

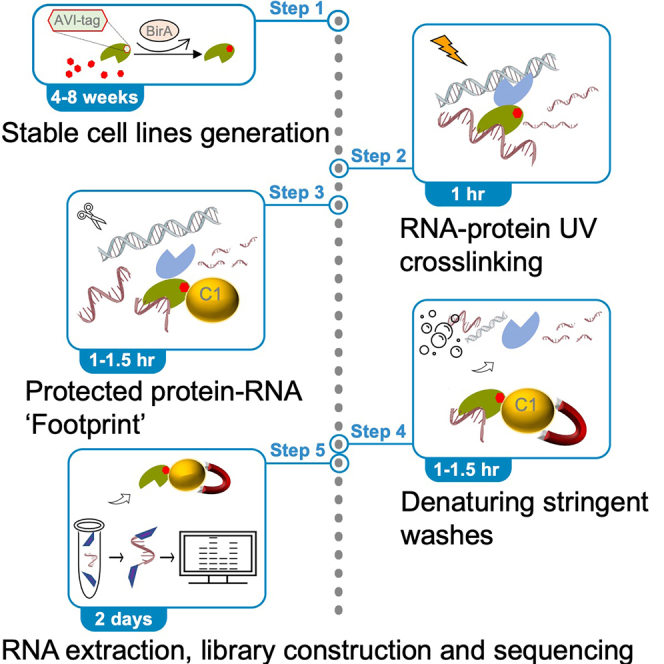
Summary
The isolation of protein-RNA complexes in the “denaturing cross-linked RNA immunoprecipitation” (dCLIP) protocol is based on biotin-tagging proteins of interest, UV cross-linking RNA to protein in vivo, RNase protection assay, and isolating RNA-protein complexes under denaturing conditions over a streptavidin column. Insofar as conventional antibody-based CLIP assays have been challenging to apply to Polycomb complexes, dCLIP has been applied successfully and yields small RNA footprints from which de novo motif analysis can be performed to identify RNA binding motifs.
For complete details on the use and execution of this protocol, please refer to Rosenberg et al. (2017).
Subject areas: Genetics, Genomics, Molecular Biology, Molecular/Chemical Probes, Protein Biochemistry, Systems biology
Graphical abstract
Highlights
-
•
dCLIP biotags a protein of interest to identify cross-linked RNA interactors in vivo
-
•
Biotin-streptavidin purification system enables denaturing washing conditions
-
•
dCLIP is successfully applied to chromatin-modifying protein complexes
-
•
dCLIP allows high-resolution mapping of RNA binding sites and de novo motif analysis
The isolation of protein-RNA complexes in the “denaturing cross-linked RNA Immunoprecipitation” (dCLIP) protocol is based on biotin-tagging proteins of interest, UV cross-linking RNA to protein in vivo, RNase protection assay, and isolating RNA-protein complexes under denaturing conditions over a streptavidin column. Insofar as conventional antibody-based CLIP assays have been challenging to apply to Polycomb complexes, dCLIP has been applied successfully and yields small RNA footprints from which de novo motif analysis can be performed to identify RNA binding motifs.
Before you begin
Prepare stable cell lines expressing BirA biotinylase and AVI-tagged protein/s of interest
We utilize the in vivo biotinylation system described previously in Kim et al. (Kim et al., 2009). Prepare 2 types of stable cell lines: (1) Parental line expressing V5-His-tagged bacterial biotin ligase BirA (BirAV5His) and (2) Experimental cell line/s using the parental line to express both bacterial biotin ligase BirA and the protein of interest fused to biotag (GLNDIFGAQKIEWHE) or a similar AVI-tag (GLNDIFEAQKIEWHE). These peptides are specifically recognized as a substrate by BirA and undergo biotinylation on the lysine residue.
Note: We used puromycin and neomycin resistance for cell line selection, but different combinations of selection markers can be utilized.
Alternatives: Generate a biotagged protein of interest by knocking-in a biotinylation tag on the N-terminus or C-terminus of the endogenous gene via CRISPR-assisted genome editing in BirA-expressing parental cell line.
Alternatives: The Strep-Tag system (IBA-Lifesciences, https://www.iba-lifesciences.com/strep-tag) can be utilized to achieve a cost-effective high-affinity binding without the requirement for expressing two transgenic proteins. Strep-Tag serves as a mimic for biotin and occupies the same binding pocket on the specifically designed streptavidin. Omitting the need to express BirA enzyme can reduce the potential background signal due to off-target BirA activity.
Timing: 4-8 Weeks
-
1.
Subclone gene/s of interest into an expression vector with biotinylation tag and selection marker for puromycin resistance
-
2.Generate parental stable cell line expressing BirA.
-
a.Grow cells to 70% confluence in T-75 flasks.
-
b.Trypsinize cells to create a single-cell suspension in PBS.
-
c.Electroporate to transfect cells with pEF1aBirAV5His vector in PBS. Use GenePulser II (Bio-Rad) according to the manufacturer’s instructions (https://www.bio-rad.com/en-us/product/gene-pulser-xcell-electroporation-systems?ID=b1a35eb3-d55c-47b3-aaf3-95e4d1d85848#fragment-doc). Use the following cell number/DNA ratio (Ratios may differ for other cell lines).
-
i.2 × 107 ES mouse cells (EL 16.7, (Lee and Lu, 1999)) with 30 μg linearized vector.
-
ii.1 × 107 HEK293 or MEF cells with 15 μg linearized vector.
-
i.
-
d.Culture cells in growth media supplemented with 300 μg/mL Geneticin reagent (G418) to select for transfected cells.
-
e.Confirm transfection and expression of recombinant BirA by PCR genotyping and Western blotting with specific antibodies. Isolate clones with the highest expression of BirA to use as a parental line.
-
a.
-
3.Generate recombinant protein cell line/s.
-
a.Electroporate to transfect parental cells (BirA expressing line) with pEF1a-FlagBiotag-Protein of Interest - PGKpuro or pBrCAG-Avi-GFP-Protein of Interest vectors carrying the protein of interest fused to biotinylation tag or AVI-tag respectively. Prepare control cell lines expressing the empty vector (pEF1a-FlagBiotag- PGKpuro) or Avi-tagged GFP only (pBrCAG-Avi-GFP). Use GenePulser II as in step 2c.Use the following cell number/DNA ratio (ratios may differ for other cell lines).
-
i.2 × 107 ES mouse cells with 30 μg linearized vector (EL 16.7, (Lee and Lu, 1999)).
-
ii.1 × 107 HEK293 or MEF cells with 15 μg linearized vector.
-
i.
