

Abstract
A combination of olanzapine and samidorphan (OLZ/SAM) was recently approved by the US Food and Drug Administration for treatment of patients with schizophrenia or bipolar I disorder. The effects of moderate hepatic impairment on the pharmacokinetics (PKs) of olanzapine and samidorphan after a single dose of OLZ/SAM were characterized in a clinical study. Physiologically‐based pharmacokinetic (PBPK) modeling was used to extend the clinical findings to predict the effects of varying degrees of hepatic impairment on the PKs of olanzapine and samidorphan. A previously developed PBPK model for OLZ/SAM was refined to recover the observed pharmacokinetic differences between individuals with moderate hepatic impairment and healthy controls. The optimized model was applied to predict changes in olanzapine and samidorphan PKs after multiple once‐daily doses of OLZ/SAM in subjects with mild, moderate, and severe hepatic impairment relative to healthy controls. Modifications to model parameters, including absorption rate constant and fraction unbound to plasma protein, were made to recover the observed change in the PKs of olanzapine and samidorphan in individuals with moderate hepatic impairment. In applying the optimized model, mild, moderate, and severe hepatic impairment were predicted to increase steady‐state total systemic exposures by 1.1‐, 1.5‐, and 1.6‐fold, respectively, for olanzapine, and by 1.2‐, 1.9‐, and 2.3‐fold, respectively, for samidorphan. PBPK modeling allowed for prediction of untested clinical scenarios of varying degrees of hepatic impairment in lieu of additional clinical studies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Hepatic metabolism plays a major role in the clearance of olanzapine and samidorphan. Findings from a clinical study indicated a modest increase in both olanzapine and samidorphan exposures in subjects with moderate hepatic impairment compared with healthy controls with normal hepatic function.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study addressed the feasibility of using physiologically‐based pharmacokinetic (PBPK) modeling to predict the effects of varying degrees of hepatic impairment on steady‐state exposures of olanzapine and samidorphan after daily administration of OLZ/SAM.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study highlights the utility of applying the PBPK model for OLZ/SAM to predict the effects of mild, moderate, and severe hepatic impairment on pharmacokinetics of OLZ and SAM in lieu of additional clinical studies.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Appropriately executed PBPK models may enhance understanding of drug exposures, supporting clinical development and regulatory decision making, while potentially reducing the need to conduct additional clinical trials.
INTRODUCTION
A combination of the antipsychotic olanzapine and the opioid receptor antagonist samidorphan1, 2, 3 (OLZ/SAM; Lybalvi, Alkermes, Inc.) was recently approved by the US Food and Drug Administration for the treatment of patients with schizophrenia or bipolar I disorder. The inclusion of samidorphan in OLZ/SAM mitigates weight gain associated with olanzapine use, without compromising the efficacy of olanzapine. The antipsychotic efficacy and weight mitigation effect of OLZ/SAM were established in separate randomized, active comparator‐ and/or placebo‐controlled, double‐blind, phase III clinical trials.4, 5 The pharmacokinetics (PKs) of olanzapien and samidorphan has been evaluated in a number of clinical studies of OLZ/SAM, in both healthy subjects6, 7, 8 and in patients with schizophrenia.9
Olanzapine is extensively metabolized in the liver, with 7% of the administered dose being excreted renally as unchanged olanzapine.10 The primary metabolic pathways of olanzapine are direct glucuronidation, via uridine 5'‐diphospho‐glucuronosyltransferase (UGT) enzymes, and cytochrome P450 (CYP)‐mediated oxidation, mainly by CYP1A2, with contributions from other CYP enzymes, particularly CYP2C8.10, 11 Samidorphan is eliminated primarily via CYP3A4‐mediated hepatic metabolism and renal excretion, with ~ 20% of the administered dose being excreted renally as unchanged samidorphan.12, 13
Because hepatic metabolism plays a major role in both olanzapine and samidorphan clearance, it is possible that impairment in hepatic function could impact the PKs of both compounds. As an initial step toward understanding the effects of liver dysfunction on PKs of olanzapine and samidorphan, a clinical study was conducted in subjects with moderate hepatic impairment (Child‐Pugh scores ranging from 7–9, class B) and healthy controls with normal hepatic function.12 Following a single oral dose of OLZ/SAM 5 mg/10 mg (5/10),12 subjects with moderate hepatic impairment had increased olanzapine and samidorphan exposures (area under the plasma concentration‐time [C‐T] curve from time 0 to infinity [AUCinf] increased 1.67‐ and 1.52‐fold, respectively, and maximum plasma concentrations [Cmax] increased 2.17‐ and 1.63‐fold, respectively) compared with healthy controls. Based on the magnitude of increase in olanzapine and samidorphan exposures observed with moderate hepatic impairment after a single dose, further evaluation of the effect of other categories of hepatic impairment under a clinical dosing paradigm (i.e., chronic daily dosing) was needed.
Therefore, to extend the findings from the clinical study of OLZ/SAM in subjects with moderate hepatic impairment12 to other untested clinical scenarios, we verified the physiologically‐based pharmacokinetic (PBPK) modeling predictions against existing clinical data and used PBPK modeling to predict the effect of mild, moderate, and severe hepatic impairment on the steady‐state PKs of olanzapine and samidorphan after multiple once‐daily administration of OLZ/SAM.
