

Abstract
Long-term complications such as radiation-induced second malignancies occur in a subset of patients following radiation-therapy, particularly relevant in pediatric patients due to the long follow-up period in case of survival. Radiation-induced gliomas (RIGs) have been reported in patients after treatment with cranial irradiation for various primary malignancies such as acute lymphoblastic leukemia (ALL) and medulloblastoma (MB). We perform comprehensive (epi-) genetic and expression profiling of RIGs arising after cranial irradiation for MB (n = 23) and ALL (n = 9). Our study reveals a unifying molecular signature for the majority of RIGs, with recurrent PDGFRA amplification and loss of CDKN2A/B and an absence of somatic hotspot mutations in genes encoding histone 3 variants or IDH1/2, uncovering diagnostic markers and potentially actionable targets.
Subject terms: Cancer genomics, CNS cancer
Radiation-induced gliomas (RIGs) have been reported in patients after treatment with cranial irradiation for various primary malignancies but their origin are still unclear. Here, the authors define the genomic, epigenetic and transcriptional landscape of 32 RIGs cases.
Introduction
Radiation therapy (RT) constitutes an essential element in the standard treatment of many cancers, improving clinical outcomes of patients with childhood malignancies including leukemia and central nervous system (CNS) tumors1. However, radiation-induced malignancies are observed post-RT in a subset of patients2,3, especially in pediatric patients due to their prolonged follow-up in case of long-term survival4,5.
Radiation-induced gliomas (RIGs) are known to arise in a subset of patients receiving cranial RT for the treatment of different primary malignancies including acute lymphoblastic leukemia (ALL) and medulloblastoma (MB), occurring after a variable latency ranging from 2.5 to 35 years after irradiation (Table 1)6–18.
Table 1.
Summary of radiation-induced gliomas reported in previous studies.
| Reference | Number of cases | Primary malignancy | Latency period (years) | Age at RIG diagnosis (years) | Gender ratio (m:f) | Death (y/number of cases) | OS (months) | PDGFRA status | CDKN2A/B status | Further genetic alterations |
|---|---|---|---|---|---|---|---|---|---|---|
| Brat et al.6 | 6 | ALL, Pineal tumors, Lymphoblastic Lymphoma, Pituary adenoma, Rhabdomyosarkoma, Craniopharyngeoma | 5–23 | 9–60 | 2:1 | N.D. | N.D. | N.D. | N.D. | – |
| Donson et al.7 | 5 | Burkitt’s Lymphoma, MB, low-grade astrocytoma, ependymoma, ALL | 3–15 | 11–23 | 3:2 | 5/5 | 1–10 | Overexpressed | N.D. | – |
| Lopez et al.20 | 12 | Craniopharyngioma, Germinoma, Medulloblastoma, Pineocytoma, ALL, Hodgkin’s Lymphoma | 4–41 | 7–48 | 8:4 | 5/7 | <24 | Amplified in 42%, mutated in one case | Loss in 33% | Amplified CDK4 in 33%, amplified MET in 17% |
| Nakao et al.13 | 4 | Pituary adenoma, Meduloblastoma, Craniopharyngioma, PNET | 22–29 | 32–55 | 3:1 | 4/4 | 11–30 | N.D. | N.D. | IDH1 and H3F3A wild type |
| Paugh et al.21 | 10 | ALL, Germinoma, MB, Ependymoma | N.A. | 8–19 | N.A. | 7/9 | 8–91 | Amplified in 50% | Loss in 50% | 1q gain (50%), 1p loss (50%), 13q loss (70%) |
| Phi et al.22 | 5 | Medulloblastoma (SHH, Group3) | 4.3–10 | 9.2–17 | 2:3 | N.D. | N.D | Missense mutation, gene fusion | N.D. | TP53 (somatic mutations or 17p loss), 7q gain (EZH2) |
| Romeike et al.16 | 7 | ALL, MB | 7–14 | 9–19 | 4:3 | 7/7 | 9–27 | N.D. | N.D | – |
| Walter et al.18 | 7 | ALL | 7–14 | 10–24 | 4:3 | 5/7 | 0.1–93 | N.D. | N.D. | – |
OS overall survival, N.D. not determined.
RIGs commonly display an aggressive clinical course associated with poor prognosis, making early and precise diagnosis (including distinguishing RIGs from recurrences of the primary tumor) crucial for optimal treatment planning7,8. This is particularly challenging in cases where putative recurrent CNS tumors are not biopsied or resected, and where material for histological or molecular confirmation is therefore lacking.
Histopathologically, most RIGs present as high-grade gliomas (HGG) reminiscent of their sporadic counterparts7,8,13,14,19, and histopathological features to distinguishing them from de-novo HGG are missing6,7. A report of morphological features of both MB and HGG within secondary tumors led to the hypothesis of transformation from MB (stem-)cells as a possible route for RIG development11, while the higher genetic homogeneity of RIGs compared to de-novo pediatric HGG is suggestive of a common origin for RIGs7. Genetic alterations including gain of chromosome arm 1q, loss of CDKN2A/B, and PDGFRA amplification or overexpression have been frequently observed in RIGs13,20–22, and—in contrast to sporadic pediatric HGG—recurrent somatic hotspot mutations in genes encoding histone 3 variants or IDH1/2 were found to be uncommon13,20–22. To date, however, most series have been relatively small, and there remains some lack of clarity about the origin of RIGs and the underlying molecular and biological mechanisms leading to their formation.
Recent studies have shown that DNA methylation profiling represents a robust and reproducible approach to classify CNS tumors into clinically meaningful entities23–26. Thereby, pediatric HGG represent a heterogeneous group of CNS tumors, clearly distinguishable from their adult counterparts21,27–29, that can be classified into distinct subgroups with characteristic genetic and epigenetic alterations and clinical associations30–33. Next-generation sequencing and transcriptomic profiling approaches represent complementary tools for molecular characterization of tumorigenic processes.
Here, we performed comprehensive molecular characterization of RIGs to detect distinctive, (epi-)genetic features which might predict or explain their formation after RT, and/or which may act as diagnostic or therapeutic markers.