-
b.Culture cells in growth media supplemented with 1 μg/mL Puromycin and 300 μg/mL Geneticin (G418) to select for transfected cells.
-
a.
Alternatives: A protein of interest fused to a mutated biotinylation tag (Furuyama and Henikoff, 2006) can be used as an alternative approach for the empty vector or GFP only controls.
Note: We recommend conducting calibration experiments with a range of antibiotic concentrations to determine the minimal concentration that kills the entire cell population within 2–3 days. In our experience, insufficient antibiotic concentration results in a large number of false-positive clones. The instructions for the “killing curve” experiment can be found elsewhere (https://www.sigmaaldrich.com/technical-documents/articles/biology/antibiotic-kill-curve.html)
-
4.
Confirm transfection and expression of recombinant proteins by PCR genotyping and Western blotting with specific antibodies to the recombinant proteins.
Note: To minimize non-specific cross-linking to RNA species. We recommend choosing clones with overall protein expression (recombinant + endogenous) that is most similar to the physiological level, as depicted in Figure 1
Figure 1.

The method for selection of clonal cell lines for dCLIP experiments
Representative Western blotting for clonal cell lines derived from mouse embryonic fibroblasts with stable expression of BirA and Avi-GFP-EZH2 or Avi-GFP alone (control). Protein extracts prepared from each clonal cell line were probed with specific antibodies against EZH2 and GAPDH (loading control) proteins. Following densitometry analysis, the intensity ratios between Avi-GFP-EZH2 and endogenous EZH2 and between total EZH2 (transgenic + endogenous) and GAPDH were computed. Note that expression of transgenic EZH2 resulted in reduced expression of the endogenous EZH2, thus leveraging a total amount of EZH2 protein between control and Avi-GFP-EZH2 expressing cells. Clones# 1C, C14 (highlighted in Bold), that exhibited total EZH2 / GAPDH ratio closest to the control cell lines, were chosen for subsequent dCLIP experiments.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ezh2 (D2C9) XP rabbit monoclonal antibody, 1:1000 dilution | Cell Signaling Technologies Cat# 5246 | RRID:AB_10694683 |
| GAPDH (14C10) rabbit monoclonal antibody, 1:5000 dilution | Cell Signaling Technologies Cat# 2118 | RRID:AB_561053 |
| Bacterial and virus strains | ||
| pEF1aBirAV5His | Dr. Stuart Orkin, Harvard Medical School | n/a |
| pEF1-Flag-Biotag | Dr. Stuart Orkin, Harvard Medical School | n/a |
| pBrCAG-Avi-GFP-IRES-Puro | Gift from Dr. Mitinori Saitou, Kyoto University, Japan | n/a |
| Chemicals, peptides, and recombinant proteins | ||
| Geneticin G418 | Life Technologies | Cat# 10131035 |
| Puromycin | Life Technologies | Cat# A1113803 |
| Roche cOmplete Mini EDTA-free Protease Inhibitor Cocktail Tablets | Sigma-Aldrich | Cat# 4693159001 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# P8340 |
| Roche Protector RNAse Inhibitor, 40U/μL | Sigma-Aldrich | Cat# 3335402001 |
| TURBO™ DNase, 2 U/μL | Life Technologies | Cat# AM2239 |
| RNase I, 10 U/μL | Thermo Fisher Scientific | Cat# EN0601 |
| SUPERaseIn RNase Inhibitor, 20 U/μL | Life Technologies | Cat# AM2694 |
| Dynabeads™ Protein G for Immunoprecipitation | Invitrogen | Cat# 10004D |
| Dynabeads™ MyOne™ Streptavidin C1 | Invitrogen | Cat# 65001 |
| ATP, [γ-32P]-6000Ci/mmol 10mCi/mL, 250 μCi | PerkinElmer | Cat# BLU002Z250UC |
| ATP solution, 100 mM | Thermo Fisher Scientific | Cat# R0441 |
| T4 Polynucleotide Kinase, 10U/μL | New England Biolabs | Cat# M0201S |
| LongAmp_Taq 2X Master Mix | New England Biolabs | Cat# M0287S |
| GlycoBlue™ Coprecipitant, 15 mg/mL | Invitrogen | Cat# AM9516 |
| SuperScript III Reverse Transcriptase | Invitrogen | Cat# 18080044 |
| Trizol Reagent | Life Technologies | Cat# 15596018 |
| Acid-Phenol:Chloroform with IAA, 125:24:1, pH 4.5 | Thermo Fisher Scientific | Cat# AM9722 |
| Novex NuPAGE™ LDS Sample Buffer (4X) | Life Technologies | Cat# NP0007 |
| Western Lightning Plus-ECL | PerkinElmer | Cat# NEL103001EA |
| Critical commercial assays | ||
| NEBNext® Small RNA Library Prep Set for Illumina® (Multiplex Compatible) | New England Biolabs | Cat# E7330S |
| Bioanalyzer High Sensitivity DNA Analysis | Agilent | Cat# 5067-4626 |
| KAPA Library Quantification Kit | Roche | Cat# KK4844 |
| Experimental models: Cell lines | ||
| ES mouse cells (EL 16.7 (129/Cas) | Lee Lab | n/a |
| Irradiated mouse embryonic fibroblast feeder cells | Lee Lab | n/a |
| HEK293 | ATCC | CRL-1573 |
| Other | ||
| Amersham Protran 0.2 μm nitrocellulose membrane | GE Healthcare | Cat# 10600011 |
| Glass microfiber filter | Whatman | Cat# 1823010 |
| Costar SpinX column | Corning | Cat# 8161 |
| Novex NuPAGE™ 4%–12%, Bis-Tris, 1.5 mm, Mini Protein Gel | Life Technologies | Cat# NP0335PK2 |
| Gene Pulser II Electroporation System | Bio-Rad | Cat# 165-2105 |
| UV Stratalinker 1800 | Stratagene | n/a |
| Thermomixer - Mixer HC | USA Scientific | Cat# 8012-0000 |
| 5prime phase-lock gel heavy 2 mL, | Fisher Scientific | Cat# 2302830 |
| TapeStation System | Agilent | Cat# G2992AA |
| DNA LoBind tube 1.5 mL | Eppendorf | Cat# 022431021 |
Materials and equipment
PBS lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS pH 7.4 (10×) | 1× | 5 mL |
| MgCl2 (1M) | 1 mM | 50 μL |
| CaCl2 (0.1M) | 0.1 mM | 50 μL |
| Sodium Deoxycholate (10%) | 0.5% | 2.5 mL |
| Nonidet-P-40 (10%) | 0.5% | 2.5 mL |
| ddH2O | N/A | 39.9 mL |
| Total | N/A | 50 mL |
Store at 4°C for up to 12 months
10 M Urea stock solution
Add 24 g urea powder to 21.3 mL ddH2O in 50 mL tube, vortex and incubate at 30°C on rotatory shaker until complete dissolving. Then, equilibrate to 21°C–24°C. Store at 21°C–24°C for up to 6 monhts.
