

Abstract
Accumulating evidence demonstrates that cancer is an oxidative stress-related disease, and oxidative stress is closely linked with heat shock proteins (HSPs). Lipid oxidative stress is derived from lipid metabolism dysregulation that is closely associated with the development and progression of malignancies. This study sought to investigate regulatory roles of HSPs in fatty acid metabolism abnormality in ovarian cancer. Pathway network analysis of 5115 mitochondrial expressed proteins in ovarian cancer revealed various lipid metabolism pathway alterations, including fatty acid degradation, fatty acid metabolism, butanoate metabolism, and propanoate metabolism. HSP60 regulated the expressions of lipid metabolism proteins in these lipid metabolism pathways, including ADH5, ECHS1, EHHADH, HIBCH, SREBP1, ACC1, and ALDH2. Further, interfering HSP60 expression inhibited migration, proliferation, and cell cycle and induced apoptosis of ovarian cancer cells in vitro. In addition, mitochondrial phosphoproteomics and immunoprecipitation-western blot experiments identified and confirmed that phosphorylation occurred at residue Ser70 in protein HSP60, which might regulate protein folding of ALDH2 and ACADS in ovarian cancers. These findings clearly demonstrated that lipid metabolism abnormality occurred in oxidative stress-related ovarian cancer and that HSP60 and its phosphorylation might regulate this lipid metabolism abnormality in ovarian cancer. It opens a novel vision in the lipid metabolism reprogramming in human ovarian cancer.
1. Introduction
In a biological context, free radical reactive oxygen species (ROS) is a normal byproduct of oxygen metabolism, which plays positive roles in cell signaling and homeostasis [1]. However, the dramatically increased ROS levels are related to numerous diseases and degenerative processes, such as autoimmune diseases, cancers, and Alzheimer's diseases [2]. Mitochondria were one of the crucial places to produce ROS during the oxidative phosphorylation process. When electrons are passed through the electron transport chain via oxidation-reduction reactions across the inner mitochondrial membrane, a few oxygen is incompletely reduced to give the superoxide radical (∙O2−) through a series of proteins, such as complex I and complex III of mitochondria [3]. In general, the superfluous ROS would have harmful effects on the cells via damage of DNA or RNA, oxidation of amino acids, damage of mitochondrial and cytoskeletal functions, lipid peroxidation, and oxidative deactivation of specific enzymes [4]. Cancer cells showed greater ROS stress compared to normal cells, partly due to increased metabolic activity, carcinogenesis stimulation, and mitochondrial malfunction [5]. In ovarian cancer, the previous studies showed that ROS could activate various oncogenic transcription factors such as NF-κB [6], AP-1 [7], and STAT3 [8], to influence cellular transformation, tumor cell proliferation, inflammation, tumor cell survival, metastasis and invasion, and angiogenesis. Moreover, molecular and cellular function analysis of ovarian cancer mitochondrial expressed proteins (mtEPs) found that many mitochondrial differentially expressed proteins (mtDEPs) were associated with ROS production, such as generation of active oxygen species, metabolism of hydrogen peroxide, and synthesis, metabolism, production, and quantity of ROS [9]. It clearly demonstrated that ovarian cancer was an ROS-related disease or oxidative stress-related disease.
Heat shock proteins (HSPs), also known as stress proteins, were a conserved mitochondrial chaperone family. The abnormal posttranslational modifications (PTMs), abundance levels, cellular localization, or mutations of HSPs would influence multiple cellular biological processes [10]. HSPs played a key role in pathophysiology and pharmacology of cancer, which was leading to cancer growth, drug resistance, local infiltration, and distant metastasis. HSPs were proven to be potential clinical biomarkers for cancer early diagnosis, follow-up surveillance, and therapeutic targets in multiple cancers [11]. HSP60, as one classical member of the HSP family, was a crucial mitochondrial chaperone and involved in the protein folding process that converts an unfolded substrate into a folded substrate to the mitochondrial matrix [12]. The domain of HSP60 protein was similar to its prokaryotic ortholog GroEL, which indicated the mechanism of HSP60 protein in cancer. In short, unfolded proteins bound to the amino acid residue of HSP60 cavity. Also, HSP60 interacted with another chaperone HSP10 and ATP, which induced conformational changes of HSP60 to change the properties of the binding sites—in turn opening or closing the HSP60 cavity. After the substrate protein completed protein folding and obtained its native conformation in several cycles, it would be released [13]. Therefore, HSP60 had a pivotal role in the misfolded processes of protein homeostasis that was associated with various diseases. Additionally, ROS was also typically seen in diseases associated with misfolded proteins and had been reported to result in apoptosis [14]. HSP60 was proven to be a mediator of ROS [15], and ROS in turn could also regulate the expression and PTMs of the HSP family [16]. For example, proteomic analysis found that HSP60 silencing increased excessive ROS and drove metabolic reprogramming to promote cell growth and tumor progression via activating NRF2-mediated oxidative stress responses [15]. ROS was also reported to regulate HSPs via the JAK/STAT pathway. Mechanistically, H2O2 induced expressions of HSP70 in a time-dependent manner by activating the HSP70 promoter with enhanced STAT binding [16]. Even though the relationship between oxidative stress and HSPs has been demonstrated in numerous studies, the molecular mechanism and the downstream signaling pathways of HSPs remain unclear.
Metabolic alterations have developed into a new hallmark in cancer researches. Evidence showed that expressions of hub molecules were changed in the lipid metabolism pathway, which raised the idea that lipid biomarkers might provide broad prospects for cancer diagnosis and that inhibition of the lipid metabolism process in cancer cells might hold therapeutic implications [17]. A variety of lipid molecules and their metabolic intermediates might participate in inflammation, cell signal transduction, cell adhesion and movement, angiogenesis, and cell proliferation. Abnormal metabolism in cancer cells had the characteristics of an uncontrolled de nova fatty acid synthesis pathway; thus, the increased lipid synthesis and decreased fatty acid degradation could generate a relatively high proliferation rate [18]. One study showed that upregulation of lipogenesis pathways was associated with poor survival outcomes in cancer, and lipid metabolism was closely related to the malignant phenotype (invasion, migration, etc.) of tumor cells in in vitro experiments [19]. Hyperlipidemia promoted hormone-related cancers (ovarian cancer, endometrial cancer, etc.). Therefore, an abnormal blood fat index might be a useful marker to evaluate some types of tumors. Cancer cells also produced a series of lipolysis substances and released them into the blood, such as TNF-α, glucocorticoid, hormone-sensitive lipase (HSL), and lipid-mobilizing factor (LMF/ZAG) [20]. HSL was detectable in serum and ascites of patients with ovarian cancer, and the activity of HSL was significantly higher (3 times) than that of the normal control based on clinical data [21]. Additionally, lipids would transfer from adipocytes to ovarian cancer cells by coculture condition, which promoted cancer cell growth [22]. All the evidence suggested that lipid disorder was closely related to tumorigenesis and tumor development. Novel applications of electrospray ionization mass spectrometry (ESI/MS) facilitated lipidomic development [23]. Lipidomics, as a novel subject, revealed altered biosynthetic pathways in various vital phenomena and cell signaling as biomarkers. Lipid metabolism disorders or reprogramming of tumors formed tumor lipidomics, which could provide potential biomarkers and therapeutic targets for cancer patients. Lipid metabolism dysregulation, as an important part of energy reprogramming, was closely associated with the development and progression in cancers. Moreover, KEGG pathway network analysis of ovarian cancer mtEPs (n = 5115) revealed that many mtEPs were significantly involved in lipid metabolism pathways, including fatty acid degradation, fatty acid metabolism, butanoate metabolism, and propanoate metabolism [24]. Ingenuity pathway analysis (IPA) of ovarian cancer mtDEPs (n = 1198) also revealed that many mtDEPs were also significantly involved in lipid metabolism pathways, including fatty acid beta-oxidation I [9]. Lipid oxidative stress was derived from lipid metabolism dysregulation that was closely associated with the development and progression of malignancies. However, the regulatory mechanisms of fatty acid metabolism abnormality in ovarian cancer remain unclear.
The previous studies demonstrated that HSP60 knockdown drove metabolic reprogramming in clear cell renal cell carcinoma to promote tumor progression and enhance mitochondrial-dependent biosynthesis [25]. In terms of lipid metabolic reprogramming, one study indicated that HSP60 might be involved in the lipid regulatory effect of whole grain highland hull-less barley (WHLB). Quantitative real-time polymerase chain reaction analysis showed that rats given with WHLB diet exhibited downregulated expression of HSP60 compared to rats given with the control high-fat diet [26]. The relationship between lipid hydroperoxide (LOOH) level and Hsp60 was investigated in sera from newborns, which proved that Hsp60 level was positively correlated with LOOH level [27]. In terms of regulatory mechanisms between HSP60 and lipid metabolism, one study demonstrated that the HSP10-HSP60 chaperoning protein folding complex played a role in the folding of medium-chain acyl-CoA dehydrogenase (MCAD), and this folding process was influenced by deacetylation of HSP10 [28]. In HEK-293 cells, both the amounts of total MCAD proteins and folded MCAD proteins were closely related to the expression levels of HSP60. HSP60 downregulation impaired both wild-type and disease-associated MCAD proteins. A similar result was observed that the mitochondrial short-chain acyl-CoA dehydrogenase (ACADS) wild type was also regulated by the HSP60/10 folding machinery [29]. Acyl-CoA dehydrogenases were important in multiple lipid metabolism pathways, and those previous researches demonstrated that HSP60/10 chaperoning proteins were crucial for the folding process of acyl-CoA dehydrogenase. Dysregulation of HSP60 expression levels did jeopardize those kinds of protein folding, leading to the alteration of fatty acid oxidation and energy supply. Additionally, a previous phosphoproteomic study found a new phosphorylation site at residue Ser70 in HSP60 in mitochondrial samples from ovarian cancer tissues, and its phosphorylation level was significantly increased in ovarian cancers compared to controls [30]. These previous findings clearly revealed the potential functions of HSP60 for mitochondrial protein folding related to lipid metabolism pathways, which further showed the roles of HSP60 in cancer development.
This present study used the integrative analysis of mtEP (n = 5115) and mitochondrial phosphoprotein (mtPP) datasets in ovarian cancers, which found that HSP60 might be related to lipid metabolism pathways in ROS-related ovarian cancers. The multiple cellular lipid metabolism signaling pathways were changed based on the mtEP dataset, which might play an important role in ovarian cancer invasion and metastasis. In addition, functional analyses demonstrated that HSP60 knockdown suppressed cell proliferation and migration and induced apoptosis and cell cycle arrest. Also, mitochondrial HSP60 was differentially phosphorylated at its residue Ser70. HSP60 was highly related to lipid metabolism and might regulate fatty acid oxidation by influencing both the expression and folding of related enzymes in the lipid metabolism pathways, and regulatory roles of phosphorylated HSP60 in lipid metabolism pathways were further investigated in ovarian cancers. The experimental flowchart is shown in Figure 1(a).
Figure 1.
