

Abstract
Introduction:
The cellular prion protein (PrPC) is well known for its pathogenic roles in prion diseases, several other neurodegenerative diseases (such as Alzheimer’s disease), and multiple types of cancer, but the beneficial aspects of PrPC and its cleavage products received much less attention.
Areas covered:
Here the authors will systematically review the literatures on the negative as well as protective aspects of PrPC and its derivatives (especially PrP N-terminal N1 peptide and shed PrP). The authors will dissect the current findings on N1 and shed PrP, including evidence for their neuroprotective effects, the categories of PrPC cleavage, and numerous cleavage enzymes involved. The authors will also discuss the protective effects and therapeutic potentials of PrPC-rich exosomes. The cited articles were obtained from extensive PubMed searches of recent literature, including peer-reviewed original articles and review articles.
Expert Opinion:
PrP and its N-terminal fragments have strong neuroprotective activities that should be explored for therapeutics and prophylactics development against prion disease, Alzheimer’s disease and a few other neurodegenerative diseases. The strategies to develop PrP-based therapeutics and prophylactics for these neurodegenerative diseases will be discussed in a companion article (Part II).
Keywords: Alpha-cleavage, ADAM, Aβ and other toxic oligomers, Alzheimer’s disease, neurodegenerative diseases, N1 peptide, neuroprotection, prion protein, shedding, therapeutics
1. Overview of cellular PrP
The cellular prion protein (PrPC) is known for its essential roles in prion diseases (PrD), a group of fatal and transmissible neurodegenerative diseases affecting humans and several mammal species1–3. Common PrD includes various forms of Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy in cattle, scrapie in goats and sheep, and chronic wasting disease in cervids (deer, elk, moose).
PrPC has been implicated in a large number of biological or pathological processes. Its diverse biological roles have been recently reviewed4–5, including neuronal survival6–8, stress protection9–14, inhibition of neuronal excitation and copper homeostasis15–19, peripheral myelin maintenance20, cellular proliferation and differentiation8, 21–24, immune function25–26, iron uptake27, and circadian rhythm28–30; but some of the implied functions are questioned4. Pathologically, PrPC is a critical player in several neurodegenerative protein-misfolding diseases. It is essential for both prion replication and prion pathogenesis in PrD31. It has also been reported to serve as the receptor for cytotoxic amyloid-β (Aβ) oligomers in Alzheimer’s disease (AD), toxic soluble aggregates of Tau in AD and a few other common neurodegenerative diseases involving Tau32–33 and α-synuclein (αSyn) oligomers that are critical in Parkinson’s disease (PD) and other synucleinopathies34–35. It is worth noting that a recent article reports no binding of PrPC to αSyn oligomers and the absence of PrPC had no effect on the toxicity of αSyn oligomers36, raising questions on the role of PrPC in αSyn toxicity. Knocking-out or knocking-down the PrP gene expression showed only limited negative effects in mice37–40 or cattle41. Goats naturally devoid of PrP also appear largely healthy42. Some modest defects such as progressive demyelinating neuropathy of the peripheral nervous system20,43, impaired hippocampus-dependent spatial-learning and long-term potentiation44, and sensitivity to oxidative stress45 were later detected in PrP-null goat and/or mice. These factors make PrPC a highly attractive target for development of prevention and therapeutics against PrD, AD, PD, and several other neurodegenerative diseases.
PrPC is a universally expressed glycoprotein that attaches to the outer layer of the cell membrane via a glycosylphosphatidylinositol (GPI) anchor, with highest expressions in the nervous system, muscles, and lymphoid tissues46–51 and expression in developing embryos52. The mature mouse PrPC is ~210 amino acids long, consisting of a flexible unstructured N-terminal domain (residues 23–120), a highly structured globular C-terminal domain (residues 121–231) with three α-helices, two β-sheets, a disulfide bond, two Asn-linked glycans, and a GPI anchor53–55 (Figure 1). PrPC undergoes various cleavages under physiological and pathological conditions, some of which are beneficial and protective. During de novo prion formation and seeded prion replication, PrPC undergoes a not well understood process of conformational changes into the misfolded aggregated prion or scrapie form (PrPSc)56–57. One study suggests that intracellular prion conversion occurs primarily in the multivesicular bodies58 that derive exosomes. PrPSc in turn leads to neural damages and eventually clinical symptoms and death in a progressive process that requires cell surface PrPC and involves the cytotoxic oligomeric PrPSc 59, but the detailed mechanisms are still unclear3. Recent studies show that prion infectivity and prion toxicity can be separated60 and high-density oligomeric PrPs found in rapidly progressive AD patient brains are neurotoxic and may contribute to pathogenesis61.
Figure 1. Diagrams of the structure and cleavages of cellular PrP.
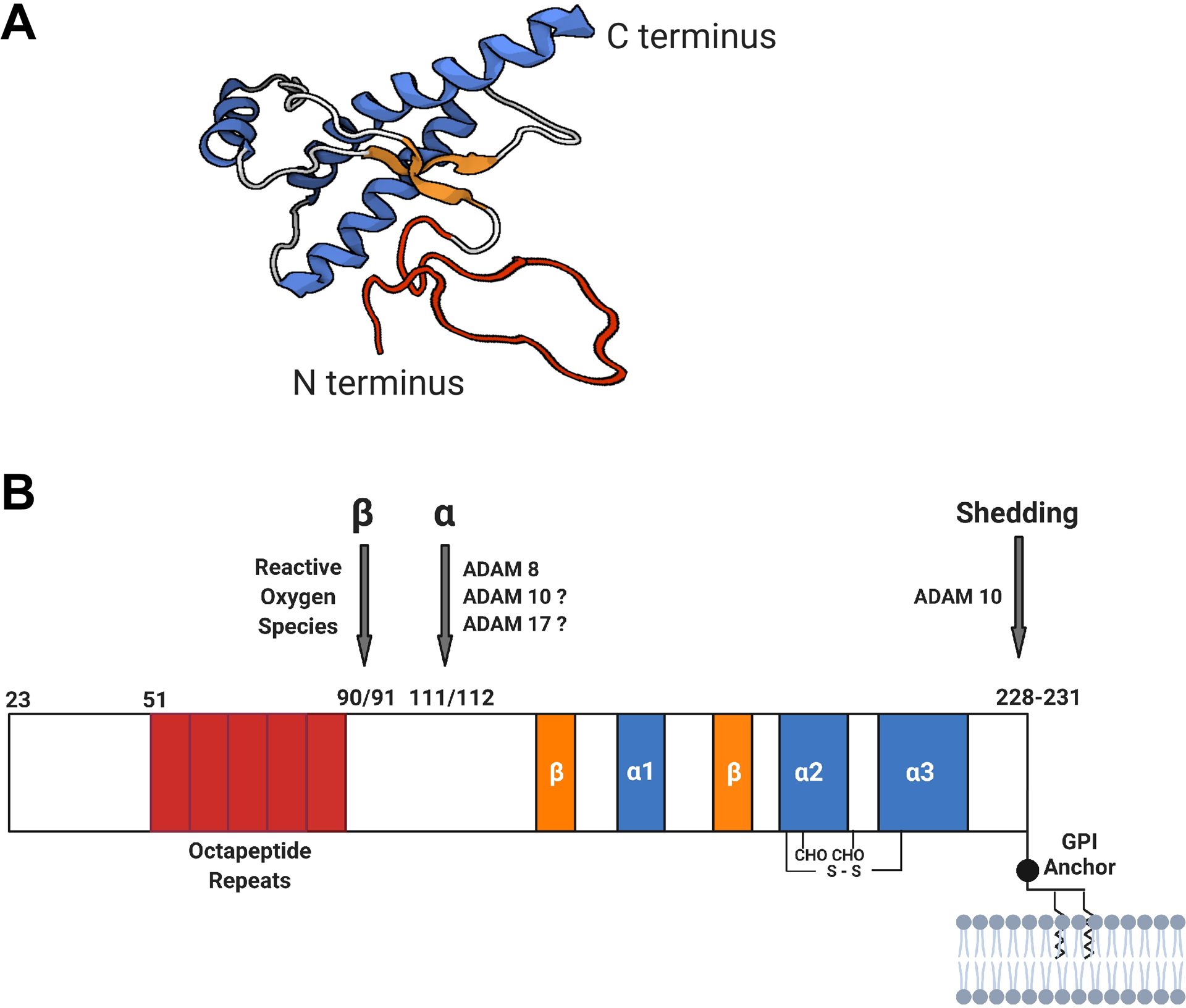
A. 3D model of human PrP structure55. The segments of different secondary structures are color coded: α-helices (blue), β-sheets (orange), loops (gray), and unstructured (red). B. Schematic diagram of PrP cleavage sites (modified from Figure 1 in ref 81). The mature PrPC is ~210 amino acids long, consisting of a flexible unstructured N-terminal domain (residues 23–120) and a highly structured globular C-terminal domain (residues 121–231) composed of three α-helices, two β-sheets, a disulfide bond, two Asn-linked glycans, and a C-terminal glycosylphosphatidylinositol (GPI) anchor. PrP can undergo cleavages at different positions: within the hydrophobic region (residues 109–120) (α-cleavage), within or at the end of the octapeptide repeats region (residues 51–91) (β-cleavage), and at or near the GPI anchor [within residues 228–231 or possibly within the GPI anchor (not depicted)] (shedding). The suspected or confirmed enzymes or molecules involved in these cleavages are marked. The β-cleavage of PrPC produces the N2 and C2 fragments that both appear to be neutral.
Here we review the various protective forms of PrPC (including its cleavage and derivative products) as well as the enzymes involved in the cleavages, with an emphasis on the N-terminal fragment derived from the PrPC α-cleavage, the shed full-length PrP derived from PrPC shedding, and PrPC-containing exosomes.