PBS-urea wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS pH 7.4 (10×) | 1× | 5 mL |
| Urea (10M) | 8 M | 40 mL |
| SDS (10%) | 0.1% | 0.5 mL |
| ddH2O | N/A | 4.5 mL |
| Total | N/A | 50 mL |
Store at 21°C–24°C for up to 12 months.
PBS-SDS wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS pH 7.4 (10×) | 1× | 5 mL |
| SDS (10%) | 2% | 10 mL |
| ddH2O | N/A | 35 mL |
| Total | N/A | 50 mL |
Store at 21°C–24°C for up to 12 months.
PBS high salt wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS pH 7.4 (10×) | 1× | 5 mL |
| NaCl (5M) | 850 mM | 8.5 mL |
| SDS (10%) | 0.1% | 0.5 mL |
| Sodium Deoxycholate (10%) | 0.5% | 2.5 mL |
| Nonidet-P-40 (10%) | 1% | 5 mL |
| ddH2O | N/A | 28.5 mL |
| Total | N/A | 50 mL |
Store at 4°C for up to 12 months
PBS low salt wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS pH 7.4 (10×) | 1× | 5 mL |
| NaCl (5M) | 150 mM | 1.5 mL |
| SDS (10%) | 0.1% | 0.5 mL |
| Sodium Deoxycholate (10%) | 0.5% | 2.5 mL |
| Nonidet-P-40 (10%) | 1% | 5 mL |
| ddH2O | N/A | 35.5 mL |
| Total | N/A | 50 mL |
Store at 4°C for up to 12 months
DNase mix: Prepared fresh
| Reagent | Final concentration | Amount |
|---|---|---|
| DNase buffer (10×) | 1× | 16 μL |
| Turbo DNase (2 u/μL) | 0.1 u/μL | 8 μL |
| Protease Inhibitor Cocktail (100×) | 1× | 1.6 μL |
| Roche Protector RNAse inhibitor (40U/ μL) | 0.4U/ μL | 1.6 μL |
| SUPERaseIn RNase Inhibitor (20 U/μL) | 0.1 U/μL | 0.8 μL |
| ddH2O | N/A | 132 μL |
| Total | N/A | 160 μL |
Low pH PNK buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 6.48 (1M) | 70 mM | 2.8 mL |
| MgCl2 (1M) | 10 mM | 0.4 mL |
| DTT (1M) | 5 mM | 0.2 mL |
| ddH2O | N/A | 36.6 mL |
| Total | N/A | 40 mL |
Store at 4°C for up to 6 months
Low-pH PNK mix: Prepared fresh
| Reagent | Final concentration | Amount |
|---|---|---|
| Low pH PNK buffer | 1× | 80 μL |
| T4 PNK 10 U/μL, | 0.24 U/ μL | 2 μL |
| Roche Protector RNAse inhibitor (40 U/ μL) | 0.95 U/ μL | 2 μL |
| Total | N/A | 84 μL |
PNK buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 7.4 (1M) | 50 mM | 2 mL |
| MgCl2 (1M) | 10 mM | 0.4 mL |
| DTT (1M) | 5 mM | 0.2 mL |
| NP40 (20%) | 0.5% | 2 mL |
| ddH2O | N/A | 35.4 mL |
| Total | N/A | 40 mL |
Store at 4°C for up to 6 months
PNK mix: Prepared fresh
| Reagent | Final concentration | Amount |
|---|---|---|
| PNK buffer | 1× | 80 μL |
| 10 mM ATP | 0.86 mM | 8 μL |
| T4 PNK 10 U/μL, | 0.32 U/ μL | 3 μL |
| Roche Protector RNAse inhibitor (40 U/ μL) | 0.86 U/ μL | 2 μL |
| Total | N/A | 93 μL |
PNK mix 2 for membrane elution: Prepared fresh
| Reagent | Final concentration | Amount |
|---|---|---|
| PNK buffer | 1× | 80 μL |
| ATP, [γ-32P] 6000 Ci/mmol, 10 mCi/mL |
0.45 μCi/μL | 4 μL |
| T4 PNK 10 U/μL, | 0.34 U/μL | 3 μL |
| Roche Protector RNAse inhibitor (40 U/ μL) | 0.9 U/μL | 2 μL |
| Total | N/A | 89 μL |
PNK wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 7.4 (1M) | 50 mM | 2.5 mL |
| NaCl (5M) | 150 mM | 1.5 mL |
| EDTA (0.5M) | 10 mM | 1 mL |
| NP40 (20%) | 0.5% | 2.5 mL |
| ddH2O | N/A | 42.5 mL |
| Total | N/A | 50 mL |
Store at 4°C for up to 12 months
Proteinase K buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 8.0 (1M) | 100 mM | 5 mL |
| NaCl (5M) | 200 mM | 2 mL |
| EDTA (0.5M) | 5 mM | 0.5 mL |
| SDS (10%) | 0.1% | 0.5 mL |
| ddH2O | N/A | 42 mL |
| Total | N/A | 50 mL |
Store at 21°C–24°C for up to 12 months.
PK solution: Prepared fresh
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 7.4 (1M) | 100 mM | 100 μL |
| NaCl (5M) | 50 mM | 10 μL |
| EDTA (0.5M) | 10 mM | 20 μL |
| Proteinase K (20 mg/mL) | 4 mg/mL | 200 μL |
| ddH2O | N/A | 670 μL |
| Total | N/A | 1 mL |
PK-urea solution: Prepared fresh
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 7.4 (1M) | 100 mM | 100 μL |
| NaCl (5M) | 50 mM | 10 μL |
| EDTA (0.5M) | 10 mM | 20 μL |
| Urea (10M) | 7 M | 700 μL |
| ddH2O | N/A | 170 μL |
| Total | N/A | 1 mL |
20× NuPage MOPS/SDS running buffer pH 7.7
| Reagent | Final concentration | Amount |
|---|---|---|
| MOPS | 1 M | 104.6 gr |
| Tris base | 1 M | 60.6 gr |
| SDS | 2% | 10 gr |
| EDTA | 20 mM | 3 gr |
| ddH2O | N/A | to 500mL |
Store at 4°C for up to 3 months
20× NuPage transfer buffer pH 7.2
| Reagent | Final concentration | Amount |
|---|---|---|
| Bicine | 0.5 M | 10.2 gr |
| Bis-Tris (free base) | 0.5 M | 13.1 gr |
| EDTA | 20 mM | 0.75 gr |
| ddH2O | N/A | to 125 mL |
Store at 4°C for up to 12 months
DNase mix 2: Prepared fresh
| Reagent | Final concentration | Amount |
|---|---|---|
| DNase buffer (10×) | 1× | 5 μL |
| Turbo DNase (2 u/μL) | 0.04 U/μL | 1 μL |
| Roche Protector RNase inhibitor (40 U/ μL) | 0.8 U/ μL | 1 μL |
| ddH2O | N/A | 43 μL |
| Total | N/A | 50 μL |
Step-by-step method details
In vivo UV cross-linking of RNA-protein complexes
This step creates a snapshot of protein-RNA interactions at a set time. Covalent cross-linking stabilizes the connection between protein and RNA residues situated in close proximity inferring the interaction between them.
