

Abstract
Background & Aims:
Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase upregulated in many tumor types and a promising target for cancer therapy. Here, we elucidated the functional role of FAK in intrahepatic cholangiocarcinoma (iCCA) development and progression.
Methods:
Expression levels and activation status of FAK were determined in human iCCA samples. The functional contribution of FAK to Akt/YAP murine iCCA initiation and progression was investigated using conditional Fak KO mice and constitutive Cre or inducible Cre mice, respectively. The oncogenic potential of FAK was further examined via overexpression of FAK in mice. In vitro cell line studies and in vivo drug treatment were applied to address the therapeutic potential of targeting FAK for iCCA treatment.
Results:
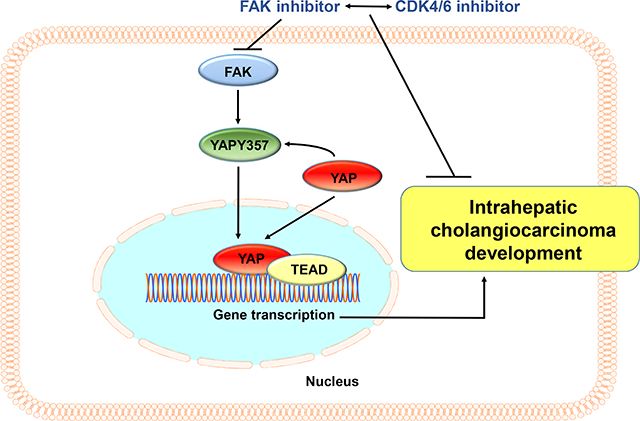
FAK was ubiquitously upregulated and activated in iCCA lesions. Ablation of FAK strongly delayed Akt/YAP driven mouse iCCA initiation. FAK overexpression synergized with activated AKT to promote iCCA development and accelerated Akt/Jag1-driven cholangiocarcinogenesis. Mechanistically, FAK was required for YAP(Y357) phosphorylation, supporting the role of FAK as a central YAP regulator in iCCA. Significantly, ablation of FAK after Akt/YAP-dependent iCCA formation strongly suppressed tumor progression in mice. Furthermore, a remarkable iCCA growth reduction was achieved when a FAK inhibitor and Palbociclib, a CDK4/6 inhibitor, were administered simultaneously in human iCCA cell lines and Akt/YAP mice.
Conclusions:
FAK activation contributes to the initiation and progression of iCCA by inducing the YAP protooncogene. Targeting FAK, either alone or in combination with anti-CDK4/6 inhibitors, may be an effective strategy for iCCA treatment.
Keywords: intrahepatic cholangiocarcinoma, FAK, YAP, targeted therapy, iCCA
Lay summary
We found that Focal Adhesion Kinase (FAK) is upregulated and activated in human and mouse intrahepatic cholangiocarcinoma (iCCA) samples. FAK promotes iCCA development, whereas deletion of FAK strongly suppresses iCCA initiation and progression. Mechanistically, we discovered that FAK regulates the YAP pathway. Combined FAK and CDK4/6 inhibitor treatment is highly detrimental for the growth of in vitro and in vivo iCCA models. This combination therapy might represent a valuable and novel treatment against human iCCA.
Graphical Abstract
Introduction
Intrahepatic cholangiocarcinoma (iCCA) is the second most frequent primary liver tumor with few treatment options.1 A detailed portrait and understanding of the molecular mechanisms leading to iCCA development and progression remain an unmet need.
Focal adhesion kinase (FAK), encoded by the PTK2 gene, is a non-receptor protein tyrosine kinase.2 Overexpression and activation of FAK are associated with the progression of many tumor types by modulating cell survival, proliferation, and invasion,3 thus representing a promising target for cancer therapy.4,5 Several FAK inhibitors have been tested in clinical trials, with preliminary results showing cytostatic effects as single agents.5 Studies on the involvement of FAK in cholangiocarcinogenesis have been conducted previously.6,7 However, comprehensive research on the role of FAK in iCCA development and progression is missing.
Yes-associated protein (YAP) is a main downstream effector of the Hippo pathway. YAP’s cellular localization controls its activity: cytoplasmic YAP is indeed recruited to the disruption complex, ubiquitinated, and degraded,8 whereas nuclear YAP induces gene transcription in association with the TEA domain transcription factor (TEAD) DNA binding proteins.8,9 Previous studies demonstrated that the nuclear-cytoplasmic translocation of YAP depends on its phosphorylation status8. Specifically, YAP S127 and S397 sites are negative regulators of YAP nuclear localization, whereas phosphorylation at the Y357 (p-YAPY357) residue stabilizes YAP and promotes nuclear localization.10 YAP plays a pivotal role in iCCA, where its overexpression and nuclear localization are ubiquitous.11 Silencing of YAP inhibits iCCA cell growth in vitro,12 and overexpression of the activated form of YAP, which is modified at 127 site (YapS127A) and cannot be targeted for proteasomal degradation, cooperates with activated AKT to promote iCCA formation in mice.11 Notably, several observations suggest that FAK mediates YAP nuclear translocation and induces its full activation.13,14 The possible molecular crosstalk between FAK and YAP has not been investigated in iCCA.
Here, we evaluated FAK expression in human iCCA and its functional contribution to cholangiocarcinogenesis using in vitro and in vivo models.
Materials and Methods
Mouse experiments
Wild-type (WT) FVB/N mice were purchased from Charles River Laboratories (Wilmington, MA). Fakf/f mice were provided by Dr. Hilary Beggs (University of California San Francisco, San Francisco, CA), and Yapf/f mice by Dr. Eric Olson (University of Texas Southwestern Medical Center, Dallas, TX). Detailed information is available in Table S1. Mice were housed and monitored under protocols approved by the Committee for Animal Research, UCSF.
