

Abstract
Cancer‐associated fibroblasts (CAFs) as a predominant cell component in the tumour microenvironment (TME) play an essential role in tumour progression. Our earlier studies revealed oxidized ATM activation in breast CAFs, which is independent of DNA double‐strand breaks (DSBs). Oxidized ATM has been found to serve as a redox sensor to maintain cellular redox homeostasis. However, whether and how oxidized ATM in breast CAFs regulates breast cancer progression remains poorly understood. In this study, we found that oxidized ATM phosphorylates BNIP3 to induce autophagosome accumulation and exosome release from hypoxic breast CAFs. Inhibition of oxidized ATM kinase by KU60019 (a small‐molecule inhibitor of activated ATM) or shRNA‐mediated knockdown of endogenous ATM or BNIP3 blocks autophagy and exosome release from hypoxic CAFs. We also show that oxidized ATM phosphorylates ATP6V1G1, a core proton pump in maintaining lysosomal acidification, leading to lysosomal dysfunction and autophagosome fusion with multi‐vesicular bodies (MVB) but not lysosomes to facilitate exosome release. Furthermore, autophagy‐associated GPR64 is enriched in hypoxic CAFs‐derived exosomes, which stimulates the non‐canonical NF‐κB signalling to upregulate MMP9 and IL‐8 in recipient breast cancer cells, enabling cancer cells to acquire enhanced invasive abilities. Collectively, these results provide novel insights into the role of stromal CAFs in promoting tumour progression and reveal a new function of oxidized ATM in regulating autophagy and exosome release.
Keywords: autophagy, cancer‐associated fibroblasts, exosomes, invasion, oxidized ATM
Abbreviations
- ACP
2Acid phosphatase 2
- Aps
Autophagosomes
- ATF2
Activating transcription factor 2
- ATG
Autophagy‐associated genes
- ATLD
Ataxia‐telangiectasia‐like disorder
- ATM
Ataxia telangiectasia‐mutated gene
- BafA1
Bafilomycin A1
- BNIP3BCL2
interacting protein 3
- BSA
Bovine serum albumin
- CAFs
Cancer‐associated fibroblasts
- CHK2
Checkpoint kinase 2
- CM
Conditioned medium
- CTTN
Cortactin
- DMEM
Dulbecco's modified eagle medium
- DSBsDNA
double‐strand breaks
- EVs
Extracellular vesicles
- FBS
Fetal bovine serum
- FLOT1
Flotillin‐1
- GPR64
Adhesion G protein‐coupled receptor G2
- H&E staining
Hematoxylin and eosin staining
- HNSCC
Head and neck squamous cell carcinoma
- HRP
Horseradish peroxidase
- IDC
Invasive ductal carcinoma
- IFI
mmunofluorescence
- IHCI
mmunohistochemistry
- IL‐8
Interleukin 8
- IPI
mmunoprecipitation
- IRIon
irradiation
- LAMP1
Lysosome‐associated membrane protein 1
- LC3B
Light chain 3B
- MEF2D
Myocyte enhancer factor 2D
- MMP9
Matrix metallopeptidase 9
- MRN
complexMre11‐Rad50‐Nbs1 complex
- MVB
Multi‐vesicular body
- NFs
Normal fibroblasts
- NTAanalysis
analysisNanoparticle tracking analysis
- OS
Oxidized stress
- siRNA
Small interfering RNA
- TEM
Transmission electron microscope
- TEM
Transmission electron microscope
- TME
Tumor microenvironment
- WB
Western blotting.
1. INTRODUCTION
Tumour progression is governed by the cross‐talk between tumour cells and the surrounding tumour microenvironment (TME). Cancer‐associated fibroblasts (CAFs), the most abundant stromal cells in the TME, have a critical role in tumour initiation, progression, and metastasis. Hypoxia is one of the most common features in the TME of solid tumours and is associated with a more aggressive phenotype including angiogenesis, drug resistance, and poor clinical outcomes (Chen et al., 2018). However, few reports have investigated the effects of hypoxic CAFs on tumour progression properties such as tumour cell invasion or metastasis abilities, which remain to be elucidated.
In previous studies, we and others revealed that a number of genes including ataxia telangiectasia‐mutated gene (ATM) are dysregulated in CAFs compared with paired normal fibroblasts (NFs) (Peng et al., 2013). Generally, the evolutionarily conserved ATM protein kinase is activated by Mre11‐Rad50‐Nbs1 complex (MRN complex) and responds to DNA double‐strand breaks (DSBs) as a DNA damage response (DDR). It coordinates the activation of cell cycle checkpoints to maintain genomic integrity or gene stability (Czornak et al., 2008). The nuclear function of ATM is well known to be tightly associated with DDR and cell cycle checkpoints through the ATM‐Chk2 and ATM‐ATR‐Chk1 pathways as well as the MRN complex. Recently, the critical cytoplasmic function of ATM (called oxidized ATM) was reported. Oxidized ATM participates in regulating oxidative stress and cell metabolism by acting as a redox sensor. It plays a crucial role in regulating cellular redox homeostasis (Bencokova et al., 2009; Shiloh & Ziv, 2013) and other biological processes. For example, our earlier studies unravelled that oxidized ATM is activated in breast CAFs under hypoxic conditions to promote aberrant proliferation of breast CAFs (Tang et al., 2015). ATM activation by oxidative stress was found to regulate IL‐8 to enhance cancer cell migration and invasion to promote metastatic potential (Chen et al., 2015). In particular, our and Moreno‐Sánchez Rafael's studies revealed that oxidized ATM can regulate glycolysis in stromal CAFs to facilitate tumour cell metastasis (Marín‐Hernández et al., 2019; Sun et al., 2019). These studies demonstrate that the function of oxidative stress‐mediated ATM is distinct from that of the classical DNA damage‐activated nuclear ATM. To date, the effects and molecular mechanisms of oxidized ATM in regulating tumour cell function are poorly characterized.
CAFs have been shown to contribute to cancer progression in various ways. Direct interactions and paracrine effects of secreted proteins are well‐known communication mechanisms between stromal cells and tumour cells. It has been shown that SDF‐1, IL32, and TGFβ work as mediators of the crosstalk between breast CAFs and breast cancer cells in promoting tumour cell proliferation and invasion (Tang et al., 2019; Wen et al., 2019). CAFs also increase tumour cell invasion, metastasis, and multidrug resistance by extracellular matrix (ECM) remodelling and metabolic‐coupling between CAFs and tumour cells (Sun et al., 2019; Tang et al., 2016; Yu et al., 2017). Recently, exosome trafficking has emerged as a critical mechanism in intercellular communications. Indeed, exosomes carry and transport molecules such as oncoproteins and oncopeptides, RNA species, lipids, DNA fragments from donors to recipient cells, resulting in profound phenotypic changes in the TME. Emerging evidence indicates that exosomes derived from tumour cells are important modulators in regulating the activation of the surrounding fibroblasts (Yeon et al., 2018) and conversely, CAF‐derived exosomes play an essential part in regulating cancer cell proliferation (Kim et al., 2020), migration (Wu et al., 2020), metabolism (Yan et al., 2018), and therapy‐resistance (Zhang et al., 2020). Notably, we observed that ATM‐expressing CAFs release more exosomes than ATM‐silenced CAFs under hypoxia and promote breast cancer cell migration and invasion. We speculate that that the oxidized ATM may regulate exosome release from CAFs and confer an increased tumour cell invasion.
Autophagy is a highly‐conserved biological process involved in the degradation of damaged organelles and proteins for cellular homeostasis (Li et al., 2015). In the TME, autophagy not only functions in enzymatic degradation for its own metabolic stability, but also mediates the cross‐talk between stromal cells and tumour cells, thereby regulating cancer cell functions. Notably, high basal levels of autophagy in head and neck squamous cell carcinoma (HNSCC)‐associated CAFs facilitate HNSCC progression via autophagy‐dependent secretion of tumour‐promoting factors (New et al., 2017). Autophagy can be induced by hypoxic stimulation, a lack of amino acids, insufficient insulin, growth factor deficiency, medicinal treatment and ionizing radiation (IR). Recently, the role of DNA damage‐activated ATM in autophagy has been uncovered. For example, chemotherapy drugs or IR‐activated ATM mainly relies on the ATM‐AMPK‐mTORC1‐ULK1/2 pathway, Beclin1/PI3KIII complexes, MAPK14 or ATM‐CHK2‐Beclin 1 axis, to enhance autophagy and influence the radiosensitivities or chemosensitivities of tumours (Liang et al., 2019). Also, DNA damage‐independent ATM may also play a critical role in autophagy. For instance, oxidized ATM caused by accumulated ROS was reported to dysregulate autophagy (mitophagy and pexophagy) and be involved in inflammation and chronic disease, such as atherosclerosis and cancer (Stagni et al., 2018). Additionally, oxidized ATM enhanced by the cholesterol in lipid droplets was found to promote the fusion of autophagosomes and lysosomes in early atherosclerosis (Le Guezennec et al., 2012). However, whether oxidized ATM controls autophagy in TME is unclear. Particularly, it is unknown whether oxidized ATM regulates autophagy in breast CAFs and the crosstalk between CAFs and breast tumour cells in cancer progression.
In this study, we reveal that activation of oxidized ATM induces autophagy by phosphorylating BNIP3 and regulates lysosome functions by phosphorylating ATP6V1G1 in hypoxic breast CAFs, leading to the fusion between autophagosomes (APs) with multi‐vesicular bodies (MVBs). This results in enrichment of autophagy‐associated GPR64 in secreted exosomes, which in turn activates non‐canonical NF‐κB signalling pathways in breast cancer cells to enhance their invasion capabilities.
2. MATERIALS AND METHODS
2.1. Clinical breast tumour samples
Human breast cancer tissues and corresponding normal breast tissues were obtained from patients undergoing surgery in the First Affiliated Hospital of Chongqing Medical University. A total of 74 patient's tissues were used for IHC staining, and the details of clinical characteristics are provided in Supplemental Table 1. Plasma samples from 58 breast cancer patients (from 42 to 69 years old) and 10 healthy women (from 43 to 67 years old) were used to isolate plasma exosomes, and detailed information for these participants is listed in Supplemental Table 2. None of the patients in our study had previously received any radiotherapy or neoadjuvant chemotherapy, and all patients underwent an informed consent process. The study was approved by the Ethics Committee of Chongqing Medical University (2017‐080).
2.2. Cell culture, treatment and preparation of conditioned medium (CM)
The culture and immortalization of paired normal fibroblasts (NFs) and cancer‐associated fibroblasts (CAFs) from breast cancer patients were previously established (Peng et al., 2013). NFs, CAFs, and breast cancer cells MDA‐MB‐231 were grown in Dulbecco's Modified Eagle Medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA), while breast cancer cells BT549 were grown in RPMI 1640 medium (Gibco, USA) with 10% FBS.
Hypoxic treatment of cells was performed in a tri‐gas incubator (Thermo, USA) flushed with a gas mixture of 1% O2, 5% CO2 and 94% nitrogen. Briefly, stromal fibroblasts were cultured in normoxia to around 90% confluency, followed by the replacement of fresh culture medium. The cells were then cultured under hypoxia for 8 h in the presence or absence of inhibitors (Selleck, USA). All hypoxic stimuli (or conditioned hypoxia treatment) mentioned in this paper refer to cells cultured in 1% O2 for 8 h to avoid DNA double‐strand breaks. For ion irradiation treatment, cells received ions from 55 MeV carbon ions at the Munich ion microbeam, combined with normoxic or hypoxic treatment in the meantime.
For preparation of conditioned medium, when cell growth reached about 80% confluency, cells were washed with PBS 3 times and an appropriate volume of fresh FBS‐free medium was added to the cells with or without inhibitor, and cultured in hypoxia for the designated time. The supernatant was then collected and centrifuged for 10 min at 300 × g as the conditioned medium (CM). CM was normalized by the ratio of CM volume (ml) to the cell numbers (when CM was collected) (LY & Baddour, 2016) and prepared for co‐culture with target cells. The volume of applied CM depended on the concentration of normalized CM to eliminate the influence of cell number differences between experimental groups on CM. To further obtain exosome‐free CM, the CM was spun down successively at 300 × g for 30 min, 2000 × g for 30 min, 10,000 × g for 30 min and 120,000 × g for 70 min to deplete exosomes from the supernatant.
2.3. RNA extraction and qRT‐PCR
Total RNA was isolated using Trizol (Invitrogen, USA) according to the manufacturer's instructions and was reverse‐transcribed to cDNA using PrimeScript RT Reagent Kit (Takara, China). Quantitative real‐time PCR (qRT‐PCR) was then performed using SYBR Premix Ex TaqTM II (Takara, China) and corresponding primers used are listed in Supplementary Table 3. All experiments were performed three times.
2.4. RNA interference, engineered fibroblasts and luciferase report assay
Lentivirus‐mediated shRNAs targeting ATM were designed and synthesized by GenePharma (Shanghai, China). All small interfering RNA (siRNA) oligonucleotides (Supplementary Table 4) were purchased from PandaBio (Chongqing, China), and transfected into cells using Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer's instructions. Knockdown efficiency of siRNA/shRNA against the targets in transfected cells was confirmed by qRT‐PCR and/or by western blotting.
pcDNA3‐Flag‐ATM plasmid was a product of Addgene. The wild type BNIP3 WT (SSHCDSPPRSQT), mutant BNIP3 S135A construct (in which SSHCDSPPRSQT (S:serine) was mutated to SSHCDSPPRAQT (A:alanine) to generate a hypophosphorylated BNIP3), and mutant BNIP3 S135D construct (in which SSHCDSPPRSQT was mutated to SSHCDSPPRDQT (D:aspartic acid) to generate a hyperphosphorylated BNIP3) were generated by GenePharma (Shanghai, China), and inserted into pcDNA3‐Flag tagged plasmid to obtain pcDNA3‐Flag‐BNIP3 (WT), pcDNA3‐Flag‐BNIP3 mutant (S135A) and pcDNA3‐Flag‐BNIP3 mutant (S135D). The constructed plasmids were transfected into endogenous BNIP3‐silenced CAFs using Lipofectamine 3000 to establish engineered CAFs.
To acquire the wild‐type luciferase reporter of Atg5 and Atg16L, the target promoter fragment (‐1100∼+100) was amplified by PCR and inserted into the pGL3‐basic reporter plasmid. Then, the FOXO3 binding site at Atg5 or Atg16L promoter was point mutated from GTAAAAAT into GTGGAAGT, or from GTGAACAT into GTAGACGT to obtain mutant reporter plasmids. pcDNA‐p300 and pcDNA‐FOXO3 plasmids were obtained from Addgene. All experiments were performed three times.
2.5. Protein extraction and Western blotting
Protein lysates were obtained according to the procedures previously described (Tang et al., 2016). Equal amounts of protein were loaded for SDS‐PAGE. After membranes were blocked with 5% non‐fat milk, primary antibodies and appropriate horseradish peroxidase (HRP)‐conjugated secondary antibodies were applied. Protein bands were visualized using the enhanced chemiluminescence system (Amersham Pharmacia Biotech, Japan). All experiments were performed three times. Protein levels were normalized as a ratio to β‐actin after calculating their grayscale intensity values and presented as a relative fold‐change to the control group, and quantification was achieved by Image‐Pro Plus software according to three‐repeated assays. The primary antibodies used are listed as follows: ATM (s1981) (1:1000, Abcam), ATM (1:1000, Abcam), KAP (s824) (1:2000, Abcam), FLOT1 (1:2000, Abcam), FOXO3 (1:1000, Abcam), p300 (1:2000, Abcam), γH2AX (1:1000, Cell Signaling Technology, CST), LC3B (1:1000, CST), p62 (1:1000, CST), BNIP3 (1:1000, CST), phospho‐(Ser/Thr) ATM/ATR substrate antibody (1:1000, CST), CD63 (1:1000, Abcam), CD81 (1:1000, CST), TSG101 (1:1000, Abcam), ATP6V1G1 (1:1000, Abcam), Rel‐A (1:1000, CST), Rel B (1:1000, CST), ATG5 (1:1000, CST), ATG16L (1:1000, CST), MMP9 (1:1000, Abcam), IL‐8 (1:1000, CST), β‐Actin (1:1000, CST), Akt (s473) (1:1000, Santa Cruz Biotech), LAMP1 (1:1000, Santa Cruz Biotech), ACP (1:1000, Abcam), GPR64 (1:1000, NOVUS), IGF2 (1:1000, Abcam), TEK (1:1000, Abcam), TLR6 (1:1000, Abcam).