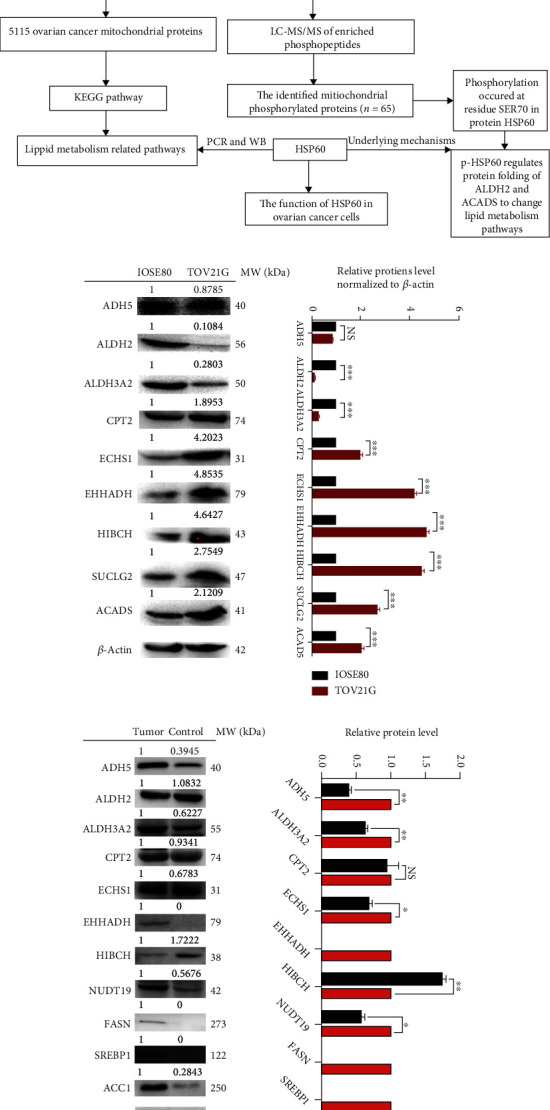
Experimental flowchart and verifications of mtDEPs in lipid metabolism pathways. (a) Experimental flowchart to identify lipid metabolism-related pathways and underlying mechanisms. (b) Western blot analysis of ADH5, ALDH2, ALDH3A2, CPT2, ECHS1, EHHADH, HIBCH, SUCLG2, and ACADS in ovarian cancer cells TOV21G compared to control cells IOSE80. (c) Western blot analysis of ADH5, ALDH2, ALDH3A2, CPT2, ECHS1, EHHADH, HIBCH, NUDT9, FASN, ACC1, and SREBP1 in lipid metabolism pathways between ovarian cancer tissues and benign ovarian disease tissues. n = 3. ∗∗∗p < 0.001. NS: no significance.
2. Material and Methods
2.1. Ovarian Cancer Mitochondrial Protein Dataset
In total, 5115 ovarian cancer mtEPs were identified by isobaric tags for relative and absolute quantitation- (iTRAQ-) based quantitative proteomics [24], including 1198 mtDEPs in human epithelial ovarian cancers relative to control ovary tissues [9].
2.2. Ovarian Cancer Phosphoprotein Dataset
In total, 67 ovarian cancer mtPPs were identified by TiO2 enrichment-based iTRAQ quantitative phosphoproteomics [30].
2.3. Pathway Network Analysis
The IPA software (https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/) was used to analyze 1198 mtDEPs for determining their molecular and cellular functions in ovarian cancers. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with DAVID Bioinformatics Resources 6.7 (https://david.ncifcrf.gov/home.jsp) were used to analyze 5115 mtEPs for determining signaling pathway networks in ovarian cancers. STRING 10.0 (http://string-db.org/cgi/input.pl) was used to construct the protein-protein interaction between the proteins in the identified lipid metabolism pathways and HSP60. PYMOL (http://www.pymol.org) was used to modularize the structures of human HSP60-ACADS and HSP60-ALDH2. Protein kinase-specific prediction (http://bioinfo.ncu.edu.cn/PKPred_Prediction.aspx) was used to analyze the upstream kinase of HSP60 (Ser70) based on a single kinase.
2.4. Western Blot Verification of mtDEPs in Lipid Metabolism Pathways in the Ovarian Cancer Cell Line
Western blot was used to verify each mtDEP in lipid metabolism pathways, including ADH5, ALDH2, ALDH3A2, CPT2, ECHS1, EHHADH, HIBCH, SUCLG2, and ACADS (BBI Life Science Corporation, HK, China), between the ovarian cancer cell TOV-21G and the normal ovarian epithelial cell IOSE80. Additionally, western blot was used to verify each mtDEP in lipid metabolism pathways, including ADH5, ALDH2, ALDH3A2, CPT2, ECHS1, EHHADH, HIBCH, NUDT9, FASN, ACC1, and SREBP1 (Abcam, UK), between ovarian cancer tissues and control ovary tissues. β-Actin was used as the internal standard.
2.5. Effects of HSP60 on Ovarian Cancer Cell Biological Behaviors
Transient transfection of the ovarian cancer cell TOV-21G was used in this study. Cells TOV-21G and IOSE80 were ordered from the Keibai Academy of Science (Nanjing, China). TOV-21G cells were cultured in 1640 medium (Thermo Fisher Scientific, USA) containing 10% fetal bovine serum (Biological Industries, South America). IOSE80 cells were cultured in DMEM (Thermo Fisher Scientific, USA) containing 10% fetal bovine serum (Biological Industries, South America). The siHSP60 and sicontrol were designed and synthesized by GenePharma (Shanghai, China). The si-RNA sequences for HSP60 were 5′-GACGAUGCCAUGCUCUUAATT-3′ (sense strand) and 5′-UUAAGAGCAUGGCAUCGUCTT-3′ (antisense strand). TOV-21G cells (2 × 105) were seeded in 6-well plates, and transient transfection was performed when cell density is up to 30-50%. TOV-21G cells in different wells were transfected with siHSP60 (20 μM) or si-control using Lipofectamine 3000 reagents (Invitrogen, USA), followed by incubation for 24-48 h. Then, the following experiments were done: (i) a wound healing assay was used to detect cell mobility between control and si-Hsp60 groups. TOV-21G cells were photographed when split with a micropipette tip (10 μl) and 36 h after being split. (ii) A CCK8 cell assay was used to detect cell proliferation of TOV-21G cells between control and si-HSP60 groups. (iii) An EdU assay was used to measure positive proliferative cells with fluorescence staining DNA synthesis between control and si-HSP60 groups. (iv) An apoptosis assay was performed with an annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (BestBio, Shanghai). Briefly, TOV-21G cells (2 × 106) in si-HSP60 and control groups were washed 3 times with PBS. Annexin V-FITC and propidium iodide (PI) were added at room temperature away from light and incubated for 20 min. Fluorescence-activated cell sorting (FACS) using a BD Accuri™ C6 (BD Biosciences, Ashland, OR, USA) was performed, and the apoptosis rate was analyzed with FlowJo (Tree Star, Ashland, OR, USA). (v) Cells (2 × 106) were washed 3 times with PBS and fixed with absolute ethyl alcohol (-20°C, 2 h). After washing with PBS, cells were coincubated with PI and 20 μl RNaseA (BestBio, Shanghai) at 37°C in a water bath for 30 min away from light. Cells were then analyzed for DNA proportion distribution with FACS in a BD Accuri™ C6 (BD Biosciences), and then, the cell cycle was analyzed with ModFit LT for Windows version 3.2. Extrinsic and intrinsic apoptosis evaluation was necessary for a better understanding of the apoptotic pathway activated in HSP-silenced cancer cells. Western blot was used to verify apoptosis-related proteins, including caspase-3, caspase-8, and caspase-9 between si-HSP60 and control groups.
2.6. qRT-PCR and Western Blot Were Used to Verify HSP60-Targeted Molecules in Lipid Metabolism Pathways
Total RNAs were isolated from three biological replicates of si-HSP60 or control groups with the TRizol reagent (Invitrogen). The mRNAs from 500 ng of total RNAs were reversely transcribed into cDNAs for quantitative real-time PCR (qRT-PCR) analysis with the SYBR Premix ExTaq kit (TaKaRa). Beta-actin was set as an internal control for gene quantification. Supplementary Table 1 contains those mRNA molecules and their corresponding primers for qRT-PCR analysis, including ADH5, ALDH2, ALDH3A2, CPT2, ECHS1, EHHADH, HIBCH, SUCLG2, and ACADS. Western blot was used to verify apoptosis-related proteins, including ADH5, ALDH3A2, CTP2, ECHS1, EHHADH, HIBCH, NUDT19, FASN, SREBP1, ACC1, ACADS, and ALDH2 in TOV-21G cells between si-HSP60 and control groups.
2.7. Validation of Phosphorylated HSP60 Using Immunoprecipitation
An anti-phospho-HSP60 (Ser70) antibody was prepared by Affinity Company (USA), and the polypeptide synthesis sequence of HSP60 was tVIIEQSWGsPK. The brief procedure was as follows: (i) polypeptide with phosphorylation at residue Ser70 in HSP60 was synthesized (12-16 amino acids, purity greater than 90%, and 10 mg), without phosphorylation modification in the other amino acid residues. (ii) KLH was coupled with cysteine of C or N terminal. (iii) Two New Zealand rabbits were immunized, and peripheral blood was tested after ten weeks. (iv) Antigen was purified by affinity chromatography for two steps to obtain 5 mg anti-phospho-HSP60 (Ser70) antibody and 1 mg anti-HSP60 antibody. (v) Mass spectrometry and ELISA were used to detect the quality of affinity-synthesized antigen.
Phosphorylated HSP60 (Ser70) between ovarian cancer cells TOV-21G and control IOSE80 was semiquantified by immunoprecipitation (IP) and western blot. Briefly, HSP60 proteins were immunoprecipitated with a mouse anti-human HSP60 monoclonal antibody (10 μg; Proteintech, USA) or a mouse anti-human IgG monoclonal antibody (5 μg; Proteintech, USA) from an amount (1 mg) of proteins extracted from the ovarian cancer cell TOV-21G and control cell IOSE80. The total proteins and IP products of the ovarian cancer cell TOV-21G and control cell IOSE80 were used to detect phosphorylated HSP60 (Ser70) by western blotting based on a rabbit anti-human phosphorylation antibody (1 : 500 dilution).
2.8. Statistical Analysis
For KEGG enrichment analysis and IPA pathway network analysis, p values were corrected by Benjamini-Hochberg multiple testing. For western blot, wound healing assay, CCK8 cell assay, EdU assay, apoptosis assay, cell cycle assay, and qRT-PCR data, data were expressed as mean ± SD with a statistically significant level of p < 0.05 using the Student t-test in SPSS 13.0 (SPSS Inc., Chicago, USA) (n = 3).
