

Abstract
Although CAR-T cells are widely used to treat cancer, efficiency of CAR-T cell cytolytic responses has not been carefully examined. We engineered CAR specific for HMW-MAA (high-molecular-weight melanoma-associated antigen) and evaluated potency of CD8+ CAR-T cells to release cytolytic granules and to kill tissue-derived melanoma cells, which express different levels of HMW-MAA. CAR-T cells efficiently killed melanoma cells expressing high level of HMW-MAA, but not melanoma cells with lower levels of HMW-MAA. The same melanoma cells presenting significantly lower level of stimulatory peptide-MHC ligand were readily lysed by T cells transduced with genes encoding α,β-TCR specific for the peptide-MHC ligand. The data suggest that higher level of targeted molecules is required to engage a larger number of CARs than TCRs to induce efficient cytolytic granule release and destruction of melanoma cells. Understanding the difference in molecular mechanisms controlling activation thresholds of CAR- versus TCR-mediated responses will contribute to improving efficiency of CAR T cells required to eliminate solid tumors presenting low levels of targeted molecules.
Keywords: T-cell receptor (TCR), T-cell bioengineering, chimeric antigen receptor (CAR), receptor regulation, immunotherapy, tumor immunology, killing efficiency of various melanoma cells by CAR- or TCR-triggered T-cells
Abbreviations: BFP, blue fluorescent protein; CAR, chimeric antigen receptor; CTL, cytotoxic T lymphocytes; GFP, green fluorescent protein; HMW-MAA, high-molecular-weight melanoma-associated antigen; LN2, liquid nitrogen; PBMC, peripheral blood mononuclear cells; scFv, single chain variable fragment; TCR, T-cell receptor
Strategies that enhance the immune response to eliminate tumor cells have evolved into the rapidly growing field of immunotherapy, which has offered promising results in treating several types of cancer (1, 2, 3, 4). While chemotherapy and radiation treatments can cause extensive off-target toxicity, properly designed immunotherapeutic interventions to combat cancer appears to be more specific and minimize damage to healthy host tissues. Engineered T cells directed against tumor cells offer an innovative immunotherapy that is rapidly becoming widely used (5).
Transduction of T cells with chimeric antigen receptors (CARs) recognizing selected cell surface molecules demonstrated successful application of this technology for treatment of hematologic malignancies (2). However, targeting tumors in tissues by CAR-T cells has proved to be challenging with significantly lower success rate and the development of adverse effects (e.g., (6)). A limited access of CAR-T cells to tumors inside tissues, tumor microenvironment, and cross-reactivity of CAR-T cells with antigens in healthy tissues are well-identified problems (7, 8, 9). However, the role of the level of targeting antigen on solid tumors and the level of CAR expression that both determine efficiency of CAR-mediated response have not been carefully evaluated.
CARs comprise an engineered fusion receptor containing an extracellular single chain variable fragment (scFv), a hinge or stalk region that is typically derived from CD8, a transmembrane domain, and intracellular signaling domains (8, 10). The scFv is engineered from variable domains of heavy and light chains of an antibody with the specificity of interest. This “chimeric moiety” will bind specifically to a desired targeting molecule on tumor cells. The hinge region of CAR is designed to allow spatial arrangement and flexibility for the scFv allowing CAR molecules to form dimers at the cell surface. This permits antigen binding at otherwise unfavorable distances and angles between targeting molecules. The transmembrane and intracellular signaling domains of CAR often contain intracellular portions of CD28 and CD3ζ or other domains of signaling and costimulatory molecules in T cells.
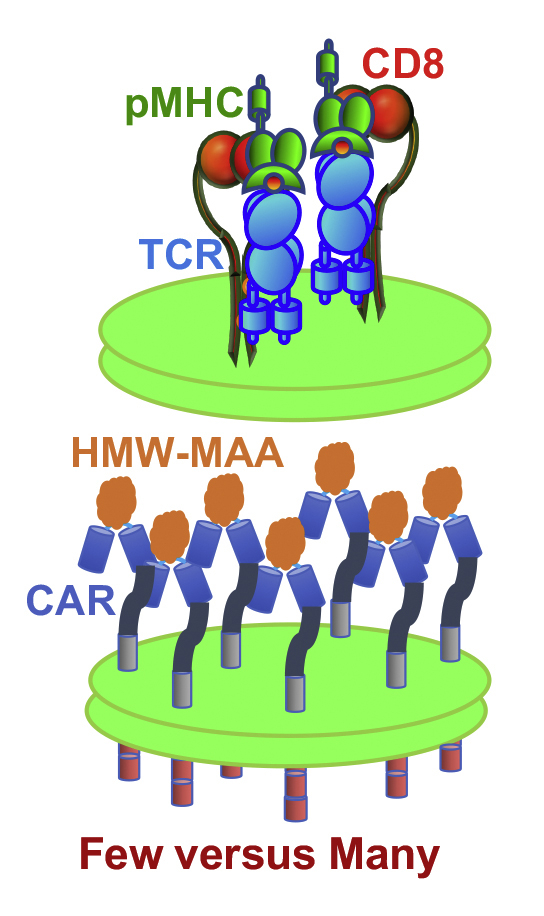
We engineered CD8+ CAR-T cells recognizing high-molecular-weight melanoma-associated antigen (HMW-MAA) that is overexpressed on the surface of up to 90% of melanoma cells (11). Also, known as chondroitin sulfate proteoglycan 4 (CSPG4), HMW-MAA plays a pivotal role in cell migration and metastasis (11). The level of HMW-MAA expression is varied on different melanoma cell lines. We compared potency of the specific lysis of these melanoma cells that express different levels of HMW-MAA by CD8+ CAR-T cells. For comparison reasons, we also tested efficacy of the same target cell destruction by T cells transduced with genes encoding α,β-TCR specific for cognate peptide-MHC (pMHC) ligands presented on the target cells at a significantly lower level. The results led us to conclude that TCR triggering of cytolytic response has a lower threshold as opposed to that required to initiate response by CAR even though both receptors were expressed on T cells at a very similar level. This suggests that higher numbers of engaged CARs than TCRs are required to achieve similar potency of cytolytic response by CAR-T and TCR-T cells.
Results
Strategy of the proposed analysis
To compare efficiency of the cytolytic effector functions of CD8+ CAR-T with that of human virus-specific CD8+ CTL, we chose widely used design of CAR (Fig. 1A) specific for HMW-MAA, which is usually overexpressed on melanoma cells. To produce CAR-T cells, we utilized cord-blood-derived T cells that contain mostly naïve T cells compared with more diverse T cells from adult individuals. Cord blood T cells possess a high proliferative capacity, allowing to readily expand these cells to produce sufficient amount of CAR-T cells required for analysis (Fig. 1B). Activated cord-blood-derived T cells were transduced with a retroviral pBMN-I vector coexpressing GFP and the C21CD28CD3ζ CAR. The killing efficiency of purified CD8+ CAR-T cells was compared with CER43 and 68A62 CTLs as well as with that of CD8+ T cells transduced with genes encoding α and β chains of D3 TCR (D3TCR-T cells) by testing ability of these cytolytic effectors to kill melanoma cell lines that display various levels of either HMW-MAA protein or cognate peptides bound to MHC class I proteins. The magnitude and sensitivity of the cytolytic activity of the effector cells and the extent of their degranulation have been compared.