Results
Clinical characteristics
Patients in our series developing a radiation-induced glioma (RIG, n = 32) were previously treated with cranial irradiation for MB (MB-RIG, n = 23) or ALL (ALL-RIG, n = 9). A variety of different RT regimens was administered, including craniospinal radiotherapy in MB patients or prophylactic cranial irradiation in ALL patients. In line with a higher probability of RIG occurrence in areas receiving a higher effective radiation dose, most MB-RIG tumors were encountered in the cerebellum (20/23, 87% vs. 3/23 in the cerebral hemisphere), the location with the highest dose in RT. ALL-RIG tumors predominantly appeared in the cerebral hemisphere (7/9, 78%, vs 3/23 in MB-RIGs, p < 0.01; Fig.1a), with only two cases arising in the posterior fossa. The gender ratio was slightly shifted towards males (male:female ratio: 1.28:1) (Fig. 1b). The latency for RIG occurrence ranged from 2 to 30 years in MB patients (n = 23, median: 5 years) and from 3–17 years in ALL patients (n = 9, median: 8 years; Fig. 1c).
Fig. 1. Clinical patient characteristics.

Tumor location for 23 MB-RIG and 9 ALL-RIG illustrated with circled numbers indicating the number of patients with RIG occurrence in the respective brain region (a). Significant differences in tumor location were observed between MB-RIG and ALL-RIG patients (p < 0.01). Gender distributions, age at RIG diagnosis (b) and latency period (c) between cranial irradiation and RIG diagnosis are shown with numbers in brackets indicating group size. MB-RIG patients displayed a wider distribution regarding the age at RIG diagnosis and latency period. RIG radiation-induced glioma, MB-RIG post-medulloblastoma RIG, ALL-RIG post-ALL RIG.
For 16/23 MB-RIG and 5/9 ALL-RIG patients with available follow-up data, we observed an aggressive clinical course that was as bad as or even slightly worse than histone 3 K27M-mutant tumors – with 13 out of 16 MB-RIG and four out of five ALL-RIG patients showing fatal outcome during the follow-up period (OS: 6 months, range: 3–10 months vs 8.5 months, range: 2–18 months in MB-RIG patients; Suppl. Table 1). One ALL-RIG (RIG_09, follow-up period: 3 months) and 3 MB-RIG patients diagnosed at the age of 14 year (RIG_22, follow-up period: 9 months), 15 years (RIG_30, follow-up period: 3 months), and 39 years (RIG_14, follow-up period: 18 months) were known to be alive at last follow-up, with the latter showing signs of progression after 12 months (Fig. 2).
Fig. 2. Kaplan–Meier curves illustrating overall survival for RIGs with representative CNS tumor entities.

PXA pleiomorphic xanthoastrocytoma, pedGBM_RTK1 pediatric glioblastoma subclass RTK1, pedGBM_RTK2 pediatric glioblastoma subclass RTK2, pedGBM_MYCN pediatric glioblastoma subclass MYCN, DMG K27 diffuse midline glioma H3 K27 mutant, GBM G34 glioblastoma, H3.3 G34 mutant.
DNA methylation profiling of radiation-induced gliomas and primary medulloblastomas
DNA methylation patterns of both MB- and ALL-RIGs were analyzed by unsupervised clustering and t-SNE alongside 120 reference samples from pediatric HGG subgroups with known molecular features. The majority of RIGs after both primary indications closely resembled those of pedGBM_RTK1 tumors (29/32, 91%; Fig. 3a, b)32. Two MB-RIG and one ALL-RIG showed a methylation pattern more similar to pleomorphic xanthoastrocytomas (PXA) (Fig. 3b). Thus, RIG DNA methylation patterns suggest a high degree of similarity (and likely common origins) between tumors, and were clearly distinct from any other HGG DNA methylation class.
Fig. 3. Molecular classification of RIGs by DNA methylation profiling.

Unsupervised analysis of 32 RIG tumors was conducted based on the 10,000 most variably methylated probes. Through a t-SNE analysis, the majority of RIG tumors form a homogenous group of tumors resembling the pediatric GBM RTK1 group. ALL-RIG and MB-RIG samples were compared with 120 well-characterized reference samples representing CNS tumors of known histological and/or molecular subtype. PXA pleiomorphic xanthoastrocytoma, pedGBM_RTK1 pediatric glioblastoma subclass RTK1, pedGBM_RTK2 pediatric glioblastoma subclass RTK2, pedGBM_MYCN pediatric glioblastoma subclass MYCN, DMG_K27 diffuse midline glioma H3 K27 mutant, GBM_G34 glioblastoma, H3.3 G34 mutant.
The 11 pre-RIG primary MBs for which DNA was available were also profiled, and found to represent all four major MB subgroups (MB-WNT: 1/11; MB-SHH: 3/11; MB-Group 3: 2/11, MB-Group 4: 5/11), with no obvious enrichment for a particular primary subtype relative to their overall incidence rate. DNA methylation and CNV profiling confirmed the distinct biological nature between the primary MB and secondary RIG using t-SNE and copy-number analyses (Fig. 4a, b). Histological staining for hematoxylin and eosin (Fig. 4c) of two paired primary medulloblastoma and radiation-induced glioma couples (MB/RIG_28, MB/RIG_29) display distinct morphological characteristics.
Fig. 4. Comparison of matched primary MB and post-radiation glioma samples.

T-SNE analysis of primary MB and post-MB RIG compared with existing MB subgroups (a) and representative copy-number profile (b) reveal distinct genome-wide methylation patterns and recurrent high-level PDGFRA amplification and CDKN2A/B deletion. Available primary MB and post-MB RIG samples (n = 11 pairs) were investigated with 100 MB reference samples representing established MB subtypes57,70. MB and RIG pairs from our series were encircled and linked with dotted lines. Histological staining for hematoxylin and eosin (c) of two paired primary medulloblastoma and radiation-induced gliomas (MB/RIG_28, MB/RIG_29) display distinct morphological characteristics. Scale bar represents 200 μm. T-SNE t-stochastic neighboring embedding, MB medulloblastoma, MB-SHH-INF medulloblastoma sonic-hedgehog infant subgroup, MB-SHH-AD medulloblastoma sonic-hedgehog adult subgroup, CNP copy-number profile.
