

Abstract
Erdafitinib is a potent oral pan‐fibroblast growth factor receptor inhibitor being developed as oncology drug for patients with alterations in the fibroblast growth factor receptor pathway. Erdafitinib binds preferentially to α1‐acid glycoprotein (AGP) and is primarily metabolized by cytochrome P450 (CYP) 2C9 and 3A4. This article describes a physiologically based pharmacokinetic (PBPK) model for erdafitinib to assess the drug–drug interaction (DDI) potential of CYP3A4 and CYP2C9 inhibitors and CYP3A4/CYP2C9 inducers on erdafitinib pharmacokinetics (PK) in patients with cancer exhibiting higher AGP levels and in populations with different CYP2C9 genotypes. Erdafitinib's DDI potential as a perpetrator for transporter inhibition and for time‐dependent inhibition and/or induction of CYP3A was also evaluated. The PBPK model incorporated input parameters from various in vitro and clinical PK studies, and the model was verified using a clinical DDI study with itraconazole and fluconazole. Erdafitinib clearance in the PBPK model consisted of multiple pathways (CYP2C9/3A4, renal, intestinal; additional hepatic clearance), making the compound less susceptible to DDIs. In poor‐metabolizing CYP2C9 populations carrying the CYP2C9*3/*3 genotype, simulations shown clinically relevant increase in erdafitinib plasma concentrations. Simulated luminal and enterocyte concentration showed potential risk of P‐glycoprotein inhibition with erdafitinib in the first 5 h after dosing, and simulations showed this interaction can be avoided by staggering erdafitinib and digoxin dosing. Other than a simulated ~ 60% exposure reduction with strong CYP3A/2C inducers such as rifampicin, other DDI liabilities were minimal and considered not clinically relevant.
STUDY HIGHLIGHTS.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Based on in vitro studies and the human mass balance study, the clearance pathways of erdafitinib were identified. The contribution of cytochrome P450 (CYP) 3A4 and 2C9 were verified in a clinical drug–drug interaction (DDI) study. Erdafitinib is highly protein bound and free fraction is dependent on α1‐acid glycoprotein (AGP) levels.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study addressed the feasibility of using a physiologically based pharmacokinetic model to evaluate the DDI potential of erdafitinib as victim and perpetrator in different populations, such as a population with cancer with varying AGP levels as well as different genotype populations for CYP2C9.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study can address questions to potential dose adjustments in patients with different genotypes, with comedications both as a victim or a perpetrator.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Increased knowledge and verification of potential dosing scenarios serves to better inform prescribers and health care providers regarding DDIs.
INTRODUCTION
Erdafitinib is an oral pan‐fibroblast growth factor receptor (FGFR) inhibitor characterized by a potent antitumor activity against FGFR‐aberrant malignancies in preclinical and clinical studies.1, 2, 3 Erdafitinib was granted a breakthrough designation by the US Food and Drug Administration (FDA) for the treatment of locally advanced or metastatic urothelial carcinoma with susceptible FGFR3 or FGFR2 genetic alterations and progression during or following at least one line of platinum‐containing chemotherapy, with subsequent accelerated approval for this indication in April 2019.4 Erdafitinib is also approved in Canada and a few other countries for the same indication. Pharmacokinetic (PK) investigations of erdafitinib within phase I clinical trials conducted in patients with cancer support dose‐proportional and time‐independent PK after single and multiple daily dosing at dose levels from 0.5–12‐mg, with a recommended maximum dose of 9‐mg given once daily (q.d.).5, 6 Erdafitinib binds preferentially to α1‐acid glycoprotein (AGP) and free fraction in plasma protein (f up) decreases with increasing AGP levels, resulting in decreased clearance.7
Per in vitro systems, erdafitinib is a substrate for cytochrome P450 (CYP) 2C9, CYP3A4, and P‐glycoprotein (Pgp), a weak time‐dependent inhibitor (TDI) and weak inducer of CYP3A4 and an inhibitor of organic cation transporter 2 (OCT2) and Pgp.4 M6 is the O‐demethylated erdafitinib and the major fecal metabolite in humans (24%). It is formed predominantly by CYP2C9 and to a small extent by CYP3A4. M8, N‐dealkylated erdafitinib, is the other important fecal metabolite in humans accounting for ~ 3% of the dose (1% in urine), which is primarily formed by CYP3A4.8 Subsequently, the role of CYP3A4 and CYP2C9 in the elimination of erdafitinib were confirmed in vivo through a fluconazole and itraconazole drug–drug interaction (DDI) trial.9
A physiologically based PK (PBPK) model was developed for erdafitinib that captured the AGP‐dependent f up and corresponding effects on total and free (unbound) exposure, along with the differential relative contributions of clearance pathways, based on in vitro data and single‐ascending dose and multiple‐ascending dose clinical and human mass balance data, and was subsequently verified using a clinical DDI study with itraconazole and fluconazole. The PBPK model was used to evaluate the DDI of perpetrator drugs: CYP3A4/CYP2C9 inhibitors and CYP3A4/CYP2C9 inducers, on erdafitinib, considering the effect of different CYP2C9 genotype populations and differences in AGP levels between healthy individuals and patients with cancer.
The DDI potential of erdafitinib as perpetrator for transporter inhibition (Pgp and OCT2/multidrug and toxin extrusion protein 2) and for time‐dependent inhibition and/or induction of CYP3A was also evaluated.
METHODS
Software
PBPK modeling was performed using the Simcyp™ v16.1 simulator (Simcyp Ltd). Characteristics of the clinical studies included in the PBPK model development, optimization and verification, and simulated trial design settings are listed in the Supplemental Information, Tables S1–S4. Input parameters of the PBPK models used are listed in Tables S5–S15. Incubation details of in vitro studies are given in Tables S16 and S17.
Model development
Figure 1 provides an overview of the strategy used for the erdafitinib PBPK model development, verification, and application. Parameters representing physicochemical properties, in vitro absorption, distribution, metabolism, and excretion (ADME) studies, and clinical total and free plasma concentration time profiles for erdafitinib were used to derive the input parameters for the erdafitinib PBPK model (Table S18). Per the PBPK model, erdafitinib clearance consisted of a combination of metabolism by CYP3A4 (20%) and CYP2C9 (39%) based on the in vitro phenotyping information, renal clearance (13%), and intestinal secretion (21%; implemented as biliary clearance) as derived from the human mass balance study. Additional hepatic metabolic clearance was estimated at 8%, derived from the minor amount of other metabolites excreted in the feces in the human mass balance study. As disease state can alter the plasma protein binding (e.g., increased AGP levels in patients with cancer),10, 11 the variability in f up was considered using protein concentration‐dependent f up. Moreover, a patient‐like virtual population was created based on the virtual healthy volunteers population in Simcyp™ v16.1 by modifying the AGP levels, reflecting the observed AGP levels in a healthy subject population (EDI1006 and EDI1007) or a patient population (EDI1001) (Table S19).6 Simulated and observed correlations between AGP levels and f up are shown in Figure S1.
