

Summary
Wilms tumor is the most widespread kidney cancer in children and frequently associated with homozygous loss of the tumor suppressor WT1. Pediatric tumorigenesis is largely inaccessible in humans. Here, we develop a human kidney organoid model for Wilms tumor formation and show that deletion of WT1 during organoid development induces overgrowth of kidney progenitor cells at the expense of differentiating glomeruli and tubules. Functional and gene expression analyses demonstrate that absence of WT1 halts progenitor cell progression at a pre-epithelialized cell state and recapitulates the transcriptional changes detected in a subgroup of Wilms tumor patients with ectopic myogenesis. By “transplanting” WT1 mutant cells into wild-type kidney organoids, we find that their propagation requires an untransformed microenvironment. This work defines the role of WT1 in kidney progenitor cell progression and tumor suppression, and establishes human kidney organoids as a phenotypic model for pediatric tumorigenesis.
Keywords: Wilms tumor, WT1, SIX2, tumor suppressor, iPS cell, human kidney organoid, kidney progenitor cell, pediatric tumor, disease modeling, mesenchymal-epithelial transition
Graphical abstract
Highlights
-
•
Loss of WT1 induces human kidney organoid hyperplasia
-
•
WT1 coordinates epithelialization and exit from the progenitor cell state
-
•
WT1 mutant organoids resemble human Wilms tumors with ectopic myogenesis
-
•
Untransformed niche cells are required for long-term propagation of WT1 mutant cells
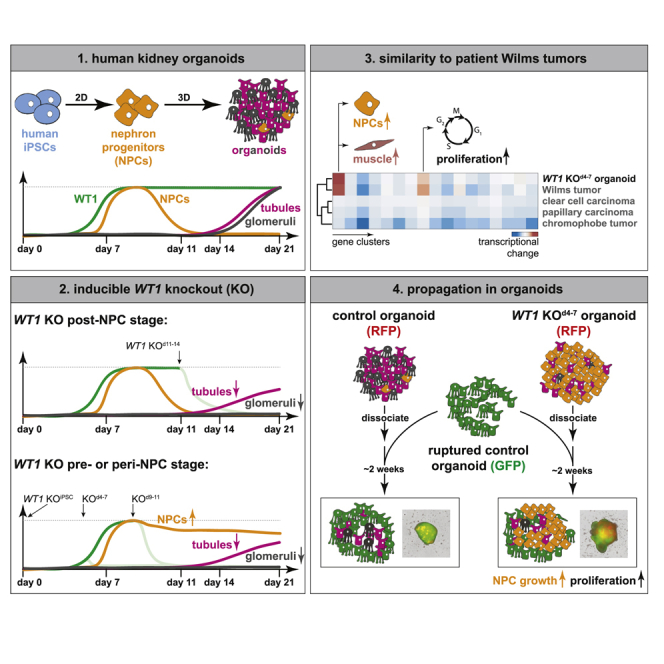
In this article, Waehle et al. develop and characterize an induced pluripotent stem cell-derived organoid model for the pediatric kidney cancer Wilms tumor using inducible knockout of the tumor suppressor WT1. WT1 knockout organoids display hyperplasia, loss of tubular and glomerular differentiation, and recapitulate the transcriptional changes detected in a subgroup of Wilms tumors patients with ectopic myogenesis.
Introduction
Tumor initiation and progression are typically studied in genetically engineered mice (Kersten et al., 2017), whose relevance to human disease is limited by species- and strain-specific mechanisms, and the artificial induction of multiple oncogenes at the same time. Most human cancer models, including cell lines grown on tissue plastic or as patient-derived xenografts in mice (Hynds et al., 2018) employ patients' cells that are derived from tumor resections or biopsies, typically at a late stage of the disease. Such systems depend on prolonged culture under non-physiological conditions that alter tumor cell properties and result in poor clinical predictiveness. Patient tumor-derived organoids, in contrast, preserve tumor heterogeneity, stages of tumor progression, and drug responses. Furthermore, engineering of cancer lesions into wild-type organoids induces cancer-specific transcriptional changes and predisposes to tumor formation upon xenotransplantation (Clevers and Tuveson, 2019). Organoids may therefore be suitable to study mechanisms of tumor initiation and progression.
Wilms tumor (WT) is the most common kidney cancer in childhood and accounts for 7% of all pediatric cancers (Treger et al., 2019). Stalled nephrogenesis is thought to be the major cause of disease. This is supported by the transcriptional similarity of WT to fetal cell types and by the function of several WT oncogenes and tumor suppressors in normal kidney development (Treger et al., 2019). WT patients show mutations in a diverse set of genes, including homozygous inactivation of the tumor suppressor Wilms tumor 1 (WT1) (Schumacher et al., 1997) and neomorphic mutations in genes encoding the kidney transcription factors (TFs) SIX1 and SIX2. Gene expression analysis has suggested that Wilms tumorigenesis initiates in distinct cell types of origin and is influenced not only by which genes or pathways are mutated but also by the developmental context in which genetic lesions occur (Gadd et al., 2012).
Here, we exploited human kidney organoids to study mechanisms of WT initiation and progression. We showed that genetic knockout (KO) of WT1 induces overgrowth of nephron progenitor cells (NPCs) at the expense of tubular and glomerular differentiation. Further characterization revealed progression into an organoid state that transcriptionally and phenotypically resembles a subset of WT patients, as well as arrest of NPC differentiation at a pre-epithelialized cell state. Our study therefore defines WT1-mediated developmental cell fate transitions that drive organogenesis and protect from hyperplasia, and establishes modeling of Wilms tumorigenesis in human kidney organoids.