METHODS
An overview of the steps underlying PBPK model development, validation, and application are presented in Figure 1.
FIGURE 1.
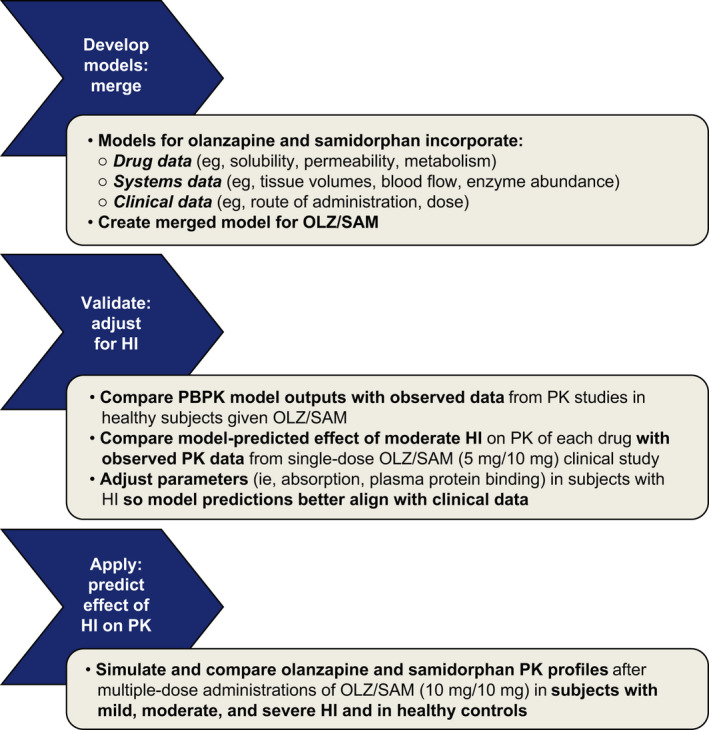
Schematic of PBPK model development, validation, and application for predicting effects of hepatic impairment on the pharmacokinetics of OLZ/SAM. HI, hepatic impairment; OLZ/SAM, combination of olanzapine and samidorphan; PBPK, physiologically‐based pharmacokinetic; PK, pharmacokinetic
Model development
The PBPK models for olanzapine (minimal model; single adjusting compartment) and for samidorphan (full PBPK model; inclusion of additional tissues) were constructed using the Simcyp Simulator (Certara; Princeton, NJ) and were previously described, including the input parameters for each.14 The two models were combined to represent the administration of olanzapine and samidorphan in combination as OLZ/SAM. This PBPK model was previously developed based on in vitro and clinical PK data and applied to evaluate potential drug‐drug interactions with OLZ/SAM.14 Here, modifications to the absorption parameters within the previously developed PBPK model and changes in plasma protein binding in subjects with hepatic impairment were required to recover the observed differences in Cmax, time to Cmax (Tmax), and AUCinf between subjects with moderate hepatic impairment and age‐ and weight‐matched healthy controls12 (described in further detail in the Model Validation section below).
Predictions of plasma drug C‐T profiles and PK parameters were performed using the Simcyp Simulator in populations of virtual individuals. Default Simcyp parameter values for creating a virtual North European Caucasian population (physiological parameters, including liver volume and blood flows, and enzyme abundances) have been described previously.15 All parameter values for the healthy volunteer population, with the exception of demographic data, were the same as the default Simcyp parameters for the White population. The following equation was used to recover the weight distributions observed for both the healthy age‐matched control subjects and subjects with moderate hepatic impairment: weight (kg) = eC1 × height + C0, with values for C0 and C1 of 2.8 and 0.0097 in men and 2.925 and 0.0097 in women, respectively. Once the value was calculated for expected weight, a random variability (coefficient of variation [CV]: 10% for both men and women) was added, assuming a normal distribution.
Three cirrhosis population models are available in the Simcyp Simulator, reflecting Child‐Pugh classes A, B, and C16 (corresponding to subjects with mild, moderate, and severe hepatic impairment, respectively). Some of the key differences between these models and the healthy volunteer population model include liver volume, enzyme abundance levels, and extent of plasma protein binding.16
The decrease in liver size with increasing severity of cirrhosis is based on measurements of the functional hepatocyte volume as a direct reflection of the functional reserve of the organ. Liver volume in subjects with mild, moderate, and severe hepatic impairment was estimated to be 0.89‐, 0.71‐, and 0.63‐fold, respectively, of that in healthy age‐matched control subjects. The abundance of CYP3A4 was estimated to be 137, 108, 56, and 31 pmol P450/mg protein in healthy controls and subjects with Child‐Pugh class A, B, and C, respectively. Similarly, the respective abundance of CYP1A2 was estimated to be 52.0, 32.9, 13.6, and 6.1 pmol P450/mg protein; the respective abundance of CYP2C8 was estimated to be 24.0, 16.6, 12.5, and 7.9 pmol P450/mg protein. It should be noted that some UGTs have been reported to be more resistant to hepatic injury.17 Thus, the UGT1A4 enzyme levels were fixed in all models at a value of 52.0 pmol/mg protein. An increase or decrease in plasma protein binding of a drug is often observed in mild, moderate, and severe hepatic impairment due to decreased levels of albumin or an increase in plasma levels of alpha acid. Data from the clinical study indicated that the fraction unbound to plasma protein (fu) for olanzapine was similar between subjects with moderate hepatic impairment and healthy control subjects.12 Therefore, two scenarios of fu were assessed: first, simulations assumed no change in the extent of plasma protein binding for either olanzapine or samidorphan in subjects with hepatic impairment (0.07 for olanzapine and 0.69 for samidorphan14), then simulations assumed Simcyp predicted values of fu for populations with mild, moderate, and severe hepatic impairment (0.08, 0.10, and 0.13 for olanzapine, respectively, and 0.71, 0.76, and 0.81 for samidorphan, respectively). For both drugs, it was assumed that the main binding protein was albumin.