3. Results
3.1. KEGG Pathway Analysis Revealed Lipid Metabolism Pathway Alterations in Ovarian Cancers
KEGG pathway analysis of 5115 mtEPs in ovarian cancers revealed various lipid metabolism pathways, including fatty acid degradation, fatty acid metabolism, butanoate metabolism, and propanoate metabolism [24] (Table 1; Supplementary Figure 1). Fatty acid degradation-related proteins were significantly increased in ovarian cancers relative to controls (fold change > 1.5), including CPT2 (fold change = 2.05, p = 1.99E − 2), ACOX1 (fold change = 1.53, p =3.40E-2), EHHADH (fold change = 1.62, p = 2.25E − 3), ECHS1 (fold change = 1.52, p = 3.56E − 3), and ECI1 (fold change = 1.64, p = 1.99E − 3). Fatty acid metabolism-related proteins were significantly increased in ovarian cancers relative to controls (fold change > 1.5), including ACAA1 (fold change = 2.70, p = 1.00E − 2), ACOX1 (fold change = 1.53, p = 3.40E − 2), CPT2 (fold change = 2.05, p = 1.99E − 2), ECHS1 (fold change = 1.52, p = 3.56E − 3), ECI1 (fold change = 1.64, p = 1.99E − 3), and PECR (fold change = 1.57, p = 4.14E − 4). Butanoate metabolism-related proteins were significantly increased in ovarian cancers relative to controls (fold change > 1.5), including EHHADH (fold change = 1.62, p = 2.25E − 3), ECHS1 (fold change = 1.52, p = 3.56E − 3), BDH1 (fold change = 1.54, p = 3.00E − 3), and HMGCS2 (fold change = 2.17, p = 2.10E − 3). Propanoate metabolism-related proteins were significantly increased in ovarian cancers relative to controls (fold change > 1.5), including GSTK1 (fold change = 2.13, p = 4.20E − 5), ACAA1 (fold change = 2.70, p = 1.00E − 2), NUDT19 (fold change = 1.55, p = 6.40E − 3), PECR (fold change = 1.57, p = 4.14E − 4), PEX13 (fold change = 1.61, p = 3.58E − 3), PEX14 (fold change = 1.99, p = 6.51E − 4), IDH2 (fold change = 2.02, p = 2.07E − 3), ABCD3 (fold change = 1.99, p = 6.51E − 4), and PHYH (fold change = 1.94, p = 7.71E − 3).
Table 1.
KEGG pathway analysis of 5115 mtEPs revealed four statistically significant lipid metabolism pathway alterations in ovarian cancer.
| Lipid metabolism pathways | Accession | Description | Gene name | Coverage | Unique peptides | Peptides | PSMs | MW (kDa) | Calc. pI | Average tumor/control | t-test p value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fatty acid degradation pathway | E7ERD7 | Long-chain fatty acid-CoA ligase 6 OS=Homo sapiens GN=ACSL6 PE=1 SV=1—[E7ERD7_HUMAN] | ACSL6 | 8.27 | 3 | 5 | 7 | 75.20 | 7.42 | 1.38 | 6.22E − 03 |
| B2RAQ8 | cDNA, FLJ95058, highly similar to Homo sapiens carnitine palmitoyltransferase 1A (liver) (CPT1A), nuclear gene encoding mitochondrial protein, mRNA OS=Homo sapiens PE=2 SV=1—[B2RAQ8_HUMAN] | CPT1A | 34.41 | 22 | 22 | 47 | 88.28 | 8.59 | 1.41 | 9.89E − 03 | |
| P23786 | Carnitine O-palmitoyltransferase 2, mitochondrial OS=Homo sapiens GN=CPT2 PE=1 SV=2—[CPT2_HUMAN] | CPT2 | 42.40 | 5 | 24 | 77 | 73.73 | 8.18 | 2.05 | 1.99E − 02 | |
| A0A024R8L7 | Acyl-coenzyme A oxidase OS=Homo sapiens GN=ACOX1 PE=3 SV=1—[A0A024R8L7_HUMAN] | ACOX1 | 29.85 | 14 | 14 | 23 | 74.64 | 7.62 | 1.53 | 3.40E − 02 | |
| D4QEZ8 | Short-chain acyl-CoA dehydrogenase OS=Homo sapiens GN=ACADS PE=2 SV=1—[D4QEZ8_HUMAN] | ACADS | 27.67 | 8 | 8 | 19 | 44.33 | 7.72 | 1.33 | 6.43E − 06 | |
| P45954 | Short/branched chain-specific acyl-CoA dehydrogenase, mitochondrial OS=Homo sapiens GN=ACADSB PE=1 SV=1—[ACDSB_HUMAN] | ACADSB | 25.93 | 8 | 8 | 15 | 47.46 | 6.99 | 1.29 | 1.06E − 02 | |
| Q08426 | Peroxisomal bifunctional enzyme OS=Homo sapiens GN=EHHADH PE=1 SV=3—[ECHP_HUMAN] | EHHADH | 33.47 | 22 | 22 | 34 | 79.44 | 9.14 | 1.62 | 2.25E − 03 | |
| P30084 | Enoyl-CoA hydratase, mitochondrial OS=Homo sapiens GN=ECHS1 PE=1 SV=4—[ECHM_HUMAN] | ECHS1 | 75.86 | 19 | 19 | 156 | 31.37 | 8.07 | 1.52 | 3.56E − 03 | |
| Q08426 | Peroxisomal bifunctional enzyme OS=Homo sapiens GN=EHHADH PE=1 SV=3—[ECHP_HUMAN] | EHHADH | 33.47 | 22 | 22 | 34 | 79.44 | 9.14 | 1.62 | 2.25E − 03 | |
|
| |||||||||||
| Fatty acid metabolism pathway | Q9P035 | Very-long-chain (3R)-3-hydroxyacyl-CoA dehydratase 3 OS=Homo sapiens GN=HACD3 PE=1 SV=2—[HACD3_HUMAN] | HACD3 | 22.93 | 7 | 7 | 26 | 43.13 | 8.94 | 1.06 | 6.65E − 2 |
| Q5VWC8 | Very long-chain (3R)-3-hydroxyacyl-CoA dehydratase 4 OS=Homo sapiens GN=HACD4 PE=1 SV=1—[HACD4_HUMAN] | HACD4 | 4.31 | 2 | 2 | 3 | 27.50 | 8.57 | 1.26 | 1.74E − 02 | |
| A0A0A0MTI6 | Elongation of very long-chain fatty acid protein OS=Homo sapiens GN=ELOVL5 PE=1 SV=1—[A0A0A0MTI6_HUMAN] | ELOVL5 | 3.05 | 1 | 1 | 2 | 30.72 | 7.52 | 1.37 | 1.94E − 03 | |
| P09110 | 3-Ketoacyl-CoA thiolase, peroxisomal OS=Homo sapiens GN=ACAA1 PE=1 SV=2—[THIK_HUMAN] | ACAA1 | 46.23 | 4 | 14 | 50 | 44.26 | 8.44 | 2.70 | 1.00E − 02 | |
| D4QEZ8 | Short-chain acyl-CoA dehydrogenase OS=Homo sapiens GN=ACADS PE=2 SV=1—[D4QEZ8_HUMAN] | ACADS | 27.67 | 8 | 8 | 19 | 44.33 | 7.72 | 1.33 | 6.43E − 06 | |
| P45954 | Short/branched chain-specific acyl-CoA dehydrogenase, mitochondrial OS=Homo sapiens GN=ACADSB PE=1 SV=1—[ACDSB_HUMAN] | ACDSB | 25.93 | 8 | 8 | 15 | 47.46 | 6.99 | 1.29 | 1.06E − 02 | |
| A0A024R8L7 | Acyl-coenzyme A oxidase OS=Homo sapiens GN=ACOX1 PE=3 SV=1—[A0A024R8L7_HUMAN] | ACOX1 | 29.85 | 14 | 14 | 23 | 74.64 | 7.62 | 1.53 | 3.40E − 02 | |
| E7ERD7 | Long-chain fatty acid-CoA ligase 6 OS=Homo sapiens GN=ACSL6 PE=1 SV=1—[E7ERD7_HUMAN] | E7ERD7 | 8.27 | 3 | 5 | 7 | 75.20 | 7.41 | 1.38 | 6.22E − 03 | |
| B2RAQ8 | cDNA, FLJ95058, highly similar to Homo sapiens carnitine palmitoyltransferase 1A (liver) (CPT1A), nuclear gene encoding mitochondrial protein, mRNA OS=Homo sapiens PE=2 SV=1—[B2RAQ8_HUMAN] | CPT1A | 34.41 | 22 | 22 | 47 | 88.28 | 8.59 | 1.41 | 9.89E − 03 | |
| P23786 | Carnitine O-palmitoyltransferase 2, mitochondrial OS=Homo sapiens GN=CPT2 PE=1 SV=2—[CPT2_HUMAN] | CPT2 | 42.40 | 5 | 24 | 77 | 73.73 | 8.18 | 2.05 | 1.99E − 02 | |
| Q08426 | Peroxisomal bifunctional enzyme OS=Homo sapiens GN=EHHADH PE=1 SV=3—[ECHP_HUMAN] | EHHADH | 33.47 | 22 | 22 | 34 | 79.44 | 9.14 | 1.62 | 2.25E − 03 | |
| P30084 | Enoyl-CoA hydratase, mitochondrial OS=Homo sapiens GN=ECHS1 PE=1 SV=4—[ECHM_HUMAN] | ECHM | 75.86 | 19 | 19 | 156 | 31.37 | 8.07 | 1.52 | 3.56E − 03 | |
| Q16836 | Hydroxyacyl-coenzyme A dehydrogenase, mitochondrial OS=Homo sapiens GN=HADH PE=1 SV=3—[HCDH_HUMAN] | HCDH | 64.01 | 16 | 16 | 69 | 34.27 | 8.85 | 1.34 | 2.11E − 02 | |
| Q8N8R3 | Mitochondrial basic amino acid transporter OS=Homo sapiens GN=SLC25A29 PE=1 SV=2—[MCATL_HUMAN] | MCATL | 20.13 | 6 | 6 | 7 | 32.04 | 8.75 | 1.07 | 4.30E − 02 | |
| Q9BY49 | Peroxisomal trans-2-enoyl-CoA reductase OS=Homo sapiens GN=PECR PE=1 SV=2—[PECR_HUMAN] | PECR | 26.40 | 7 | 7 | 10 | 32.52 | 8.81 | 1.57 | 4.14E − 04 | |
| Q86SK9 | Stearoyl-CoA desaturase 5 OS=Homo sapiens GN=SCD5 PE=1 SV=2—[SCD5_HUMAN] | SCD5 | 9.70 | 2 | 2 | 4 | 37.59 | 9.61 | 0.77 | 1.05E − 02 | |
|
| |||||||||||
| Butanoate metabolism pathway | D4QEZ8 | Short-chain acyl-CoA dehydrogenase OS=Homo sapiens GN=ACADS PE=2 SV=1—[D4QEZ8_HUMAN] | ACADS | 27.67 | 8 | 8 | 19 | 44.33 | 7.72 | 1.33 | 6.43E − 06 |
| Q08426 | Peroxisomal bifunctional enzyme OS=Homo sapiens GN=EHHADH PE=1 SV=3—[ECHP_HUMAN] | EHHADH | 33.47 | 22 | 22 | 34 | 79.44 | 9.14 | 1.62 | 2.25E − 03 | |
| P30084 | Enoyl-CoA hydratase, mitochondrial OS=Homo sapiens GN=ECHS1 PE=1 SV=4—[ECHM_HUMAN] | ECHS1 | 75.86 | 19 | 19 | 156 | 31.37 | 8.07 | 1.52 | 3.56E − 03 | |
| Q16836 | Hydroxyacyl-coenzyme A dehydrogenase, mitochondrial OS=Homo sapiens GN=HADH PE=1 SV=3—[HCDH_HUMAN] | HADH | 64.01 | 16 | 16 | 69 | 34.27 | 8.85 | 1.34 | 2.11E − 02 | |