Figure 1.

Production and testing of CD8+ CAR-T cells specific for high-molecular-weight melanoma-associated antigen.A, the structure of CAR; the CAR was inserted into retroviral vector containing GFP. B, transduction of cord blood derived T cells with CAR caring retrovirus and comparison CD8+ CAR-T cell and CTL functional activities.
Generation of HMW-MAA-specific CAR-T cells and D3TCR-T cells recognizing SL9 peptide in association with HLA-A2
We transduced activated T cells derived from cord blood with a HMW-MAA-specific CAR. The transduction yielded 5% GFP+ cells. High-expressing GFP cells were sorted out using flow cytometry. Additional round of stimulation was performed as needed to expand the transduced cells and CD8+ CAR-T cells were isolated by negative immunomagnetic sorting. The purity of isolated CD8+ CAR-T cells was confirmed by flow cytometry (Fig. 2A). The CAR-T cells were stained with fluorescent-labeled penta-His antibody to estimate the level of expression of His-tagged CAR. Using calibrating fluorescent beads, we determined an average of 12,700 CAR receptors per CD8+ T cell (Fig. S1A). To produce D3TCR-T cells specific for HIV Gag-derived SLYNTVATL (SL9) peptide in association with HLA-A2, α and β D3TCR genes were transduced into activated freshly isolated T cells using similar vector containing BFP allowing to select high-expressing BFP T cells by flow cytometry sorting. After additional stimulation and expansion of transduced T cells, CD8+ T cells were selected by negative immunomagnetic sorting, and purity of CD8+ T cells was characterized by flow cytometry (Fig. 2B). The level of D3TCR expression was measured by staining of CD8+ T cells with fluorescent labeled anti-Vβ5.1 antibody and subsequent comparison of the fluorescence intensity with calibrating fluorescent beads as described for CAR-T cells. An average of 11,900 D3TCR was expressed per CD8+ T cell.
Figure 2.

Level of CAR and D3TCR expression transduced into CD8+ T cells.A, CAR expression on CAR-transduced (right) or control (left) CD8+ T cells. PBMCs were transduced with GFP-labeled pBMN-I retrovirus containing CAR or with a mock vector and GFP positive cells were sorted by Flow Cytometry. CD8+ T cells were isolated from stimulated GFP-positive T cells by magnetic sorting. Expression of CAR on CD8+GFP+ T cells was confirmed by staining with anti-His-tag Alexa Fluor 647 antibody (see Fig. S1). Representative GFP and Alexa Fluor 647 dot-plot is shown (N = 3). CAR expression level was equal 12,700 ± 2900 receptors per cell (mean ± SD) as determined from three independent experiments. B, D3TCR expression on CD8+ T cells transduced with pMSCV-IRES-Blue FP vector or with mock vector. BFP positive cells were sorted by flow cytometry, and CD8+ T cells were purified by magnetic sorting. The TCR expression was determined from comparison of fluorescence intensity of Vβ5.1 antibody bound to TCR on CD8+BFP+ cells with fluorescence intensity of calibrating fluorescent beads. The level of D3TCR expression was determined to be 11,900 ± 900 receptors per cell.
The level of expression of targeting HMW-MAA on melanoma tumor cells is widely varied
We utilized fluorescent labeled HMW-MAA-specific antibodies and calibrating fluorescent beads to evaluate the level of HMW-MAA expression on different melanoma cell lines. We found that skin melanoma cells express higher surface levels of HMW-MAA as compared with uveal melanoma cells (see Table 1 and Fig. S2). Skin A375 melanoma cells were estimated to present 280,000 HMW-MAA molecules per cell. The patient-derived skin melanoma cell line CM006 was found to express 120,000 HMW-MAA molecules per cell. In contrast, patient-derived uveal melanoma cell lines UM004 and UM002B displayed ∼52,000 and ∼4000 molecules per cell, respectively.
Table 1.
Expression of HMW-MAA on a surface of melanoma cells
| Melanoma cell line | Melanoma type | HMW/MAA/cella |
|---|---|---|
| A375 | Skin | 280,000 ± 50,000 |
| CM006 | Skin | 119,000 ± 31,000 |
| UM004 | Uveal | 52,000 ± 24,000 |
| UM002B | Uveal | 4000 ± 1700 |
Mean ± SD calculated from three independent experiments.
Target antigen expression level strongly influences CAR-mediated cytolytic activity and release of cytolytic granules
We first tested efficiency of CD8+ CAR-T cells to kill skin and uveal melanoma tumor cells expressing different levels of HMW-MAA. CD8+ CAR-T cells efficiently lysed the high-expressing A375 cells (∼280,000 HMW-MAA molecules/cell) and CM006 cells (∼120,000 HMW-MAA molecules/cell) even at low effector-to-target ratios (E:T = 5:1) (Fig. 3). CD8+ CAR-T cells lysed specifically the low-expressing cell line UM004 (∼52,000 HMW-MAA molecules/cell) but the killing was marginal. The killing of UM002B (∼4000 HMW-MAA molecules/cell) was still detectable at high E:T ratios.
Figure 3.

Destruction of melanoma cells that express various levels of HMW-MAA by CD8+CAR-T cells. Cytolytic activity by CD8+ CAR-T cells against melanoma cell line A375 (A), CM006 (B), UM004 (C), and UM002B (D) is shown. Estimated numbers of HMW-MAA molecules per each melanoma cell surface are indicated. CD8+ T cells transduced with “empty” vector were utilized as control effector cells. All assays were performed in triplicates. Means and SD are calculated for each point, and the error bars are indicated for each mean value. p-values were calculated by Student’s t test, and p-values for statistically significant results are indicated for each point (∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05). Data shown are representative of 3 to 5 independent experiments.
To reiterate the observed difference in specific lysis of melanoma cells by CD8+ CAR-T cells, we analyzed the release of cytolytic granules by the effector cells in response to recognition of the same melanoma cells. To this end, the effector cells were cocultured with the melanoma cells at various density of the target cells, and the degranulation marked by surface expression of CD107a was evaluated. The recognition of A375 target cells that display the highest number of HMW-MAA per cell led to detectable expression of CD107a marker on the surface of CD8+ CAR-T cells (Fig. 4A). The appearance of CD107a on the same effector cells in response to recognition of CM006, UM004, or UM002B was below detectable level (Fig. S3).
Figure 4.