FIGURE 1.
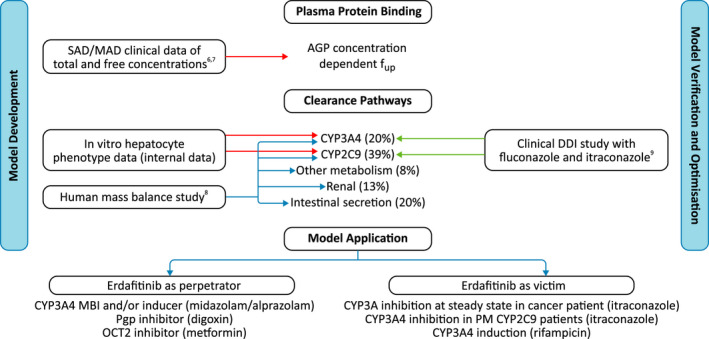
Overview of the physiologically based pharmacokinetic model development, verification, and application strategy.6, 7, 8, 9 AGP, α1‐acid glycoprotein; CYP, cytochrome P450; DDI, drug–drug interaction; fup, free fraction in plasma protein; MAD, multiple‐ascending dose; MBI, mechanism‐based inactivation; OCT2, organic cation transporter 2; Pgp, P‐glycoprotein; PM, poor metabolizer; SAD, single‐ascending dose
Complete absorption of erdafitinib was assessed based on the human ADME study with 14C‐erdafitinib (12‐mg).8 At the end of the sampling period, the total amount of radioactivity recovered ranged from 79.2% to 93.1% of the administered dose. The majority of total radioactivity was recovered in feces (58.7%–75.1%). The average total radioactivity recovered in urine was 16.2%–24.8% of the administered dose (accounting for unchanged drug and metabolites) and can be assigned to absorbed erdafitinib that was systemically available before urinary elimination. Metabolite profiling of pooled feces indicated M6 and M8 resulting from O‐demethylation and N‐demethylation of erdafitinib contributed to 24% and 3.23%, respectively, of the dose in the feces and can be accounted for as absorbed erdafitinib that was systemically metabolized and eliminated in the feces.4, 6 A slow elimination of drug and recovery of unchanged drug in the feces over extended days (17% recovered over ~ 16 days with < 5% of the dose recovered within the first 3 days), which is beyond the normal gastrointestinal transit time, suggesting a slow disposition of absorbed erdafitinib. Due to lack of evidence of direct conjugate formation in in vitro human hepatocyte incubations and rat biliary excretion study, the possibility of biliary‐excreted conjugated drug is unlikely. Therefore, the unchanged drug recovered in feces must first have been absorbed and subsequently secreted in the intestine directly or eliminated via the bile. Overall, the data obtained in the mass balance study indicate that erdafitinib is ≥ 94% absorbed from the gastrointestinal tract. Therefore, the absorption of erdafitinib was considered complete following oral administration.
Model verification
In addition to the in vitro data, relevant data from three clinical studies were used to build and verify the PBPK model for the relative contribution of CYP pathways and for potential involvement of Pgp in the overall clearance of the compound. The first study was the aforementioned first‐in‐human phase I study,6 which allowed verification of the PBPK model for simulation of the PK of total and free erdafitinib in a patient population following single and multiple dosing. The second was a human mass balance study in eight healthy individuals receiving a single dose of 12‐mg erdafitinib, which allowed to determine the intestinal secretion and percent bioavailability.8 During the early clinical development, efficacious dose was considered in the range of 9–12‐mg q.d., therefore a 12‐mg dose was used in the mass balance study.8 The third study was a DDI clinical study assessing the effect of multiple doses of 400‐mg q.d. fluconazole (a moderate inhibitor of CYP2C9 and a weak inhibitor of CYP3A) and 200‐mg q.d. itraconazole capsule (a strong inhibitor of CYP3A412 and an inhibitor of Pgp13 as well) on the PK of a single 4‐mg oral dose of erdafitinib in 16 healthy adults in a parallel design.8 In addition, both total and free concentrations were compared with clinically observed concentrations.
Prespecified acceptance criteria to evaluate the ability of the PBPK model to predict the observed data were based on the methods described by Abduljalil et al.14 for the PK parameters, and for DDI predictions the method based on Guest et al.15 was applied, considering the magnitude of the observed interaction. Although a Simcyp verified (SV)‐itraconazole PBPK model is available in the Simcyp compound library, several shortcomings have been previously described by Ke et al.,16 including mismatch in simulated and observed PK for the hydroxyitraconazole metabolite. An updated PBPK model, taking these shortcomings into account, was published by Chen et al.17 and is used in this article. The SV‐fluconazole file from the Simcyp™ v16.1 library was used, a file for which verification was done for the CYP3A4 inhibition properties with modifications to the CYP2C9 inhibition constant (Ki) value from 7.92 µM to 20.4 µM18 implemented to accurately capture the CYP2C9 inhibition properties.
Model application
Modified virtual populations with specific input parameters for poor metabolizer (PM) phenotypes for CYP2C9 were created, as described in Tables S20 and S21, to accurately simulate the effects of CYP2C9 genotypes observed in CYP2C9 PM with *2/*2, *2/*3, and *3/*3 genotypes. Relative enzyme abundance was back‐calculated from published studies using tolbutamide as a CYP2C9 probe substrate.19, 20, 21 For each virtual population, the coefficients of variance were further modified based on the published data by Chiba et al.22 These CYP2C9 PM virtual populations were used to investigate a worst‐case scenario with CYP3A4 inhibition in a PM CYP2C9*3/*3 genotype population.