-
1.
Grow cells to full confluence in 15 cm tissue culture dishes. Prepare one 15 cm confluent plate per one dCLIP pull-down. We recommend using 2 plates per sample type since, as described below, we use two RNase concentrations per every clonal cell line.
-
2.
Wash cells with 10 mL ice-cold PBS.
-
3.
Irradiate cells covered in 5 mL ice-cold PBS in opened lid dish using 254 nm wavelength.
Note: Expose monolayer cells to 150 mJ/cm2 and ES cell colonies to 400 mJ/cm2
-
4.
Add 5 mL of ice-cold PBS and scrape cells into 15 mL tubes.
-
5.
Centrifuge to pellet cells 5 min, 1,000 g at 4°C. Discard supernatant.
Pause point: Shock-freeze cell pellets in liquid nitrogen and store at −80°C.
CRITICAL: We strongly advise conducting a small-scale dCLIP experiment prior to entering a large-scale library preparation step (See Small-scale dCLIP protocol, (Rosenberg et al., 2017)). The small-scale protocol is similar to the membrane elution protocol described below with the following modifications. The lysate from one cross-linked 15 cm plate is divided into four equal portions that are treated with No RNase, 100 u/mL, 10 u/mL, and 2u/mL RNase respectively. The washing steps are conducted with 500 μl volumes and the 3′-end dephosphorylation step is omitted. The volumes of DNase mix and PNK mix are downscaled 4-fold. Finally, only 32P-labeled ATP is utilized at the phosphorylation step. A successful small-scale experiment for Chromobox Protein Homolog 7 (CBX7) protein is shown in Figure 2A. Notice the clear pattern of radiolabeled RNA fragments cross-linked to CBX7 protein in dCLIP-CBX7 ( Figure 2A) compared to extremely high background signal which masked the specific pattern of CBX7 cross-linked RNAs in conventional CLIP-CBX7 (Figures 2B and 2C). Another important control for small-scale experiments is a No UV control, where a dCLIP protocol is conducted on cells not treated with UV, and thus are presumed to lack a radioactive signal emanating from cross-linked RNA fragments, shown in Figure 2D (Rosenberg et al., 2021). In Figure 3, we provide an example of a small-scale dCLIP experiment for Embryonic Ectoderm Development (EED) protein that did not exhibit a detectable dCLIP signal (Rosenberg et al., 2021) despite being recognized as a potential RNA-binding protein in other studies (Long et al., 2017). For that reason, we did not proceed further to a large-scale dCLIP experiment with EED protein.
Figure 2.

dCLIP vs Conventional CLIP of Polycomb proteins in mouse ES cells
(A) Representative autoradiography dCLIP experiment. The red arrowhead indicates the Bio-tagged-CBX7 signal in two clonal cell lines, 3E and 6F, expressing nearly physiological levels of Bio-tagged-CBX7. The excised membrane area is marked in white.
(B) Representative autoradiography of CLIP experiment using specific antibodies against CBX7. Rabbit IgG was used as a control. The expected size of CBX7 protein is marked with a red arrowhead. A high background signal was visible at 40 kDa level and was not cleared by up to 1M salt washes.
(C) Representative autoradiography of CLIP experiment with anti-HA tag antibody. The 6C and 12D clonal cell lines express HA-tagged-CBX7. The red arrowhead marks HA-CBX7 related signal. Note the presence of strong background signal of anti-HA antibody, similar to anti-CBX7 antibody in (B).
(D) Representative dCLIP experiments for EZH2. Left panel, autoradiography of dCLIP experiment. Right panel, Western blot with the anti-GFP antibody. Red arrows, GFP-Biotagged-EZH2 signal. Ezh2-4A and Ezh2-5A are two clonal 16.7 mES cell lines expressing nearly physiological levels of GFP-Biotagged-EZH2. Green arrowhead – Avi-GFP protein.
Figure 3.

Embryonic Ectoderm Development (EED) protein as an example for protein without dCLIP-detectable RNA binding
(A) Representative dCLIP experiment with N-terminally – Flag-Biotagged EED protein. Left panel, autoradiography of dCLIP experiment. Right panel, Western blot with anti-FLAG antibody. Red arrow, FLAG-Biotagged-EED signal.
(B) Representative dCLIP experiment with C-terminally – HA-Biotagged EED protein. Left panel, autoradiography of dCLIP experiment. Right panel, Western blot with the anti-HA antibody. Red arrow, HAG-Biotagged-EED signal. mEed-4H and mEed-2G are two clonal cell lines expressing nearly physiological levels of FLAG-Biotagged-EED. mEed-11B and mEed-12B are two clonal cell lines nearly expressing physiological levels of HA-Biotagged-EED. Note the lack of EED-specific dCLIP signal in both panels.
Protected protein-RNA “footprint” preparation by partial RNase digestion
To determine the sequence of RNA binding sites in the RNA-protein complexes, RNA molecules covalently bound to the protein of interest are trimmed to a short size range to create a characteristic RNA ‘footprint’.
-
6.
Pre-thaw 2 cell pellets per sample type at 4°C and resuspend each pellet in 1.25 mL of ice-cold PBS-lysis buffer supplemented with 1 tablet of Complete-mini EDTA-free tablet, 40 U/mL Protector RNase inhibitor, 1mM Dithiothreitol (DTT).
-
7.
Rotate 25 min at 4°C.
-
8.
Spin down and add 25 μL TurboDNase 2 U/μL. Roughly estimate the sample volume.
-
9.
Add RNase I 100 U/μL to a final concentration of 10 U/mL and 2 U/mL (See Critical note above)
-
10.
Incubate 15 min in a 37°C. Gently mix samples every 5 min.
