ prediction accuracy and precision results are shown in Table 3, and the error distributions obtained with those models are illustrated in Figure 3.
TABLE 3.
Different models’ clearance prediction performance
| Model | MPE % (95% CI) | p | MAPE % (95% CI) | p | RMSE % (95% CI) | p | P25% | p | < P25% | p | > P25% | p |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| New model | −0.5 (−3.1–2.0) | Reference | 18.7 (17.0–20.5) | Reference | 27.2 (23.0–31.5) | Reference | 81 | Reference | 10 | Reference | 9 | Reference |
| Nava | 5.7 (2.7–8.7) | < 0.001 | 20.0 (17.7–22.3) | 0.710 | 29.9 (25.1–34.8) | 0.710 | 80 | 1.000 | 6 | 0.125 | 14 | 0.063 |
| McCune | −9.8 (−12.5 to −7.2) | < 0.001 | 22.4 (20.6–24.1) | < 0.001 | 31.3 (26.8–35.9) | < 0.001 | 68 | 0.002 | 25 | < 0.001 | 7 | 0.500 |
| Bartelink | −16.7 (−18.9 to −14.5) | < 0.001 | 24.2 (22.8–25.6) | < 0.001 | 36.9 (31.6–42.2) | < 0.001 | 47 | < 0.001 | 46 | < 0.001 | 7 | 0.500 |
| Long‐Boyle | 4.5 (0.2–8.8) | 0.286 | 29.7 (26.5–32.9) | < 0.001 | 43.1 (36.2–50.0) | < 0.001 | 54 | < 0.001 | 22 | 0.012 | 24 | < 0.001 |
| Paci | −16.4 (−18.8 to −16.9) | < 0.001 | 25.5 (23.9–27.0) | < 0.001 | 35.8 (30.9–40.8) | < 0.001 | 47 | < 0.001 | 46 | < 0.001 | 7 | 0.500 |
Abbreviations: <P25% and >P25%, percentage of predicted clearances below and above ±25% deviation interval from observed clearances, respectively; MAPE, mean absolute prediction error; MPE, mean prediction error; P25%, percentage of predicted clearances within the acceptable ±25% deviation from the observed clearances; RMSE, root mean square prediction error.
p Values for MAPE, MPE, and RMSE were determined using the Wilcoxon signed‐rank test. p Values for P25%, < P25%, and > P25% were determined using the McNemar test for related samples. Significance for both tests was set as p = 0.01 after Bonferroni correction for five pairwise comparisons.
The acceptability interval was set as ±25% deviation from the observed first‐dose clearance, which corresponds to the difference between the center value of the target therapeutic area under the curve for 6 h (19.7 mg·h/L), with the upper and lower ranges of the therapeutic window (14.8–24.6 mg·h/L).
FIGURE 3.
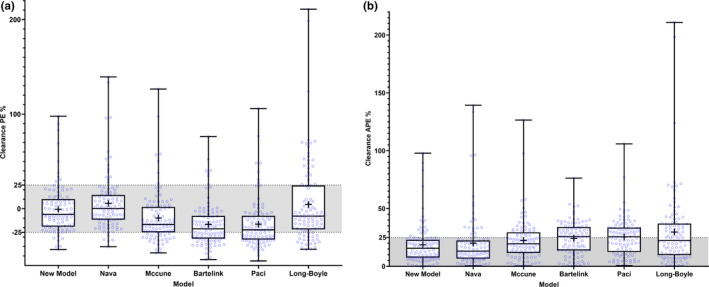
(a) Accuracy and (b) precision of the model‐predicted first‐dose clearances obtained with the new glutathione‐s‐transferase A1‐based and fludarabine‐based models and other tested models. The shaded area represents the acceptance interval in terms of bias and precision. The central lines in the boxplots represent the median values. The plus signs represent the mean values. The bottom and top edges of the boxplots indicate the 25th and 75th percentiles of predicted clearance errors. The whiskers represent the full range of prediction errors. The new model exhibits better accuracy (mean prediction error closer to 0) than every model except for the Long‐Boyle model (see Table 3). The new model exhibits better precision (lowest mean absolute prediction error, error distribution within the 25% acceptance limit) than every model except for the Nava model. APE, absolute prediction error; PE, prediction error
The CLs obtained with the new model showed minimal bias. The new model showed better prediction accuracy than the other models—significantly better than all but the Long‐Boyle model.18 MAPE results provided acceptable prediction precision with the new model—within the limits of acceptance, set at ±25%. RMSE slightly deviated from the acceptance threshold (RMSE %, 27.2%; 95% CI, 23.1%–31.4%). The new model's CL prediction was significantly more precise than all except the Nava model.27 The new model resulted in 81% of the predicted CL values being within the acceptable ±25% deviation from the observed CL.
