

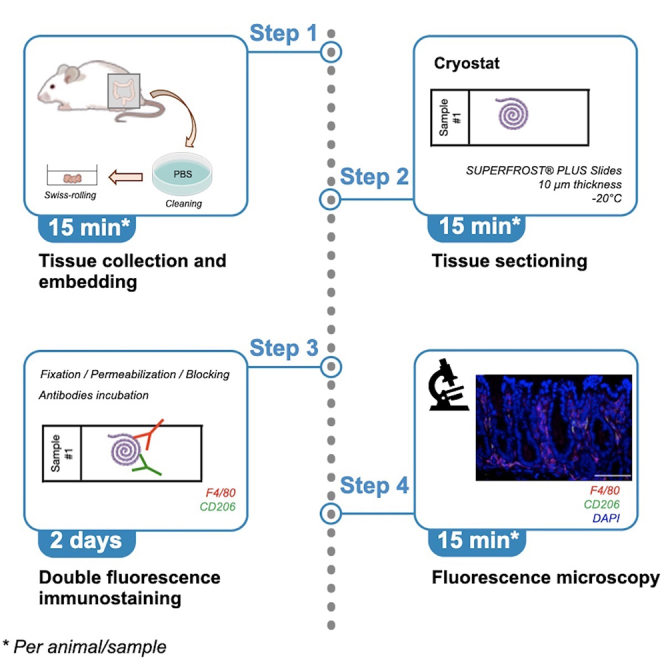
Summary
We recently characterized the association between DNA damage and immunoresponse in vivo in colonic mucosa of mice infected with a Salmonella Typhimurium strain expressing a genotoxin, known as typhoid toxin. In this protocol, we describe how to assess the extent and features of infiltrating macrophages by double immunofluorescence. Total macrophage population was determined using an F4/80 antibody, whereas the specific M2-like population was assessed using a CD206 antibody.
For complete details on the use and execution of this protocol, please refer to Martin et al. (2021).
Subject areas: Cell-based assays, Microscopy, In situ hybridization
Graphical abstract
Highlights
-
•
Collection and OCT-embedding of mice colon
-
•
Detailed description and optimization of double immunofluorescence
-
•
Characterization of total macrophage population and specific M2-like population
We recently characterized the association between DNA damage and immunoresponse in vivo in colonic mucosa of mice infected with a Salmonella Typhimurium strain expressing a genotoxin, known as typhoid toxin. In this protocol, we describe how to assess the extent and features of infiltrating macrophages by double immunofluorescence. Total macrophage population was determined using an F4/80 antibody, whereas the specific M2-like population was assessed using a CD206 antibody.
Before you begin
Preparation and sectioning of murine colon for immunofluorescence
Timing: 2 days
-
1.
During the necropsies, colons have been collected and prepared for histology following the Swiss-roll technique by rolling up the intestine lengthwise. The colon is removed from the abdomen and cleaned with 1× PBS (no Mg2+, no Ca2+) from the colonic content. The colon is rolled up longitudinally with the help of a toothpick from the distal to the proximal end (Moolenbeek and Ruitenberg, 1981). Immediately insert the rolled colon in a cryo-mold containing cryo-embedding medium (OCT). Then, cover completely the tissue by adding more OCT. The cryo-mold should be placed immediately on dry ice. Frozen tissue blocks can be stored at −80°C, for long-term storage or prior sectioning.
-
2.
The tissue sectioning is performed using a Cryostat at the temperature of −20°C.
-
3.
Cut sections at 10 μm thickness and mount them onto SUPERFROST® PLUS Microscope Adhesion Slides (positively charged).
-
4.
Keep the sections for 1 h on dry ice to retain tissue adherence and store slides at −80°C until usage.
The protocol below describes the specific steps for immunofluorescence (IF) on colon. However, it can be used also for other tissues (e.g., spleen, liver). This protocol has been done in fresh frozen tissues and its application to Formalin-Fixed Paraffin-Embedded (FFPE) might require further optimization.
Preparation of solutions for IF
Permeabilization buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Bovine Serum Albumin (BSA) | 1% (w/v) | 400 mg |
| Fetal Bovine Serum (FBS) | 2% (v/v) | 800 μL |
| Triton X-100 (1%) | 0.1% (v/v) | 4 mL |
| PBS | ∼ 35 mL | |
| Total | n/a | 40 mL |
Note: Permeabilization buffer can be stored at +4°C for at least 1 month.