Due to reduced renal blood flow and increased renal resistance, there is a progressive reduction in renal function with increasing severity of liver disease. Serum creatinine values describing renal function in liver cirrhosis within Simcyp were back‐calculated from typically reported glomerular filtration rate values for each of the Child‐Pugh classes.
Finally, gastrointestinal symptoms are common in cirrhosis, and their pathophysiology probably involves factors related to liver disease severity, psychological distress, and gut dysfunction (e.g., gastric sensorimotor dysfunction and delayed gut transit).18 Gastric emptying and small bowel transit have generally been shown to be prolonged in subjects with hepatic impairment.18 In the Simcyp models, the gastric emptying time was increased from the healthy control value of 0.4 h to 0.48 h, 0.55 h, and 0.6 h for subjects with mild, moderate, and severe hepatic impairment, respectively.
Model validation
The PBPK model for OLZ/SAM was initially validated by comparing model‐simulated plasma C‐T profiles and PK parameters of olanzapine and samidorphan with observed clinical data following a single dose of OLZ/SAM 10 mg/10 mg (10/10) in healthy subjects,6, 8 and following multiple once‐daily doses of OLZ/SAM 10/10 for 14 days in patients with schizophrenia.6, 8 Initial model validation was previously reported.14 The model was further validated by comparing model‐simulated C‐T profiles and the PK parameters of olanzapine and samidorphan with observed data from the clinical study that evaluated the effects of moderate hepatic impairment on the PKs of olanzapine and samidorphan after a single dose of OLZ/SAM 5/10.12
Virtual trials were generated to match subject demographics (i.e., age and sex) and treatment characteristics of the clinical study12: 10 virtual trials of 10 subjects with moderate hepatic impairment (20% women; aged 49–70 years), and 10 trials of 10 healthy controls with normal hepatic function (27% women; aged 55–67 years). All virtual subjects were nonsmokers and received a single dose of OLZ/SAM 5/10 in the fasted state.
Model application
The optimized model was applied to predict hepatic impairment–induced changes in steady‐state systemic exposures of olanzapine and samidorphan after multiple once‐daily doses of OLZ/SAM 10/10 for 14 days. Each simulation consisted of 10 virtual trials of 10 subjects each (20% female; aged 49–70 years) in the following groups: (1) subjects with mild hepatic impairment (Child‐Pugh class A), (2) subjects with moderate hepatic impairment (Child‐Pugh class B); (3) subjects with severe hepatic impairment (Child‐Pugh class C); and (4) healthy control subjects with normal hepatic function. All virtual subjects were nonsmokers.
RESULTS
Model validation
A comparison of model‐predicted PK parameters with observed data following a single dose of OLZ/SAM 5/10 in subjects with moderate hepatic impairment revealed that the original PBPK model14 did not capture the observed shortening of Tmax, resulting in an underprediction of Cmax for both olanzapine and samidorphan. The results of automated sensitivity analyses identified a relationship between the absorption rate constant (ka) and the predicted Tmax for olanzapine and samidorphan in subjects with moderate hepatic impairment (Figure 2a,b, respectively). A ka value of 4 h−1 for olanzapine and 1.6 h−1 for samidorphan were required to recover the observed shortening of Tmax values in subjects with moderate hepatic impairment. In addition, simulations assuming an increased fu in subjects with hepatic impairment (0.10 for olanzapine and 0.76 for samidorphan) compared with healthy controls (0.07 for olanzapine and 0.69 for samidorphan) resulted in predicted Cmax and/or AUCinf ratios (moderate hepatic impairment/healthy controls) that were more consistent with the observed data in the clinical study.12
FIGURE 2.