Figure 4 illustrates the different models’ ability to achieve the target AUCs. As expected from the CL prediction results, the first doses predicted by our new model resulted in 81% of the predicted AUCs being within the therapeutic window. Although the Nava model achieved a similar result, the other models had significantly lower percentages of AUCs within the therapeutic window.
FIGURE 4.
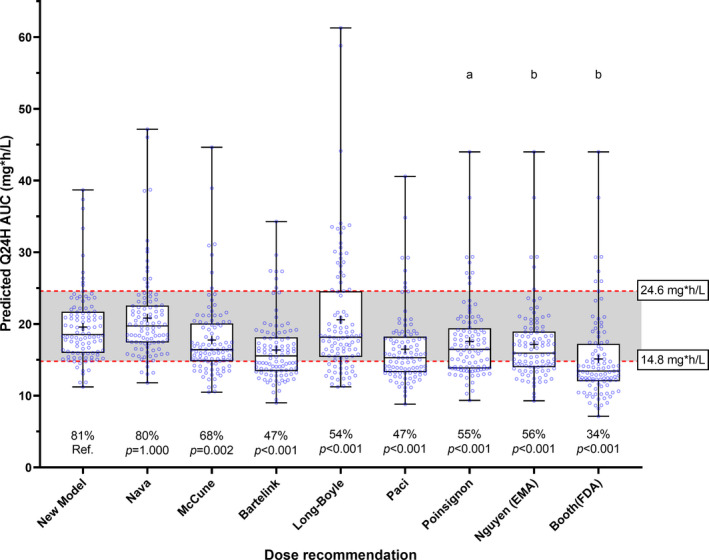
Box plots of simulated AUCs with predicted q24h doses using the evaluated dosing guidelines on the external validation data set. The boxplot's central line represents the median. The plus sign represents the mean value. The bottom and top edges of the boxplots indicate the 25th and 75th percentiles. The whiskers represent the full range of predicted AUCs. The shaded area represents the conventional therapeutic window of busulfan. The percentage of patients within the target window and the p values from the pairwise comparison with the McNemar test for related samples are displayed below the boxes. aDose calculation based on nomogram, with target AUC fixed by the author as 19.7 mg·h/L for q24h doses. bDose calculation based on nomogram, with target AUC fixed by the author as 18.5 mg·h/L for q24h doses. AUC, area under the curve; EMA, European Medicines Agency's dose recommendation; FDA, U.S. Food and Drug Administration's dose reccomendation; q24h, every 24 h dosing; Ref., reference.
DISCUSSION
This article describes a PopPK study in a cohort of 402 pediatric patients who received i.v. Bu, taking into consideration the genetic marker for GSTA1 metabolic capacity. Previous large multicentric PopPK studies were performed in mixed pediatric populations.16, 17, 21, 33 The McCune et al.16 model also included adult patients and can therefore be applied in a wider population. However, these studies did not account for markers for GSTA1 metabolic capacity, which were reported in several studies to significantly influence Bu PK.15, 25, 26, 27
A base model using an age‐dependent allometric scaling of actual BW explained Bu CL adequately. Bartelink et al.33 used a similar approach to describe Bu CL with a varying allometric scaling exponent according to a patient's BW instead. Including the patient's age is crucial to describe the maturation of Bu's and other drugs’ metabolism, as several studies have reported.16, 18, 39, 40 In the present study, patient age better predicted Bu CL when the child's metabolic maturation was taken into account, including the prenatal period (PMA in years),39, 41 although we inferred PMA by adding a fixed value of 40 weeks (the usual gestational period) to patients’ postnatal age. Age‐dependent allometric scaling of BW has been previously used to describe the PK of other drugs, with fixed allometric exponents according to five age cutoffs.34 To the best of our knowledge, no other PopPK model used an allometric scaling exponent equation that varies with PMA. This strategy might become reliable for future pediatric PopPK studies of other drugs.