Note: As Triton X-100 is very dense, we recommend to prepare an initial diluted solution in PBS. In our case, we prepared a 1% solution of Triton X-100. Note to adjust the volume of Triton X-100 added to the final buffer depending on the percentage of the prepared solution.
Note: In order to ensure that BSA is properly dissolved, store the BSA solution in PBS at +4°C for 1 hour. Avoid vortex the solution before BSA is dissolved. Strong shaking prior complete dissolution would result in excessive formation of foam.
CRITICAL: Triton X-100 is considered as harmful if swallowed (H302), may cause skin irritation (H315), and causes serious eye damage (H319). Therefore, it should be manipulated by wearing appropriate recommended PPE (personal protection equipment), such as skin and eye/face protection.
Blocking buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Bovine Serum Albumin | 1% (w/v) | 400 mg |
| Fetal Bovine Serum (FBS) | 2% (v/v) | 800 μL |
| PBS | ∼ 39 mL | |
| Total | n/a | 40 mL |
Note: Blocking buffer can be stored at +4°C for at least 1 month.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal Rat Anti-F4/80 IgG (1:100) | Bio-Rad | Cat# MCA497G; RRID:AB_872005 |
| Polyclonal Goat Anti-CD206 (1:500) | R&D system | Cat# AF2535; RRID:AB_2063012 |
| Donkey Anti-Goat IgG (H+L) Alexa Fluor® 488 (1:1000) | Thermo Fisher Scientific | Cat# A32814, RRID:AB_2762838 |
| Goat Anti-Rat IgG (H+L) Alexa Fluor® 555 (1:1000) | Thermo Fisher Scientific | Cat# A-21434; RRID:AB_2535855 |
| Chemicals, peptides, and recombinant proteins | ||
| Triton® X-100 | Serva | Cat# 39795 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A7906 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | Cat# 10270-106 |
| Acetone | Sigma-Aldrich | Cat# 17124 |
| VECTASHIELD® Antifade Mounting Medium with DAPI | Vector Lab | Cat# H-1200-10 |
| Tissue-Tek OCT | Sakura Finetek | Cat# 4583 |
| Software and algorithms | ||
| ImageJ/Fiji | https://imagej.net/Fiji/Downloads | https://10.1038/nmeth.2019 |
| Microsoft Excel | microsoft.com | n/a |
| GraphPad Prism | https://www.graphpad.com | RRID:SCR_002798 |
| NIS-Elements AR 3.2 | Nikon | RRID:SCR_014329 |
| Other | ||
| SUPERFROST® Plus Microscope Adhesion Slides | Thermo Scientific | Cat# 10149870 |
| Tissue-Tek Cryomolds (15 mm × 15mm) | Sakura Finetek | Cat# 4566 |
| Cryostat | Leica | Cat# CM-3050S |
| Coverslips (24×32 mm) | Menzel-Gläser | Cat# 4610014 |
| Humidified chamber StainTray™ 10 slides staining system | Simport Scientific | Cat# M918-2 |
| EASYDIP™ Slide Staining Jars | Simport Scientific | Cat# M900-12B |
| Lab soaker VERSI-DRY® Nalgene® | Thermo Scientific | Cat# 52857-106 |
| PAP pen for immunostaining | Sigma-Aldrich | Cat# Z672548-1EA |
| Nikon Eclipse 90i | Nikon | RRID:SCR_020335 |
Materials and equipment
Alternatives:
Home-made humidified chambers can be used as long as they can maintain the humid environment (i.e., they properly close and humidity is maintained by soaking wipes with water and avoid drying out) and they properly protect slides from light after incubation with secondary antibodies (Figure 1).
Figure 1.

Examples of humidified chambers
Home-made (A) and commercially available (B) from Simport Scientific.
Step-by-step method details
Immunofluorescence staining specific for M2-like and total macrophage populations
The main purpose of this step is to obtain the binding between the first primary antibodies specific for i) the membrane receptor CD206, which is expressed mainly in the surface of macrophages with a M2-like phenotype; ii) the pan murine macrophage marker F4/80. This allows the characterization of the immune response profile in the studied tissues.
In order to succeed in the staining procedure, the steps of fixation, permeabilization and blocking of unspecific sites are crucial.
-
1.Remove the selected slides from −80°C and allow them to reach room temperature (20°C–25°C) for 20 min.
-
a.Draw 2–4 times the hydrophobic barrier around the sections using the hydrophobic marker PAP Pen and let the barrier dry completely for 5 min at room temperature (20°C–25°C). Do not draw too close to the tissue section and do not touch it while drawing the hydrophobic barrier.