Degranulation efficiency of CD8+CAR-T cells induced by A375 melanoma cell line.A, percent of responding effector cells against A375 cells was determined by measuring appearance of CD107a on the surface of responding cells. B, the extent of cytolytic granule release by effector cells in response to recognition of A375 cells was quantified by measuring serine esterase activity in extracellular medium. The results are representative of at least three independent experiments. Each experimental value represents the mean of triplicate. Standard deviations are indicated for each point as error bars. p-values were calculated by Student’s t test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
In addition to measuring the appearance of CD107a on the surface of the effector cells during coculturing with tumor cells, we also evaluated serine esterase activity of granzymes, a component of cytolytic granules, released to the culture supernatant. The amount of the released granzymes was measured from changes in adsorption spectrum of converted by the granzyme substrate added to the extracellular medium (see Experimental procedures for details). CD8+ CAR-T cells released detectable amount of granules followed by recognition of A375 target cells (Fig. 4B). The amount of released granules by CD8+ CAR-T cells in response to recognition of CM006, UM004, and UM002B target cells was undetectable (Fig. S3).
Collectively, CD8+ CAR-T cells only released measurable amounts of granules in response to tumor cells expressing high levels of targeting antigen HMW-MAA on the cell surface. However, the amount of released granules was significantly diminished compared with previously observed for both CD4+ and CD8+ CTL stimulated via TCR (12).
CTLs effectively lyse melanoma cells and release cytolytic granules even when presented with a low level of cognate pMHC ligands
To determine whether melanoma tumor cells possess any intrinsic resistance to cytolytic response exercised by CD8+ CAR-T cells, we exploited the human Flu-specific cloned CTL CER43 to examine killing of the same HLA-A2 melanoma cells sensitized with various concentrations of cognate Flu-derived peptide GL9 to generate GL9-HLA-A2 ligands on the cell surface recognizable by CER43 CD8+ CTL. Indeed, the CTL specifically lysed each of the melanoma cell lines even when the lines were sensitized at very low peptide concentration resulting in significantly lower level of GL9-HLA-A2 ligands on the surface of target cells as compared with HMW-MAA antigens (Fig. 5A and Table 2). Similar data were produced with HIV-specific CTL clone 68A62 that recognizes ILKEPVHGV (IV9) peptide in the context of HLA-A2 (Fig. S4).
Figure 5.

CER43 CD8 T cell clone cytolytic response and degranulation efficiency induced by melanoma cell lines presenting cognate pMHC ligands at various levels. Cytolytic activity (A), appearance of CD107a (B), and serine protease release (C) were measured for each indicated target cell line sensitized with cognate peptide. Peptide concentration required for 50% of maximal lysis (SD50) of each target cell line is indicated by an arrow. Each experimental point was tested in triplicates in at least three independent experiments. Representative experiments are shown, and mean values with standard deviation are indicated for each experimental point. p-values that are statistically different from corresponding value obtained with target cells not pulsed with peptide are indicated (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001, two-tailed Student’s t test).
Table 2.
Number of GL9-HLA-A2 complexes per melanoma cell at antigenic peptide concentration required for half maximal cell lysis (SD50) by CER-43 CTL
| Melanoma cell line | Melanoma type | Total HLA-A2/cella | GL9 concentration at SD50b, M | GL9-HLA-A2/cell at SD50c |
|---|---|---|---|---|
| A375 | Skin | 77,400 ± 5860 | 1 × 10−10 | <48 |
| CM006 | Skin | 68,700 ± 9480 | 2 × 10−8 | <7579 |
| UM004 | Uveal | 82,500 ± 8664 | 2 × 10−9 | <1010 |
| UM002B | Uveal | 3000 ± 1400 | 3 × 10−9 | <93 |
Mean ± SD calculated from three independent experiments; There are statistically significant difference in HLA-A2 expression level between UM002B cells and A375 (p < 0.01), CM006 (p < 0.01), and UM004 (p < 0.001) cell lines as determined by one-way ANOVA followed by Tukey post hoc test. There was no statistically significant difference between A375, CM006, and UM004 lines.
Determined from cytolytic assay with CER-43 CTL and GL9-sensitized melanoma cells.
Number of pMHC ligands per cell calculated in the assumption that all cell surface MHCs are receptive for peptide loading. The occupancy of the “empty” HLA-A2 molecules is calculated using the equation: α = KeqC/(1 + KeqC), where α is fraction of HLA-A2 molecules on a cell surface occupied by GL9 peptide, K equilibrium binding constant for interaction between peptide and MHC (Keq = 6.2 × 106 L/M) (48); C is peptide concentration at SD50. Actual number of GL9-HLA-A2 per cell is lower than those indicated because the calculation performed in the assumption that all HLA-A2 molecules on the cell surface are receptive for peptide loading.
We also tested the extent of CER43 CTL degranulation in response to TCR engagement by cognate GL9-HLA-A2 ligand on the surface of the melanoma cell lines. CER43 CTLs were cocultured with target cells presenting varying levels of cognate pMHC ligands and percent of degranulating CTL was determined by measuring upregulation of CD107a on the cell surface. In parallel, serine esterase release was also quantified. The fraction of CD107a expressing CTL and percent of released granules in response to the CTL stimulation with the melanoma cell lines were significantly higher than those observed for CD8+ CAR-T cells targeting the same melanoma cells even at significantly lower level of stimulatory pMHC ligands on target cells (Figs. 4 and 5, B and C).
The observed killing of the melanoma cells by CER43 CTL and 68A62 CTL as well as by CD8+ CAR-T cells was completely inhibited in the presence of EGTA (Fig. S5) indicating that the killing induced by all tested effector cells was granule-mediated.
These data suggest that failure of CD8+ CAR-T cells to exert cytolytic activity against melanoma is attributed to inability of CARs to induce sufficient release of cytolytic granules. This is despite high level of expression of targeted molecule on melanoma cells recognizable by CARs with a high affinity (2 × 107 L/M) (13) as opposed to lower affinity of TCR for cognate pMHC (2 × 105 L/M) (14). However, CAR expression of engineered T cells is substantially lower (≈12,700 CARs per cell) than TCR expression on CTL (30,000–60,000 TCR per cell depending on a day after stimulation) (Anikeeva and Sykulev, unpublished results).
D3TCR-T cells effectively lyse melanoma cells
To directly compare efficiency of CAR-mediated and TCR-mediated killing of target cells, we engineered D3TCR-T cells, which expressed ≈12,000 D3TCR per cells, very similar to average number of CAR per T cell (Fig. 2). We also determined the number of cognate pMHC on A375 and UM004 cells. To this end, we counted the number of fluorescent labeled peptide AF647-IV9 bound to accessible cell surface HLA-A2 molecules on the melanoma cells by comparing the fluorescence intensity with that of calibrated fluorescent beads (see Experimental procedures for details) (Fig. S6). We previously exploited this peptide (15) and showed that labeled and unlabeled peptides bound equally well to cell surface HLA-A2. The binding affinity of ILKEPVHGL (IV9) peptide to HLA-A2 has been determined (KD = 2 × 10−7 M) (16) and was similar to estimated affinities of SL9 and GL9 peptides for HLA-A2 (Fig. S7). We found that at the peptide concentration 10−7 M in the extracellular medium, 1100 and 1400 peptide molecules per cell are presented by HLA-A2 on A375 and UM004 melanoma cells, respectively. At this peptide concentration, killing capacity of A375 and UM004 by 68A62 CTLs at E:T = 5:1 approaches its maximum (Fig. S4).
