

Abstract
The identification and application of the Von Hippel-Lindau (VHL) gene is a seminal breakthrough in kidney cancer research. VHL and its protein pVHL are the root cause of most kidney cancers, and the cascading pathway below them is crucial for understanding hypoxia, in addition to the aforementioned tumorigenesis routes and treatments. We reviewed the history and functions of VHL/pVHL and Hypoxia-inducible factor (HIF), their well-known activities under low-oxygen environments as an E3 ubiquitin ligase and as a transcription factor, respectively, as well as their non-canonical functions revealed recently. Additionally, we discussed are how their dysregulation promotes tumorigenesis: beginning with chromosome 3 p-arm (3p) loss/epigenetic methylation, followed by two-allele knockout, before the loss of complimentary tumor suppressor genes leads cells down predictable oncological paths. This pathway can ultimately determine the grade, outcome, and severity of the deadliest genitourinary cancer. We finish by investigating current and proposed schemes to therapeutically treat clear cell renal cell carcinoma (ccRCC) by manipulating the hypoxic pathway utilizing Vascular Endothelial Growth Factor (VEGF) inhibitors, mammalian target of rapamycin complex 1 (mTORC1) inhibitors, small molecule HIF inhibitors, immune checkpoint blockade therapy, and synthetic lethality.
Keywords: VHL, HIF, ccRCC, kidney cancer, hypoxia
Kidney cancer is among the ten most common in both men and women, with ~75,000 new cases and ~15,000 deaths every year in the United States, ~67,000 cases and ~23,000 deaths in China, and ~430,000 cases and ~180,000 deaths globally (Chen, Wanqing et al., 2016; 2020; Sung et al., 2021). Clear cell renal cell carcinoma (ccRCC) is by far the predominant type, making up to 80% of all renal cell carcinoma cases. Von Hippel-Lindau (VHL), a crucial hypoxia regulator, has a towering influence in current research: the inactivation of VHL occurs in ~90% of ccRCC tumors. This first silencing acts as a truncal, initial mutation from which others follow determining the subtype and form (Nickerson et al., 2008; Elias et al., 2020). The initial inactivation of VHL is caused mostly by the loss (typically via chromothripsis) of the short arm of chromosome 3, found in over 90% of patients ((Nickerson et al., 2008; Mitchell et al., 2018). This short arm (3p) carries three other two-hit tumor suppressor genes (TSG)s as well: polybromo 1 (PBRM1), SET domain containing 2 (SETD2), and BRCA1-associated protein 1 (BAP1). VHL and (optionally, one or more of) the other three are fully silenced when the other allele succumbs to point mutations in 60–70% of cases, or epigenetic silencing (Dalgliesh et al., 2010). The application of VHL to ccRCC is an instructive story of biology’s unifying aspect: the gene is named after a separate disease named von Hippel-Lindau syndrome, characterized by the formation of tumors in a number of different organs: e.g. retinal/craniospinal haemangioblastomas, pancreatic tumors, and epididymal cystadenomas (Lonser et al., 2003). The gene responsible for the autosomal dominant disorder was narrowed down to the 3p region in 1988 before being definitively found in 1993 (Latif et al., 1993). Given that the leading cause of death in VHL disease is metastatic ccRCC, curiosity turned to the gene’s cancer connections (Maher et al., 1990). At the time, an unqualified mechanism of action was absent, but the correlative power between VHL and ccRCC was strong: inactivation on both alleles was ubiquitous in ccRCC patients, and the reintroduction of a functional VHL into tumor cells in vivo was sufficient to suppress the ability to form tumors in mice (Iliopoulos et al., 1995). Thus, at the turn of the millennium, it became increasingly obvious that VHL and ccRCC were linked, and the only remaining question was how.
The VHL-HIF Canonical Signaling Axis
A fundamental part of this question was so importantly and elegantly resolved that the Nobel Committee at the Karolinkska Instituet determined to award the Nobel Prize in Physiology or Medicine to Sir Peter J. Ratcliffe, William G. Kaelin Jr., and Gregg L. Semenza ‘for their discoveries of how cells sense and adapt to oxygen availability’ on the Erythropoietin-VHL-HIF axis in 2019 (Zhang et al., 2019).
VHL codes for pVHL, which, after binding to elongin B and C, is linked to RING-box protein 1 and cullin-2 (Kaelin, 2002) (Fig. 1). This complex is an (E3) ubiquitin ligase that binds to hypoxia-inducible factor (HIF) α and ubiquitinates it, following which it is degraded in the proteasome (Kaelin, 2002). Thus, a decrease in pVHL leads to an increase in HIF (due to a decrease in degradation), which possesses three alpha isoforms that dimerize with a shared beta subunit. The isoforms vary in locality, function, and inhibitory pathways; HIF1α is universally expressed among cell types while HIF2α is cell-type specific, the isoforms have some overlap but also regulate some different genes each, and some pockets in the structure are conserved but not all of them are (Hara et al., 2001; Raval et al., 2005; Wu et al., 2015). The sensitivity of this pathway to hypoxia is that prior to recognition by the E3 ubiquitin ligase complex, prolyl hydroxylases of the EglN (Egl nine homolog 1; aka proyl hydroxylase domain-containing protein (PHD)) family hydroxylate proline sites on the HIFα subunits (Kaelin, 2002). This prolyl hydroxylation is crucial to recognition by the E3 ubiquitin ligase. However, these hydroxylases require oxygen as a co-substrate; under hypoxia, their activity is decreased, preventing hydroxylation and recognition by the pVHL degradation complex, and resultingly increasing HIF activity. HIF1 and 2 induce the transcription of hundreds of target genes, all with the goal of steering the cell successfully through the oxygen-poor environment (Semenza, 2007). HIF-3 is much less understood than its two counterparts, with prior evidence suggesting it can act as a negative regulator of HIF-1/2 and recent studies indicating it functions as a complement leading to increased activation of HIF genes (Wu et al., 2015). Leading evidence suggests that while HIF-1α overexpression inhibits ccRCC, HIF-2α overexpression drives ccRCC. Whilst expression of HIF-1α in VHL–/– renal adenocarcinoma cell lines delayed tumor growth, overexpressing HIF-2α is both necessary and sufficient for such growth (Raval et al., 2005; Shen et al., 2011).
Fig. 1. The VHL-pVHL-E3-ccRCC axis.