-
a.
-
2.Fixation
-
a.Place the slides in a slide-holder and cover completely the sections (∼400 μL) with cold acetone (stored at −20°C). Let it dry completely for 10 min at room temperature (20°C–25°C).Note: this step should be done under the chemical hood.
-
b.Rehydrate the tissue in 1× PBS for 10 min by placing the slides in a slide staining jar.
-
c.Place the slides in a slide staining jar and wash three times in 1× PBS under gentle shaking, for 5 min each wash (Methods video S1).Methods video S1. Washing the slides, related to steps 2c, 3c, 5e, 6e, and 7eDownload video file (5.4MB, mp4)
-
a.
-
3.Permeabilization
-
a.Remove excess PBS from the slides using a wipe (Methods video S2). Do not touch the sections.
-
b.Cover the sections with 100 μL permeabilization buffer and incubate for 30 min in a humidified chamber containing ddH2O at room temperature (20°C–25°C).
-
c.Place the slides in a slide staining jar and wash three times in 1× PBS under gentle shaking, for 5 min each wash (Methods video S1).
-
a.
-
4.Blocking
-
a.Remove excess PBS from the slides using a wipe (Methods video S2). Do not touch the sections.
-
b.Cover the sections with 100 μL blocking buffer and incubate for 30 min in a humidified chamber at room temperature (20°C–25°C).
-
a.
-
5.Addition of first primary antibody
-
a.Dilute the goat anti CD206 antibody (1:500 v/v) in blocking buffer.
-
b.Gently vortex the solution for 2 s at moderate speed.
-
c.Remove excess blocking solution from the slides using a wipe (Methods video S2). Do not touch the sections.
-
d.Cover the sections with 100 μL blocking buffer supplemented with anti-CD206 and incubate for 12–16 h in a humidified chamber at +4°C.
-
e.Place the slides in a slide staining jar and wash three times in 1× PBS under gentle shaking, for 5 min each wash. (Methods video S1).
-
a.
Figure 2.

Designing of hydrophobic barrier and coverage of the sections
(A) The hydrophobic barrier retains the liquid solution on the top of the sections. The tissue sections must be always covered.
(B) The presence of the hydrophobic barrier allows to perform different stainings in tissues located in the same slide.
Note: The usage of hydrophobic marker is strongly recommended to avoid the spreading of the staining solution throughout the slide. Moreover, the hydrophobic barrier can be useful to avoid cross-contamination, if different staining solutions are used next to each other in the same slide (Figure 2B)
-
6.Addition of second primary antibody
-
a.Dilute the rat anti-F4/80 antibody (1:100 v/v) in blocking buffer.
-
b.Gently vortex the solution for 2 s at moderate speed.
-
c.Remove excess PBS from the slides using a wipe (Methods video S2). Do not touch the sections.
-
d.Cover the sections with 100 μL blocking buffer supplemented with anti-F4/80 antibody and incubate for 1 h in a humidified chamber at room temperature (20°C–25°C).
-
e.Place the slides in a slide staining jar and wash three times in 1× PBS under gentle shaking, for 5 min each wash. (Methods video S1).
-
a.
-
7.Addition of secondary fluorescent-conjugated antibodies
-
a.Dilute 1:1000 (v/v) the Alexa Fluor® 555-conjugated anti-rat and Alexa Fluor® 488- conjugated anti-goat secondary antibodies in blocking buffer.
-
b.Gently vortex the solution for 2 s at moderate speed.
-
c.Remove excess PBS from the slides using a wipe (Methods video S2). Do not touch the sections.
-
d.Cover the sections with 100 μL blocking buffer supplemented with secondary antibodies and incubate for 1 h in a humidified chamber at room temperature (20°C–25°C).
-
e.Place the slides in a slide staining jar and wash three times in 1× PBS under gentle shaking, for 5 min each wash. (Methods video S1).
-
a.
-
8.Mounting
-
a.Remove excess of 1× PBS from the slides using a wipe (Methods video S2). Do not touch the sections.
-
b.Place one drop of VECTASHIELD® Antifade Mounting Medium with DAPI on the tissue section.
-
c.Lay a microscope glass coverslip on the tissue section.
-
d.Remove excess of VECTASHIELD® Antifade Mounting Medium with DAPI at the edges of the slide, avoiding to press the glass coverslip.
-
e.Fix the coverslip at the perimeters with nail polish.