We evaluated specific lysis of melanoma cells by engineered D3TCR-T cells and compared with the ability of CAR-T cells to kill the same target cells. 51Cr-labeled A375 and UM004 cells were combined with D3TCR-T cells at various E:T ratios in the presence of SL9 peptide added to the extracellular media at 10−7 M concentration; the peptide was kept in the media during the assay. Figure 6 shows that D3TCR-T cells efficiently lysed both A375 and UM004 cells. Even though lysis of A375 cells by CAR-T cells (see Fig. 3A) was comparable to that induced by D3TCR-T cells, the level of HMW-MAA molecules per target cells was ∼200 times higher than the level of cognate pMHC on these cells recognizable by D3TCR-T cells. UM004 melanoma cells that display 52,000 HMW-MAA molecules per cell were killed very poorly by CAR T cells (see Fig. 3C) as opposed to more efficient killing of these cells by D3TCR-T cells that responded to 1400 cognate pMHC ligands per target cell. It has to be noted that average level of CAR and D3TCR receptors on engineered T cells was very similar, ∼12,700 and ∼11,900 receptors per cells, respectively, but intrinsic affinity of these receptors for cognate ligands is 2 × 107 L/M and 2.4 × 105 L/M, correspondingly (13, 14). These data suggest that more CARs need to be engaged as compared with D3TCRs to initiate comparable T-cell cytolytic response. Thus, TCR appears to be more superior to CAR in triggering cytolytic response by CD8+ T cells.
Figure 6.

Specific lysis of A375 and UM004 melanoma cells by D3TCR-T cells. The level of expression of targeted SL9-HLA-A2 ligands per cell recognizable by D3TCR-T cells as well as E:T ratios are indicated. Percent of specific lysis of the target cells sensitized with 10−7 M of SLYNYVATL (SL) peptide is indicated by closed circles. Extent of the target cell lysis sensitized with irrelevant peptide GILGFVFTL (GL9) is shown by open circles. p-values were calculated by Student’s t test, and p-values for statistically significant results are indicated for each point (∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05). Data shown are representative of three independent experiments.
Discussion
The major finding from described experiments is that comparison of the potency of two different receptors to trigger cytolytic activity revealed superiority of TCR over CAR. Our data clearly demonstrated that efficient killing of melanoma cells can be achieved only at high level of HMW-MAA antigen expression on melanoma cells. In contrast, CTL and TCR-T cells efficiently destroy melanoma presenting significantly lower level of cognate pMHC ligands. Further support for the difference between the two cytolytic effectors attacking tumor cells is provided by striking disparity in the appearance of CD107a on CTL and CAR-T cells indicative of membrane fusion events of specialized lysosomes containing perforin and granzyme with the cell membrane on the effector cells. In total, 60 to 70% of CTL attacking melanoma cells upregulate CD107a as opposed to only 20% of responding CAR-T cells that are capable to do so (see Figs. 4A and 5B). Consistent with this, CTLs could release up to 50% of all available cytolytic granules, while CAR-T cells excreted only 20% at maximum (Figs. 4B and 5C). Efficient killing of melanoma cells was also observed by primary CD8+ CTL derived from melanoma lesions (17).
To provide additional evidence, we evaluated potency of killing by engineered T cells transduced with TCR that was expressed on T cells at the same level as CAR was. The affinity of TCR was significantly lower than that of CAR for the cognate ligands recognizable by the two receptors. At the same time, the level of HMW-MAA ligands on melanoma cells targeted by CAR-T cells was considerably higher as opposed to the level of pMHC ligands recognized by TCR-T cells. Nevertheless, TCR-T cells demonstrated more efficient response than CAR-T cells against the same melanoma cells (Figs. 3 and 6). These data clearly point to inefficient use of the available cellular machinery for granule delivery by T cells in response to CAR triggering. Molecular basis of this disparity remains to be elucidated.
Because the level of antigen expression on target cells influences the magnitude and sensitivity of T cell responses (18, 19), it is expected that tumor-specific response of engineered cytolytic effectors would also depend on the level of targeting molecules on tumor cells. Indeed, tumor cells strategically downregulate MHC proteins or mutate tumor-associated antigens (TAA) to evade destruction by immune cells (20, 21, 22, 23, 24).
Although level of targeting molecules presented on tumor cells is clearly essential, the origin of transformed cells appears to be an important factor that also determines the sensitivity to cytolytic granule-mediated attack. Lymphoblasmoid cell lines are generally known to be “conventional targets” to study T-cell cytolytic responses because these cells are more sensitive to granule-mediated lysis than tissue-derived tumor cells (25). Not surprisingly, initial success of targeting lymphoblastic malignancies by CAR-T cells (26) led to a dramatic increase in the popularity of CAR T cells engineering. In another example, erythroleukemia cells K562 transduced with CD19 that is expressed at relatively lower level could still be efficiently killed by CD19-specific CAR-T cells (27). It has been shown that erythrocyte membrane is very sensitive to cytolytic granules (28) explaining that K562 cells could be readily destroyed even at a relatively low level of targeting molecules expression. Thus, the nature of tumor cells appears to be another important factor determining the susceptibility of their killing.
One way to improve the sensitivity of responsiveness of engineered T cells is to endow T cells with high-affinity TCR (29). However, antigen recognition by high-affinity TCRs does not always induce an optimal T-cell response and often mediates off-target toxicity or apoptosis of the T cells (30, 31). This led to a notion that T cells having a moderate TCR affinity could be more effective. In fact, we have previously shown that affinity of TCR on T cells approaches a “ceiling” (18), which is sufficient to initiate T-cell response even to a few stimulatory peptide-MHC ligands on target cells (32), and further increase in TCR affinity does not improve the sensitivity of the response.
While our data show that efficient cytolytic response of CD8+ CAR-T cells is limited by expression level of targeted molecules on melanoma cells (Table 1 and Fig. 3), much fewer activating pMHC ligands on the same target cells are needed to initiate cytolysis by the CTLs (Table 2 and Fig. S2) and TCR-T cells (Fig. 6). This suggests that significantly more CARs than TCRs are required to be engaged to initiate sufficient cytolytic activity by CD8+ CAR-T cells. Consistent with this, variations of the expression level of TCR controlled by inducible promotor in TCR transgenic mice revealed that even as low as 1/20th the normal TCR numbers on the T cells was still sufficient to initiate T-cell response with no indication of phenotypic skewing (33). Most likely, signal amplification induced by TCR is much more potent than that triggered by CAR. This is in accord with previously findings showing that T cells transduced with αβ TCR genes demonstrated more superior cytolytic activity compared with the T cells endowed with CAR containing variable domains of the same TCR (34). In addition, it has been recently shown that CAR mediates very inefficient recruitment of ZAP70 that leads to significantly lower sensitivity of CAR-T cell responses to target cells (35).