| Q08426 | Peroxisomal bifunctional enzyme OS=Homo sapiens GN=EHHADH PE=1 SV=3—[ECHP_HUMAN] | EHHADH | 33.47 | 22 | 22 | 34 | 79.44 | 9.14 | 1.62 | 2.25E − 03 | |
| Q02338 | D-Beta-hydroxybutyrate dehydrogenase, mitochondrial OS=Homo sapiens GN=BDH1 PE=1 SV=3—[BDH_HUMAN] | BDH1 | 41.98 | 11 | 11 | 23 | 38.13 | 8.95 | 1.54 | 2.99E − 03 | |
| Q9BUT1 | 3-Hydroxybutyrate dehydrogenase type 2 OS=Homo sapiens GN=BDH2 PE=1 SV=2—[BDH2_HUMAN] | BDH2 | 17.14 | 4 | 4 | 6 | 26.71 | 7.65 | 0.51 | 1.81E − 04 | |
| Q86V21 | Acetoacetyl-CoA synthetase OS=Homo sapiens GN=AACS PE=1 SV=1—[AACS_HUMAN] | AACS | 2.68 | 2 | 2 | 2 | 75.10 | 6.24 | 1.35 | 5.30E − 04 | |
| P54868 | Hydroxymethylglutaryl-CoA synthase, mitochondrial OS=Homo sapiens GN=HMGCS2 PE=1 SV=1—[HMCS2_HUMAN] | HMGCS2 | 12.80 | 7 | 7 | 11 | 56.60 | 8.16 | 2.17 | 2.10E − 03 | |
| B1AK13 | 3-Hydroxymethyl-3-methylglutaryl-coenzyme A lyase (hydroxymethylglutaricaciduria), isoform CRA_b OS=Homo sapiens GN=HMGCL PE=2 SV=1—[B1AK13_HUMAN] | HMGCL | 53.00 | 13 | 14 | 49 | 31.71 | 7.61 | 1.33 | 2.91E − 03 | |
|
| |||||||||||
| Propanoate metabolism pathway | Q7Z5G3 | Acetyl-coenzyme A synthetase OS=Homo sapiens GN=ACSS1 PE=2 SV=1—[Q7Z5G3_HUMAN] | ACSS1 | 17.32 | 10 | 10 | 11 | 74.61 | 7.23 | 0.82 | 1.17E − 02 |
| A0A0B4J1R2 | Acyl-CoA synthetase short-chain family member 3, mitochondrial OS=Homo sapiens GN=ACSS3 PE=1 SV=1—[A0A0B4J1R2_HUMAN] | ACSS3 | 42.48 | 23 | 23 | 62 | 74.54 | 8.63 | 0.83 | 8.45E − 04 | |
| Q6NVY1 | 3-Hydroxyisobutyryl-CoA hydrolase, mitochondrial OS=Homo sapiens GN=HIBCH PE=1 SV=2—[HIBCH_HUMAN] | HIBCH | 34.46 | 14 | 14 | 29 | 43.45 | 8.19 | 1.58 | 2.50E − 03 | |
| Q08426 | Peroxisomal bifunctional enzyme OS=Homo sapiens GN=EHHADH PE=1 SV=3—[ECHP_HUMAN] | EHHADH | 33.47 | 22 | 22 | 34 | 79.44 | 9.14 | 1.62 | 2.25E − 03 | |
| P30084 | Enoyl-CoA hydratase, mitochondrial OS=Homo sapiens GN=ECHS1 PE=1 SV=4—[ECHM_HUMAN] | ECHS1 | 75.86 | 19 | 19 | 156 | 31.37 | 8.07 | 1.52 | 3.56E − 03 | |
| Q9NTX5 | Ethylmalonyl-CoA decarboxylase OS=Homo sapiens GN=ECHDC1 PE=1 SV=2—[ECHD1_HUMAN] | ECHDC1 | 9.45 | 3 | 3 | 10 | 33.68 | 8.21 | 1.69 | 3.67E − 03 | |
| Q96I99 | Succinate-CoA ligase (GDP-forming) subunit beta, mitochondrial OS=Homo sapiens GN=SUCLG2 PE=1 SV=2—[SUCB2_HUMAN] | SUCLG2 | 44.91 | 19 | 20 | 115 | 46.48 | 6.39 | 1.71 | 8.17E − 04 | |
| Q96I99 | Succinate-CoA ligase (GDP-forming) subunit beta, mitochondrial OS=Homo sapiens GN=SUCLG2 PE=1 SV=2—[SUCB2_HUMAN] | SUCLG2 | 44.91 | 19 | 20 | 115 | 46.48 | 6.39 | 1.71 | 8.17E − 04 | |
| Q96PE7 | Methylmalonyl-CoA epimerase, mitochondrial OS=Homo sapiens GN=MCEE PE=1 SV=1—[MCEE_HUMAN] | MCEE | 38.07 | 5 | 5 | 8 | 18.74 | 9.09 | 1.22 | 4.87E − 02 | |
To validate mtDEPs in these lipid metabolism pathways derived from iTRAQ quantitative proteomics, mtDEPs were further examined by western blotting in ovarian cancer cells TOV21G and control cells IOSE80 (Figure 1(b)), including ADH5 (fold change = 0.85, p = 0.0682), ALDH2 (fold change = 0.12, p < 0.0001), and ALDH3A2 (fold change = 0.30, p < 0.0001) enriched in the fatty acid degradation pathway; CPT2 (fold change = 1.99, p < 0.0001) enriched in fatty acid degradation and fatty acid metabolism pathways; ECHS1 (fold change = 4.15, p < 0.0001) enriched in fatty acid degradation, butanoate metabolism, and propanoate metabolism pathways; EHHADH (fold change = 4.67, p < 0.0001) enriched in fatty acid degradation, fatty acid metabolism, butanoate metabolism, and propanoate metabolism pathways; HIBCH (fold change = 4.48, p < 0.0001) and SUCLG2 (fold change = 2.68, p < 0.0001) enriched in the propanoate metabolism pathway; and ACADS (fold change = 2.04, p < 0.0001) enriched in fatty acid degradation, fatty acid metabolism, and butanoate metabolism pathways. These western blotting results in cell models had a good consistency with the results of iTRAQ quantitative mitochondrial proteomics in ovarian cancer tissues.
Moreover, mtDEPs were also examined by western blotting between ovarian cancer tissues and normal control tissues (Figure 1(c)), including ADH5, ALDH2, ALDH3A2, CPT2, ECHS1, EHHADH, HIBCH, and NUDT9 enriched in metabolism pathways. Further, the key enzymes of fatty acid synthesis metabolism were significantly expressed between ovarian cancer tissues and normal control tissues, including FASN, ACC1, and SREBP1. The most of these western blotting results in tissues had a good consistency with the results of iTRAQ quantitative mitochondrial proteomics in ovarian cancer tissues. It indicated that lipid metabolism might be disordered in ovarian cancer.
3.2. si-HSP60 Inhibited In Vitro Migration and Proliferation of Ovarian Cancer Cells
The knockdown of HSP60 was established in the TOV-21G cell line model with more than 60% inhibition rate at the inhibition level. The anticancer ability of si-HSP60 was measured with the wound healing assay, CCK8 assay, and EdU assay between si-HSP60-transfected and control TOV-21G cell groups. Downregulated expression of HSP60 significantly slowed wound healing (Figures 2(a) and 2(b)), decreased the number of EdU-positive cells (Figures 2(c) and 2(d)), and inhibited ovarian cancer cell growth (Figure 2(e)), when compared to the control group. These results clearly demonstrated that si-HSP60 significantly inhibited in vitro migration and proliferation of ovarian cancer cells.
Figure 2.
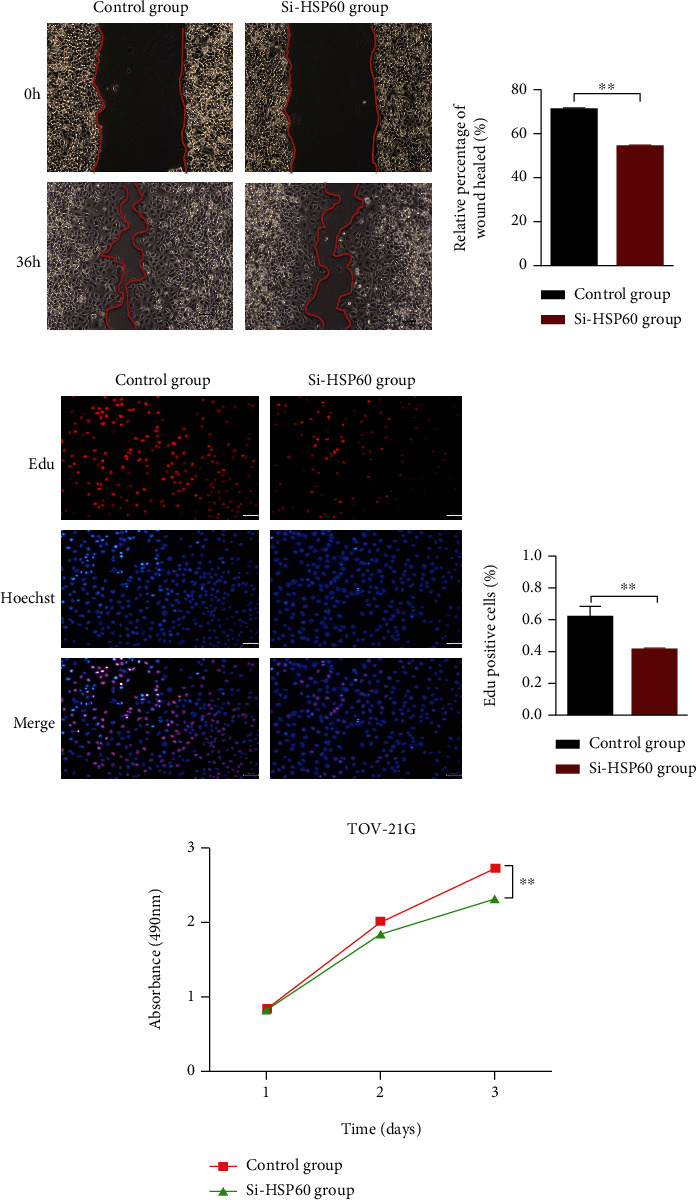
The si-HSP60 inhibited the migration and proliferation of ovarian cancer cells in vitro. (a) Cell migration was measured by the wound healing assay in TOV-21G cells transfected with si-HSP60 and control sequences (n = 3). (b) The histogram of cell migration results with the wound healing assay in TOV-21G cells transfected with si-HSP60 and control sequences (n = 3). (c) EdU cell proliferation test of TOV-21G transfected with si-HSP60 and control sequences (n = 3). (d) The histogram of EdU cell proliferation test of TOV-21G transfected with si-HSP60 and control sequences (n = 3). (e) CCK8 cell proliferation test of TOV-21G transfected with si-HSP60 and control sequences (n = 3). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
3.3. si-HSP60 Inhibited Cell Cycle Progression and Promoted Cell Apoptosis of Ovarian Cancer Cells
To explore the mechanism that si-HSP60 inhibited ovarian cancer cell proliferation, the apoptosis was measured by FACS between si-HSP60-ransfected TOV-21G and control TOV-21G that were stained with PI and annexin V. The results showed a higher ratio of cell apoptosis in the si-HSP60 transfection group compared to the control group (Figures 3(a) and 3(b)). Moreover, western blots for caspase-3, caspase-8, and caspase-9 were used to evaluate whether the extrinsic or intrinsic apoptosis pathway was activated. The results showed that caspase-3 and caspase-8 in the extrinsic apoptotic pathway were activated in HSP-silenced cancer cells (Figures 3(c) and 3(d)). Furthermore, the different distributions of the cell cycle were analyzed by FACS after transfection with si-HSP60. The results found that G0/G1 stage arrest was observed in the si-HSP60 transfection group compared to the control group; namely, the number of cells at the G0/G1 stage was increased and the number of cells at the S stage was decreased in the si-HSP60 group relative to the control group (Figures 3(e) and 3(f)). These data strongly demonstrated that HSP60 inhibited cell proliferation by blocking cell cycle progression and promoting its apoptosis.