-
a.
Pause point: The mounted slides can be stored at +4°C for one week under protection from light prior imaging.
Note: Fluorophore-conjugated secondary antibodies tend to form precipitates. It is recommended to vortex them prior usage to reduce the risk of fluorescent clamps in the staining procedure, which would interfere with the imaging process.
Microscopy imaging
-
9.
Adjust the exposure time and gain of each channel blue (DAPI), green (CD206) and red (F4/80) to satisfy the dynamic range of the experiment.
Note: The adjustment should be done from the sample having the highest intensity.
-
10.
Once the settings are selected, acquire pictures of at least 5 representative fields from each sample on individual blue (DAPI), red (F4/80) and green (CD206) channels.
-
11.
Multi-channel images can be merged or separated, based on the visualization purposes. ImageJ/FIJI can be utilized for merging or separating the channels of the different pictures.
Note: Fluorescent staining are not permanent. Therefore, it is recommended to acquire the images of all the samples in the same day to relate any variation of the signal intensity to biological relevance and not to fluorescence stability.
Expected outcomes
The described method allows a straightforward staining and quantification of total and M2-macrophages in colonic murine tissues in order to assess the intestinal immunological environment. As expected, staining of M2-like macrophages (CD206-positive cells) must co-localize with F4/80 staining and both stainings must be localized at the cell membrane.
Quantification and statistical analysis
Quantitative results were obtained upon microscopy image and processing (Figure 3 and Table 1). Quantification was done using the available tools in Fiji/ImageJ. Total number of cells was counted based on the DAPI nuclear staining with the help of the following macro for simplifying and automating cell nuclei counting:
run("Set Measurements...", "area mean min center display redirect=None
decimal=0");
setSlice(1);
run("Duplicate...", "title=dapi");
run("Mean...", "radius=2");
run("Subtract Background...", "rolling=20");
setAutoThreshold("Mean");
//run("Threshold...");
setAutoThreshold("Mean dark");
setOption("BlackBackground", false);
run("Convert to Mask");
run("Watershed");
setAutoThreshold("Mean");
run("Analyze Particles...", "size=50-Infinity pixel add");
close();
setSlice(1);
roiManager("Show All with labels");
roiManager("Show All");
roiManager("Measure");
Figure 3.

Quantitative analysis using Image J
(A) Detected cell nuclei after using the above mentioned macro. A separate table with the total number accompanies this image.
(B) Quantification of F4/80-positive cells and the corresponding table for counting (red fluorescence).
(C) Quantification of F4/80-CD206-double positive cells and the corresponding table for counting (green fluorescence).
Scale bar: 50 μm.
Table 1.
Example of total and M2-like macrophages quantification
| Mouse ID | Picture | Total nuclei | F4/80 pos | % F4/80 | CD206 pos (M2) | % M2 | % M2 over F4/80 |
|---|---|---|---|---|---|---|---|
| A | 1 | 581 | 44 | 7.573 | 21 | 3.614 | 47.727 |
| 2 | 775 | 71 | 9.161 | 28 | 3.613 | 39.437 | |
| 3 | 650 | 41 | 6.308 | 24 | 3.692 | 58.536 | |
| … |
Both F4/80 and CD206 stainings must be located at the cell membrane and any hints of nuclear staining should not be considered as positive (Figure 4). Manual quantification of F4/80-positive cells (total macrophages) and of F4/80-CD206-double positive cells (M2 macrophages) was done by using the cell counter plugin included in ImageJ. No macro was used for positive stained cells. An example of the quantification is shown in Table 1. Data can be plotted as bar graphs showing the percentage of M2-like macrophages over the total percentage of macrophages (mean ± SEM). The significance of differences between two experimental groups (e.g., mice infected with the control and the genotoxin Salmonella strain) is determined by the Student t-test. The significance of differences between three (or more) experimental groups is determined by ANOVA with Fisher’s LSD post-test. p values <0.05 were considered significant. For a representative quantification of each experimental condition, we suggest to acquire 5 images of different fields across the organ, for a total of ∼2500 nuclei per mouse.
Figure 4.

Representative image of F4/80-CD206-double positive cells (M2 macrophages)
Arrow shows the proper staining located at membrane while arrowhead depicts false positive nuclear staining. Scale bar: 10 μm.