RIGs display recurrent PDGFRA amplification and loss of CDKN2A/B
Amplification of PDGFRA (6/9 ALL-RIG; 11/23 MB-RIG) and loss of CDKN2A/B (4/9 ALL-RIG; 17/23 MB-RIG) represented the most common copy-number alterations (CNAs) in RIGs, with 2/8 ALL-RIG and 8/23 MB-RIG patients exhibiting co-occurrence of both (Fig. 5). This is in keeping with the methylation profile of these tumors matching to the pedGBM_RTK1 group, where both changes are also well-known characteristic features. Indeed, the overall frequency of PDGFRA amplification in the RIG cohort (17/32, 55%) is higher than in an unselected pedHGG cohort, but is not significantly higher than that reported in the pedGBM_RTK1 subgroup (33%, p = 0.14)32. Amplification of MET (9/32, 28%; previously also linked to MET fusion in pediatric GBMs34) and CDK4 (5/32, 16%) were frequently encountered in our cohort. Additional broader chromosomal CNAs included gain of chromosome arm 1q (16/32, 50%), 1p deletion (19/32, 59%), 6q deletion (18/32, 56%), 13q deletion (23/32, 72%), and 14q deletion (16/32, 45%) (Fig. 5). A more focused analysis was performed to illustrate the potential discrepancies between RIGs (n = 32) and the non-RIG pedGBM_RTK1 tumors (n = 20) regarding their respective characteristic copy-number changes, with both groups exhibiting a similar pattern of chromosomal gains and losses (Suppl. Fig. 1a, c). The number of chromosomal breakpoints in RIGs and non-RIG pedGBM_RTK1 tumors were further visually determined to estimate the extent of genomic rearrangement caused by double-strand DNA breaks. Overall, RIGs display a significantly wider range and a higher number of chromosomal breakpoints (median: 38.5, range: 4–134) compared to non-RIG pedGBM_RTK1 tumors (median: 23, range: 13–51; two-sided t-test: p < 0.04) (Suppl. Fig. 1 d).
Fig. 5. Oncoplot illustrating the genomic landscape in RIGs.

Summary of recurrent, characteristic genetic alterations (focal copy-number alterations, mutations) identified through DNA methylation arrays and whole-exome/gene panel sequencing. MGMT O6-methylguanine–DNA methyltransferase. ALL acute lymphoblastic leukemia, MB medulloblastoma.
Genomic profile of radiation-induced gliomas
RNA sequencing (n = 9), gene panel (n = 5), and whole-exome (n = 13) sequencing with matched blood samples were performed for a subset of RIG samples. None of the RIGs harbored somatic hotspot mutations in genes encoding histone 3 variants, IDH1/2, BRAF, or in the TERT promoter region. Four out of 18 tested samples (22%) exhibited somatic mutations in the TP53 gene. Additional somatic mutations included CBL, PDGFRA, NTRK2, EGFR, RAF1, ATRX, and BCOR (Suppl. Table 1). One patient (RIG_25) harbored a TP53 germline splice site mutation. One patient (RIG_14) was clinically diagnosed with Li-Fraumeni syndrome, but tumor material was insufficient for molecular testing. Further germline alterations associated with cancer predisposition syndromes were absent in the remainder of cases with germline material available (n = 13).
Analysis of RNA sequencing data (n = 9) revealed relevant gene fusions including PTPRZ1:MET (RIG_11, RIG_25), CAPZA2:MET (RIG_09), FYCO1:RAF1 (RIG_08), and GFAP1:NTRK2 (RIG_13) (Fig. 6). Interestingly, the FYCO1:RAF1 and GKAP1:NTRK2 fusions were detected in two of the three tumors from the PXA-like subgroup based on DNA methylation profiling (Fig. 3). Both rearrangements are predicted to lead to a constitutive activation of the kinase domain, via loss of the N-terminal regulatory domain (RAF1) or constitutive dimerization (NTRK2). Deriving from a complex structural rearrangement, PRPRZ1:MET fusion proteins harbor an almost full length MET protein driven by the highly active PTPRZ1 promoter. All MET-fused cases (RIG_09, RIG_11, RIG_25) demonstrated amplification of MET on copy-number profiles derived from DNA methylation arrays. While all four genetic rearrangements, which result in aberrant MAPK/ERK pathway activation, were reported in high-grade gliomas, none of them were previously discovered in RIGs34–36.
Fig. 6. Alternative genetic rearrangements leading to aberrant MAPK/ERK pathway signaling.

Partial deletions of chromosomal arm 3p induce formation of a FYCO1:RAF1 fusion (a), with occurrence of a GKAP1:NTRK2 fusion (d) emerging through inversion of a chromosomal arm 9q segment. Both rearrangements are predicted to lead to constitutive activation of the kinase domain (KD). Induced by complex rearrangements, CAPZA2:MET (b) and PTPRZ1:MET fusion (c, e) contained almost the full length MET protein, with overexpression driven by the highly active CAPZA2 or PTPRZ1 promoter.
Transcriptional profiling of radiation-induced gliomas
Array-based gene expression analysis was performed on seven RIG samples as well as 24 additional tumors representing the H3/IDH-wildtype pediatric HGG pedGBM_RTK1 (n = 5), pedGBM_RTK2 (n = 3), pediatric HGG MYCN (n = 8)32, and PXA (n = 8) subgroups. Unsupervised hierarchical clustering analysis based on the 100 most differentially expressed genes recapitulated the methylation-defined subgrouping, with the largest fraction of the RIGs forming a homogenous expression pattern, resembling pedGBM_RTK1 tumors. Two cases (RIG_08, RIG_13), previously categorized into the PXA methylation group, also displayed an expression profile similar to PXAs (Fig. 7). The level of expression glioma-characteristic genes (e.g. CD34, GFAP, OLIG2, MAP2, MKI67, RBFOX3, SOX2, SYN; Suppl. Fig. 2) between RIGs and sporadic H3/IDH-wildtype pediatric HGGs did not reveal any notable differences, supporting the conclusion of a general similarity in their pathway activation.
Fig. 7. Heatmap representing the expression levels of the 100 most significant, differentially expressed genes comparing RIG samples with reference groups (PXA, pedGBM_RTK1, pedGBM_RTK2, and pedGBM_MYCN).