In addition, we observed an 9% decrease in CL on the subsequent days of Bu administration, supporting earlier evidence from Bartelink et al.33 (12% decrease) and McCune et al.16 (6.8% decrease from 6 h–36 h after Bu therapy initiation and 8.1% decrease from 36 h onward). In a recent study, Marsit et al.42 reported a Bu CL decrease on later days of conditioning in the majority of patients, with an 8% median decrease on Day 2, and a 15% median decrease on Day 3 compared with Day 1 of conditioning. In the latter study, 20% of patients showed no significant variation or an increase in Bu CL. Hence, this variability should be considered in models for Bayesian dose estimation following Bu TDM.16 This intraindividual CL decrease could be explained by glutathione depletion during the course Bu conditioning.43, 44 Inconsistencies in the administration of Bu, such as noncontrolled infusion durations and rates, or omission of central venous catheter flushing between dose administrations, could also add to this observation. However, as several studies have consistently reported this phenomenon,16, 17, 33, 42, 43, 45 and considering Bu first‐order elimination implying a CL independent of the dose, this observed CL decrease is likely to have a physiological explanation. Further mechanistic investigations should be performed to better understand the factors leading to this CL decrease.
The present study confirmed the finding of our previous model27—the influence of GSTA1 genetics on Bu CL—in a larger data set that also considered patients receiving Flu. Using the three GSTA1 metabolic groups based on the complete GSTA1 promoter haplotypes composed of six single nucleotide polymorphisms, as described in Supplemental Materials S3 and S4, was found to be a better genetic covariate than the incomplete haplotypes only based on the single nucleotide polymorphisms −52G/A rs3957356 or −69C/T rs3957357 (*A and *B haplotypes). Two other PopPK studies have assessed the influence of GSTA1 on Bu PK in pediatric patients, both only testing incomplete haplotypes of GSTA1, which resulted in conflicting results. Although Zwaveling et al.46 did not find any significant influence of GSTA1 on Bu CL, Yuan et al.47 reported a 19% lower CL with *A*B haplotypes compared with *A*A in Chinese children. These conflicting results could derive from the high prevalence *B1b haplotypes in East Asian populations, which is associated with the lowest GSTA1 expression potential.30 Patients carrying at least one *B1b are considered poor metabolizers in our classification. Supported by the previous experimental and clinical evidence,15, 26, 27, 30 the grouping with the complete haplotype of GSTA1 promoter should therefore be considered for the classification of patients according to their GSTA1 metabolic capacity affecting Bu CL.30
The PopPK analysis also showed that the coadministration of Flu significantly decreased Bu CL. A previous PopPK model in a Japanese pediatric population with primary immunodeficiencies have reported a 6% lower CL in patients receiving Flu.48 Another study have reported a CL lowered by 20% in patients that received clofarabine in addition to Flu and Bu.45 Interestingly, clofarabine and Flu are both purine analogs with very similar molecular structures, probably explaining the more important Bu CL decrease when coadministered with both of these drugs. We confirmed the effect of Flu on Bu CL in a large, multicentric, pediatric cohort, considering the patients’ GSTA1 genetics. The clinical significance of this drug–drug interaction (DDI) is still unclear.29 Because of Bu's narrow therapeutic window, even this small effect size should be considered in dose recommendations,48 especially because Flu is increasingly used in reduced‐toxicity regimens and that treatment‐related toxicities have also been related with this association in patients overexposed to Bu.49 The underlying mechanism of the observed Flu–Bu DDI and the varying effect of this interaction across different GSTA1 metabolic groups have yet to be elucidated. Bu and Flu are metabolized along two distinct pathways.23, 50 However, we cannot exclude that Flu might inhibit GST enzymes, thus diminishing Bu CL. The present study only assessed Flu's DDIs with Bu because Flu is the only chemotherapeutic agent in our cohort that is administered on the same days as Bu. Bu DDIs with supportive care drugs were not evaluated in this study. The previous PopPK analyses that addressed this question19, 33 found no drugs significantly influencing Bu PK despite several reported DDIs with Bu.23