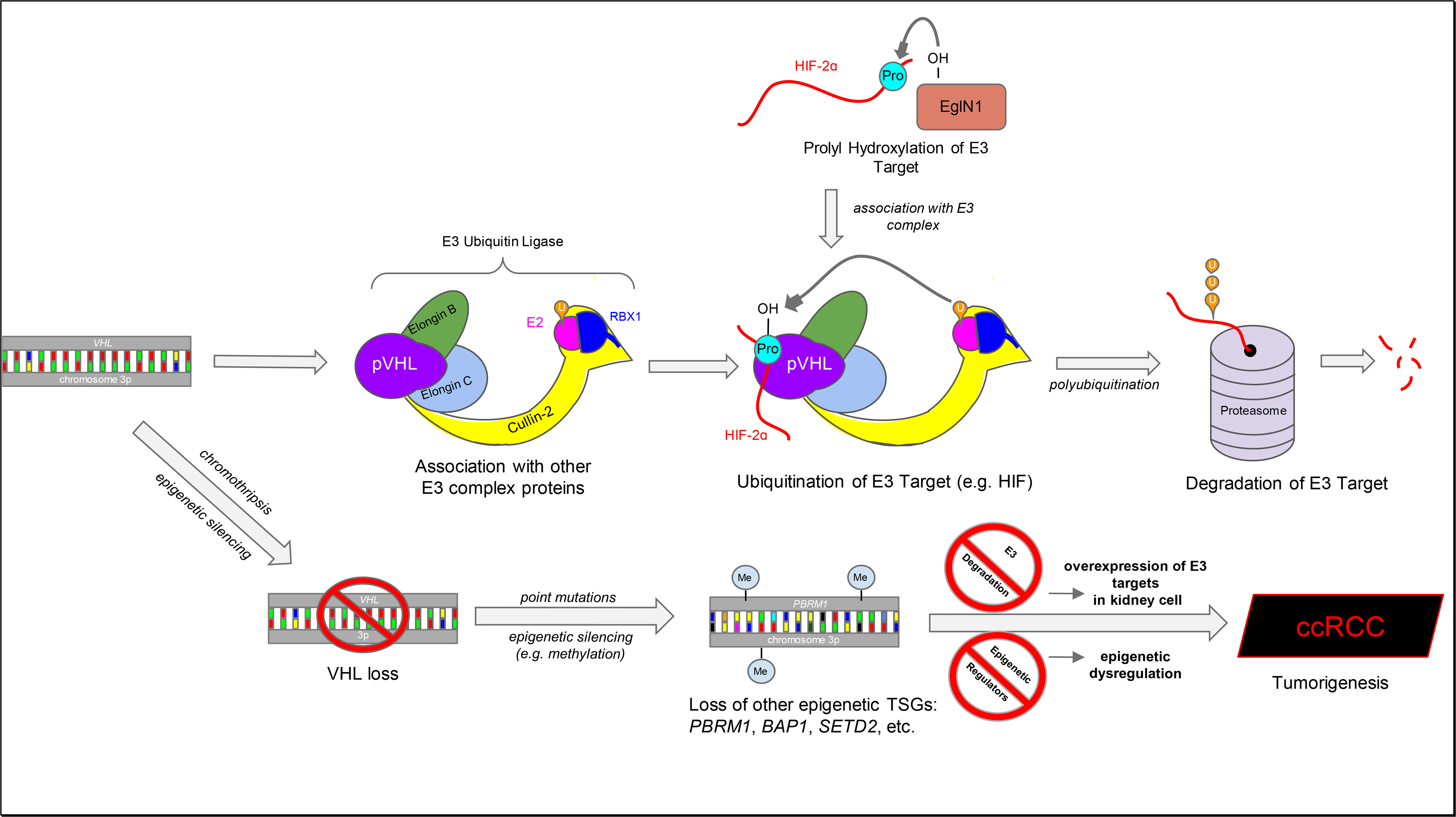
The VHL gene is transcribed into the pVHL protein, which associates with several other proteins to form the E3 Ubiquitin Ligase complex. When a hydroxylated substrate is recognized, it is polyubiquitinated, which is a signal leading to degradation into oligonucleotides in the proteasome. This proline hydroxylation is crucial for recognition by the E3 complex, and the use of oxygen as a co-substrate in this step makes it sensitive to oxygen concentration. Under hypoxia (where oxygen is absent) or following VHL loss, this complex is no longer available to degrade its substrates in the usual way. When VHL loss (leading to overexpression of E3 complex substrates) is combined with the loss of other tumor-suppressing genes relating to epigenetic regulation, this leads kidney cells down a sure path leading to tumorigenesis and ccRCC. (p)VHL, von Hippel-Lindau tumor suppressor (protein); E2, Ubiquitin-conjugating enzyme; RBX1, RING-box protein 1; HIF-2, Hypoxia-inducible factor 2; EglN1, Egl nine homolog 1; TSG, tumor-suppressor gene; PBRM1, polybromo 1; BAP1, BRCA1-associated protein; SETD2, SET domain containing 2; ccRCC, clear cell renal cell carcinoma; U, ubiquitin; Pro, proline; Me, methyl group.
VHL-HIF signaling and the HIF-mediated transcriptional response are far more important than their connections to cancer when defective alone, as these series of regulation are part of the normal, well-functioning response to a hypoxic stimulus. The VHL-HIF pathway is now being investigated as a therapeutic target and finding treatments for a disease which, despite being the deadliest genitourinary cancer, until 2005 only had one United States Food & Drug Administration-approved (FDA) drug with which to combat it (interleukin-2).
VHL Tumorigenesis as a Truncal Mutation
VHL loss is required but not solely sufficient for ccRCC (Wang et al., 2014). Parallel targeted sequencing enabled scientists to find three more tumor-suppressing genes (i.e. PBRM1, SETD2, and BAP1) on chromosome 3p, often deleted in tandem with VHL. Each of these genes encode proteins that happen to be epigenetic-related-PBRM1 is a nucleosome remodeling complex component, SETD2 is a histone methyltransferase, and BAP1 is a histone deubiquitinase. All three genes, in addition to VHL itself, reside within a very select region of 3p (Moore et al., 2012). With heterozygous VHL mutations leading to a proclivity towards tumorigenesis in humans, VHL possesses all the hallmarks of a Knudson two-hit suppressor gene for oncogenesis (Zbar et al., 1987; Crossey et al., 1994). In mice, ccRCC seems to require VHL and Pbrm1/Bap1 deficiencies, as inactivation of any single gene was not sufficient (Wang et al., 2014; Turajlic et al., 2018b). Of these genes, PBRM1 is mutated in 45–55% of human ccRCC cases, SETD2 20– 40%, and BAP1 14–19%. Interestingly, PBRM1 and BAP1 appear to be mutated together less than random chance would indicate, leaving them with a degree of mutual exclusivity (Gu et al., 2017). The different epigenetic genes mutated lead to different outcomes: BAP1-absent tumors possess worse patient survival, higher grade, and develop faster than PBRM1-absent tumors (Kapur et al., 2013; Turajlic et al., 2018a). PBRM1-loss tumors can be converted into tumors of a higher grade by inactivating tuberous sclerosis complex 1 (TSC1), often mutated in ccRCC and a regulator of mammalian target of rapamycin complex 1 (mTORC1) (Kucejova et al., 2011; Gu et al., 2017). The mutually exclusive PBRM1 and BAP1 mutations also develop separately; SETD2 mutations are typically found only in tumors that lack already lack PBRM1, and not BAP1 (Peña-Llopis et al., 2013). Other branches from the truncal PBRM1 mutation include modifications to the mTOR pathway (TSC1, PTEN, PIK3CA) and chromatin-modification genes by somatic copy number alterations/driver mutations, deletion of chromosome 9 and 14, and loss of TP53 (Elias et al., 2020). In addition, other mutations have been characterized to increase the belligerence and metastasis of PBRM1-loss tumors along mTOR, driver SCNA, and SETD2 pathways (Turajlic et al., 2018a; Turajlic et al., 2018b).
The pVHL-HIF Regulatory Pathway
The pathways that descend from HIF are too complex and lengthy to fully mention here; the HIF system regulates hundreds of genes (Semenza, 2007). Out of all the complexity, one observation that has posed exceptionally relevant to clinical treatment is that, as a result of HIF signaling, ccRCC seems to have the largest expression of Vascular endothelial growth factor A (VEGFA) mRNA of all epithelial tumors (Jubb et al., 2004). Current therapeutic strategies aim to target every step of this pathway, from pVHL directly, to its substrate HIF, to HIF transcription targets such as VEGF. Other transcription/ translation factors also work in HIF-dependent and -independent avenues during hypoxia to maintain oxygen homeostasis. For example, Nuclear Factor-κB and HIF are known to induce each other, Activator Protein 1 cooperates with HIF, and p53 and MYC Proto-Oncogene act in a mixed role as antagonist and cooperator of HIF (Kenneth and Rocha, 2008). HIF has also been discovered to interact with chromatin-modifying proteins/complexes and exhibits control over translation (Kenneth and Rocha, 2008). On the other hand, Notch signaling appears to act independently of HIF in pVHL-loss human ccRCC cells grown in vivo (Sjölund et al., 2008).