2.6. Immunoprecipitation‐Western blotting (IP‐WB) assay
Co‐IP assay was conducted according to established procedures (Yu et al., 2017). In brief, KU60019‐treated CAF, CAFs/shATM, and their control cells were pretreated with hypoxia before the assay. Whole cell lysates were then mixed with phospho‐(Ser/Thr) ATM/ATR substrate antibody and Protein A/G Magnetic Beads (Selleck, USA) at 4 °C overnight. After washing with lysis buffer, the released protein complex was used for Western blotting analysis using indicated antibodies. All experiments were performed three times.
2.7. Immunofluorescence (IF)
Cells were grown on glass coverslips in 6‐well plates. After the specific inhibitor and hypoxic treatment, cells were fixed with paraformaldehyde, treated with Triton X‐100, blocked with goat serum, and incubated with primary antibody LC3B/p62 (1:100, CST) and FITC/CY3‐labelled secondary antibody (1:200, Abclonal Tech, China), followed by nuclear staining with DAPI. Images were captured from at least ten random vision fields using a Nikon Eclipse 80i microscope, and the average fluorescence intensity per 103 cells was quantified with Image‐Pro Plus software. All experiments were performed three times.
2.8. Breast tissue samples and immunohistochemistry (IHC)
Detailed procedures for IHC have been described previously (Du et al., 2017). Briefly, after dewaxing, dehydration, and blocking endogenous peroxidase and non‐specific binding sites, the tissue sections were incubated with primary antibody against activated ATM (s1981) (1:100, Abcam), followed by the biotinylated secondary antibody, HRP‐labelled avidin‐biotin reagents and HRP substrate solution treatment. After counterstaining with haematoxylin, images were captured using a Nikon Eclipse 80i microscope (Eclipse 80i, Tokyo, Japan). The images were recorded as follows: 0 (positive rate < 25%), 1 (positive rate: 25%–50%), 2 (positive rate: 51%–75%), and 3 (positive rate > 75%). IHC staining intensities (I) were scored as: 0 (negative), 1+ (weak), 2+ (moderate), 3+ (strong). Results were evaluated separately by two pathologists who were blinded to the experiment. All experiments were performed three times.
2.9. Transwell invasion assay
Transwell invasion assay was performed using 8 μm pore chambers coated with Matrigel (1:7.5, Millipore, USA). Cells along with normalized conditioned medium or normalized extracellular vesicles were seeded in the upper chamber and allowed to invade towards the lower chamber. The working concentration of chosen exosomes depended on the physiological ratio of CAFs to cancer cells in tumour, which was maintained between 1 and 10 CAFs per cancer cell (LY & Baddour, 2016). The invaded cells were then stained with purple crystal dye to allow precise counting of invaded cells. All experiments were performed three times.
2.10. miRNA and mRNA microarray analysis
Total RNA was extracted from paired breast NFs and CAFs using Trizol (Invitrogen, USA) according to the manufacturer's instructions. Probe synthesis and hybridization to SurePrint Human miRNA Microarrays were performed on the Agilent 2100 Bioanalyzer Platform (Biotechnology, Shanghai, China). Analysis of microarray data was conducted using GeneSpring GX (Agilent) and online SBC Analysis System based on R language statistics. T‐test analysis comparisons were employed for differential genetic expression analysis between groups.
2.11. Phosphoproteomics analysis and bioinformatics analysis
Paired breast NFs and CAFs were grown under normoxia until about 80% confluency. After treatment under hypoxia for 8 h, cells were collected for SDT lysis and protein extraction. FASP enzymolysis, phosphopeptide enrichment, label‐free analysis by Maxquant and LC‐MS/MS analysis were performed by PTM‐BIO Company (Shanghai, China). Subsequent protein clustering analysis and signalling pathway enrichment analysis were achieved using DAVID online software (Version. 6.8) (Huang et al., 2009a, 2009b).
2.12. Subcellular structure
Cells were treated with hypoxia or inhibitors for around 8 h. After centrifugation, the collected cell sediment was fixed using 4% glutaraldehyde, and was further processed into ultra‐thin slices of 60 nm in thickness, followed by observation for target subcellular structures like autophagosomes, lysosomes, and multi‐vesicle bodies using transmission electron microscope (TEM) (Tecnai G2 Spirit, ThermoFisher Scientific, USA). All experiments were performed three times.
2.13. Exosome isolation, quantification, characterization, and transmission electron microscope (TEM) analysis
Exosomes were isolated by differential ultracentrifugation (Zeng et al., 2018). For exosome collection from FBS‐free medium, FBS‐free fresh medium was added to the plate when CAFs reached 80%–90% confluency and cultured under hypoxia with or without inhibitor for 12 h, and CM was then collected. For exosome collection from patient plasma, 7 ml of venous blood from each participant were collected. CM or plasma was centrifuged at 300 × g for 10 min, 3000 × g for 20 min and 10 000 × g for 30 min at 4°C to remove cellular debris and intracellular organelles. Further ultracentrifugation was set at 100 000 × g for 70 min at 4 °C (XE‐100, Beckman Coulter, USA), and then washed in PBS using the same ultracentrifugation conditions. The pellet was re‐suspended in ∼100 μl of PBS for nanoparticle tracking analysis (NTA), transmission electron microscope (TEM) assays or used in co‐culture with breast cancer cells.
At the designated CM collection time, cell numbers were counted to allow the appropriate correction of volume of CM supplied to exosome isolation. The protein content of isolated exosomes was measured using BCA Kit (Beyotime BioTech, China), and normalized to 1 × 106 cells to show the amount as microgram per 1 × 106 cells. For cell treatment, 2 μg exosomes (equivalent to those collected from 5 × 106 CAFs) were added to 2 × 105 cancer cells.
The morphology of exosomes was visualized by TEM analysis. The isolated exosomes were fixed on grids with 2% glutaraldehyde, and stained with 2% phosphotungstic acid for 10 min at room temperature. Images were obtained using an electron microscope H‐7600 (Hitachi, Tokyo, Japan).
The size distribution and concentration of exosomes were assessed by NTA. Exosomes resuspended in PBS at a concentration of 10 ng/ml were injected into the sample chamber of ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany), and the exosome particles were measured based on Brownian motion and the diffusion coefficient. Data analysis was performed with the corresponding software, ZetaView 8.02.28. All experiments were performed three times.
2.14. Fluorescent labelling and transfer of exosomes
Isolated exosomes were labelled with PKH67 (Thermo Scientific, USA) at a working concentration of 10 mM and incubated for 20 min at 37°C. The labelled exosomes were washed with PBS and centrifuged again at 12000 × g for 70 min at 4°C. Breast cancer cells were incubated with PKH67‐labeled exosomes for 12 h at a concentration of 20 mg/ml in a 24‐well format, and internalized exosomes in breast cancer cells were observed by confocal microscopy.
2.15. In vitro kinase assay
pcDNA3‐Flag‐ATM, pcDNA3‐Flag‐BNIP3 WT or pcDNA3‐Flag‐S135A mutant were transfected into 293T cells using Lipofectamine 3000. 293T cells transfected with pcDNA3‐Flag‐BNIP3 WT or mutant were cultured in normoxia, while 293T cells with pcDNA3‐Flag‐ATM were cultured in normoxia or hypoxia and treated with or without 5 μM KU60019, respectively. All experiments were performed three times.
Flag‐ATM and Flag‐BNIP3 (WT and S135A mutant) were immunopurified from whole cell lysates with Anti‐FLAG® M2 Magnetic Beads (M8823, Sigma) (Zhang et al., 2014). Kinase reactions were carried out using the system including purified ATM and purified wild type or mutant BNIP3 in kinase buffer provided by Amplite Universal Fluorimetric Kinase Assay Kit *Red Fluorescence* (AAT Bioquest). ADP products were measured using the Kinase Assay Kit following the manufacturer's protocols, using a NOVOstar microplate reader (BMG LabTech) at Ex540/Em590 for 25 min.
2.16. Lysosomal pH measurement
CAFs and engineered CAFs were pre‐treated with hypoxia or inhibitors for a designated time, then incubated with 100 μg/ml of Lysosensor yellow‐blue Dextran for 12 h. The cells were washed twice with PBS and re‐suspended using high‐K+ solutions (containing 1 mM MgCl2, 5 mM CaCl2, 20 mM NaCl, 100 mM KCl, 5 mM potassium citrate and 5 mM HEPES). Lysosensor fluorescence emission was measured at 460 nm and 540 nm and the ratio was converted to pH based on a standard curve previously established. All experiments were performed three times.
2.17. Orthotopic xenografts and metastasis assay
Animal experiments were authorized by The Animal Care Ethics Committees of Chongqing Medical University. A bioluminescent MAD/MB‐231/Luc cell line was generated with a vector encoding a luciferase gene. Four‐week‐old female nude mice (n = 5/group) were given subcutaneous injection of engineered CAFs (1 × 106) mixed with MDA‐MB‐231 breast cancer cells (1 × 106). Tumour size was assessed every 3 days. At the end of animal experiments, mouse lung was prepared for haematoxylin and eosin (H&E) staining and IF staining of luciferase protein for a subsequent blinded evaluation of metastatic foci. All experiments were performed three times.
2.18. Statistical analysis
All experiments were performed three times in parallel and data were presented as Mean ± SD. Statistical analyses were performed using GraphPad Prism 7.00. For data comparison within two experimental groups, the non‐parametric Mann‐Whitney U test was used to evaluate the differences of the mean ranks. For data comparison within multiple groups, Kruskal‐Wallis test was firstly performed to assess the overall difference among groups. If differences existed, multiple pairwise comparisons of the means were performed by Mann‐Whitney U test. Statistical difference was considered when P ≤ 0.05
3. RESULTS
3.1. Oxidized ATM in breast CAFs promotes breast cancer cell invasion via enhancing exosome release
We previously found aberrant activation of DNA damage‐independent ATM signalling in breast CAFs (Peng et al., 2013). The expression of ATM was further evaluated in paired primary NFs and CAFs isolated from breast cancer patients who had not received any chemotherapy. These primary NFs and CAFs displayed spindle‐like morphology (Peng et al., 2013) (data not shown) and the CAFs expressed the well‐established biomarker alpha‐SMA (α‐SMA) (Hanley et al., 2018) (Figure 1a). As expected, the expression of ATM in CAFs was higher than that in paired NFs (Figure 1a, Figure S1A), and phosphorylated ATM (s1981), which is often used as a marker of activated ATM (Lang et al., 2018; Sun et al., 2019), also exhibited a higher level in CAFs than in NFs (Figure 1a). The expression of activated ATM (s1981) in stromal CAFs was further confirmed by immunohistochemistry in tumour tissues. There was negative or weak staining of ATM (s1981) in normal breast stromal fibroblasts (N = 35), while a moderate intensity of ATM (s1981) staining was observed in invasive ductal carcinoma (IDC) without metastasis (N = 53) and a strong ATM (s1981) staining in IDC with metastasis (Figure 1b‐c, N = 21), indicating a positive correlation between stromal activated ATM levels and metastatic breast cancer. Recently, it has been reported that ATM activation can be induced by oxidized stress (OS), which is independent of DNA double‐strand breaks (DSBs) (Bencokova et al., 2009). Therefore, isolated CAFs were then treated with hypoxia, which is a common feature of solid tumour tissues. As expected, ATM (s1981) in CAFs was obviously increased by hypoxia in a time‐dependent manner (Figure 1d, left panels). Activated ATM (ATM s1981) was induced by hypoxia after 8 h without an increase in γH2AX (s139) or KAP (s824), two known markers of DNA DSBs. DNA damage was increased by hypoxia from around 12 to 36 h. CAFs were therefore cultured in hypoxia for around 8–10 h to avoid DSBs. To further confirm that DNA damage‐independent oxidized ATM can be induced by hypoxia, ion irradiation (IR, which leads to DNA damage) was used to treat CAFs as a positive control. Hypoxia treatment for 8–10 h increased oxidized ATM levels but failed to enhance IR‐induced ATM (s1981), γH2AX (s139), and KAP (s824) levels (Figure 1d, right panels). There was no significant change in ATM RNA expression between hypoxia‐ or IR‐treated and control CAFs (Figure S1B). Taken together, hypoxia treatment can efficiently stimulate the activation of DNA damage‐independent ATM (oxidized ATM) in breast CAFs and oxidized ATM activation in stromal CAFs potentially correlates with invasion and metastasis of breast cancer.
FIGURE 1.
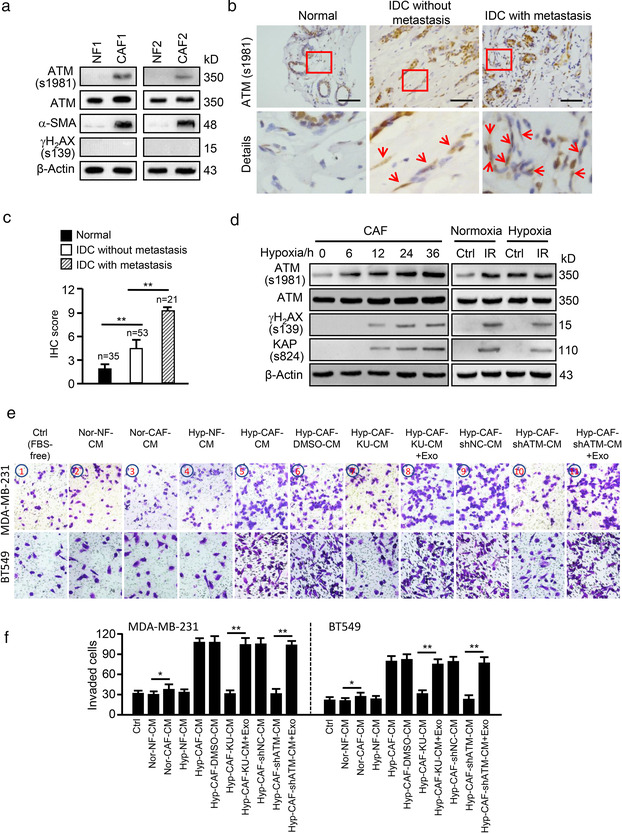
Hypoxia‐mediated oxidized ATM in breast CAFs promotes breast cancer cell invasion. (a) Protein levels of ATM, ATM (s1981), α‐SAM and γH2AX (s139) were detected by western blotting in paired primary NFs and CAFs from two patients. (b) Representative images of activated ATM (oxidized ATM) proteins in stromal fibroblasts of clinical breast tumour tissues (Scale bar, 200 μm; Red arrow, oxidized ATM). (c) Quantification of oxidized ATM in stromal fibroblasts of BC tissues. (d) Western blotting to determine activated ATM, total ATM, γH2AX (s139) and KAP (s824) protein levels in CAFs under hypoxia treatment for the indicated hours or in CAFs exposed to 0.5 Gy ray irradiation (IR) as a control group. (e) MDA‐MB‐231 and BT549 cells were co‐cultured with the CM from normoxic or hypoxic NFs and CAFs; or CM from hypoxic CAFs treated with or without KU60019 (KU:KU60019), or CM from ATM wild type (shNC) or silenced (shATM) hypoxic CAFs. Exosomes isolated from hypoxic CAF (Hyp‐CAF‐Exo: Exo) were added to the co‐culture system, and cell invasion potentials were evaluated by Transwell assay. Ctrl: Tumour cell + FBS‐free medium. (f) Quantification of invaded cells in (e) (*P < 0.05; **P < 0.01.)