To translate in vitro observations regarding the inhibitory effects of erdafitinib on OCT2 into clinical relevance, DDI simulations were conducted by including the in vitro transporter inhibition data into the erdafitinib PBPK model using transporter substrate metformin (OCT2 and multidrug and toxin extrusion protein) as a sensitive DDI victim substrate. Although erdafitinib is a potent Pgp inhibitor in vitro, Pgp inhibition at a systemic level can be excluded at the clinically relevant doses {concentration resulting in 50% inhibition [IC50 = 0.67 µM] > R (50*[maximum concentration (Cmax)free] = 0.57 µM)} based on the FDA and European Medicines Agency guidelines.23, 24 However, at the intestinal levels, it cannot be excluded that the IC50 for erdafitinib is < 0.1*dose (9‐mg erdafitinib)/250 ml (= 8.1 µM), which is the requisite for a Pgp inhibition of potential clinical significance according to the guidance documents.23, 24 To evaluate the impact of Pgp inhibition more dynamically during transit through the gastrointestinal tract, simulated luminal and enterocyte concentrations were used to determine the time frame where erdafitinib concentrations are higher than the unbound extracellular IC50 and the unbound intracellular IC50 for Pgp inhibition, respectively. DDI simulations with digoxin as a probe substrate of intestinal and hepatic Pgp with coadministration of erdafitinib and with staggered dosing were used to further evaluate the Pgp DDI potential (sensitivity analysis of IC50 values is shown in Figure S2). Verified PBPK models for rifampicin and digoxin were available in the Simcyp™ v16.1 compound library and were used in conjunction with the derived erdafitinib PBPK model. The SV‐metformin file from the Simcyp™ v16.1 library was used with the modification described by Burt et al.,25 and some adjustments toward the relative activity factors of the transporters were done based on in vitro data (on file) to be able to adequately describe the PK of the compound using the settings of Simcyp™ (Table S22).25, 26, 27, 28, 29, 30
RESULTS
Erdafitinib PBPK model performance verification
The PK parameters and concentration‐time plots of the observed and simulated data for erdafitinib lowest (4‐mg) and highest (9‐mg) clinical dose per its approved indication in patients with cancer are presented in Table 1 and Figure 2. Per visual predictive checks and the area under plasma concentration versus time curve (AUC) simulated/AUC observed and Cmax simulated/Cmax observed ratios, the simulated profiles matched the observed data both after single dosing and at steady state.
TABLE 1.
Simulated (n = 96) and observed single dose (n = 9) and steady‐state (n = 6) pharmacokinetic parameters for erdafitinib after 4‐mg and 9‐mg single doses followed by 9‐mg q.d. dosing from Day 4 onward in patients with cancer
| Patients | Simulated | Observed (EDI1001) | ||||||
|---|---|---|---|---|---|---|---|---|
| Single dose | q.d. dosing | Single dose | q.d. dosing | |||||
| Cmax (ng/ml) | AUC0–72 (ng·h/ml) | Cmax (ng/ml) | AUCtau (ng·h/ml) | Cmax (ng/ml) | AUC0–72 (ng·h/ml) | Cmax (ng/ml) | AUCtau (ng·h/ml) | |
| 4‐mg dose | N = 96 | N = 7 | N = 5 | |||||
| Mean | 190 | 7898 | 580 | 11,970 | 182 | 6743 | 675 | 13,516 |
| Geometric mean | 179 | 7408 | 543 | 10,896 | 163 | 6022 | 630 | 12,064 |
| Median | 174 | 7383 | 576 | 11,911 | 173 | 6360 | 569 | 13,516 |
| Standard deviation | 69.7 | 2722 | 211 | 4827 | 77.1 | 3318 | 267 | 8619 |
| Ratio predicted/observeda | 1.10 | 1.23 | 0.86 | 0.90 | ||||
| Success criteriab | 0.55–1.83 | 0.72–1.39 | 0.48–2.07 | 0.33–3.04 | ||||
| 9‐mg dose | N = 96 | N = 9 | N = 6 | |||||
| Mean | 428 | 17,664 | 1282 | 26,371 | 431 | 17,564 | 2018 | 39,587 |
| Geometric mean | 403 | 16,560 | 1201 | 23,994 | 390 | 15,091 | 1911 | 36,608 |
| Median | 390 | 16,522 | 1270 | 26,209 | 389 | 16,053 | 1765 | 30,594 |
| Standard deviation | 157 | 6095 | 464 | 10,642 | 177 | 9364 | 744 | 17,575 |
| Ratio predicted/observeda | 1.03 | 1.10 | 0.63 | 0.66 | ||||
| Success criteriab | 0.65–1.54 | 0.49–2.03 | 0.54–1.86 | 0.48–2.09 | ||||
Abbreviations: AUC0–72, area under the curve from time 0 to 72 h; AUCtau, area under the curve from time 0 to the end of the dosing period; Cmax, maximum plasma concentration; q.d., once daily.
Ratios of predicted geometric mean value and observed mean value.
Success criteria defined on observed data as described by Abduljalil et al.14
FIGURE 2.