-
11.Spin down, add 48 U SUPERaseIN, and adjust the SDS concentration to a final 0.1% to terminate RNase I activity.Note: SUPERaseIN is used in this step for its ability to inhibit RNase I, compared to other common RNase inhibitors. 0.1% SDS is also intended to inactivate RNase I according to manufacturer instructions.Optional: We observed that pulling down proteins that are tightly bound to chromatin, such as EZH1 and JARID2 is facilitated by using a higher concentration of SDS.
-
a.In step 11, adjust the SDS concentration to 1% SDS instead of 0.1% and rotate samples for 5 min at 21°C–24°C.
-
b.Continue with steps 12–15 at 21°C–24°C instead of 4°C.
-
a.
-
12.
Centrifuge samples twice, 10 min, 21,130g at 4°C to remove residual cell debris.
-
13.
Transfer supernatant to new 2 mL tubes.
Note: In the next steps magnetic Protein G Dynabeads (G-beads) are used for pre-clearing of the lysate from the proteins and nucleic acids binding non-specifically to magnetic beads whereas magnetic Dynabeads MyOne Streptavidin C1 (C1-beads) are used for isolating the RNA-biotinylated protein complexes from cell lysates. Prior to use, thoroughly mix beads and aliquot the volume needed for the total number of samples. Prepare 80 μL beads per sample and use a magnetic separator to separate beads from the supernatant. All bead washes are done with ice-cold buffer at 21°C–24°C.
-
14.Pre-clearing of cell lysates.
-
a.Aliquot the volume G-beads needed for the total number of samples. Remove supernatant.
-
b.Wash G-beads twice with 1 mL of PBS-lysis buffer.
-
c.Resuspend G-beads in 150 μL PBS-lysis buffer per sample multiplied by the number of samples.
-
d.Transfer 150 μL G-beads to 2 mL tubes. Remove supernatant.
-
e.Add cell lysates to pre-clearing beads. Mix well and rotate 1 h at 4°C.
-
a.
-
15.Pull-down biotinylated proteins with C1-beads.
-
a.Wash C1-beads 3 times in 1 mL PBS-lysis buffer + 0.5% BSA.
-
b.Resuspend C1-beads in 150 μL PBS lysis buffer + 0.5% BSA per sample multiplied by the number of samples.
-
c.Transfer 150 μL C1-beads to 2 mL tubes. Remove supernatant.
-
d.Capture pre-clearing G-beads (step 14e) on the magnetic separator and transfer the supernatant to C1-beads tubes.
-
e.Mix well and rotate for 2 h at 4°C.
-
a.
Denaturing stringent washes
Using the biotin-streptavidin high interaction strength property (Kd=10−15 M, (Weber et al., 1989)), the dCLIP protocol was uniquely designed to enable extremely stringent washes. The washing step efficiently removes non-covalently bound RNA species and proteins thus significantly reducing the background signal commonly observed in conventional CLIP protocols that rely on antibody-antigen interactions ( Figure 2).
Note: All wash steps are done in 1.2 mL volume rotating for 5 minutes. Use a magnetic separator to capture the beads and discard the supernatant. The wash buffers do not include RNase inhibitors and rely on the presence of 0.1% SDS (or higher concentration) which irreversibly inactivate RNase I (See manufacturer’s instructions, https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FMAN0012009_RNase_I_UG.pdf&title=VXNlciBHdWlkZTogUk5hc2UgSQ==).
-
16.
Wash samples twice in PBS-Urea wash buffer at 21°C–24°C.
-
17.
Wash samples twice PBS-SDS wash buffer at 21°C–24°C.
-
18.
Wash once in PBS high-salt wash buffer at 4°C.
-
19.
Wash once in PBS low-salt wash buffer at 4°C.
DNase treatment
The dCLIP protocol distinctively includes three DNase treatment steps. The first DNase treatment in step 8 promotes the dissociation of proteins tightly bound to chromatin such as CBX7 (El Messaoudi-Aubert et al., 2010). The second DNase treatment, similar to conventional CLIP protocols, aims to remove all DNA linked to RNA-protein of interest complexes since the UV crosslinking step crosslinks nucleic acid residues, RNA and DNA alike, to associated proteins. In the third DNase treatment, added here, the naked RNA molecules at the end of the precipitation step are treated to remove any DNA contaminates immediately prior to library construction (Darnell, 2012).
-
20.
Resuspend the beads in 800 μL 1xDNase buffer and transfer to 1.5 mL tubes.
Note: This step aims to remove the remaining detergents used in previous wash steps that might inhibit the DNase activity.
-
21.
Rotate 5 min at 4°C. Capture C1-beads and discard the supernatant.
-
22.
Resuspend the C1-beads in 160 μL of DNase mix and rotate 30 min at 37°C.
-
23.
Spin down briefly to collect the liquid retained in the lid back to the main volume, capture C1-beads, and discard the supernatant.
-
24.
Wash C1-beads once in PBS low-salt wash buffer. Rotate 5 min at 4°C.
3′-end dephosphorylation, 5′-end phosphorylation, and RNA extraction
The 3′-end dephosphorylation and 5′-phosphorylation (steps 25 and 26) prepare the RNA fragments for proper ligation of 5′ and 3′ adapters in the library construction section.
In our experience, there’s a significantly higher background due to non-specific RNA binding when undifferentiated mouse ES colonies RNA is eluted directly from beads, as shown in Figure 4.
Note: All washes are done for 5 min, rotating at 21°C–24°C.
-
25.3′ end dephosphorylation
-
a.Wash C1-beads once in 1 mL Low pH PNK buffer. Discard supernatant.
-
b.Incubate C1-beads in 80 μL Low-pH-PNK mix 20 min at 37°C, shaking at 1,000 rpm for 15 s every 2 min.
-
c.Capture C1-beads and discard the supernatant.
-
a.
Note: If choosing the Membrane RNA elution option use the Optional steps to replace steps 26 and 27.
-
26.RNA 5′ end phosphorylation.
-
a.Wash C1-beads once in 1 mL PNK buffer. Discard supernatant.
-
b.Incubate C1-beads in 80 μL PNK mix 20 min at 37°C, shaking samples at 1,000 rpm for 15 s every 2 min
-
c.Capture C1-beads and discard the supernatant.Optional: RNA 5′ end phosphorylation for membrane RNA elution.
All washes are done for 5 min, rotating at 4°C.Note: Since RNA 5′ end phosphorylation for membrane RNA elution involves radioactive material, the work should be performed according to the safety regulations specified by the institution or facility where the work is conducted. -
d.Wash C1-beads once in 1 mL of PNK buffer.