Figure 3.
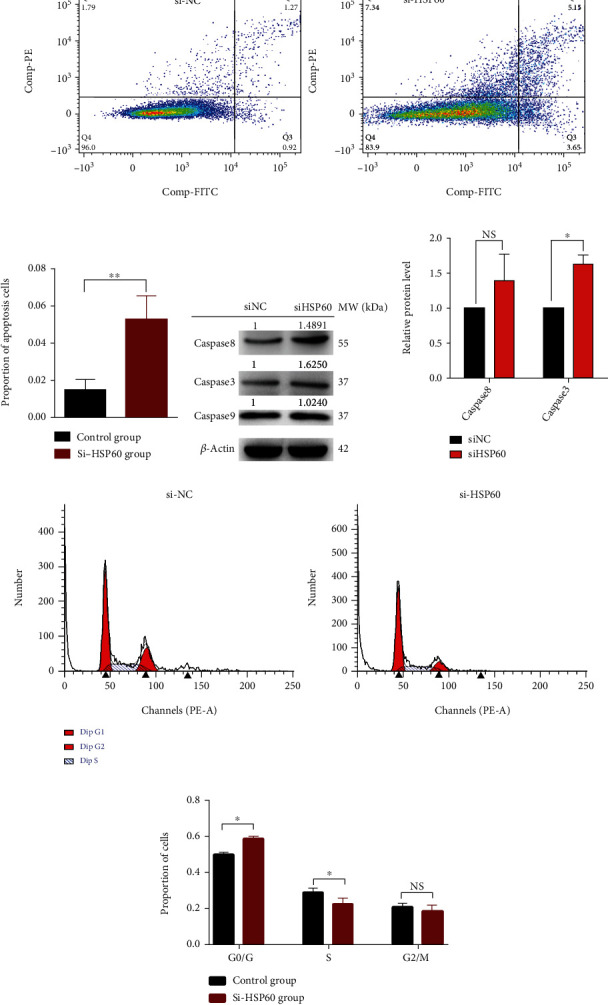
The si-HSP60 promoted apoptosis and inhibited the cell cycle of ovarian cancer cells in vitro. (a) Apoptosis percentage of TOV-21G cells transfected with si-HSP60 and control sequences, measured by fluorescence-activated cell sorting (FACS) (n = 3). (b) The histogram of apoptosis percentage of TOV-21G cells transfected with si-HSP60 and control sequences, measured by fluorescence-activated cell sorting (FACS) (n = 3). (c) Western blots for caspase-3, caspase-8, and caspase-9 to evaluate extrinsic and intrinsic apoptosis pathways. (d) The histogram of caspase-3 and caspase-8 expression between si-HSP60 and control sequence groups. (e) Differences in cell cycle distributions of TOV-21G transfected with si-HSP60 and control sequences, measured by FACS (n = 3). (f) The histogram of cell cycle distributions of TOV-21G transfected with si-HSP60 and control sequences, measured by FACS (n = 3). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
3.4. HSP60 Might Regulate the Expressions or Folding of Important Proteins in Lipid Metabolism Pathways in Ovarian Cancers
HSP60 was reported to be used for survival prediction in advanced serous ovarian cancer [31]. Mitochondrial molecular chaperones like HSP60 regulated folding of mitochondrial matrix proteins, such as MCAD and ACADS. It provided a basis for characterization of mitochondria involved in folding and degradation of ACD proteins and lipid metabolism [29]. It was often considered that the HSP60 protein domain was similar to its prokaryotic ortholog GroEL (Figure 4(a)). The HSP10-HSP60 chaperoning protein folding complex played a role in protein folding. Briefly, unfolded proteins bound to the amino acid residue of HSP60 cavity. The mechanism of mitochondrial protein folding within the HSP60-HSP10 complex was investigated in Escherichia coli where the GroEL-GroES protein complex was exploited. After the substrate protein bound to the amino acid residue of HSP60 cavity, completed protein folding, and obtained its native conformation in several cycles, it would be released [11] (Figure 4(b)). Additionally, cd03344 (GroEL) was a member of the superfamily cl02777 (chaperonin_like), and the evolutionary relationship between cd03344 (GroEL) and cd03343 (HSP60) was near (Figure 4(c)). The iTRAQ quantitative proteomics identified the significantly upregulated expression of total HSP60 (fold change = 1.43, p = 3.81E − 2), and quantitative phosphoproteomics identified the significantly increased phosphorylation level at residue Ser70 in HSP60 (p-HSP60) (fold change = 2.34, p = 1.70E − 3), in ovarian cancer tissues relative to control ovary tissues (Figure 4(d)).
Figure 4.
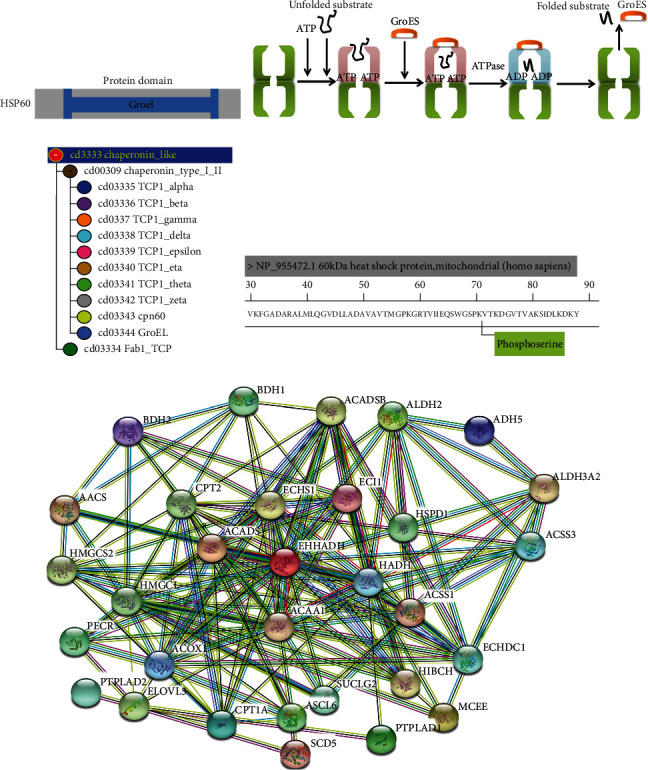
HSP60 associated with lipid metabolism pathways. (a) The drawing of the HSP60 protein domain. (b) The mechanism of mitochondrial protein folding within the HSP60-HSP10 complex. Modified from Horwich [56], with permission from Nature Publishers, copyright 2011. (c) Evolutionary relationship of the superfamily cl02777. CPN60 is the alias of HSP60. (d) HSP60 phosphorylation at residue Ser70. (e) Protein-protein interaction (PPI) in lipid metabolism pathways constructed with the STRING network. HSPD1 is the alias of HSP60.
The protein-protein interaction (PPI) network between the identified proteins in lipid metabolism pathways (Table 1) and HSP60 was constructed in ovarian cancer with the STRING network. This PPI network clearly demonstrated that HSP60 closely interacted with proteins HADH, ACAA1, ECHS1, ACADSB, ACOX1, PECR, CPT2, HMGCS2, HMGCL, BDH, CPT1A, ACADS, ECHDC1, ACSL6, ALDH3A2, ACSS1, ACSS3, ADH5, AACS, HIBCH, ECI1, SUCLG2, ALDH2, ELOVL5, HSPD1, PTPLAD2, EHHADH, MCEE, BDH2, SCD5, and PTPLAD1 in four lipid metabolism pathways, including fatty acid degradation, fatty acid metabolism, butanoate metabolism, and propanoate metabolism (Figure 4(e)), which indicated that HSP60 might play a role in the regulation of lipid metabolism. Further, after si-HSP60 or si-RNA negative control were transfected into TOV-21G cells, qRT-PCR analysis found that si-HSP60 significantly inhibited the expressions of potential target genes at the mRNA level when compared to controls, including ADH5 (fold change = 0.69, p < 0.001), ALDH3A2 (fold change = 0.62, p < 0.001), ECHS1 (fold change = 0.81, p = 0.0048), EHHADH (fold change = 0.61, p < 0.001), and HIBCH (fold change = 0.60, p < 0.001) in lipid metabolism pathways (Figure 5(a)). Moreover, western blot analysis found that si-HSP60 significantly inhibited the expressions of potential target genes in the protein level when compared to controls, including ADH5, ALDH2, ECHS1, EHHADH, and HIBCH in lipid metabolism pathways. Additionally, the key enzymes of fatty acid synthesis metabolism were also significantly expressed between si-HSP60 and control groups, including ACC1 and SREBP1 (Figures 5(b) and 5(c)).
Figure 5.
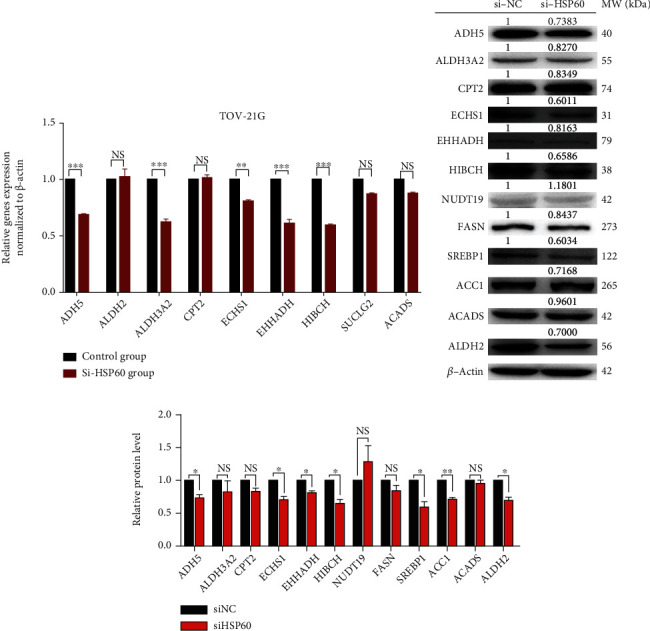
HSP60 regulation in lipid metabolism pathways. (a) The mRNA expressions of potential target genes of HSP60 were verified by qRT-PCR in ovarian cancer cells TO-21G with and without transfection with si-HSP60. (b) The protein expressions of potential target genes of HSP60 were verified by WB in ovarian cancer cells TO-21G with and without transfection with si-HSP60. (c) The histogram of protein expressions of potential target genes of HSP60 in ovarian cancer cells TO-21G with and without transfection with si-HSP60. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Even though some of these potential target genes (ALDH2, CPT2, SUCLG2, and ACADS) in the lipid metabolism pathway did not have mRNA expression changes in ovarian cancers, HSP60-regulated protein folding might be possible for those lipid metabolism-related proteins, based on the mechanism of the HSP10-HSP60 chaperoning protein folding complex. PDB documents of ALDH2, CPT2, SUCLG2, ACADS, and HSP60 were obtained from the RCSB PDB website (http://www.rcsb.org) and were used to construct the structures of human HSP60-ALDH2, HSP60-CPT2, HSP60-SUCLG2, and HSP60-ACADS complexes, with PYMOL. The results indicated that HSP60-ALDH2 and HSP60-ACADS might exist in binding sites (Hsp60:4PJ1, ALDH2:3N80, and ACADS:2VIG) (Figures 6(a) and 6(b)). ALDH2 participated in the metabolism or detoxification of various exogenous and endogenous aliphatic and aromatic aldehydes, which was a member of the aldehyde dehydrogenase 2 family and affected lipid peroxidation [32]. ACADS encoded a tetrameric mitochondrial flavoprotein, which was a member of the acyl-CoA dehydrogenase family. This enzyme catalyzed the initial step of the mitochondrial fatty acid beta-oxidation pathway [33].