Results
WT1 deletion inhibits NPC epithelialization and differentiation and induces organoid hyperplasia
We generated kidney organoids using an adaptation (Ungricht et al., 2021) of a two-step differentiation protocol (Morizane et al., 2015) (Figure S1A). Organoids contained podocytes that express WT1, PODXL, and NPHS1, and epithelial tubule cells that express high levels of EPCAM (EPCAMhigh) with distal and proximal segments additionally labeled by CDH1 and LTL, respectively (Figures S1B and S1C). For KO of WT1 we infected induced pluripotent stem cells (iPSCs) harboring a doxycycline (DOX)-inducible Cas9 protein (WT29-iCas9) (Figures S1D and S1E) (Ungricht et al., 2021) with lentiviruses driving expression of a WT1-specific guide RNA (gRNA1) and a red fluorescent protein (RFP). We induced genome editing by adding DOX at different stages of kidney organoid differentiation: in iPSCs prior to differentiation (KOiPSC), during intermediate mesoderm specification (KOd4-7), during NPC differentiation (KOd9-11), and during nephrogenesis (KOd11-14). To determine KO efficiency and kinetics, we quantified WT1 protein by flow cytometry. This showed that KOiPSC and KOd4-7 impaired the steep increase of WT1-expressing cells between day (d) 5 and d6 of organoid formation, resulting in an approximate 80% and 70% reduction of the WT1-positive cell fraction by d9, respectively (Figures S1F and S1G). Although not as efficient, loss of WT1 upon KOd9-11 and KOd11-14, similar to KOd4-7, occurred between 24 h and 48 h after Cas9 induction.
At d21, control and WT1 mutant organoids were of similar size, except for those derived from KOiPSC cells, which were considerably smaller (Figure S2A). Culture until d30 showed that KOiPSC organoids remained small, that KOd4-7 and KOd9-11 organoids overgrew, and that growth of KOd11-14 organoids was similar to controls. Profiling with cell-type-specific markers at d21 (Figures 1A, 1B, and S2B) revealed that SIX2-positive (SIX2pos) cells were abundant in KOiPSC and KOd4-7 organoids (50% and 38% of all cells, respectively), and absent from KOd11-14 organoids similar to controls. In addition, the proliferation marker KI67 was elevated, in particular upon KOiPSC and KOd4-7. Many KI67-positive (KI67pos) cells co-expressed SIX2, suggesting overgrowth of NPCs. KOd9-11 organoids displayed intermediate phenotypes that frequently did not differ significantly from controls.
Figure 1.

Loss of WT1 induces NPC overgrowth
(A) Quantification of subpopulations in un-edited, KOiPSC, KOd4-7, KOd9-11, and KOd11-14 d21 organoids by flow cytometry of indicated markers. Data are presented as mean percentage of positive cells ± SD derived from n = 5 (un-edited, KOiPSC, KOd4-7) or n = 2 (KOd9-11, KOd11-14) independent experiments. Two-sided Student's t test; p value: ns, > 0.05; ∗≤0.05; ∗∗≤0.01; ∗∗∗≤0.001; ∗∗∗∗≤0.0001.
(B) Staining of indicated markers in representative d21 organoids as in (A). Scale bar: 100 μm. White boxed regions are shown as blow-ups. Scale bar: 25 μm.
(C) Flow cytometry-based quantification of EPCAMhigh, EPCAMmid, and EPCAMlow populations (gating shown in Figure S2G) in organoids of indicated time points and genotypes. Data are shown as mean percentage of positive cells ± SD derived from n = 3 (d7); n = 4 (d9); n = 2 (d10); n = 5 (un-edited, KOiPSC, KOd4-7; d21) or n = 2 (KOd9-11, KOd11-14; d21) independent experiments. Two-sided student's t test; p value: ns > 0.05; ∗≤0.05; ∗∗≤0.01; asterisks are placed above the respective populations (EPCAMhigh and EPCAMmid).
PODXL- and NPHS1-expressing podocytes were strongly reduced in all WT1 mutant organoids, including KOd11-14 organoids (Figure 1B). Strikingly, the few detectable PODXL-positive cells co-expressed WT1, demonstrating that they are descendants of un-edited or heterozygous mutant NPCs or of NPCs harboring in-frame WT1 mutations. EPCAMhigh and LTL-positive (LTLpos) tubules were reduced, indicating impaired formation of tubules (Figures 1B and 1C). We estimate that approximately 15% of WT1-deficient cells expressed EPCAMhigh (Figure S2C). This was lower than un-edited control organoid cells (30%), confirming reduced epithelialization. Defective nephrogenesis was also observed in KOd11-14 organoids, in which exit from the SIX2pos state was unperturbed, indicating that WT1 drives NPC, and proximal tubule and podocyte differentiation through successive mechanisms. We sought to validate these findings with two independent WT1 gRNAs and found a reduction of WT1 and EPCAMhigh, maintenance of SIX2, elevation of KI67, and co-expression of SIX2 and KI67 in KOiPSC organoids. Notably, phenotypic strength scaled with the KO efficiency of the respective gRNA, demonstrating target specificity (Figures S2D and S2E).