2. Beneficial PrP forms and protective PrP processing
There are three main types of PrPC processing: α-cleavage, β-cleavage, and shedding (Figure 1B). The α-cleavage occurs at the 110/111 or 111/112 peptide bond of the hydrophobic central region of PrP, resulting in the membrane-attached C-terminal C1 fragment62 and the N-terminal N1 peptide (88–89 amino acid residues) released to the extracellular space. The β-cleavage cuts towards the C-terminal end of the octapeptide repeat region, creating the membrane-attached C2 fragment and releasing the N-terminal N2 fragment62–64. Shedding of PrP is achieved through cleavages near the C-terminus of PrPC (residues 228–231) or within the GPI anchor, releasing anchorless full length PrPC from the cell surface65–68.
As will be discussed in detail below, α-cleavage and shedding of PrPC and the resulting PrP peptides or anchorless PrP protein are beneficial (Figure 2). The β-cleavage of PrPC also seems to be cytoprotective. The β-cleavage of PrPC was reported to involve reactive oxygen species (ROS), and it is thought to be an early and critical event in protection against oxidative stress, since cells expressing PrP mutants incapable of β-cleavage had increased sensitivity to oxidative stress when challenged with H2O2 and Cu2+ 69. The C-terminal β-cleavage product (C2) does not appear to have significant protective effects, but one recent report shows that both N1 and N2 reduce reactive oxygen species and lead to decreased growth and differentiation of murine neural stem cells70. Recombinant ADAM8 has also been reported to cut within the octapeptide repeat region of recombinant PrP in vitro and this cleavage is influenced by Cu2+ and Zn2+ 71, but its biological relevance remains to be seen. Wik et al.72 suggested that suppression of β-cleavage may make more PrPC available for the protective α-cleavage and shedding processes.
Figure 2. Protective activities of PrP and its N-terminal fragment released from cell surface by α-cleavage or shedding.
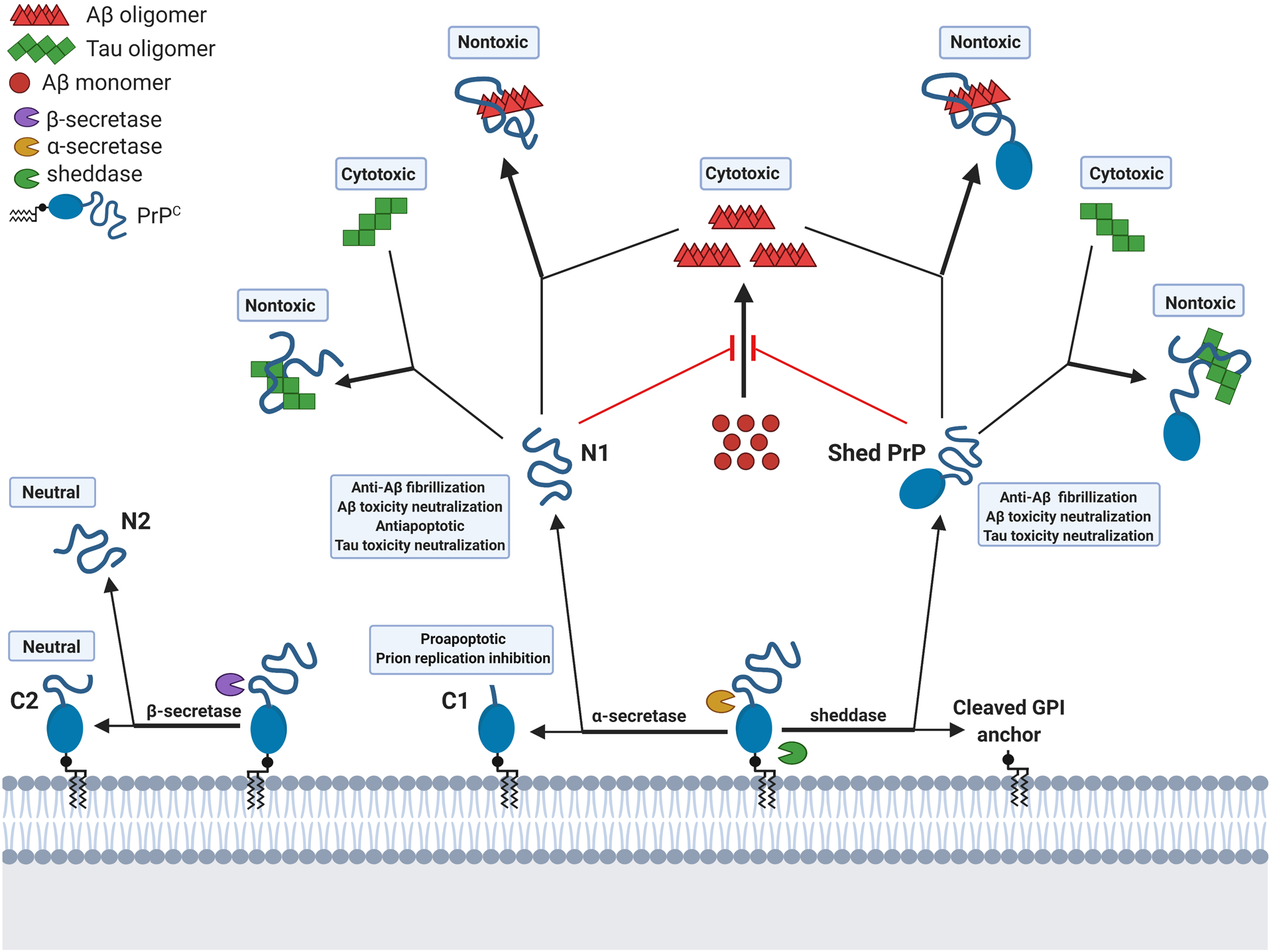
The N1 peptide, the N-terminal fragment of PrP released to the extracellular space by α-cleavage of PrPC, can prevent Aβ fibrillization, neutralize Aβ and Tau toxicity through binding to the neurotoxic Aβ or Tau oligomers, and prevent stress-induced apoptosis. Shed PrP is shown (or expected to show) similar protective activities. Both N1 and shed PrP can associate with other toxic oligomeric misfolded proteins such as αSyn in Parkinson’s disease (not depicted). C1, the C-terminal fragment from α-cleavage of PrPC, is reported to inhibit prion replication and seems to be proapoptotic, although transgenic mice overexpressing C1 appear normal.
2.1. α-Cleavage of PrP and the cleavage products
The α-cleavage appears to be the most beneficial PrPC processing. It cleaves at the central hydrophobic region of PrPC to produce the N-terminal N1 fragment and the C-terminal C1 fragment (Figure 1B), leading to reduction of the amount of cell surface PrPC that is not only the substrate for prion replication but also a key mediator of toxicity in prion diseases, AD, and other neurodegenerative diseases. Of the two fragments produced, N1 is neuroprotective73–74 (see details in section 2.4) and murine C1 inhibits prion replication in a dominant-negative fashion mice75–76. A recent report shows that a highly conserved seven residue deletion (Δ190–196) of the bovine C1 fragment leads to spontaneous prions from the mutant C1 in the RK13 rabbit kidney cells77, but such spontaneous mutant C1 prions failed to infect cells expressing wild type PrP77, further confirming the resistance of wild type C1 to prion conversion. Nevertheless, the prion convertibility of C1 from humans and other species has not been investigated. C1 also has a negative side: it has been reported to upregulate the transcription and activity of p53 and potentiate caspase 3 activation induced by staurosporin (a proapoptotic molecule) in HEK293 cells78. Consistent with these observations, we found that p53 is involved in the primary myopathy in a mouse model induced to overexpress wild type PrP and accumulate high levels of C1 in the skeletal muscles79–80. Fortunately, the negative activities of C1 may be counteracted by N1 through down-regulating p53 expression73. Moreover, transgenic mice overexpressing PrP(Δ23–111) that corresponds to GPI-anchored C1 did not appear to exhibit neurological deficits or histological lesions75, consistent with the notion that C1 may be pro-apoptotic only under apoptotic stimuli and is nontoxic under normal conditions81. The α-cleavage of PrPC also has additional biological functions: Bremer et al. reported that the α-cleavage of PrPC is essential for myelin maintenance in peripheral nerves after examining four independent PrP-knockout mouse strains20.
The α-cleavage of PrP was first discovered as the cleavage of the hydrophobic region at the 110/111 or 111/112 peptide bond, resulting in a GPI-anchored C1 fragment62. A later study using TSM1 neurons and HEK293 cells overexpressing wild-type (wt) PrPC and 3F4-tagged murine PrPC revealed that cellular PrP can be cleaved to release a soluble 11–12-kDa N-terminal peptide termed N182. This α-cleavage occurs within the hydrophobic central domain of PrP (residues 105–120) since its deletion abolishes α-cleavage and partial deletion led to reduction in α-cleavage proportional to the size of the deletions83. However, the cleavage site itself is unexpectedly tolerant to sequence variations as long as overall hydrophobicity is maintained83. The sequence variation tolerance of the α-cleavage site may be an evolutionarily advantageous feature, as the α-cleavage of PrPC confers protection to neurons and other cell types.
Two studies suggest that the α-cleavage occurs late in the secretory pathway and several enzymes may be involved, most notably ADAM9, ADAM10, and ADAM1784–85. Inhibition, overexpression, or knock-out of ADAM10 or ADAM17 in cultured cells revealed that ADAM10 contributes to constitutive N1 production while ADAM17 (also named TACE) mainly participates in regulated N1 formation85. A later report found that reducing endogenous ADAM9 expression using antisense cDNA lowered N1 secretion and co-expression of ADAM10 and ADAM9 led to enhanced α-cleavage in human PrP-expressing fibroblasts84. However, transient transfection of ADAM9 into primary fibroblasts derived from ADAM10 KO mice failed to increase N1 production, suggesting that ADAM9 likely indirectly regulates the α-cleavage by modulating ADAM10 activity through shedding of the ADAM10 ectodomain. Forced PrP dimerization has also been reported to enhance the α-cleavage of mouse PrPC 86, but it is still unclear whether physiological PrP dimers confer the same effect.