To further explore the effects of oxidized ATM in hypoxic CAFs on breast cancer cell invasion, a co‐culture system was employed. Firstly, we found that tumour cells cultured with conditioned medium (CM) from normoxic CAFs (Nor‐CAF‐CM) had a promoting effect on tumour cell invasion of MDA‐MB‐231 and BT549 breast cancer cells compared with CM from normoxic NFs (Nor‐NF‐CM) (Figure S1C‐S1D, 3rd vs. 2nd image/histogram on the left). Then, we wondered whether CAFs in hypoxia have increased ability to induce invasion of breast cancer cells. Thus, we evaluated the invasion abilities of tumour cell cultured with CM derived from hypoxic CAFs (Hyp‐CAF‐CM) and hypoxic NFs (Hyp‐NF‐CM). Interestingly, CM from hypoxic CAFs (Hyp‐CAF‐CM) increased cell invasion of breast cancer cells to a greater extent than the CM from normoxic CAFs (Nor‐CAF‐CM) (Figure S1C‐S1D, 5th vs. 3rd image/histogram on the left). However, the CM from hypoxic NFs (Hyp‐NF‐CM) had similar effects on tumour cell invasion in comparison with the CM from normoxic NFs (Nor‐NF‐CM) (Figure S1C‐S1D, 4th vs. 2nd image/histogram on the left), suggesting that CAFs under hypoxia have increased ability to promote breast cancer cell invasion. Next, the role of oxidized ATM in CAFs on tumour cell invasion was assessed. Treatment with KU60019 (Figure S1E), an ATM‐specific inhibitor (Jachimowicz et al., 2019; Sun et al., 2019), to inhibit activation of oxidized ATM, or lentivirus‐mediated ATM shRNAs (Figure S1F) to silence ATM expression in hypoxic CAFs, obviously attenuated the invasion abilities of breast cancer cells compared with their controls (Figure S1C‐S1D, 7th vs. 6th image/histogram in the middle, and 9th vs. 8th image/histogram on the right), revealing that oxidized ATM is necessary for CAF‐CM in promoting tumour cell invasion.
As there are secretory soluble factors and exosomes in CM, we explored whether these secreted soluble factors or exosomes in CM from hypoxic CAFs played a critical role in oxidized ATM‐mediated tumour cell invasion. Thus, we separated exosomes and secreted soluble factors (CMExo−, or called exosome‐free CM) from CM via ultracentrifugation. The isolated exosomes of CAFs were assessed via nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM), and Western blotting. As shown in Figure S2A‐S2C, the exosomes were approximately 100 nm in diameter (Figure S2A) with a typical cup‐shaped morphology (Figure S2B), and expressed exosome‐specific protein markers CD63, CD81, TSG101 and flotillin‐1 (FLOT1) rather than GRP94 (a negative marker of exosomes) (Figure S2C). We cultured exosome‐free CM (CMExo−) or exosomes (Exo) with tumour cells separately, and found that only 12%–22% more cells invaded in the group with CMExo− from normoxic CAFs (Nor‐CAF‐CMExo−) than the group with CMExo− from normoxic NFs (Nor‐NF‐CMExo−) (P > 0.05) (Figure S2D‐S2Ea/b, 3rd vs. 1st image/histogram on the left), while exosomes isolated from normoxic CAFs (Nor‐CAF‐Exo) had more ability to increase tumour cell invasion by about 23%–33% in comparison to the exosomes or exosome‐free CM (CMExo−) from normoxic NFs (P < 0.05) (Figure S2D‐S2Ea/b, 4th vs. 2nd or 1st image/histogram on the left). Furthermore, we compared the effects of exosomes and secretory soluble factors of CAF's CM on tumour cell invasion under hypoxic conditions. We found that secreted soluble factors in CM (CMExo−) from hypoxic CAFs (Hyp‐CAF‐CMExo−) increased the invasive ability of tumour cell about 1.68‐2.05 times compared with CMExo− from hypoxic NFs (Hyp‐NF‐CMExo−, P < 0.05) or normoxic NFs (Nor‐NF‐CMExo−, P < 0.05) (Figure S2D‐S2Ea/b, 7th vs. 5th or 1st image/histogram), while exosomes released from hypoxic CAFs (Hyp‐CAF‐Exo) are about 2.0‐2.46 times more potent on inducing cancer cell invasion than the CMExo− or exosomes from hypoxic NFs (Hyp‐NF‐Exo, P < 0.01) or normoxic NFs (Nor‐NF‐Exo, P < 0.01) (Figure S2D‐S2Ea/b, 8th vs. 6th or 4th image/histogram in the middle). These data revealed that exosomes derived from hypoxic or normoxic CAFs play more prominent role in promoting breast cancer cell invasion than secreted soluble factors in CM. To understand the role of oxidized ATM in exosome‐mediated tumour cell invasion under hypoxia, KU60019 and shRNA strategies were employed. Upon inhibiting activated oxidized ATM by KU60019 or silencing ATM in hypoxic CAFs, secreted soluble factors in CM (Hyp‐CAF Ku‐CMExo−, Hyp‐CAF shATM‐CMExo−, P < 0.05) reduced the invasion ability of tumour cells by about 37%–39.5% in comparison with their controls (Hyp‐CAF DMSO‐CMExo−, Hyp‐CAF shNC‐CMExo−) (Figure S2D‐S2Ea/b, 11th vs. 9th, and 15th vs. 13th image/histogram). More importantly, exosomes released from these ATM‐inhibited hypoxic CAFs (Hyp‐CAF Ku‐Exo, Hyp‐CAF shATM‐Exo) decreased tumour cell invasive ability by 47.7%–50.8% compared with corresponding controls (Hyp‐CAF DMSO‐Exo, Hyp‐CAF shNC‐Exo, P < 0.01) (Figure S2D‐S2E, 12th vs. 10th, and 16th vs. 14th image/histogram on the right), suggesting that oxidized ATM in CAFs can promote tumour cell invasion mainly via exosome release. In addition, pharmacologically blocking exosome release from hypoxic CAFs (Figure S2F) by GW4869, a non‐competitive N‐SMase inhibitor (Ge et al., 2021) with non‐detectable effect on the viability of CAFs (Figure S2G), reduced the effect of Nor‐CAF‐CM on tumour cell invasion compared with Nor‐NF‐CM (Figure S2H‐S2I, 1st vs. 2nd or 3rd, 2nd or 3rd vs. 4th image/histogram on the left). Especially, the reduced effect of hypoxic CAFs‐derived CM caused by GW4869 treatment (Hyp‐CAF‐GW‐CM) on tumour cell invasion was more obvious compared with its control CM (Hyp‐CAF‐DMSO‐CM or Hyp‐CAF‐CM) (Figure S2H‐S2I, 5th or 6th vs. 7th image/histogram on the right).
Next, we assessed exosome content and found that normoxic CAFs could release more exosomes than normoxic NFs, and hypoxia treatment significantly further increased exosome release from CAFs compared with normoxic CAFs (Figure S3A). Inactivation of oxidized ATM by KU60019 or shRNA effectively reduced the amount of released exosomes (Figure S3B), suggesting hypoxia‐stimulated activation of oxidized ATM plays a crucial role in CAF exosome release. To further confirm the role of oxidized ATM‐mediated exosome release from hypoxic CAFs in promoting tumour cell invasion, a rescue assay was carried out. As shown in Figure 1e‐f (7th and 8th, and 10th and 11th image/histogram), addition of exosomes from hypoxic CAFs (labelled as Hyp‐CAF‐Exo) to the co‐culture system rescued the invasion potentials of MDA‐MB‐231 and BT549 cells co‐cultured with CM from KU60019‐treated CAFs or ATM‐silenced CAFs. To determine whether CAF‐derived exosomes are internalized by breast cancer cells, exosomes purified from CAF‐CM were labelled with PKH67 and incubated with breast cancer cells for 12 h. We observed that PKH67‐labeled exosomes were internalized by MDA‐MB‐231 and BT549 cells (Figure S3C). Taken together, these data indicate that oxidized ATM facilitates exosome release from hypoxic breast CAFs to enhance invasion of breast cancer cells.
3.2. Oxidized ATM stimulates autophagy in hypoxic CAFs
ATM exerts its physiological functions by phosphorylating target proteins at specific sites (Shiloh & Ziv, 2013). To identify the potential target proteins of oxidized ATM in hypoxic CAFs, phosphoproteomics analysis of hypoxic NFs and CAFs was conducted using high‐resolution LC‐MS/MS analysis (Figure 2a). A total of 3,057 phosphosites in 1,186 phosphoproteins were successfully identified. A quantification ratio of more than 1.5 was considered as upregulation and less than 0.67 was considered as downregulation. A total of 222 differentially‐regulated phosphoproteins between hypoxic CAFs and NFs were identified (Supplementary list 1). Among these proteins, 121 phosphosites in 47 phosphoproteins were predicted to be the direct target of oxidized ATM on the basis of a consensus ATM substrate motif (SQ/TQ) (Figure 2b). All the changed phosphoproteins as direct and indirect targets of oxidized ATM are listed in Supplementary list 2. Some direct target proteins of oxidized ATM are listed in Figure 2c. To reveal the role of these phosphoproteins of oxidized ATM in biological processes and corresponding signalling pathways, we conducted bioinformatics analyses. As shown in Figure 2d, the phosphoproteins in hypoxic CAFs were found to be involved in various biological processes including autophagy, response to oxygen levels, vesicle transportation, and extracellular exosomes. After reviewing literatures on these biological processes, autophagy signalling attracted our attention.
FIGURE 2.
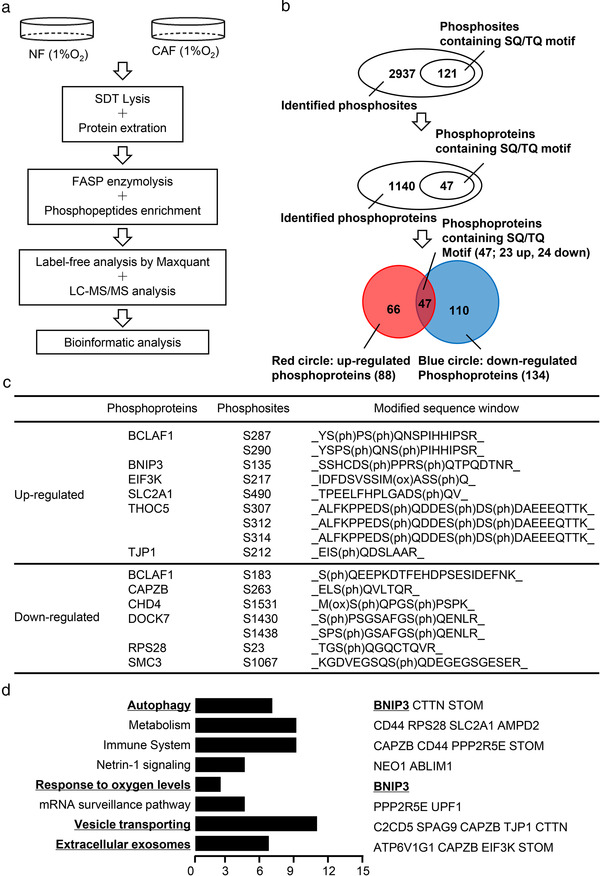
Analysis of oxidized ATM‐mediated biological processes in hypoxic CAFs. (a) Schematic diagram of phosphoproteomics procedures between paired NFs and CAFs under hypoxia treatment. (b) Phosphoproteomics analysis to identify phosphoproteins and phosphosites in hypoxic CAFs. (c) Representative identified phosphoproteins containing SQ/TQ motif in hypoxic CAFs are shown. (d) Biological processes enrichment analysis of identified phosphoproteins with SQ/TQ motif dysregulated in hypoxic CAFs. Part of the representative proteins associated with the indicated biological process are shown in the right panel
In order to define the relationship between oxidized ATM and autophagy in hypoxic breast CAFs, LC3B II and p62, two protein markers of typical autophagy, were detected in normoxic and hypoxic CAFs. In comparison with normoxic CAFs, higher levels of LC3B II protein expression (Figure 3a) and dot accumulation (Figure 3b (white arrow)) were observed in the cytoplasm of hypoxic CAFs, which were reduced when oxidized ATM was inhibited by KU60019 or shRNA (Figure 3a, and Figure 3b (white arrow)). In typical autophagy, autophagosomes are fused with lysosomes and lead to p62 degradation. Interestingly, we found that hypoxia treatment resulted in a moderate decrease in p62 protein levels compared with normoxic CAFs, and KU60019 treatment of CAFs caused a modest increase in p62 in hypoxic CAFs (Figure 3a, and Figure 3c (white arrow)), indicating an atypical autophagy in hypoxic CAFs, in which only a proportion of autophagosomes may fuse with lysosomes. In line with these results, more autophagosomes were also observed in hypoxic CAFs than in normoxic CAFs, and KU60019 could substantially reduce them in hypoxic CAFs as observed by TEM (Figure 3d, red arrow). Taken together, these results suggest that oxidized ATM induced by hypoxia treatment stimulates autophagosome accumulation in breast CAFs.
FIGURE 3.
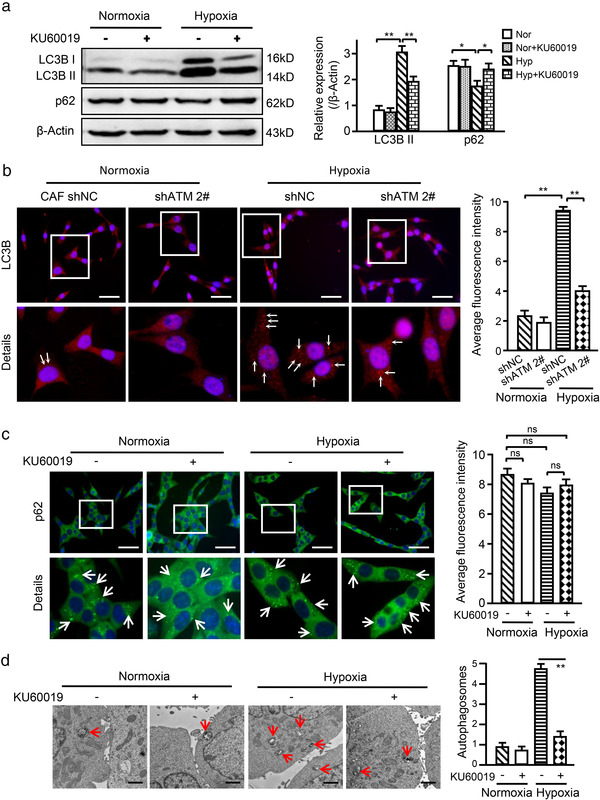
Oxidized ATM stimulates autophagy in CAFs. (a) Western blotting to test autophagy protein markers LC3B II and p62 in normoxic and hypoxic CAFs treated with or without KU60019 (5 μM, 8 h). β‐Actin was used as a loading control. Protein levels normalized as a ratio to β‐actin after grayscale intensity quantification and presented as a fold‐change relative to the control group are shown in the right panel. (b) Immunofluorescence staining to assess the autophagy protein marker LC3B in normoxic and hypoxic CAFs with or without shRNA against ATM, and the relative quantification of LC3B based on the average fluorescence intensity per cell is shown in the right panel (Scale bar, 200 μm; White arrow showing the dot‐like LC3B protein particles). (c) Immunofluorescence staining to assess autophagy protein marker p62 in normoxic and hypoxic CAFs treated with or without KU60019 (5 μM) for 8 h, and the relative quantification of p62 based on the average fluorescence intensity per cell is shown in the right panel (Scale bar, 200 μm; White arrow showing the dot‐like p62 protein particles). (d) Autophagosomes in normoxic and hypoxic CAFs with or without KU60019 treatment (5 μM, 8 h) were observed by transmission electron microscope and the relative quantification of autophagosomes is shown in the right panel (Scale bar, 2 μm; Red arrow showing the typical autophagosomes). (ns, not significant; *P < 0.05; **P < 0.01.)