Finally, a comparison with other available Bu dosing algorithms found that our new PopPK model provided improved accuracy and precision in Bu CL prediction (Figure 2). To avoid any bias in that assessment, it was performed on an external representative data set not used in the model's development. Despite the new model also considering similar GSTA1 metabolic groups,27 it provided significantly better CL prediction accuracy (mean prediction error) than Nava's model, which did not consider patients receiving Flu. Using the new model on the external representative data set of 100 patients, 10 first‐dose AUCs were below the conventional therapeutic window (14.8–24.6 mg·h/L for 6 h) and nine were above it. Among these 19 patients outside the desired range, only eight predicted AUCs deviated from the therapeutic window by more than 15%. The model proposed in this study is thus preferable to other models whenever a patient's GSTA1 genotype is available.
In conclusion, we developed and externally validated a PopPK model for i.v. Bu, which considered and included GSTA1 genetics, DDI with Flu, and patients’ BW and age to predict Bu's PK parameters. The model resulted in a dose recommendation algorithm that could accurately predict Bu exposure in more than 80% of pediatric patients. Coupled with efficient TDM, this model could limit the variability in patients’ Bu exposure and clinical outcomes. This new model should also aid the development of a Bayesian model for Bu PK in an attempt to eliminate TDM and make Bu conditioning available to patients with limited access to accredited laboratories for PK analysis. As future work should be focused on the implementation of model‐informed dosing in clinical practice, the feasibility of routinely including GSTA1 genotyping in Bu dose recommendations will be assessed in an international, multicentric, prospective randomized trial (NCT04822532).
MODEL CODE AVAILABILITY
The model code is provided in the Supplemental Material File S11.
CONFLICTS OF INTEREST
The authors declared no competing interests for this work. Dr. Nastya Kassir was employed by Certara in the past. She is now employed by Genentech/Roche.
AUTHOR CONTRIBUTIONS
K.B.H., T.N., C.R.S.U., and M.A. wrote the manuscript. K.B.H., T.N., H.B., M.K., C.R.S.U., and M.A. designed the research. K.B.H., T.N., C.R.S.U., and M.A. performed the research. K.B.H., T.N., Y.T., C.E.N., Y.D., N.K., V.L., R.G.M.B., P.J.S., H.B., M.K., C.R.S.U., and M.A. analyzed the data. Y.T., C.E.N., and Y.D. contributed with new reagents/analytical tools.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank the patients and their parents for consenting to participate in this study. We also thank Mohamed Aziz Rezgui, Rodolpho Lo Piccolo, Denis Marino, Mary Khoshbeen‐Boudal, and Fanny Muet for their valuable contributions to this work.
Ben Hassine K, Nava T, Théoret Y, et al. Precision dosing of intravenous busulfan in pediatric hematopoietic stem cell transplantation: Results from a multicenter population pharmacokinetic study. CPT Pharmacometrics Syst Pharmacol. 2021;10:1043–1056. 10.1002/psp4.12683
Ben Hassine and Nava contributed equally to this work.
Funding Information
This study was funded by the Swiss National Science Foundation (Grant 153389), the CANSEARCH Foundation, the Oak Foundation (Grant OCAY‐17‐642), and Fondation Charles–Bruneau. Christa Nath is supported by the Leukaemia Research and Support Fund at The Children's Hospital at Westmead.
Data Availability Statement
The data set analyzed in this study is available from the corresponding author upon reasonable request.