While quantifying EPCAM expression, we found an expansion of EPCAMmid cells in KOiPSC and KOd4-7 d21 organoids (Figures 1C, S2C, S2F, and S2G). A proportion of these cells expressed SIX2 (33% and 26%, respectively). KOd11-14 organoids, in contrast, were indistinguishable from controls. Immunofluorescence confirmed expression of EPCAM in SIX2pos cells at lower levels than in fully differentiated tubules and not confined to the basolateral membrane (Figure S2H). Consistent with an epithelializing intermediate state, these structures also expressed low levels of CDH1 (Figure S2H). EPCAMmid cells emerged as early as d7 and preceded the formation of EPCAMhigh cells (Figure 1C). These cells are therefore likely equivalent to EPCAMdim cells in the human fetal kidney (Pode-Shakked et al., 2017), which are kidney progenitor intermediates undergoing a mesenchymal-epithelial transition (MET) and bridging the differentiation of EPCAMlow NPCs into EPCAMhigh renal vesicle cells. The frequency of EPCAMmid cells, in particular of those co-expressing SIX2, increased steadily in developing KOiPSC and KOd4-7 organoids (Figure S2I), suggesting that WT1 promotes progression beyond a pre-epithelial SIX2pos/EPCAMmid NPC transition state, but not the initiation of MET.
We noted increased SIX2 protein levels in NPCs at d9 (Figure S2J). To test if this is causal for progenitor overgrowth, we generated iPSCs with a DOX-inducible mCherry::T2A::SIX2 transgene and induced SIX2 by adding DOX from d7 and d9 onwards, thus initiating overexpression in NPCs and during nephrogenesis, respectively (Figures S2K and S2L). The former resulted in smaller organoids that, compared with uninduced controls, had fewer EPCAMhigh and LTLpos tubules and WT1-positive glomeruli (Figures S2M and S2N). Quantitative PCR (qPCR) showed that SIX2 was expressed roughly 30-fold higher in d7-induced NPCs than in uninduced controls (Figure S2K). In both, d7- and d9-induced organoids, however, KI67 expression was unchanged. SIX2, despite heterogeneous and mosaic expression, was detectable in differentiated EPCAMhigh and LTLpos tubule cells at d21 (Figure S2N).
Taken together, these observations suggest that the removal of WT1 impairs NPC progression and leads to organoid hyperplasia. We found that over-activation of SIX2 was not sufficient to recapitulate these phenotypes (Figures S2K–S2N), arguing for additional WT1 targets. Tubule and podocyte differentiation defects in KOd11-14 organoids indicate roles of WT1 in nephrogenesis that are independent of SIX2 silencing and consistent with WT1 activating podocyte-specific genes (Kann et al., 2015) and promoting epithelial tubule maturation (Berry et al., 2015).
WT1 drives developmental transcription
We decided to define the transcriptional changes induced by absence of WT1, and performed RNA sequencing (RNA-seq) of KOiPSC and KOd4-7 cells and respective controls at different time points during organoid development (Figure S3A). Inspection of cell state-specific markers (Figures S3B–S3D) revealed consistency between transcriptional and phenotypic changes: (1) NPC markers were induced by d5 and declined after d9 in control organoids but persisted upon KO of WT1; (2) markers of differentiation intermediates, and of mature podocytes and proximal tubules, were induced by d9 and d21 in un-edited controls, respectively, which was impaired in mutant organoids; and (3) transcriptional dynamics of epithelial-mesenchymal transition (EMT) signature genes, including EPCAM and CDH1, were perturbed in KO organoids.
To systematically define WT1-dependent gene regulation, we performed k-means clustering of transcripts that change significantly during organoid development and/or in KO organoids (Figures 2A and 2B Table S1). This identified 17 gene clusters, of which some were induced or repressed during organoid development and unchanged in mutants (clusters 5, 7, 11, and 14), while others were deregulated in KO organoids, in particular at d21 (clusters 1, 8, and 13). Notably, we did not identify clusters that were mis-regulated across all time points, suggesting that WT1's target genes depend on the developmental context. Also, expression defects in KOiPSC and KOd4-7 organoids, despite distinct growth rates (Figure S2A), were very similar, and indistinguishable at d21.
Figure 2.

Absence of WT1 impairs developmental transcription
(A) k-means clustering of 7,626 genes significantly changing in any of the shown contrasts. Left: time course of wild-type organoid development; log2-fold expression changes relative to iPSCs. Right: changes in KOiPSC or KOd4-7 cells at indicated time points; log2-fold expression changes relative to un-edited control at each time point. log2 FC = log2-fold change.
(B) Quantification of mRNA log2-fold expression changes of indicated gene clusters and in contrasts as specified in (A).
(C) Gene set overlap significance scores (see supplemental experimental procedures) of 17 gene clusters with fetal kidney cell type gene sets as defined in Lindström et al. (2018). Z scores are color coded. Diff, differentiate.
(D) t-SNE maps of scRNA-seq data from week 16 human fetal kidney (Hochane et al., 2019). Expression levels of indicated clusters and gene sets as in (C) are color-coded. prol, proliferation; Prox, proximal.