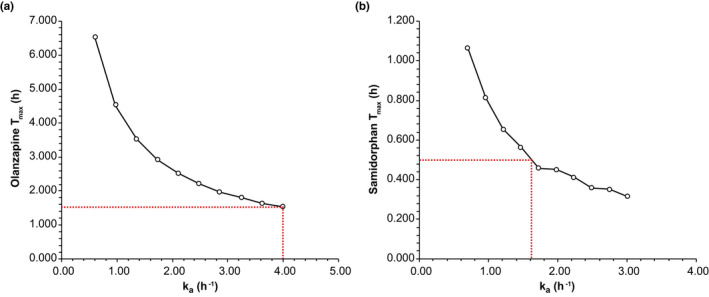
Sensitivity analyses depicting the relationship between the absorption rate constant (ka) and the predicted Tmax of (a) olanzapine or (b) samidorphan in subjects with moderate hepatic impairment. ka, first order absorption rate constant; Tmax, time to maximum plasma concentration
Following modification of the ka and fu values for subjects with moderate hepatic impairment, model‐simulated plasma C‐T profiles and PK parameters for olanzapine and samidorphan in healthy controls and in subjects with moderate hepatic impairment were reasonably consistent with observed data (Table 1 and Figure 3a–d).12 The only exception was the mean Cmax value for olanzapine in individuals with moderate hepatic impairment; for this parameter, the model underpredicted the observed data by 46% (Table 1). However, whereas the predicted mean Cmax values for the 10 virtual trials of 10 individuals with moderate hepatic impairment ranged from 4.1 to 7.0 ng/ml, the predicted minimum to maximum Cmax values for the population of 100 virtual individuals spanned the range of observed Cmax values (observed Cmax range: 5.3–17.7 ng/ml; predicted individual Cmax range: 1.9–20.9 ng/ml). Based on geometric mean data (Table 2), the model predicted a 1.51‐fold increase in the AUCinf of olanzapine in subjects with moderate hepatic impairment compared with healthy controls, which was consistent with the 1.67‐fold increase observed in the clinical study.12 Similarly, the model predicted a 1.79‐fold increase in the AUCinf of samidorphan in subjects with moderate hepatic impairment, consistent with the 1.52‐fold increase observed in the clinical study.12 The observed 2.17‐fold increase in olanzapine Cmax was underpredicted by the PBPK model (1.14‐fold), whereas the observed 1.63‐fold increase in samidorphan Cmax was well‐predicted by the PBPK model (1.61‐fold).12
TABLE 1.
Comparison of model‐predicted and observed arithmetic mean PK parameter values for olanzapine and samidorphan after a single dose of OLZ/SAM 5 mg/10 mg in healthy control subjects with normal hepatic function and subjects with moderate hepatic impairment (model validation)
| Parameter | Olanzapine | Samidorphan | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmax, ng/ml |
Tmax,a h |
AUCinf, ng h/ml | t½, h | CL/F, L/h | Cmax, ng/ml |
Tmax, a h |
AUCinf, ng h/ml | t½, h | CL/F, L/h | |
| Single dose of OLZ/SAM 5 mg/10 mg in healthy controls with normal hepatic function | ||||||||||
| Predicted mean (n = 100) | 4.9 | 6.1 | 243 | 31.0 | 24.2 | 33.9 | 0.9 | 286 | 12.7 | 37.5 |
| Range of predicted mean across virtual trials (n = 10) | 3.8–5.7 | 6.0–8.6 | 200–297 | 26.7–37.0 | 19.4–29.0 | 30.9–37.4 | 0.9–1.0 | 243–321 | 11.7–13.8 | 33.9–45.0 |
| Observed mean (n = 10)12 | 4.9 | 7.0 | 275 | 51.9 | 22.2 | 30.8 | 1.0 | 263 | 9.0 | 39.8 |
| Range (n = 10) | 3.0–7.4 | 2.0–12.0 | 146–620 | 33.5–96.3 | 8.1–34.2 | 19.1–42.8 | 0.5–3 | 199–412 | 7.9–10.3 | 24.3–50.3 |
| Ratio of predicted to observed | 1.00 | 0.87 | 0.89 | 0.60 | 1.09 | 1.10 | 0.91 | 1.09 | 1.42 | 0.94 |
| Single dose of OLZ/SAM 5 mg/10 mg in subjects with moderate hepatic impairment | ||||||||||
| Predicted mean (n = 100) | 5.8 | 1.6 | 378 | 47.3 | 16.6 | 55.6 | 0.5 | 527 | 14.9 | 21.7 |
| Range of predicted mean across virtual trials (n = 10) | 4.1–7.0 | 1.3–2.4 | 290–516 | 37.8–58.8 | 12.0–22.2 | 50.1–60.3 | 0.5–0.6 | 397–623 | 13.0–16.9 | 17.3–32.0 |
| Observed mean (n = 10)12 | 10.6 | 1.5 | 424 | 53.3 | 12.2 | 50.7 | 0.5 | 408 | 11.9 | 25.5 |
| Range (n = 10) | 5.3–17.7 | 0.5–4 | 311–547 | 37.3–95.3 | 9.14–16.1 | 31.4–71.3 | 0.5–1 | 309–550 | 8.8–15.3 | 18.2–32.4 |
| Ratio of predicted to observed | 0.54 | 1.06 | 0.89 | 0.91 | 1.36 | 1.10 | 1.01 | 1.29 | 1.25 | 0.85 |
Abbreviations: AUCinf, area under the concentration‐time curve from 0 to infinity; CL/F, apparent total clearance of the compound from plasma after oral administration; Cmax, maximum plasma concentration; OLZ/SAM, combination of olanzapine and samidorphan; PK, pharmacokinetic; t½, elimination half‐life; Tmax, time to maximum plasma concentration.