As in all things, the pathway is more complicated than just HIF; pVHL has more substrates than hypoxia-inducible factor alone. For instance, Zinc fingers and homeoboxes 2 (ZHX2), Scm-like with four malignant brain tumor domains (SFMBT1), have been found to be substrates of pVHL while TANK binding kinase 1 (TBK1) was found to be a pVHL adaptor protein (Zhang et al., 2018; Hu et al., 2020; Liu, X. et al., 2020). pVHL interacts with ZHX2 and SFMBT1, like for HIF, in a process leading to ubiquitination and degradation (for instance, after SFMBT1 proline hydroxylation by PHD2). TBK1 is dephosphorylated and rendered inactive in the presence of pVHL, which is aptly similar to the previously reported finding of Akt1 phosphorylation regulation by pVHL following proline hydroxylation (Guo et al., 2016). When pVHL is suppressed, these three proteins become overactive and oncogenic. ZHX2 depletion decreased the expression of NF-κB genes (IL6, IL8, CCL2) and several genes related to invasion, metastasis, metabolism, and anti-apoptosis (Zhang et al., 2018). SFMBT1 had previously been found to be a transcriptional repressor in the LSD1 demethylase complex but was discovered to activate sphingosine kinase 1 (SPHK1), leading to tumorigenesis in vivo and in vitro. TBK1 was formerly known to interact with stimulator of interferon genes (STING) in the presence of virus, triggering TBK1 phosphorylation and subsequently, the type I IFN immune response (Liu et al., 2015). This immune signaling protein was later investigated for its role in cancer, and it was established that the molecule phosphorylates AKT and p62 with oncogenic results (Hu et al., 2020). TBK1 was additionally later found to be a target for conducting the strategy of synthetic lethality in VHL-loss kidney cancer (Hu et al., 2020). All of these aforementioned substrates act in a fully HIF-independent manner to promote ccRCC tumorigenesis.
Other substrates or binding partners which can be ubiquitinated and degraded or display altered activity upon pVHL binding include euchromatic histone-lysine methyltransferase 2, actin cross-linker filamin A, erythropoietin receptor, transcription factor B-Myb, ceramide kinase like protein, N-Myc downstream regulated gene 3, Card9, Akt1, and BIM-EL (Na et al., 2003; Chen et al., 2015; Lee et al., 2015; Guo et al., 2016; Heir et al., 2016; Okumura et al., 2016; Segura et al., 2016; Casciello et al., 2017; Yin et al., 2017). The plethora of functions these substrates reminds us that the cell’s response to hypoxia and cancer-caused dysregulation is broad and complex. While pVHL is more famously a contributor to the ubiquitin ligase complex, the evidence above suggests it may have other functions and can act as an adaptor protein. The search for more potential pVHL substrates, HIF target genes, and a more complete knowledge of these complex interconnected relationships will be crucial to understanding hypoxia and defeating ccRCC.
The Modern and Multi-pronged Mechanisms to Therapeutically Address ccRCC
ccRCC has posed quite the foe, given that the disease is shockingly common yet deadly, resistant to chemotherapy, and lacked therapeutic options before 2005 (Choueiri and Motzer, 2017). However, on account of the knowledge recently gained and summarized above, researchers have at last begun to exploit these interconnected genes and proteins with the cancer’s destruction in mind.
The scope of these drugs’ effects should be firmly laid out; while the following treatments do extend progression-free survival (PFS) and often have an admirable partial response (PR) rate, they are rarely cures – complete responses are few and far between. The prolonging of life and improvements to quality of life nonetheless remain invaluable for patients and their loved ones. Given that, at first, the only tool at medicine’s arsenal was interleukin-2, we certainly have come a long way. Subsequently, a second flurry of activity followed, with the FDA accepting six new drugs in four years. Now, given that cabozantinib, nivolumab, and lenvatinib with everolimus have been shown to be better than everolimus alone, and with the sunitinib comparison trials below also held in mind, it feels as if we have entered a third generation of safe and effective ccRCC treatments (Molina et al., 2014; Motzer et al., 2015a; Motzer et al., 2015b; Choueiri et al., 2016; Choueiri et al., 2017).
Anti-VEGF Therapy
The first modern therapies to be approved by the Food & Drug Administration took advantage of the extremely high VEGFA expression in ccRCC tumors by inhibiting the VEGF receptor. VEGFA promotes angiogenesis and cell growth; inhibiting it or its receptor could therefore slow uncontrolled proliferation of ccRCC. Sorafenib, a tyrosine-kinase inhibitor (TKI) targeting the VEGF receptors, was the first of these treatments to be approved, heralding a new, second age of ccRCC therapeutics (See Table 1 for drug clinical profiles) (Escudier et al., 2007). The TKI sunitinib and bevacizumab (a monoclonal antibody targeting VEGFA) were approved thereafter (Motzer et al., 2007; Summers et al., 2010). Two more TKIs – axitinib and pazopanib – have also been approved for the treatment of ccRCC, with evidence suggesting that, in comparison to sunitinib, the latter is safer and has better quality-of-life for patients, and the former has a better PFS (Motzer et al., 2013a; Motzer et al., 2013b). Other more recently approved TKIs include cabozantinib and lenvatinib (Motzer et al., 2015a; Choueiri et al., 2016). These six TKI drugs (all excluding monoclonal antibody bevacizumab) inhibit additional receptors (Table 1); a strategy targeting several receptors can often be more comprehensive and effective while reducing the possibility of tumor resistance. For example, cabozantinib also inhibits the hepatocyte growth factor receptor (HGFR/c-Met) while a series of converging factors in ccRCC heavily depend upon and utilize this pathway. MET protein is overexpressed and phosphorylated by VHL-deficient cells, induces tumor growth following oncogenesis (which it promotes), is implicated in resistance to the commonly-used drug sunitinib, and is necessary for ccRCC survival (Nakaigawa et al., 2006; Bommi-Reddy et al., 2008; Zhou et al., 2016). This approach of targeting multiple vulnerable, hyper-dependent pathways would later be the basis of synthetic lethality targeting.
Table 1.
Selected studies demonstrating clinical profiles of ccRCC treatments.
| Trial ID | Compound / FDA Approval Date | Target(s) | n / Phase of Trial | ORR (in %) | OS (Median in months) | PFS |
|---|---|---|---|---|---|---|
| Rapalogs | ||||||
| NCT00065468 | Temsirolimus (2007) ^ | mTORC1 | 626/III | 8.6 v 4.8 | 10.9 v 7.3 | 3.8 v 1.9 |
| NCT00510068 | Everolimus (2009) | mTORC1 | 410/III | NR v 8.8 | NR v 8.8 | 4.0 v 1.9 |
| VEGF Inhibitors | ||||||
| NCT00073307 | Sorafenib (2005) | VEGFR, PDFGR, Flt-3, B-Raf | 902/III | 10 v 2 | 17.8 v 15.2 | 5.5 v 2.8 |
| NCT00083889 | Sunitinib (2006) ^ | VEGFR, PDFGR, c-Raf | 750/III | 47 v 12 | 26.4 v 21.8 | 11 v 5 |
| NCT00738530 | Bevacizumab (2009)* | VEGFA | 649/III | 30 v 12 | 23.3 v 21.3 | 10.2 v 5.4 |
| NCT00334282 | Pazopanib (2009) | VEGFR, PDGFR, c-Kit, FGFR | 435/III | 30 v 3 | 22.9 v 20.5 | 9.2 v 4.2 |
| NCT00678392 | Axitinib (2012) ^ | VEGFR, PDGFR, c-Kit | 723/III | 19 v 9 | 20.1 v 19.2 | 8.3 v 5.7 |
| NCT01865747 | Cabozantinib (2016) ^ | VEGFR, c-Met, AXL | 658/III | 17 v 3 | 21.4 v 16.5 | 7.4 v 3.8 |
| NCT01136733 | Lenvatinib (2016)* ^ | VEGFR, FGFR | 153/II | 43 v 6 | 18.5 v 16.5 | 14.6 v 5.5 |
| Immune Checkpoint Inhibitors | ||||||
| NCT02231749 | Nivolumab (2018) ^ | PD-1 | 821/III | 23 v 4 | 25.8 v 19.7 | 4.2 v 4.5 |
| NCT02231749 | Ipilimumab (2018)* ^ | CTLA-4 | 1096/III | 42 v 27 | NR v 26.0 | 11.6 v 8.4 |
| NCT02853331 | Pembrolizumab (2019)* ^ | PD-1 | 861/III | 59.3 v 35.7 | NR v NR | 15.1 v 11.1 |
| NCT02684006 | Avelumab (2019)* ^ | PD-L1 | 886/III | 51.4 v 25.7 | NR v NR | 13.8 v 8.4 |
| HIF-inhibitors | ||||||
| NCT02293980 | PT-2385 | HIF-2α | 51/I | 14 v NR | NR v NR | NR v NR |
| NCT03401788 | MK-6482 | HIF-2α | 61/II | 27.9 v NR | NR v NR | NR v NR |
| RO7070179 | ARO-HIF2 | HIF-2α | 8/Ib | NR v NR | NR v NR | NR v NR |
Asterisked drugs are not monotreatments; comparison treatment is placebo unless marked ^.