Figure 6.
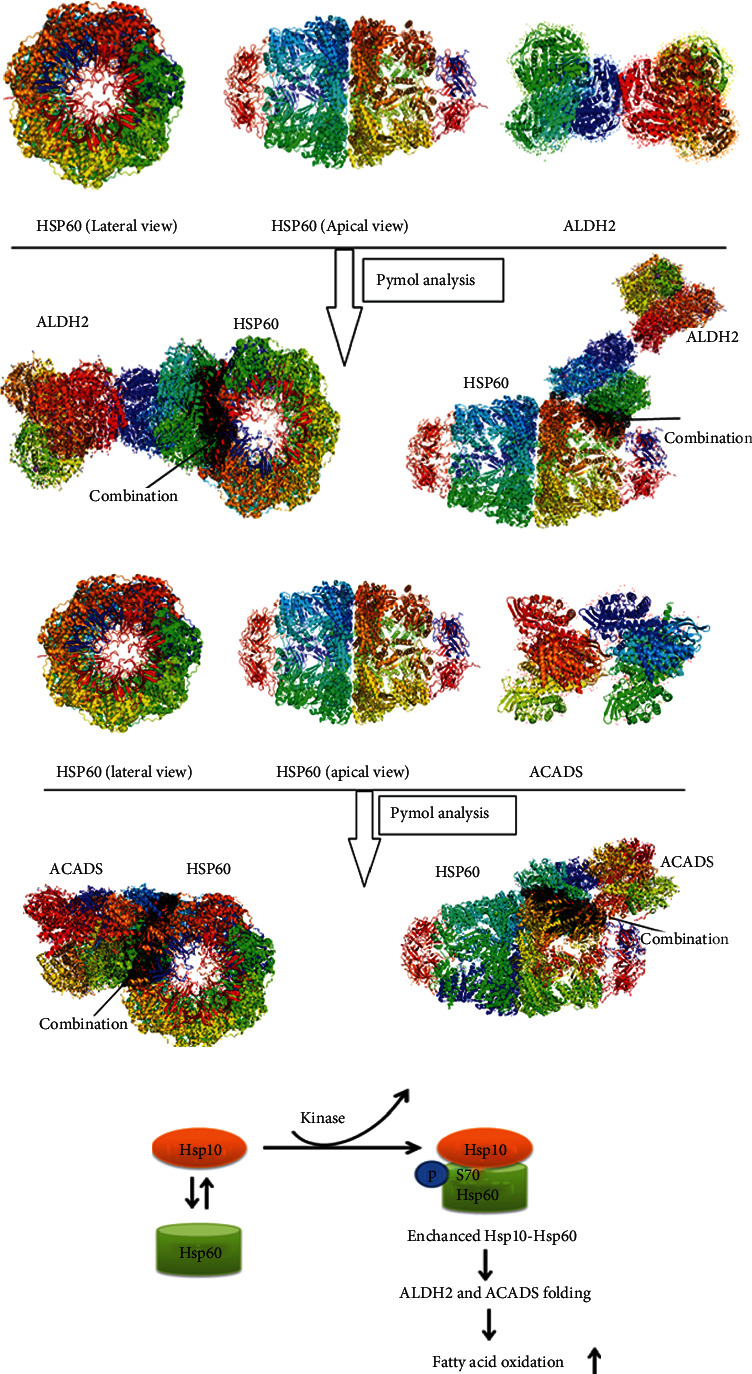
Spatial structural analysis of the molecular mechanism that HSP60 regulates lipid metabolism pathways. (a) HSP60-ALDH2 existed in binding sites with PYMOL based on PDB documents. (b) HSP60-ACADS existed in binding sites with PYMOL based on PDB documents. (c) The hypothesis that protein kinase phosphorylates HSP60, and the activated HSP60 in turn regulates protein folding of ALDH2 and ACADS in lipid metabolism pathways.
The LC-MS/MS spectrum from the phosphorylated peptide TVIIEQSWGS∗PK (m/z = 678.36865, S∗ = phosphorylated serine residue) derived from HSP60 (Swiss-Prot no. B3GQS7) was shown with a high-quality MS/MS spectrum, excellent signal-to-noise (S/N) ratio, and extensive product-ion b-ion and y-ion series (b1, b2, b3, b4, b5, b6, b7, y1, y2, y3, and y4) (Figure 7(a)). The phosphorylation site was localized at amino acid residue Ser70 in HSP60, and the phosphorylation level was significantly increased in ovarian cancers compared to controls (ratio of T/N = 2.34; p = 3.45E − 03). HSP60 immunoprecipitation in combination with a p-HSP60 immunoblot was used to semiquantify the p-HSP60 level in ovarian cancer cells TOV-21G, which was higher than control cells IOSE80 (Figure 7(b)). The immunoblot result was immune-based semiquantitative measurement and was consistent with rigorous MS/MS-based quantitative mitochondrial phosphoproteome data in ovarian cancers. Thereby, we assume that protein kinase phosphorylates HSP60, and the activated HSP60 in turn regulates protein folding of ALDH2 and ACADS to change lipid metabolism pathways (Figure 6(c)).
Figure 7.
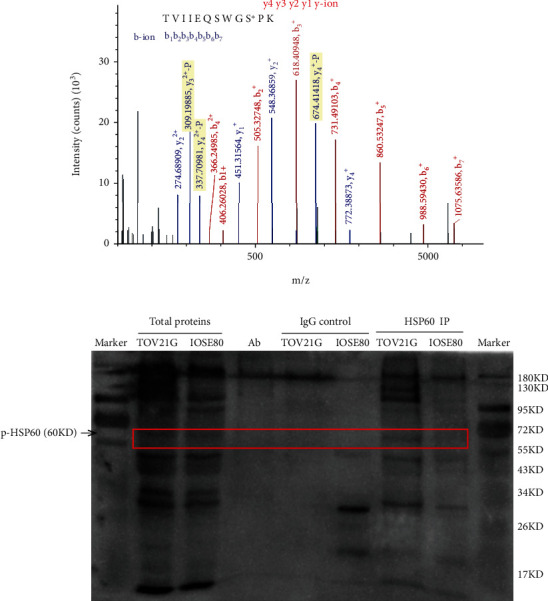
Analysis of HSP60 phosphorylation at residue Ser70 in human ovarian cancer tissue mitochondrial samples with mass spectrometry and in ovarian cancer cell TOV-21G with immunoprecipitation and western blot. (a) The MS/MS spectrum of phosphopeptide TVIIEQSWGS∗PK (S∗ = phosphoserine) derived from HSP60 in ovarian cancer tissues. (b) HSP60 was immunoprecipitated with an anti-HSP60 antibody, followed by phosphorylation analysis of immunoprecipitated HSP60 with western blot against the anti-phosphoserine antibody at residue Ser70 of HSP60 in ovarian cancer cells TOV-21G.
4. Discussion
During the last decade, a great attention has been focused on metabolic alterations of cancer. Studies on “Warburg effect” and “the reverse Warburg effect” have set the pioneering work of glycometabolism functions in cancer [34]. Despite metabolic plasticity being one of the hallmarks described in human disease, the process of glycometabolism disorder was considered conditioned and inefficient for neoplastic transformation. Lipid metabolic reprogramming is less known. Lipid metabolism disorders are associated with an increase in plasma lipids such as low-density lipoprotein, very low-density lipoprotein, and triglycerides which most commonly lead to cardiovascular diseases. They are recently becoming more important in the peculiarities of tumor cell metabolism [35]. Interestingly, the results demonstrated that ovarian cancer cells exhibit an increased dependence on lipid metabolism, such as biosynthesis of unsaturated fatty acids, butanoate metabolism, fatty acid degradation, fatty acid metabolism, propanoate metabolism, and peroxisome pathway, which might play an important role in invasion and metastasis of ovarian cancer. These findings are in agreement with those obtained from other literature. For instance, lipids located in cell membranes (particularly cholesterol and sphingolipids) form lipid rafts, which contain an array of signaling proteins and receptors involved in the cancer-relevant signaling pathways [36]. One study revealed that upregulation of genes (ABCA1, ACSL1, AGPAT1, and SCD) related to lipogenesis and cholesterol synthesis pathways was associated with poor survival outcomes in colorectal cancer patients [19]. Indeed, the coculture of adipocytes and ovarian cancer cells results in promoting ovarian cancer cell growth [22]. The Cancer Genome Atlas provides a system view of metabolic heterogeneity (including lipid metabolism) within and across cancer types and identifies pathway cross-talk, suggesting related predictive, therapeutic, and prognostic utilities [35]. Moreover, the present study on the basis of lipid metabolism in ovarian cancer suggests potential biomarkers, including BDH2, ADH5, PRKCDBP, ECHS1, ACOX1, BDH1, NUDT19, PXMP4, PECR, HIBCH, PEX13, EHHADH, ECI1, ECHDC1, SUCLG2, ABCD3, PHYH, PEX14, IDH2, CPT2, GSTK1, HMGCS2, and ACAA1 in lipid metabolism pathways. Some of them have been reported as biomarkers in other types of cancers, such as GSTK1, IDH2, CAVIN3, HMGCS2, and ACOX1. GSTK1, which is known as a tumor marker, showed some kind of relationship with cancer cell proliferation [37]. In fact, GSTK1 was predominantly expressed in many types of tumor cells and regarded as its marker protein, including oral premalignant and malignant lesions [38] and human erythroleukemia [39]. IDH2 was also reported as a diagnostic and prognostic serum biomarker for non-small-cell lung cancer [40]. Lipid metabolism was closely associated with the occurrence and development of cancer. Therapeutic targeting in lipid metabolism pathways or key related molecules would be challenges and opportunities for ovarian cancer treatment.