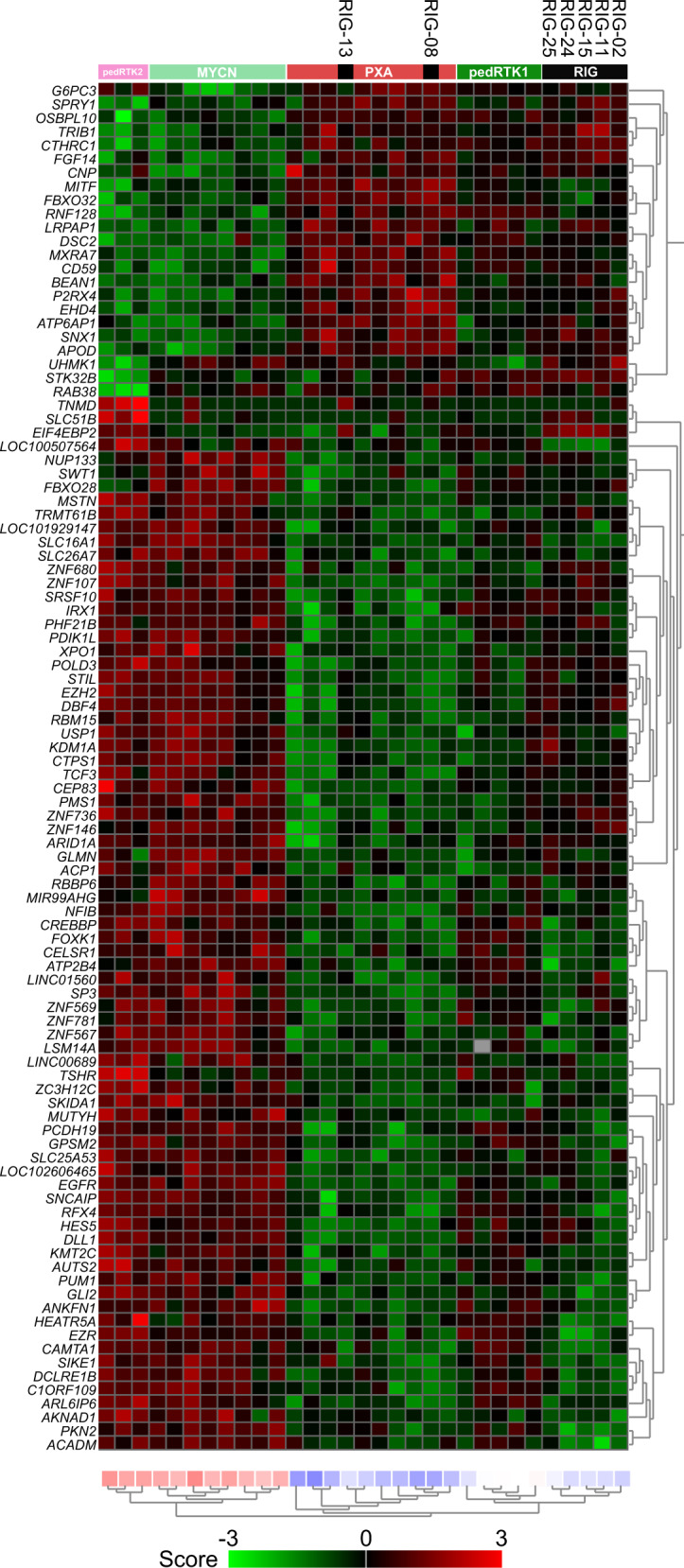
Unsupervised hierarchical clustering reveals that gene expression profiling reflects our aforementioned findings based on DNA methylation profiling, with most RIGs resembling the pedGBM_RTK1 subgroup and a small subset more similar to PXAs. Each column represents one sample and each lane represents one gene. Expression levels are indicated by a color scale as indicated. PXA pleiomorphic xanthoastrocytoma, pedGBM_RTK1 pediatric glioblastoma subclass RTK1, pedGBM_RTK2 pediatric glioblastoma subclass RTK2, pedGBM_MYCN pediatric glioblastoma subclass MYCN.
Discussion
RIGs represent a fatal long-term side effect of cranial irradiation2,3,19,37–43. A better understanding of the occurrence of these tumors and their molecular background is therefore of important clinical relevance. Our study supports the findings that RIGs harbor recurrent genetic alterations converging on an aberrant activation of the MAPK/ERK pathway (particularly via PDGFRA) together with loss of cell cycle control, facilitating tumorigenesis7,20,35,36. Importantly, we show that RIGs harbor a largely homogenous genetic and epigenetic profile, closely resembling sporadic pediatric GBM RTK1 tumors32. This suggests that both radiation-induced and sporadic pedGBM_RTK1 tumors might share a common cell of origin, which could be particularly vulnerable to ionizing radiation. However, the outcome of RIG patients is particularly poor, and worse than what has previously been described for sporadically occurring PDGFRA-amplified, 9p21-deleted pediatric GBM RTK1 tumors32. The discrepancy could suggest that additional oncogenic mechanisms might play a role during RIG formation, e.g. the trend towards greater genomic instability in the RIG cohort, as indicated by the elevated frequency of chromosomal breakpoints compared to their sporadic counterparts, potentially caused by radiation-induced DNA double-strand breaks. On the other hand, the RIG patient cohort also typically represents children with a long history of often aggressive prior treatment for their primary malignancy, which means that certain treatment options typically available at first diagnosis of GBM (e.g. radiotherapy) may no longer be an option for RIG patients44,45. It is also anticipated that the general health status of RIG patients is likely to be worse than that of primary-diagnosis counterparts. Further, some RIGs arising after MB were treated with an MB relapse protocol on the assumption that the new lesion was a recurrence of the primary tumor in the absence of biopsy confirmation, posing a possible risk that these protocols may be less efficacious against high-grade glioma than upfront GBM protocols. Thus, there are a number of key clinical reasons why an inferior survival might be expected, in addition to molecular mechanistic differences.
A small subset of RIGs with a more PXA-like profile harbored potentially druggable alterations in RAF1 and NTRK2, and it will be of interest to investigate the precise characteristics of this group in larger cohorts. Our findings also do not at present suggest a substantial contribution of germline alterations in known tumor-associated genes in the occurrence of RIGs. However, further comprehensive germline analyses with matched blood samples are required to elucidate in full the potential role of hereditary predisposition in RIG formation.