We have previously found that the kinetics of degranulation, which is regulated by the kinetics of Ca2+ signaling, is the major factor controlling the efficiency of cytolytic activity (36, 37). The kinetics of accumulation and sustained level of intracellular calcium controls functioning of dynein motors that moves cytolytic granules along microtubules toward the MTOC (36, 38). The MTOC polarization and its close juxtaposition to the target cell-contacting membrane are a second important factor that controls efficiency of cytolytic granule delivery by CTL (38, 39). CAR engagement does not lead to complete MTOC polarization and results in initial rapid intracellular Ca2+ accumulation, which then abruptly declined (40). Thus, the difference in the kinetics of calcium mobilization induced by CAR or TCR implies that CAR-mediated release of cytolytic granules is transient as opposed to TCR-induced degranulation. Indeed, T cells stimulated through CAR demonstrated inefficient granules release (see Fig. 4B). Swift and sustained TCR signaling can mediate rapid granule release and efficient killing of target cells at lower effector-to-target ratio and scarce density of epitopes on the target cell (41). Transient calcium signaling could also cause rapid detachment of CD8+ CAR-T cells from target cells (40) that is counterproductive to efficient attack by effector cells. Consistent with this, LFA-1 plays an essential role in killing of target cells by CTL (42), while the role of LFA-1 in cytolytic activity by CAR-T cells is not entirely clear.
We conclude that CD8+ CAR-T cells are less sensitive than CTL and TCR-T cells in the detection and killing of tumor cells. Thus, higher level of targeted molecules on tumor cells resulting in a greater number of engaged CARs than TCRs is required for successful attack of tumor cells. Understanding mechanisms responsible for this disparity is warranted as it will provide basis to advance engineering of CAR-T cells that could be exploited to fight solid tumors that present low levels of targeted molecules.
Experimental procedures
Melanoma cell lines and packaging cell line
Uveal and skin melanoma cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS, 1% L-Glutamine, 1% Penicillin, 1% HEPES, and 1% non-essential amino acids (complete RPMI medium). Human skin melanoma cell line CM006 and uveal melanoma cell lines UM004 and UM002B were established and characterized in Dr Takami Sato laboratory. The A375 human malignant skin melanoma cell line was supplied by ATCC and was maintained in DMEM containing 10% FBS, 1% L-Glutamine, and 1% Penicillin. The Phoenix amphotropic packaging cell line was purchased from ATCC and was cultured in DMEM supplemented with 10% FBS, 1% L-Glutamine, and 1% Penicillin. The cells were utilized for generation of retrovirus for transduction.
Cytotoxic lymphocytes
The CD8+ CTL clone CER43 for Flu-derived peptide GILGFVFTL was kindly provided by Dr Antonio Lanzavecchia and the HIV-specific CD8+ CTL clone 68A62 that recognizes the reverse transcriptase-derived peptide ILKEPVHGV (IV9) was a gift from Dr Bruce Walker. Both clones were stimulated and maintained in culture as previously described (12, 36, 42, 43).
Antibodies and calibrating beads
Hybridoma OKT3 producing mouse antibodies specific for human CD3ε was purchased from ATCC. Hybridoma PA2.1 expressing antibody specific for the α2 helix of HLA-A2 was a gift of Herman Eisen, Massachusetts Institute of Technology. OKT-3 and PA2.1 antibodies were purified from hybridoma cultural supernatant using affinity chromatography with protein A. Purified PA2.1 antibody was labeled with Alexa Fluor 488 in correspondence with manufacturer instruction (Molecular Probes, Thermo Fisher Scientific). Stimulatory antibody against CD28 (clone 9.3) was purchased from GeneTex. Antibody specific for CD107a PE (clone H4A3) was obtained from BD Pharmingen. Anti-His tag antibody was purchased from Qiagen. Anti-TCR Vβ5.1 antibody was supplied by Miltenyi Biotec. The following antibodies were used for quantification assays: anti-6-Histidine Tag Alexa Fluor 647 (clone AD1.1.10; Novus Biological), anti-HMW-MAA (NG2) Alexa Fluor 488 (clone 9.2.27, eBioscience), anti-TCR Vβ5.1 (clone REA1062, Miltenyi Biotec). Alexa Fluor 488, R-PE, and anti-Alexa Fluor 647 calibrating beads from Bangs Labs Quantum MESF kits were exploited to measure the number of HMW-MAA, CAR, and TCR molecules on target and effector cells, respectively.
Purification and stimulation of human T cells
Human peripheral blood mononucleated cells (PBMCs) were purified by Ficoll-Hypaque density centrifugation from decoded cord blood samples provided by the Department of Gynecology at Thomas Jefferson University. Ficoll-Hypaque (GE Healthcare) was added to a mixture of blood and DPBS at 1:2 ratio and centrifuged for 30 min @ 2200 rpm with no brake, at 18 °C. The PBMC layer was extracted and washed three times with RPMI medium. The PBMC mixture was incubated in 10 cm Petri dish for 1 h at 37 °C to remove adherent cells, and nonadherent cells were collected. T25 flask was precoated with 10 μg/ml anti-CD3 and 1 μg/ml anti-CD28 antibodies in DPBS buffer. The subsequent PBMC population was incubated in the flask in the presence of 100 U/ml of human IL2 to activate T cells.
Generation of HMW-MAA specific CAR and HIV Gag p1777–85-specific D3 TCR constructs, production of retrovirus, and transduction of effector cells
To produce the CAR construct, we utilized human single chain antibody scFvC21 against HMW-MAA antigen, which were isolated from a semisynthetic phage display scFv antibody library (13, 44). His6 tag was introduced at N-terminal end. Gene encoding VL and VH single-chain fragment was fused with genes encoding CD8 alpha hinge region, CD28 transmembrane and signaling domains, and CD3ζ signaling domain and cloned into the pBMN-I-GFP retroviral vector (a gift from Dr Garry Nolan). To generate D3 TCR retroviral construct, D3 TCR-alpha and D3 TCR-beta genes (14) were fused using 2A sequence and cloned into pMSCV-IRES-Blue FP vector. pMSCV-IRES-Blue FP was a gift from Dario Vignali (Addgene plasmid # 52115). To generate retrovirus, Phoenix-Ampho cells were transfected with the recombinant plasmid using Lipofectamine LTX with Plus reagent (Life Technology). Supernatants containing the retrovirus were collected 48 and 72 h later. Activated human T cells (mixture of CD4+ and CD8+ T cells) were combined with recombinant virus and Lipofectamine LTX with Plus reagent. The mixture was centrifuged at 700g for 90 min, and the cells were incubated overnight at 37 °C in IL-2 containing complete media. Next day the same procedure was repeated with freshly collected virus supernatant. Transduction efficiency was measured by flow cytometry using GFP or BFP as a marker. The cells were grown for at least 6 days before sorting.
Purification of CD8+ CAR- and D3TCR-T cells
GFP+ T cells were sorted using a BD Fortessa Flow Cytometer to isolate CAR-expressing T cells. The resultant heterogeneous CD4+ and CD8+GFP+ T cell population expressing high levels of GFP was expanded and then was subjected to negative immunomagnetic sorting (Miltenyi Biotech) to purify CD8+ CAR-T cells. Cell surface expression of CAR was verified with biotinylated antibody against His6 Tag (Novus Biologicals). CD8+BFR+D3TCR-T cells were isolated in a similar manner. Cell surface expression of D3 TCR was verified by staining with R-PE labeled antibody against TCR Vβ5.1 (Miltenyi Biotec).