Because of the differentiation defects in WT1 KO organoids, we wondered if any of the gene clusters reflect cell-type-specific transcription. We made use of published gene sets that discriminate cell types of the human fetal kidney (Lindström et al., 2018) and calculated gene set overlap enrichments (see supplemental experimental procedures). This identified gene clusters 1, 8, 11, 13, and 17 to be most similar to markers of relevant cell types in the developing kidney (Figure 2C). Notably, cluster 11 was not changed, and cluster 17 only transiently deregulated in KO organoids (Figure 2A, Table S1). We validated cell type specificity of clusters 1, 8, and 13 genes by visualizing their expression in t-distributed stochastic neighbor embedding (t-SNE) maps of single-cell RNA-seq (scRNA-seq) of the week 16 human fetal kidney (Hochane et al., 2019). This confirmed co-expression with gene sets of kidney progenitor cells (M1), proliferating intermediates (M12 and M13), and podocytes (M6) and proximal tubule cells (M9), respectively (Figure 2D).
Collectively, the transcriptional defects correlate with the phenotypic persistence of kidney progenitor cells (cluster 1) and reduced tubular and glomerular formation (cluster 13) in mutant organoids. The specificity of the former to WT1 depletion before d11 (Figures 1A, 1B, S2A, S2B, S2C, S2F) is consistent with WT1 acting in and driving differentiation of NPCs. Although we cannot exclude that upregulation of cluster 8 genes in mutant organoids reflects growth of a proliferating transit amplifying cell population, we note (1) that cluster 8 genes are downregulated specifically at d21, suggesting cell type-independent transcription (Figures 2B and S3E); and (2) that a significant proportion of SIX2pos cells co-express KI67 (Figure 1A), whose encoding gene MKI67 is a cluster 8 gene (Table S1). Upregulation of cluster 8 genes is therefore likely due to NPC hyperproliferation rather than the persistence of an additional transit amplifying cell population.
WT1 KO organoids recapitulate transcriptional changes in Wilms tumors
To explore the similarity of WT1 KO kidney organoids and kidney tumors, we compared transcriptional changes in mutant d21 organoids with mean gene expression changes in WT (Gadd et al., 2017), kidney chromophobe tumor (KICH) (Davis et al., 2014), kidney renal papillary cell carcinoma (KIRP) (Network et al., 2016), and kidney renal clear cell carcinoma (KIRC) (Creighton et al., 2013) patients. This showed that the magnitude and the directionality of cluster 1 and 8 deregulation was conserved in WT but not KICH, KIRP, or KIRC tumors (Figure 3A). Pairwise comparison of transcriptome-wide changes confirmed that changes in mutant d21 organoids correlate most strongly with those of WT patients (Figure 3B). WT1 KO kidney organoids therefore resemble WT patient tissue, despite being derived in vitro and consisting of nephron cell types only (Morizane et al., 2015).
Figure 3.

WT1 KO organoids recapitulate the transcriptional changes of a Wilms tumor patient subgroup
(A) Unsupervised clustering of mean expression changes of clusters 1–17 in organoids and kidney tumors. Mean log2-fold expression changes in KOiPSC and KOd4-7 organoids relative to un-edited controls and in patient tumors relative to control tissue were used.
(B) Pearson correlation coefficients and unsupervised clustering of pairwise comparisons between scaled mean log2-fold expression changes as described in (A).
(C) Unsupervised clustering of mean expression changes of WTPGCs 1–8 in organoids and kidney tumors. Scaled mean log2-fold expression changes as described in (A) were used. myo_subset and non-myo_subset are the subsets of WT patients defined in Figure S3G.
(D) Unsupervised clustering of scaled mean log2-fold expression changes of WTPGC 3 genes as in (C). Gadd_1/2/3/4 are mean-centered scaled expression changes of WT patient subsets defined in Gadd et al. (2012). Cluster containing muscle genes is highlighted.
(E) Unsupervised clustering of log2-fold expression changes of cluster 1 genes in individual WT patients relative to corresponding normal tissue. Clusters containing NPC and muscle genes are highlighted.
Using gene ontology (GO) analysis (Table S1) we found enrichment of cell cycle terms in cluster 8, while cluster 1, despite containing key NPC TFs such as SIX1 and SIX2, was highly enriched for muscle-related processes (Figure S3F). Notably, WT1 mutations are associated with ectopic myogenesis in mice and in WT patients (Berry et al., 2015; Gadd et al, 2012, 2017; Miyagawa et al., 1998). To test if WT1 KO organoids resemble this patient subset, we first stratified WT patients by gene expression. Using k-means clustering (Figure S3G, Table S2), we identified several WT patient gene clusters (WTPGCs), including WTPGCs 6 and 8, that were deregulated in all 126 WT patients (Gadd et al., 2017) and enriched for developmental and cell cycle terms, respectively (Figure S3H, Table S2). WTPGC 3, in contrast, was upregulated in a subgroup of 15 patients only (myo_subset), enriched for muscle GO terms (Figure S3H), induced in previously described myogenic S2 and S4 WT patient subgroups (Gadd et al., 2012) (Figure S3I), and upregulated in d21 KO organoids (Figures 3C and 3D). Vice versa, unsupervised clustering of cluster 1 genes in WT patients (Figure 3E) identified NPC TFs upregulated in all samples, while the myogenic TFs MYOD1 and MYOG, and the skeletal muscle genes TNNC2 and MYLPF, were specifically dysregulated in the myo_subset of patients. We therefore conclude that transcriptional changes in WT1 mutant kidney organoids recapitulate those of a myogenic subset of WT patients.