RIG following MB of various subtypes as well as after ALL displayed converging fingerprints, despite the diversity of applied radiation protocols (MB-RIGs arose mostly at the site of local boost, while ALL-RIGs occurred throughout the cranial radiation field). All of the four main molecular MB subgroups were represented in the primary lesions, with no recurrent alterations observed in the primary MBs. Our findings unfortunately suggest that no conclusions can currently be drawn regarding predictive features in the patient or primary tumor for assessing the probability of subsequent RIG formation. An important question to address in future will be the precise role of radiotherapy protocols (craniospinal vs local boost, fractionation schema, photons/protons/heavy ions) in determining the risk of secondary malignancy46–50.
The ALL-RIGs included in our series were all diagnosed with their second tumor between 9 and 18 years of age, suggesting a particularly vulnerable but relatively narrow time frame for RIG occurrence after ALL. If confirmed in larger series, this could be an important variable for the planning of surveillance in this patient population. In contrast, both the age at diagnosis and latency after the primary tumor were much more varied for medulloblastoma. The earliest RIG occurred after a latency period of only 2 years following treatment for MB – within a timeframe which could easily allow misidentification as a recurrence of the primary tumor. Molecular characterization of 31 clinically-presumed relapse MBs within the prospective INFORM study51 revealed that 6/31 ‘relapsed’ MBs (19%) were in fact secondary HGG (unpublished data). In light of their aggressive clinical course and fundamentally different biology, it is essential to distinguish newly-arising RIGs from true recurrences in order to adjust treatment planning as early as possible to increase treatment efficacy. Thus, biopsy of recurrent lesions after MB is clearly warranted and should be considered whenever surgically feasible. Furthermore, the observation that a subset of prognostically favorable MBs (e.g. the MB-WNT subgroup) are also at risk of developing RIGs, highlights the urgency of current efforts to further examine the application of alternative treatment modalities (e.g. proton/heavy ion beam therapy) or the general de-intensification of radiation therapy in these patients47,52–58.
In summary, our findings demonstrate that RIGs are an aggressive, relatively homogenous group of CNS tumors with recurrent amplification of PDGFRA and loss of CDKN2A/B in the absence of somatic H3/IDH hotspot mutations. Our study uncovers possible similarities in origins with the pedGBM_RTK1 group of sporadic HGG, which will be important for further understanding the mechanisms by which these secondary tumors arise.
Methods
Patient population and tumor samples
Criteria for patient selection adapted from Cahan et al.59,60 included (1) a glioma emerging from the previously irradiated field, and (2) a non-glial primary malignancy, in this series MB (n = 23) and ALL (n = 9), to exclude potential tumor progression/recurrence not directly related to radiotherapy61–63.
This study is covered by the ethical approval of the Heidelberg University’s medical faculty. Additional clinical data and tumor material were collected by international collaborating centers according to local ethical and institutional review board approval and collected at the German Cancer Research Center (DKFZ, Heidelberg, Germany). Informed consent for molecular profiling was obtained from all patients and their legal representatives.
DNA methylation profiling
The Illumina Infinium HumanMethylation450 (450k) array and Illumina Infinium MethylationEPIC (EPIC) array were used to obtain genome-wide DNA methylation profiles, according to the manufacturer’s instructions (Illumina, San Diego, USA). Data were generated at the Genomics and Proteomics Core Facility of the DKFZ (Heidelberg, Germany) and St. Jude Children’s Research Hospital (Memphis, USA). DNA methylation data was generated from both fresh-frozen and formalin-fixed paraffin-embedded (FFPE) tissue samples. On-chip quality metrics of all samples were carefully controlled.
Copy-number variation (CNV) analysis from 450k and EPIC methylation array data was performed using the conumee Bioconductor package version 1.12.0.
All computational analyses were performed in R version 3.4.4 (R Development Core Team, 2019). Raw signal intensities were obtained from IDAT-files using the minfi Bioconductor package version 1.24.0. Illumina EPIC and 450k samples were merged to a combined data set by selecting the intersection of probes present on both arrays (combineArrays function, minfi). Each sample was individually normalized by performing a background correction (shifting of the 5th percentile of negative control probe intensities to 0) and a dye-bias correction (scaling of the mean of normalization control probe intensities to 10,000) for both color channels. Subsequently, a correction for the type of material tissue (FFPE/frozen) and array (450k/EPIC) was performed by fitting univariate, linear models to the log2-transformed intensity values (removeBatchEffect function, limma package version 3.34.5). The methylated and unmethylated signals were corrected individually. Beta-values were calculated from the retransformed intensities using an offset of 100 (as recommended by Illumina).
Before further analysis, the following filtering criteria were applied: Removal of probes targeting the X and Y chromosomes (n = 11,551), removal of probes containing a single-nucleotide polymorphism (dbSNP132 Common) within five base pairs of and including the targeted CpG-site (n = 7998), probes not mapping uniquely to the human reference genome (hg19) allowing for one mismatch (n = 3965), and 450k array probes not included on the EPIC array. In total, 428,230 probes were kept for downstream analysis64–66.
To perform unsupervised dimension reduction, the remaining probes were used to calculate the 1-variance weighted Pearson correlation between the samples. The resulting distance matrix was used as input for t-SNE analysis (t-Distributed Stochastic Neighbor Embedding; Rtsne package version 0.13). The following non-default parameters were applied: theta = 0, pca = F, max_iter = 2500 perplexity = 20.
To perform unsupervised hierarchical clustering, the 10.000 probes with highest standard deviation were selected to calculate the Euclidean distance between samples, followed by applying Wards linkage method for sample clustering. In the heatmap, representation probes were reordered by complete linkage hierarchical clustering of the Euclidean distance between probes.
To evaluate focal amplifications and deletions and chromosomal gains and losses, we visually inspected copy-number profiles of each case. Candidate genes and their 3′ and 5′ intergenic neighborhood were further investigated using the Integrative Genomic Viewer (IGV) for the presence of breakpoints, as an indication for potential gene fusions.