3.3. Oxidized ATM phosphorylates BNIP3 at serine135 to induce autophagy and exosome release from CAFs
To understand how oxidized ATM regulates autophagy and exosome release of hypoxic breast CAFs, we further analysed our phosphoproteomic data. Phosphorylated BCL2 interacting protein 3 (BNIP3) caught our attention. BNIP3 was found to be involved in autophagy signalling and response to oxygen levels (Figure 2d), suggesting that BNIP3 may play a role in hypoxia‐induced autophagy in hypoxic CAFs. BNIP3 was phosphorylated by oxidized ATM at serine 135 (s135), confirmed by mass spectrometry analysis (Figure 4a). Also, the levels of p‐BNIP3 were higher in CAFs compared with NFs, and hypoxia further enhanced the levels of p‐BNIP3 in CAFs (Figure S4A). To experimentally determine whether oxidized ATM can phosphorylate BNIP3 at s135, co‐immunoprecipitation (CO‐IP) assay was firstly performed using phospho‐(Ser/Thr) ATM/ATR substrate antibody. Using western blotting with a BNIP antibody, decreased phosphorylated BNIP3 was observed in the immunoprecipitates in response to KU60019 treatment or in ATM‐knockdown hypoxic CAFs (Figure S4B). In addition, using a BNIP3 antibody in the CO‐IP assay and phosphorylated BNIP3 in western blotting, we again detected decreased p‐BNIP3 in these co‐immunoprecipitates from KU60019‐treated or ATM‐silenced hypoxic CAFs (Figure 4b), suggesting an activated ATM‐dependent phosphorylation of BNIP3 in hypoxic CAFs. To further evaluate the kinase activity of oxidized ATM in phosphorylating BNIP3 at s135, we mutated the s135 of BNIP3 from S (serine) to A (alanine) to acquire a hypophosphorylated BNIP3 (BNIP3 S135A mutant), or mutated s135 of BNIP3 from S (serine) to D (aspartic acid) to produce a hyperphosphorylated BNIP3 (BNIP3 S135D mutant). HEK293T cells were transfected with pcDNA3‐Flag‐ATM or pcDNA3‐Flag‐BNIP3 WT (WT) or pcDNA3‐Flag‐BNIP3 S135A mutant (Mut) construct. In vitro kinase activity assays based on ATP depletion and ADP formation showed that ADP formation was increased in the mixture of activated ATM and wild type BNIP3; BINP3 S135A mutant or KU60019 treatment resulted in a significantly decreased ADP amount (Figure 4c). Correspondingly, phospho‐BNIP3 was detected with wild type ATM and wild type BNIP3 in HEK293T cells under hypoxia treatment (Figure 4d). These findings support that oxidized ATM phosphorylates BNIP3 at s135 in hypoxic CAFs.
FIGURE 4.
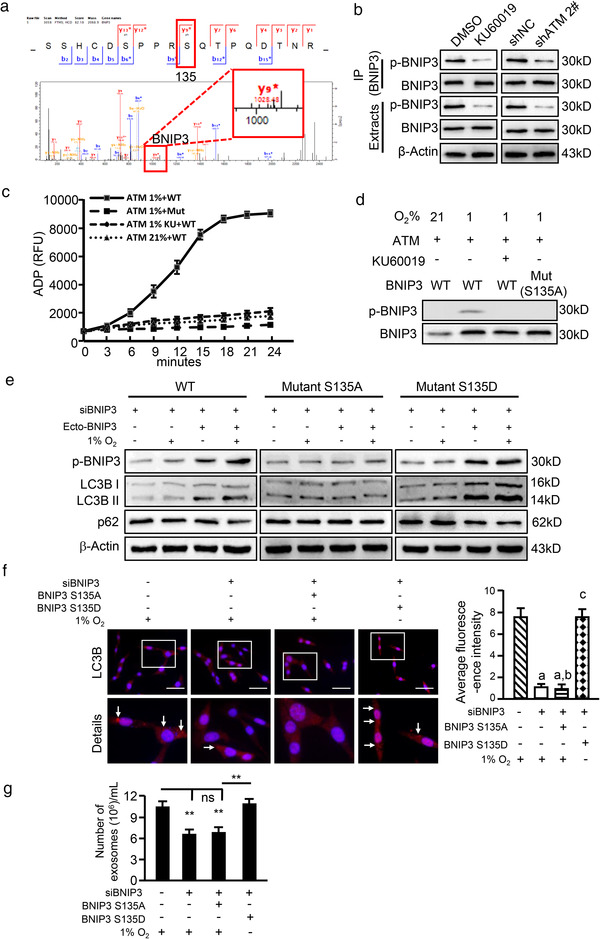
Oxidized ATM phosphorylates BNIP3 at s135 to induce autophagy and exosome release from CAFs. (a) The spectrum of phosphorylated BNIP3 protein identified by LC/MS‐MS. (b) Hypoxic CAFs were treated with or without KU60019, or ATM was knocked down by specific shRNA in CAFs. Cell extracts were immunoprecipitated with BNIP3 antibody, followed by immunoblotting with specific antibody against phosphorylated BNIP3, and the phosphorylated BNIP3 in the immunoprecipitates is shown. (c) In vitro kinase assay was performed using purified ATM kinase mixed with purified WT or mutant BNIP3. ADP products were detected by Fluorimetric Protein Kinase Assay (WT, BNIP3 WT; Mut, BNIP3 S135A mutant; KU, oxidized ATM inhibitor KU60019). (d) 293T cells with pcDNA3‐Flag‐BNIP3 WT and BNIP3 S135A mutant were cultured under normoxia or hypoxia and treated with or without KU60019 as shown, the phosphorylated BNIP3 in these cells was measured by western blotting. (e) Endogenous BNIP3 was knocked down in immortalized CAFs, which were then transfected with ectopic WT, mutant BNIP3 (S135A, S135D). The cells were cultured in normoxia and hypoxia, phosphorylated BNIP3 and autophagy markers were evaluated by western blotting. (f) CAFs were cultured in hypoxia, the autophagy‐associated marker LC3B in engineered hypoxic CAFs was viewed by immunofluorescence staining, and the relative quantification of LC3B based on the average fluorescence intensity per cell is shown in the right panel (Scale bar, 200 μm; white arrow, accumulation of dot‐like LC3B protein particles; a, P < 0.01, CAFs with silenced BNIP3 or endogenous BNIP3‐silenced CAFs transfected with BNIP3 S135A mutant vs. CAFs with control vector; b, P > 0.05, endogenous BNIP3‐silenced CAFs transfected with BNIP3 S135A mutant vs. BNIP3‐silenced CAFs; c, P < 0.01, CAFs with BNIP3 S135D mutant vs. BNIP3‐silenced CAFs). (G) Exosome concentrations from the indicated engineered hypoxic CAFs described in Figure 4f were determined by nanoparticle tracking analysis (NTA). (**P < 0.01; ns, not significant)
Next, we examined whether oxidized ATM‐mediated phosphorylation of BNIP3 regulates enhanced autophagy and exosome release. Knockdown of endogenous BNIP3 in hypoxic CAFs caused a significant decrease in autophagy‐associated marker LC3B II, but only a slight increase in p62 (Figure S4C). Furthermore, transfection of BNIP3 S135D mutant into BNIP3‐silenced hypoxic CAFs resulted in a remarkably enhanced LC3B II, but mild or slight decrease in p62 compared with the control CAFs (Figure S4D). In addition, we found that BNIP3‐silenced CAFs stably‐transfected with ectopic BNIP3 (WT) under hypoxia had a significantly enhanced LC3B II and weakly reduced p62 in comparison with the control CAFs (Figure 4e, left panel); BNIP3 S135A mutant failed to impact autophagy in response to hypoxia stimulation (Figure 4e, middle panel). In contrast, BNIP3 S135D mutant (hyperphosphorylated BNIP3) led to a significantly increased LC3B II and slightly reduced p62, independent of hypoxia stimulation (Figure 4e, right panel). In agreement with these findings, knockdown of endogenous BNIP3 or transfection of ectopic BNIP3 S135A mutant into the endogenous BNIP3‐knockdown CAFs significantly decreased LC3B staining dots under hypoxia; in contrast, ectopic BNIP3 S135D mutant in endogenous BNIP3‐knockdown CAFs notably rescued LC3B staining dots under normoxia conditions (Figure 4f, white arrow). These results suggest that phosphorylated BNIP3 at s135 can induce autophagy.
Next, we asked whether phosphorylation of BNIP3 at s135 is responsible for exosome release. BNIP3‐engineered CAFs were treated with hypoxia and exosomes were isolated for further analysis. As shown in Figure 4g, knockdown of endogenous BNIP3 or stable transfection of ectopic BNIP3 S135A mutant into the BNIP3‐knockdown hypoxic CAFs significantly reduced the number of released exosomes. However, stable transfection of BNIP3 S135D mutant into BNIP3‐knockdown normoxic CAFs could rescue the ability of CAFs in releasing exosomes. Together, these data suggest that phosphorylation of BNIP3 at s135 plays an essential role in exosome release in hypoxic CAFs. Meanwhile, we found that knockdown of BNIP3 or stable transfection of BNIP3 S135A mutant into BNIP3‐silenced hypoxic CAFs abolished the pro‐invasion potential of exosomes derived from these hypoxic CAFs for breast cancer cells; whereas transfection of BNIP3 S135D into BNIP3‐knockdown normoxic CAFs partially rescued the pro‐invasion ability of CAF‐derived exosomes (Figure S4E). Taken together, these data support that oxidized ATM phosphorylates BNIP3 at s135 to induce autophagy and exosome release from hypoxic CAFs.
3.4. Phosphorylated BNIP3 upregulates Atg5 and Atg16L to induce autophagy and exosome release
Next, we asked how phosphorylated BNIP3 is involved in autophagy and exosome release. By analysing our previous mRNA sequence data from primary CAFs and NFs, we found sets of upregulated autophagy‐associated genes (e.g., Atg5, Atg7 and Atg16L) (Figure 5a), which are necessary for autophagosome and exosome formation, and several forehead box transcription factors (e.g., FOXO3, FOXH1, FOXB2) (data not shown) in hypoxic CAFs. Interestingly, Atg5 and Atg16L were increased in hypoxic CAFs compared with normoxic CAFs, and loss of ATM decreased Atg5 and Atg16L expression under hypoxia (Figure 5b). Transfection of wild type BNIP3 or BNIP3 S135D mutant, but not BNIP3 S135A mutant, prevented the decrease in Atg5 and Atg16L caused by endogenous BNIP3 knockdown in hypoxic CAFs; while Atg7 and Atg12 remained unchanged in these CAFs (Figure 5b), indicating that oxidized ATM enhanced Atg5 and Atg16L via p‐BNIP3 (s135) in hypoxic CAFs. Using informatics analysis, we found that there is a binding site of the transcription factor FOXO3 in the promoters of Atg5 or Atg16L (Figure 5c‐d). Notably, it has been reported that FOXO3 regulates Atg5 and Atg16L in adult neural stem cells (Audesse et al., 2019). Hypoxia stimulation or ectopic FOXO3 overexpression obviously increased the luciferase activities driven by the Atg5 or Atg16L promoter, whereas FOXO3 silencing led to a notable decrease in Atg5 and Atg16L reporter activities (Figure 5e), suggesting FOXO3 is a potential transcription factor for Atg5 and Atg16L expression. Phospho‐BNIP3 was reported to interact with p300 (Thompson et al., 2015) and p300 was also predicted to bind to FOXO3. To validate the predicted interaction between phospho‐BNIP3 with p300 and FOXO3, IP‐Western blotting was employed. As shown in Figure S5A, BNIP3 S135D mutant had a stronger ability to interact with p300 and FOXO3, and hypoxia‐stimulated phosphorylation of wild type BNIP3 (BNIP3 WT) rather than BNIP3 S135A mutant enhanced the interaction among p300, p‐BNIP3 and FOXO3. Furthermore, we introduced p300, FOXO3 and BNIP3 (WT, S135A or S135D) into 293T cells, and found that BNIP3 S135D or WT BNIP3 and p300 facilitated the increase of Atg5 and Atg16L transcriptional levels by FOXO3 under hypoxia (Figure S5B). To evaluate whether the increased Atg5 and Atg16L promote autophagy, Atg5 and Atg16L were knocked down in breast CAFs (Figure S5C‐S5F). Hypoxia exposure increased autophagy‐associated marker LC3B II and slightly decreased p62 levels in CAFs with Atg5 and Atg16L, and the two markers remained unaltered in Atg5 or Atg16L‐silenced hypoxic CAFs compared with their corresponding normoxic CAFs (Figure 5f). These data indicate that Atg5 and Atg16L are essential for oxidized ATM‐p‐BNIP3 signalling in triggering autophagy in hypoxic CAFs. Accordingly, phospho‐BNIP3/p300/FOXO3‐mediated Atg5 and Atg16L expression promoted exosome release in hypoxic CAFs, suppression of autophagy by silencing Atg5 or Atg16L led to a remarkable reduction of exosome amount (Figure 5g) and correspondingly reduced cancer cell invasion (Figure S5G). Together, these data support that the phospho‐BNIP3/p300/FOXO3 complex positively regulates Atg5 and Atg16L expression involved in autophagy and exosome release of hypoxic CAFs, which promote breast cancer cell invasion.
FIGURE 5.
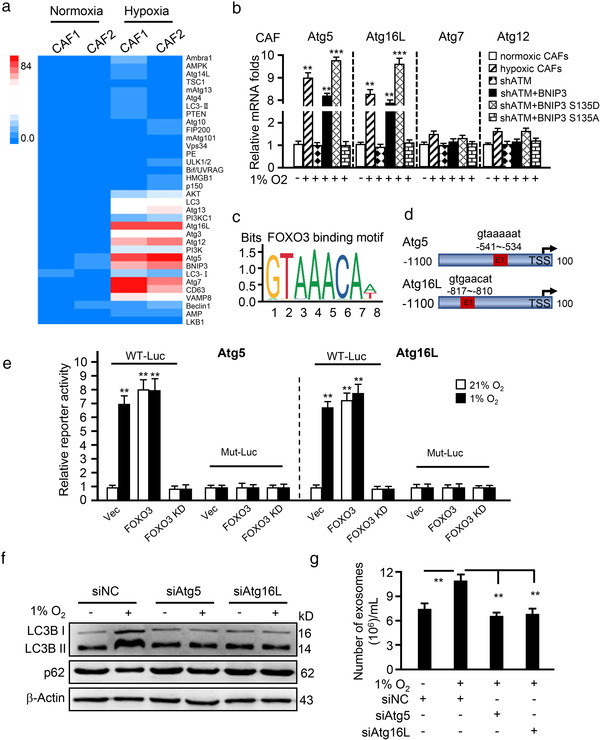
Phosphorylated BNIP3 upregulates Atg5 and Atg16L to induce autophagy and exosome release. (a) Geometric mean‐centred, hierarchical cluster heat‐map from microarray data of normoxic CAFs and hypoxic CAFs, and targets of interest are shown. (b) qRT‐PCR to test Atg5, Atg16L, Atg7 and Atg12 expression in normoxic CAFs, hypoxic CAFs, endogenous ATM‐silenced CAFs (shATM), and endogenous ATM‐silenced CAFs transfected with ectopic wild type BNIP3 (shATM+BNIP3) or BNIP3 mutant (S135D mutant, shATM+BNIP3 S135D; or S135A mutant, shATM+BNIP3 S135A). (c, d) Schematic diagram shows the FOXO3 binding motif (c) and binding sites in ATG5 and ATG16L promoter (d). (e) The Atg5 or Atg16L wild type LUC reporter (with FOXO3 binding site, WT‐Luc) or mutant LUC reporter (FOXO3 binding site being mutated, Mut‐Luc) were transfected into CAFs (control CAFs), engineered CAFs with or without FOXO3 and cultured at indicated condition for 30 h, the luciferase activities of Atg5 and Atg16L were measured. **P < 0.01: compared with vector (Vec) group in normoxia (21%). (f) Western blotting to determine autophagy protein markers LC3B II and p62 in normoxia and hypoxic CAFs with or without silenced‐Atg5 or Atg16L. (g) Concentration of exosomes isolated from normoxic or hypoxic CAFs with silenced‐Atg5 or Atg16L was evaluated by NTA assay. (**P < 0.01; ***P < 0.001.)
3.5. Oxidized ATM impairs lysosomal acidification
The above findings suggest that oxidized ATM promotes non‐canonical autophagy and exosome release in CAFs, and raise the question of whether increased autophagosomes fuel exosome release in hypoxic CAFs. Thus, we evaluated whether autophagic flux changes might contribute to autophagosome accumulation in hypoxic CAFs. Hypoxic CAFs were treated with KU60019 and/or bafilomycin A1 (BafA1), an inhibitor of lysosomal fusion with autophagosomes that degrades the autophagy contents (Mauthe et al., 2018). Increased LC3B II was found in response to BafA1 treatment (Figure 6a, left panel), and KU60019 attenuated the BafA1‐mediated increase in LC3B II (Figure 6a, right panel). p62 remained unchanged under BafA1 or combined BafA1 and KU0019 treatment, indicating that there are dysfunctional lysosomes in CAFs caused by oxidized ATM. To further validate this finding, lysosomal pH and lysosome‐associated membrane protein 1 (LAMP1) as well as acid phosphatase 2 (ACP2) (Arrant et al., 2018) levels in CAFs were measured. Lysosomal pH in hypoxic CAFs was slightly higher at around 4.8 than that in control CAFs at around 4.0; however, inhibiting oxidized ATM with KU60019 attenuated the increase in lysosomal pH levels (Figure 6b). Furthermore, lower levels of lysosome‐associated LAMP1 and ACP2 were detected in hypoxic CAFs compared with normoxic CAFs, and inhibition of oxidized ATM by KU60019 partially rescued LAMP1 and ACP2 levels in hypoxic CAFs (Figure 6c). Correspondingly, ATM silencing decreased lysosomal pH levels to maintain an acidic environment (Figure 6d) and enhance LAMP1 and ACP2 expression (Figure 6e). These findings demonstrate that oxidized ATM impairs lysosomal acidification and leads to lysosomal dysfunction in hypoxic CAFs.