Median.
FIGURE 3.
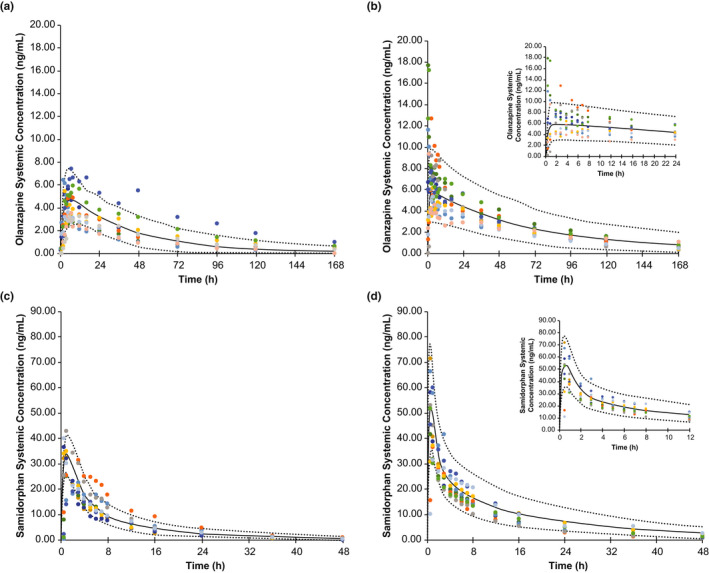
Model validation: observed and simulated concentrations of olanzapine in (a) healthy controls and (b) subjects with moderate hepatic impairment, and of samidorphan in (c) healthy controls and (d) subjects with moderate hepatic impairment, following a single dose of OLZ/SAM 5 mg/10 mg. The solid line represents the mean data for the simulated population (n = 100), and the dashed lines are the 5th and 95th percentiles of the population. Observed data (n = 1012) are presented as symbols; each subject is indicated by a distinct color. The insets in (b) and (d) depict early time points in greater detail. OLZ/SAM, combination of olanzapine and samidorphan
TABLE 2.
Comparison of model‐predicted and observed geometric mean Cmax and AUC values for olanzapine and samidorphan after a single dose of OLZ/SAM 5 mg/10 mg in healthy control subjects with normal hepatic function and subjects with moderate hepatic impairment (model validation)
| Parameter |
Cmax (ng/ml) |
AUCinf (ng h/ml) |
||||
|---|---|---|---|---|---|---|
| Healthy control | Moderate HI | Ratioa | Healthy control | Moderate HI | Ratioa | |
| Olanzapine | ||||||
| Observed12 | 5.2 | 11.2 | 2.17 | 276 | 462 | 1.67 |
| Simulated | 4.6 | 5.3 | 1.14 | 225 | 338 | 1.51 |
| Samidorphan | ||||||
| Observed12 | 29.6 | 48.1 | 1.63 | 278 | 422 | 1.52 |
| Simulated | 33.5 | 54.0 | 1.61 | 277 | 495 | 1.79 |
Abbreviations: AUCinf, area under the plasma drug concentration‐time curve from time 0 to infinity; Cmax, maximum plasma concentration; HI, hepatic impairment; OLZ/SAM, combination of olanzapine and samidorphan.
Moderate HI relative to age‐matched controls with normal hepatic function.
Model application
Simulated steady‐state plasma C‐T profiles of olanzapine and samidorphan in subjects with mild, moderate, or severe hepatic impairment compared with healthy, age‐matched controls after once‐daily administration of OLZ/SAM 10/10 for 14 days are depicted in Figure 4a,b.
FIGURE 4.
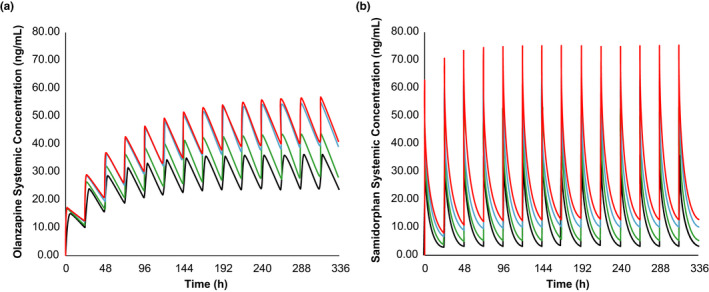
Model application: simulated plasma concentrations of olanzapine (a) and samidorphan (b) after once‐daily doses of OLZ/SAM 10 mg/10 mg for 14 days. The lines represent the mean data for simulated populations (n = 100) of subjects with normal hepatic function (black) and subjects with mild hepatic impairment (green), moderate hepatic impairment (blue) and severe hepatic impairment (red). Simulations assume reduced plasma protein binding in individuals with hepatic impairment. For olanzapine, the mean fu values were 0.07, 0.08, 0.10, and 0.13 for subjects with normal hepatic function and for those with mild, moderate and severe hepatic impairment, respectively; for samidorphan, the mean fu values were 0.69, 0.71, 0.76, and 0.81 for subjects with normal hepatic function and for those with mild, moderate and severe hepatic impairment, respectively. fu, fraction unbound in plasma; OLZ/SAM, combination of olanzapine and samidorphan
Model‐predicted steady‐state Cmax (Cmax,ss) and AUC (AUCss) of olanzapine and samidorphan in subjects with mild, moderate, or severe hepatic impairment compared with healthy controls are presented in Table 3. PBPK predictions indicated that exposures of olanzapine and samidorphan increased as the severity of hepatic impairment increased. The increase in Cmax,ss was predicted to be less than 1.5‐fold for both olanzapine and samidorphan in subjects with mild hepatic impairment compared with subjects with normal hepatic function. Increases in Cmax,ss of 1.5‐ to 1.6‐fold for olanzapine and 1.8‐ to 2.1‐fold for samidorphan were predicted in subjects with moderate to severe hepatic impairment compared with healthy controls. The increase in AUCss was predicted to be up to 1.6‐fold for olanzapine and up to 2.3‐fold for samidorphan in subjects with severe hepatic impairment compared with healthy controls.