Quantification of cell surface receptors
Fluorescent labeled beads from Bangs Labs Quantum MESF kits were used to estimate the number of HMW-MAA and HLA-A2 on target cells as well as CAR and D3 TCR molecules on transduced T cells. Corresponding fluorescent beads were placed into individual 5 ml polystyrene flow cytometry tubes containing 400 μl of FACS buffer (DPBS +2% FBS). Fluorescent labeled antibodies specific for HMW-MAA or HLA-A2 were used to stain target cells. Anti-His tag antibody was utilized to stain CAR on the T cell surface. Fluorescent intensity associated with the stained cells was measured by flow cytometry and related to the fluorescent intensity of the calibrating beads according to the instructions provided by Bangs Labs.
The number of IV9-HLA-A2 complexes on A375 and UM004 cells was directly measured using IV9 variant ILKEPVHCV that was stoichiometrically labeled with Alexa Fluor 647 at the penultimate Cysteine (15). The peptide labeling at the Cysteine does not impair the peptide binding to HLA-A2 (45). A375 and UM004 melanoma cells were pulsed with various concentrations of ILKEPVHC(Alexa Fluor 647)V (IV9-AF647) peptide for 1 h at 37 °C in the presence or absence of 100-fold excess of unlabeled IV9 peptide. The cells were washed free of unbound peptide and associated with the cell fluorescence was measured by flow cytometry. The numbers of the peptides bound to the cell surface HLA-A2 were determined using calibrating Alexa Fluor 647 beads. The number of IV9-HLA-A2 complexes on the cell surface was calculated as the difference between total numbers of bound IV9-AF647 peptides and numbers of the labeled peptides bound to the cell surface in the presence of 100-fold excess of cold peptide.
The number of stimulatory GILGFVFTL-HLA-A2 complexes on the same target cells was measured at the peptide concentration required to induce half maximal lysis (SD50) (19, 46, 47). The number of pMHC ligands per cell were calculated using law of mass action: α = KeqC/(1 + KeqC), where α is fraction of HLA-A2 molecules on a cell surface occupied by GL9 peptide, K equilibrium binding constant for interaction between peptide and MHC (Keq = 6.2 × 106 for HLA-A2 and GL9 interaction (48)); C is peptide concentration at SD50. SLYNTVATL (SL9) peptide from HIV-1 p17 Gag(77–85) (46) was synthesized by the Protein/peptide Core facility of Massachusetts General Hospital. Custom synthesis, purification (more than 95%), and characterization of ILKEPVHC(Alexa647)V peptide were performed by ProImmune Ltd. The peptide GILGFVFTL (GL9) from matrix protein of Flu virus (49) was synthesized by Research Genetics, Inc and the peptide purity (>96%) was confirmed by HPLC and mass spectrometric analysis. ILKEPVHGV (IV9) peptide from HIV reverse transcriptase (46) was a gift from Herman Eisen.
Chromium release assay
Specific lysis of the target cells by cytotoxic lymphocytes was tested as previously described (19, 32). 1 × 106 melanoma target cells were labeled with 200 μCi of 51Cr (PerkinElmer) for 1 h, washed free of unreacted chromium, and were plated at 5 × 103 cells/well in a round-bottomed 96-well plates. Engineered cytolytic effectors were then added to the plates at various effector-to-target ratio, and the plates were incubated for 4 h at 37 °C in a CO2 incubator. To block granzyme/perforin-mediated pathway of target cell killing, cytolytic assay was performed in the presence of 2 mM EGTA.
Cytolytic potency of the human CTLs (CER43 and 68A62) was examined against the same HLA-A2+ melanoma target cells sensitized with GL9 or IV9 cognate peptides. Various amounts of the peptide in 50 μl of Dulbecco’s PBS were combined with 100 μl of 51Cr-labeled target cells and 2.5 × 104 CTL in 50 μl of complete medium in round-bottomed 96-well plates at an effector-to-target cell ratio of 5:1. The plates were incubated for 4 h at 37 °C in a CO2 incubator.
To measure cytolytic response of D3TCR-T cells, 1 × 106 A375 or UM004 melanoma target cells were labeled with Cr51 as above and combined with D3TCR-T cells in 96-well plates at various E:T ratios in the presence of either cognate (SL9) or noncognate (GL) peptides added to the extracellular media at 10−7 M.
51Cr release in the supernatant was quantified by automatic Gamma Counter (PerkinElmer). Percent specific lysis was calculated from the average of triplicates as follows: 100 × ([51Cr experimentally released − spontaneous release]/[maximum release in 0.1% NP40 − spontaneous release]).
CD107a granule release assay
Degranulation of CAR-T cells and CTLs was measured by appearance of CD107a on the surface of responding cytolytic effectors, as previously described (50). Effector cells in complete RPMI 1640 media containing PE-labeled anti-CD107a antibodies were combined with target cells at various densities in flat bottom 96-well plates. The plates were incubated for 4 h at 37 °C in CO2 incubator. Effector cells were then harvested and analyzed for appearance CD107a on the surface by flow cytometry using PE-labeled anti-human CD107a antibodies (BD Pharmingen). The percent of responding effector cells was determined.
Serine esterase release assay
The extent of degranulation of the effector cells responding to the target cells was evaluated by measuring serine-esterase activity of granzymes, a component of released cytolytic granules as previous described with some modification (12). The cytolytic effectors were combined with live target cells at various densities as above. Following incubation, plates were spun down for 5 min at 1300 rpm. In total, 100 μl of supernatant was removed from each well and placed into a fresh 96-well flat bottom plate; 50 μl of 2 mM 5,5′-dithio-bis-[2-nitrobenzoic acid] (DTNB or Ellman’s Reagent) in assay buffer (0.15 M NaCl, 0.01 M Hepes) was added to each well. The plate was incubated at room temperature for 15 min, and then 50 μl of 0.8 mM benzyloxycarbonyl-L-lysine thiobenzyl ester (BLT), a substrate for serine esterases, was added in assay buffer to each well followed by incubation for 30 min at 37 °C. The substrate digestion resulted in the color change of absorbed light measured at 405 nm by automatic ELISA plate reader. An equal number of effector cells were also lysed using two liquid nitrogen freeze–thaw cycles to determine maximal release of serine esterases. Percent released granules was calculated from the average of triplicates as follows: 100 × ([experimental granule release − spontaneous release]/[total release after LN2-mediated lysis − spontaneous release]).
Data availability
All data presented in the manuscript could be shared upon request.