FIGURE 6.
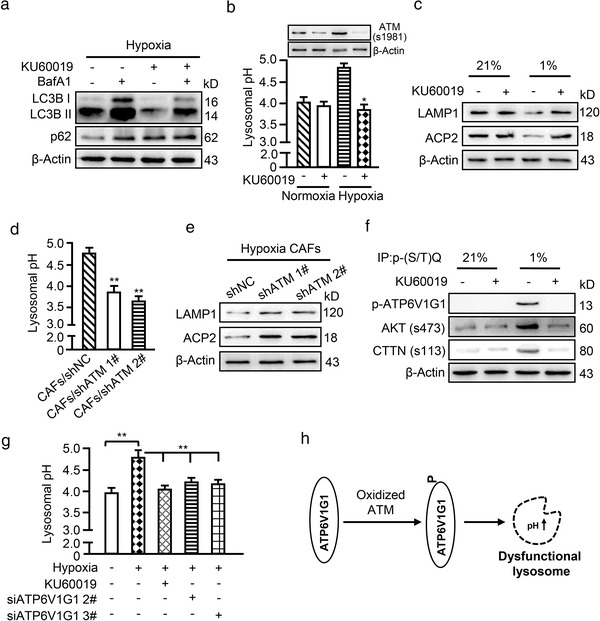
Oxidized ATM impairs lysosomal acidification. (a) Western blotting to test autophagy‐associated protein markers LC3B II and p62 in hypoxic CAFs treated with BafA1 (100 nM) and KU60019 (5 μM) for 8 h, alone or in combination. (b, c) Lysosomal pH (b) and lysosome‐related protein markers LAMP1 and ACP2 (c) were determined for normoxic and hypoxic CAFs with or without KU60019 treatment. The corresponding expression of oxidized ATM (s1981) is shown. (d, e) Lysosomal pH (d) and lysosome‐related protein markers LAMP1 and ACP2 (e) were measured for hypoxic CAFs with or without endogenous ATM. (f) CAFs were treated with or without KU60019 and cell extracts were immunoprecipitated with p‐(S/T)Q antibody, followed by immunoblotting with antibody against anti‐ATP6V1G1, and two known targets of ATM (phosphorylated AKT and phosphorylated CTTN). (g) Lysosomal pH was tested for hypoxic CAFs treated with or without KU60019 and siRNA specifically against ATP6V1G1. (h) Schematic diagram to show oxidized ATM phosphorylates ATP6V1G1 to impair lysosomal acidification. (*P < 0.05; **P < 0.01.)
ATP6V1G1 is an important subunit of proton pump in lysosomes and is responsible for maintenance of lysosomal acidification (Meo‐Evoli et al., 2015). Recently, ATP6V1G1 was reported to be a phosphorylated target of ATM (Kang et al., 2017) and was also identified in our phosphoproteomics (Supplementary list 1). Indeed, using an antibody specifically against the phospho‐ATM/ATR substrates for immunoprecipitations, we detected phosphorylated ATP6V1G1 and two known targets of ATM (phosphorylated AKT and phosphorylated cortactin (CTTN) (Lang et al., 2018)) in the immunoprecipitates of hypoxic CAFs, which were attenuated by KU60019 (Figure 6f). Correspondingly, lysosomal pH was increased under hypoxic conditions and use of KU60019 or silencing ATP6V1G1 (Figure S5H) decreased lysosomal pH of hypoxic CAFs (Figure 6g). Taken together, these data support that oxidized ATM is involved in the impairment of lysosomal acidification and lysosomal dysfunction via phosphorylation of ATP6V1G1 (Figure 6h).
3.6. Oxidized ATM promotes the fusion of autophagosome (AP) and multi‐vesicular body (MVB) in hypoxic CAFs
A previous study showed that autophagosomes can either fuse with lysosomes or with MVBs, thus modulating exosome release (Baixauli et al., 2014). We asked whether the increased exosome release is attributed to oxidized ATM‐induced lysosomal dysfunction. To test this hypothesis, we observed the cellular structures of autophagosomes and MVBs, as well as their fusion in hypoxic CAFs. As shown in Figure 7a, some single autophagosomes or MVBs with typical spherical membrane structure were observed in normoxic CAFs (Figure 7A‐b and 7b) but not normoxic NFs (Figure 7A‐a and 7b). Notably, more multiple autophagic vesicles and MVBs containing proteins or intraluminal vesicles were observed in CAFs treated with hypoxia (Figure 7A‐c/d and 7b). More interestingly, accumulation of MVBs accompanied with autophagosomes occurred near the cellular membrane; in particular, some MVBs were fused with autophagosome (Figure 7a‐e/f). The average diameter of AP and MVB ranged from 200 nm to 400 nm and the fused AP‐MVB was 400 nm to 600 nm in diameter (Figure 7c). These data suggest there is a multitudinous fusion between autophagosomes and MVBs in hypoxic CAFs.
FIGURE 7.
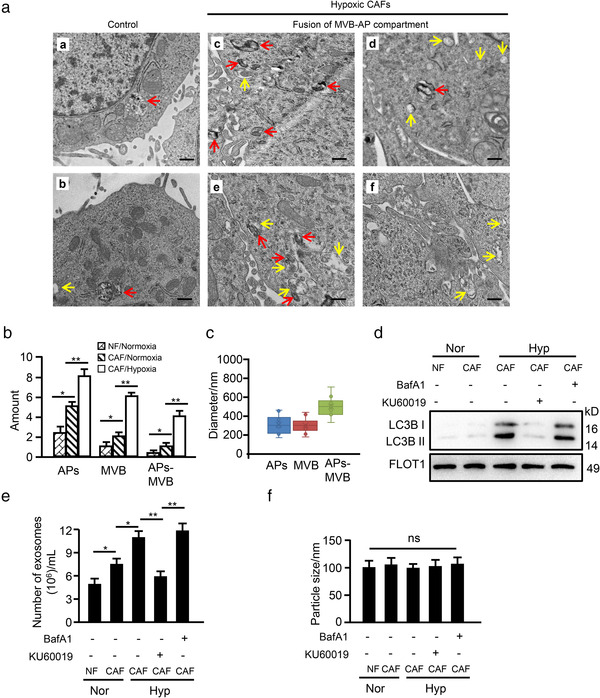
Oxidized ATM promotes autophagosomal fusion with multivesicular bodies (MVB) in hypoxic CAFs. (a) NFs and CAFs were exposed to normoxia (a‐b) or hypoxia (c‐f) for 8 h and ultrastructure was assessed by TEM (Red arrows indicate autophagosomes which are spherical structures with double layer membranes; yellow arrows indicate multivesicular bodies which contain membrane‐bound intraluminal vesicles). (b, c) The amount (b) and size (c) of the autophagosomes and multi‐vesicular bodies derived from hypoxic CAFs and control cells were quantified. (d) Western blotting to evaluate autophagy protein markers LC3B in exosomes derived from control cells and hypoxic CAFs treated with or without BafA1 (100 nM) or KU60019 (5 μM) alone or in combination. (e‐f) Particle concentration (e) and size (f) from the cells in (d) detected by NTA assay. (*P < 0.05; **P < 0.01; ns, not significant)
It has been reported that the AP‐MVB fusion compartment acts as a positive regulator of the release of autophagy‐associated components via exosomes to maintain cellular homeostasis (Wang et al., 2019). To confirm exosomes originating from AP‐MVBs in hypoxic CAFs, we detected autophagy‐associated protein LC3B (a processed membrane protein of autophagosome (Ramkumar et al., 2017)) in the released exosomes by western blotting. As expected, exosomal LC3B from normoxic CAFs was higher than that from normoxic NFs, and hypoxia treatment significantly increased LC3B levels in exosomes from CAFs; inhibition of oxidized ATM by KU60019 notably reduced LC3B levels in exosomes, while blockage of autophagosome‐lysosome fusion by BafA1 further increased LC3B levels of exosomes from CAFs (Figure 7d), supporting an enhanced AP‐MVB fusion in hypoxic CAFs. Correspondingly, hypoxia‐stimulated activation of oxidized ATM promoted exosome release, while inhibition of oxidized ATM by KU60019 decreased exosome amount. In addition, inhibiting autophagosome‐lysosome fusion using BafA1 further increased exosome release (Figure 7e). However, the particle size of isolated exosomes among these groups remained unchanged (Figure 7f). These data demonstrate that exosomes derived from hypoxic CAFs are closely related with multitudinous fusion of autophagosomes (APs) and multi‐vesicular bodies (MVBs), which facilitates exosome release from hypoxic CAFs.
3.7. GPR64 of exosomes derived from hypoxic CAFs stimulates non‐canonical NF‐κB signalling in tumour cells to boost cell invasion
To determine whether exosome contents are responsible for hypoxic CAF‐mediated breast cancer cell invasion, we analysed the exosome database from ExoCarta (http://www.exocarta.org) (Keerthikumar et al., 2016) including the identified genes/proteins in exosomes, and the database from GEO including autophagy‐related genes/proteins profiles. We obtained 66 candidate genes/proteins in hypoxic CAF‐derived exosomes (Figure 8a) by merging the common genes/proteins in the two databases with our previous mRNA profiles of CAFs (Peng et al., 2013). These proteins might mediate the effect of exosomes from hypoxic CAFs on cancer cell invasion. The candidate genes are listed in Supplementary List 3. Twelve proteins among these 66 candidate genes/proteins are potentially related to tumour invasion and metastasis (Supplemental Table 5). qRT‐PCR, confirmed that 4 genes (e.g., IGF2, GPR64, TEK and TLR6) among these 12 genes were highly expressed in hypoxic CAFs (Figure S6A). Among these four genes, the adhesion G protein‐coupled receptor G2 (ADGRG2, also known as GPR64), was the most highly expressed in exosomes from normoxic CAFs (Nor‐CAF‐Exo) compared with normoxic NFs (Nor‐NF‐Exo), and was further increased in exosomes from hypoxic CAFs (Hyp‐CAF‐Exo) (Figure S6B). Thus, we speculated that exosomal GPR64 might mediate the effect of oxidized ATM on tumour cell adhesion and migration. Indeed, knockdown of GPR64 in CAFs (Figure S7A‐S7B) had no effect on exosome release and particle size (Figure S7C), but significantly mitigated the promoting effect of exosomes isolated from hypoxic CAFs on breast cancer cell invasion (Figure 8b and Figure S7D). Moreover, as shown in Figure 8c‐d, inhibiting oxidized ATM activation using KU60019 or impeding autophagy with siRNA against ATG5 significantly reduced GPR64 levels in hypoxic CAF‐derived exosomes but not in the control exosomes. Furthermore, knockdown of ATM in CAFs or transfection of ectopic BNIP3 S135A mutant into ATM‐silenced CAFs reduced GPR64 levels in exosomes (Figure 8e, middle panel), while expression of ectopic BNIP3 S135D mutant in ATM‐knockdown CAFs restored the GPR64 levels in exosomes (Figure 8e, right panel). Collectively, these data support that oxidized ATM‐induced autophagy in hypoxic CAFs enriches GPR64 in released exosomes to contribute to breast cancer cell invasion.
FIGURE 8.
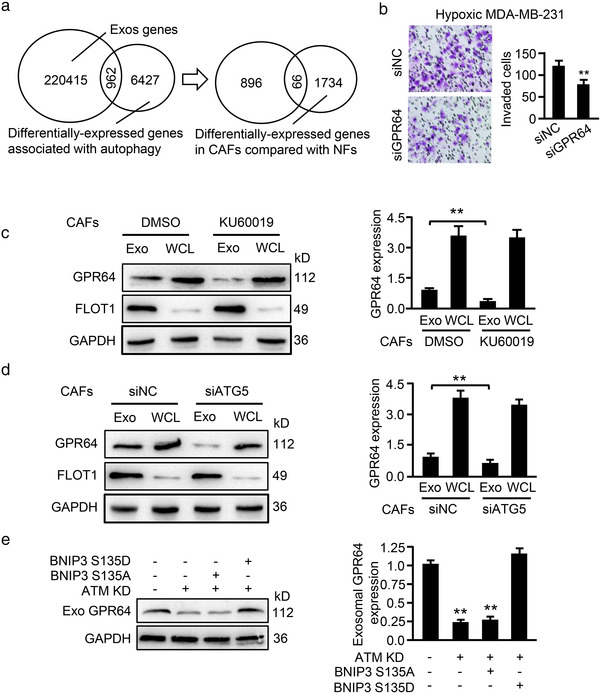
Oxidized ATM enriches autophagy‐associated GPR64 in exosomes. (a) Venn diagram showing autophagy‐related proteins in exosomes derived from hypoxic CAFs. (b) Exosomes isolated from GPR64‐silenced hypoxic CAFs and control hypoxic CAFs were added to breast cancer cells and invasion abilities of MDA‐MB‐231 were evaluated by Transwell assay. Representative images of invaded cancer cells and their quantification are shown in the right panel. (c‐d) Western blotting to detect GPR64 expression in exosomes and WCL derived from hypoxic CAFs with or without KU60019 treatment (c), or from Atg5‐knocked down hypoxic CAFs and control hypoxic CAFs (d). FLOT1 was used as the protein marker of exosomes, and GAPDH was used as the loading control. Relative quantification is shown in the right panel (Exo, exosomes; WCL, whole‐cell lysate). (e) Western blotting to determine GPR64 levels in exosomes isolated from control hypoxic CAFs, ATM‐silenced hypoxic CAFs, and endogenous ATM‐silenced hypoxic CAFs transfected with ectopic BNIP3 mutant S135A and S135D (**P < 0.01)
It has been shown that GPR64 can activate the NF‐κB signalling pathways (Peeters et al., 2015). Canonical NF‐κB signalling is defined by an increased Rel A nuclear location or DNA binding activity. Rel B, on the other hand, is associated with the non‐canonical NF‐κB signalling pathway (Kabacaoglu et al., 2019). Thus, expression of Rel A and Rel B was measured. Inhibition of oxidized ATM by KU60019 or silencing ATM in hypoxic CAFs caused a marked drop in Rel B but not Rel A levels in cocultured cancer cells (Figure 9a). Similarly, exosomes from BNIP3‐knocked down or KU60019‐treated hypoxic CAFs clearly decreased Rel B levels (Figure 9b, middle panels); exosomes from hypoxic CAFs with ectopic BNIP3 S135D mutant with KU60019 treatment rescued Rel B expression in breast cancer cells (Figure 9b, right panel). In line with these findings, knockdown of ATG5 or GPR64 in hypoxic CAFs significantly downregulated Rel B expression in tumour cells, while Rel A protein remained unchanged (Figure 9c). Matrix metallopeptidase 9 (MMP9) and interleukin 8 (IL‐8) are the known secreted factors regulated by non‐canonical NF‐κB signalling and involved in tumour cell invasion (Qin et al., 2018). Exosomes from GPR64‐knocked down or control hypoxic CAFs were co‐cultured with Rel B‐silenced (Figure S7E‐S7F) or control breast cancer cells. MMP9 and IL‐8 expression and tumour cell invasion abilities were assessed. As shown in Figure 9d, knockdown of GPR64 in CAFs or Rel B in cancer cells significantly decreased MMP9 and IL‐8 levels in breast cancer cells, which correspondingly led to reduced cell invasion abilities of MDA‐MB‐231 (Figure 9e, middle panels) and BT549 cells (Figure S7G, middle panels). On the other hand, overexpression of Rel B in tumour cells rescued MMP9 and IL‐8 expression attenuated by exosomes from GPR64‐knockdown hypoxic CAFs (Figure 9d, right panel), and correspondingly increased tumour cell invasion (Figure 9e and Figure S7G, right panel).
FIGURE 9.