Gene expression profiling
Tumor samples with sufficient high-quality RNA were analyzed on the Affymetrix GeneChip Human Genome U133 Plus (v.2.0) Array (Affymetrix, Santa Clara, USA) at the Microarray Department of the University of Amsterdam, the Netherlands or the Genomics ad Proteomics Core Facility of the German Cancer Research Center (DKFZ). Subsequent library preparation, hybridization, and quality control was conducted following the manufacturer’s guideline. Expression data were normalized using the MAS5.0 algorithm. Identification of significantly differentially expressed genes between subgroups (test: anova; correction for multiple testing: false discovery rate, transform: z-score) and unsupervised hierarchical clustering were performed within the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl).
Next-generation gene panel, whole-exome, low-coverage whole genome, and RNA sequencing
For the detection of single-nucleotide variations (SNVs), small insertions and deletions (indels), and gene fusions, a subset of tumor samples (n = 5) were analyzed via a customized enrichment/hybrid-capture-based next-generation sequencing gene panel67. DNA from FFPE tissue was extracted using the Promega Maxwell device (Promega), sheared on Covaris M220 (Covaris), according to the manufacturer’s guidelines. Following a successful quality control using a Bioanalyzer 2100 (Agilent), sequencing was performed on a NextSeq 500 instrument (Illumina). Paired-end sequencing was applied to increase the detection sensitivity of duplicates and possible gene fusions.
For whole exome (WES; n = 13) and RNA sequencing (n = 9), library preparation, sequencing and data processing were conducted following the pipeline established in the INFORM study51. In brief, library preparation for WES was performed using the Agilent SureSelectXT Human V5 kit. Prepared libraries were sequenced together with a tumor cDNA library (poly(A) + RNA, Illumina TruSeq RNA Kit v2) on an Illumina HiSeq. The 1000 Genomes phase 2 human reference assembly (NCBI build 37.1) was selected for mapping the sequencing reads using BWA (version 0.6.2). For detection of SNVs and indels, custom pipelines were used68. In brief, the computational analysis was performed using SAMtools mpileup and bctools version 0.1.19 to detect somatic variants. To annotate variants, ANNOVAR was applied with Gencode version 17. All high confidence coding or splice site germline variants in a selected panel of cancer predisposition genes were extracted using a custom Python script. To call short insertions or deletions (InDels) in tumor and control blood BAM files, the extracted data was annotated using Platypus (version 0.5.2/0.7.4 with parameters genInDels = 1, genSNPs = 0, ploidy = 2, nIndividuals = 2). Poor genotype quality and low variant counts in the tumor were excluded for the subsequent analysis. RNA sequencing data were analyzed with deFuse to detect gene fusions69.
Potentially relevant somatic and germline alterations were manually assessed and cross-examined through various databases (http://www.ncbi.nlm.nih.gov/SNP build 135, http://www.1000genomes.org, http://exac.broadinstitute.org, https://cancer.sanger.ac.uk/cosmic, https://www.ncbi.nlm.nih.gov/clinvar/).
Statistics
Overall survival (OS) and progression-free survival (PFS) were analyzed by Kaplan–Meier analysis and tested for significant differences using a log-rank test. Binary and categorical patient characteristics between different subgroups were compared by a two-sided Fisher’s exact test. P-values < 0.05 were considered significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
This study was supported by the German Consortium for Translational Cancer Research (DKTK), the Shantella Foundation (Vaduz, Liechtenstein), ‘Ein Herz für Kinder’, German Cancer Aid (DKH), Deutsche Kinderkrebsstiftung (DKKS), the German Federal Ministry of Science and Research (BMBF), the Ministry of Research and the Arts of the State of Baden-Württemberg and a generous donation from the Scheu family. M.Y.D. was supported by fellowships from the Mildred Scheel doctoral program of the German Cancer Aid and the German National Scholarship Foundation. A. K. is supported by the Helmholtz Association Research Grant (Germany). M.R. is supported by RSF Research Grant 18-45-06012. For excellent technical support and expertize we thank the Genomics and Proteomics Core Facility (GPCF) and Omics, IT and Data Management Core Facility (ODCF) of the German Cancer Research Center (DKFZ).
Source data
Author contributions
D.T.W.J. and S.M.P. conceived the original idea and jointly supervised this study. M.Y.D., M.S., D.Stu., E.P., S.M.P. and D.T.W.J. wrote the manuscript with support from all authors. S.T., C.K., A.M.D., A.L.G., C.J., J.S., M.E., M.U.S., B.C.J., C.M.v.T., A.W., A.G., M.R., J.E., T.M., O.W., F.S., D.R., D.Su., J.Z., A.K., and A.v.D. provided samples and clinical data. M.Y.D., D.Stu., E.P., and A.W. prepared and processed the samples. M.S., D.Sti., and G.P.B. performed the computational processing of raw data. M.Y.D., D.Stu., E.P., S.M.P., and D.T.W.J. performed the data analysis and interpretation. All authors discussed the results and contributed to the final version of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
Methylation array data from this study have been deposited in GEO: GSE131482. Gene expression array were deposited in GEO: GSE168457. DNA sequencing data are available from the European Genome-Phenome Archive (EGA) under accession EGAS00001005243. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review informationNature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Maximilian Y. Deng, Dominik Sturm, Elke Pfaff.
These authors jointly supervised this work: Stefan M. Pfister, David T. W. Jones.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-25708-y.