Statistical analysis
Data were analyzed using the Prism 8.0 software (GraphPad, San Diego, CA) and presented as Means ± SD. A two-tailed unpaired t-test was conducted to compare two groups that achieved Gaussian distribution, or a non-parametric test was performed when the sample size was small. Welch correction and linear regression were applied when necessary. Kaplan–Meier method and log-rank test were used for survival analysis. P values < 0.05 were considered statistically significant.
Additional detailed materials and methods are available in the Supporting document.
Results
FAK is upregulated and activated in human and Akt/YAP iCCA lesions
First, we analyzed the levels of the PTK2 gene, encoding FAK, in the TCGA CHOL dataset.15 Among 36 iCCA analyzed, 3 samples demonstrated PTK2 amplification, and 1 harbored a missense mutation (H1033N) (Supplementary Figure 1A). PTK2 mRNA levels were upregulated in iCCA samples compared to the surrounding non-tumorous liver tissues (Figure 1A). The results were independently validated in the NCI iCCA dataset (Figure 1A),15,16, and our collection of human iCCA samples (Figure 2B). Using median expression as the cut-off, higher PTK2 mRNA expression correlated with a lower iCCA survival rate (Figure 1C and Supplementary Tables 2–4). This association remained strongly significant after multivariate Cox regression analysis (Supplementary Table 5), supporting PTK2 mRNA levels as a possible independent prognostic factor for iCCA. Using immunohistochemistry, positive immunoreactivity for total FAK and activated/phosphorylated (p-)FAKY397 was detected in most iCCA samples (45/50 and 42/50, respectively). In contrast, a faint/absent staining for total and p-FAKY397 characterized the non-tumorous surrounding livers (Supplementary Figures 2 and 3). Consistently, we retrieved the FAK activation signature from KEGG (FOCAL),17 and found that activation of the FAK signaling pathway is evident based on the TCGA CHOL dataset (Figure 1B).
Figure 1. FAK is activated in human iCCA patients and Akt/YAP-induced iCCA mouse lesions.

(A) PTK2 mRNA levels in human intrahepatic cholangiocarcinoma (iCCA) from The Cancer Genome Atlas (TCGA) and National Cancer Institute (NCI) datasets. (B) Quantitative real-time RT-PCR analysis of PTK2 expression in iCCA (n = 50) and corresponding non-tumorous surrounding liver tissues (ST; n = 50) from our cohort. (C) Kaplan–Meier survival curves of human iCCA specimens from our cohort with high and low PTK2 mRNA levels, showing the unfavorable outcome of patients with elevated expression of this gene. (D) GSEA analysis of FAK pathway-related proteins in iCCA from TCGA. (E-F) FAK activation in the Akt/YAP-induced iCCA mouse model.
Figure 2. Deletion of FAK suppresses iCCA development in Akt/YAP mice.

(A) Study design. (B) Survival analysis of Fakf/f mice bearing Akt/YAP/pCMV (n =15) and Akt/YAP/Cre (n = 10) tumors. (C) Liver weight of Akt/YAP/pCMV and Akt/YAP/Cre mice at 10 weeks post hydrodynamic injection. (D) Gross image and H&E staining of Akt/YAP/pCMV and Akt/YAP/Cre mouse livers. (E) Western blot analysis of iCCA tissues from Akt/YAP-pCMV and Cre Fakf/f mice. GAPDH was the loading control. (F) H&E and IHC staining of CK19 and HNF-4α in Akt/YAP/pCMV and Akt/YAP/Cre Fakf/f mice, respectively. CK19 staining was quantified as the percentage of the positive staining area of the whole tumor section area. HNF-4α positive cells were quantified as HNF-4α index. (G) Relative mRNA expressions of AFP and GPC3 were analyzed using the −ΔΔCt method and presented as mean ± SD. (H) Immunohistochemistry of Ki-67 and C-C-3 was performed in Akt/YAP/pCMV and Akt/YAP/Cre Fakf/f mice. Ki-67 and C-C-3 positive cells were quantified using the Image J. Tukey–Kramer test: at least P < 0.005; a, vs. Akt/YAP/pCMV; b, vs. Akt/YAP/Cre. Abbreviations: C-C-3, Cleaved Caspase 3; T, tumor; ST, surrounding tissue. Scale bar: 100 μm for 200×.
Previously, we established a mouse iCCA model induced by hydrodynamic transfection of activated forms of AKT (myr-AKT) and YAP (YAPS127A) (Akt/YAP).11 Using immunohistochemistry, p-FAKY397 was readily detected in Akt/YAP iCCA lesions (Figure 1C). We performed RNASeq analysis of Akt/YAP iCCA (Figure 1D). Gene expression signature analysis confirmed FAK cascade’s activation in Akt/YAP mouse iCCA (Figure 1D).
Altogether, the data demonstrate FAK activation in human and mouse iCCA.
FAK is required for iCCA growth in vitro and tumor initiation in vivo
We found that FAK is expressed in KKU-M213 and HuCC-T1 human iCCA cell lines. Culturing these cells on collagen triggered increased p-FAKY397 levels (Supplementary Figure 4A). PTK2 was silenced in the two cell lines using shPTK2, which effectively decreased FAK protein expression (Supplementary Figure 4B). FAK loss significantly suppressed the clonogenic capacity of both iCCA cell lines cultured in the two-dimensional culture matrix (Supplementary Figure 4B). Notably, shPTK2 decreased iCCA cell growth in Collagen I 3D growth condition (Supplementary Figure 4C). Mechanistically, FAK ablation induced the expression of apoptosis-related protein cleaved caspase 3 (Supplementary Figure 4D) but did not affect proliferation-associated proteins, including p-Rb, PCNA, and Cyclin A/D1/E. Accordingly, PTK2 silencing via specific small-interfering RNA resulted in a mild decline of proliferation but a robust induction of apoptosis in KKU-M213 and HuCCT-1 cells (Supplementary Figure 5A–C). These data suggest FAK is required for human iCCA cell growth in vitro, mainly via apoptosis suppression.