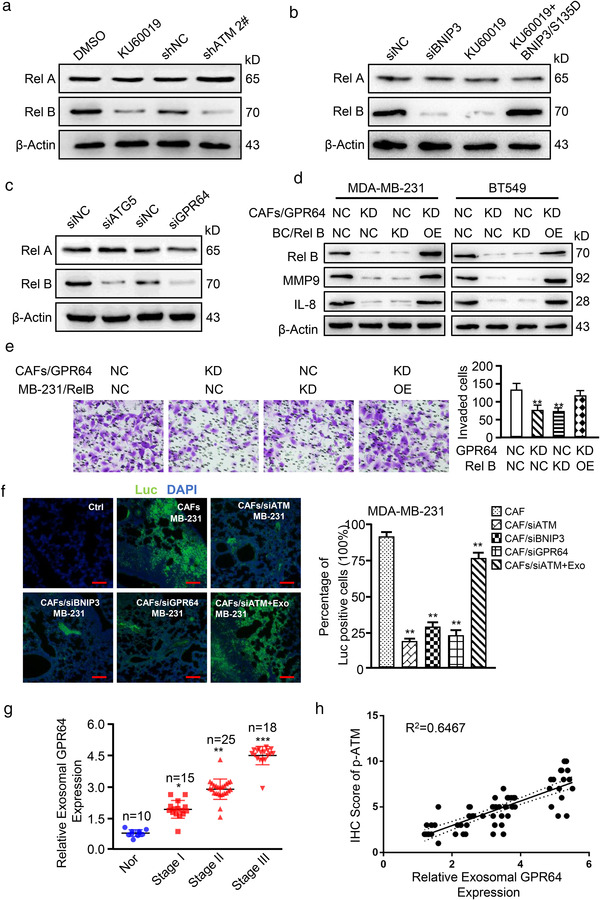
Exosomal GPR64 from hypoxic CAFs stimulates non‐canonical NF‐κB signaling in tumour cells to boost cell invasion. (a‐c) Breast cancer cells MDA‐MB‐231 were co‐cultured with exosomes derived from indicated CAFs. Rel A and Rel B levels in MDA‐MB‐231 were determined by Western blotting. (a) Exosomes from hypoxic CAFs treated with KU60019 (5 μM), ATM‐silenced CAFs or control CAFs; (b) Exosomes from BNIP3‐silenced hypoxic CAFs, hypoxic CAFs treated with KU60019, endogenous BNIP3‐silenced CAFs transfected with ectopic BNIP3 S135D mutant under KU60019 treatment, and control CAFs; (c) Exosomes from hypoxic CAFs with Atg5‐silenced, or GPR64‐silenced, and their control CAFs. (d, e) MDA‐MB‐231 (labelled as MB‐231) with Rel B‐silenced (KD), Rel B‐overexpressed (OE) or control cells were co‐cultured with exosomes isolated from GPR64‐silenced CAFs or control CAFs. MMP9 and IL‐8 levels in MDA‐MB‐231 were measured by Western blotting (d); and tumour cell invasion was evaluated using Transwell assay (e) (KD, knocked down; OE, overexpressed). (f) The indicated CAFs mixed with MDA‐MB‐231/Luc or Rel B‐silenced MDA‐MB‐231/Luc were co‐injected into the mammary pad of nude mice. The representative images of pulmonary metastases assessed by IF. Ctrl: normal lung; Luc: metastatic breast cancer cells were stained with antibody specifically against luciferase as green colour; DAPI: parenchymal cells were stained with DAPI as blue colour; MB‐231: MDA‐MB‐231 cells (Scale bar, 40 μm.). (g) Relative exosomal GPR64 level in plasma of breast cancer patients at the time of diagnosis and healthy controls (Nor) was determined in a colorimetric assay. Data are shown as mean ± SD. (h) Correlation between the relative exosomal GPR64 level (via western blotting) and relative p‐ATM expression (via IHC assay) is shown. (*P < 0.05; **P < 0.01; ***P < 0.001.)
To extend our findings, parental CAFs or engineered CAFs (with ATM‐silenced, BNIP3‐silenced, or GPR64‐silenced) mixed with MDA‐MB‐231 were co‐injected into nude mice to test the abilities of tumour invasion and metastasis in vivo. Compared with the mice injected with CAFs and MDA‐MB‐231, the mice injected with the mixture of engineered CAFs (siATM, siBNIP3, or siGPR64) and MDA‐MB‐231 had less metastases in lung (Figure 9f). In addition, supplementation with Hyp‐CAF‐Exo, which is enriched in GPR64, to the mice injected with the mixture of ATM‐knocked down CAFs (CAF/siATM) and MDA‐MB‐231 could partially rescue tumour metastasis (Figure 9f). Suppression of ATM, BNIP3, or GPR64 led to reduction of exosomal GPR64 expression (Figure S7H), and Rel B, MMP9 and IL‐8 expression in tumours (Figure S7I). Administering Hyp‐CAF‐Exo to the mice injected with the mixture of ATM‐knockdown CAFs and MDA‐MB‐231 could partially rescue the Rel B, MMP9 and IL‐8 expression in tumours (Figure S7I). To further investigate the clinical relevance of our findings, we assessed exosomal GPR64 expression in plasma samples obtained from 58 breast cancer patients who had not received any clinical treatment. Exosomal GPR64 was higher in plasma samples of breast cancer patients compared with the healthy women, and enhanced GPR64 was associated with tumour progression (Figure 9g) and tumour grade (P = 0.014), T (P = 0.002), N (P = 0.04) and TNM (P = 0.001) stages (Supplementary Table 6). Consistently, there was a positive correlation between p‐ATM and exosomal GPR64 in patients with breast cancer (Figure 9h), suggesting exosomal GPR64 in breast tumours correlates with poor prognosis.
In summary, our work demonstrates that DSB‐independent ATM activation (oxidized ATM), induced by hypoxia, can promote exosome release through phosphorylating BNIP3 to regulate AP accumulation and through phosphorylating ATP6V1G1 to induce lysosomal dysfunction, thus leading to fusion of APs and MVBs in hypoxic CAFs. Furthermore, autophagy‐associated GPR64, enriched in released exosomes, stimulates the activation of non‐canonical NF‐κB signalling in tumour cells and contributes to breast cancer malignant invasion and metastasis (Figure 10).
FIGURE 10.
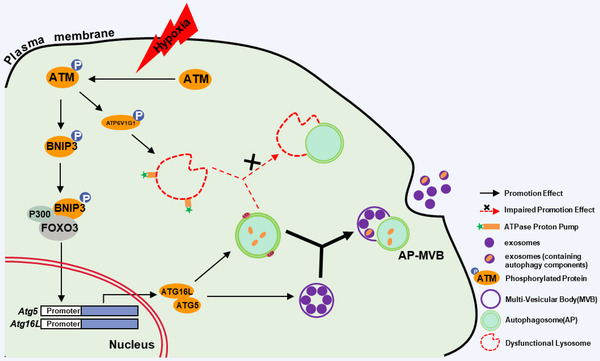
A schematic diagram for the role of oxidized ATM in hypoxic CAFs in inducing autophagy and EVs release. Oxidized ATM under hypoxia phosphorylates BNIP3 and ATP6V1G1. The phosphorylated BNIP3 increases the expression of ATG5 and ATG16L by recruiting P300 and FOXO3 to the promoter of Atg5 and Atg16L to promote formation of autophagosomes (APs) and multi‐vesicular bodies (MVBs). The phosphorylated ATP6V1G1 induces enhanced lysosomal pH to lead to dysfunctional lysosomes and impair fusion of APs and lysosome. APs tend to fuse with MVBs to form AP‐MVB rather than with dysfunctional lysosomes. The fusion of APs with MVB facilitates exosome release and related GPR64 enrichment in exosomes, thus fuelling the invasion of breast cancer cells
4. DISCUSSION
ATM is recognized as an important modulator of response to DSBs and DNA damage repair through phosphorylation of specific substrates such as activating transcription factor 2 (ATF2), checkpoint kinase 2 (CHK2) and myocyte enhancer factor 2D (MEF2D) (Bhoumik et al., 2005; Matsuoka et al., 2000). In addition, ATM is known as a redox sensor (namely DNA damage‐independent ATM or oxidized ATM) and can be activated by oxidative stress stimulators to maintain cellular redox homeostasis (Shiloh & Ziv, 2013). For example, oxidized ATM is activated by oxidized stress in fibroblasts derived from ataxia‐telangiectasia‐like disorder (ATLD) patents with mutated Mre11, which exhibits impairment in ATM activation via DSB pathways (Guo et al., 2010). Oxidized stress‐stimulated activation of ATM contributed to cancer cell migration and invasion via upregulation of IL‐8 (Chen et al., 2015). Here, we provided evidence for a novel function of oxidized ATM in hypoxic breast CAFs to promote breast cancer pathological invasion through autophagy‐associated enrichment of a specific protein (GPR64) in secreted exosomes.
The classical ATM protein kinase conducts its functions generally through phosphorylating target proteins at specific sites (Bennetzen et al., 2010). DSB‐dependent ATM phosphorylates ATF2 at serine 490 and 498 leading to its co‐localization with MRN complex and γH2AX in DNA repair foci and plays a role in S phase checkpoint control and DNA damage response (Bhoumik et al., 2005). Oxidized ATM induced by insulin treatment phosphorylates Akt at serine 473, resulting in the activation of insulin signalling and insulin resistance in muscle cells (Halaby et al., 2008). In this study, oxidized ATM‐mediated phosphoproteins identified by phosphoproteomics analysis may participate in various biological processes including autophagy, vesicle transportation, glycolysis and response to oxygen levels (Figure 2d). BNIP3, one of the candidate phosphoproteins, was found to be involved in oxidized ATM‐mediated autophagy of CAFs. BNIP3 has been demonstrated to trigger autophagy and removal of damaged mitochondria in cardiac myocytes (Hamacher‐Brady et al., 2007). The phosphorylation of BNIP3 at serine residues 17 and 24 in the LC3‐interacting domain positively regulates the binding ability of BNIP3 to specific Atg8 members LC3B and GATE‐16 in autophagy (Zhu et al., 2013). Here, we demonstrated that oxidized ATM phosphorylates BNIP3 at s135 and the phosphorylated BNIP3 plays an essential role in autophagy of CAFs. Ectopic expression of BNIP3 S135D (a hyper‐phosphorylated BNIP3 mutant), rather than S135A mutant (a hypo‐phosphorylated BNIP3 mutant), in the endogenous BNIP3‐silenced CAFs directly induced an enhanced autophagy phenotype, which provides strong evidence for the positive regulating autophagy of CAFs by phospho‐BNIP3 (s135).
Generally, autophagosomes can either fuse with lysosomes for degradation or with MVBs for exosome release and the balance between the two processes might be regulated by cellular status (Baixauli et al., 2014). Interestingly, we found that oxidized ATM‐mediated autophagosomes tend to fuse with MVBs to facilitate exosome release rather than to fuse with lysosomes in hypoxic CAFs. Mechanically, we reveal that oxidized ATM induces lysosomal dysfunctions through phosphorylation of ATP6V1G1, a key subunit of proton pump to maintain lysosomal acidification (Kang et al., 2017). Phosphorylated ATP6V1G1 resulted in increased pH in the lysosomes of hypoxic CAFs, and attenuation of oxidized ATM activity restored reacidification of the lysosomes. Autophagosomes have been reported to fuse with MVBs (AP‐MVBs) and the AP‐MVB compartment was confirmed to modulate exosome release (Baixauli et al., 2014), which might act as a regulator to maintain cellular homeostasis via autophagy‐associated component release. On one hand, activation of oxidized ATM under hypoxia induces autophagosome accumulation, and on the other hand results in lysosomal dysfunction, thus leading to the fusion between autophagosomes and MVBs and promoting autophagy‐associated component release via autophagy‐MVB‐exosome pathways.
Emerging evidence also implies an essential role of autophagy‐mediated crosstalk between stromal CAFs and the malignant progression of cancer cells. Autophagy in HNSCC‐associated CAFs was proved to promote HNSCC progression via modulating IL‐6 and IL‐8 (New et al., 2017). Further, autophagy pathways were found to regulate the biogenesis and secretion of exosomes including exosome amount, size and contained products (Baixauli et al., 2014). For example, previous findings have supported that autophagy stimulated the co‐localization of α‐synuclein with SCAMP5 in secreted exosomes (Yang et al., 2017) and modulated prion protein release via exosomal pathways (Abdulrahman et al., 2018). Here, we demonstrated that oxidized ATM‐mediated autophagy in stromal CAFs could affect exosome release and the abundance of proteins in exosomes without alteration of particle size. Oxidized ATM‐mediated autophagy in CAFs promoted the recruitment of some specific proteins into secreted exosomes and correspondingly, the inhibition of oxidized ATM activity or impeding autophagy decreased the enrichment of GPR64 in released exosomes, suggesting a positive effect of oxidized ATM‐mediated autophagy on the abundance of proteins in exosomes.
Several lines of evidence have revealed that exosomes are involved in stromal CAF‐induced malignant progression of cancer cells, such as tumorigenesis (Richards et al., 2017), cell migration (Wu et al., 2020), therapy resistance (Zhang et al., 2020) and EMT (Fiori et al., 2019). Exosomes favour tumour malignant progression through the carried oncogenic miRNA, proteins, lipids and metabolites (Hoshino et al., 2015). We found that GPR64, an enriched protein in hypoxic CAF‐derived EVs, plays a pivotal role in facilitating breast cancer invasion. The accumulation of GPR64 in CAF‐released EVs activated the non‐canonical NF‐κB signalling in breast cancer cells to fuel cell invasion. Inactivation of oxidized ATM, interfering with phosphorylation of BNIP3, or silencing GPR64 expression in CAFs significantly decreased GPR64 levels in hypoxic CAF‐derived EVs and attenuated cancer cell invasion. Our studies reveal that enrichment of a specific protein (GPR64) in hypoxic CAF‐derived EVs could be modulated by autophagy and play an essential role in cell‐cell communication.
5. CONCLUSION
In conclusion, ATM kinase, a regulator of DNA damage repair, can also be activated by non‐DNA damage in breast CAFs. Further analysis implied that autophagy and exosome secretion are involved in the mechanism by which oxidized ATM in CAFs promotes breast cancer cell invasion and metastasis. The current study also demonstrates that oxidized ATM can enrich GPR64 in hypoxic CAF‐derived exosomes through autophagy regulation and provides novel insight into the role of breast CAF‐associated oxidized ATM in cancer cell invasion.
Future studies need to further investigate how oxidized ATM‐induced autophagy facilitates GPR64 enrichment in exosomes. Overall, the present study provides a new tumour microenvironment‐related regulatory mechanism for breast cancer cell invasion and metastasis, and may help develop new strategies for breast cancer treatment.
CONFLICT OF INTEREST
We declare no conflict of interest in this work.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
Ethical approval was given by the Medical Ethics Committee of Chongqing Medical University. Animal experiments were permitted by the Animal Ethics Committees of Chongqing Medical University. All animal work was conducted according to the approved protocols and the institutional animal welfare guidelines.
CONSENT FOR PUBLICATION
All authors are aware of and agree to the content of the paper and their being listed as a co‐author of the paper.
AVAILABILITY OF SUPPORTING DATA
The supporting data and materials are provided in Supplementary Figures and Tables.
AUTHOR CONTRIBUTIONS
Meixi Peng, Lei Xi, Shuiqing Liu and Manran Liu designed this study; Lei Xi, Meixi Peng, Shuiqing Liu, Yongcan Liu, Xueying Wan, Yixuan Hou, LP, and Shanchun Chen performed the experiments; Yilu Qin and Huan Zeng analysed the data; Meixi Peng, Lei Xi and Shuiqing Liu drew the figures and tables; Lei Xi and Meixi Peng drafted the initial manuscript; Xiaojiang Cui, Yong Teng and Manran Liu revised this manuscript; all authors read and approved the final manuscript.