Niche signals propagate WT1 mutant NPCs
The persistence of SIX2pos cells and NPC transcription in WT1 KO organoids may result from a block or a delay in developmental progression. We reasoned that serial passaging in differentiation-promoting conditions would discriminate between the two by enforcing the commitment of delayed but not of blocked mutant cells. To do so, we aggregated single cells derived from KOd4-7 d21 organoids and exposed them to organoid-forming conditions for 12d (corresponding to d9–d21 of iPSC differentiation), and repeated this procedure for up to four passages (Figure 4A). Since niche signals affect tumor growth and progression (Hanahan and Coussens, 2012), we also tested the role of environment signaling in passaging of SIX2pos cells. We therefore added defined ratios (0%, 10%, 25%, 75%, and 90%) of green fluorescent protein (GFP)-expressing wild-type d9 NPCs to dissociated KOd4-7 organoids during the aggregation step at the beginning of each passage, expecting that the wild-type kidney structures formed by these NPCs would provide niche signals to KO cells. Quantifying the percentage of GFP-expressing cells in chimeric organoids (Figure 4B) showed stable contribution of KO cells over passages, demonstrating that KOd4-7 d21 organoid cells grew as fast as wild-type d9 NPCs. Un-edited control d21 organoid cells proliferated less (Figure S4A). The mutant cell type composition, in contrast, varied across passages and mixing ratios (Figures 4B, 4C, and S4B): in the absence (0%) or presence of 10% and 25% wild-type cells, the fraction of SIX2pos cells increased from approximately 30% to 60% after the first passage, but gradually declined during further passaging. Un-edited control cells, in contrast, did not gain SIX2 expression (Figure S4C). In the presence of 75% wild-type cells, the percentage of SIX2pos cells remained at 30% over passages for at least 60d. Presence of 90% wild-type cells stabilized SIX2-expression in 10% of KOd4-7 cells after the first passage. Therefore, signals by wild-type cells, particularly at a wild-type/mutant cell ratio of 3:1, support self-renewal of KOd4-7 SIX2pos cells. At and below mixing ratios of 1:3, SIX2pos cells were lost during passaging. This was not accompanied by an induction of EPCAMhigh cells (Figure 4B) and therefore not due to overt tubular differentiation.
Figure 4.

Non-cell-autonomous regulation of self-renewal and heterogeneity
(A) Schematic of the experimental flow.
(B) Quantification of indicated cell populations in chimeric organoids after passaging with indicated ratios of wild-type GFP-expressing d9 NPCs and at indicated passages. p0 indicates KOd4-7 d21 organoids that were used as starting material for passaging. Data are shown as mean percentage ± SD from n = 1 or 2 independent experiments. Percentage of all cells is relative to all cells of chimeric organoids, and percentage of KOd4-7 cells is relative to all mutant cells, therefore excluding wild-type host cells.
(C) Staining at p3 of indicated markers and mixing ratios as outlined in (A). Scale bar: 100 μm.
(D) Images of organoids obtained after adding 12,500 RFP-positive un-edited d21 organoid cells (top) and KOd4-7 d21 organoid cells (bottom) to ruptured wild-type GFP-expressing organoids. Images were recorded at indicated time points using an Incucyte system. Scale bar: 1 mm.
Passaging in the presence of wild-type NPCs is not comparable with growth in mature tissues, such as during tumor progression in patients or in patient-derived xenograft mouse models. We therefore decided to use ruptured GFP-expressing d21 organoids as a differentiated cell substrate for passaging of WT1 mutant cells (Figure 4A). We estimated that d21 organoids contain 150,000–200,000 cells, to which we added 12,500 and 25,000 cells of RFP-expressing control or mutant single cells, corresponding to a mixing ratio of approximately 80%–90%. KOd4-7 d21 cells expanded visibly (Figure 4D), expressed SIX2 and KI67, and caused chimeric organoid overgrowth (Figures 4D, S4D, and S4E). In contrast, un-edited d21 cells contributed poorly to organoids. To exclude that this is because of poor survival upon dissociation or reaggregation, we used un-edited d9 NPCs as control, and found that they integrated as well as KOd4-7 d21 cells and contributed efficiently to EPCAM-positive epithelia. However, these cells did not induce organoid overgrowth or maintain SIX2 expression (Figures S4D–S4F). Passaging in d21 kidney organoids therefore enables persistence of SIX2 and proliferation of WT1 mutant cells. Notably this did not require extrinsic CHIR and FGF9 (Figures S4G–S4I), and is therefore independent of differentiation- and growth-promoting culture conditions.
We conclude that kidney organoids can be exploited for tumor cell transplantation and growth in a developmental (d9 NPCs) and mature (d21 organoids) tissue context. Our observations suggest cell-autonomous and non-cell-autonomous mechanisms that drive the ectopic growth of mutant NPCs, specifically a developmental block induced by absence of WT1 and pro-self-renewal signals from a wild-type niche environment.
Discussion
WT1 mutations in humans predispose to familial forms of WT and are found in 10%–20% of sporadic WT patients (Hastie, 2017; Treger et al., 2019). Loss of WT1 in humans is thought to transform immature kidney progenitor cells (Treger et al., 2019). However, direct evidence for a role of WT1 in human NPC development is elusive. Using timed deletion in human iPSC-derived kidney organoids, we identified two successive functions of WT1: driving exit from the NPC state and MET, and podocyte and tubule differentiation.