To unravel the functional significance of FAK in the regulation of Akt/YAP-driven cholangiocarcinogenesis in vivo, we simultaneously deleted FAK while overexpressing AKT and YAP oncogenes in the mouse liver using FAK conditional KO (Fakf/f) mice. In brief, Akt and YAP with pCMV-Cre (Akt/YAP/Cre) plasmids were injected into Fakf/f mice. As a control, additional Fakf/f mice were injected with Akt/YAP and pCMV empty vector (Akt/YAP/pCMV) (Figure 2A). In accordance with previous findings, 26 all control Akt/YAP/pCMV mice developed large abdominal masses and required euthanasia by 6–10 weeks post-injection. In striking contrast, loss of FAK strongly delayed Akt/YAP-driven liver tumor development. However, eventually all Akt/YAP/Cre injected Fakf/f mice became moribund and were harvested by 20 weeks post-injection (Figure 2B).
Grossly, when two cohorts of mice were harvested at 10 weeks post-injection, the liver tumor burden, estimated using total liver weight, was significantly lower in the Akt/YAP/Cre group (Figure 2C and 2D). Histologically, numerous tumor lesions occupied most of the liver parenchyma in Akt/YAP/pCMV mice at 10 weeks post-injection. At this time point, only small tumor lesions were appreciable instead in Akt/YAP/Cre mouse livers (Figure 2D). Western blot analysis confirmed FAK loss in Akt/YAP/Cre mice (Figure 2E).
Morphologically, Akt/YAP/pCMV tumors were classified as pure iCCA, as also substantiated by positive immunoreactivity for the biliary marker CK19 and negative immunolabeling for the hepatocellular marker HNF-4α (Figure 2F). Intriguingly, lesions consisted primarily of hepatic adenoma or HCC, but not iCCA, in Akt/YAP/Cre liver tissues. Even at 20 weeks post-injection, when large tumor lesions were present in Akt/YAP/Cre mouse livers, most lesions were HCC-like. This observation was confirmed by CK19 and HNF-4α immunohistochemistry (Figure 2F) and by qRT-PCR analysis of alpha-fetoprotein (Afp) and Glypican 3 (Gpc3) HCC markers (Figure 2G).
Loss of FAK did not affect liver tumor cell proliferation, as indicated by equal expression levels of PCNA and Cyclin A/B1/D1/E and Ki-67 immunohistochemistry in Akt/YAP/pCMV and Akt/YAP/Cre tumor lesions (Figure 2E and 2H). In contrast, cleaved caspase-3 levels significantly increased in Akt/YAP/Cre tumor tissues (Figure 2H), revealing augmented apoptosis in Akt/YAP lesions depleted of FAK.
Overall, the data indicate FAK requirement for iCCA in vitro growth and survival and Akt/YAP-induced iCCA formation in vivo. In these mice, loss of FAK delays tumor growth and alters the tumor phenotype.
FAK depletion represses YAP nuclear retention both in vitro and in vivo
Next, we investigated the mechanisms whereby FAK contributes to cholangiocarcinogenesis. YAP and one of its downstream effectors, the NOTCH pathway, play a pivotal role in biliary cell fate determination.11,18 Previous studies suggested FAK as a driver of YAP nuclear translocation.13,14 It is worth noting that in Akt/YAP induced iCCA model, we used the YAP (S127A) construct. There are additional phosphorylation sites of YAP, including YAPY357, which is necessary for YAP full activation.19
First, we tested whether FAK regulates YAP/NOTCH in vitro. Silencing of FAK in KKU-M213 and HuCC-T1 cells decreased nuclear YAP accumulation (Supplementary Figures 6A and 6B). Both shPTK2 and PND1186, a small FAK inhibitor, repressed p-YAPY357 expression and decreased the levels of YAP target genes, as well as the downstream NOTCH pathway in these cells (Supplementary Figures 6 and 7). Other YAP phosphorylation sites, including p-YAPS127 and p-YAPS397, did not show consistent changes.
In Akt/YAP mouse iCCA lesions, FAK deficiency did not affect the AKT or ERK signaling (Figure 3A and Supplementary Figure 8). Equivalent levels of ectopically expressed human YAP1 mRNA were detected in both cohorts of mouse liver tissues (Figure 3B), indicating the successful expression of the human YAP1 gene in these mice. In contrast, Akt/YAP/Cre tumors exhibited significantly lower total YAP and p-YAPY357 levels (Figure 3A and Supplementary Figure 8). Immunohistochemistry revealed intense nuclear YAP staining in Akt/YAP/pCMV iCCA lesions. In Akt/YAP/Cre liver tumors, YAP expression was weaker, and both nucleus and cytoplasm staining was appreciable, with a significantly lower percentage of nuclear-only positive staining (Figure 3C). A decrease of YAP downstream targets, including SOX9, NOTCH2, and Jagged1 proteins, also occurred (Figure 3A and Supplementary Figure 8). Moreover, downregulation of canonical YAP targets (Figure 3D) and NOTCH targets (Figure 3E) mRNA expression was observed in Akt/YAP/Cre tumor tissues.