-
e.Incubate C1-beads in 80 μL PNK mix 2–10 min at 37°C.
-
f.Add 8 μL 10 mM ATP and incubate 20 min at 37°C.
-
g.Capture C1-beads and discard the supernatant.
-
h.Wash C1-beads 3 times with 1 mL of ice-cold PNK wash buffer.
-
i.Incubate C1-beads in 85 μL 1xSDS sample buffer 5 min at 95°C (80 mM Tris pH 6.8, 2% SDS, 100 mM DTT, 10% Glycerol)Optional: The 1xSDS sample buffer can be substituted with 85 μL NuPage 3xLDS sample buffer. Incubate at 80°C for 10 min. 1xNuPage-LDS buffer is not sufficient to disrupt streptavidin – biotin interactions at 80°C thus precluding an elution of protein-RNA complexes.Pause point: Store samples at −20°C
-
j.To continue reheat samples for 10 min at 80°C or 5 min 95°C (depending on the sample buffer used)
-
k.Skip steps 27 and 28 and continue with the ‘Optional: Membrane RNA elution’ steps.
-
a.
-
27.RNA extraction from C1-beads
-
a.Wash C1-beads twice with 1 mL PBS-Urea wash buffer
-
b.Wash C1-beads once with 1 mL Proteinase K buffer
-
c.Incubate C1-beads in 200 μL Proteinase K buffer supplemented with 1 mg/mL Proteinase K, 30 min at 55°C, rotating.
-
a.
-
28.RNA precipitationOptional: Membrane RNA elution
-
a.RNA transfer to nitrocellulose membrane
-
i.Load 40 μL per lane in two adjacent lanes on 1.5 mm NuPage 4%–12% Bis-Tris gradient gels. Run gels in NuPage MOPS/SDS running buffer on 200 V.
-
ii.Transfer samples to nitrocellulose membrane for 1.5 h, 140 mA at 4°C in NuPage 1× transfer buffer + 12% methanol.
-
iii.Expose the membrane to a phosphoimager screen.
-
i.
-
b.RNA elution from the membraneCRITICAL: The excised membrane pieces containing protein-RNA complexes should start in the area at least 10 kda above the size of protein of interest (corresponding to a protein of interest covalently bound to RNA fragments larger than 30 bp. Excise a similar area from control samples (Illustrated in Figure 2A, white box).
-
i.Excise membrane pieces (two lanes/sample). Cut into small pieces of 0.5–2 mm with a clean blade on the surface of sterile plastic 100 mm dish, and transfer to 1.5 mL low-binding 1.5 mL tubes.
-
ii.Incubate membrane pieces in 200 μL PK solution for 20 min at 55°C with constant shaking at 1,200 rpm in Thermomixer.
-
iii.Add 200 μL of PK-urea solution to membrane pieces. Incubate 20 min at 55°C with constant vortexing at 1,200 rpm.
-
iv.Spin the samples 1 min at maximum speed to pellet membrane pieces and transfer supernatant to phase-lock tubes.
-
v.Proceed to RNA precipitation steps 28 c-o.
-
vi.Dissolve the RNA in 25 μL DNase mix 2. Then combine samples of the same cell type that were treated with different RNase concentrations (see step 9) to a final volume of 50 μL.
-
vii.Proceed to steps 28 q-s.
-
i.
Note: At this step samples from the same cell type (see step 9) that were treated with different RNase concentrations are combined at a final volume of 400 μL.-
c.Centrifuge phase-lock gel 2 mL tubes, 30 s 16,000 g to pellet gel.
-
d.Load 400 μL eluted RNA samples.
-
e.Add 400 μL acidic phenol-chloroform and shake vigorously.
-
f.Centrifuge 5 min 16,000 g at 21°C–24°C.
-
g.Repeat steps c-d.
-
h.Transfer the upper aqueous phase to a new low-binding 1.5 mL tube.
-
i.Add 40 μL 3M Sodium Acetate, 1 μL Glycoblue, and 1 mL 100% ethanol,CRITICAL: Adding GlycoBlue is essential for visualizing the small RNA pellet before the elution step. The RNA pellet can easily detach from the tube’s low-binding surface during steps involving the removal of the supernatant.
-
j.Mix well and incubate 16 h at −20°C.
-
k.Centrifuge 30 min 13,523 g at 4°C. Discard supernatant.
-
l.Wash pellets in 1 mL 75% ethanol.
-
m.Centrifuge 10 min 13,523 g at 4°C. Discard supernatant.
-
n.Spin down and manually remove the remaining ethanol with a pipettor and 20 μL tip without lifting the pellet.
-
o.Air dry pellets 5–10 min at 21°C–24°C or 5 min in the laminar flow hood.
-
p.Dissolve pellets in 50 μL DNase mix 2 .
-
q.Incubate 30 min at 37°C.
-
r.Add 950 μL Trizol reagent and follow manufacturer instructions for RNA extraction. Add 0.5 μL GlycoBlue to the precipitation step.
-
s.Elute RNA pellets in 8 μL nuclease-free ddH2O.
-
a.
Figure 4.

Comparison between bead elution and membrane elution dCLIP methods
dCLIP libraries for CBX7 were prepared in Day 0 undifferentiated ES cells growing in colonies on feeder layer (A) and differentiating Day 7 ES cells growing in monolayer (B). The libraries were sequenced, aligned to reference mouse genome and the RNA binding profiles for CBX7-expressing cells vs control cells were generated and visualized in Integrative Genomic Viewer. Dusp9 RNA dCLIP profile in Day 0 cells (A) and Day 7 cells (B) were examined to assess differences between two RNA extraction methods – elution directly from beads vs. SDS-PAGE purification, nitrocellulose membrane transfer, and elution of RNA from the membrane. Note that control cells exhibited a very high background signal after beads elution protocol had been applied in Day 0 cells but not in Day 7 cells. RING1 And YY1 Binding Protein (RYBP) protein served as a control for dCLIP specificity since, compared to CBX7, RYBP is predicted to possess low RNA binding activity (Tavares et al., 2012).
Library construction
Library construction is the first step of NextGen sequencing. In this step, multiplex adapters are added to uniquely barcode each RNA molecule and the samples are prepared to enable a high-throughput short-read sequencing on the Illumina platform.
-
29.For library preparation use Multiplex Compatible NEBNext Small RNA Library Prep according to manufacturer instructions with the following modifications (https://www.neb.com/protocols/2018/03/27/protocol-for-use-with-nebnext-small-rna-library-prep-set-for-illumina-e7300-e7580-e7560-e7330):
-
a.Use 7 μL RNA for library preparation.