Limitations
The major limitations of this protocol are related to the preservation of the tissue prior sectioning in order to maintain the proper architecture. Therefore, it is essential to immediately freeze down the Swiss rolls once they are prepared during the necropsy and to ensure that both the colon and the tissue sections slides are properly stored at −80°C until use. Furthermore, it is very important to avoid sections from drying out during staining and make sure that the tissues are always covered by the solutions during the whole procedure.
Troubleshooting
Successful performance of this staining mainly relies on the quality of the tissue sections and the use of proper fixation and blocking solutions.
Problem 1
Tissue sections are not properly prepared during cutting at the Cryostat (e.g., tissue sections are not stretched and flattened prior adhesion to the positively charged slide) (Before you begin, steps 2 to 4).
Potential solution
When recovering the tissue section to the microscope adhesion slide, ensure that: i) the section is properly flattened; ii) place the microscope adhesion slide (on the positive charged side, indicated by “+”) parallel to the Cryostat platform. Once the tissue section is attached to the microscope adhesion slide, do not press down nor move the slide onto the Cryostat platform but place it immediately on dry ice to favor a better adhesion.
Problem 2
Incorrect solution used for fixation or blocking steps results in unspecific staining and high background (Figure 5) (Steps 2 and 4).
Figure 5.

Example of M2-like macrophage unspecific staining (shown by arrowheads)
Scale bar: 50 μm.
Potential solution
The problem of unspecific staining may rise especially due to incorrect procedure in fixation or in blocking.
For a proper fixation for this staining (step 2), store the acetone solution at −20°C for at least 24–48 h before carrying out the immunofluorescence assay. The acetone will evaporate during the incubation time. Therefore, it is important to not exceed the incubation time of fixation to avoid the sections to dry out for long time, which would lead to unspecific fluorescent signal.
The blocking step (step 4) is also crucial to mitigate the unspecific binding. To further improve the saturation of unspecific binding sites, it is recommended to increase the concentration of BSA from 1% to 2% (w/v) in the blocking solution.
If the staining still does not satisfy the expectations, we suggest to employ normal serum at 1%–5% (v/v) dissolved in 1× PBS with incubation for 10 min at room temperature (20°C–25°C).
Note: Serum can be employed from the same species where the secondary antibodies were raised. Do not use the same species as the primary antibodies (e.g., if the primary antibody was raised in rabbit and the secondary in goat, the goat serum might be employed to saturate unspecific binding sites but rabbit serum should not be used).
Problem 3
Precipitates of secondary antibodies or traces of dirt are seen under microscopy all over the colonic tisue (step 7) (Figure 6).
Figure 6.

Example of precipitates of secondary antibodies or traces of dirt
Scale bar: 50 μm.
Potential solution
Ensure to properly mix secondary antibodies with fluorophores before use and/or replace and prepare a new solution if the one available has been stored for long-term. Furthermore, we suggest to increase the number of washes with 1× PBS after incubation with secondary fluorescent-conjugated antibodies to ensure the complete removal of traces of precipitates, dust and/or dirt.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Océane CB Martin (oceane.martin@u-bordeaux.fr).
Materials availability
This study did not generate any specific material/reagent.
Acknowledgments
This investigation was supported by grants from the Swedish Cancer Society (CAN 2017/315 and 20 0699 PjF), the Swedish Research Council (2018–02521), the Kempestiftelserna (JCK-1826), the Cancer Research Foundation in Northern Sweden (AMP20- 993 and AMP 17–884), and Umeå University (to T.F.). We acknowledge the Biochemical Imaging Center Umeå (BICU) at Umeå University and the National Microscopy Infrastructure (NMI) (VR-RFI 2016-00968) for providing assistance with microscopy.
Author contributions
O.C.B.M. and T.F. designed the experiments. M.L.C., A.B., and O.C.B.M. performed the experiments. All authors contributed to the protocol writing.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100833.
Data and code availability
Original data for figures are available upon request to the lead contact.
References
- Martin O.C.B., Bergonzini A., Lopez Chiloeches M., Paparouna E., Butter D., Theodorou S.D.P., Haykal M.M., Boutet-Robinet E., Tebaldi T., Wakeham A. Influence of the microenvironment on modulation of the host response by typhoid toxin. Cell Rep. 2021;35:108931. doi: 10.1016/j.celrep.2021.108931. [DOI] [PubMed] [Google Scholar]
- Moolenbeek C., Ruitenberg E.J. The "Swiss roll": a simple technique for histological studies of the rodent intestine. Lab. Anim. 1981;15:57–59. doi: 10.1258/002367781780958577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data for figures are available upon request to the lead contact.