Some reports cast doubts on the role of ADAM9, ADAM10 or ADAM17 in the α-cleavage of PrPC. Taylor et al.68 reported that the overexpression of ADAM9, ADAM10 or ADAM17, or the siRNA knockdown of ADAM9 and ADAM10 in murine PrPC expressing HEK cells did not alter the amount of C1 fragment relative to full-length PrPC, indicating a lack of influence of these three ADAMs on the α-cleavage. This was corroborated by a later study from Wik and colleagues72 showing that the addition of σ-phenanthroline (a general ADAM inhibitor) and TAPI-1 (an ADAM17 inhibitor) to bovine PrPC-expressing baby hamster kidney-21 (BHK) cells did not reduce α-cleavage by measuring the kinetics of the generation of the C1 fragment. It is worth noting these two studies both quantified C1 to measure α-cleavage activity. In addition, Altmeppen et al.87 showed that the α-cleavage of PrPC as measured by both C1 and N1 levels was not changed in primary neurons derived from neuron-specific ADAM10-KO mice and concluded that ADAM10 is not involved in the α-cleavage of PrPC. On the other hand, McDonald et al.71 showed that recombinant ADAM10 and ADAM17 can both cleave recombinant murine PrP in vitro at the 119/120 peptide bond (termed α3-cleavage), which is 9–10 residues downstream of the normal α-cleavage site, but such α3-cleavage has not been confirmed in cells or animals.
We have reported that ADAM8 is the primary α-cleavage enzyme for PrPC in muscles based on in vitro experiments using recombinant PrP and recombinant ADAM8 as well as data from cultured muscle cell line (C2C12) and muscle tissues of PrP transgenic mice and ADAM8 knockout mice88. We found that ADAM8 protein level positively correlates with C1 production in the skeletal muscles of mice and in the C2C12 myoblast cell line, and that recombinant ADAM8 directly cleaved recombinant PrP to generate the C1 fragment in vitro. ADAM8 also contributes to the α-cleavage of PrPC in the brain, although not as the primary α-secretase (Liang and Kong, unpublished data). McDonald et al.71 studied PrP α-cleavage with recombinant mouse PrP and recombinant ADAM8, ADAM10 and ADAM17. They found that, in addition to cleaving at the previously reported α-cleavage site (109/110) (termed α1-cleavage), ADAM8 can also cut within the octapeptide repeats and at 116/117 (termed α2-cleavage), and the cleavage site preference is influenced by Cu2+ and Zn2+. The novel ADAM8 cleavage sites have not been confirmed by studies in cells or animals.
The mechanisms underlying the contradicting results concerning the roles of ADAM9, ADAM10 and ADAM17 in α-cleavage of PrPC are unclear. Cissé and colleagues84 and Vincent and colleagues85 (2001) both concluded that ADAM10 and ADAM17 are implicated in the α-cleavage, based on their data using the SAF-32 antibody (recognizing PrP amino acids [aa] 79–92) to detect N1. In contrast, Taylor and colleagues68 used the 6H4 antibody (recognizing PrP aa144–152) to detect C1 and reached the opposite conclusion, i.e., ADAM9, ADAM10, and ADAM17 are not involved. On the other hand, Altmeppen and colleagues87 used the POM2 antibody (recognizing PrP octarepeat region) to detect N1 and used the POM1 antibody (recognizing PrP C-terminal aa121–230) to detect C1 and concluded that ADAM10 is not involved in α-cleavage. N1 is easily degraded and difficult to quantify whereas C1 is much more stable and easier to measure accurately. If the unusual α-cleavages by ADAM8, ADAM10 and ADAM17 reported by McDonald et al.71 can be confirmed in cell and animal studies, it will establish the roles of ADAM10 and ADAM17 in the α-cleavage of PrPC and help reconcile the conflicting findings in the other cell and animal studies utilizing various monoclonal anti-PrP antibodies. Concurrent measurements of both N1 and C1 with appropriate anti-PrP antibodies, or revisiting the N1 measurements under stringent conditions, may also help resolve the controversy.
Alternatively, it is conceivable that, when one α-cleavage enzyme (such as ADAM10) is knocked down, the very limited impact on overall α-cleavage could be due to compensation from another α-cleavage enzyme(s) that cleaves at the same site or a slightly different location in the hydrophobic region. In addition, enhancing the activity of one α-cleavage enzyme may fail to augment overall α-cleavage because another α-cleavage enzyme(s) may be downregulated concurrently.
In addition, it is entirely possible, even expected, that the enzyme(s) responsible for the α-cleavage of PrPC may vary with the cell types and their tissue environment. Further investigations that measure both N1 and C1 in different cell types in vitro and in vivo will be necessary to fully identify all the enzymes involved in the crucial α-cleavage of PrPC in each cell type.
2.2. Shedding of PrP and extracellular full-length PrP
Shedding of PrPC from the cell surface increases extracellular PrP levels with a concurrent decrease of cell surface PrPC, both leading to protection against the toxicity of misfolded protein oligomers in AD and other diseases. PrPC shedding is the process in which cell surface PrPC is cleaved near the GPI anchor by a secretase-like protease enzyme termed “sheddase” or at the GPI anchor by phospholipases, releasing an anchorless full-length PrPC molecule into the extracellular space87. Mass spectrometric analysis shows that this shedding cleavage takes place just three amino acids away from the GPI anchor, between residues 228/22987. Several studies suggest that ADAM9 and ADAM10 are involved in PrP shedding68,71,87,89.
Using recombinant mouse PrP and recombinant ADAM8, ADAM10 and ADAM17, McDonald et al.71 demonstrated that ADAM10 can cleave PrP near its C-terminal end in test tubes, but ADAM8 and ADAM17 showed no shedding activities. Through overexpression and siRNA knockdown experiments in HEK cells, Taylor and colleagues68 found that ADAM9 and ADAM10, but not ADAM17, are implicated in PrP shedding. Overexpression of ADAM10 alone led to a significant increase in the amount of shed PrP in the conditioned cell culture medium, and co-overexpression of ADAM9 and ADAM10 led to even more shed PrP, while overexpression of ADAM17 had no significant effect on PrP shedding. Moreover, siRNA knockdown of endogenous ADAM enzymes found that the knockdown of either ADAM9 or ADAM10 reduced the amount of shed PrP in the conditioned medium. In contrast, when ADAM10 is knocked down, ADAM9 overexpression did not affect shedding of PrP, indicating that the effect of ADAM9 on PrP shedding is achieved through modulating the activity of ADAM10. In addition, Altmeppen and colleagues87 found that there was a 77% decrease of shed PrPC in the culture medium of primary neurons derived from neuron-specific ADAM10 KO mice when compared to the littermate controls. Expression of ADAM10 from a plasmid in the ADAM10-KO neurons restored PrPC shedding. Together these findings confirm that ADAM10 is the primary sheddase for PrPC and ADAM9 affects PrPC shedding through modulating ADAM10.
2.3. Full length PrP
2.3.1. Protective effects of full length PrP
Full length PrPC has been reported to possess protective effects in vitro and in vivo. Cell surface PrPC enhances neuronal survival through activation of the cAMP-dependent PKA pathway6–8. Cell surface PrPC serves as a trophic receptor that activates cAMP/protein kinase A (PKA) to protect against anisomycin-induced cell death in cell cultures of retinal explants6 and staurosporine-induced cell death in primary hippocampal slices8. The protective effect of PrPC is also corroborated by Chen and colleagues7, who observed increased apoptosis of primary cerebellar neurons from PrP-null mice compared to primary cerebellar neurons from wt mice. PrPC was also reported to be necessary for recombinant stress-inducible protein 1 (STI1)-induced neuritogenesis through activating the MAPK pathway in primary hippocampal slices8.
Full length PrPC has also been observed to be protective under cellular stress. PrPC levels were found to be elevated in neuronal soma in the gray matter in ischemic or hypoxic human or mouse brains and the absence of PrPC led to bigger infarct in ischemic mouse brains10. An independent study confirmed this report and found that these protective effects required the N-terminal octarepeat region of PrPC 11. In addition, PrPC has been reported to enhance the cell viability through Fyn kinase activation in immortalized hippocampal neuronal cells exposed to hydrogen peroxide12 and in neuronal cells (SH-SY5Y and N2a) treated with kainite (an excitotoxin)13.
Axonal PrPC that can undergo α-cleavage appears to play an important role in peripheral myelin maintenance, since axon morphometry analysis demonstrated that all four independent PrPC-deficient mouse strains experience chronic demyelinating polyneuropathy20. In addition, α-cleavage of PrPC was found to be elevated in ScN2a neuroblastoma cells treated with soluble αSyn aggregates, which may have contributed to the observed enhanced clearance of the toxic αSyn aggregates90.
PrPC monomers can also form dimers13,91 under physiological conditions, through interactions of the hydrophobic central domain86,92. PrP dimerization seems to be beneficial through promoting PrPC α-cleavage86, mediating stress protective effects13, and inhibiting prion replication in a dominant-negative manner93.
Transgenic expression of a dimeric PrP-immunoglobulin Fcγ fusion protein was also shown to delay PrPSc accumulation and disease onset in wildtype mice intracerebrally or intraperitoneally inoculated with prions94.
2.3.2. The negative roles of full-length PrPC
Given the protective effects of PrPC, it is tempting to adopt overexpression of full-length PrPC as a therapeutic approach. However, this strategy carries multiple risks, since PrPC plays a negative role in several neurodegenerative diseases (such as PrD and AD) as well as in several types of cancer and other diseases.