TABLE 3.
Model‐predicted geometric mean steady‐state Cmax and AUC values for olanzapine and samidorphan after once‐daily doses of OLZ/SAM 10 mg/10 mg for 14 days in subjects with hepatic impairment compared with age‐matched healthy controls (model application)
| Variable | Cmax,ss, ng/ml | AUCss, ng h/ml | ||||||
|---|---|---|---|---|---|---|---|---|
| Healthy control |
Mild HI |
Moderate HI |
Severe HI |
Healthy control |
Mild HI |
Moderate HI |
Severe HI |
|
| Olanzapine | 34.4 | 40.9 | 51.2 | 53.2 | 688 | 783 | 1031 | 1077 |
| Ratioa | — | 1.19 | 1.49 | 1.55 | — | 1.14 | 1.50 | 1.57 |
| Samidorphan | 35.2 | 51.8 | 63.0 | 74.0 | 261 | 321 | 495 | 604 |
| Ratioa | — | 1.47 | 1.79 | 2.10 | — | 1.23 | 1.90 | 2.32 |
Abbreviations: AUCss, area under the plasma drug concentration‐time curve at steady state; Cmax,ss maximum plasma concentration at steady state; HI, hepatic impairment; OLZ/SAM, combination of olanzapine and samidorphan.
Relative to age‐matched controls with normal hepatic function.
DISCUSSION
In the current study, a previously developed and validated PBPK model for OLZ/SAM14 was modified and further optimized using the observed data from a clinical study that evaluated the effects of moderate hepatic impairment on the PKs of olanzapine and samidorphan after a single dose of OLZ/SAM 5/10.12 The model was applied to further evaluate the effects of varying degrees of hepatic impairment on steady‐state olanzapine and samidorphan exposures under the clinical dosing regimen (i.e., chronic daily dosing at the therapeutic dose of OLZ/SAM 10/10) and highlights the utility of PBPK modeling to extend available clinical findings to additional untested clinical scenarios, especially in populations where conducting trials can be challenging (e.g., subjects with hepatic impairment). Ultimately, this may reduce the number of patients needed to characterize new pharmacologic entities, and may be associated with investigational time and cost savings.
Chronic liver disease produces a number of physiologic effects that have subsequent impact on drug PKs. Liver disease progression is associated with scar tissue deposition that disrupts the normal liver architecture and reduces the number of functional hepatocytes.16, 19 These changes may directly affect the clearance of drugs by altering liver size, enzyme expression, plasma protein binding, and hepatic blood flow. In addition, gastrointestinal symptoms are common in liver disease.18 The PBPK model used here to simulate PK profiles of olanzapine and samidorphan in subjects with varying degrees of liver deterioration took into account these altered processes (i.e., changes in liver size and enzyme abundance and slower gastrointestinal emptying).
Two primary findings related to the current PBPK modeling warrant further discussion. First, the median Tmax of 1.5 h for olanzapine and 0.5 h for samidorphan were observed in subjects with moderate hepatic impairment compared with 7.0 h for olanzapine and 1.0 h for samidorphan in healthy control subjects with normal hepatic function.12 A shortening of Tmax in individuals with hepatic impairment relative to those with normal hepatic function also was observed with the drug bosutinib, and was ascribed to decreased gastrointestinal absorption due to congestion and reduced blood flow to the intestinal mucosa.20 When combined with the Simcyp moderate hepatic impairment (Child‐Pugh‐B) population model, the previously developed PBPK model for OLZ/SAM did not capture the observed shortening of Tmax in subjects with moderate hepatic impairment, resulting in an underprediction of Cmax for both olanzapine and samidorphan. Subsequent sensitivity analyses revealed that modifications to the absorption rate constant ka were required to recover the observed olanzapine and samidorphan Tmax and Cmax values in subjects with hepatic impairment (Table 1).