Supporting information
Supplementary Figure 1. Effects of hypoxic CAFs on BT549 cell invasion. (A) qRT‐PCR to test ATM expression in paired NFs and CAFs isolated from breast cancer patients. β‐actin is the internal control. (B) Quantification of Figure 1D is shown. (C) MDA‐MB‐231 and BT549 cells were co‐cultured with the CM from normoxic or hypoxic NFs and CAFs; or CM from hypoxic CAFs with or without shATM, and treated with or without KU60019 (5 μM; KU:KU60019). Transwell assay was used to detect cell invasion of breast cancer cells. Ctrl: Tumour cells cultured alone. (D) Quantification of invaded cells from (C) is shown. (E) Western blotting to test expression of ATM, phosphorylated ATM (s1981) and AKT (s473) in CAFs treated with different concentration of KU60019. (F) Lentivirus‐mediated knockdown efficiencies of ATM in breast CAFs were determined by western blotting. (*P < 0.05; **P < 0.01; ns, not significant). Supplementary Figure 2. Continued from Supplementary Figure 1. Effects of hypoxic CAFs on BT549 cell invasion. (A) The size distribution and concentration of exosomes were detected by nanoparticle tracking analysis (NTA). (B) Transmission electron microscopy of exosomes derived from hypoxic CAFs (Scale bar, 200 nm). (C) Western blotting to test exosome markers CD63, CD81, TSG101 and FLOT1. (D) Tumour cells were cultured with CM containing secreted factors (CMExo−) or exosomes of CAFs or NFs. Transwell assay was used to detect cell invasion of breast cancer cells. (E) Quantification of invaded cells from (D) is shown. (F‐G) Normoxic (Nor) or hypoxic (Hyp) CAFs treated with or without GW4869, and exosome concentration (F) or cell viability (G) was detected by NTA assay or determined using Annexin V‐FITC kit and flow cytometric assay at the time point of conditioned medium collected. The percentage of viable cells was calculated. (H) Tumour cells were cultured with CM from NF or CAF treated with or without GW4869 (GW), and cell invasion was detected via Transwell assay. (I) Quantification of invaded cells from (H) is shown. (*P < 0.05; ** P < 0.01; ns, not significant). Supplementary Figure 3. Continued from Supplementary Figure 1. Effects of hypoxic CAFs on BT549 cell invasion. (A) Size concentration of exosomes from normoxic NFs and CAFs, and hypoxic CAFs measured by NTA assay. (B) NTA to assess size concentration of exosomes isolated from hypoxic CAFs treated with KU60019 or ATM‐silenced hypoxic CAFs. (C) BT549 and MDA‐MB‐231 cells were incubated with PKH67‐labeled exosomes (20 μg/ml) derived from hypoxic CAF for 12 h, and the green signal of labelled exosomes in tumour cells was detected by confocal microscopy. (*P < 0.05; **P < 0.01.). Supplementary Figure 4. Phosphorylation of BNIP3 at s135 promotes autophagy and tumour cell invasion. (A) Protein expression levels of BNIP3 and p‐BNIP3 in paired primary NFs and CAFs were detected by western blotting. (B) CAFs were cultured in conditioned hypoxia with KU60019 treatment, or ATM in CAFs was knocked down by shRNA, and cell extracts were immunoprecipitated with p‐(S/T)Q antibody, followed by immunoblotting with the BNIP3 antibody to recognize p‐BNIP3 in co‐immunoprecipitates, and other indicated antibodies for extracts. (C) Primary CAFs isolated from breast tumour tissues infected with lentivirus vector encoding siBNIP3 and cultured in hypoxia, the extracts were used for immunoblotting against autophagy markers (LC3BI, LC3BII and p62), oxidized ATM (s1981), ATM and BNIP3. (D) Hypoxic primary CAFs with or without ectopic mutant BNIP3 (S135D) and treated with KU60019, western blotting was used to evaluate expression of autophagy markers (LC3BI, LC3BII and p62) and phospho‐BNIP3 at s135. (E) Breast cancer cells MDA‐MB‐231 and BT549 were co‐cultured with exosomes derived from indicated engineered hypoxic CAFs and Transwell assay was performed to detect cancer cell invasive ability. Representative images of invaded cells, and their relative quantification are shown (*P < 0.05; **P < 0.01.). Supplementary Figure 5. Binding interaction of phospho‐BNIP3 with p300 and FOXO3. (A) P300 was immunoprecipitated from endogenous BNIP3‐silenced hypoxic CAFs, endogenous BNIP3‐silenced CAFs transfected with ectopic WT BNIP3, S135A or S135D mutant, and western blotting probed for phosphorylated BNIP3 and FOXO3. (B) The effects of p300/FOXO3/phosphorylated BNIP3 on luciferase activities in 293T cells were measured under hypoxia. (C‐D) siRNA‐mediated knockdown efficiency of Atg5 in breast CAFs was determined by qRT‐PCR (C) and western blotting (D). The histogram in the right panel shows the relative quantifications of western blots in (D). (E‐F) siRNA‐mediated knockdown efficiencies of Atg16L in breast CAFs were assessed by qRT‐PCR (E) and western blotting (F). The relative quantifications of western blots in (F) are shown in the right panel. (G) Breast cancer cells MDA‐MB‐231 and BT549 were co‐cultured with exosomes derived from control hypoxic CAFs and Atg5‐silenced hypoxic CAFs. Transwell assay was performed to detect cancer cell invasive ability. Representative images of invaded cancer cells are shown, and the relative quantifications of invaded cells were calculated. (H) siRNA‐mediated knockdown efficiencies of ATP6V1G1 in breast CAFs were determined by western blotting assay. (*P < 0.05; ** P < 0.01.). Supplementary Figure 6. Prediction and verification of GPR64. (A) Expression levels of 17 genes potentially related to cancer invasion were verified by qRT‐PCR in indicated NF and CAF. (B) Levels of 4 candidate proteins in the indicated exosomes were evaluated by western blotting. Supplementary Figure 7. Effects of Rel B on cell invasion of breast cancer BT549 cells. (A, B) Knockdown efficiency of GPR64 in breast CAFs was determined by qRT‐PCR (A) and western blotting (B), and the quantifications of western blots in (B) are shown in the right panel. (C) Concentration and particle size of isolated particles from hypoxic CAFs/siNC and CAFs/siGPR64 were measured by nanoparticle tracking analysis (NTA). (D) Exosomes isolated from GPR64‐silenced hypoxic CAFs and control hypoxic CAFs were added to breast cancer BT549 cells, and cell invasion abilities were evaluated by Transwell assay. Representative images of invaded cancer cells are shown and the quantifications of invaded cells were calculated. (E‐F) siRNA‐mediated knockdown efficiencies of Rel B in breast cancer MDA‐MB‐231 cells were determined by qRT‐PCR (E) and western blotting (F). The relative quantifications of western blots are shown in the right panel. (G) BT549 cells with Rel B‐silenced, Rel B‐overexpressed or control cells were co‐cultured with exosomes isolated from GPR64‐silenced CAFs or control CAFs, and tumour cell invasion was evaluated using Transwell assay. Representative images of invaded cancer cells are shown and their relative quantifications are displayed in the right histogram (NC, negative control; KD, knocked down; OE, overexpression). (H) Expression of exosomal GPR64 in the blood of mice was detected by western blotting. (I) Levels of Rel B, MMP9, and IL‐8 in mouse tumours were measured by western blotting. (*P < 0.05, **P < 0.01; ns, not significant).
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGEMENTS
We acknowledge Dr. Anthea Hammond for editorial assistance. This work was supported in part by National key projects of Ministry of Science and Technology of China (MOST 2018YFE0113700), National Natural Science Foundation of China (NSFC81472476, NSFC31671481, and NSFC 31171336), and by Innovation Research Group in Colleges and Universities Program of Chongqing Municipal Education Commission (No. CXQT20012) to Manran Liu. And also supported by the outstanding Postgraduate Fund of Chongqing Medical University (BJRC202021) for Yongcan Liu.
Xi, L., Peng, M., Liu, S., Liu, Y., Wan, X., Hou, Y., Qin, Y., Yang, L., Chen, S., Zeng, H., Teng, Y., Cui, X., & Liu, M. (2021). Hypoxia‐stimulated ATM activation regulates autophagy‐associated exosome release from cancer‐associated fibroblasts to promote cancer cell invasion. Journal of Extracellular Vesicles, 10, e12146. 10.1002/jev2.12146
Lei Xi, Meixi Peng, Shuiqing Liu, Yongcan Liu and Xueying Wan contributed equally to this study.
REFERENCES
- Abdulrahman, B. A., Abdelaziz, D. H., & Schatzl, H. M. (2018). Autophagy regulates exosomal release of prions in neuronal cells. The Journal of biological chemistry, 293(23), 8956–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrant, A. E., Onyilo, V. C., Unger, D. E., & Roberson, E. D. (2018). Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid lipofuscinosis. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 38(9), 2341–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audesse, A. J., Dhakal, S., Hassell, L.‐A., Gardell, Z., Nemtsova, Y., & Webb, A. E. (2019). FOXO3 directly regulates an autophagy network to functionally regulate proteostasis in adult neural stem cells. Plos Genetics, 15(4), e1008097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baixauli, F., Lopez‐Otin, C., & Mittelbrunn, M. (2014). Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Frontiers in Immunology, 5, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencokova, Z., Kaufmann, M. R., Pires, I. M., Lecane, P. S., Giaccia, A. J., & Hammond, E. M. (2009). ATM activation and signaling under hypoxic conditions. Molecular and Cellular Biology, 29(2), 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen, M. V., Larsen, D. H., Bunkenborg, J., Bartek, J., Lukas, J., & Andersen, J. S. (2010). Site‐specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Molecular and Cellular Proteomics, 9, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoumik, A., Takahashi, S., Breitweiser, W., Shiloh, Y., Jones, N., & Ronai, Z. (2005). ATM‐dependent phosphorylation of ATF2 is required for the DNA damage response. Molecular Cell, 18(5), 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.‐T., Liu, C.‐C., Yu, J.‐S., Li, H.‐H., & Lai, M.‐C. (2018). Integrated omics profiling identifies hypoxia‐regulated genes in HCT116 colon cancer cells. Journal of proteomics, 188, 139–151. [DOI] [PubMed] [Google Scholar]
- Chen, W.‐T., Ebelt, N. D., Stracker, T. H., Xhemalce, B., Van Den Berg, C. L., & Miller, K. M. (2015). ATM regulation of IL‐8 links oxidative stress to cancer cell migration and invasion. Elife, 4. 10.7554/eLife.07270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czornak, K., Chughtai, S., & Chrzanowska, K. H. (2008). Mystery of DNA repair: The role of the MRN complex and ATM kinase in DNA damage repair. Journal of Applied Genetics, 49(4), 383–396. [DOI] [PubMed] [Google Scholar]
- Du, Y.‐E., Tu, G., Yang, G., Li, G., Yang, D., Lang, L., Xi, L., Sun, K., Chen, Y., Shu, K., Liao, H., Liu, M., & Hou, Y. (2017). MiR‐205/YAP1 in Activated Fibroblasts of Breast Tumor Promotes VEGF‐independent Angiogenesis through STAT3 Signaling. Theranostics, 7(16), 3972–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori, M. E., Di Franco, S., Villanova, L., Bianca, P., Stassi, G., & De Maria, R. (2019). Cancer‐associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Molecular cancer, 18(1), 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, X., Meng, Q., Wei, L., Liu, J., Li, M., Liang, X., Lin, F., Zhang, Y., Li, Y., Liu, Z., Fan, H., & Zhou, X. (2021). Myocardial ischemia‐reperfusion induced cardiac extracellular vesicles harbour proinflammatory features and aggravate heart injury. Journal of extracellular vesicles, 10(4), e12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., Kozlov, S., Lavin, M. F., Person, M. D., & Paull, T. T. (2010). ATM Activation by Oxidative Stress. Science, 330(6003), 517–521. [DOI] [PubMed] [Google Scholar]
- Halaby, M.‐J., Hibma, J. C., He, J., & Yang, D.‐Q. (2008). ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cellular Signalling, 20(8), 1555–1563. [DOI] [PubMed] [Google Scholar]
- Hamacher‐Brady, A., Brady, N. R., Logue, S. E., Sayen, M. R., Jinno, M., Kirshenbaum, L. A., Gottlieb, R. A., & Gustafsson, Å. B. (2007). Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death and Differentiation, 14(1), 146–157. [DOI] [PubMed] [Google Scholar]
- Hanley, C. J., Mellone, M., Ford, K., Thirdborough, S. M., Mellows, T., Frampton, S. J., Smith, D. M., Harden, E., Szyndralewiez, C., Bullock, M., Noble, F., Moutasim, K. A., King, E. V., Vijayanand, P., Mirnezami, A. H., Underwood, T. J., Ottensmeier, C. H., & Thomas, G. J. (2018). Targeting the myofibroblastic cancer‐associated fibroblast phenotype through inhibition of NOX4. Journal of the National Cancer Institute, 110(1), 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongyun Zhao, L. Y., Baddour, J. & Achreja, A. (2016). Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLIFE, 5, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino, A., Costa‐Silva, B., Shen, T.‐L., Rodrigues, G., Hashimoto, A., Tesic Mark, M., Molina, H., Kohsaka, S., Di Giannatale, A., Ceder, S., Singh, S., Williams, C., Soplop, N., Uryu, K., Pharmer, L., King, T., Bojmar, L., Davies, A. E., Ararso, Y.…Lyden, D. (2015). Tumour exosome integrins determine organotropic metastasis. Nature, 527(7578), 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, D. W, Sherman, B. T., & Lempicki, R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research, 37(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, D. W., Sherman, B. T., & Lempicki, R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4(1), 44–57. [DOI] [PubMed] [Google Scholar]
- Jachimowicz, R. D., Beleggia, F., Isensee, J., Velpula, B. B., Goergens, J., Bustos, M. A., Doll, M. A., Shenoy, A., Checa‐Rodriguez, C., Wiederstein, J. L., Baranes‐Bachar, K., Bartenhagen, C., Hertwig, F., Teper, N., Nishi, T., Schmitt, A., Distelmaier, F., Lüdecke, H.‐J., Albrecht, B.…Shiloh, Y. (2019). UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell, 176(3), 505–519.e22. [DOI] [PubMed] [Google Scholar]
- Kabacaoglu, D., Ruess, D. A., Ai, J., & Algül, H. (2019). NF‐κB/Rel transcription factors in pancreatic cancer: Focusing on RelA, c‐Rel, and RelB. Cancers, 11(7), 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H. T., Park, J. T., Choi, K., Kim, Y., Choi, H. J. C., Jung, C. W., Lee, Y.‐S., & Park, S. C. (2017). Chemical screening identifies ATM as a target for alleviating senescence. Nature Chemical Biology, 13(6), 616–623. [DOI] [PubMed] [Google Scholar]
- Keerthikumar, S., Chisanga, D., Ariyaratne, D., Al Saffar, H., Anand, S., Zhao, K., Samuel, M., Pathan, M., Jois, M., Chilamkurti, N., Gangoda, L., & Mathivanan, S. (2016). ExoCarta: A web‐based compendium of exosomal cargo. Journal of molecular biology, 428(4), 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. E., Kim, B. G., Jang, Y., Kang, S., Lee, J. H., & Cho, N. H. (2020). The stromal loss of miR‐4516 promotes the FOSL1‐dependent proliferation and malignancy of triple negative breast cancer. Cancer Letters, 469, 256–265. [DOI] [PubMed] [Google Scholar]
- Lang, L., Hou, Y., Chen, Y., Tu, G., Tao, J., Yang, D., Xi, L., Fu, L., Sun, K., Yin, J., Chen, R., Peng, M., Liu, S., & Liu, M. (2018). ATM‐mediated phosphorylation of cortactin involved in actin polymerization promotes breast cancer cells migration and invasion. Cellular Physiology and Biochemistry, 51(6), 2972–2988. [DOI] [PubMed] [Google Scholar]
- Le Guezennec, X., Brichkina, A., Huang, Y.‐F., Kostromina, E., Han, W., & Bulavin, D. V. (2012). Wip1‐dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metabolism, 16(1), 68–80. [DOI] [PubMed] [Google Scholar]
- Li, W.‐D., Hu, N., Lei, F.‐R., Wei, S., Rong, J.‐J., Zhuang, H., & Li, X.‐Q. (2015). Autophagy inhibits endothelial progenitor cells migration via the regulation of MMP2, MMP9 and uPA under normoxia condition. Biochemical and Biophysical Research Communications, 466(3), 376–380. [DOI] [PubMed] [Google Scholar]