PrPC has been reported to be a critical toxicity mediator for Aβ aggregates in AD. PrPC binds to synthetic or AD brain-derived Aβ and shows the highest affinity for its oligomeric form over monomers and fibrils95–97. The crucial sites for PrPC-Aβ interaction in solutions in vitro have been determined as the N-terminal residues 23–27 and the ~92–110 region of PrPC 98. To mediate Aβ toxicity, PrPC binds to metabotropic glutamate receptor 5 (mGluR5) on the cell surface to form co-receptors for Aβ oligomers99 and mediate Aβ toxicity100. The PrPC-mGluR5 co-receptor triggers toxic signaling pathways upon binding with Aβ oligomers, activating the Fyn kinase (a member of the Src family kinases)99,101, which in turn causes activation of NMDAR through phosphorylation of its NR2B subunit, leading to calcium influx and cell death101.
Moreover, Aβ-induced lactate dehydrogenase (LDH) release and dendritic spine loss also require the PrPC-Aβ oligomer-mGluR5 complex in mouse hippocampal neuronal cultures99, 101. In addition, PrPC is implicated in Aβ-induced impairment of long-term potentiation (LTP)35,95, but this is disputed by other reports that found no significant effect of PrPC levels on Aβ-induced LTP inhibition in PrP null mice102 or cells ablated or overexpressing PrP103. The reasons for this discrepancy are unclear, but could be due to differences in the models (cell types, mouse strains) and reagents (Aβ preparations) used.
PrPC also seems to mediate αSyn oligomer toxicity through the same pathway as that for Aβ oligomers104. Cell surface PrPC was reported to preferentially bind to soluble aggregates or shorter fibrils of αSyn over their monomeric and large fibrillary forms35,90 and αSyn oligomer binding is dependent on similar PrP regions (aa23–31 and aa92–110)35 implicated in Aβ oligomer binding. PrPC has also been implicated in the internalization of recombinant αSyn fibrils by cultured N2a cells and mouse primary hippocampal neurons. The rate of αSyn uptake was significantly lower in PrP-null cells90,105,106 and in PrP-null mice90, and there was less αSyn aggregation, astroglial activation and loss of dopaminergic neurons in the brains of PrP-null mice after intracerebral inoculation with recombinant αSyn fibrils90. In addition, PrP-null mice did not exhibit αSyn-induced LTP inhibition and treatment with an anti-PrP antibody reversed the αSyn-induced LTP defect in hippocampal slices from a transgenic mouse model of PD104. These observations further validate the role of cell surface PrPC as a critical mediator of αSyn toxicity.
Cell surface PrPC seems to play a similar role in Tau aggregate-induced toxicity that requires the same N-terminal PrP regions (aa23–31 and aa92–110) used by Aβ and αSyn oligomers35,107–108. Recombinant PrP is reported to show a higher affinity for soluble recombinant Tau aggregates than for Tau monomers or end-stage fibrils in vitro35. Again, PrPC seems to mediate the internalization and toxicity of Tau aggregates, but the mechanisms are less well understood. The uptake of recombinant Tau K18 amyloids was reported to be greatly reduced in N2a cells ablated for PrPC 108, and soluble Tau aggregates-induced neurotoxicity is abolished when PrP gene is ablated, knocked-down, or when neurons are pre-treated with anti-PrP blocking antibodies in mouse primary neurons35. PrPC is also involved in toxicity induced by soluble Tau aggregates in vivo109. Intracerebroventricular administration of anti-PrP antibody 6D11 (epitope aa95–105) prior to intrahippocampal injection with recombinant Tau aggregates or human AD brain-derived Tau abrogated the inhibition of LTP in male rats109.
Elevated PrPC levels are associated with several cancers110–114. For instance, Pan and colleagues110 reported that high levels of PrPC found in gastric cancer tissues enhanced invasion and metastasis, probably via the PrPC N-terminal region-mediated activation of MMP11 through the MEK/ERK pathway. Furthermore, the increased PrPC expression in cancer cells seems to be directly correlated with shorter survival for pancreatic cancer patients112.
Elevated PrPC levels will also increase the susceptibility and sensitivity to prions since PrPC is the essential substrate for prion replication and necessary for prion pathogenesis. Moreover, too much PrPC may worsen AD, PD and other neurodegenerative diseases because PrPC is a key receptor for the toxic Aβ, Tau, and αSyn oligomers34–35,95, 97,100–101,107–109,115–116.
In addition, overexpression of wild type full-length PrP alone can cause diseases. Transgenic mice overexpressing wt PrP from hamster or mice exhibited spontaneous degeneration of muscles and peripheral and central nervous systems117. Similarly, transgenic mice overexpressing wt mouse PrPC ten-fold developed a progressive neurological illness with tremor, paralysis of the hind limbs, and abnormal posture, accompanied by accumulation of non-transmissible but neurotoxic PrP aggregates, and significant granule neuron degeneration and synaptic terminal enlargement118. We also found that muscle-specific overexpression of wild type PrPC led to a primary myopathy that is associated with muscular accumulation of the C1 fragment80 and involves the p53 pro-apoptotic pathway81. Moreover, TSM1 neuronal cells and HEK293 kidney cells overexpressing full-length PrPC were found to be more prone to apoptosis after treatment with proapoptotic agents119.
In summary, overexpression of full-length PrPC has many serious caveats, which makes it a very risky therapeutic strategy.
2.4. N1 peptide
2.4.1. Protective effects of N1 peptide
The extracellular N1 peptide, one of the α-cleavage products of PrPC, has been shown to be broadly protective to cells from toxic molecules, including reactive oxygen species and multiple toxic protein oligomers implicated in AD, PD and other diseases, and might ultimately enhance neuronal viability.
In 2009, Guillot-Sestier and colleagues73 first demonstrated that recombinant N1 fragment, but not the N2 fragment, shows cytoprotective function. They found that the N1 peptide show antiapoptotic effect through lowering caspase 3 activation in a dose-dependent fashion in mouse primary cortical neurons treated with staurosporin, and it protects against apoptosis through lowering p53 activity, as seen by a 46% reduction in mRNA transcription in HEK293 cells and 91% reduction in rat retinal cells under pressure-induced ischemia.
Recombinant N1 also protects primary cultured neurons against toxicity and cell death triggered by Aβ oligomers in conditioned medium from HEK293 cells overexpressing mutant APPs associated with familial AD or extracts from AD brain tissues120. A later report found that recombinant N1 binds to Aβ oligomers with high affinity and inhibits fibrillization of Aβ monomers and the association between recombinant N1 and Aβ oligomers is dependent on two positively charged regions (aa23–31 and aa95–105) of N1 and the intervening region (aa32–94)121.
The mechanism of N1 neutralization of Aβ toxicity has been studied. Béland and colleagues122 reported that coincubation of recombinant N1 and Aβ oligomers secreted by CHO-7PA2 cells causes a conformational change in Aβ oligomers that turns Aβ oligomers into insoluble, amorphous Aβ aggregates that cannot assemble with other Aβ, thereby removing Aβ oligomers from the normal fibrillization pathway. N1 also co-immunoprecipitated with Aβ in the guanidine-extractable fraction from postmortem AD brain tissues, suggesting that N1 may coaggregate with Aβ in vivo as well122. By measuring the ratio of deglycosylated full length PrPC and C1 in postmortem AD patient brains, Béland and colleagues122 also observed an increase in α-cleavage of PrPC, supporting the idea that the PrPC α-cleavage is an endogenous neuroprotective mechanism against AD. Recombinant full-length PrP and N1 may also inhibit Aβ oligomerization and neutralize the toxicity of pre-existing Aβ oligomers, possibly by blocking Aβ oligomers from binding with cell surface PrPC in hippocampal slices and neurons from wt and PrP-null mice123. Preincubation of these recombinant PrP forms with preformed Aβ oligomers before treating rat hippocampal slices was shown to decrease LDH release and restore LTP, further suggesting a protective effect of recombinant full-length PrP and N1 against Aβ123. Furthermore, preincubation of recombinant N1 with Aβ oligomers reduced the loss of synaptic markers in primary neurons and rescued Aβ oligomer–induced behavior deficits in C57/BL6 mice after intracerebroventricular injection123.
In a very recent study, recombinant N1 induced secretion of the Cxcl10 cytokine and enhanced the metabolism and morphology of microglia that required direct cell-to-cell contact in a neuron-microglia co-culture system, suggesting that N1 may also improve neuronal cell viability124.
2.4.2. Potential caveats of N1 peptide
There are seemingly significant yet misinformed concerns on the safety of using N1 as a therapeutic agent. There are a few reports suggesting that the N-terminal region of PrP is toxic. Sonati et al.125 reported that the anti-prion antibodies induce toxicity in mice and in cerebellar organotypic cultured slices where the N-terminal flexible tail of cell-surface PrPC plays an important role. The same group subsequently showed that transgenic mice expressing an internally truncated PrP protein (Δ141–225), in which the N-terminal region (aa 1–140) and the GPI-anchor signal peptide (aa 232–254) are retained, developed a progressive, inexorably lethal neurodegeneration morphologically and biochemically similar to that triggered by antibodies against the PrP globular domain126. In this case, the toxic PrP is a GPI-anchored cell surface protein that contains the flexible N-terminal tail (aa23–128) and part of the 1st α-helix of PrPC. In addition, when the PrP N-terminal tail (aa23–109) is expressed as a GPI-anchored GFP fusion protein (aa1-109-GFP-GPI anchor), it is toxic by inducing ionic currents127. Moreover, antibodies targeting epitopes in the C-terminal domain of PrPC induce ionic currents and cause degeneration of dendrites on murine hippocampal neurons that are dependent on the presence of the N-terminal tail of PrPC, and there is intramolecular docking between N- and C-terminal domains of PrPC 127. These data support an intramolecular auto-inhibitory mechanism for PrP toxicity, but all the evidences for the toxicity of PrP N-terminal fragments are based on GPI-anchored PrP forms located on the cell surface. This cell surface PrP location allows for ionic currents or other toxic signaling to damage the host cells. There is no evidence suggesting that PrP N1 and similar N-terminal fragments are toxic when they exist as free molecules in the extracellular space, such as after α-cleavage of PrPC, expressed as a secreted peptide, or administered as a recombinant peptide.