-
b.Dilute all adapters and primers 12-fold, since the initial amount of eluted RNA in the dCLIP protocol is significantly less than the initial amount specified by the manufacturer.
-
c.Use 20 μL of adapter-ligated RNA for cDNA preparation. Take 8 μL for the No-RT negative controls.Note: Out of convenience, we replaced the kit’s M-MuLV reverse transcriptase and Murine RNase inhibitor, with SuperScript III Reverse Transcriptase (Life Technologies) and Protector RNase inhibitor (Roche) respectively.Pause point: Store the cDNA at −20°C .
-
d.Amplify cDNA with Illumina multiplexed primers for 25 cycles using LongAmp_Taq 2X Master Mix.Note: No-RT negative controls were amplified for 35 cycles to control for contamination.Pause point: Store the amplified DNA at −20°C .
-
e.PCR product isolation and size selection.
-
i.Run PCR products on 6% TBE-acrylamide gel.
-
ii.Excise gel pieces ranging from 150 bp to 600 bp. Avoid cutting close to the adapter dimers area (Figure 5A).
-
iii.Transfer to 1.5 mL low-binding tubes with 400 μL Gel elution buffer (NEB) and crush using a 1 mL syringe plunger.
-
iv.Rotate overnight at 21°C–24°C to elute the PCR products from a gel.Optional: Elute DNA for 2 hrs at 37°C, or 4 hrs at 30°C.
-
v.Spin down and load the sup on a glass microfiber filter placed in a Costar SpinX tube to remove the remaining gel pieces. Centrifuge 1 min 15,871 g at 21°C–24°C.
-
vi.Follow Ethanol precipitation steps 28 g-m.Note: long 16 h incubation after adding ethanol is not necessary in this step. 1 h incubation on −20°C is sufficient.
-
vii.Resuspend pellets in 12 μL of nuclease-free water.
-
i.
-
a.
Note: AMPure beads can be used for size selection and isolation of amplified DNA, although we have not tried it in our hands for dCLIP protocol. Follow the manufacturer’s recommendations for the appropriate beads to sample ratios (https://www.beckman.com/reagents/genomic/cleanup-and-size-selection/pcr/bead-ratio).
-
30.Library QC
-
a.Run 1 μL of each sample on a High Sensitivity DNA chip or Tapestation to determine sample size distribution. A typical size profile is shown in Figure 5B.
-
b.Use the KAPA Illumina Library Quantification kit to quantify the PCR products.
-
a.
-
31.
Pool 3 to 5 samples to one multiplexed sample. Combine equivalent amounts of 1.5 nM–2 nM of each sample.
Note: To obtain sufficient coverage, we used one lane of Illumina HiSeq 2000 apparatus for library sequencing to render 200–240 million 50 bp paired-end reads.
Figure 5.

Representative size profile for amplified dCLIP library
(A and B) Size selection of amplified dCLIP libraries for CBX7 is shown in (A). Amplified libraries were subjected to electrophoresis on 6% TBE-acrylamide gels and the area corresponding to the white rectangle was excised from each lane to avoid contamination with adapter dimers. PCR products were eluted and analyzed on the Bioanalyzer using a High Sensitivity DNA chip (B).
(C) Following alignment of the sequencing reads obtained from dCLIP samples, strand-specific enriched peaks (depicting RNA-binding sites) from three replicate dCLIP libraries for CBX7 were pooled, and overlapped peaks were merged into long enrichment regions in a strand-specific manner. Length distribution frequency of the enriched dCLIP peaks, as well as mean, median, and standard deviation were calculated.
Expected outcomes
A successful outcome of dCLIP experiment is shown in Figures 2, 4, and 5. First, the clear radioactive signal at and above the size of protein of interest, compared to control must be obtained, as demonstrated in Figures 2A and 2D for CBX7 and EZH2 proteins respectively. The unsuccessful small-scale dCLIP experiment is shown in Figure 3 for EED protein, and in this case, we do not recommend proceeding further. The RNase-protection step in dCLIP protocol enables mapping of RNA-binding sites with superior resolution and accuracy (Figures 4 and 5C). A similar high resolution was achieved with additional proteins (Rosenberg et al., 2021).
Quantification and statistical analysis
For protein expression analysis presented in Figure 1, protein bands were developed using Western Lightning Plus -ECL Kit (Perkin-Elmer), and the signal intensity was analyzed using Chemidoc MP Imaging System (Bio-Rad) and ImageLab Ver. 5.2.1 software (Bio-Rad). Exposures were captured on different time points using ChemiDoc cumulative signal option to avoid signal saturation. Densitometry analysis was conducted using ImageLab software (Bio-Rad). All dCLIP experiments were performed in at least n=2 biological replicates (two separate clonal cell lines). No replicates were excluded from analyses presented, and all attempts to replicate the data were successful.
Limitations
The dCLIP RNA library is represented by non-abortive reverse transcription products only. Protein-RNA covalent bonds created by UV cross-linking are resistant to Proteinase K treatment, resulting in protein residues or small peptides remaining at cross-linking points in eluted RNA. In course of reverse transcription, reverse-transcriptase can either pass protein-RNA cross-linking sites uninterrupted, bypass a protein-RNA cross-linking sites by introducing a random nucleotide, or be stalled and fall off, resulting in abortive cDNA species (Darnell, 2010; Konig et al., 2011).
Small RNA library protocol employed in the dCLIP method relies on adapter ligation to both sides of RNA fragment. Thus, for dCLIP fragments to be amplified from both adapters after cDNA synthesis, reverse transcription should go into completion. Lastly, contamination by natively biotinylated proteins that interact directly with streptavidin beads may occur.
Troubleshooting
Problem 1
Inefficient pull-down of biotinylated protein of interest in step 14.
Potential solution
The efficient pull-down of AVI-tagged protein depends on (1) the accessibility of the AVI-tag to the BirA biotin ligase inside the cells and (2) the accessibility of the biotinylated tag to the streptavidin beads during the pull-down step. First, the AVI tag can be fused either to the N-terminus or the C-terminus of the protein of interest, with subsequent assessment of pull-down efficiency. To improve the accessibility of streptavidin to the biotinylated tag, we suggest increasing the SDS concentration to 1% at the end of the RNase digestion step. This should increase protein unfolding resulting in improved accessibility to the biotinylated protein tag.
Problem 2
A high background signal in control dCLIP samples compared to dCLIP experimental samples following dCLIP, beads elution, library sequencing, and mapping to the reference genome (Figure 4A, beads-eluted samples of EL16.7 Day 0 cells).