Supporting information
This article contains supporting information.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We apologize that we were not able to quote many significant publications in the field since our goal was to emphasize particular issues without being comprehensive. We would like to thank Mariusz Wasik and Kerry Campbell for critical reading of the manuscript and helpful discussion. We thank Bruce Walker and Antonio Lanzavecchia for supplying human CD8+ T-cell clones. We also thank the Flow Cytometry Facility of the Sidney Kimmel Cancer Center for excellent technical assistance. We would like to acknowledge help and support of all members of Takami Sato and Yuri Sykulev laboratories. We also would like to acknowledge Dario Vignali for offering pMSCV-IRES-Blue FP retroviral vector and Garry Nolan for providing pBMN-I-GFP retroviral vector pMSCV-IRES-Blue FP.
Author contributions
N. A., S. P., N. W. M., T. S., and Y. S. conceptualization; N. A., S. P., N. W. M., M. T., T. S., and Y. S. data curation; N. A., S. P., N. W. M., M. T., T. S., and Y. S. formal analysis; N. A., T. S., and Y. S. funding acquisition; S. P. methodology; N. A., N. W. M., M. T., and Y. S. writing—original draft: T. S. and Y. S. writing—review and editing.
Funding and additional information
This work was supported by The Dean’s Transformational Science Award Grant (Thomas Jefferson University, United States) to T. S. and Y. S. and by the grant from Hasumi International Research Foundation (Japan) to T. S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Edited by Peter Cresswell
Supporting information
Supplemental data.
References
- 1.Gubin M.M., Zhang X., Schuster H., Caron E., Ward J.P., Noguchi T., Ivanova Y., Hundal J., Arthur C.D., Krebber W.J., Mulder G.E., Toebes M., Vesely M.D., Lam S.S., Korman A.J. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuster S.J., Svoboda J., Chong E.A., Nasta S.D., Mato A.R., Anak O., Brogdon J.L., Pruteanu-Malinici I., Bhoj V., Landsburg D., Wasik M., Levine B.L., Lacey S.F., Melenhorst J.J., Porter D.L. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017;377:2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 4.Amara S., Tiriveedhi V. The five immune forces impacting DNA-based cancer immunotherapeutic strategy. Int. J. Mol. Sci. 2017;18:650. doi: 10.3390/ijms18030650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feins S., Kong W., Williams E.F., Milone M.C., Fraietta J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019;94:S3–S9. doi: 10.1002/ajh.25418. [DOI] [PubMed] [Google Scholar]
- 6.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scarfo I., Maus M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother. Cancer. 2017;5:28. doi: 10.1186/s40425-017-0230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez M., Moon E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma S., Li X., Wang X., Cheng L., Li Z., Zhang C., Ye Z., Qian Q. Current progress in CAR-T cell therapy for solid tumors. Int. J. Biol. Sci. 2019;15:2548–2560. doi: 10.7150/ijbs.34213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang C., Liu J., Zhong J.F., Zhang X. Engineering CAR-T cells. Biomark. Res. 2017;5:22. doi: 10.1186/s40364-017-0102-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang C.C., Campoli M., Luo W., Zhao W., Zaenker K.S., Ferrone S. Immunotherapy of melanoma targeting human high molecular weight melanoma-associated antigen: Potential role of nonimmunological mechanisms. Ann. N. Y. Acad. Sci. 2004;1028:340–350. doi: 10.1196/annals.1322.040. [DOI] [PubMed] [Google Scholar]
- 12.Beal A.M., Anikeeva N., Varma R., Cameron T.O., Norris P.J., Dustin M.L., Sykulev Y. Protein kinase C theta regulates stability of the peripheral adhesion ring junction and contributes to the sensitivity of target cell lysis by CTL. J. Immunol. 2008;181:4815–4824. doi: 10.4049/jimmunol.181.7.4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X., Katayama A., Wang Y., Yu L., Favoino E., Sakakura K., Favole A., Tsuchikawa T., Silver S., Watkins S.C., Kageshita T., Ferrone S. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011;71:7410–7422. doi: 10.1158/0008-5472.CAN-10-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anikeeva N., Lebedeva T., Krogsgaard M., Tetin S.Y., Martinez-Hackert E., Kalams S.A., Davis M.M., Sykulev Y. Distinct molecular mechanisms account for the specificity of two different T-cell receptors. Biochemistry. 2003;42:4709–4716. doi: 10.1021/bi026864+. [DOI] [PubMed] [Google Scholar]
- 15.Anikeeva N., Gakamsky D., Scholler J., Sykulev Y. Evidence that the density of self peptide-MHC ligands regulates T-cell receptor signaling. PLoS One. 2012;7 doi: 10.1371/journal.pone.0041466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sidney J., del Guercio M.F., Southwood S., Hermanson G., Maewal A., Appella E., Sette A. The HLA-A∗0207 peptide binding repertoire is limited to a subset of the A∗0201 repertoire. Hum. Immunol. 1997;58:12–20. doi: 10.1016/s0198-8859(97)00206-1. [DOI] [PubMed] [Google Scholar]
- 17.Ioannidou K., Randin O., Semilietof A., Maby-El Hajjami H., Baumgaertner P., Vanhecke D., Speiser D.E. Low avidity T cells do not hinder high avidity T cell responses against melanoma. Front. Immunol. 2019;10:2115. doi: 10.3389/fimmu.2019.02115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sykulev Y., Cohen R.J., Eisen H.N. The law of mass action governs antigen-stimulated cytolytic activity of CD8+ cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 1995;92:11990–11992. doi: 10.1073/pnas.92.26.11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sykulev Y., Vugmeyster Y., Brunmark A., Ploegh H., Eisen H. Peptide antagonism and T cell receptor interactions with peptide-MHC complexes. Immunity. 1998;9:475–483. doi: 10.1016/s1074-7613(00)80631-7. [DOI] [PubMed] [Google Scholar]
- 20.Seliger B., Cabrera T., Garrido F., Ferrone S. HLA class I antigen abnormalities and immune escape by malignant cells. Semin. Cancer Biol. 2002;12:3–13. doi: 10.1006/scbi.2001.0404. [DOI] [PubMed] [Google Scholar]
- 21.Ruiz-Cabello F., Cabrera T., Lopez-Nevot M.A., Garrido F. Impaired surface antigen presentation in tumors: Implications for T cell-based immunotherapy. Semin. Cancer Biol. 2002;12:15–24. doi: 10.1006/scbi.2001.0406. [DOI] [PubMed] [Google Scholar]