The significance of the reported p53 suppression by N173 needs to be investigated. It is interesting to note that p53 binds directly to the promoter region of the PrP gene to activate its transcription128. We have reported the association of enhanced muscular p53 activity in a primary myopathy associated with dramatic overexpression of PrP and elevated PrP α-cleavage in the muscles of an inducible mouse model79–80.
3. PrP shedding, shed PrP, and extracellular full-length PrP
The nearly full-length shed anchorless PrP is believed to be protective as well129, but no study has directly investigated the effect of shed PrP. There are several reports implying that the process of PrP shedding itself is beneficial since it reduces cell surface PrPC levels87,130, thereby decreasing levels of the essential substrate for prion replication and a critical receptor for several toxic protein aggregates as described above.
3.1. Beneficial effects of shed PrP
Several studies using recombinant full-length PrP have demonstrated its protective effect, and the potential benefits of shed PrP have been reviewed in depth129. Recombinant full-length PrP has been reported to induce neuritogenesis. Incubation of full-length recombinant hamster or mouse PrP with embryonic rat hippocampal neurons was reported to increase the development of synapses and length of neuronal networks131. Likewise, in a hippocampal slice culture system, soluble full-length recombinant mouse PrP induced a concentration-dependent neurite outgrowth and rapid growth cone turning towards the source of PrP, both requiring the presence of cell surface GPI-anchored PrPC 132. Recombinant full-length PrP also inhibits both fibrillization of Aβ1–42 monomers and Aβ aggregate toxicity in cultured neurons133 and neutralizes Aβ oligomer toxicity in cultured slices of AD mouse brains121.
Recombinant full-length PrP has also been reported to bind to toxic Tau oligomers and aSyn oligomers35. Although experimental evidence is still lacking, it is very likely recombinant full-length PrP will also neutralize the toxicity of Tau oligomers and αSyn oligomers. It is worth noting that recombinant full-length PrP is not an ideal substitute for shed PrPC since recombinant PrP generated from bacteria does not have the O-linked glycans that shed PrPC possesses.
Enhanced shedding of cell surface PrPC has been implicated in protection against prions. Heiseke et al.130 reported that, in cultured murine N2a neuronal cells, overexpression of a sorting nexin (SNX33) increased shedding of PrPC, reduced the cell surface PrPC level, and lowered PrPSc formation by ~60% in persistently scrapie infected cells and inhibited scrapie infection in naïve cells. Transgenic overexpression of ADAM10, the primary PrPC sheddase, led to reduced full-length PrPC levels and prolonged survival after prion infection in mice134, and neuron-specific knockout of ADAM10 led to elevated PrPC levels, increased PrPSc accumulation and reduced incubation time following prion infection135. However, there is one report arguing that enhanced shedding of PrPC may not reduce prion replication in cultured scrapie-infected cells68. In persistently scrapie-infected ScN2a mouse neuroblastoma cells, suppressing ADAM10 with an inhibitor did not change the amount of protease-resistant PrPSc. Moreover, overexpression of ADAM10 from a plasmid failed to alter the percentage of protease-resistant PrPSc-positive cells, and the cell surface PrPC levels were not found to be altered in these cells68. Further investigation is needed to clarify this issue.
The mechanism of PrPSc accumulation inhibition by PrPC shedding is not well understood, but cell surface PrP levels, which can be reduced by enhanced PrP shedding, are known to be inversely correlated with incubation times after prion infection in mouse models135–137. Reduction of cell surface PrPC levels through PrP shedding in peripheral tissues may also slow neuroinvasion. PrPC expression in the follicular dendritic cells of the spleen has been shown to be critical for neuroinvasion after peripheral prion inoculations138–139. In addition, transgenic mice expressing only GPI-anchorless PrP in an endogenous PrP-null background exhibited slow and infrequent CNS neuroinvasion after peripheral prion inoculations140, which is consistent with a potential protective effect of increased PrP shedding.
3.2. Potential caveats of shed PrP
Shed PrP exists in extracellular space, which should mimic transgenically expressed anchorless PrP or administered recombinant full-length PrP to confer protection against toxic oligomeric aggregates of amyloidogenic proteins by sequestering these toxic molecules in the extracellular space. However, enhancing PrPC shedding may also lead to negative biological activities in several ways.
First, shed PrP may serve as a good substrate for prion replication. Mice co-expressing anchorless PrPC and GPI-anchored PrPC have been reported to show accelerated prion disease141 after prion inoculation. Extensive prion spread142–143 and very high levels of infectious prions144–145 were also found in transgenic mice expressing anchorless PrP after prion inoculation. Second, ADAM10 is the sheddase for several important cell surface proteins146, so enhancing ADAM10 activity may have negative consequences for cancer147–150, Fragile X Syndrome151, and Huntington’s Disease152. Third, treatment with recombinant full-length PrP, and by extension, enhancement of PrPC shedding, may risk inducing inflammation in the CNS. Full-length recombinant human PrP induced the release of inflammatory mediators IL-6 and CCL2 in primary human astrocytes153 and of CCL2, CXCL-12, and IL-8 in both uninfected and HIV-infected human primary monocytes154. CCL2 and TNF-α were shown to enhance the activity of ADAM10 (the PrPC sheddase) in human astrocytes infected with HIV154, and soluble PrPC levels were elevated in the CSF of HIV-infected people with cognitive impairment153. These data suggest a positive feedback loop whereby extracellular PrP induces the release of cytokines, which augments ADAM10 activity that leads to elevated extracellular PrP levels through enhanced shedding of PrPC.
4. Exosomal PrP
4.1. Protective effects of exosomal PrP
PrPC on exosomes has also been shown to be protective against Aβ toxicity155–158. Exosomal PrP isolated from N2a cells was found to bind preferentially to smaller synthetic Aβ42 aggregates in a PrPC-dependent manner in vitro158. Moreover, exosomal surface PrPC derived from hippocampal cells and injected intracerebroventricularly into the brain of rats was reported to bind to Aβ-derived diffusible ligands (ADDLs) prepared from synthetic Aβ1–42 in vivo157. The mechanism through which exosomal PrPC binds to Aβ has not been reported, but it likely depends on the same PrP regions (aa23–31 and aa95–105) that are crucial for recombinant N1 binding to Aβ121. Exosomal PrPC has also been reported to protect against Aβ-related toxicity. First, preincubation of synthetic Aβ42 with PrP-containing exosomes accelerated Aβ42 aggregation into its fibrillar form, reduced synthetic Aβ uptake, and abolished Aβ42-induced apoptosis158. It is possible that, like extracellular recombinant PrP and N1, exosomal PrPC may also inhibit the formation of toxic oligomeric Aβ and promote the formation of fibrillary Aβ. Second, intracerebroventricular injection of PrPC-containing exosomes rescued the LTP inhibition induced by ADDLs or AD brain extracts in the rat brains157. Third, exosomes isolated from murine N2a cells or mouse primary cortical neurons expressing PrPC enhanced the clearance of endogenous Aβ aggregates after intrahippocampal injection in an APP transgenic model, likely through exosome binding with the aggregates that upregulate internalization and degradation of the aggregates by microglia155–156. Continuous intraventricular administration of N2a cell-derived exosomes also ameliorated synaptic dysfunction in an APP transgenic mouse model, as seen by higher synaptophysin immunoreactivities compared to vehicle-treated mice155.
Since full-length recombinant and neuronal PrPs have also been shown to bind to Tau and αSyn oligomers35, exosomal PrPs can be expected to confer protection similarly by sequestering the toxic Tau/αSyn oligomers in the extracellular space, thereby hindering the toxic signaling through preventing Tau/αSyn oligomers from binding with cell surface PrPC in the CNS of patients suffering from various tauopathies and synucleinopathies.
4.2. Potential caveats of exosomal PrP
Exosomes may be a powerful vehicle to harness and deliver the protective powers of PrPC, but using exosomal PrPC for therapeutic purposes may carry the same risks as using shed PrP or recombinant full-length PrP, such as facilitating the replication and spread of prions. Exosomes have been shown to associate with PrPSc in the culture media of prion infected cells159. Exosomes derived from prion-infected neuronal cell lines were reported to initiate prion propagation in uninfected non-neuronal cells and induce PrD when injected into mice160, and blood-derived exosomes from prion-infected mice were infectious when injected into the Tga20 mice that overexpress mouse PrPC 161. Although there is no direct study yet, exosomal PrPC might also induce CNS inflammation in a positive feedback loop involving ADAM10, as has been shown for recombinant full-length PrP. More work needs to be done to examine other biological activities that exosomal PrPC may possess. In addition, exosomes contain many other molecules on its surface and its internal space, and some of them may induce toxic signaling or alter the recipient cell functions after being internalized162–165.
5. Conclusion
In summary, extracellular forms of PrP, including shed full-length PrP, exosomal full-length PrP, the N1 peptide released by PrPC α-cleavage from cell surface, or externally administered recombinant full-length PrP or N1 peptide, have shown protective effect against certain stresses and several toxic molecules that are critical in various neurodegenerative diseases, including PrD, AD, PD, and other tauopathies (such as progressive supranuclear palsy and frontotemporal lobar degeneration) and synucleinopathies (such as dementia with Lewy bodies, multiple system atrophy, and Lewy body variant of AD). These observations suggest that extracellular forms of PrP have great prophylactic and therapeutic potentials (Figure 2). We hypothesize that elevating the extracellular levels of one or more of these protective PrP forms is a promising broad-spectrum strategy against these devastating neurodegenerative diseases. A companion article (Part II) explores and evaluates this strategy for prophylactic and therapeutic purposes.