References
- 1.Taylor RE, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J. Clin. Oncol. 2003;21:1581–91. doi: 10.1200/JCO.2003.05.116. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong GT, et al. Reduction in late mortality among 5-year survivors of childhood cancer. New Engl. J. Med. 2016;374:833–42. doi: 10.1056/NEJMoa1510795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Travis LB, et al. Second malignant neoplasms and cardiovascular disease following radiotherapy. J. Natl Cancer Inst. 2012;104:357–70. doi: 10.1093/jnci/djr533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christopherson KM, et al. Late toxicity following craniospinal radiation for early-stage medulloblastoma. Acta Oncol. 2014;53:471–80. doi: 10.3109/0284186X.2013.862596. [DOI] [PubMed] [Google Scholar]
- 5.Relling MV, et al. High incidence of secondary brain tumours after radiotherapy and antimetabolites. Lancet. 1999;354:34–9. doi: 10.1016/S0140-6736(98)11079-6. [DOI] [PubMed] [Google Scholar]
- 6.Brat DJ, et al. Molecular genetic alterations in radiation-induced astrocytomas. Am. J. Pathol. 1999;154:1431–8. doi: 10.1016/S0002-9440(10)65397-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donson AM, et al. Unique molecular characteristics of radiation-induced glioblastoma. J. Neuropathol. Exp. Neurol. 2007;66:740–9. doi: 10.1097/nen.0b013e3181257190. [DOI] [PubMed] [Google Scholar]
- 8.Elsamadicy AA, Babu R, Kirkpatrick JP, Adamson DC. Radiation-induced malignant gliomas: a current review. World Neurosurg. 2015;83:530–42. doi: 10.1016/j.wneu.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Gessi M, et al. Radiation-induced glioblastoma in a medulloblastoma patient: a case report with molecular features. Neuropathology. 2008;28:633–9. doi: 10.1111/j.1440-1789.2008.00900.x. [DOI] [PubMed] [Google Scholar]
- 10.Hamasaki K, Nakamura H, Ueda Y, Makino K, Kuratsu J. Radiation-induced glioblastoma occurring 35 years after radiation therapy for medulloblastoma: case report. Brain Tumor Pathol. 2010;27:39–43. doi: 10.1007/s10014-009-0258-8. [DOI] [PubMed] [Google Scholar]
- 11.Madden JR, et al. Radiation-induced glioblastoma multiforme in children treated for medulloblastoma with characteristics of both medulloblastoma and glioblastoma multiforme. J. Pediatr. Hematol. Oncol. 2010;32:e272–8. doi: 10.1097/MPH.0b013e3181e51403. [DOI] [PubMed] [Google Scholar]
- 12.Menon G, Nair S, Rajesh BJ, Rao BR, Radhakrishnan VV. Malignant astrocytoma following radiotherapy for craniopharyngioma. J. Cancer Res. Ther. 2007;3:50–2. doi: 10.4103/0973-1482.31974. [DOI] [PubMed] [Google Scholar]
- 13.Nakao T, et al. Radiation-induced gliomas: a report of four cases and analysis of molecular biomarkers. Brain Tumor Pathol. 2017;34:149–154. doi: 10.1007/s10014-017-0292-x. [DOI] [PubMed] [Google Scholar]
- 14.Ng I, Tan CL, Yeo TT, Vellayappan B. Rapidly fatal radiation-induced glioblastoma. Cureus. 2017;9:e1336. doi: 10.7759/cureus.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prasad G, Haas-Kogan DA. Radiation-induced gliomas. Expert Rev. Neurother. 2009;9:1511–7. doi: 10.1586/ern.09.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romeike BF, Kim YJ, Steudel WI, Graf N. Diffuse high-grade gliomas as second malignant neoplasms after radio-chemotherapy for pediatric malignancies. Childs Nerv. Syst. 2007;23:185–93. doi: 10.1007/s00381-006-0199-z. [DOI] [PubMed] [Google Scholar]
- 17.Salvati M, et al. Radio-induced gliomas: 20-year experience and critical review of the pathology. J. Neurooncol. 2008;89:169–77. doi: 10.1007/s11060-008-9565-x. [DOI] [PubMed] [Google Scholar]
- 18.Walter AW, et al. Secondary brain tumors in children treated for acute lymphoblastic leukemia at St Jude Children’s Research Hospital. J. Clin. Oncol. 1998;16:3761–7. doi: 10.1200/JCO.1998.16.12.3761. [DOI] [PubMed] [Google Scholar]
- 19.Yamanaka, R., Hayano, A. & Kanayama, T. Radiation-induced gliomas: a comprehensive review and meta-analysis. Neurosurg. Rev.41, 719–731 (2016). [DOI] [PubMed]
- 20.Lopez, G. Y. et al. The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol.137, 139–150 (2019). [DOI] [PMC free article] [PubMed]
- 21.Paugh BS, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010;28:3061–8. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phi, J. H. et al. Genomic analysis reveals secondary glioblastoma after radiotherapy in a subset of recurrent medulloblastomas. Acta Neuropathol135, 939–53 (2018). [DOI] [PubMed]
- 23.Capper D, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–474. doi: 10.1038/nature26000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hovestadt V, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510:537–41. doi: 10.1038/nature13268. [DOI] [PubMed] [Google Scholar]
- 25.Pajtler KW, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27:728–43. doi: 10.1016/j.ccell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sturm D, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 2016;164:1060–1072. doi: 10.1016/j.cell.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones C, Perryman L, Hargrave D. Paediatric and adult malignant glioma: close relatives or distant cousins? Nat. Rev. Clin. Oncol. 2012;9:400–13. doi: 10.1038/nrclinonc.2012.87. [DOI] [PubMed] [Google Scholar]
- 28.Northcott PA, Pfister SM, Jones DT. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol. 2015;16:e293–302. doi: 10.1016/S1470-2045(14)71206-9. [DOI] [PubMed] [Google Scholar]
- 29.Sturm D, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat. Rev. Cancer. 2014;14:92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buczkowicz P, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014;46:451–6. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones, C. & Baker, S. J. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer14, 10.1038/nrc3811 (2014). [DOI] [PMC free article] [PubMed]
- 32.Korshunov A, et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017;134:507–516. doi: 10.1007/s00401-017-1710-1. [DOI] [PubMed] [Google Scholar]
- 33.Sturm D, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 34.International Cancer Genome Consortium PedBrain Tumor Project. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016;22:1314–1320. doi: 10.1038/nm.4204. [DOI] [PubMed] [Google Scholar]
- 35.Jones DT, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 2013;45:927–32. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones DT, et al. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28:2119–23. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agnihotri S, et al. Therapeutic radiation for childhood cancer drives structural aberrations of NF2 in meningiomas. Nat. Commun. 2017;8:186. doi: 10.1038/s41467-017-00174-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhuller KS, et al. Late mortality, secondary malignancy and hospitalisation in teenage and young adult survivors of Hodgkin lymphoma: report of the Childhood/Adolescent/Young Adult Cancer Survivors Research Program and the BC Cancer Agency Centre for Lymphoid Cancer. Br. J. Haematol. 2016;172:757–68. doi: 10.1111/bjh.13903. [DOI] [PubMed] [Google Scholar]