HSPs, stimulated by heat shock or other stressors, constitute a large protein family implicated in protein folding and maturation. HSPs have an impact on cellular proliferation, differentiation, and carcinogenesis, whose major family members include HSP27, HSP60, HSP10, HSP40, HSP90, HSP70, and large HSPs. A review article addressed the comprehensive roles of major HSPs in cancer development and pharmacology, which summarized HSP family members in cancer progression, metastasis, diagnosis, treatment, and drug resistance [11]. For instance, HSP27 was identified as phosphorylated at residues Ser15, Ser78, and Ser82, which might regulate the functions of human HSP27, including regulating p53 signaling, inducing the expression of phosphatase and tensin homolog (PTEN), and interaction with cytochrome c [41]. High expression of multiple HSP40 family members was observed in gastric cancer, lung cancer, colorectal cancer, and cervical cancer, and HSP40 had dual roles in both procancer processes and anticancer [42]. This present study detected the expression of total HSP60 and phosphorylated HSP60 in ovarian cancer and control tissues, which showed that total HSP60 and phosphorylated HSP60 were both upregulated in ovarian cancers. In cells with increased or phosphorylated HSP60 levels, the amounts of total mitochondrial short-chain acyl-CoA dehydrogenase (ACADS) proteins and folded ACADS were increased, which might influence mitochondrial protein folding and lipid metabolism. One speculated that HSP60 was involved in the folding of ACADS variant proteins and alerting ACADS enzyme activity, which in turn regulated the lipid metabolism pathway and fatty acid oxidation. HSP60 was mainly located in the mitochondria where it combined as the HSP60/HSP10 complex [43]. Moreover, HSP60 not only was located in mitochondria but also has been found widespread in the body, such as in the cytosol, extracellular space, cell surface, and peripheral blood [44]. Overexpression of HSP60 proved poor prognosis in various carcinomas, including advanced ovarian cancer, head and neck cancer, non-small-cell lung cancer, prostate cancer, gastric cancer, neuroblastoma, bronchial cancer, and colorectal cancer [11]. Moreover, increased HSP60 expression was correlated with chemotherapy drug resistance, and inhibition of HSP60 expression somehow reversed drug resistance [45]. Various mechanisms of HSP60 associated with procarcinogenesis were reported, including inhibiting intracellular protein clusterin, interacting with cyclophilin D, and being involved in the protein nuclear factor-kB pathway [11]. The most exciting feature was that a variety of anticancer drugs were approved by the FDA to target HSPs for cancer treatment, such as sorafenib (Nexavar) [46] and Ruxolitinib (Jakafi) [47]. Overall, our findings were consistent with previous researches, and it was the first time to integrate quantitative mitochondrial proteomics and quantitative mitochondrial phosphoproteomics in human ovarian cancers to explore the relationship between HSP60 and lipid metabolism.
PTM is one of the most common forms in living cells and is a common reason for the formation of human proteoforms [48]. Approximately thirty percent proteins encoded were evaluated to be phosphorylated during their biological cycle [49]. Protein phosphorylation was a reversible process regulating cellular biology characteristics such as cell proliferation, division, migration, and differentiation through highly dynamic or signaling pathways containing phosphorylase activity [50]. So much evidence had reported that various phosphorylation phenotypes induced different pathological features. Researchers had investigated the different phosphorylation level in cancer cells, discussing its correlation with clinicopathologic features and prognosis of carcinoma. Phosphorylation was associated with histopathology grade and the depth of tissue invasion [51]. Mass spectrometry has been widely used to identify and quantify protein PTMs in specific pathways and proteomes [52]. However, few phosphoproteomic approaches detected the subcellular localization of phosphoproteins, which was a significant factor to understand the characterization of the subcellular phosphoproteome in regulating biological processes [53]. The increasing number of identified mitochondrial phosphoproteins, phosphatases, and kinases in recent years suggested that reversible protein phosphorylation played an important part in the control of mitochondrial processes. In addition, many mitochondrial phosphoproteins probably still remain to be identified. HSP60 was also phosphorylated at tyrosine residue (Tyr227) by an activated form of Src kinase, which correlated with proteasomal degradation of HSP60. Phosphorylation and subsequent transient degradation of mitochondrial HSP60 resulted in the inhibition of premature import of nonstructural protein into mitochondria, thereby delaying early apoptosis [54]. HSP60 phosphorylation might not only influence the function or molecular conformation of itself but also regulate other protein folding or cellular protein movement. HSP60 functioned in the regulation of the cellular process in various types of cancer cells to influence tissue growth, which could serve as a target for anticancer therapy [55]. It might be effective to develop specific locus targeting HSP60 in ovarian cancers.
5. Conclusion
Abnormal ROS metabolism was significantly found in ovarian cancers based on mitochondrial proteomic data. The phosphorylation regulation of HSP60 that influences lipid metabolism demonstrated molecular mechanisms of oxidative stress-related diseases. Understanding the mechanisms that cause differences in HSP60 expression levels in ovarian cancer may be useful in the development of therapeutic approaches for pharmaceutical, clinical, and biotechnological applications. The present study integrated a large-scale quantitative mitochondrial proteomic data and quantitative mitochondrial phosphoproteomic data, which found a certain number of proteins with the potential biomarkers, drug targets, and a novel vision in the lipid metabolism biomechanism of human ovarian cancer. Further studies would be helpful to identify new biomarkers and potential drug targets of ovarian cancer.
Acknowledgments
The authors acknowledge the financial support from the Shandong First Medical University Talent Introduction Funds (to X.Z.), the Hunan Provincial Hundred Talent Plan (to X.Z.), the National Natural Science Foundation of China (82172866), and the Academic Promotion Program of Shandong First Medical University (2019ZL002).
Abbreviations
- ACADS:
Acyl-CoA dehydrogenase
- ESI:
Electrospray ionization
- FACS:
Fluorescence-activated cell sorting
- HSL:
Hormone-sensitive lipase
- HSPs:
Heat shock proteins
- IPA:
Ingenuity pathway analysis
- iTRAQ:
Isobaric tags for relative and absolute quantitation
- KEGG:
Kyoto Encyclopedia of Genes and Genomes
- LMF:
Lipid-mobilizing factor
- MCAD:
Medium-chain acyl-CoA dehydrogenase
- MS:
Mass spectrometry
- mtDEPs:
Mitochondrial differentially expressed proteins
- mtEPs:
Mitochondrial expressed proteins
- OC:
Ovarian cancer
- PPI:
Protein-protein interaction
- PTMs:
Posttranslational modifications
- qRT-PCR:
Quantitative real-time PCR
- ROS:
Reactive oxygen species.
Data Availability
All the data used in this study are collected in this article and supplemental materials.
Ethical Approval
All the patients were informed about the purposes of the study. All investigations conformed to the principles outlined in the Declaration of Helsinki and were performed with permission by the responsible Medical Ethics Committee of Xiangya Hospital of Central South University, China, and Medical Ethics Committee of Shandong First Medical University, China.
Consent
All the patients have signed their “consent of the patient” form.
Conflicts of Interest
The authors have declared that no competing interests exist.
Authors' Contributions
Na.L. analyzed the data, designed and performed the experiments, prepared the figures and tables, and designed and wrote the manuscript. Nannan L. performed the western blot experiments based on the si-SHP60-transfected TOV-21G cell model and analyzed the western blot data of ovarian cancer tissues. Y.Z. and Q.L. performed the western blot experiments of ovarian cancer tissues. S.W., S.Z., B.L., and J.Y. participated in partial experiments. L.S. and L.X. collected the tissue samples with clinical diagnosis. X.Z. conceived the concept, designed the experiments and manuscript, instructed the experiments, supervised the results, coordinated, wrote and critically revised the manuscript, and was responsible for its financial supports and the corresponding works. All authors approved the final manuscript.
Supplementary Materials
Supplemental Table 1: the list of RNA molecules that have been assessed on the cell lines.
Supplementary Figure 1: KEGG pathway analysis of 5115 ovarian cancer mitochondrial proteins (mtEPs) revealed four lipid metabolism pathway alterations in human ovarian cancers.
References
- 1.Day J. J., Sweatt J. D. DNA methylation and memory formation. Nature Neuroscience. 2010;13(11):1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belhadj Slimen I., Najar T., Ghram A., Dabbebi H., Ben Mrad M., Abdrabbah M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. International Journal of Hyperthermia. 2014;30(7):513–523. doi: 10.3109/02656736.2014.971446. [DOI] [PubMed] [Google Scholar]
- 3.Li X., Fang P., Mai J., Choi E. T., Wang H., Yang X. F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. Journal of Hematology & Oncology. 2013;6(1):p. 19. doi: 10.1186/1756-8722-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang S., Lian G. ROS and diseases: role in metabolism and energy supply. Molecular and Cellular Biochemistry. 2020;467(1-2):1–12. doi: 10.1007/s11010-019-03667-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris I. S., DeNicola G. M. The complex interplay between antioxidants and ROS in cancer. Trends in Cell Biology. 2020;30(6):440–451. doi: 10.1016/j.tcb.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Kim M., Park W. H., Lee S., et al. Oligonol, a low molecular weight polyphenol, enhances apoptotic cell death in ovarian cancer cells via suppressing NF-κB activation. Nutrition and Cancer-an International Journal. 2019;71(1):141–148. doi: 10.1080/01635581.2018.1557215. [DOI] [PubMed] [Google Scholar]
- 7.Zhou G., Peng F., Zhong Y., Chen Y., Tang M., Li D. Rhein suppresses matrix metalloproteinase production by regulating the Rac1/ROS/MAPK/AP-1 pathway in human ovarian carcinoma cells. International Journal of Oncology. 2017;50(3):933–941. doi: 10.3892/ijo.2017.3853. [DOI] [PubMed] [Google Scholar]
- 8.Seo J. M., Park S., Kim J. H. Leukotriene B4 Receptor-2 Promotes Invasiveness and Metastasis of Ovarian Cancer Cells through Signal Transducer and Activator of Transcription 3 (STAT3)-dependent Up-regulation of Matrix Metalloproteinase. Journal of Biological Chemistry. 2012;287(17):13840–13849. doi: 10.1074/jbc.M111.317131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N., Zhan X. Signaling pathway network alterations in human ovarian cancers identified with quantitative mitochondrial proteomics. Epma Journal. 2019;10(2):153–172. doi: 10.1007/s13167-019-00170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimas D. T., Perlepe C. D., Sergentanis T. N., et al. The prognostic significance of Hsp70/Hsp90 expression in breast cancer: a systematic review and meta-analysis. Anticancer Research. 2018;38(3):1551–1562. doi: 10.21873/anticanres.12384. [DOI] [PubMed] [Google Scholar]