The second, later, function is reminiscent of the role of mouse Wt1 in glomerulus and renal tubule development (Berry et al., 2015; Kann et al., 2015), suggesting that WT1 promotes lineage progression through conserved target genes, such as podocyte-specifying TFs (Kann et al., 2015). The first, earlier, function of WT1 appears less conserved in organoids, although it remains to be determined if this is due to actual species-specific mechanisms or different experimental settings: (1) early deletion of Wt1 in mice results in an expansion of mesenchymal cells, but not in ectopic NPC marker expression or tumor formation (Berry et al., 2015; Huang et al., 2016), while KO of WT1 in organoids was sufficient for SIX2pos cell overgrowth. (2) Early deletion of Wt1 in mice blocks MET and, consequentially, formation of CDH1-expressing tubule cells (Berry et al., 2015). Accumulation of SIX2pos cells co-expressing EPCAMmid in KO organoids indicates that human WT1 coordinates NPC differentiation and epithelialization, too. However, the perturbed transcriptional induction of MET genes WNT4, CDH1, EPCAM, and other EMT signature genes, such as Claudins, at d11/12 was rescued by d21 (Figure S3D), and KO cells differentiated into EPCAMhigh-expressing tubule cells, although with reduced efficiency (Figure S2C). Importantly, formation of EPCAMhigh cells was impaired not only in KOiPSC and KOd4-7 but also in KOd11-14 organoids, in which exit from the NPC state and EPCAMmid regulation was unperturbed. Tubular differentiation defects in KO organoids are therefore due to WT1's role in epithelial maturation rather than its earlier function in activating MET. Notably, epithelial elements are also found in WT with WT1 lesions (Schumacher et al., 1997), and WT1 mutations can be detected in epithelial cells of patient-derived organoids (Calandrini et al., 2020).
In addition to perturbing kidney development, early loss of WT1 causes upregulation of muscle genes both in mouse (Berry et al., 2015) and in human kidney organoids. This indicates a conserved role of WT1 in repressing competing paraxial mesoderm fates, and suggests that expression of myogenic genes in a subset of WT patients (Gadd et al, 2012, 2017; Miyagawa et al., 1998) reflects the developmental history rather than plasticity of the tumors (Shukrun et al., 2014).
In mouse, constitutive WT1 deletion causes apoptosis of the metanephric mesenchyme (Kreidberg et al., 1993), while NPC induction is largely unperturbed in KOiPSC and KOd4-7 organoids (Figure S3B). Although this difference may be because of species-specific WT1 functions or because of non-cell-autonomous rescue in organoids by unrecombined wild-type cells, we speculate that in vitro culture conditions compensate for WT1 deficiency: Deletion of FGF receptor 1 (Fgfr1) and Fgfr2, and of Fgf20 and Fgf9 in mice causes phenotypes that are reminiscent of loss of WT1 (Barak et al., 2012; Poladia et al., 2006). WT1 directly binds to and regulates the transcription of several Fgf genes, and treatment of WT1 mutant embryonic kidneys with recombinant FGF20 rescues cell death (Motamedi et al., 2014). The extrinsic FGF9 that is added between d7 and d14 of organoid formation may therefore suppress the pro-apoptotic effect of WT1 loss and expose WT1's oncogenic role in developmentally more advanced NPCs.
Passaging in organoids showed that wild-type cells provide niche signals that maintain proliferation and expansion of mutant SIX2pos cells in the presence of differentiation-promoting cues. In particular, we were not able to propagate mutant SIX2pos cells in isolation. Future studies will be required to dissect how cell-to-cell contact, secreted signals, and spatial organization establish a growth-permissive environment. It is conceivable that WT1 ablation in developing organoids resembles cancer initiation in an intact tissue context, while cell mixing recapitulates subsequent aspects of tumor growth in patients. We note that primary WT biopsies can be repeatedly passaged as spheroids (Wegert et al., 2020) and organoids (Calandrini et al., 2020) in three dimensions. Although growth conditions differ, it is possible that WT1 KO kidney organoids recapitulate early stages of the disease, and that further tumor progression leads to independence of niche signals.
Taken together, we here establish a human kidney organoid-based tumorigenesis model by inducible deletion of WT1, complementing the current toolbox of pre-clinical WT models. We show that WT1 drives exit from the human NPC state and, consequentially, prevents NPC hyperplasia, and that loss of WT1 recapitulates defects observed in a clinically relevant patient subset. Overall, our work motivates the use of human kidney organoids to study pediatric kidney tumorigenesis. Tumor organoids provide access to the cancer cell type of origin, to crosstalk between different oncogenes and tumor suppressors, to interactions with the niche environment, and, eventually, to organotypic and phenotypic platforms for drug discovery and development.
Experimental procedures
Kidney organoids
iPSCs were differentiated into NPCs for 9 days on Laminin (Biolamina #LN521)-coated six-well-plates and, after single cell-dissociation using TrypLE Express (Thermo Fisher #12604013), transferred into ultra-low-attachment 96-well plates (Thermo Fisher #174925) for organoid formation in suspension culture (Morizane et al., 2015; Ungricht et al., 2021). SIX2 and Cas9 were induced by adding 1 μg/mL and 0.2 μg/mL DOX (Clontech #631311), respectively. Chimeric organoids were generated by aggregating single-cell suspensions of WT1 KO d21 organoids with GFP-expressing WT29-iCas9 d9 NPCs or with ruptured GFP-expressing WT29-iCas9 d21 organoids. See supplemental experimental procedures for details.