REFERENCES
- 1.Slattery JT, Sanders JE, Buckner CD, et al. Graft‐rejection and toxicity following bone marrow transplantation in relation to busulfan pharmacokinetics. Bone Marrow Transplant. 1995;16:31‐42. [PubMed] [Google Scholar]
- 2.Slattery JT, Risler LJ. Therapeutic monitoring of busulfan in hematopoietic stem cell transplantation. Ther Drug Monit. 1998;20:543‐549. [DOI] [PubMed] [Google Scholar]
- 3.Dix SP, Wingard JR, Mullins RE, et al. Association of busulfan area under the curve with veno‐occlusive disease following BMT. Bone Marrow Transplant. 1996;17:225‐230. [PubMed] [Google Scholar]
- 4.McCune JS, Gooley T, Gibbs JP, et al. Busulfan concentration and graft rejection in pediatric patients undergoing hematopoietic stem cell transplantation. Bone Marrow Transplant. 2002;30:167‐173. [DOI] [PubMed] [Google Scholar]
- 5.Bolinger AM, Zangwill AB, Slattery JT, et al. Target dose adjustment of busulfan in pediatric patients undergoing bone marrow transplantation. Bone Marrow Transplant. 2001;28:1013‐1018. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen L, Leger F, Lennon S, Puozzo C. Intravenous busulfan in adults prior to haematopoietic stem cell transplantation: a population pharmacokinetic study. Cancer Chemother Pharmacol. 2006;57:191‐198. [DOI] [PubMed] [Google Scholar]
- 7.Copelan EA, Bechtel TP, Avalos BR, et al. Busulfan levels are influenced by prior treatment and are associated with hepatic veno‐occlusive disease and early mortality but not with delayed complications following marrow transplantation. Bone Marrow Transplant. 2001;27:1121‐1124. [DOI] [PubMed] [Google Scholar]
- 8.Güngör T, Teira P, Slatter M, et al. Reduced‐intensity conditioning and HLA‐matched haemopoietic stem‐cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. The Lancet. 2014;383:436‐448. [DOI] [PubMed] [Google Scholar]
- 9.Bartelink IH, Lalmohamed A, van Reij EML, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol. 2016;3:e526‐e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCune JS, Holmberg LA. Busulfan in hematopoietic stem cell transplant setting. Expert Opin Drug Metab Toxicol. 2009;5:957‐969. [DOI] [PubMed] [Google Scholar]
- 11.Ciurea SO, Andersson BS. Busulfan in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2009;15:523‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer J, McCune JS, Perales MA, et al. Personalizing busulfan‐based conditioning: considerations from the American Society for Blood and Marrow Transplantation Practice Guidelines Committee. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2016;22:1915‐1925. [DOI] [PubMed] [Google Scholar]
- 13.Malär R, Sjöö F, Rentsch K, Hassan M, Güngör T. Therapeutic drug monitoring is essential for intravenous busulfan therapy in pediatric hematopoietic stem cell recipients. Pediatr Transplant. 2011;15:580‐588. [DOI] [PubMed] [Google Scholar]
- 14.Booth BP, Rahman A, Dagher R, et al. Population pharmacokinetic‐based dosing of intravenous busulfan in pediatric patients. J Clin Pharmacol. 2007;47:101‐111. [DOI] [PubMed] [Google Scholar]
- 15.Nava T, Rezgui MA, Uppugunduri CRS, et al. GSTA1 genetic variants and conditioning regimen: missing key factors in dosing guidelines of busulfan in pediatric hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23:1918‐1924. [DOI] [PubMed] [Google Scholar]
- 16.McCune JS, Bemer MJ, Barrett JS, et al. Busulfan in infant to adult hematopoietic cell transplant recipients: a population pharmacokinetic model for initial and Bayesian dose personalization. Clin Cancer Res. 2014;20:754‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartelink IH, van Kersten C, Boelens JJ, et al. Predictive performance of a busulfan pharmacokinetic model in children and young adults. Ther Drug Monit. 2012;34:574‐583. [DOI] [PubMed] [Google Scholar]