6. Expert opinion
The cellular PrP has been confirmed or implicated in many biological and pathological processes. On the one hand, it is suggested to play critical biological roles such as in development, myelin maintenance and cellular protection. On the other hand, it promotes cancer, is essential for prion replication and pathogenesis, and seems to be a mediator for toxicity through serving as a key receptor for several toxic oligomers of misfolded proteins (Aβ, Tau, and αSyn) in common neurodegenerative diseases, such as AD and other tauopathies as well as PD and other synucleinopathies. Cellular PrP is also subject to several types of regulated cleavage processing, including α-cleavage, β-cleavage and shedding, all appear to be protective at varying degrees. Unfortunately, PrP is still generally perceived as a “bad” protein by most and the positive side of PrP is often ignored.
Here we highlight the positive aspects and beneficial forms of PrP through reviewing the extensive literature on posttranslational PrP cleavages and the PrP cleavage products as well as the biological and pathological effects of cellular PrP and its cleavage products when localized on the cell surface or in the extracellular space. It is clear that most of the pathological effects of PrP are associated with cell surface full-length PrP and several PrP forms are protective when presented in the extracellular space. These protective PrP forms include the N1 peptide (the N-terminal PrP fragment derived from α-cleavage of cell surface PrP), the shed PrP (derived from shedding of cell surface PrP, primarily by ADAM10 in the brain), and exosomal PrP, all of which exist in the extracellular space. These extracellular beneficial PrP forms protect against the toxicity of misfolded proteins through sequestering the toxic oligomeric forms in the extracellular space and prevent the initiation of toxic signaling cascades that ultimately lead to cytotoxicity and neurodegeneration. In the case of N1, it can also directly reduce the toxic Aβ oligomers by inhibiting its formation and promoting its further aggregation into large fibrils that are much less toxic. The potential caveats of these beneficial PrP forms are also discussed, which mostly involve the risks of activating inflammation and facilitating the replication and spread of prions (and other misfolded protein aggregates in the case of exosomes). The N1 peptide and possibly other PrP N-terminal peptides do not have these drawbacks, making it a prime candidate for broad spectrum PrP-based therapeutics and prophylactics against PrD, AD, PD and other related neurodegenerative diseases.
The α-cleavage of PrP appears to be the most beneficial PrP processing since the two products (N1 and C1) are both protective and the negative effect of C1 is counteracted by N1, and the cleavage process itself reduces the level of cell surface PrP that is a substrate for prion replication and a critical mediator of toxicity in PrD, AD and several other neurodegenerative diseases. Therefore, enhancing PrP α-cleavage is a promising strategy to develop treatments against these diseases. Given the essential roles of cell surface PrP in prion replication and pathogenesis, the RNA interference strategy targeting cellular PrP mRNAs has been tried with great success in treating prion diseases in mouse and cell models166–170. Since cellular PrP also mediates the toxicity of Aβ, Tau, and αSyn oligomers, it is reasonable to expect similar success through knocking down cellular PrP expression. Taking it one step further, simultaneous elevation of a PrP N-terminal peptide and knocking-down of the cellular PrP should be an even better strategy to develop safe and effective treatments and prevention for these neurodegenerative diseases. The various PrP-based strategies to develop broad spectrum treatments and prevention of PrD, AD, PD and a few other neurodegenerative diseases are covered in depth in the companion paper (Part II).
Article highlights.
The full length cellular prion protein (PrPC) has diverse biological roles such as neuronal survival, stress protection, neuronal excitation, peripheral myelin maintenance, and cellular proliferation and differentiation, yet PrP-null animals are largely normal.
PrPC is essential for prion diseases and serves as a key common receptor for a few toxic protein oligomers in Alzheimer’s disease (AD) and a few other common neurodegenerative diseases involving Tau or α-synuclein (αSyn).
PrPC can undergo various cleavages under physiological and pathological conditions, some of which have been shown to be protective, most notably α-cleavage and shedding.
Extracellular forms of PrP such as shed full-length PrP, exosomal full-length PrP, the N1 peptide released by PrPC α-cleavage from cell surface (PrP-N), or recombinant PrP forms have been shown to be protective against toxic stressors.
The N1 peptide derived from α-cleavage of cell surface PrP and recombinant N1 can protect against the toxicity of Aβ oligomers by inhibiting Aβ oligomer formation, hindering toxic signaling, and promoting oligomer aggregation into larger fibrils that are less toxic.
Knocking down PrPC expression while elevating the extracellular level of the PrP N-terminal peptide should have excellent prophylactic and therapeutic potential against several neurodegenerative diseases including PrD and AD, which will be explored in depth in the companion paper (Dexter and Kong, 2021).
Acknowledgements
The figures were created with BioRender.com.
Funding
This work was partially supported by NIH R01 NS109532 and a research grant from CJD Foundation.
Footnotes
Declaration of interests
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982;216:136–44 [DOI] [PubMed] [Google Scholar]
- 2.Colby DW and Prusiner S. B. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sigurdson CJ, Bartz JC, Glatzel M. Cellular and Molecular Mechanisms of Prion Disease. Annu Rev Pathol. 2019Jan 24;14:497–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castle AR, Gill AC. Physiological Functions of the Cellular Prion Protein. Front Mol Biosci. 2017;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linden R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front Mol Neurosci. 2017;10:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiarini LB, Freitas AR, Zanata SM, et al. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21:3317–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen S, Mangé A, Dong L, et al. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci, 2003;22:227–33 [DOI] [PubMed] [Google Scholar]
- 8.Lopes MH, Hajj GN, Muras AG, et al. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci. 2005;25:11330–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown DR, Nicholas RSJ, Canevari L Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res 2002;67, 211–24 [DOI] [PubMed] [Google Scholar]
- 10.McLennan NF, Brennan PM, McNeill A, et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol. 2004;165:227–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitteregger G, Vosko M, Krebs B, et al. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007April;17:174–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krebs B, Wiebelitz A, Balitzki-Korte B, et al. Cellular prion protein modulates the intracellular calcium response to hydrogen peroxide. J Neurochem. 2007;100:358–67 [DOI] [PubMed] [Google Scholar]
- 13.Rambold AS, Müller V, Ron U, et al. Stress-protective signaling of prion protein is corrupted by scrapie prions. EMBO J. 2008;27:1974–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang B, Cowden D, Zhang F, et al. Prion Protein Protects against Renal Ischemia/Reperfusion Injury. PLoS One. 2015;10:e0136923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khosravani H, Zhang Y, Tsutsui S, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008May5; 181:551–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beraldo FH, Arantes CP, Santos TG, et al. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J Biol Chem. 2019;285:36542–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carulla P, Bribián A, Rangel A, et al. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol Biol Cell. 2011;22:3041–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alfaidy N, Chauvet S, Donadio-Andrei S, et al. Prion protein expression and functional importance in developmental angiogenesis: role in oxidative stress and copper homeostasis. Antioxid Redox Signal. 2013;18:400–11 [DOI] [PubMed] [Google Scholar]
- 19.Gasperini L, Meneghetti E, Pastore B, et al. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal 2015;22, 772–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bremer J, Baumann F, Tiberi C, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–18 [DOI] [PubMed] [Google Scholar]
- 21.Santuccione A, Sytnyk V, Leshchyns’ka I, et al. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005April25; 169:341–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehrabian M, Brethour D, MacIsaac S, et al. CRISPR-Cas9-based knockout of the prion protein and its effect on the proteome. PLoS One. 2014;9:e114594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehrabian M, Brethour D, Wang H, et al. The prion protein controls polysialylation of neural cell adhesion molecule 1 during cellular morphogenesis. PLoS One. 2015;10:e0133741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehrabian M, Brethour D, Williams D, et al. Prion Protein Deficiency Causes Diverse Proteome Shifts in Cell Models That Escape Detection in Brain Tissue. PLoS One. 2016;11:e0156779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haddon DJ, Hughes MR, Antignano F, et al. Prion protein expression and release by mast cells after activation. J. Infect. Dis 2009; 200, 827–31 [DOI] [PubMed] [Google Scholar]
- 26.Chatterjea D, Martinov T Mast cells: versatile gatekeepers of pain. Mol. Immunol 2015;63, 38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tripathi AK, Haldar S, Qian J, et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic Biol Med. 2015;84:322–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tobler I, Deboer T, Fischer M Sleep and sleep regulation in normal and prion protein-deficient mice. J. Neurosci 1997;17, 1869–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tobler I, Gaus SE, Deboer T, et al. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996;380, 639–42 [DOI] [PubMed] [Google Scholar]
- 30.Cagampang FRA, Whatley SA, Mitchell AL, et al. Circadian regulation of prion protein messenger RNA in the rat forebrain: a widespread and synchronous rhythm. Neuroscience 1999;91, 1201–4 [DOI] [PubMed] [Google Scholar]
- 31.Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol. 2006;4:765–75 [DOI] [PubMed] [Google Scholar]