Figure 3. YAP signaling inactivation in iCCA from FAK conditional knockout Akt/YAP mice.

(A) Western blot analysis of lysates from Akt/YAP/pCMV and Akt/YAP/Cre Fakf/f mice. (B) Levels of human Yap1 mRNA in Akt/YAP/pCMV and Akt/YAP/Cre mice. (C) Knockout of FAK reduced YAP nuclear translocation as shown by immunohistochemistry and the percentage of positive nuclear staining. (D) YAP and (E) NOTCH signaling inactivation at the transcriptional level, as assessed by qPCR. Tukey–Kramer test: at least P < 0.05; a, vs Akt/YAP-pCMV; b, vs Akt/YAP-Cre.
We further investigated the FAK and YAP interplay in human iCCA specimens. Using immunohistochemistry, we detected the concomitant activation of FAK and YAP (as indicated by its nuclear accumulation) in most iCCA samples (37/50, 74%) (Supplementary Figure 2). Furthermore, a direct correlation between the mRNA levels of PTK2 and YAP targets (CTGF, CYR61, NOTCH2, and JAG1), but not YAP mRNA, was detected (Supplementary Figure 9). The TCGA CHOL dataset revealed similar data (Supplementary Figure 10).
Overall, FAK ablation represses YAP activation by inhibiting p-YAPY357, leading to decreased YAP nuclear localization and activation. Simultaneous activation of FAK and YAP occurs in most human iCCA.
FAK overexpression synergizes with activated AKT to induce iCCA formation in mice
Next, we determined FAK oncogenic potential in vivo. Overexpression of FAK alone did not result in tumor development, consistent with previous findings.20 Given that activated AKT synergizes with YAP to induce iCCA development, and FAK can activate YAP, we hypothesized that overexpression of FAK and AKT might trigger cholangiocarcinogenesis. To test this hypothesis, FAK and myr-AKT plasmids were co-injected into the mouse liver (Akt/Fak mice). Previously, we showed that overexpression of myr-AKT results in tumor development by 28 weeks post plasmid injection.21 In Akt/Fak mice (Figure 4A), liver tumor nodules were detectable by ~15 weeks post-injection (Figure 4B). All mice developed a significant tumor burden and became moribund by 18–19 weeks post-injection (Figures 4B and 4C). Histologically, different from Akt mice, characterized almost exclusively by hepatocellular lesions,21 Akt/Fak tumors displayed a solid and ductular phenotype resembling human iCCA (Figure 4B). All tumor cells were positive for CK19 and Ki-67 (Figure 4D). Western blotting confirmed that AKT and FAK pathways were successfully activated in Akt/Fak iCCAs (Figure 4E). Intense cytoplasmic and robust nuclear immunoreactivity for YAP was detected in Akt/Fak iCCA lesions (Figure 4D). Similarly, protein levels of YAP and related downstream targets Jagged1 and NOTCH2 were significantly induced (Figure 4E). The qRT-PCR analysis revealed that YAP target genes, including Ctgf, Cyr61, and Axl, and the NOTCH pathway’s target genes (Hes1 and HeyL), were upregulated in Akt/Fak tumors (Figure 4F).
Figure 4. Co-expression of FAK and AKT leads to iCCA formation in mice.

(A) Study design. (B) Gross image and H&E staining of Akt/Fak mouse livers. (C) Liver weight of Akt/Fak mice at 18–19 weeks post-injection. (D) Immunohistochemistry of CK19, Ki-67, and YAP in Akt/Fak mice. (E) Western blot analysis of FVB mice and Akt/Fak mouse model liver tissues. (F) YAP and NOTCH signaling activation at the transcriptional level, as assessed by qPCR. Tukey–Kramer test: at least P < 0.05; a, vs FVB WT; b, vs Akt.
Altogether, the present findings indicate that FAK overexpression cooperates with activated AKT to promote iCCA development, an event accompanied by YAP activation, in the mouse liver.
FAK overexpression accelerates Akt/Jag1-induced iCCA development in mice
Previously, we established an iCCA model by co-expressing myr-AKT and the Notch ligand Jag1 in the mouse liver (Akt/Jag1 mice).22 Akt/Jag1 co-expression induces cystic-like iCCA lesions over long latency, characterized by low levels of YAP.22 We hypothesized that overexpression of FAK might accelerate Akt/Jag1-driven cholangiocarcinogenesis by activating YAP. Thus, FAK was overexpressed in AKT/Jag1 mice (Akt/Jag1/Fak mice). Additional AKT/Jag1 mice were injected with the pT3-EF1α empty vector as control (Akt/Jag1/PT3) (Figure 5A). Mice were harvested 10 weeks post-injection (Figure 5B). At this time point, liver tumor burden, estimated using total liver weight, was significantly higher in Akt/Jag1/Fak than in Akt/Jag1/PT3 mice (Figure 5B). Both gross images and histological analysis revealed only small cystic nodules in Akt/Jag1/PT3 mouse livers (Figure 5C), consistent with previous data.22 In Akt/Jag1/Fak mice, tumors showed solid and ductal phenotypes, similar to those described in the Akt/YAP iCCA model (Figure 5C).11 Tumor cells in both mouse cohorts were CK19 (+), with higher CK19 (+) areas in Akt/Jag1/Fak mice, supporting the higher iCCA burden (Figure 5C).
Figure 5. FAK accelerates tumor development in Akt/Jag 1 mice.