ORR, overall response rate; OS, overall survival; PFS, progression-free survival; NR, not reached/not available.
HIF-2a Inhibition
HIF-2α was long thought to be “undruggable” as it lacked a ligand-binding domain, but recent groundbreaking crystallography found the existence of several pockets in the protein that a small molecule could fit into to disrupt its dimerization with HIF-1β (Scheuermann et al., 2009; Wu et al., 2015). If a small molecule inhibitor could be found, this could hypothetically inhibit not just a target like VEGFA, but the entirety of HIF-2α-mediated transcriptional activity. Screens of hundreds of thousands of compounds and drug fragments in screens yielded a class of similarly structured molecules with allosteric inhibitory ability (Scheuermann et al., 2013). The general characteristics of the ligands discovered found from this study were used later in a mixture of intentional modification and structure selectivity to create compound PT2385, the first HIF-targeting small-molecule inhibitor to face a human clinical trial (Wehn et al., 2018). The safety profile was favorable, with none of the 51 patients choosing to leave the experiment as a result of severe adverse effects or a dose-limiting event (Courtney et al., 2018). The overall response rate was 14%; the disease control rate was 66%. There was a significant difference in PFS between those who had a trough concentration of PT2385 over 0.5 μg/mL 12 hours after receiving the oral dose on day 15 as opposed to those that did not, with those with the higher concentration faring better. This, another study on glioblastoma, and a combination study with nivolumab illustrated a frustratingly high drug exposure variability due to differing metabolism of the drug betwixt patients, with a higher exposure being correlated with better PFS (Rini et al., 2019a; Strowd et al., 2019). This variability left some patients underexposed and made decisions around dosage of PT2385 more difficult.
PT2977 (later known as MK-6482, Belzutifan) was developed to improve upon this first step, with higher HIF pocket affinity, less lipophilicity, less affinity for serum proteins, and lower glucoronidation (shown to be the primary divergent metabolic path causing exposure variation) (Xu et al., 2019). Despite only a small structural change, the pharmacokinetics were far improved, and a phase 3 trial for the molecule is currently in the recruiting phase (NCT04195750 & NCT04586231). A phase I/II and a phase II study of PT2977 suggest similar safety levels as PT2385, along with promising efficacy rates even in pretreated cases. ORR was 24% (I/II) and 28% (II), the disease control rate was 80% (I/II), 67% (I/II) and 87% (II) of patients had tumor shrinkage, the 12-month PFS rate was 98% (II), and 81% had responses greater than six months (I/II) (Choueiri et al., 2020b; Jonasch et al., 2020). Other clinical trials for PT2977 and PT2385 are ongoing.
One drawback to the small-molecule inhibitor approach is that the mechanism of action of this technique depends on the structure of HIF-2, which can change due to de novo mutation. Indeed, two mutations have been found – one on HIF-2α decreasing small-molecule affinity and the other on HIF-1β increasing affinity for dimerization with HIF-2α – that convey a level of acquired resistance to these treatments (Chen, Wenfang et al., 2016). Both of these mutations have been found in tumorgrafts following exposure to PT-2399 and the former mutation in two human patients during a PT2385 clinical trial (Chen, Wenfang et al., 2016; Courtney et al., 2020). However, several more pockets have been found in HIF-2α, which presents an opportunity for combination treatments and the development of additional inhibitors (Wu et al., 2015; Wu, D. et al., 2019). While acquired resistance to these inhibitors is unfortunate, we must remember to compare them against the existing treatments today; notably, acquired resistance to sunitinib developed within 60 days in mice tumorgrafts but took over 120 days for PT-2399, representing significant progress in ccRCC therapeutics (Chen et al., 2016).
Another mechanism to directly target HIF-2α utilizes siRNA. Arrowhead Pharmaceuticals develops siRNA to target ligands found only in ccRCC. This RNAi clinical mechanism has been known for some time, but one of the largest challenges was engineering a manner to deliver the siRNA effectively to the target (Bobbin and Rossi, 2016). In a strategy known as Dynamic PolyConjugates, researchers can link the targeting siRNA with a vehicle that targets the cancer cells by recognizing the intermembrane proteins αvβ3 and αvβ5 commonly overexpressed in ccRCC, enabling the effective delivery of siRNA. This method inhibited tumor growth in mouse xenografts, and, if successful, could help overcome acquired resistance via HIF-2α/ HIF-1β structural changes and decrease toxicity by seeking out ccRCC ligands directly (Wong et al., 2018). A small phase Ib trial was conducted utilizing these techniques with mixed results on advanced hepatocellular carcinoma (Wu et al., 2019). On account of failing to meet the primary endpoint of HIF-1α mRNA silencing after a single dose, the trial was ended early. On the other hand, one of the nine patients exhibited a PR enduring 72 weeks following the study start date. Another phase 1b trial is currently recruiting patients (NCT04169711). Hopefully, this creative delivery mechanism that targets ccRCC and evades one mechanism of tumor resistance will eventually provide better outcomes to patients.
Immunotherapeutics
Yet another approach has been to use immune-checkpoint inhibitors to restore T cell antitumor activity; this line of attack has led to a recent flurry of FDA-approved drugs within the past few years. CTLA-4 is a T cell protein receptor that transmits inhibitory signals to nearby T cells, promoting immunotolerance. Ligand PD-L1, expressed in tumors, acts upon T cell surface receptor PD-1 contributing to immunosuppression and tumor survival – not surprisingly, both of these receptors are upregulated in cancers. Current immunotherapies use monoclonal antibodies to target these T cell receptors and ligands: nivolumab and pembrolizumab targeting PD-1, avelumab targeting PD-L1, and ipilimumab targeting CTLA-4. Nivolumab and pembrolizumab were both approved as singular treatments for RCC following studies comparing them to a placebo, but ipilimumab was approved in combination with nivolumab after being shown to be more effective than sunitinib (Motzer et al., 2015b; Tykodi et al., 2019; Motzer et al., 2020). Avelumab also followed this latter path, being accepted with axitinib following a comparison trial to sunitinib (Choueiri et al., 2020a).
The connection between the effectiveness of immunotherapy and the varying forms of ccRCC caused by different mutations is currently under dispute. While it is logical that tumors with separate paths to tumorigenesis and different expression of genes and proteins would be commensurately distinctly prone or invulnerable to immunotherapy, the jury is still out as to which is true for PBRM1-loss ccRCC. While one team of researchers found that loss of this gene defined a resistance to immune checkpoint inhibitor (ICI) therapy, another laboratory found the opposite was true, and that clinical benefits to anti-PD-1 therapy was correlated with loss of the same gene (Miao et al., 2018; Liu, X.-D. et al., 2020). More work remains to be done to provide specific, targeted therapy to the various strains of cancer previous research has elucidated.
mTORC Inhibition
Other trials have aimed to inhibit mTORC1, which regulates cell survival and proliferation and is one of the most frequently dysregulated pathways in human cancers (Xu et al., 2014). The drugs termsirolimus and everolimus act as analogs of rapamycin (rapalogs) and inhibit mTORC1. Both drugs have been successful over interferon-α treatment (a common previous standard of treatment), were approved by the FDA, and represent some of the first few bold steps taken after 2005 (Hudes et al., 2007; Motzer et al., 2008).