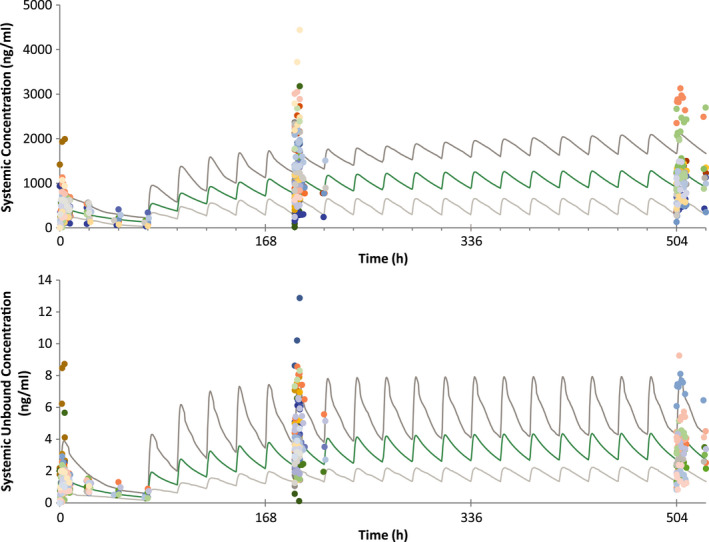
Overlay linear plots of simulated mean and 90% prediction interval (green and gray lines) and observed (colored symbols represent individual concentration data points, and each color represents one subject) plasma concentration time profiles of erdafitinib (top, total concentration in ng/ml; bottom, free concentration in ng/ml) after 9‐mg single dose followed by 9‐mg q.d. dosing from Day 4 in patients with cancer (study EDI1001)6, 7 q.d., once daily
Simulated and observed PK parameters for erdafitinib in healthy subjects (EDI1007)8 with the extensive metabolizer (EM) CYP2C9 genotype, when administered alone or with itraconazole 200‐mg q.d. or fluconazole 400‐mg q.d., are summarized in Table 2. The results showed an increase in Cmax and area under the curve to infinite time (AUCinf) ratios of 1.21 and 1.49, respectively, when erdafitinib was administered with fluconazole and of 1.04 and 1.34, respectively, when administered with itraconazole. No significant effect of included CYP2C9 genotype (*1/*1, *1/*2, *1/*3) was observed; geometric mean AUCinf ratios in the *1*1 subgroup with fluconazole was 1.52 versus 1.48 in a combined *1*1/*1*2 subgroup and with itraconazole this was 1.23 versus 1.34, respectively. The minor DDI effect observed with codosing of strong inhibitors such as fluconazole and itraconazole confirmed that other clearance pathways than CYP3A and CYP2C9 metabolism are also playing a role, such as the potential intestinal secretion. Pgp is not likely involved in the active secretion process of erdafitinib based on the observation that similar relative total amount of erdafitinib (Pgp substrate) and metabolite M6 (not a Pgp substrate) is excreted in urine after dosing erdafitinib with itraconazole (Pgp inhibitor). Considering intersubject and intertrial variability, the differences between observed and simulated ratios were small (Figure 3). In addition, in contrast to the simulations, the clinical DDI trial was not a cross‐over study and differences between trials should be considered as well. If differences between virtual trials are considered, for example, in the virtual trial 2, an erdafitinib geometric mean AUC in the presence of itraconazole of 11,091 ng/ml·h was simulated, which is 1.32‐fold higher as the erdafitinib geometric mean exposure of 8401 ng/ml·h without itraconazole obtained in virtual trial 5 (similar to the observed calculated interaction between itraconazole and erdafitinib of 1.34‐fold; Table 2).
TABLE 2.
Simulated (n = 96) and observed (n = 15, 16 or 17) pharmacokinetic parameters for erdafitinib following dosing of itraconazole 200‐mg q.d. or fluconazole 400‐mg q.d. from Days 1 to 11 and on Day 5 a single oral dosing of erdafitinib 4‐mg in healthy subjects (EDI1007) with the EM CYP2C9 genotype
| Simulated (n = 96 subjects) | Observed (n = 15, EDI‐1007; CYP2C9 *1/*1 + *1/*2) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmax (ng/ml) | AUCinf (ng·h/ml) | Cmax ratioa | AUC ratioa | Cmax (ng/ml) | AUCinf (ng·h/ml) | Cmax ratioa | AUC ratioa | ||
| No inhibitor | Cohort A (n = 17) | ||||||||
| Mean | 133 | 9087 | – | – | 138 | 9145 | – | – | |
| Geometric mean | 125 | 8653 | – | – | 134 | 8777 | – | – | |
| Standard deviation | 48.6 | 2851 | – | – | 34.9 | 2523 | – | – | |
| Ratio predicted/observedb | 0.93 | 0.99 | Success criteriac | 0.77‐1.30 | 0.75‐1.34 | ||||
| With 400‐mg q.d. fluconazole | Cohort B (n = 15 or 16) | ||||||||
| Mean | 146 | 14,852 | 1.04 | 1.66 | 177 | 14,233 | – | – | |
| Geometric mean | 137 | 14,265 | 1.04 | 1.65 | 162 | 13,060 | 1.21 | 1.49 | |
| Standard deviation | 50.2 | 4189 | 0.01 | 0.22 | 75.7 | 6087 | – | – | |
| Ratio predicted/observedb | 0.85 | 1.09 | Success criteriac | 0.61‐1.63 | 0.61‐1.63 | ||||
| DDI acceptance ranged | 1.03‐1.42 | 1.12‐1.98 | |||||||
| With 200‐mg q.d. itraconazole | Cohort C (n = 16 or 17) | ||||||||
| Mean | 132 | 10,973 | 1.02 | 1.18 | 145 | 12,228 | – | – | |
| Geometric mean | 125 | 10,376 | 1.02 | 1.18 | 140 | 11,785 | 1.04 | 1.34 | |
| Standard deviation | 43.9 | 3648 | 0.01 | 0.05 | 37.2 | 3203 | – | – | |
| Ratio predicted/observedb | 0.89 | 0.88 | Success criteriac | 0.76‐1.32 | 0.75‐1.33 | ||||
| DDI acceptance ranged | 1.00‐1.08 | 1.07‐1.00 | |||||||
Abbreviations: AUC, area under plasma concentration versus time curve; AUCinf, area under the curve to infinite time; Cmax, maximum plasma concentration; CYP, cytochrome P450; DDI, drug–drug interaction; EM, extensive metabolizer; q.d., once daily.
Simulated ratios expressing the fold increase and calculated as follows: parameter without inhibitor/with inhibitor.
Ratios of predicted geometric mean value and observed mean value.
Success criteria defined on observed data as described by Abduljalil et al.14
Model acceptance criteria based on Guest et al.15 used to determine an acceptance range dependent on the observed magnitude of DDI.
FIGURE 3.