(A) Study design. (B) Akt/Jag1/pT3 and Akt/Jag1/Fak mice’s liver weight at ~10 weeks post hydrodynamic injection. (C) Gross image, H&E, CK19, and (D) Ki-67 staining in Akt/Jag1/PT3 and Akt/Jag1/Fak mice. Ki-67 positive cells were quantified using Image J software. (E) Representative Western Blot analysis of relative pathways in Akt/Jag1/PT3 and Akt/Jag1/Fak mice. (F) Immunohistochemistry of YAP in Akt/Jag1/PT3 and Akt/Jag1/Fak mice. (G) YAP and NOTCH signaling activation at the transcriptional level was assessed by qPCR. (H) Study design in Yapf/f mice. (I-J) Liver weight (I) and gross image, H&E, CK19, and Ki-67 staining (J) of Akt/Jag1/Fak-pCMV and -Cre Yapf/f mice ~9 weeks post-injection. Tukey–Kramer test: at least P < 0.005; (G) a, vs WT; b, vs Akt/Jag1/Fak/pCMV, (I) a, vs Akt/Jag1/Fak/pCMV.
Mechanistically, FAK overexpression triggered increased iCCA cell proliferation (Figured 5D), with augmented expression of proliferation-related proteins, including CCND1 and PCNA (Figure 5E). Western blotting confirmed increased total and activated/phosphorylated FAK levels in Akt/Jag1/Fak tumor lesions (Figure 5E). Notably, while Akt/Jag1/PT3 iCCA cells exhibited YAP cytoplasmic staining, intense cytoplasmic and nuclear YAP immunoreactivity characterized Akt/Jag1/Fak iCCA (Figure 5F). Nuclear accumulation of YAP was paralleled by increased p-YAPY357 and NOTCH2 levels (Figure 5E). Moreover, the qRT-PCR analysis revealed an increased expression of YAP and NOTCH target genes in Akt/Jag1/Fak tumors (Figure 5G).
To determine whether Akt/Jag1/Fak-driven iCCA development is YAP dependent, Akt/Jag1/Fak/pCMV (control) and Akt/Jag1/Fak/Cre plasmids were injected in Yapf/f mice (Figure 5H). Nine weeks post-injection, all mice were harvested. Strikingly, Yap ablation completely suppressed iCCA formation in Akt/Jag1/Fak mice at this time point (Figures 5I and 5J). While Akt/Jag1/Fak/Cre injected Yapf/f mice were healthy with no macroscopic liver nodules, all Akt/Jag1/Fak/pCMV injected mice exhibited a high tumor burden. Histological analysis revealed iCCA lesions in Akt/Jag1/Fak/pCMV injected mouse livers. In contrast, no malignant lesions developed in Akt/Jag1/Fak/Cre livers, where hepatocytes with enlarged clear cytoplasm owing to increased fat and glycogen storage induced by AKT activation were detected (Figure 5J).21
Overall, the data indicate that FAK overexpression accelerates Akt/Jag1-driven cholangiocarcinogenesis in a YAP-dependent manner.
iCCA progression in vivo requires an intact FAK
To assess the therapeutic potential of targeting FAK for iCCA treatment, we determined whether FAK ablation after Akt/YAP iCCA formation triggers tumor regression. Thus, we generated a sleeping beauty transposase-based vector with Tamoxifen-inducible Cre, i.e., CreERT2, under the control of the CK19 promoter (pT3-CK19pro-CreERT2) (Supplementary Figure 11A). The pT3-CK19pro-CreERT2 plasmid together with AKT and YAP constructs was injected into Fakf/f mice. Mice were aged until ~3.5 to 4 weeks post-injection when iCCA tumors are appreciable on the mouse surface (Figure 6A and Supplementary Figure 11B). A group of mice was harvested as a pretreatment cohort, and additional mice were either intraperitoneally injected with corn oil (control) or Tamoxifen (Supplementary Table 6). Tamoxifen administration allows the activation of the Cre recombinase and the subsequent deletion of FAK only in CK19(+) iCCA tumor cells. Significantly, deletion of FAK in Akt/YAP lesions profoundly inhibited tumor progression (Figure 6B). Indeed, while all corn oil-injected mice required euthanasia by 5–10 weeks post hydrodynamic injection, Tamoxifen-treated mice survived considerably longer (Figure 6B). In total, 10 Akt/YAP/CreERT2 mice were treated with Tamoxifen (Supplementary Table 6). Four mice (Group 1) developed a significant liver tumor burden and required euthanasia by 11–15 weeks post-injection (Figure 6C and Supplementary Figure 11B). Additional 6 mice (Group 2) appeared to be healthy, with 3 exhibiting low tumor burden when harvested at 10 weeks post-injection. The remaining 3 mice demonstrated low tumor burden even 17–20 weeks after hydrodynamic injection (Figures 6B and 6C). Upon dissection, limited tumor burden was observed in this mouse cohort. Indeed, few tumors were detected, and most lesions consisted of small cell clusters (Figure 6D and Supplementary Figure 11B). When assessing FAK by Western blotting, we found similar FAK levels in control and Tamoxifen-treated Group 1 mice (Figure 6E). In contrast, FAK protein was significantly decreased in Group 2 mice (Figure 6E). The results indicate that Tamoxifen failed to induce sufficiently elevated Cre activity in Group 1 mice, leading to the continuous growth of the iCCA lesions retaining FAK expression. To rule out the possibility that Tamoxifen treatment was responsible for the impaired tumor progression, we injected FVB/N mice with Akt/YAP/CreERT2 and repeated the experiments (Supplementary Figure 11C). As expected, Tamoxifen treatment did not trigger tumor growth inhibition in wild-type mice.
Figure 6. FAK downregulation after tumor formation using the CK19-CreERT2 Tamoxifen system inhibits tumor progression.