There are significant structural and functional differences between mTORC1 and its related protein complex, mTORC2, which both contain mTOR. For instance, current mTORC1 inhibitors are not effective against mTORC2, and while HIF-1α expression relies upon both complexes, HIF-2α is regulated by mTORC2 alone. Trials attempting to target both mTORC1 and 2 have generally failed due to overly high toxicity, which is all too unfortunate because inactivation of mTORC1 results in the loss of negative feedback inhibition of mTORC2 (Santoni et al., 2014).
Combinatorial Treatments & Synthetic Lethality
This idea of combining treatments as in the mTORC1-mTORC2 or ICB-ICB (immune checkpoint blockade) studies above is hardly new but is occasionally like a siren’s song – alluring yet deadly. The advantages of this strategy could potentially be vast, as attacking ccRCC in multiple ways could increase effectiveness, decrease the possibility of resistance, and yet yield no significant increase in toxicity by spreading the load across several separate systems. Another primary matter of consideration is giving thought to how the pathways stemming from both drugs might interact with each other. The challenge of implementing this stratagem so far has been managing the toxicity. For instance, the administration of drugs that inhibit both mTOR and VEGF was too toxic, and blocking VEGF with both bevacizumab and sunitinib led to peril in its patients (Feldman et al., 2009; Flaherty et al., 2015). Additionally, the original testing of termsirolimus with and without interferon-α found no significant difference in survival (Hudes et al., 2007). On the other hand, there is broad reason to think that using combination therapy to treat ccRCC has a bright future. The FDA approved a dual treatment of lenvatinib (inhibitor of VEGFR and FGFR) and everolimus (mTORC1 inhibitor), which is more effective than everolimus alone, even as the two drugs must be dosed at lower concentrations together than they would have to be if administered separately (Molina et al., 2014; Motzer et al., 2015a). Five of the FDA-approved drugs for ccRCC are approved only as a part of combination treatment (Table 1). Moreover, five separate combinations of ICB/VEGF inhibitors have greater benefits in regards to PFS, overall survival (OS), and response rates, depending on the combination, compared to sunitinib alone (Motzer et al., 2018; Motzer et al., 2019; Rini et al., 2019b; Choueiri et al., 2021; Motzer et al., 2021).
Several more studies are currently underway to continue pushing this paradigm by utilizing a small-molecule HIF-2α inhibitor and a tyrosine kinase inhibitor at once, looking at the combination of PT2385 and nivolumab, PT2385 and cabozantinib, and PT2977 and cabozantinib (NCT02293980, NCT03634540). The campaign to find more effective and new combinations of already established and verified drugs lives on and will continue to be a valuable resource in oncology.
Synthetic lethality - the approach of looking for and targeting genes which have become essential for a cell after VHL loss - is another leading strategy in fighting kidney cancer. This plan is appealing for its cytotoxicity, specificity, and its capacity to evade acquired ccRCC resistance. At this time, several compounds have been found to be selectively deadly to VHL-absent ccRCC. STF-62247 was found in a compound screen, and is selectively lethal to in vivo and in vitro ccRCC cells via a HIF-independent autophagy-inducing mechanism (Turcotte et al., 2008). The authors also discovered another compound, STF-31, which possessed the same synthetic lethality and decreased tumor growth in mice in vivo (Chan et al., 2011). STF-31 directly binds and inhibits glucose transport protein, GLUT1 (encoded by the gene SLC2A1s). GLUT1 is overexpressed in ccRCCs, and SLC2A1s is upregulated by HIF factors, leading to a differential dependency on glycolysis (Chan et al., 2011).
Thompson et al. conducted another screen to find that inhibitors of ROCK1 had a cytotoxic effect inhibiting in vivo mouse tumor growth in VHL-deficient ccRCC but only under hypoxia, suggesting that it utilizes a HIF-dependent mechanism of action (Thompson et al., 2017). These same researchers have also separately found that some statins (HMG-CoA reductase inhibitors) are lethally synthetic due to their inhibition of mevalonate synthesis (Thompson et al., 2018).
A third team conducted lethality screens in fly and human cancer lines and discovered that the inhibition of both CDK4 and CDK6, in combination with VHL, possessed a synthetic relationship independent of HIFs (even though HIF-2α is known to induce CDK partner cyclin D1) (Nicholson et al., 2019). When a CDK4/6 dual inhibitor (palbociclib) was combined with HIF-2α small-molecule inhibitor PT-2399, they operated synergistically and by some metrics outperformed the use of either drug individually in mice xenografts (Nicholson et al., 2019). Previously, this team had found that shRNA inhibition of CDK6 alone, MET, and MAP2K1/MEK1 each preferentially inhibited two different VHL-deficient ccRCC lines in a partially HIF-independent fashion, and a dual CDK4/6 inhibitor limited in vitro growth in these lines (Bommi-Reddy et al., 2008). This follows multiple studies showing inhibitors ademaciclib and palbociclib had deleterious effects on ccRCC, although the particular mechanism of action was not yet definitively proven (Nicholson et al., 2019). This same Dana-Farber Cancer Institute group has also discovered that HIF drives many histone lysine demethylases and is hyperdependent on some portions of this change in expression; loss of EZH1 – a H3K27 methyltransferase – was synthetically lethal (Chakraborty et al., 2017).
Omacetaxine mepusuccinate (homoharringtonine) had already been approved by the FDA to treat chronic myeloid leukaemia before it was found to be synthetically lethal with ccRCC, making it an especially interesting compound to consider (Wolff et al., 2015).
Yet another screen found that the selenocysteine biosynthesis pathway was dysregulated in ccRCC; five of six genes in the pathway had depleted sgRNAs targeting them in VHL-absent ccRCC compared to cells with VHL (Sun et al., 2019). The researchers also found that the DNA damage response (DDR) was a key dysregulated player in cancer; loss of pVHL leads to genetic instability and the upregulation of DDR to repair and maintain the cancer cell’s DNA. Knockout of several genes in both the selenocsysteine and DDR pathways were found to be synthetically lethal (Sun et al., 2019). Further studies have found that DNA repair and the SWI/SNF chromatin remodeling complex inhibitors are synthetically lethal to PBRM1-deficient cancers, adding another layer of complexity and discernment to the treating of various strains of kidney cancer (Sasaki and Ogiwara, 2020; Chabanon et al., 2021).
Lastly, we have found that TBK1 is hyper-activated in ccRCC, independently of HIF, upon pVHL loss or under hypoxic conditions, and tumors from patients revealed that TBK1 phosphorylation is notably increased (Hu et al., 2020). Conversely, a direct interaction with pVHL mediated by EglN-mediated hydroxylation was established to lead to lower levels of phosphorylation observed in the wild-type. The loss of TBK1 via targeted sgRNAs or shRNAs led to decreased cell proliferation and growth defects in VHL-null cells while leaving VHL-proficient cells unaffected – the hallmark of synthetic lethality. TBK1 was previously known to be an innate immune function actor, but the mechanism of action in this case was not related to these pathways. TBK1 was discovered to phosphorylate and contribute to stabilization of p62/SQSTM1 (an oncogene overexpressed in kidney cancer), a crucial protein for cell proliferation. This and many other synthetic lethal targets will hopefully lead to better outcomes for patients, laying low tumor cells while leaving normal cells unscathed. In the mean while, the strategy of finding lethal partners for ccRCC will continue to consist of the repetitive methods described above (screening tens of thousands of compounds) until computational technology acquires the ability to meaningfully induce possible drug candidates (Murali et al., 2021).