Potential solution
To minimize the background signal we highly recommend conducting the membrane elution option. When choosing this option one needs to take all required precautions that involve working with radioactive isotopes as specified by radiation safety regulations. Resolving the protein-RNA complexes on gel aids the isolation of a sample subset that corresponds to the RNA-protein of interest complexes (Figure 2A). Furthermore, only RNA species associated with proteins are expected to bind to a nitrocellulose membrane. Non-covalent bound RNAs will diffuse to the surrounding buffer during the transfer step resulting in higher sample purity.
Problem 3
An excessive amount of adapter dimers in the amplified library – step 29d, Figure 5A.
Potential solution
One of the main reasons for an excessive amount of adapter dimers is the substantial loss of input RNA throughout the procedure. Consequently, we consistently utilized GlycoBlue reagent to stain RNA pellets in order to avoid removing a pellet during a supernatant collection after ethanol precipitation and centrifugation steps. Also, avoid using low-binding tubes during RNA extraction and 3rd DNAse digestion steps because of the very loose attachment of RNA pellets to the sides.
Problem 4
Contamination with bacterial ribosomal RNA during the RNA elution from membrane step.
Potential solution
We and others noticed that some nitrocellulose membrane brands are the source of bacterial ribosomal RNA contamination after membrane extraction (Figure 5A. blank sample, (Van Nostrand et al., 2017)). We highly recommend using the nitrocellulose membranes brands recommended by Van Nostrand and colleagues to reduce a background signal (Amersham Protran 0.2 μm nitrocellulose membrane).
Problem 5
Small-scale dCLIP experiment renders very weak radioactive signal compared to No UV control.
Potential solution
While a very weak dCLIP signal compared to No UV control can serve as evidence that the protein of interest is an extremely weak RNA binder, it is important to confirm that there is no technical failure on the execution side, especially for inexperienced users. Hence, to establish the dCLIP protocol in the lab, we recommend choosing a known RNA-binding protein for pilot dCLIP experiments. Even a conventional CLIP experiment with a specific antibody could suffice. In our case, we conducted a conventional CLIP assay with MOV10 RNA helicase and confirmed that we had been able to obtain a strong radioactive signal at the desired location before continuing to the dCLIP protocol.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jeannie T. Lee (lee@molbio.mgh.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Acknowledgments
We thank the lab members for helpful advice and many important discussions. We also give special thanks to U. Kim and A. Maisonet for excellent technical assistance with deep sequencing. This work was funded by the HHMI and NIH R01-HD097665 to J.T.L.
Author contributions
M.R., V.K.M., and J.T.L. conceived the idea of utilizing in vivo biotinylation to improve the conventional CLIP protocol. M.R. designed the dCLIP method, with V.K.M.’s initial involvement. B.K. aligned sequencing data. R.B. devised the bioinformatics pipeline and performed the computational analysis for the identification of consensus motifs. M.R., V.L., and J.T.L. drafted figures and wrote the manuscript.
Declaration of interests
A patent related to dCLIP has been filed by the MGH. J.T.L. is a cofounder of Translate Bio and Fulcrum and an Advisor to Skyhawk Therapeutics.
Data and code availability
This study did not generate a new dataset or code for analysis.
References
- Darnell R. CLIP (cross-linking and immunoprecipitation) identification of RNAs bound by a specific protein. Cold Spring Harb Protoc. 2012;2012:1146–1160. doi: 10.1101/pdb.prot072132. [DOI] [PubMed] [Google Scholar]
- Darnell R.B. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA. 2010;1:266–286. doi: 10.1002/wrna.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Messaoudi-Aubert S., Nicholls J., Maertens G.N., Brookes S., Bernstein E., Peters G. Role for the MOV10 RNA helicase in polycomb-mediated repression of the INK4a tumor suppressor. Nat. Struct. Mol. Biol. 2010;17:862–868. doi: 10.1038/nsmb.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama T., Henikoff S. Biotin-tag affinity purification of a centromeric nucleosome assembly complex. Cell Cycle. 2006;5:1269–1274. doi: 10.4161/cc.5.12.2889. [DOI] [PubMed] [Google Scholar]
- Kim J., Cantor A.B., Orkin S.H., Wang J. Use of in vivo biotinylation to study protein-protein and protein-DNA interactions in mouse embryonic stem cells. Nat. Protoc. 2009;4:506–517. doi: 10.1038/nprot.2009.23. [DOI] [PubMed] [Google Scholar]
- Konig J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. iCLIP--transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J. Vis. Exp. 2011 doi: 10.3791/2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.T., Lu N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell. 1999;99:47–57. doi: 10.1016/s0092-8674(00)80061-6. [DOI] [PubMed] [Google Scholar]
- Long Y., Bolanos B., Gong L., Liu W., Goodrich K.J., Yang X., Chen S., Gooding A.R., Maegley K.A., Gajiwala K.S. Conserved RNA-binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. Elife. 2017;6:e31558. doi: 10.7554/eLife.31558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy S.L., Reijo Pera R.A. Preparation of mouse embryonic fibroblast feeder cells for human embryonic stem cell culture. CSH Protoc. 2008;2008 doi: 10.1101/pdb.prot5041. pdb prot5041. [DOI] [PubMed] [Google Scholar]
- Rosenberg M., Blum R., Kesner B., Aeby E., Garant J.M., Szanto A., Lee J.T. Motif-driven interactions between RNA and PRC2 are rheostats that regulate transcription elongation. Nat. Struct. Mol. Biol. 2021;28:103–117. doi: 10.1038/s41594-020-00535-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg M., Blum R., Kesner B., Maier V.K., Szanto A., Lee J.T. Denaturing CLIP, dCLIP, pipeline identifies discrete RNA footprints on chromatin-associated proteins and reveals that CBX7 targets 3' UTRs to regulate mRNA expression. Cell Syst. 2017;5:368–385.e15. doi: 10.1016/j.cels.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares L., Dimitrova E., Oxley D., Webster J., Poot R., Demmers J., Bezstarosti K., Taylor S., Ura H., Koide H. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012;148:664–678. doi: 10.1016/j.cell.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand E.L., Nguyen T.B., Gelboin-Burkhart C., Wang R., Blue S.M., Pratt G.A., Louie A.L., Yeo G.W. Robust, cost-effective profiling of RNA binding protein targets with single-end enhanced crosslinking and immunoprecipitation (seCLIP) Methods Mol. Biol. 2017;1648:177–200. doi: 10.1007/978-1-4939-7204-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber P.C., Ohlendorf D.H., Wendoloski J.J., Salemme F.R. Structural origins of high-affinity biotin binding to streptavidin. Science. 1989;243:85–88. doi: 10.1126/science.2911722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate a new dataset or code for analysis.