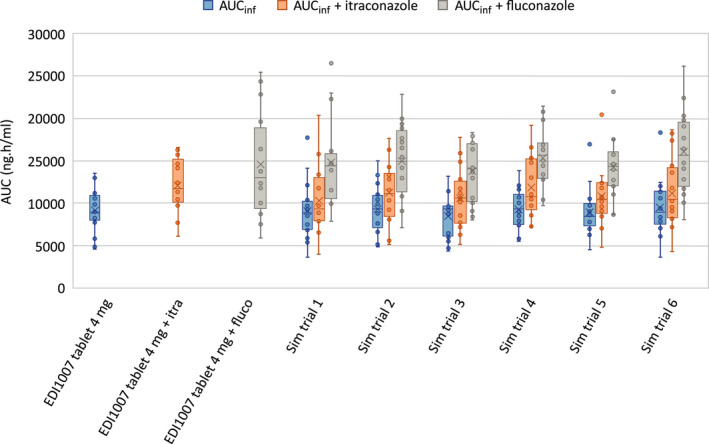
Box plot of AUCinf values in healthy subjects after dosing erdafitinib alone or in combination with itraconazole 200‐mg once daily or fluconazole 400‐mg once daily as observed in EDI10079 in healthy subjects with EM CYP2C9 genotypes compared with simulated AUCinf values in six simulated trials in healthy subjects with EM CYP2C9 genotypes. AUCinf, area under the curve to infinite time; CYP, cytochrome P450; EM, extensive metabolizer; fluco, fluconazole; itra, itraconazole; sim, simulation
DDI simulations with erdafitinib as the victim
Following verification of the model, additional prospective DDI simulations were performed to evaluate the DDI potential as victim in different genotype populations and the effect of increased AGP levels in patients with cancer. DDI simulations in virtual patients with erdafitinib 9‐mg as the victim are summarized in Table S23. When coadministered with itraconazole 200‐mg q.d. or fluconazole 400‐mg q.d., erdafitinib exposure was increased with respect to AUC and Cmax by 1.13 and 1.11 or 1.61 and 1.50‐fold, respectively. In the PM CYP2C9 genotype *3/*3, when erdafitinib was dosed with concomitant itraconazole 200‐mg q.d., the AUC and Cmax ratios increased up to 1.79 and 1.73 (worst‐case scenario), respectively; corresponding increases were 1.32 and 1.30, respectively, for CYP2C9 genotype *2/*2 and 1.46 and 1.44, respectively, for CYP2C9 genotype *2/*3 (Figure 4). Simulation of induction with rifampicin, a strong inducer of CYP3A4 and moderator inducer of CYP2C9, resulted in ~ 60% decrease in erdafitinib AUC and Cmax (Figures 4 and 5).
FIGURE 4.
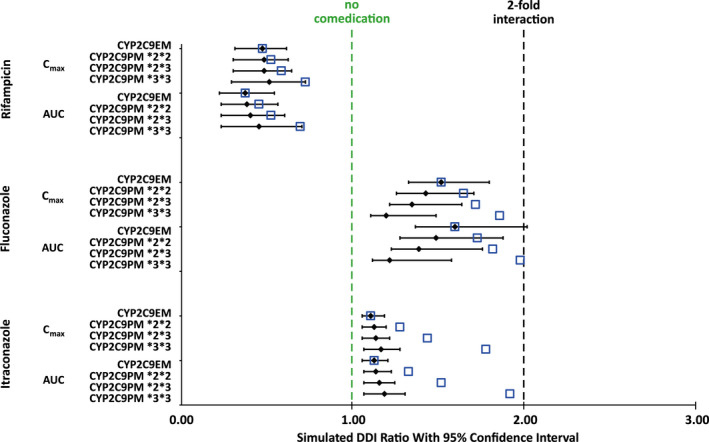
Simulated AUC ratios and Cmax ratios for erdafitinib following 9‐mg q.d. dose in the presence of comedication in CYP2C9 EM and different PM healthy subjects. The black bars represent the ratios expressing the geometric mean of the fold increase and the 95% confidence intervals calculated by dividing the parameter with inhibitor by the parameter without inhibitor for the same phenotype. The blue open squares represent the median ratios of the simulated DDI when comparing the distinct populations to a population of CYP2C9 EM without comedication. AUC, area under plasma concentration versus time curve; Cmax, maximum plasma concentration; CYP, cytochrome P450; DDI, drug–drug interaction; EM, extensive metabolizer; PM, poor metabolizer; q.d., once daily
FIGURE 5.
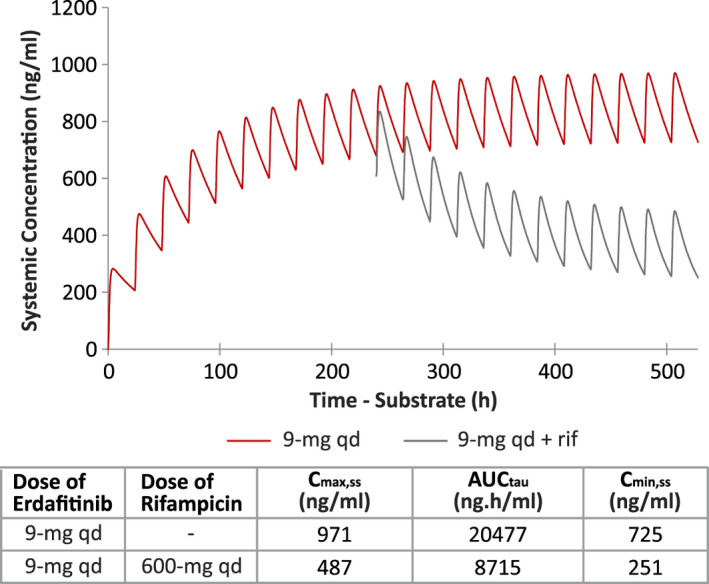
Mean linear plot of simulated erdafitinib plasma concentration‐time profiles following 9‐mg q.d. dosing of erdafitinib with and without rifampicin 600‐mg q.d. to compensate the decrease in exposure due to induction. AUCtau, area under the curve from time 0 to the end of the dosing period; Cmax.ss, maximum steady state plasma drug concentration; Cmin.ss, minimum steady‐state plasma drug concentration; q.d., once daily; rif, rifampicin
DDI simulations with erdafitinib as the perpetrator
Simulated midazolam and alprazolam Cmax and AUC ratios with and without erdafitinib 9‐mg q.d. are summarized in Table S24. Erdafitinib increased the exposure of midazolam by 1.5‐fold in a worst‐case scenario (i.e., f u,gut, fraction unbound in gut = 1), supporting that it is a weak TDI of CYP3A4. The higher ratios for prediction using f u,gut = 1 versus f u,gut = f up support that a large portion of the simulated inhibiting effect is attributed to inhibition in the gut. The minor in vitro induction effect did not result in any decrease of exposure of midazolam. Moreover, based on the data generated for erythromycin in the same experiment, the TDI effect of erdafitinib is likely to be overestimated (Table S25).
As a Pgp inhibitor, simulated luminal and enterocyte concentration after erdafitinib 9‐mg showed a potential risk of Pgp inhibition in the first 5 h after dosing only in the different parts of the intestinal tract (Figure S3). This resulted in an increase of digoxin Cmax values by 1.45‐fold when dosed together (Figure S4). However, the potential concern for a Pgp‐mediated DDI was alleviated by dose staggering of erdafitinib and digoxin by at least 6 h.