Second, although the fraction of olanzapine unbound to plasma protein (fu) observed in the clinical study was similar in subjects with moderate hepatic impairment and healthy controls,12 the PBPK model–predicted AUC that assumed an increased fu with moderate hepatic impairment (fu = 0.10 for olanzapine; fu = 0.76 for samidorphan) compared with healthy controls with normal hepatic function (fu = 0.07 for olanzapine and fu = 0.69 for samidorphan14) was more consistent with the observed data. Therefore, simulations were carried out assuming an increase in fu for populations with mild, moderate, and severe hepatic impairment (i.e., 0.08, 0.10, and 0.13, respectively, for olanzapine, and 0.71, 0.76, and 0.81, respectively, for samidorphan).
Following modification of ka and fu, the predicted 1.51‐fold increase in geometric mean AUC for olanzapine in subjects with moderate hepatic impairment was reasonably consistent with the 1.67‐fold increase observed in clinical study.12 Similarly, for samidorphan, the predicted 1.79‐fold increase in geometric mean AUC in subjects with moderate hepatic impairment was reasonably consistent with the 1.52‐fold increase observed in the clinical study.12 Modification to ka and fu values had minimal impact on the predicted effect of moderate hepatic impairment on the AUCs of olanzapine and samidorphan, and, therefore, is not expected to impact the predictability of the model in untested clinical scenarios (e.g., patients with mild or severe hepatic impairment).
The use of PBPK modeling as a translational tool has increased in recent years owing to a number of factors.21 Progress has been achieved in understanding the systems biology underlying PBPK models (e.g., enzyme and transporter function in organs is now better defined), commercial software platforms have become available, and PBPK models allow existing clinical data to be extended to unstudied populations without the cost and difficulty of conducting and coordinating additional clinical trials, sparing patients from potential adverse events. Findings from appropriately executed PBPK models may enhance the understanding of drug exposures and support preclinical and clinical development and regulatory decision making.22 The findings presented here are strengthened by the well‐characterized PKs of olanzapine and samidorphan from clinical trials,6, 7, 8, 9 whereas study limitations include those common to PBPK models in general,22 such as the possibility that in vitro assays may not fully represent in vivo pathways or potential knowledge gaps (e.g., use scaling factors to calculate abundance; parameter non‐identifiability). In addition, the effects of hepatic impairment on the PKs of olanzapine and samidorphan were evaluated in nonsmokers in the current model, and smoking is associated with an increase in abundance of CYP1A2.23 However, because olanzapine exposure is known to be lower in smokers compared with nonsmokers, and samidorphan exposure is not affected by smoking,14, 24 the effect of hepatic impairment evaluated based on nonsmokers is a more conservative estimate of the higher drug exposure levels due to hepatic impairment.
In conclusion, a previously developed PBPK model for OLZ/SAM was optimized using existing clinical data and applied to assess the impact of varying degrees of hepatic impairment on the PKs of olanzapine and samidorphan in lieu of additional clinical studies. PBPK modeling indicated that mild hepatic impairment would have minimal impact on steady‐state exposures of olanzapine and samidorphan, and moderate to severe hepatic impairment would result in up to 1.6‐fold and 2.3‐fold increases in total exposure (AUC) of olanzapine and samidorphan, respectively.
CONFLICTS OF INTEREST
L.S. is an employee of Alkermes, Inc. L.v.M. was an employee of Alkermes, Inc., at the time of this study. Z.B. and K.R.Y. are employees of Certara UK Limited, Simcyp Division.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript and designed the research. L.S., Z.B., and K.R.Y. conducted the research. Z.B and K.R.Y. analyzed the data. All authors participated in data interpretation.
DISCLAIMER
As an Associate Editor for CPT: Pharmacometrics & Systems Pharmacology, Karen Rowland Yeo was not involved in the review or decision process for this paper.
ACKNOWLEDGEMENTS
The authors thank Mark S. Todtenkopf, PhD, who assisted in the preparation and proofreading of the manuscript. Medical writing and editorial support were provided by Gina Daniel, PhD, and John H. Simmons, MD, of Peloton Advantage, LLC, an OPEN Health company, and funded by Alkermes, Inc.
Sun L, Barter Z, von Moltke L, Rowland Yeo K. Using physiologically‐based pharmacokinetic modeling for predicting the effects of hepatic impairment on the pharmacokinetics of olanzapine and samidorphan given as a combination tablet. CPT Pharmacometrics Syst Pharmacol. 2021;10:1071–1080. 10.1002/psp4.12675
Funding information
This study was sponsored by Alkermes, Inc.