- Liang, N., He, Q., Liu, X., & Sun, H. (2019). Multifaceted roles of ATM in autophagy: From nonselective autophagy to selective autophagy. Cell Biochemistry and Function, 37(3), 177–184. [DOI] [PubMed] [Google Scholar]
- Marín‐Hernández, Á., Rodríguez‐Enríquez, S., & Moreno‐Sánchez, R. (2019). Oxidized ATM protein kinase is a new signal transduction player that regulates glycolysis in CAFs as well as tumor growth and metastasis. EBioMedicine, 41, 24–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, S., Rotman, G., Ogawa, A., Shiloh, Y., Tamai, K., & Elledge, S. J. (2000). Ataxia telangiectasia‐mutated phosphorylates Chk2 in vivo and in vitro. Proceedings of the National Academy of Sciences of the United States of America, 97(19), 10389–10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauthe, M., Orhon, I., Rocchi, C., Zhou, X., Luhr, M., Hijlkema, K.‐J., Coppes, R. P., Engedal, N., Mari, M., & Reggiori, F. (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome‐lysosome fusion. Autophagy, 14(8), 1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meo‐Evoli, N., Almacellas, E., Massucci, F. A., Gentilella, A., Ambrosio, S., Kozma, S. C., Thomas, G., & Tauler, A. (2015). V‐ATPase: A master effector of E2F1‐mediated lysosomal trafficking, mTORC1 activation and autophagy. Oncotarget, 6(29), 28057–28070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New, J., Arnold, L., Ananth, M., Alvi, S., Thornton, M., Werner, L., Tawfik, O., Dai, H., Shnayder, Y., Kakarala, K., Tsue, T. T., Girod, D. A., Ding, W.‐X., Anant, S., & Thomas, S. M. (2017). Secretory autophagy in cancer‐associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer research, 77(23), 6679–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters, M. C., Fokkelman, M., Boogaard, B., Egerod, K. L., Van De Water, B., Ijzerman, A. P., & Schwartz, T. W. (2015). The adhesion G protein‐coupled receptor G2 (ADGRG2/GPR64) constitutively activates SRE and NFκB and is involved in cell adhesion and migration. Cellular Signaling, 27, 2579–2588. [DOI] [PubMed] [Google Scholar]
- Peng, Q., Zhao, L., Hou, Y., Sun, Y., Wang, L., Luo, H., Peng, H., & Liu, M. (2013). Biological characteristics and genetic heterogeneity between carcinoma‐associated fibroblasts and their paired normal fibroblasts in human breast cancer. Plos One, 8(4), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, H., Zhou, J., Xu, J., Cheng, L., Tang, Z., Ma, H., & Guo, F. (2018). The nuclear transcription factor RelB functions as an oncogene in human lung adenocarcinoma SPC‐A1 cells. Cancer Cell International, 18, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkumar, A., Murthy, D., Raja, D. A., Singh, A., Krishnan, A., Khanna, S., Vats, A., Thukral, L., Sharma, P., Sivasubbu, S., Rani, R., Natarajan, V. T., & Gokhale, R. S. (2017). Classical autophagy proteins LC3B and ATG4B facilitate melanosome movement on cytoskeletal tracks. Autophagy, 13(8), 1331–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, K. E., Zeleniak, A. E., Fishel, M. L., Wu, J., Littlepage, L. E., & Hill, R. (2017). Cancer‐associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene, 36(13), 1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh, Y., & Ziv, Y. (2013). The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nature Reviews Molecular Cell Biology, 14(4), 197–210. [PubMed] [Google Scholar]
- Stagni, V., Cirotti, C., & Barilà, D. (2018). Ataxia‐telangiectasia mutated kinase in the control of oxidative stress, mitochondria, and autophagy in cancer: A maestro with a large orchestra. Frontiers in Oncology, 8, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, K., Tang, S., Hou, Y., Xi, L., Chen, Y., Yin, J., Peng, M., Zhao, M., Cui, X., & Liu, M. (2019). Oxidized ATM‐mediated glycolysis enhancement in breast cancer‐associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine, 41, 370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, S., Hou, Y., Zhang, H., Tu, G., Yang, L., Sun, Y., Lang, L., Tang, X., Du, Y.‐E., Zhou, M., Yu, T., Xu, L., Wen, S., Liu, C., & Liu, M. (2015). Oxidized ATM promotes abnormal proliferation of breast CAFs through maintaining intracellular redox homeostasis and activating the PI3K‐AKT, MEK‐ERK, and Wnt‐beta‐catenin signaling pathways. Cell Cycle, 14(12), 1908–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X., Hou, Y., Yang, G., Wang, X., Tang, S., Du, Y.‐E., Yang, L., Yu, T., Zhang, H., Zhou, M., Wen, S., Xu, L., & Liu, M. (2016). Stromal miR‐200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell death and differentiation, 23(1), 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X., Tu, G., Yang, G., Wang, X., Kang, L., Yang, L., Zeng, H., Wan, X., Qiao, Y., Cui, X., Liu, M., & Hou, Y. (2019). Autocrine TGF‐β1/miR‐200s/miR‐221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer letters, 452, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. W., Wei, J., Appau, K., Wang, H., Yu, H., Spiga, M. G., Graham, R. M., & Webster, K. A. (2015). Bnip3 binds and activates p300: Possible role in cardiac transcription and myocyte morphology. Plos One, 10(8), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K., Wei, Y., Liu, W., Liu, L., Guo, Z., Fan, C., Wang, L., Hu, J., & Li, B. (2019). Mechanical stress‐dependent autophagy component release via extracellular nanovesicles in tumor cells. ACS Nano, 13(4), 4589–4602. [DOI] [PubMed] [Google Scholar]
- Wen, S., Hou, Y., Fu, L., Xi, L., Yang, D., Zhao, M., Qin, Y., Sun, K., Teng, Y., & Liu, M. (2019). Cancer‐associated fibroblast (CAF)‐derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3‐p38 MAPK signalling. Cancer letters, 442, 320–332. [DOI] [PubMed] [Google Scholar]
- Wu, H.‐J., Hao, M., Yeo, S. K., & Guan, J.‐L. (2020). FAK signaling in cancer‐associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs‐mediated intercellular communication. Oncogene, 39(12), 2539–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, W., Wu, X., Zhou, W., Fong, M. Y., Cao, M., Liu, J., Liu, X., Chen, C.‐H., Fadare, O., Pizzo, D. P., Wu, J., Liu, L., Liu, X., Chin, A. R., Ren, X., Chen, Y., Locasale, J. W., & Wang, S. E. (2018). Cancer‐cell‐secreted exosomal miR‐105 promotes tumour growth through the MYC‐dependent metabolic reprogramming of stromal cells. Nature Cell Biology, 20(5), 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., Qin, M., Bao, P., Xu, W., & Xu, J. (2017). Secretory carrier membrane protein 5 is an autophagy inhibitor that promotes the secretion of alpha‐synuclein via exosome. Plos One, 12(7), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeon, J. H., Jeong, H. E., Seo, H., Cho, S., Kim, K., Na, D., Chung, S., Park, J., Choi, N., & Kang, J. Y. (2018). Cancer‐derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer‐associated fibroblasts. Acta biomaterialia, 76, 146–153. [DOI] [PubMed] [Google Scholar]
- Yu, T., Yang, G., Hou, Y., Tang, X., Wu, C., Wu, X.‐A., Guo, L., Zhu, Q., Luo, H., Du, Y.‐E., Wen, S., Xu, L., Yin, J., Tu, G., & Liu, M. (2017). Cytoplasmic GPER translocation in cancer‐associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene, 36(15), 2131–2145. [DOI] [PubMed] [Google Scholar]
- Zeng, Z., Li, Y., Pan, Y., Lan, X., Song, F., Sun, J., Zhou, K., Liu, X., Ren, X., Wang, F., Hu, J., Zhu, X., Yang, W., Liao, W., Li, G., Ding, Y., & Liang, L. (2018). Cancer‐derived exosomal miR‐25‐3p promotes pre‐metastatic niche formation by inducing vascular permeability and angiogenesis. Nature Communications, 9(1), 5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Deng, T., Liu, R., Ning, T., Yang, H., Liu, D., Zhang, Q., Lin, D., Ge, S., Bai, M., Wang, X., Zhang, L., Li, H., Yang, Y., Ji, Z., Wang, H., Ying, G., & Ba, Y. (2020). CAF secreted miR‐522 suppresses ferroptosis and promotes acquired chemo‐resistance in gastric cancer. Molecular Cancer [Electronic Resource], 19(1), 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P., Wei, Y., Wang, L., Debeb, B. G., Yuan, Y., Zhang, J., Yuan, J., Wang, M., Chen, D., Sun, Y., Woodward, W. A., Liu, Y., Dean, D. C., Liang, H., Hu, Y., Ang, K. K., Hung, M.‐C., Chen, J., & Ma, L. (2014). ATM‐mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nature Cell Biology, 16(9), 864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y., Massen, S., Terenzio, M., Lang, V., Chen‐Lindner, S., Eils, R., Novak, I., Dikic, I., Hamacher‐Brady, A., & Brady, N. R. (2013). Modulation of serines 17 and 24 in the LC3‐interacting region of Bnip3 determines pro‐survival mitophagy versus apoptosis. The Journal of biological chemistry, 288(2), 1099–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Effects of hypoxic CAFs on BT549 cell invasion. (A) qRT‐PCR to test ATM expression in paired NFs and CAFs isolated from breast cancer patients. β‐actin is the internal control. (B) Quantification of Figure 1D is shown. (C) MDA‐MB‐231 and BT549 cells were co‐cultured with the CM from normoxic or hypoxic NFs and CAFs; or CM from hypoxic CAFs with or without shATM, and treated with or without KU60019 (5 μM; KU:KU60019). Transwell assay was used to detect cell invasion of breast cancer cells. Ctrl: Tumour cells cultured alone. (D) Quantification of invaded cells from (C) is shown. (E) Western blotting to test expression of ATM, phosphorylated ATM (s1981) and AKT (s473) in CAFs treated with different concentration of KU60019. (F) Lentivirus‐mediated knockdown efficiencies of ATM in breast CAFs were determined by western blotting. (*P < 0.05; **P < 0.01; ns, not significant). Supplementary Figure 2. Continued from Supplementary Figure 1. Effects of hypoxic CAFs on BT549 cell invasion. (A) The size distribution and concentration of exosomes were detected by nanoparticle tracking analysis (NTA). (B) Transmission electron microscopy of exosomes derived from hypoxic CAFs (Scale bar, 200 nm). (C) Western blotting to test exosome markers CD63, CD81, TSG101 and FLOT1. (D) Tumour cells were cultured with CM containing secreted factors (CMExo−) or exosomes of CAFs or NFs. Transwell assay was used to detect cell invasion of breast cancer cells. (E) Quantification of invaded cells from (D) is shown. (F‐G) Normoxic (Nor) or hypoxic (Hyp) CAFs treated with or without GW4869, and exosome concentration (F) or cell viability (G) was detected by NTA assay or determined using Annexin V‐FITC kit and flow cytometric assay at the time point of conditioned medium collected. The percentage of viable cells was calculated. (H) Tumour cells were cultured with CM from NF or CAF treated with or without GW4869 (GW), and cell invasion was detected via Transwell assay. (I) Quantification of invaded cells from (H) is shown. (*P < 0.05; ** P < 0.01; ns, not significant). Supplementary Figure 3. Continued from Supplementary Figure 1. Effects of hypoxic CAFs on BT549 cell invasion. (A) Size concentration of exosomes from normoxic NFs and CAFs, and hypoxic CAFs measured by NTA assay. (B) NTA to assess size concentration of exosomes isolated from hypoxic CAFs treated with KU60019 or ATM‐silenced hypoxic CAFs. (C) BT549 and MDA‐MB‐231 cells were incubated with PKH67‐labeled exosomes (20 μg/ml) derived from hypoxic CAF for 12 h, and the green signal of labelled exosomes in tumour cells was detected by confocal microscopy. (*P < 0.05; **P < 0.01.). Supplementary Figure 4. Phosphorylation of BNIP3 at s135 promotes autophagy and tumour cell invasion. (A) Protein expression levels of BNIP3 and p‐BNIP3 in paired primary NFs and CAFs were detected by western blotting. (B) CAFs were cultured in conditioned hypoxia with KU60019 treatment, or ATM in CAFs was knocked down by shRNA, and cell extracts were immunoprecipitated with p‐(S/T)Q antibody, followed by immunoblotting with the BNIP3 antibody to recognize p‐BNIP3 in co‐immunoprecipitates, and other indicated antibodies for extracts. (C) Primary CAFs isolated from breast tumour tissues infected with lentivirus vector encoding siBNIP3 and cultured in hypoxia, the extracts were used for immunoblotting against autophagy markers (LC3BI, LC3BII and p62), oxidized ATM (s1981), ATM and BNIP3. (D) Hypoxic primary CAFs with or without ectopic mutant BNIP3 (S135D) and treated with KU60019, western blotting was used to evaluate expression of autophagy markers (LC3BI, LC3BII and p62) and phospho‐BNIP3 at s135. (E) Breast cancer cells MDA‐MB‐231 and BT549 were co‐cultured with exosomes derived from indicated engineered hypoxic CAFs and Transwell assay was performed to detect cancer cell invasive ability. Representative images of invaded cells, and their relative quantification are shown (*P < 0.05; **P < 0.01.). Supplementary Figure 5. Binding interaction of phospho‐BNIP3 with p300 and FOXO3. (A) P300 was immunoprecipitated from endogenous BNIP3‐silenced hypoxic CAFs, endogenous BNIP3‐silenced CAFs transfected with ectopic WT BNIP3, S135A or S135D mutant, and western blotting probed for phosphorylated BNIP3 and FOXO3. (B) The effects of p300/FOXO3/phosphorylated BNIP3 on luciferase activities in 293T cells were measured under hypoxia. (C‐D) siRNA‐mediated knockdown efficiency of Atg5 in breast CAFs was determined by qRT‐PCR (C) and western blotting (D). The histogram in the right panel shows the relative quantifications of western blots in (D). (E‐F) siRNA‐mediated knockdown efficiencies of Atg16L in breast CAFs were assessed by qRT‐PCR (E) and western blotting (F). The relative quantifications of western blots in (F) are shown in the right panel. (G) Breast cancer cells MDA‐MB‐231 and BT549 were co‐cultured with exosomes derived from control hypoxic CAFs and Atg5‐silenced hypoxic CAFs. Transwell assay was performed to detect cancer cell invasive ability. Representative images of invaded cancer cells are shown, and the relative quantifications of invaded cells were calculated. (H) siRNA‐mediated knockdown efficiencies of ATP6V1G1 in breast CAFs were determined by western blotting assay. (*P < 0.05; ** P < 0.01.). Supplementary Figure 6. Prediction and verification of GPR64. (A) Expression levels of 17 genes potentially related to cancer invasion were verified by qRT‐PCR in indicated NF and CAF. (B) Levels of 4 candidate proteins in the indicated exosomes were evaluated by western blotting. Supplementary Figure 7. Effects of Rel B on cell invasion of breast cancer BT549 cells. (A, B) Knockdown efficiency of GPR64 in breast CAFs was determined by qRT‐PCR (A) and western blotting (B), and the quantifications of western blots in (B) are shown in the right panel. (C) Concentration and particle size of isolated particles from hypoxic CAFs/siNC and CAFs/siGPR64 were measured by nanoparticle tracking analysis (NTA). (D) Exosomes isolated from GPR64‐silenced hypoxic CAFs and control hypoxic CAFs were added to breast cancer BT549 cells, and cell invasion abilities were evaluated by Transwell assay. Representative images of invaded cancer cells are shown and the quantifications of invaded cells were calculated. (E‐F) siRNA‐mediated knockdown efficiencies of Rel B in breast cancer MDA‐MB‐231 cells were determined by qRT‐PCR (E) and western blotting (F). The relative quantifications of western blots are shown in the right panel. (G) BT549 cells with Rel B‐silenced, Rel B‐overexpressed or control cells were co‐cultured with exosomes isolated from GPR64‐silenced CAFs or control CAFs, and tumour cell invasion was evaluated using Transwell assay. Representative images of invaded cancer cells are shown and their relative quantifications are displayed in the right histogram (NC, negative control; KD, knocked down; OE, overexpression). (H) Expression of exosomal GPR64 in the blood of mice was detected by western blotting. (I) Levels of Rel B, MMP9, and IL‐8 in mouse tumours were measured by western blotting. (*P < 0.05, **P < 0.01; ns, not significant).
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The supporting data and materials are provided in Supplementary Figures and Tables.