- 18.Long‐Boyle JR, Savic R, Yan S, et al. Population pharmacokinetics of busulfan in pediatric and young adult patients undergoing hematopoietic cell transplant: a model‐based dosing algorithm for personalized therapy and implementation into routine clinical use. Ther Drug Monit. 2015;37:236‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paci A, Vassal G, Moshous D, et al. Pharmacokinetic behavior and appraisal of intravenous busulfan dosing in infants and older children: the results of a population pharmacokinetic study from a large pediatric cohort undergoing hematopoietic stem‐cell transplantation. Ther Drug Monit. 2012;34:198‐208. [DOI] [PubMed] [Google Scholar]
- 20.Trame MN, Bergstrand M, Karlsson MO, Boos J, Hempel G. Population pharmacokinetics of busulfan in children: increased evidence for body surface area and allometric body weight dosing of busulfan in children. Clin Cancer Res. 2011;17:6867‐6877. [DOI] [PubMed] [Google Scholar]
- 21.Poinsignon V, Faivre L, Nguyen L, et al. New dosing nomogram and population pharmacokinetic model for young and very young children receiving busulfan for hematopoietic stem cell transplantation conditioning. Pediatr Blood Cancer. 2020, 67:e28603. [DOI] [PubMed] [Google Scholar]
- 22.Zao JH, Schechter T, Liu WJ, et al. Performance of busulfan dosing guidelines for pediatric hematopoietic stem cell transplant conditioning. Biol Blood Marrow Transplant. 2015;21:1471‐1478. [DOI] [PubMed] [Google Scholar]
- 23.Myers AL, Kawedia JD, Champlin RE, et al. Clarifying busulfan metabolism and drug interactions to support new therapeutic drug monitoring strategies: a comprehensive review. Expert Opin Drug Metab Toxicol. 2017;13:901‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huezo‐Diaz P, Uppugunduri CRS, Tyagi AK, Krajinovic M, Ansari M. Pharmacogenetic aspects of drug metabolizing enzymes in busulfan based conditioning prior to allogenic hematopoietic stem cell transplantation in children. Current Drug Metabolism. 2014;15:251‐264. 10.2174/1389200215666140202214012 [DOI] [PubMed] [Google Scholar]
- 25.Ansari M, Lauzon‐Joset JF, Vachon MF, et al. Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplant. 2010;45:261‐267. [DOI] [PubMed] [Google Scholar]
- 26.Ansari M, Huezo‐Diaz Curtis P, Uppugunduri CRS, et al. GSTA1 diplotypes affect busulfan clearance and toxicity in children undergoing allogeneic hematopoietic stem cell transplantation: a multicenter study. Oncotarget. 2017;8:90852‐90867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nava T, Kassir N, Rezgui MA, et al. Incorporation of GSTA1 genetic variations into a population pharmacokinetic model for IV busulfan in paediatric hematopoietic stem cell transplantation: GSTA1‐based busulfan population pharmacokinetic model in children. Br J Clin Pharmacol. 2018;84:1494‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Castro FA, Lanchote VL, Voltarelli JC, Colturato VAR, Simões BP. Influence of fludarabine on the pharmacokinetics of oral busulfan during pretransplant conditioning for hematopoietic stem cell transplantation. J Clin Pharmacol. 2013;53:1205‐1211. [DOI] [PubMed] [Google Scholar]
- 29.Yeh RF, Pawlikowski MA, Blough DK, et al. Accurate targeting of daily intravenous busulfan with 8‐hour blood sampling in hospitalized adult hematopoietic cell transplant recipients. Biol Blood Marrow Transplant. 2012;18:265‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mlakar V, Huezo‐Diaz Curtis P, Armengol M, et al. The analysis of GSTA1 promoter genetic and functional diversity of human populations. Sci Rep. 2021;11:5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holford NHG, Anderson BJ. Allometric size: The scientific theory and extension to normal fat mass. Eur J Pharm Sci. 2017;109S:S59‐S64. [DOI] [PubMed] [Google Scholar]
- 32.Anderson BJ, Holford NH. What is the best size predictor for dose in the obese child? Paediatr Anaesth. 2017;27:1176‐1184. [DOI] [PubMed] [Google Scholar]