- 22.Cabrera T., Lopez-Nevot M.A., Gaforio J.J., Ruiz-Cabello F., Garrido F. Analysis of HLA expression in human tumor tissues. Cancer Immunol. Immunother. 2003;52:1–9. doi: 10.1007/s00262-002-0332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 24.Vesely M.D., Schreiber R.D. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 2013;1284:1–5. doi: 10.1111/nyas.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caramalho I., Faroudi M., Padovan E., Muller S., Valitutti S. Visualizing CTL/melanoma cell interactions: Multiple hits must be delivered for tumour cell annihilation. J. Cell. Mol. Med. 2009;13:3834–3846. doi: 10.1111/j.1582-4934.2008.00586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hay K.A., Turtle C.J. Chimeric antigen receptor (CAR) T cells: Lessons learned from targeting of CD19 in B-cell malignancies. Drugs. 2017;77:237–245. doi: 10.1007/s40265-017-0690-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pillai V., Muralidharan K., Meng W., Bagashev A., Oldridge D.A., Rosenthal J., Van Arnam J., Melenhorst J.J., Mohan D., DiNofia A.M., Luo M., Cherian S., Fromm J.R., Wertheim G., Thomas-Tikhonenko A. CAR T-cell therapy is effective for CD19-dim B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy. Blood Adv. 2019;3:3539–3549. doi: 10.1182/bloodadvances.2019000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henkart P.A., Millard P.J., Reynolds C.W., Henkart M.P. Cytolytic activity of purified cytoplasmic granules from cytotoxic rat large granular lymphocyte tumors. J. Exp. Med. 1984;160:75–93. doi: 10.1084/jem.160.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stone J.D., Harris D.T., Kranz D.M. TCR affinity for p/MHC formed by tumor antigens that are self-proteins: Impact on efficacy and toxicity. Curr. Opin. Immunol. 2015;33:16–22. doi: 10.1016/j.coi.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linette G.P., Stadtmauer E.A., Maus M.V., Rapoport A.P., Levine B.L., Emery L., Litzky L., Bagg A., Carreno B.M., Cimino P.J., Binder-Scholl G.K., Smethurst D.P., Gerry A.B., Pumphrey N.J., Bennett A.D. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engels B., Chervin A.S., Sant A.J., Kranz D.M., Schreiber H. Long-term persistence of CD4(+) but rapid disappearance of CD8(+) T cells expressing an MHC class I-restricted TCR of nanomolar affinity. Mol. Ther. 2012;20:652–660. doi: 10.1038/mt.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sykulev Y., Joo M., Vturina I., Tsomides T.J., Eisen H.N. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity. 1996;4:565–571. doi: 10.1016/s1074-7613(00)80483-5. [DOI] [PubMed] [Google Scholar]
- 33.Labrecque N., Whitfield L.S., Obst R., Waltzinger C., Benoist C., Mathis D. How much TCR does a T cell need? Immunity. 2001;15:71–82. doi: 10.1016/s1074-7613(01)00170-4. [DOI] [PubMed] [Google Scholar]
- 34.Harris D.T., Hager M.V., Smith S.N., Cai Q., Stone J.D., Kruger P., Lever M., Dushek O., Schmitt T.M., Greenberg P.D., Kranz D.M. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J. Immunol. 2018;200:1088–1100. doi: 10.4049/jimmunol.1700236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gudipati V., Rydzek J., Doel-Perez I., Goncalves V.D.R., Scharf L., Konigsberger S., Lobner E., Kunert R., Einsele H., Stockinger H., Hudecek M., Huppa J.B. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat. Immunol. 2020;21:848–856. doi: 10.1038/s41590-020-0719-0. [DOI] [PubMed] [Google Scholar]
- 36.Beal A.M., Anikeeva N., Varma R., Cameron T.O., Vasiliver-Shamis G., Norris P.J., Dustin M.L., Sykulev Y. Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity. 2009;31:632–642. doi: 10.1016/j.immuni.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sykulev Y. T cell receptor signaling kinetics takes the stage. Sci. Signal. 2010;3 doi: 10.1126/scisignal.3153pe50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Combs J., Kim S.J., Tan S., Ligon L.A., Holzbaur E.L., Kuhn J., Poenie M. Recruitment of dynein to the Jurkat immunological synapse. Proc. Natl. Acad. Sci. U. S. A. 2006;103:14883–14888. doi: 10.1073/pnas.0600914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stinchcombe J.C., Majorovits E., Bossi G., Fuller S., Griffiths G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006;443:462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- 40.Davenport A.J., Cross R.S., Watson K.A., Liao Y., Shi W., Prince H.M., Beavis P.A., Trapani J.A., Kershaw M.H., Ritchie D.S., Darcy P.K., Neeson P.J., Jenkins M.R. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E2068–E2076. doi: 10.1073/pnas.1716266115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anikeeva N., Sykulev Y. Mechanisms controlling granule-mediated cytolytic activity of cytotoxic T lymphocytes. Immunol. Res. 2011;51:183–194. doi: 10.1007/s12026-011-8252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anikeeva N., Somersalo K., Sims T.N., Thomas V.K., Dustin M.L., Sykulev Y. Distinct role of lymphocyte function-associated antigen-1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 2005;102:6437–6442. doi: 10.1073/pnas.0502467102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anikeeva N., Lebedeva T., Clapp A.R., Goldman E.R., Dustin M.L., Mattoussi H., Sykulev Y. Quantum dot/peptide-MHC biosensors reveal strong CD8-dependent cooperation between self and viral antigens that augment the T cell response. Proc. Natl. Acad. Sci. U. S. A. 2006;103:16846–16851. doi: 10.1073/pnas.0607771103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X., Ferrone S. International Bureau of World Intellectual Property Organization; 2010. Fully Human Antibodies to High Molecular Weight-Melanoma Associated Antigen and Uses Thereof. Article Number WO 2010/045495 A2. [Google Scholar]
- 45.Gakamsky D.M., Davis D.M., Strominger J.L., Pecht I. Assembly and dissociation of human leukocyte antigen (HLA)-A2 studied by real-time fluorescence resonance energy transfer. Biochemistry. 2000;39:11163–11169. doi: 10.1021/bi000763z. [DOI] [PubMed] [Google Scholar]
- 46.Tsomides T.J., Walker B.D., Eisen H.N. An optimal viral peptide recognized by CD8+ T cells binds very tightly to the restricting class I major histocompatibility complex protein on intact cells but not to the purified class I protein. Proc. Natl. Acad. Sci. U. S. A. 1991;88:11276–11280. doi: 10.1073/pnas.88.24.11276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sykulev Y., Brunmark A., Jackson M., Cohen R.J., Peterson P.A., Eisen H.N. Kinetics and affinity of reactions between an antigen-specific T-cell receptor and peptide-MHC complexes. Immunity. 1994;1:15–22. doi: 10.1016/1074-7613(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 48.Scotto M., Afonso G., Osterbye T., Larger E., Luce S., Raverdy C., Novelli G., Bruno G., Gonfroy-Leymarie C., Launay O., Lemonnier F.A., Buus S., Carel J.C., Boitard C., Mallone R. HLA-B7-restricted islet epitopes are differentially recognized in type 1 diabetic children and adults and form weak peptide-HLA complexes. Diabetes. 2012;61:2546–2555. doi: 10.2337/db12-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gotch F., Rothbard J., Howland K., Townsend A., McMichael A. Cytotoxic T lymphocytes recognize a fragment of influenza virus matrix protein in association with HLA-A2. Nature. 1987;326:881–882. doi: 10.1038/326881a0. [DOI] [PubMed] [Google Scholar]
- 50.Steblyanko M., Anikeeva N., Campbell K.S., Keen J.H., Sykulev Y. Integrins influence the size and dynamics of signaling microclusters in a Pyk2-dependent manner. J. Biol. Chem. 2015;290:11833–11842. doi: 10.1074/jbc.M114.614719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in the manuscript could be shared upon request.