Flow cytometry
Single cells were fixed using the BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences #554714) according to the manufacturer's instructions with the exception that 0.4% bovine albumin fraction V solution (Thermo Fisher #15260037) in PBS was added before centrifugation. Cells were incubated with primary antibodies in permeabilization buffer at 4°C for 60 min, washed three times, incubated with secondary antibodies at 4°C for 60 min, washed three times, and resuspended in permeabilization buffer. Flow cytometry was performed on a BD LSRFortessa or BD LRSII, and data analyzed using FlowJo software.
Immunofluorescence
Kidney organoids were washed with PBS twice and fixed with 4% paraformaldehyde in PBS for 20 min at 4°C. After washing three times with PBS, organoids were resuspended in 50% sucrose (Sigma #84097) in PBS and stored at 4°C. Organoids were embedded in a 7.5% gelatin solution (Millipore #48722 dissolved in 10% sucrose in PBS) overnight at 4°C and mounted with Q Path Tissue OCT Medium (VWR #0011243) to generate frozen blocks that were cut into 10–14-μm sections using a Leica CM3050S cryostat. Sections were stained (see supplemental experimental procedures), images acquired on Zeiss LSM710 and LSM900 scanning head confocal microscopes, and processed with Fiji software.
Data availability
The accession number for the RNA-seq data reported in this study is ArrayExpress : E-MTAB-9957.
Author contributions
V.W., R.U., P.S.H., and J.B. conceived the study. V.W. designed and performed all experiments and analyzed the data. R.U. adapted the organoid protocol, generated the WT29-iCas9 and GFP-expressing WT29-iCas9 lines, and performed CDH1 staining of WT1 KO organoids. J.B. performed computational analyses. J.B. and P.S.H. supervised the study. V.W. and J.B. wrote the manuscript with input from all authors.
Conflicts of interest
The authors declare no competing interests.
Acknowledgments
S.A. Smallwood and team (FMI) for library preparation and sequencing; P. Papasaikas and M.B. Stadler (FMI) for help with computational analysis; L. Gelman (FMI) for help with confocal microscopy; M. Rittirsch for technical assistance; L. Morelli (Novartis) for help with virus production and infection; M. Mueller (Novartis) for providing the WT29 iPSC line; and H. Grosshans, S. Gasser (FMI), and S. Lienkamp (University of Zurich) for comments on the manuscript. V.W. acknowledges support by a Boehringer Ingelheim Fonds PhD fellowship, and J.B. from the Novartis Research Foundation.
Published: August 26, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.07.023.
Supplemental information
References
- Barak H., Huh S.-H., Chen S., Jeanpierre C., Martinovic J., Parisot M., Bole-Feysot C., Nitschké P., Salomon R., Antignac C., et al. FGF9 and FGF20 maintain the Stemness of nephron progenitors in mice and man. Dev. Cell. 2012;22:1191–1207. doi: 10.1016/j.devcel.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry R.L., Ozdemir D.D., Aronow B., Lindström N.O., Dudnakova T., Thornburn A., Perry P., Baldock R., Armit C., Joshi A., et al. Deducing the stage of origin of Wilms’ tumours from a developmental series of Wt1-mutant mice. Dis. Model Mech. 2015;8:903–917. doi: 10.1242/dmm.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandrini C., Schutgens F., Oka R., Margaritis T., Candelli T., Mathijsen L., Ammerlaan C., van Ineveld R.L., Derakhshan S., de Haan S., et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat. Commun. 2020;11:1310. doi: 10.1038/s41467-020-15155-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H., Tuveson D.A. Organoid models for cancer research. Annu. Rev. Cancer Biol. 2019;3:223–234. [Google Scholar]
- Creighton C.J., Morgan M., Gunaratne P.H., Wheeler D.A., Gibbs R.A., Robertson A.G., Chu A., Beroukhim R., Cibulskis K., Signoretti S., et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis C.F., Ricketts C.J., Wang M., Yang L., Cherniack A.D., Shen H., Buhay C., Kang H., Kim S.C., Fahey C.C., et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd S., Huff V., Huang C.-C., Ruteshouser E.C., Dome J.S., Grundy P.E., Breslow N., Jennings L., Green D.M., Beckwith J.B., et al. Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children’s Oncology Group study. Neoplasia. 2012;14:742–756. doi: 10.1593/neo.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd S., Huff V., Walz A.L., Ooms A.H.A.G., Armstrong A.E., Gerhard D.S., Smith M.A., Auvil J.M.G., Meerzaman D., Chen Q.-R., et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat. Genet. 2017;49:1487–1494. doi: 10.1038/ng.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Coussens L.M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Hastie N.D. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development. 2017;144:2862–2872. doi: 10.1242/dev.153163. [DOI] [PubMed] [Google Scholar]