- 33.Bartelink I, Boelens JJ, Bredius RGM, et al. Body weight‐dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients. Clin Pharmacokinet. 2012;51:331‐345. [DOI] [PubMed] [Google Scholar]
- 34.Mahmood I. Prediction of drug clearance in children: a review of different methodologies. Expert Opin Drug Metab Toxicol. 2015;11:573‐–587.. [DOI] [PubMed] [Google Scholar]
- 35.Holford NHG, Ma SC, Anderson BJ. Prediction of morphine dose in humans. Pediatr Anesth. 2012;22:209‐222. [DOI] [PubMed] [Google Scholar]
- 36.Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm. 1981;9:503‐512. [DOI] [PubMed] [Google Scholar]
- 37.McCune JS, Quinones CM, Ritchie J, et al. Harmonization of busulfan plasma exposure unit (BPEU): a community‐initiated consensus statement. Biol Blood Marrow Transplant. 2019;25:1890‐1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keizer RJ, Jansen RS, Rosing H, et al. Incorporation of concentration data below the limit of quantification in population pharmacokinetic analyses. Pharmacol. Res. Perspect. 2015;3. 10.1002/prp2.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson BJ, Holford NHG. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24:25‐36. [DOI] [PubMed] [Google Scholar]
- 40.Savic RM, Cowan MJ, Dvorak CC, et al. Effect of weight and maturation on busulfan clearance in infants and small children undergoing hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2013;19:1608‐1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson BJ, Holford NHGMechanism‐based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303‐332. [DOI] [PubMed] [Google Scholar]
- 42.Marsit H, Philippe M, Neely M, et al. Intra‐individual pharmacokinetic variability of intravenous busulfan in hematopoietic stem cell‐transplanted children. Clin Pharmacokinet. 2020;59:1049‐1061. [DOI] [PubMed] [Google Scholar]
- 43.Langenhorst JB, Boss J, van Kestern C, et al. A semi‐mechanistic model based on glutathione depletion to describe intra‐individual reduction in busulfan clearance. Br J Clin Pharmacol. 2020;86:1499‐1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Almog S, Kurnik D, Shimoni A, et al. Linearity and stability of intravenous busulfan pharmacokinetics and the role of glutathione in busulfan elimination. Biol Blood Marrow Transplant. 2011;17:117‐123. [DOI] [PubMed] [Google Scholar]
- 45.Shukla P, Goswami S, Keizer RJ, et al. Assessment of a model‐informed precision dosing platform use in routine clinical care for personalized busulfan therapy in the pediatric hematopoietic cell transplantation (HCT) population. Front. Pharmacol. 2020;11. 10.3389/fphar.2020.00888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zwaveling J, Press RR, Bredius RGM, et al. Glutathione S‐transferase polymorphisms are not associated with population pharmacokinetic parameters of busulfan in pediatric patients. Ther Drug Monitor. 2008;30:504‐510. [DOI] [PubMed] [Google Scholar]
- 47.Yuan J, Sun N, Feng X, et al. Optimization of busulfan dosing regimen in pediatric patients using a population pharmacokinetic model incorporating GST mutations. Pharmacogenomics Pers. Med. 2021;14:253‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishiwata Y, Nagata M, Tsuge K, et al. Population pharmacokinetics of intravenous busulfan in Japanese pediatric patients with primary immunodeficiency diseases. J Clin Pharmacol. 2018;58:327‐331. [DOI] [PubMed] [Google Scholar]
- 49.Perkins JB, Kim J, Anasetti C, et al. Maximally tolerated busulfan systemic exposure in combination with fludarabine as conditioning before allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2012;18:1099‐1107. [DOI] [PubMed] [Google Scholar]
- 50.Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet. 2002;41:93‐103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data set analyzed in this study is available from the corresponding author upon reasonable request.