- 39.Inskip PD, et al. Radiation-related new primary solid cancers in the childhood cancer survivor study: comparative radiation dose response and modification of treatment effects. Int. J. Radiat. Oncol. Biol. Phys. 2016;94:800–7. doi: 10.1016/j.ijrobp.2015.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morton, L. M. et al. Genome-Wide Association Study to identify susceptibility loci that modify radiation-related risk for breast cancer after childhood cancer. J. Natl Cancer Inst.109, djx058 (2017). [DOI] [PMC free article] [PubMed]
- 41.Schwartz B, et al. Risk of second bone sarcoma following childhood cancer: role of radiation therapy treatment. Radiat. Environ. Biophys. 2014;53:381–90. doi: 10.1007/s00411-013-0510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Dijk IW, et al. Risk of symptomatic stroke after radiation therapy for childhood cancer: a long-term follow-up cohort analysis. Int. J. Radiat. Oncol. Biol. Phys. 2016;96:597–605. doi: 10.1016/j.ijrobp.2016.03.049. [DOI] [PubMed] [Google Scholar]
- 43.Winther JF, et al. Childhood cancer survivor cohorts in Europe. Acta Oncol. 2015;54:655–68. doi: 10.3109/0284186X.2015.1008648. [DOI] [PubMed] [Google Scholar]
- 44.Bakst RL, et al. Reirradiation for recurrent medulloblastoma. Cancer. 2011;117:4977–82. doi: 10.1002/cncr.26148. [DOI] [PubMed] [Google Scholar]
- 45.Wetmore C, et al. Reirradiation of recurrent medulloblastoma: does clinical benefit outweigh risk for toxicity? Cancer. 2014;120:3731–7. doi: 10.1002/cncr.28907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konig L, et al. Secondary malignancy risk following proton vs. X-ray treatment of mediastinal malignant lymphoma: a comparative modeling study of thoracic organ-specific cancer risk. Front. Oncol. 2020;10:989. doi: 10.3389/fonc.2020.00989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newhauser WD, et al. The risk of developing a second cancer after receiving craniospinal proton irradiation. Phys. Med. Biol. 2009;54:2277–91. doi: 10.1088/0031-9155/54/8/002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dasu A, Toma-Dasu I, Olofsson J, Karlsson M. The use of risk estimation models for the induction of secondary cancers following radiotherapy. Acta Oncol. 2005;44:339–47. doi: 10.1080/02841860510029833. [DOI] [PubMed] [Google Scholar]
- 49.Schneider U, Zwahlen D, Ross D, Kaser-Hotz B. Estimation of radiation-induced cancer from three-dimensional dose distributions: Concept of organ equivalent dose. Int. J. Radiat. Oncol. Biol. Phys. 2005;61:1510–5. doi: 10.1016/j.ijrobp.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Q, et al. Secondary cancer risk after radiation therapy for breast cancer with different radiotherapy techniques. Sci. Rep. 2020;10:1220. doi: 10.1038/s41598-020-58134-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Worst BC, et al. Next-generation personalised medicine for high-risk paediatric cancer patients - The INFORM pilot study. Eur. J. Cancer. 2016;65:91–101. doi: 10.1016/j.ejca.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 52.Harrabi SB, et al. Dosimetric advantages of proton therapy over conventional radiotherapy with photons in young patients and adults with low-grade glioma. Strahlenther. Onkol. 2016;192:759–769. doi: 10.1007/s00066-016-1005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones DT, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–5. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Louis, D. N. et al. WHO Classification of Tumours of the Central Nervous System, (International Agency for Research on Cancer, 2016).
- 55.Merchant TE, et al. Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function. Pediatr. Blood Cancer. 2008;51:110–7. doi: 10.1002/pbc.21530. [DOI] [PubMed] [Google Scholar]
- 56.Northcott PA, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor MD, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–72. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yock TI, et al. Long-term toxic effects of proton radiotherapy for paediatric medulloblastoma: a phase 2 single-arm study. Lancet Oncol. 2016;17:287–298. doi: 10.1016/S1470-2045(15)00167-9. [DOI] [PubMed] [Google Scholar]
- 59.Cahan WG, Woodard HQ, et al. Sarcoma arising in irradiated bone; report of 11 cases. Cancer. 1948;1:3–29. doi: 10.1002/1097-0142(194805)1:1<3::AID-CNCR2820010103>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 60.Kleinschmidt-Demasters BK, Kang JS, Lillehei KO. The burden of radiation-induced central nervous system tumors: a single institution s experience. J. Neuropathol. Exp. Neurol. 2006;65:204–16. doi: 10.1097/01.jnen.0000205146.62081.29. [DOI] [PubMed] [Google Scholar]
- 61.Mistry M, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J. Clin. Oncol. 2015;33:1015–22. doi: 10.1200/JCO.2014.58.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krieger MD, Gonzalez-Gomez I, Levy ML, McComb JG. Recurrence patterns and anaplastic change in a long-term study of pilocytic astrocytomas. Pediatr. Neurosurg. 1997;27:1–11. doi: 10.1159/000121218. [DOI] [PubMed] [Google Scholar]
- 63.Broniscer A, et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J. Clin. Oncol. 2007;25:682–9. doi: 10.1200/JCO.2006.06.8213. [DOI] [PubMed] [Google Scholar]
- 64.Cavalli FMG, et al. Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol. 2018;136:227–237. doi: 10.1007/s00401-018-1888-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pajtler KW, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018;136:211–226. doi: 10.1007/s00401-018-1877-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deng MY, et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol. 2018;136:239–253. doi: 10.1007/s00401-018-1865-4. [DOI] [PubMed] [Google Scholar]
- 67.Sahm F, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016;131:903–10. doi: 10.1007/s00401-015-1519-8. [DOI] [PubMed] [Google Scholar]
- 68.Alioto TS, et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015;6:10001. doi: 10.1038/ncomms10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McPherson A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramaswamy V, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016;131:821–31. doi: 10.1007/s00401-016-1569-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Methylation array data from this study have been deposited in GEO: GSE131482. Gene expression array were deposited in GEO: GSE168457. DNA sequencing data are available from the European Genome-Phenome Archive (EGA) under accession EGAS00001005243. Source data are provided with this paper.