To evaluate the potential inhibition of OCT2 in vivo, DDI simulations using both the IC50 and the IC50/2 values of erdafitinib were done, and the results showed (Table S26) no relevant clinical interaction for the OCT2 substrate metformin (Figure S5) at the maximum clinical erdafitinib dose (9‐mg q.d.). The use of in vitro determined IC50 values was shown to predict the potential DDI on 250‐mg metformin of two other known positive control inhibitors of OCT2, cimetidine and pyrimethamine. Both of the positive control inhibitors were also included in the in vitro assay with erdafitinib (Tables S5, S22, S25, S27, S28 and Figures S6–S10). Additional sensitivity analysis on the in vitro assay determined that the IC50 value for erdafitinib was performed by simulating the interaction with a 30‐fold lower IC50 value,31 resulting in mild inhibition of metformin exposure (Table S26).
DISCUSSION
The erdafitinib PBPK model is presented to provide an integrated assessment of the DDI potential of this drug, which was recently approved by the FDA and a few other countries for the treatment of locally advanced or metastatic urothelial carcinoma with susceptible FGFR3 or FGFR2 genetic alterations and progression during or following at least one line of platinum‐containing chemotherapy. These modeling results together with clinical DDI studies were used to guide the clinical development and the drug‐labeling recommendations.
The purpose of the modeling and simulation performed in this study was to assess the DDI potential of erdafitinib as a victim for CYP3A inhibitors, CYP2C9 inhibitors, Pgp transporter inhibitors, and CYP3A4/CYP2C9 inducers in healthy subjects, in patients, and in poor CYP2C9 metabolizing populations. Moreover, the DDI potential of erdafitinib as a perpetrator for transporter inhibition and for time‐dependent inhibition of CYP3A was also assessed. Given that erdafitinib is highly bound to human plasma proteins, primarily to AGP, coupled with clinical observations of higher AGP levels and high variation in f up in patients with cancer relative to healthy subjects, a PBPK model was developed using a specific population with cancer that reflected the difference in AGP. Except for AGP, all physiological parameters remained unchanged in the PBPK model. The good agreement between observed and simulated concentrations of both total and unbound erdafitinib supported the accuracy of the PBPK model. In addition, the observed differences in total exposure in patients with cancer versus healthy subjects (~ 1.4‐fold) and similarity of free exposure in both populations were well captured in the PBPK model. Thus, the fold‐change in AUC ratio obtained from erdafitinib DDI simulations in virtual patients are applicable to healthy subjects and vice versa. The DDI ratios observed in the clinical study EDI1007,8 dosing erdafitinib in combination with the CYP2C9 and CYP3A4 inhibitor fluconazole (400‐mg q.d.), were too low to explain the clearance pathways by CYP‐mediated metabolism only. Considering complete oral absorption of erdafitinib based on its high solubility–high permeability Biopharmaceutics Classification System Class 1 characteristics as well as the prolonged excretion of parent compound in feces up to even 1 week after dosing,11 an intestinal secretion pathway seemed plausible. Intestinal secretion was also shown with other drugs (e.g., talinolol32). Potentially, a fraction of the erdafitinib dose excreted into intestine may be recirculated via reabsorption, which is unknown, and no separate factor was considered in the current PBPK simulations. Recirculation is not a clearance mechanism, but is more a distribution phenomenon. The fraction of the systemic clearance that is intestinally secreted derived from the human mass balance study was included in the PBPK model. Hence, the net effect of intestinal secretion including possible reabsorption is indirectly accounted for in the PBPK model. Hypothetically, to incorporate recirculation into the model, the intrinsic intestinal secretion clearance (biliary intrinsic clearance in the model) will have to be increased to maintain the same net effect of this pathway. As a result, to maintain the same observed oral clearance, the intrinsic clearance of the CYP pathways needs to decrease. Overall, the fractions of the systemic clearance, cleared via metabolic pathways or via intestinal secretion pathway will have to stay the same and consequently assumptions used in the PBPK model are unlikely to impact DDI predictions. Active involvement of Pgp in systemic clearance seems unlikely based on the observed lack of change in renal excretion of unchanged erdafitinib (Pgp substrate) if dosed in combination with the CYP3A4 and Pgp inhibitor itraconazole (200‐mg q.d.). In addition, the DDI ratio after dosing erdafitinib in combination with itraconazole was small and could be mainly attributed to inhibition of CYP3A4 only. This suggested that Pgp is unlikely to play an important role in the clearance of erdafitinib, and the mechanism of intestinal secretion is unclear. In the PBPK model, metabolism by CYP3A4 (20%) or CYP2C9 (39%) accounted for > 50% of erdafitinib clearance.
The PBPK model was used to further evaluate the DDI potential in a patient population. Simulated DDI with itraconazole 200‐mg q.d. or fluconazole 400‐mg q.d. in patients dosed at 9‐mg q.d. resulted in increases in exposure in AUC and Cmax of 1.13 and 1.11 or 1.61 and 1.50‐fold, respectively. In poor CYP2C9 metabolizers, if dosed with concomitant strong CYP3A4 inhibitors, the DDI ratios could go up to 1.79 and 1.73 for AUC and Cmax, respectively, for CYP2C9*3*3, with smaller increases of 1.32 and 1.30, respectively, for CYP2C9*2/*2 and 1.46 and 1.44, respectively, for CYP2C9*2/*3. Of note, in the DDI clinical study,8 CYP2C9 PM genotypes were excluded; therefore, these populations could not be verified. Simulation with a strong inducer of CYP3A4 and CYP2C9, such as rifampicin, resulted in ~ 60% decrease in erdafitinib AUC and Cmax in EM as well as in PM subjects; however, the absolute difference in erdafitinib exposure between PM and EM subjects dosed with concomitant inducer rifampicin was smaller due to the higher exposure in PM subjects. Therefore, simulated dose adjustments in EM subjects are indicative for the necessary dose increase to obtain similar plasma exposure. However, there was no clinical experience, nor are there clinical plans to test higher erdafitinib dosing regimens under induction scenarios.
As a Pgp inhibitor, simulated luminal and enterocyte concentration after dosing 9‐mg erdafitinib showed a potential risk of Pgp inhibition in the first 5 h after dosing only in the different parts of the intestinal tract. This resulted in a simulated increase of digoxin Cmax values by 1.45‐fold if dosed together. The simulated interaction could be avoided by at least 6‐h dose staggering between erdafitinib and digoxin. All other DDI liabilities are minimal and thus considered not clinically relevant. With erdafitinib, plasma concentrations were simulated to be higher in patients with the CYP2C9*3/*3 genotype; those patients known or suspected to have this genotype should be monitored for increased adverse reactions.