(A) Study design. (B) Survival analysis of Fakf/f mice bearing Akt/YAP/CreERT2 tumors treated with Tamoxifen or vehicle (Control). (C) Liver weight, (D) Gross image, H&E staining, HNF-4α, and CK19 immunohistochemistry of livers from pretreatment, control, and Tamoxifen groups. Arrows indicate tiny lesions consisting of small cell clusters, which were typically conserved in Tamoxifen-treated mice. (E) FAK Western blot analysis was performed in Fakf/f mice bearing Akt/YAP/CreERT2 tumors sensitized with Tamoxifen (Group2 in C) or not (Group1 in C). (F) Large areas of necrosis (N) in livers from Group2 mice, as shown at two magnifications. Abbreviations: T, tumor; ST, surrounding tissue.
We focused our analysis on Group 2 mice, demonstrating efficient Cre-mediated FAK deletion. Histological analysis and CK19 immunohistochemistry showed that Akt/YAP mice after Tamoxifen treatment displayed a lower tumor burden than pretreatment mice (Figure 6D). FAK ablation after iCCA formation did not result in HCC-like conversion of the lesions (Figure 6D). Notably, large areas of necrosis were detected only in Tamoxifen-treated livers (Figure 6F). YAP immunohistochemistry revealed nuclear YAP staining in iCCA lesions, indicating the preservation of YAP activity in these residual iCCA lesions after FAK deletion (Supplementary Figure 11D).
Altogether, the present data demonstrate that FAK ablation in already formed iCCA inhibits tumor progression, implying FAK as a potentially relevant therapeutic target in iCCA.
Combined FAK and CDK4/6 inhibition is detrimental for iCCA growth in vitro and in vivo
To explore FAK as a therapeutic target for iCCA treatment, we treated the KKU-M213 and HuCC-T1 cells with PND1186, a FAK inhibitor. PND1186 effectively inhibited the cell growth of both cell lines with an IC50 of 4.47μM and 11.1μM, respectively (Supplementary Figures 12A). We reasoned that FAK inhibitors might have limited efficacy for cancer treatment as single agents. Based on our RNASeq analysis of Akt/YAP murine iCCA treated with various drugs, Oxaliplatin/Gemcitabine combination11, MLN012811, and Palbociclib23 treatment had no substantial influence on FAK pathway enrichment in Akt/YAP tumors (Supplementary Figures 1B and 1C). Therefore, FAK inhibitors may synergize with one of these drugs, leading to more substantial tumor growth suppression. Previously, we showed the CDK4/6 pathway activation in mouse and human iCCA. The CDK4/6 inhibitor Palbociclib profoundly suppressed iCCA cell proliferation in vitro.23 Moreover, most human iCCA samples examined (35/50, 70%) displayed concomitant immunoreactivity for activated FAK and phosphorylated/inactivated (p-)pRb, a reliable marker of CDK4/6 activity and response to CDK4/6 inhibitors (Supplementary Figure 13).24 We hypothesized that FAK and anti-CDK4/6 inhibitors might synergize against iCCA. Thus, KKU-M213 and HuCC-T1 cells were treated with Palbociclib and PND1186, either alone or in combination. Compared to single treatments, concomitant administration of Palbociclib and PND1186 to iCCA cells induced decreased cell viability (Supplementary Figures 12B). The combination index (CI) was calculated,25 and all CI values were less than 1 in Palbociclib and PND1186 treatment groups (Supplementary Figure 12C and D), indicating a synergistic anti-tumor activity of Palbociclib/PND1186 co-administration. This assumption was confirmed by assessing proliferation and apoptosis rates in the same cell lines. Indeed, the Palbociclib/PND1186-treated cohort showed the highest reduction in proliferation and massive apoptosis in KKU-M213 and HuCC-T1 cells (Supplementary Figure 14A–D). Notably, PND1186 alone displayed a significantly higher pro-apoptotic potential than Palbociclib in both cell lines (Supplementary Figure 14C and D).
Subsequently, we tested the therapeutic potential of FAK inhibitor, either alone or in combination with Palbociclib, in the Akt/YAP iCCA model. We selected VS-6063 (defactinib) as the FAK inhibitor since it is orally bioavailable and currently tested in clinical trials for cancer therapy.26 Treatment with Palbociclib (100mg/kg) and VS-6063 (50mg/kg) was well-tolerated in mice. Akt/YAP tumor-bearing mice were randomly separated into 5 cohorts (Figure 7A). The first group was harvested 4.5 weeks post hydrodynamic injection as the pretreatment group. All mice exhibited moderate iCCA liver tumor burden at this point, with an average liver weight of ~2.5g. The remaining mice were treated with vehicle, VS-6063, Palbociclib, or VS-6063/Palbociclib for 3 weeks. All mice developed large tumors in the vehicle treatment cohort and were euthanized by 6.5 to 7.5 weeks post-injection (Figure 7B). VS-6063 or Palbociclib treated mice exhibited a slower but still progressive tumor growth, indicated by the lower tumor burden than the vehicle cohort but higher tumor burden than the pretreatment cohort (Figure 7C). Strikingly, VS-6063/Palbociclib combination therapy exhibited a strong anti-neoplastic effect with the lowest liver weight (Figure 7C). No differences in liver weight between the pretreatment and combination therapy groups were detected (Figure 7C). Histologically, iCCA lesions were identified in all cohorts (Figure 7C). Using CK19-positive tumor area as a second measurement of iCCA tumor burden in mice, we confirmed that either VS-6063 or Palbociclib treatment resulted in slower but progressive disease. Combined VS-6063/Palbociclib was effective against Akt/YAP iCCA, leading to decreased tumor burden in mice compared with single treatment groups and the pretreatment cohort (Figure 7C and Supplementary Figure 15). Similar effects were detected when looking at tumor area percentage. Indeed, the combination treatment reduced the tumor burden significantly even compared with the pretreatment cohort (Figure 7C, Supplementary Figure 15).