REFERENCES
- 1.Wentland MP, Lou R, Lu Q, et al. Syntheses of novel high affinity ligands for opioid receptors. Bioorg Med Chem Lett. 2009;19:2289‐2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bidlack JM, Knapp BI, Deaver DR, et al. In vitro pharmacological characterization of buprenorphine, samidorphan, and combinations being developed as an adjunctive treatment of major depressive disorder. J Pharmacol Exp Ther. 2018;367:267‐281. [DOI] [PubMed] [Google Scholar]
- 3.Shram MJ, Silverman B, Ehrich E, Sellers EM, Turncliff R. Use of remifentanil in a novel clinical paradigm to characterize onset and duration of opioid blockade by samidorphan, a potent mu‐receptor antagonist. J Clin Psychopharmacol. 2015;35:242‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potkin SG, Kunovac J, Silverman BL, et al. Efficacy and safety of a combination of olanzapine and samidorphan in adult patients with an acute exacerbation of schizophrenia: outcomes from the randomized, phase 3 ENLIGHTEN‐1 study. J Clin Psychiatry. 2020;81:19m12769. [DOI] [PubMed] [Google Scholar]
- 5.Correll CU, Newcomer JW, Silverman B, et al. Effects of olanzapine combined with samidorphan on weight gain in schizophrenia: a 24‐week phase 3 study. Am J Psychiatry. 2020;177:1168‐1178. [DOI] [PubMed] [Google Scholar]
- 6.Sun L, McDonnell D, Liu J, von Moltke L. Bioequivalence of olanzapine given in combination with samidorphan as a bilayer tablet (ALKS 3831) compared with olanzapine‐alone tablets: results from a randomized, crossover relative bioavailability study. Clin Pharmacol Drug Dev. 2019;8:459‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun L, McDonnell D, Yu M, Kumar V, von Moltke L. A phase I open‐label study to evaluate the effects of rifampin on the pharmacokinetics of olanzapine and samidorphan administered in combination in healthy human subjects. Clin Drug Investig. 2019;39:477‐484. [DOI] [PubMed] [Google Scholar]
- 8.Sun L, McDonnell D, Liu J, von Moltke L. Effect of food on the pharmacokinetics of a combination of olanzapine and samidorphan. Clin Pharmacol Drug Dev. 2019;8:503‐510. [DOI] [PubMed] [Google Scholar]
- 9.Sun L, McDonnell D, von Moltke L. Pharmacokinetics and short‐term safety of ALKS 3831, a fixed‐dose combination of olanzapine and samidorphan, in adult subjects with schizophrenia. Clin Ther. 2018;40:1845‐1854. [DOI] [PubMed] [Google Scholar]
- 10.Kassahun K, Mattiuz E, Nyhart E Jr, et al. Disposition and biotransformation of the antipsychotic agent olanzapine in humans. Drug Metab Dispos. 1997;25:81‐93. [PubMed] [Google Scholar]
- 11.Korprasertthaworn P, Polasek TM, Sorich MJ, et al. In vitro characterization of the human liver microsomal kinetics and reaction phenotyping of olanzapine metabolism. Drug Metab Dispos. 2015;43:1806‐1814. [DOI] [PubMed] [Google Scholar]
- 12.Sun L, Yagoda S, Du Y, Von Moltke L. Effect of hepatic and renal impairment on the pharmacokinetics of olanzapine and samidorphan given in combination as a bilayer tablet. Drug Des Devel Ther. 2019;13:2941‐2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turncliff R, DiPetrillo L, Silverman B, Ehrich E. Single‐ and multiple‐dose pharmacokinetics of samidorphan, a novel opioid antagonist, in healthy volunteers. Clin Ther. 2015;37:338‐348. [DOI] [PubMed] [Google Scholar]
- 14.Sun L, von Moltke L, Rowland YK. Physiologically‐based pharmacokinetic modeling for predicting drug interactions of a combination of olanzapine and samidorphan. CPT Pharmacometrics Syst Pharmacol. 2020;9:106‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howgate EM, Rowland Yeo K, Proctor NJ, Tucker GT, Rostami‐Hodjegan A. Prediction of in vivo drug clearance from in vitro data. I: impact of inter‐individual variability. Xenobiotica. 2006;36:473‐497. [DOI] [PubMed] [Google Scholar]
- 16.Johnson TN, Boussery K, Rowland‐Yeo K, Tucker GT, Rostami‐Hodjegan A. A semi‐mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin Pharmacokinet. 2010;49:189‐206. [DOI] [PubMed] [Google Scholar]
- 17.Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64:1147‐1161. [DOI] [PubMed] [Google Scholar]
- 18.Kalaitzakis E. Gastrointestinal dysfunction in liver cirrhosis. World J Gastroenterol. 2014;20:14686‐14695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLean AJ, Morgan DJ. Clinical pharmacokinetics in patients with liver disease. Clin Pharmacokinet. 1991;21:42‐69. [DOI] [PubMed] [Google Scholar]
- 20.Ono C, Hsyu PH, Abbas R, Loi CM, Yamazaki S. Application of physiologically based pharmacokinetic modeling to the understanding of bosutinib pharmacokinetics: prediction of drug‐drug and drug‐disease interactions. Drug Metab Dispos. 2017;45:390‐398. [DOI] [PubMed] [Google Scholar]
- 21.Jamei M. Recent advances in development and application of physiologically‐based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters SA, Dolgos H. Requirements to establishing confidence in physiologically based pharmacokinetic (PBPK) models and overcoming some of the challenges to meeting them. Clin Pharmacokinet. 2019;58:1355‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plowchalk DR, Rowland Yeo K. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Clin Pharmacol. 2012;68:951‐960. [DOI] [PubMed] [Google Scholar]
- 24.Haslemo T, Eikeseth PH, Tanum L, Molden E, Refsum H. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Pharmacol. 2006;62:1049‐1053. [DOI] [PubMed] [Google Scholar]