Conclusion
Our ability to treat kidney cancer has only truly advanced insofar as our knowledge of the underlying causes and impacts of the disease has greatly improved. The implications of this work lie far beyond a single type of renal cell carcinoma; hypoxia is both a normal response to a common deficiency and a condition found in most solid cancers. These common, ubiquitous cellular pathways have proven connotations to other cancers and dysfunctions and will continue to be a deserved subject of research going forward. With all of the excitement to be found in the last decade in treating this former apex predator, we remain cautiously optimistic that this coming decade will lead to more interesting discoveries and better outcomes for patients using the strategies and therapeutics described above.
Acknowledgments
The authors would like to extend thanks unto the fellow members of the Zhang lab. We regret the non-inclusion of other extraordinary studies not summarized or cited in this review due to space limitations. Research from the Zhang lab is supported by the Cancer Prevention and Research Institute of Texas (CPRIT, RR190058), the American Cancer Society (ACS) Research Scholar Award (RSG-18-059-01-TBE), the National Cancer Institute (R01CA211732), and the Department of Defense Kidney Cancer Research Program (KCRP) Idea Development Award (W81XWH1910813).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 2020. Cancer Facts & Figures 2020. American Cancer Society, Atlanta. [Google Scholar]
- Bobbin ML, Rossi JJ, 2016. RNA interference (RNAi)-based therapeutics: delivering on the promise? Annual review of pharmacology and toxicology 56, 103–122. [DOI] [PubMed] [Google Scholar]
- Bommi-Reddy A, Almeciga I, Sawyer J, Geisen C, Li W, Harlow E, Kaelin WG, Grueneberg DA, 2008. Kinase requirements in human cells: III. Altered kinase requirements in VHL−/− cancer cells detected in a pilot synthetic lethal screen. Proceedings of the National Academy of Sciences 105(43), 16484–16489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciello F, Al-Ejeh F, Kelly G, Brennan DJ, Ngiow SF, Young A, Stoll T, Windloch K, Hill MM, Smyth MJ, 2017. G9a drives hypoxia-mediated gene repression for breast cancer cell survival and tumorigenesis. Proceedings of the National Academy of Sciences 114(27), 7077–7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabanon RM, Morel D, Eychenne T, Colmet-Daage L, Bajrami I, Dorvault N, Garrido M, Meisenberg C, Lamb A, Ngo C, 2021. PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Research. [DOI] [PubMed]
- Chakraborty AA, Nakamura E, Qi J, Creech A, Jaffe JD, Paulk J, Novak JS, Nagulapalli K, McBrayer SK, Cowley GS, 2017. HIF activation causes synthetic lethality between the VHL tumor suppressor and the EZH1 histone methyltransferase. Science translational medicine 9(398). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi J-T, Wu J, Solow-Cordero DE, 2011. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Science translational medicine 3(94), 94ra70–94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Liu F, Li H, Archacki S, Gao M, Liu Y, Liao S, Huang M, Wang J, Yu S, 2015. pVHL interacts with Ceramide kinase like (CERKL) protein and ubiquitinates it for oxygen dependent proteasomal degradation. Cellular signaling 27(11), 2314–2323. [DOI] [PubMed] [Google Scholar]
- Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, 2016. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539(7627), 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zheng R, Baade PD., Zhang S., Zeng H.,Bray F.,Jemal A.,Yu XQ., He J., 2016. Cancer statistics in China, 2015. CA: a cancer journal for clinicians 66(2), 115–132. [DOI] [PubMed] [Google Scholar]
- Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, Hammers HJ, Donskov F, Roth BJ, Peltola K, 2016. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. The Lancet Oncology 17(7), 917–927. [DOI] [PubMed] [Google Scholar]
- Choueiri TK, Halabi S, Sanford BL, Hahn O, Michaelson MD, Walsh MK, Feldman DR, Olencki T, Picus J, Small EJ, 2017. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: the alliance A031203 CABOSUN trial. Journal of clinical oncology 35(6), 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choueiri TK, Motzer RJ, 2017. Systemic therapy for metastatic renal-cell carcinoma. New England Journal of Medicine 376(4), 354–366. [DOI] [PubMed] [Google Scholar]
- Choueiri TK, Motzer RJ, Rini BI, Haanen J, Campbell M, Venugopal B, Kollmannsberger C, Gravis-Mescam G, Uemura M, Lee J, 2020a. Updated efficacy results from the JAVELIN Renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Annals of Oncology 31(8), 1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choueiri TK, Plimack ER, Bauer TM, Merchan JR, Papadopoulos KP, McDermott DF, Michaelson MD, Appleman LJ, Thamake S, Zojwalla NJ, 2020b. Phase I/II study of the oral HIF-2 α inhibitor MK-6482 in patients with advanced clear cell renal cell carcinoma (RCC). American Society of Clinical Oncology. [Google Scholar]
- Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, Oyervides Juárez VM, Hsieh JJ, Basso U, Shah AY, 2021. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. New England Journal of Medicine 384(9), 829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, Zojwalla NJ, Lowe AM, Wang K, Wallace EM, 2018. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. Journal of clinical oncology 36(9), 867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Ma Y, de Leon AD, Christie A, Xie Z, Woolford L, Singla N, Joyce A, Hill H, Madhuranthakam AJ, 2020. HIF-2 complex dissociation, target inhibition, and acquired resistance with PT2385, a first-in-class HIF-2 inhibitor, in patients with clear cell renal cell carcinoma. Clinical Cancer Research 26(4), 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossey PA, Foster K, Richards FM, Phipps ME, Latif F, Tory K, Jones MH, Bentley E, Kumar R, Lerman MI, 1994. Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours. Human genetics 93(1), 53–58. [DOI] [PubMed] [Google Scholar]
- Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, 2010. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463(7279), 360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias R, Zhang Q, Brugarolas J, 2020. The von Hippel-Lindau Tumor Suppressor Gene: Implications and Therapeutic Opportunities. The Cancer Journal 26(5), 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, 2007. Sorafenib in advanced clear-cell renal-cell carcinoma. New England Journal of Medicine 356(2), 125–134. [DOI] [PubMed] [Google Scholar]
- Feldman DR, Baum MS, Ginsberg MS, Hassoun H, Flombaum CD, Velasco S, Fischer P, Ronnen E, Ishill N, Patil S, 2009. Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma. Journal of clinical oncology 27(9),1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Manola JB, Pins M, McDermott DF, Atkins MB, Dutcher JJ, George DJ, Margolin KA, DiPaola RS, 2015. Best: a randomized phase ii study of vascular endothelial growth factor, RAF kinase, and mammalian target of rapamycin combination targeted therapy with bevacizumab, sorafenib, and temsirolimus in advanced renal cell carcinoma—a trial of the ECOG– ACRIN cancer research group (E2804). Journal of clinical oncology 33(21), 2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y-F, Cohn S, Christie A, McKenzie T, Wolff N, Do QN, Madhuranthakam AJ, Pedrosa I, Wang T, Dey A, 2017. Modeling renal cell carcinoma in mice: Bap1 and Pbrm1 inactivation drive tumor grade. Cancer discovery 7(8), 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Chakraborty AA, Liu P, Gan W, Zheng X, Inuzuka H, Wang B, Zhang J, Zhang L, Yuan M, 2016. pVHL suppresses kinase activity of Akt in a proline-hydroxylation–dependent manner. Science 353(6302), 929–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N, 2001. Expression and characterization of hypoxia-inducible factor (HIF)-3α in human kidney: suppression of HIF-mediated gene expression by HIF-3α. Biochemical and biophysical research communications 287(4), 808–813. [DOI] [PubMed] [Google Scholar]
- Heir P, Srikumar T, Bikopoulos G, Bunda S, Poon BP, Lee JE, Raught B, Ohh M, 2016. Oxygen-dependent regulation of erythropoietin receptor turnover and signaling. Journal of Biological Chemistry 291(14), 7357–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Xie H, Liu X, Potjewyd F, James LI, Wilkerson EM, Herring LE, Xie L, Chen X, Cabrera JC, 2020. TBK1 is a synthetic lethal target in cancer with VHL loss. Cancer discovery 10(3), 460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, 2007. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. New England Journal of Medicine 356(22), 2271–2281. [DOI] [PubMed] [Google Scholar]
- Iliopoulos O, Kibel A, Gray S, Kaelin WG, 1995. Tumour suppression by the human von Hippel-Lindau gene product. Nature medicine 1(8), 822–826. [DOI] [PubMed] [Google Scholar]
- Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan V, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ, 2020. Phase II study of the oral HIF-2α inhibitor MK-6482 for Von Hippel-Lindau disease–associated renal cell carcinoma. American Society of Clinical Oncology.