- 11.Wu J., Liu T., Rios Z., Mei Q., Lin X., Cao S. Heat shock proteins and cancer. Trends in Pharmacological Sciences. 2017;38(3):226–256. doi: 10.1016/j.tips.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Voos W., Jaworek W., Wilkening A., Bruderek M. Protein quality control at the mitochondrion. Essays in Biochemistry. 2016;60(2):213–225. doi: 10.1042/EBC20160009. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen K. L., Cowan N. J. A single ring is sufficient for productive chaperonin-mediated folding in vivo. Molecular Cell. 1998;2(1):93–99. doi: 10.1016/S1097-2765(00)80117-3. [DOI] [PubMed] [Google Scholar]
- 14.Chen L., Brewer M. D., Guo L., Wang R., Jiang P., Yang X. Enhanced degradation of misfolded proteins promotes tumorigenesis. Cell Reports. 2017;18(13):3143–3154. doi: 10.1016/j.celrep.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandal J. P., Shiue C.-N., Chen Y.-C., et al. PKCδ mediates mitochondrial ROS generation and oxidation of HSP60 to relieve RKIP inhibition on MAPK pathway for HCC progression. Free Radical Biology & Medicine. 2021;163:69–87. doi: 10.1016/j.freeradbiomed.2020.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Madamanchi N. R., Li S., Patterson C., Runge M. S. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arteriosclerosis Thrombosis and Vascular Biology. 2001;21(3):321–326. doi: 10.1161/01.ATV.21.3.321. [DOI] [PubMed] [Google Scholar]
- 17.Mancini R., Noto A., Pisanu M. E., de Vitis C., Maugeri-Saccà M., Ciliberto G. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene. 2018;37(18):2367–2378. doi: 10.1038/s41388-018-0141-3. [DOI] [PubMed] [Google Scholar]
- 18.Munir R., Lisec J., Swinnen J. V., Zaidi N. Lipid metabolism in cancer cells under metabolic stress. British Journal of Cancer. 2019;21(2019):1090–1098. doi: 10.1038/s41416-019-0451-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vargas T., Moreno-Rubio J., Herranz J., et al. ColoLipidGene: signature of lipid metabolism-related genes to predict prognosis in stage-II colon cancer patients. Oncotarget. 2015;6(9):7348–7363. doi: 10.18632/oncotarget.3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yue S., Li J., Lee S. Y., et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metabolism. 2014;19(3):393–406. doi: 10.1016/j.cmet.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gercel-Taylor C., Doering D. L., Kraemer F. B., Taylor D. D. Aberrations in normal systemic lipid metabolism in ovarian cancer patients. Gynecologic Oncology. 1996;60(1):35–41. doi: 10.1006/gyno.1996.0008. [DOI] [PubMed] [Google Scholar]
- 22.Nieman K. M., Kenny H. A., Penicka C. V., et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature Medicine. 2011;17(11):1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffiths W. J., Wang Y. An update on oxysterol biochemistry: new discoveries in lipidomics. Biochemical and Biophysical Research Communications. 2018;50(4):617–622. doi: 10.1016/j.bbrc.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li N., Li H., Cao L., Zhan X. Quantitative analysis of the mitochondrial proteome in human ovarian carcinomas. Endocrine-Related Cancer. 2018;25(10):909–931. doi: 10.1530/ERC-18-0243. [DOI] [PubMed] [Google Scholar]
- 25.Teng R., Liu Z., Tang H., et al. HSP60 silencing promotes Warburg-like phenotypes and switches the mitochondrial function from ATP production to biosynthesis in ccRCC cells. Redox Biol. 2019;24(101218):p. 101218. doi: 10.1016/j.redox.2019.101218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia X., Li G., Xing Y., Ding Y., Ren T., Kan J. Antioxidant activity of whole grain highland hull-less barley and its effect on liver protein expression profiles in rats fed with high-fat diets. European Journal of Nutrition. 2018;57(6):2201–2208. doi: 10.1007/s00394-017-1494-z. [DOI] [PubMed] [Google Scholar]
- 27.Giuffrè M., Rizzo M., Scaturro G., et al. Oxidative stress markers at birth: analyses of a neonatal population. Acta Histochemica. 2015;117(4-5):486–491. doi: 10.1016/j.acthis.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 28.Lu Z., Chen Y., Aponte A. M., Battaglia V., Gucek M., Sack M. N. Prolonged Fasting Identifies Heat Shock Protein 10 as a Sirtuin 3 Substrate: Journal of Biological Chemistry. 2015;290(4):2466–2476. doi: 10.1074/jbc.M114.606228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corydon T. J., Hansen J., Bross P., Jensen T. G. Down-regulation of Hsp60 expression by RNAi impairs folding of medium-chain acyl-CoA dehydrogenase wild-type and disease-associated proteins. Molecular Genetics and Metabolism. 2005;85(4):260–270. doi: 10.1016/j.ymgme.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Li N., Qian S., Li B., Zhan X. Quantitative analysis of the human ovarian carcinoma mitochondrial phosphoproteome. Aging. 2019;11(16):6449–6468. doi: 10.18632/aging.102199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hjerpe E., Egyhazi S., Carlson J., et al. HSP60 predicts survival in advanced serous ovarian cancer. International Journal of Gynecological Cancer. 2013;23(3):448–455. doi: 10.1097/IGC.0b013e318284308b. [DOI] [PubMed] [Google Scholar]
- 32.Wei S., Zhang L., Bailu wang, et al. ALDH2 deficiency inhibits ox-LDL induced foam cell formation via suppressing CD36 expression. Biochemical and Biophysical Research Communications. 2019;512(1):41–48. doi: 10.1016/j.bbrc.2019.02.012. [DOI] [PubMed] [Google Scholar]
- 33.Dessein A. F., Fontaine M., Joncquel-Chevalier Curt M., et al. Fluxomic evidence for impaired contribution of short-chain acyl-CoA dehydrogenase to mitochondrial palmitate β-oxidation in symptomatic patients with _ACADS_ gene susceptibility variants. Clinica Chimica Acta. 2017;471(101-106):101–106. doi: 10.1016/j.cca.2017.05.026. [DOI] [PubMed] [Google Scholar]
- 34.Diehl K., Dinges L. A., Helm O., et al. Nuclear factor E2-related factor-2 has a differential impact on MCT1 and MCT4 lactate carrier expression in colonic epithelial cells: a condition favoring metabolic symbiosis between colorectal cancer and stromal cells. Oncogene. 2018;37(1):39–51. doi: 10.1038/onc.2017.299. [DOI] [PubMed] [Google Scholar]
- 35.Merino Salvador M., Gómez de Cedrón M., Moreno Rubio J., et al. Lipid metabolism and lung cancer. Critical Reviews in Oncology Hematology. 2017;112(31-40):31–40. doi: 10.1016/j.critrevonc.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Mollinedo F., Gajate C. Lipid rafts as major platforms for signaling regulation in cancer. Advances in Biological Regulation. 2015;57(130-46):130–146. doi: 10.1016/j.jbior.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Makino T., Ishikawa K., Igarashi I., Yamoto T., Manabe S., Nakayama H. Relationship between GST Yp induction and hepatocyte proliferation in rats treated with phase II drug metabolizing enzyme inducers. Toxicologic Pathology. 2008;36(3):420–427. doi: 10.1177/0192623308315359. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L., Xiao Y., Priddy R. Increase in placental glutathione S-transferase in human oral epithelial dysplastic lesions and squamous cell carcinomas. Journal of Oral Pathology & Medicine. 1994;23(2):75–79. doi: 10.1111/j.1600-0714.1994.tb00260.x. [DOI] [PubMed] [Google Scholar]
- 39.Kalinina E. V., Novichkova M. D., Shcherbak N. P., Saprin A. N. Inhibitory effect of progesterone P1-1 on glutathione-s-transferase and its antiproliferative effect on human erythroleukemia K562 cells. Voprosy Onkologii. 2000;46(1):68–73. [PubMed] [Google Scholar]
- 40.Li J. J., Li R., Wang W., et al. IDH2 is a novel diagnostic and prognostic serum biomarker for non-small-cell lung cancer. Molecular Oncology. 2018;12(5):602–610. doi: 10.1002/1878-0261.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cayado-Gutiérrez N., Moncalero V. L., Rosales E. M., et al. Downregulation of Hsp27 (HSPB1) in MCF-7 human breast cancer cells induces upregulation of PTEN. Cell Stress & Chaperones. 2013;18(2):243–249. doi: 10.1007/s12192-012-0367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castle P. E., Ashfaq R., Ansari F., Muller C. Y. Immunohistochemical evaluation of heat shock proteins in normal and preinvasive lesions of the cervix. Cancer Letter. 2005;229(2):245–252. doi: 10.1016/j.canlet.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 43.Cappello F., Conway de Macario E., Marasà L., Zummo G., Macario A. J. Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biology & Therapy. 2008;7(6):801–809. doi: 10.4161/cbt.7.6.6281. [DOI] [PubMed] [Google Scholar]
- 44.Wong C. S., Wong V. W., Chan C. M., et al. Identification of 5-fluorouracil response proteins in colorectal carcinoma cell line SW480 by two-dimensional electrophoresis and MALDI-TOF mass spectrometry. Oncology Report. 2008;20(1):89–98. [PubMed] [Google Scholar]
- 45.Roberts J. L., Tavallai M., Nourbakhsh A., et al. GRP78/Dna K is a target for Nexavar/Stivarga/Votrient in the treatment of human malignancies, viral infections and bacterial diseases. Journal of Cell Physiology. 2015;230(10):2552–2578. doi: 10.1002/jcp.25014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tavallai M., Booth L., Roberts J. L., Poklepovic A., Dent P. Rationally repurposing ruxolitinib (Jakafi®) as a solid tumor therapeutic. Frontiers in Oncology. 2016;6(142) doi: 10.3389/fonc.2016.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhan X., Yang H., Peng F., et al. How many proteins can be identified in a 2DE gel spot within an analysis of a complex human cancer tissue proteome? Electrophoresis. 2018;39(7):965–980. doi: 10.1002/elps.201700330. [DOI] [PubMed] [Google Scholar]
- 48.Olsen J. V., Blagoev B., Gnad F., et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 49.Pearlman S. M., Serber Z., Ferrell J. E., Jr. A mechanism for the evolution of phosphorylation sites. Cell. 2011;147(4):934–946. doi: 10.1016/j.cell.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rojo F., Najera L., Lirola J., et al. 4E-binding protein 1, a cell signaling hallmark in breast cancer that correlates with pathologic grade and prognosis. Clinical Cancer Research. 2007;13(1):81–89. doi: 10.1158/1078-0432.CCR-06-1560. [DOI] [PubMed] [Google Scholar]
- 51.Peng F., Li J., Guo T., et al. Nitroproteins in human astrocytomas discovered by gel electrophoresis and tandem mass spectrometry. Journal of the American Society for Mass Spectrometry. 2015;26(12):2062–2076. doi: 10.1007/s13361-015-1270-3. [DOI] [PubMed] [Google Scholar]
- 52.Guo T., Wang X., Li M., et al. Identification of Glioblastoma Phosphotyrosine-Containing Proteins with Two- Dimensional Western Blotting and Tandem Mass Spectrometry. BioMed Research International. 2015;2015:21. doi: 10.1155/2015/134050.134050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Renvoise M., Bonhomme L., Davanture M., Zivy M., Lemaire C. Phosphoproteomic analysis of isolated mitochondria in yeast. Methods in Molecular Biology. 2017;1636(283-299) doi: 10.1007/978-1-4939-7154-1_18. [DOI] [PubMed] [Google Scholar]
- 54.Chattopadhyay S., Mukherjee A., Patra U., et al. Tyrosine phosphorylation modulates mitochondrial chaperonin Hsp60 and delays rotavirus NSP4-mediated apoptotic signaling in host cells. Cell Microbiology. 2017;19(3) doi: 10.1111/cmi.12670. [DOI] [PubMed] [Google Scholar]
- 55.Huang Y. H., Lin K. H., Yu J. S., et al. Targeting HSP60 by subcutaneous injections of jetPEI/HSP60-shRNA destabilizes cytoplasmic survivin and inhibits hepatocellular carcinoma growth. Molecular Carcinogenesis. 2018;57(9):1087–1101. doi: 10.1002/mc.22827. [DOI] [PubMed] [Google Scholar]
- 56.Horwich A. L. Protein folding in the cell: an inside story. Nature Medicine. 2011;17(10):1211–1216. doi: 10.1038/nm.2468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: the list of RNA molecules that have been assessed on the cell lines.
Supplementary Figure 1: KEGG pathway analysis of 5115 ovarian cancer mitochondrial proteins (mtEPs) revealed four lipid metabolism pathway alterations in human ovarian cancers.
Data Availability Statement
All the data used in this study are collected in this article and supplemental materials.