Overall, the PBPK modeling together with conducted clinical DDI studies supports the DDI risks that are captured in the approved product labeling for erdafitinib. Per the labeling, coadministration of erdafitinib with a moderate CYP2C9 or strong CYP3A4 inhibitor will increase erdafitinib exposure and may lead to increased drug‐related toxicity, whereas erdafitinib exposure will potentially be reduced with strong CYP2C9 or CYP3A4 inducers. Accordingly, the use of alternative agents with no or minimal enzyme inhibition or induction potential should be considered. Otherwise, when concurrent use is unavoidable, erdafitinib dose adjustments should be considered based on adverse event monitoring during use with moderate CYP2C9 or strong CYP3A4 inhibitors accordingly to prescription label recommendations. Due to the risk of decreased antitumor activity, concurrent use of erdafitinib with strong CYP2C9 or CYP3A4 inducers should be avoided if the dose regimen cannot be changed. Regarding Pgp substrates, the product labeling highlights the potential increase in their systemic exposure if administered concurrently with erdafitinib and that, when concurrent use is unavoidable, drugs with a narrow therapeutic index should be taken at least 6 h before or after erdafitinib to minimize the potential for interactions. In general, the sponsor received positive feedback from the health authorities (HAs) in the application of PBPK DDI simulations for drug development and supporting label recommendations for the product. In most situations, HAs accepted the PBPK simulations, and in some other areas recommended a cautious language for the label. For example, HAs accepted dosing recommendations for erdafitinib concomitant use with Pgp substrates and various CYP2C9 genotypes based on PBPK simulations. For the effect of CYP3A4/2C9 inducers on erdafitinib PK, a cautionary language for potential loss of erdafitinib exposures was recommended based on PBPK simulations. Although erdafitinib simulations to study the CYP3A TDI effect on CYP3A substrates (midazolam, alprazolam) and inhibition of OCT2 substrate (metformin) were suitably tested with appropriate sensitivity analysis, there were some uncertainties observed in predicted interactions with known positive control inhibitors for these substrates (overprediction of erythromycin [CYP3A TDI] interaction with midazolam but not alprazolam, slight underprediction of inhibitory effect of cimetidine and pyrimethamine on metformin). Therefore, clinical DDI studies are planned to confirm the simulated interaction results with CYP3A and OCT2 substrates and to provide label recommendations as appropriate. Thus, the PBPK modeling together with conducted clinical DDI studies supports the DDI risks that are captured in the approved product labeling for erdafitinib. Furthermore, erdafitinib is being individually dose titrated in the clinical setting based on a quantitatively determined biomarker strategy. This biomarker‐based dose titration further optimizes dose on an individual level in addition to the DDI guidance.
CONCLUSIONS
The multiple pathways involved in the clearance of erdafitinib make the compound less susceptible to DDIs. Overall, aside from interactions involving strong CYP3A4 or moderate 2C9 inhibitors and CYP3A/2C inducers as well as Pgp substrates, all other DDI liabilities are minimal and thus considered not clinically relevant. Dose staggering (~ 6 h) between erdafitinib dosing and narrow therapeutic index Pgp substrates can minimize DDIs at the intestinal level.
DISCLAIMER
All authors contributed to the data interpretation and review of this manuscript and confirm that they have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. All authors meet ICMJE criteria and all those who fulfilled those criteria are listed as authors. All authors had access to the study data, provided direction and comments on the manuscript, made the final decision about where to publish these data and approved submission to this journal.
CONFLICT OF INTEREST
All authors are employees of Janssen Research & Development, LLC, and are shareholders in the parent company (Johnson & Johnson).
AUTHOR CONTRIBUTIONS
L.D.Z., J.S., F.J., I.P., L.Y.L., M.M., and R.N.V.S.M. designed the research work. L.D.Z., P.V., I.G., E.S., and I.W. performed the research work. L.D.Z., J.S., I.P., and R.N.V.S.M. analyzed the data. L.D.Z., J.S., F.J., I.P., M.M., and R.N.V.S.M. wrote the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Laurie Orloski (independent medical writer) and Vaibhav Deshpande, PhD (SIRO Clinpharm Pvt. Ltd., India) for writing and editorial assistance in the development of this article. Harry Ma, PhD (Janssen Global Services, LLC) provided additional editorial support for this manuscript.
De Zwart L, Snoeys J, Jacobs F, et al. Prediction of the drug–drug interaction potential of the α1‐acid glycoprotein bound, CYP3A4/CYP2C9 metabolized oncology drug, erdafitinib. CPT Pharmacometrics Syst Pharmacol. 2021;10:1107–1118. 10.1002/psp4.12682
Funding information
The work presented in this manuscript was sponsored by Janssen Research & Development, LLC.
REFERENCES
- 1.Bahleda R, Italiano A, Hierro C, et al. Multicenter phase I study of erdafitinib (JNJ‐42756493), oral pan‐fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res. 2019;25:4888‐4897. [DOI] [PubMed] [Google Scholar]
- 2.Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381:338‐348. [DOI] [PubMed] [Google Scholar]
- 3.Perera TPS, Jovcheva E, Mevellec L, et al. Discovery and pharmacological characterization of JNJ‐42756493 (Erdafitinib), a functionally selective small‐molecule FGFR family inhibitor. Mol Cancer Ther. 2017;16:1010‐1020. [DOI] [PubMed] [Google Scholar]
- 4.BALVERSATM . Prescribing information. Horsham, PA: Janssen Products, LP; 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212018s000lbl.pdf. Accessed December 15, 2020.