- 32.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, et al. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castillo-Carranza DL, Gerson JE, Sengupta U, et al. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J. Alzheimers Dis 2014;40, S97–S111 [DOI] [PubMed] [Google Scholar]
- 34.Salazar SV, Strittmatter SM. Cellular prion protein as a receptor for amyloid-β oligomers in Alzheimer’s disease. Biochem Biophys Res Commun. 2017;483:1143–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corbett GT, Wang Z, Hong W, et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020;139:503–26 [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Cellular PrP is a key receptor mediating the toxicity of soluble aggregates (oligomers) of Aβ, Tau, and αSyn
- 36.La Vitola P, Beeg M, Balducci C, Santamaria G, Restelli E, Colombo L, Caldinelli L, Pollegioni L, Gobbi M, Chiesa R, Forloni G. Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain. 2019;142(2):249–254 [DOI] [PubMed] [Google Scholar]
- 37.Büeler H, Fischer M, Lang Y, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein, Nature. 1992;356:577–82 [DOI] [PubMed] [Google Scholar]; **Mice devoid of PrP appear normal
- 38.Manson JC, Clarke AR, Hooper ML, et al. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–7 [DOI] [PubMed] [Google Scholar]
- 39.Weissmann C, Flechsig E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull. 2003;66:43–60 [DOI] [PubMed] [Google Scholar]
- 40.Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: a phenotype under challenge. Prion. 2007;1:83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richt JA, Kasinathan P, Hamir AN, et al. Production of cattle lacking prion protein. Nature Biotechnology. 2007;25:132–8 [DOI] [PMC free article] [PubMed] [Google Scholar]; *Cattle devoid of PrP is largely normal
- 42.Benestad SL, Austbø L, Tranulis MA, Espenes A, Olsaker I. Healthy goats naturally devoid of prion protein. Vet Res. 2012;43:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skedsmo FS, Malachin G, Våge DI, Hammervold MM, Salvesen Ø, Ersdal C, Ranheim B, Stafsnes MH, Bartosova Z, Bruheim P, Jäderlund KH, Matiasek K, Espenes A, Tranulis MA. Demyelinating polyneuropathy in goats lacking prion protein. FASEB J. 2020;34:2359–75 [DOI] [PubMed] [Google Scholar]
- 44.Criado JR, Sánchez-Alavez M, Conti B, Giacchino JL, Wills DN, Henriksen SJ, et al. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol Dis. 2005;19:255–65 [DOI] [PubMed] [Google Scholar]
- 45.Wulf MA, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bendheim PE, Brown HR, Rudelli RD, Scala LJ, Goller NL, Wen GY, Kascsak RJ, Cashman NR, Bolton DC. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology. 1992;42:149–56 [DOI] [PubMed] [Google Scholar]
- 47.Lainé J, Marc ME, Sy MS, Axelrad H. Cellular and subcellular morphological localization of normal prion protein in rodent cerebellum. Eur J Neurosci. 2001;14:47–56 [DOI] [PubMed] [Google Scholar]
- 48.Liu T, Li R, Wong BS, Liu D, Pan T, Petersen RB, Gambetti P, Sy MS. Normal cellular prion protein is preferentially expressed on subpopulations of murine hemopoietic cells. J Immunol. 2001a;166:3733–42 [DOI] [PubMed] [Google Scholar]
- 49.Liu T, Zwingman T, Li R, Pan T, Wong BS, Petersen RB, Gambetti P, Herrup K, Sy MS. Differential expression of cellular prion protein in mouse brain as detected with multiple anti-PrP monoclonal antibodies. Brain Res. 2001b;896:118–29 [DOI] [PubMed] [Google Scholar]
- 50.Miele G, Alejo Blanco AR, Baybutt H, Horvat S, Manson J, Clinton M. 2003. Embryonic activation and developmental expression of the murine prion protein gene. Gene Expr. 11:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shmakov AN, McLennan NF, McBride P, Farquhar CF, Bode J, Rennison KA, Ghosh S. Cellular prion protein is expressed in the human enteric nervous system. Nat Med. 2000;6:840–1 [DOI] [PubMed] [Google Scholar]
- 52.Tremblay P, Bouzamondo-Bernstein E, Heinrich C, Prusiner SB & DeArmond SJ Developmental expression of PrP in the post-implantation embryo. Brain Res 2007;1139, 60–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riek R, Hornemann S, Wider G, et al. NMR structure of the mouse prion protein domain PrP(121–231). Nature. 1996;382:180–2 [DOI] [PubMed] [Google Scholar]
- 54.Riek R, Hornemann S, Wider G, et al. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231). FEBS Lett. 1997;413:282–8 [DOI] [PubMed] [Google Scholar]
- 55.Zahn R, Liu A, Lührs T, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97:145–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pan KM, Baldwin M, Nguyen J, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A. 1993;90:10962–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marín-Moreno A, Fernández-Borges N, Espinosa JC, et al. Transmission and Replication of Prions. Prog Mol Biol Transl Sci. 2017;150:181–201 [DOI] [PubMed] [Google Scholar]
- 58.Yim YI, Park BC, Yadavalli R, et al. The multivesicular body is the major internal site of prion conversion. J. Cell Sci 2015;128:1434–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang P, Lian F, Wen Y, et al. Prion protein oligomer and its neurotoxicity. Acta Biochim Biophys Sin (Shanghai). 2013;45:442–51 [DOI] [PubMed] [Google Scholar]
- 60.Benilova I, Reilly M, Terry C, et al. Highly infectious prions are not directly neurotoxic. Proc Natl Acad Sci U S A. 2020;117:23815–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shafiq M, Zafar S, Younas N, et al. Prion protein oligomers cause neuronal cytoskeletal damage in rapidly progressive Alzheimer’s disease. Mol Neurodegener. 2021;16:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen SG, Teplow DB, Parchi P, et al. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270:19173–80 [DOI] [PubMed] [Google Scholar]
- 63.Jimenez-Huete A, Lievens PM, Vidal R, et al. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues Am. J. Pathol, 1998;153:1561–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mange A, Beranger F, Peoc’h K, et al. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell, 2004;96:125–32 [DOI] [PubMed] [Google Scholar]
- 65.Tagliavini F, Prelli F, Porro M, et al. A soluble form of prion protein in human cerebrospinal fluid: implications for prion-related encephalopathies. Biochem Biophys Res Commun 1992;184:1398–404 [DOI] [PubMed] [Google Scholar]
- 66.Borchelt DR, Rogers M, Stahl N, et al. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology. 1993;3:319–29 [DOI] [PubMed] [Google Scholar]
- 67.Parizek P, Roeckl C, Weber J, et al. Similar turnover and shedding of the cellular prion protein in primary lymphoid and neuronal cells. J Biol. Chem 2001;276:44627–32 [DOI] [PubMed] [Google Scholar]
- 68.Taylor DR, Parkin ET, Cocklin SL, et al. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem. 2009;284:22590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watt NT, Hooper NM. Reactive oxygen species (ROS)-mediated beta-cleavage of the prion protein in the mechanism of the cellular response to oxidative stress. Biochem Soc Trans. 2005;33:1123–5 [DOI] [PubMed] [Google Scholar]
- 70.Collins SJ, Tumpach C, Groveman BR, Drew SC, Haigh CL. Prion protein cleavage fragments regulate adult neural stem cell quiescence through redox modulation of mitochondrial fission and SOD2 expression. Cell Mol Life Sci. 2018;75:3231–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDonald AJ, Dibble JP, Evans EG, et al. A new paradigm for enzymatic control of α-cleavage and β-cleavage of the prion protein. J Biol Chem. 2014a;289:803–13 [DOI] [PMC free article] [PubMed] [Google Scholar]; *Proteolytic activities of recombinant ADAM8, ADAM10 and ADAM 17 against PrP in vitro
- 72.Wik L, Klingeborn M, Willander H, et al. Separate mechanisms act concurrently to shed and release the prion protein from the cell. Prion. 2012; 6:498–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guillot-Sestier MV, Sunyach C, Druon C, et al. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem. 2009; 284:35973–86 [DOI] [PMC free article] [PubMed] [Google Scholar]; **First report on the neuroprotective effect of N1 in vitro and in vivo
- 74.Guillot-Sestier MV, Checler F. Cellular prion and its catabolites in the brain: production and function. Curr Mol Med. 2012;12:304–15 [DOI] [PubMed] [Google Scholar]
- 75.Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem. 2011December23;286:44234–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lewis V, Hill AF, Haigh CL, et al. Increased proportions of C1 truncated prion protein protect against cellular M1000 prion infection. J Neuropathol Exp Neurol. 2009;68:1125–35 [DOI] [PubMed] [Google Scholar]
- 77.Munoz-Montesino C, Larkem D, Barbereau C, et al. A seven-residue deletion in PrP leads to generation of a spontaneous prion formed from C-terminal C1 fragment of PrP. J Biol Chem. 2020;295:14025–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sunyach C, Cisse MA, da Costa CA, et al. The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent sTaurosporine-induced caspase-3 activation. J Biol Chem. 2007;282:1956–63 [DOI] [PubMed] [Google Scholar]
- 79.Huang S, Liang J, Zheng M, et al. Regulated over-expression of PrP in the skeletal muscles leads to myopathy in transgenic mice. Proc Natl Acad Sci USA. 2007;104:6800–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liang J, Parchaliuk D, Medina S, et al. Booth SA. Activation of p53-regulated pro-apoptotic signaling pathways in PrP-mediated myopathy. BMC Genomics. 2009;10:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liang J, Kong Q. α-Cleavage of cellular prion protein. Prion, 2012;6:453–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vincent B, Paitel E, Frobert Y, et al. Phorbol ester-regulated cleavage of normal prion protein in HEK293 human cells and murine neurons. J Biol Chem. 2000;275:35612–6 [DOI] [PubMed] [Google Scholar]
- 83.Oliveira-Martins JB, Yusa S, Calella AM, et al. Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS One. 2010;5:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cissé MA, Sunyach C, Lefranc-Jullien S, et al. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J. Biol. Chem 2005;280:40624–31. [DOI] [PubMed] [Google Scholar]
- 85.Vincent B, Paitel E, Saftig P, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001;276:37743–6 [DOI] [PubMed] [Google Scholar]
- 86.Béland M, Motard J, Barbarin A, et al. PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J Neurosci. 2012;32:13255–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Altmeppen HC, Prox J, Puig B, et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener. 2011;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]; **ADAM10 is the primary PrP sheddase and appears not involved in PrP α-cleavage
- 88.Liang J, Wang W, Sorensen D, et al. Cellular prion protein regulates its own α-cleavage through ADAM8 in skeletal muscle. J Biol Chem. 2012b;287:16510–20 [DOI] [PMC free article] [PubMed] [Google Scholar]; **ADAM8 is the primary α-cleavage enzyme for PrP in muscles and the only non-controversial PrP α-cleavage enzyme