Figure 7. Combining FAK inhibitor VS-6063 and Palbociclib has potent anti-neoplastic activity in iCCA lesions from Akt/YAP mice.

(A) Study design. (B) Survival curve, (C) Gross images and H&E, Ki-67, CK19, and C-C-3 staining of livers from pretreatment, vehicle-, VS-6063-, Palbociclib-, and VS-6063/Palbociclib-treated Akt/YAP mice. Asterisks indicate necrotic areas. H&E, CK19: Magnification ×40; scale bar =500μm. Ki-67, C-C-3: Magnification ×200; scale bar =100μm. CK19 positive staining, tumor area on H&E-stained slides, Ki-67, and C-C-3–positive cells were counted and quantified by Image J software. Tukey–Kramer test: at least P < 0.05. a, vs. Pretreatment; b, vs. Vehicle; c, vs. Palbociclib; d, vs. VS-6063. Abbreviations: C-C-3, cleaved caspase 3.
At the cellular level, VS-6063 or Palbociclib treatment mildly decreased Ki-67(+) positive cells, whereas the combination treatment more potently suppressed tumor cell proliferation (Figure 7C). In terms of apoptosis, VS-6063 administration induced apoptosis in iCCA lesions, and the combination treatment resulted in a further increase in cell death. Previous studies suggest the FAK modulates cancer-associated fibroblasts (CAFs) and fibrosis during tumor progression27. Using immunostaining with the anti-Vimentin antibody, we found that VS-6063 treatment did not affect CAFs in the mouse lesions (Supplementary Figure 16).
At the molecular level, VS-6063 was effective in suppressing p-FAK, Palbociclib strongly inhibited p-Rb, and the combination treatment increased the apoptosis marker cleaved caspase-3. Notably, consistent with the in vitro and in vivo data, VS-6063-mediated FAK inhibition downregulated the levels of YAP-related effectors (JAG1, NOTCH2, and HES1) (Supplementary Figure 17).
Overall, the data indicate that FAK targeting combined with CDK4/6 inhibitors induces tumor regression in Akt/YAP mice and might be an effective therapy for human iCCA.
Discussion
Here, we demonstrate that FAK is strongly activated in most human iCCA specimens. The precise mechanisms responsible for FAK activation in iCCA remain undefined. iCCA is a tumor characterized by a high desmoplastic reaction, whose components functionally interact with iCCA cells.28 Thus, it would be essential to determine whether the tumor microenvironment induces the activation of FAK in iCCA and, if so, which is the primary cell type responsible for this event.
To investigate FAK functional role in iCCA development, we applied the Akt/YAP iCCA murine model,11 as we detected FAK activation in Akt/YAP iCCA lesions. In this model, two sets of experiments were conducted. In the first set, we deleted FAK during tumor initiation in Akt/YAP mice. FAK deletion resulted in a significant delay of Akt/YAP-dependent iCCA formation. In the second set of experiments, the cholangiocyte-specific inducible Cre system combined with conditional Fak KO mice was employed. This strategy allowed FAK deletion in Akt/YAP iCCA after tumor formation to investigate FAK requirement for iCCA progression. Our study shows that FAK deletion after iCCA initiation significantly inhibited Akt/YAP-driven iCCA development. Besides, we demonstrated that FAK ablation promoted apoptosis. Further studies are required to address how FAK overexpression suppresses tumor cell apoptosis. These findings support the crucial role of FAK during both iCCA initiation and progression.
Mechanistically, we discovered that FAK induces YAP phosphorylation at the Y357 residue, thus promoting YAP activation in iCCA cell lines and mouse models. Notably, FAK also caused the increase of p-YAPS397 levels, implying inhibition of the YAP pathway. This contradicting observation might be the result of multiple feedback mechanisms in response to YAP activation.29 Moreover, we discovered that FAK ablation does not affect Akt/Nicd-driven cholangiocarcinogenesis as YAP lies upstream of NOTCH (Supplementary Figure 18). Also, it is worth noting that when deletion of Fak occurred in already formed Akt/YAP iCCA tumors, YAP activity was preserved in the residual iCCA tumor nodules. Presumably, additional FAK-independent mechanisms maintain YAP activation and sustain the proliferation of iCCA cells in Fak-deleted tumor cells.
The present findings have important translational implications. Indeed, we demonstrated that FAK inhibitors combined with CDK4/6 inhibitors potently inhibited iCCA growth both in vitro and in vivo. The relevance of this drug combination strategy is further supported by the simultaneous activation of FAK and CDK4/6 cascades in over 70% of human iCCA samples. The results open the door for testing this combination therapy for iCCA treatment in clinical trials.
Supplementary Material
Highlights.
FAK is activated in human and mouse intrahepatic cholangiocarcinoma (iCCA) samples.
Overexpression of FAK promotes cholangiocarcinogenesis, whereas deletion of FAK strongly suppresses iCCA initiation and progression.
FAK modulates YAP nuclear localization via phosphorylating YAP at the Y357 residue to promote iCCA development.
FAK is a potential therapeutic target for human iCCA treatment.
Acknowledgments
We thank Dr. Hilary Beggs at the University of California, San Francisco, for Fakf/f mice; and Dr. Eric Olson from the University of Texas Southwestern Medical Center, for Yapf/f mice.
Financial Support Statement: This work was supported by NIH grants R01CA190606 and R03CA288375 to XC; R01CA228483 to XC, TC, and RFS; R01CA197128 to WQ; and P30DK026743 for UCSF Liver Center.