- Jubb A, Pham T, Hanby A, Frantz G, Peale F, Wu T, Koeppen H, Hillan K, 2004. Expression of vascular endothelial growth factor, hypoxia inducible factor 1α, and carbonic anhydrase IX in human tumours. Journal of clinical pathology 57(5), 504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, 2002. Molecular basis of the VHL hereditary cancer syndrome. Nature Reviews Cancer 2(9), 673–682. [DOI] [PubMed] [Google Scholar]
- Kapur P, Peña-Llopis S, Christie A, Zhrebker L, Pavía-Jiménez A, Rathmell WK, Xie X-J, Brugarolas J, 2013. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. The lancet oncology 14(2), 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneth NS, Rocha S, 2008. Regulation of gene expression by hypoxia. Biochemical Journal 414(1), 19–29. [DOI] [PubMed] [Google Scholar]
- Kucejova B, Peña-Llopis S, Yamasaki T, Sivanand S, Tran TAT, Alexander S, Wolff NC, Lotan Y, Xie X-J, Kabbani W, 2011. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Molecular cancer research 9(9), 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latif F, Tory K, Gnarra J, Yao M, Duh F-M, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, 1993. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260(5112), 1317–1320. [DOI] [PubMed] [Google Scholar]
- Lee DC, Sohn HA, Park Z-Y, Oh S, Kang YK, Lee K.-m, Kang M., Jang YJ., Yang S-J., Hong YK., 2015. A lactate-induced response to hypoxia. Cell 161(3), 595–609. [DOI] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu Y-T, Grishin NV, 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347(6227). [DOI] [PubMed] [Google Scholar]
- Liu X-D, Kong W, Peterson CB, McGrail DJ, Hoang A, Zhang X, Lam T, Pilie PG, Zhu H, Beckermann KE, 2020. PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nature communications 11(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Simon JM, Xie H, Hu L, Wang J, Zurlo G, Fan C, Ptacek TS, Herring L, Tan X, 2020. Genome-wide screening identifies SFMBT1 as an oncogenic driver in cancer with VHL loss. Molecular cell 77(6), 1294–1306. e1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH, 2003. von Hippel-Lindau disease. The Lancet 361(9374), 2059–2067. [DOI] [PubMed] [Google Scholar]
- Maher E, Yates J, Harries R, Benjamin C, Harris R, Moore A, Ferguson-Smith M, 1990. Clinical features and natural history of von Hippel-Lindau disease. QJM: An International Journal of Medicine 77(2), 1151–1163. [DOI] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bossé D, Wankowicz SM, Cullen D, 2018. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359(6377), 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JH, O’Brien T, Martincorena I, Tarpey P, Angelopoulos N, Yates LR, 2018. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell 173(3), 611–623. e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina AM, Hutson TE, Larkin J, Gold AM, Wood K, Carter D, Motzer R, Michaelson MD, 2014. A phase 1b clinical trial of the multi-targeted tyrosine kinase inhibitor lenvatinib (E7080) in combination with everolimus for treatment of metastatic renal cell carcinoma (RCC). Cancer chemotherapy pharmacology 73(1), 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore L, Jaeger E, Nickerson M, Brennan P, De Vries S, Roy R, Toro J, Li H, Karami S, Lenz P, 2012. Genomic copy number alterations in clear cell renal carcinoma: associations with case characteristics and mechanisms of VHL gene inactivation. Oncogenesis 1(6), e14–e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer R, Alekseev B, Rha S-Y, Porta C, Eto M, Powles T, Grünwald V, Hutson TE, Kopyltsov E, Méndez-Vidal MJ, 2021. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. New England Journal of Medicine. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Escudier B, McDermott DF, Frontera OA, Melichar B, Powles T, Donskov F, Plimack ER, Barthélémy P, Hammers HJ, 2020. Survival outcomes and independent response assessment with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma: 42-month follow-up of a randomized phase 3 clinical trial. Journal for immunotherapy of cancer 8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, 2008. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. The Lancet 372(9637), 449–456. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S, Oudard S, Gore ME, Tarazi J, Hariharan S, 2013a. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. The Lancet Oncology 14(6), 552–562. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, Nathan P, Staehler M, de Souza P, Merchan JR, 2013b. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. New England Journal of Medicine 369(8), 722–731. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, Jassem J, Zolnierek J, Maroto JP, Mellado B, 2015a. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. New England Journal of Medicine 16(15), 1473–1482. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, 2007. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. New England Journal of Medicine 356(2), 115–124. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, Venugopal B, Kollmannsberger C, Negrier S, Uemura M, 2019. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. New England Journal of Medicine 380(12), 1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S, Logan TF, 2015b. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. Journal of clinical oncology 33(13), 1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Tannir NM, McDermott DF, Frontera OA, Melichar B, Choueiri TK, Plimack ER, Barthélémy P, Porta C, George S, 2018. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. New England Journal of Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali VS, Cobanoglu DA, Hsieh M, Zinn M, Malladi VS, Gesell J, Williams NS, Welf ES, Raj GV, Cobanoglu MC, 2021. Cancer drug discovery as a low rank tensor completion problem. bioRxiv.