- 5.Nishina T, Takahashi S, Iwasawa R, et al. Safety, pharmacokinetic, and pharmacodynamics of erdafitinib, a pan‐fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor, in patients with advanced or refractory solid tumors. Invest New Drugs. 2018;36:424‐434. [DOI] [PubMed] [Google Scholar]
- 6.Tabernero J, Bahleda R, Dienstmann R, et al. Phase I dose‐escalation study of JNJ‐42756493, an oral pan‐fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2015;33:3401‐3408. [DOI] [PubMed] [Google Scholar]
- 7.Li LY, Guo Y, Gonzalez M, Ouellet D. Effect of plasma protein binding on the pharmacokinetics of erdafitinib: results of an integrated cross‐study analysis. J Clin Pharmacol. 2020;60:391‐399. [DOI] [PubMed] [Google Scholar]
- 8.Scheers E, Borgmans C, Keung C, et al. Metabolism and disposition in rats, dogs, and humans of erdafitinib, an orally administered potent pan‐fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor. Xenobiotica. 2021;51:177‐193. [DOI] [PubMed] [Google Scholar]
- 9.Poggesi I, Li LY, Jiao J, et al. Effect of fluconazole and itraconazole on the pharmacokinetics of erdafitinib in healthy adults: a randomized, open‐label, drug‐drug interaction study. Eur J Drug Metab Pharmacokinet. 2020;45:101‐111. [DOI] [PubMed] [Google Scholar]
- 10.Kremer JM, Wilting J, Janssen LH. Drug binding to human alpha‐1‐acid glycoprotein in health and disease. Pharmacol Rev. 1988;40:1‐47. [PubMed] [Google Scholar]
- 11.Duche JC, Urien S, Simon N, et al. Expression of the genetic variants of human alpha‐1‐acid glycoprotein in cancer. Clin Biochem. 2000;33:197‐202. [DOI] [PubMed] [Google Scholar]
- 12.Vermeer LM, Isringhausen CD, Ogilvie BW, Buckley DB. Evaluation of ketoconazole and its alternative clinical CYP3A4/5 inhibitors as inhibitors of drug transporters: the in vitro effects of ketoconazole, ritonavir, clarithromycin, and itraconazole on 13 clinically‐relevant drug transporters. Drug Metab Dispos. 2016;44:453‐459. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu M, Uno T, Sugawara K, Tateishi T. Effects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P‐glycoprotein. Br J Clin Pharmacol. 2006;61:538‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abduljalil K, Cain T, Humphries H, Rostami‐Hodjegan A. Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: an analysis based on in vivo observations. Drug Metab Dispos. 2014;42:1478‐1484. [DOI] [PubMed] [Google Scholar]
- 15.Guest EJ, Aarons L, Houston JB, Rostami‐Hodjegan A, Galetin A. Critique of the two‐fold measure of prediction success for ratios: application for the assessment of drug‐drug interactions. Drug Metab Dispos. 2011;39:170‐173. [DOI] [PubMed] [Google Scholar]
- 16.Ke AB, Zamek‐Gliszczynski MJ, Higgins JW, Hall SD. Itraconazole and clarithromycin as ketoconazole alternatives for clinical CYP3A inhibition studies. Clin Pharmacol Ther. 2014;95:473‐476. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Ma F, Lu T, et al. Development of a physiologically based pharmacokinetic model for itraconazole pharmacokinetics and drug‐drug interaction prediction. Clin Pharmacokinet. 2016;55:735‐749. [DOI] [PubMed] [Google Scholar]
- 18.Neal JM, Kunze KL, Levy RH, O'Reilly RA, Trager WF. Kiiv, an in vivo parameter for predicting the magnitude of a drug interaction arising from competitive enzyme inhibition. Drug Metab Dispos. 2003;31:1043‐1048. [DOI] [PubMed] [Google Scholar]
- 19.Jetter A, Kinzig‐Schippers M, Skott A, et al. Cytochrome P450 2C9 phenotyping using low‐dose tolbutamide. Eur J Clin Pharmacol. 2004;60:165‐171. [DOI] [PubMed] [Google Scholar]
- 20.Kirchheiner J, Bauer S, Meineke I, et al. Impact of CYP2C9 and CYP2C19 polymorphisms on tolbutamide kinetics and the insulin and glucose response in healthy volunteers. Pharmacogenetics. 2002;12:101‐109. [DOI] [PubMed] [Google Scholar]
- 21.Vormfelde SV, Brockmoller J, Bauer S, et al. Relative impact of genotype and enzyme induction on the metabolic capacity of CYP2C9 in healthy volunteers. Clin Pharmacol Ther. 2009;86:54‐61. [DOI] [PubMed] [Google Scholar]
- 22.Chiba K, Shimizu K, Kato M, et al. Estimation of interindividual variability of pharmacokinetics of CYP2C9 substrates in humans. J Pharm Sci. 2017;106:2695‐2703. [DOI] [PubMed] [Google Scholar]
- 23.US Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research . In vitro drug interaction studies—cytochrome p450 enzyme‐ and transporter‐mediated drug interactions guidance for industry. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/vitro‐drug‐interaction‐studies‐cytochrome‐p450‐enzyme‐and‐transporter‐mediated‐drug‐interactions. Published 2020. Accessed December 15, 2020.
- 24.European Medicines Agency, Committee for Human Medicinal Products . Guideline on the investigation of drug interactions. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐drug‐interactions‐revision‐1_en.pdf. Published 2012. Accessed December 15, 2020.
- 25.Burt HJ, Neuhoff S, Almond L, et al. Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug‐drug interactions. Eur J Pharm Sci. 2016;88:70‐82. [DOI] [PubMed] [Google Scholar]
- 26.Gusler G, Gorsline J, Levy G, et al. Pharmacokinetics of metformin gastric‐retentive tablets in healthy volunteers. J Clin Pharmacol. 2001;41:655‐661. [DOI] [PubMed] [Google Scholar]
- 27.Pentikainen PJ, Neuvonen PJ, Penttila A. Pharmacokinetics of metformin after intravenous and oral administration to man. Eur J Clin Pharmacol. 1979;16:195‐202. [DOI] [PubMed] [Google Scholar]
- 28.Sirtori CR, Franceschini G, Galli‐Kienle M, et al. Disposition of metformin (N, N‐dimethylbiguanide) in man. Clin Pharmacol Ther. 1978;24:683‐693. [DOI] [PubMed] [Google Scholar]
- 29.Tucker GT, Casey C, Phillips PJ, et al. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol. 1981;12:235‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caille G, Lacasse Y, Raymond M, et al. Bioavailability of metformin in tablet form using a new high pressure liquid chromatography assay method. Biopharm Drug Dispos. 1993;14:257‐263. [DOI] [PubMed] [Google Scholar]
- 31.European Medicines Agency, Committee for Human Medicinal Products . Guideline on the reporting of physiologically based pharmacokinetic modelling and simulation. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐reporting‐physiologically‐based‐pharmacokinetic‐pbpk‐modelling‐simulation_en.pdf. Published 2016. Accessed December 15, 2020.
- 32.Gramatte T, Oertel R. Intestinal secretion of intravenous talinolol is inhibited by luminal R‐verapamil. Clin Pharmacol Ther. 1999;66:239‐245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material