List of Abbreviations:
- AFP
α-fetoprotein
- AKT
v-akt murine thymoma viral oncogene homolog
- CAF
cancer-associated fibroblasts
- Chemo
Oxaliplatin/Gemcitabine combination
- FAK
Focal adhesion kinase
- FOCAL
focal adhesion pathway
- Gpc3
Glypican 3
- H&E
Hematoxylin-Eosin staining
- HNF4α
Hepatocyte nuclear factor 4α
- HTVi
Hydrodynamic tail vein injection
- iCCA
intrahepatic cholangiocarcinoma
- KO
Knockout
- MLN
MLN0128
- MP
MLN0128/Palbociclib combination
- Palbo
Palbociclib
- Pre
Pretreatment
- qRT-PCR
quantitative reverse transcription PCR
- NCI
National Cancer Institute
- SB
Sleeping beauty
- TCGA
The Cancer Genome Atlas
- TEAD
TEA domain transcription factor
- YAP
Yes-associated protein
- WT
Wild-type
- IHC
immunohistochemistry
- W.P.I.
weeks post-injection
- Sac
sacrifice
Footnotes
Competing interests: The authors have no conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability:
The data that support the findings of this study are included within the article and its supplementary materials.
References
Author names in bold designate shared co-first authorship.
- 1.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nature reviews Clinical oncology 2018;15:95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nature reviews Cancer 2014;14:598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panera N, Crudele A, Romito I, Gnani D, Alisi A. Focal Adhesion Kinase: Insight into Molecular Roles and Functions in Hepatocellular Carcinoma. International journal of molecular sciences 2017;18;99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacology & therapeutics 2015;146:132–149. [DOI] [PubMed] [Google Scholar]
- 5.Mohanty A, Pharaon RR, Nam A, Salgia S, Kulkarni P, Massarelli E. FAK-targeted and combination therapies for the treatment of cancer: an overview of phase I and II clinical trials. Expert opinion on investigational drugs 2020;29:399–409. [DOI] [PubMed] [Google Scholar]
- 6.Mon NN, Hasegawa H, Thant AA, Huang P, Tanimura Y, Senga T, et al. A role for focal adhesion kinase signaling in tumor necrosis factor-alpha-dependent matrix metalloproteinase-9 production in a cholangiocarcinoma cell line, CCKS1. Cancer research 2006;66:6778–6784. [DOI] [PubMed] [Google Scholar]
- 7.Pongchairerk U, Guan JL, Leardkamolkarn V. Focal adhesion kinase and Src phosphorylations in HGF-induced proliferation and invasion of human cholangiocarcinoma cell line, HuCCA-1. World journal of gastroenterology 2005;11:5845–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rausch V, Hansen CG. The Hippo Pathway, YAP/TAZ, and the Plasma Membrane. Trends in cell biology 2020;30:32–48. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen CDK, Yi C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends in cancer 2019;5:283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes & development 2010;24:72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, Song X, Cao D, Xu Z, Fan B, Che L, et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. Journal of hepatology 2017;67:1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugihara T, Isomoto H, Gores G, Smoot R. YAP and the Hippo pathway in cholangiocarcinoma. 2019;54:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lachowski D, Cortes E, Robinson B, Rice A, Rombouts K, Del Río Hernández AE. FAK controls the mechanical activation of YAP, a transcriptional regulator required for durotaxis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2018;32:1099–1107. [DOI] [PubMed] [Google Scholar]
- 14.Kim NG, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. The Journal of cell biology 2015;210:503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell reports 2017;18:2780–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012;142:1021–1031 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao X, Chen Y, Chen M, Wang S, Wen X, Zhang S. Identification of key candidate genes and biological pathways in bladder cancer. PeerJ 2018;6:e6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S, Zhou D. Role of the transcriptional coactivators YAP/TAZ in liver cancer. Current opinion in cell biology 2019;61:64–71. [DOI] [PubMed] [Google Scholar]
- 19.Li B, He J, Lv H, Liu Y, Lv X, Zhang C, et al. c-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. The Journal of clinical investigation 2019;129:1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shang N, Wang H, Bank T, Perera A, Joyce C, Kuffel G, et al. Focal Adhesion Kinase and β-Catenin Cooperate to Induce Hepatocellular Carcinoma. Hepatology 2019;70:1631–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011;140:1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Che L, Fan B, Pilo MG, Xu Z, Liu Y, Cigliano A, et al. Jagged 1 is a major Notch ligand along cholangiocarcinoma development in mice and humans. Oncogenesis 2016;5:e274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song X, Liu X, Wang H, Wang J, Qiao Y, Cigliano A, et al. Combined CDK4/6 and Pan-mTOR Inhibition Is Synergistic Against Intrahepatic Cholangiocarcinoma. Clinical cancer research 2019;25:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schettini F, De Santo I, Rea CG, De Placido P, Formisano L, Giuliano M, et al. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Frontiers in oncology 2018;8:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews 2006;58:621–681. [DOI] [PubMed] [Google Scholar]
- 26.Gerber DE, Camidge DR, Morgensztern D, Cetnar J, Kelly RJ, Ramalingam SS, et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung cancer (Amsterdam, Netherlands) 2020;139:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang H, Liu X, Knolhoff BL, Hegde S. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut 2020;69:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaquero J, Aoudjehane L, Fouassier L. Cancer-associated fibroblasts in cholangiocarcinoma. Current opinion in gastroenterology 2020;36:63–69. [DOI] [PubMed] [Google Scholar]
- 29.Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes & development 2015;29:1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are included within the article and its supplementary materials.