- Na X, Duan HO, Messing EM, Schoen SR, Ryan CK, di SanťAgnese PA, Golemis EA, Wu G, 2003. Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel—Lindau protein. The EMBO journal 22(16), 4249–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaigawa N, Yao M, Baba M, Kato S, Kishida T, Hattori K, Nagashima Y, Kubota Y, 2006. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer research 66(7), 3699–3705. [DOI] [PubMed] [Google Scholar]
- Nicholson HE, Tariq Z, Housden BE, Jennings RB, Stransky LA, Perrimon N, Signoretti S, Harris IS, Endress JE, Kaelin WG, 2019. HIF-independent synthetic lethality between CDK4/6 inhibition and VHL loss across species. Science signaling 12(601). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, 2008. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clinical cancer research 14(15), 4726–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura F, Uematsu K, Byrne SD, Hirano M, Joo-Okumura A, Nishikimi A, Shuin T, Fukui Y, Nakatsukasa K, Kamura T, 2016. Parallel regulation of von Hippel-Lindau disease by pVHL-mediated degradation of B-Myb and hypoxia-inducible factor α. Molecular cellular biology 36(12), 1803–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S, Christie A, Xie X-J, Brugarolas J, 2013. Cooperation and antagonism among cancer genes: the renal cancer paradigm. Cancer research 73(14), 4173–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li J-L, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ, 2005. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Molecular cellular biology 25(13), 5675–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini BI, Appleman LJ, Figlin RA, Plimack ER, Merchan JR, Wang K, Thamake S, Zojwalla NJ, Choueiri TK, McDermott DF, 2019a. Results from a phase I expansion cohort of the first-in-class oral HIF-2α inhibitor PT2385 in combination with nivolumab in patients with previously treated advanced RCC. Journal of clinical oncology. [Google Scholar]
- Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, Pouliot F, Alekseev B, Soulières D, Melichar B, 2019b. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. New England Journal of Medicine 380(12), 1116–1127. [DOI] [PubMed] [Google Scholar]
- Santoni M, Pantano F, Amantini C, Nabissi M, Conti A, Burattini L, Zoccoli A, Berardi R, Santoni G, Tonini G, 2014. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochimica et Biophysica Acta -Reviews on Cancer 1845(2), 221–231. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Ogiwara H, 2020. Synthetic lethal therapy based on targeting the vulnerability of SWI/SNF chromatin remodeling complex- deficient cancers. Cancer science 111(3), 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann TH, Li Q, Ma H-W, Key J, Zhang L, Chen R, Garcia JA, Naidoo J, Longgood J, Frantz DE, 2013. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nature chemical biology 9(4), 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, Gardner KH, 2009. Artificial ligand binding within the HIF2α PAS-B domain of the HIF2 transcription factor. Proceedings of the National Academy of Sciences 106(2), 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura I, Lange C, Knevels E, Moskalyuk A, Pulizzi R, Eelen G, Chaze T, Tudor C, Boulegue C, Holt M, 2016. The oxygen sensor PHD2 controls dendritic spines and synapses via modification of filamin A. Cell reports 14(11), 2653–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, 2007. Hypoxia-inducible factor 1 (HIF-1) pathway. Science's STKE 2007(407), cm8–cm8. [DOI] [PubMed] [Google Scholar]
- Shen C, Beroukhim R, Schumacher SE, Zhou J, Chang M, Signoretti S, Kaelin WG, 2011. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer discovery 1(3), 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjölund J, Johansson M, Manna S, Norin C, Pietras A, Beckman S, Nilsson E, Ljungberg B, Axelson H, 2008. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. The Journal of clinical investigation 118(1), 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strowd RE, Ellingson BM, Wen PY, Ahluwalia MS, Piotrowski AF, Desai AS, Clarke JL, Lieberman FS, Desideri S, Nabors LB, 2019. Safety and activity of a first-in-class oral HIF2-alpha inhibitor, PT2385, in patients with first recurrent glioblastoma (GBM). Journal of clinical oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers J, Cohen MH, Keegan P, Pazdur R, 2010. FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma. The oncologist 15(1), 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Petiwala S, Lu C, Hutti JE, Hu M, Hu M, Domanus MH, Mitra D, Addo SN, Miller CP, 2019. VHL synthetic lethality signatures uncovered by genotype-specific CRISPR-Cas9 screens. The CRISPR journal 2(4), 230–245. [DOI] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F, 2021. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians n/a(n/a). 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Thompson JM, Alvarez A, Singha MK, Pavesic MW, Nguyen QH, Nelson LJ, Fruman DA, Razorenova OV, 2018. Targeting the mevalonate pathway suppresses VHL-deficient CC-RCC through an HIF-dependent mechanism. Molecular cancer therapeutics 17(8), 1781–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JM, Nguyen QH, Singh M, Pavesic MW, Nesterenko I, Nelson LJ, Liao AC, Razorenova OV, 2017. Rho-associated kinase 1 inhibition is synthetically lethal with von Hippel-Lindau deficiency in clear cell renal cell carcinoma. Oncogene 36(8), 1080–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, Nicol D, O’Brien T, Larkin J, Horswell S, 2018a. Tracking cancer evolution reveals constrained routes to metastases: TRACERx renal. Cell 173(3), 581–594. e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, O’Brien T, Lopez JI, Watkins TB, Nicol D, 2018b. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell 173(3), 595–610. e511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ, 2008. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer cell 14(1), 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tykodi SS, Donskov F, Lee J-L, Szczylik C, Malik J, Alekseev BY, Larkin JM, Matveev VB, Gafanov R, Tomczak P, 2019. First-line pembrolizumab (pembro) monotherapy in advanced clear cell renal cell carcinoma (ccRCC): Updated results for KEYNOTE-427 cohort A. Journal of clinical oncology. [Google Scholar]
- Wang S-S, Gu Y-F, Wolff N, Stefanius K, Christie A, Dey A, Hammer RE, Xie X-J, Rakheja D, Pedrosa I, 2014. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proceedings of the National Academy of Sciences 111(46), 16538–16543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehn PM, Rizzi JP, Dixon DD, Grina JA, Schlachter ST, Wang B, Xu R, Yang H, Du X, Han G, 2018. Design and activity of specific hypoxia-inducible factor-2α (HIF-2α) inhibitors for the treatment of clear cell renal cell carcinoma: discovery of clinical candidate (S)-3-((2, 2-difluoro-1-hydroxy-7-(methylsulfonyl)-2, 3-dihydro-1 H-inden-4-yl) oxy)-5-fluorobenzonitrile (PT2385). Journal of medicinal chemistry 61(21), 9691–9721. [DOI] [PubMed] [Google Scholar]
- Wolff NC, Pavia-Jimenez A, Tcheuyap VT, Alexander S, Vishwanath M, Christie A, Xie X-J, Williams NS, Kapur P, Posner B, 2015. High-throughput simultaneous screen and counterscreen identifies homoharringtonine as synthetic lethal with von Hippel-Lindau loss in renal cell carcinoma. Oncotarget 6(19), 16951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SC, Cheng W, Hamilton H, Nicholas AL, Wakefield DH, Almeida A, Blokhin AV, Carlson J, Neal ZC, Subbotin V, 2018. HIF2α-targeted RNAi therapeutic inhibits clear cell renal cell carcinoma. Molecular cancer therapeutics 17(1), 140–149. [DOI] [PubMed] [Google Scholar]
- Wu D, Potluri N, Lu J, Kim Y, Rastinejad F, 2015. Structural integration in hypoxia-inducible factors. Nature 524(7565), 303–308. [DOI] [PubMed] [Google Scholar]
- Wu D, Su X, Lu J, Li S, Hood BL, Vasile S, Potluri N, Diao X, Kim Y, Khorasanizadeh S, 2019. Bidirectional modulation of HIF-2 activity through chemical ligands. Nature chemical biology 15(4), 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Contratto M, Shanbhogue KP, Manji GA, O’Neil BH, Noonan A, Tudor R, Lee R, 2019. Evaluation of a locked nucleic acid form of antisense oligo targeting HIF-1α in advanced hepatocellular carcinoma. World journal of clinical oncology 10(3), 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Liu P, Wei W, 2014. mTOR signaling in tumorigenesis. Biochimica et Biophysica Acta Reviews on Cancer 1846(2), 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, Wang B, Wehn PM, Yang H, Dixon DD, 2019. 3-[(1 S, 2 S, 3 R)-2, 3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl] oxy-5-fluorobenzonitrile (PT2977), a hypoxia-inducible factor 2α (HIF-2α) inhibitor for the treatment of clear cell Renal cell carcinoma. Journal of medicinal chemistry. [DOI] [PubMed] [Google Scholar]
- Yin H, Zheng L, Liu W, Zhang D, Li W, Yuan L, 2017. Rootletin prevents Cep68 from VHL-mediated proteasomal degradation to maintain centrosome cohesion. Biochimica et Biophysica Acta - Molecular Cell Research 1864(4), 645–654. [DOI] [PubMed] [Google Scholar]
- Zbar B, Brauch H, Talmadge C, Linehan M, 1987. Loss of alleles of loci on the short arm of chromosome 3 in renal cell carcinoma. Nature 327(6124), 721–724. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wu T, Simon J, Takada M, Saito R, Fan C, Liu X-D, Jonasch E, Xie L, Chen X, 2018. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 361(6399), 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Yan Q, Yang H, Wei W, 2019. Oxygen sensing and adaptability won the 2019 Nobel Prize in Physiology or medicine. Genes & diseases 6(4), 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Liu X-D, Sun M, Zhang X, German P, Bai S, Ding Z, Tannir N, Wood CG, Matin SF, 2016. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 35(21), 2687–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]