- Hochane M., van den Berg P.R., Fan X., Bérenger-Currias N., Adegeest E., Bialecka M., Nieveen M., Menschaart M., Chuva de Sousa Lopes S.M., Semrau S. Single-cell transcriptomics reveals gene expression dynamics of human fetal kidney development. PLoS Biol. 2019;17 doi: 10.1371/journal.pbio.3000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Mokkapati S., Hu Q., Ruteshouser E.C., Hicks M.J., Huff V. Nephron progenitor but not stromal progenitor cells give rise to Wilms tumors in mouse models with β-catenin activation or Wt1 ablation and Igf2 upregulation. Neoplasia. 2016;18:71–81. doi: 10.1016/j.neo.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynds R.E., Vladimirou E., Janes Sam.M. The secret lives of cancer cell lines. Dis. Model Mech. 2018;11 doi: 10.1242/dmm.037366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann M., Ettou S., Jung Y.L., Lenz M.O., Taglienti M.E., Park P.J., Schermer B., Benzing T., Kreidberg J.A. Genome-wide analysis of Wilms’ tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. J. Am. Soc. Nephrol. 2015;26:2097–2104. doi: 10.1681/ASN.2014090940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten K., de Visser K.E., van Miltenburg M.H., Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. Embo Mol. Med. 2017;9:137–153. doi: 10.15252/emmm.201606857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreidberg J.A., Sariola H., Loring J.M., Maeda M., Pelletier J., Housman D., Jaenisch R. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- Lindström N.O., Brandine G.D.S., Tran T., Ransick A., Suh G., Guo J., Kim A.D., Parvez R.K., Ruffins S.W., Rutledge E.A., et al. Progressive recruitment of mesenchymal progenitors reveals a time-dependent process of cell fate acquisition in mouse and human nephrogenesis. Dev. Cell. 2018;45:651–660. doi: 10.1016/j.devcel.2018.05.010. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa K., Kent J., Moore A., Charlieu J.-P., Little M.H., Williamson K.A., Kelsey A., Brown K.W., Hassam S., Briner J., et al. Loss of WT1 function leads to ectopic myogenesis in Wilms’ tumour. Nat. Genet. 1998;18:15–17. doi: 10.1038/ng0198-15. [DOI] [PubMed] [Google Scholar]
- Morizane R., Lam A.Q., Freedman B.S., Kishi S., Valerius M.T., Bonventre J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015;33:1193–1200. doi: 10.1038/nbt.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamedi F.J., Badro D.A., Clarkson M., Lecca M.R., Bradford S.T., Buske F.A., Saar K., Hübner N., Brändli A.W., Schedl A. WT1 controls antagonistic FGF and BMP-pSMAD pathways in early renal progenitors. Nat. Commun. 2014;5:4444. doi: 10.1038/ncomms5444. [DOI] [PubMed] [Google Scholar]
- Network C.G.A.R., Linehan W.M., Spellman P.T., Ricketts C.J., Creighton C.J., Fei S.S., Davis C., Wheeler D.A., Murray B.A., Schmidt L., et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 2016;374:135–145. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pode-Shakked N., Gershon R., Tam G., Omer D., Gnatek Y., Kanter I., Oriel S., Katz G., Harari-Steinberg O., Kalisky T., et al. Evidence of in vitro preservation of human nephrogenesis at the single-cell level. Stem Cell Reports. 2017;9:279–291. doi: 10.1016/j.stemcr.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poladia D.P., Kish K., Kutay B., Hains D., Kegg H., Zhao H., Bates C.M. Role of fibroblast growth factor receptors 1 and 2 in the metanephric mesenchyme. Dev. Biol. 2006;291:325–339. doi: 10.1016/j.ydbio.2005.12.034. [DOI] [PubMed] [Google Scholar]
- Schumacher V., Schneider S., Figge A., Wildhardt G., Harms D., Schmidt D., Weirich A., Ludwig R., Royer-Pokora B. Correlation of germ-line mutations and two-hit inactivation of the WT1 gene with Wilms tumors of stromal–predominant histology. Proc. Natl. Acad. Sci. U S A. 1997;94:3972–3977. doi: 10.1073/pnas.94.8.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukrun R., Pode-Shakked N., Pleniceanu O., Omer D., Vax E., Peer E., Pri-Chen S., Jacob J., Hu Q., Harari-Steinberg O., et al. Wilms’ tumor blastemal stem cells dedifferentiate to propagate the tumor bulk. Stem Cell Reports. 2014;3:24–33. doi: 10.1016/j.stemcr.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treger T.D., Chowdhury T., Pritchard-Jones K., Behjati S. The genetic changes of Wilms tumour. Nat. Rev. Nephrol. 2019;15:240–251. doi: 10.1038/s41581-019-0112-0. [DOI] [PubMed] [Google Scholar]
- Ungricht R., Guibbal L., Lasbennes M.-C., Orsini V., Beibel M., Waldt A., Cuttat R., Carbone W., Basler A., Roma G., et al. Genome-wide screening in human kidney organoids identifies novel aspects of nephrogenesis. bioRxiv. 2021 doi: 10.1101/2021.05.26.445745. [DOI] [PubMed] [Google Scholar]
- Wegert J., Zauter L., Appenzeller S., Otto C., Bausenwein S., Vokuhl C., Ernestus K., Furtwängler R., Graf N., Gessler M. High-risk blastemal Wilms tumor can be modeled by 3D spheroid cultures in vitro. Oncogene. 2020;39:849–861. doi: 10.1038/s41388-019-1027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this study is ArrayExpress : E-MTAB-9957.