- 89.McDonald AJ, Millhauser GL. PrP overdrive: does inhibition of α-cleavage contribute to PrP(C) toxicity and prion disease? Prion. 2014b;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aulić S, Masperone L, Narkiewicz J, et al. α-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci Rep. 2017;7:10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Priola SA, Caughey B, Wehrly K, et al. A 60-kDa prion protein (PrP) with properties of both the normal and scrapie-associated forms of PrP. J Biol Chem. 1995;270:3299–305 [DOI] [PubMed] [Google Scholar]
- 92.Sangeetham SB, Huszár K, Bencsura P, et al. Interrogating the Dimerization Interface of the Prion Protein Via Site-Specific Mutations to p-Benzoyl-L-Phenylalanine. J Mol Biol. 2018;430:2784–801 [DOI] [PubMed] [Google Scholar]
- 93.Engelke AD, Gonsberg A, Thapa S, et al. Dimerization of the cellular prion protein inhibits propagation of scrapie prions. J Biol Chem. 2018;293:8020–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meier P, Genoud N, Prinz M, et al. Soluble dimeric prion protein binds PrP(Sc) in vivo and antagonizes prion disease. Cell 2003; 113:49–60 [DOI] [PubMed] [Google Scholar]
- 95.Laurén J, Gimbel DA, Nygaard HB, et al. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–32 [DOI] [PMC free article] [PubMed] [Google Scholar]; **First report on cellular PrP as a mediator of the toxicity of Aβ oligomers
- 96.Balducci C, Beeg M, Stravalaci M, et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dohler F, Sepulveda-Falla D, Krasemann S, et al. High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer’s disease. Brain. 2014;137:873–86 [DOI] [PubMed] [Google Scholar]
- 98.Chen S, Yadav SP, and Surewicz WK Interaction between human prion protein and amyloid- (A) oligomers. Role of N-terminal residues. J. Biol. Chem 2010;285:26377–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Um JW, Kaufman AC, Kostylev M, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron. 2013;79:887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haas LT, Salazar SV, Kostylev MA, et al. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain. 2016;139:526–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Um JW, Nygaard HB, Heiss JK, et al. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15:1227–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kessels HW, Nguyen LN, Nabavi S, et al. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–E5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Calella AM, Farinelli M, Nuvolone M, et al. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010;2:306–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ferreira DG, Temido-Ferreira M, Vicente Miranda H, et al. alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci 2017;20:1569–79 [DOI] [PubMed] [Google Scholar]
- 105.De Cecco E, Legname G. The role of the prion protein in the internalization of α-synuclein amyloids. Prion. 2018;12:23–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Urrea L, Segura-Feliu M, Masuda-Suzukake M, et al. Involvement of Cellular Prion Protein in α-Synuclein Transport in Neurons [published correction appears in Mol Neurobiol. 2017 May 5;:].Mol Neurobiol. 2018;55:1847–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gomes LA, Hipp SA, Rijal Upadhaya A, et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019;138:913–41 [DOI] [PubMed] [Google Scholar]
- 108.De Cecco E, Celauro L, Vanni S, et al. The uptake of Tau amyloid fibrils is facilitated by the cellular prion protein and hampers prion propagation in cultured cells [published online ahead of print, 2020 May 11].J Neurochem. 2020;10.1111. [DOI] [PubMed] [Google Scholar]
- 109.Ondrejcak T, Klyubin I, Corbett GT, et al. Cellular prion protein mediates the disruption of hippocampal synaptic plasticity by soluble Tau in vivo. J Neurosci. 2018; 38:595–10606 [DOI] [PMC free article] [PubMed] [Google Scholar]; **First report on cellular PrP as a mediator of the toxicity of soluble Tau oligomers in vivo
- 110.Pan Y, Zhao L, Liang J, et al. Cellular prion protein promotes invasion and metastasis of gastric cancer. FASEB J. 2006;20:1886–8 [DOI] [PubMed] [Google Scholar]
- 111.Han H, Bearss DJ, Browne LW, Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res. 2002;62:2890–6 [PubMed] [Google Scholar]
- 112.Li C, Yu S, Nakamura F, et al. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J Clin Invest. 2009;119:2725–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sy MS, Li C, Yu S, et al. The fatal attraction between pro-prion and filamin A: prion as a marker in human cancers. Biomark Med. 2010;4:453–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Antony H, Wiegmans AP, Wei MQ, et al. Potential roles for prions and protein-only inheritance in cancer. Cancer Metastasis Rev. 2012;31:1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shafiei SS, Guerrero-Muñoz MJ, Castillo-Carranza DL. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front Aging Neurosci. 2017;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Smith LM, Kostylev MA, Lee S, et al. Systematic and standardized comparison of reported amyloid-β receptors for sufficiency, affinity, and Alzheimer’s disease relevance. J Biol Chem. 2019;294:6042–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Westaway D, DeArmond SJ, Cayetano-Canlas J, et al. Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wt prion proteins. Cell. 1994;76:117–29 [DOI] [PubMed] [Google Scholar]
- 118.Chiesa R, Piccardo P, Biasini E, et al. Aggregated, wt prion protein causes neurological dysfunction and synaptic abnormalities. J Neurosci. 2008;28:13258–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Paitel E, Fahraeus R, Checler F. Cellular prion protein sensitizes neurons to apoptotic stimuli through Mdm2-regulated and p53-dependent caspase 3-like activation. J Biol Chem. 2003;278:10061–6 [DOI] [PubMed] [Google Scholar]
- 120.Guillot-Sestier MV, Sunyach C, Ferreira ST, et al. α-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid β (Aβ)-associated cell death. J Biol Chem. 2011;287:5021–32 [DOI] [PMC free article] [PubMed] [Google Scholar]; **First report on N1 protection against the cytotoxicity of soluble Aβ oligomers
- 121.Fluharty BR, Biasini E, Stravalaci M, et al. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J Biol Chem. 2013;288:7857–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Béland M, Bédard M, Tremblay G, et al. Aβ induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol Aging. 2014;35:1537–48 [DOI] [PubMed] [Google Scholar]
- 123.Scott-McKean JJ, Surewicz K, Choi JK, et al. Soluble prion protein and its N-terminal fragment prevent impairment of synaptic plasticity by Aβ oligomers: Implications for novel therapeutic strategy in Alzheimer’s disease. Neurobiol Dis. 2016;91:124–31 [DOI] [PMC free article] [PubMed] [Google Scholar]; *Both recombinant PrP and recombinant N1 neutralize the toxicity of Aβ oligomers
- 124.Carroll JA, Groveman BR, Williams K, et al. Prion protein N1 cleavage peptides stimulate microglial interaction with surrounding cells. Sci Rep. 2020;10:6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sonati T, Reimann RR, Falsig J, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501:102–6 [DOI] [PubMed] [Google Scholar]
- 126.Dametto P, Lakkaraju AK, Bridel C, et al. Neurodegeneration and unfolded-protein response in mice expressing a membrane-tethered flexible tail of PrP. PLoS One. 2015;10:e0117412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu B, McDonald AJ, Markham K, et al. The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife. 2017;6:e23473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vincent B, Sunyach C, Orzechowski HD, et al. p53-Dependent transcriptional control of cellular prion by presenilins. J Neurosci. 2009;29:6752–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Glatzel M, Linsenmeier L, Dohler F, et al. Shedding light on prion disease. Prion. 2015;9:244–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heiseke A, Schöbel S, Lichtenthaler SF, et al. The Novel Sorting Nexin SNX33 Interferes with Cellular PrPSc Formation by Modulation of PrPc Shedding. Traffic, 2008;9:1116–29 [DOI] [PubMed] [Google Scholar]
- 131.Kanaani J, Prusiner SB, Diacovo J, et al. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. Journal of Neurochemistry, 2005;95:1373–86 [DOI] [PubMed] [Google Scholar]
- 132.Amin L, Xuan T. A. Nguyen, Rolle IG, et al. Characterization of prion protein function by focal neurite stimulation Cell Sci. 2016;129:3878–91 [DOI] [PubMed] [Google Scholar]
- 133.Nieznanski K, Choi JK, Chen S, et al. Soluble prion protein inhibits amyloid-β (Aβ) fibrillization and toxicity. J Biol Chem. 2012;287:33104–8 [DOI] [PMC free article] [PubMed] [Google Scholar]; *Recombinant PrP and recombinant N1 inhibit the formation of toxic Aβ oligomers in vitro
- 134.Endres K, Mitteregger G, Kojro E, et al. Influence of ADAM10 on prion protein processing and scrapie infectiosity in vivo. Neurobiol Dis 2009;36:233–41 [DOI] [PubMed] [Google Scholar]
- 135.Altmeppen HC, Prox J, Krasemann S, et al. The sheddase ADAM10 is a potent modulator of prion disease. eLife, 2015;4,e04260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Watts JC, Giles K, Patel S, et al. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 2014;10: e1003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Minikel EV, Zhao HT, Le J, et al. Prion protein lowering is a disease-modifying therapy across prion disease stages, strains and endpoints. Nucleic Acids Res. 2020;48:10615–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Glatzel M, Aguzzi A. PrP(C) expression in the peripheral nervous system is a determinant of prion neuroinvasion. J Gen Virol. 2000;81:2813–21 [DOI] [PubMed] [Google Scholar]
- 139.McCulloch L, Brown KL, Bradford BM, et al. Follicular dendritic cell-specific prion protein (PrP) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog. 2011;7:e1002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Klingeborn M, Race B, Meade-White KD, et al. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol. 2011;85:1484–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chesebro B, Trifilo M, Race R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005;308:1435–9 [DOI] [PubMed] [Google Scholar]
- 142.Rangel A, Race B, Klingeborn M, et al. Unusual cerebral vascular prion protein amyloid distribution in scrapie-infected transgenic mice expressing anchorless prion protein. Acta Neuropathol Commun 2013; 1:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rangel A, Race B, Phillips K, et al. Distinct patterns of spread of prion infection in brains of mice expressing anchorless or anchored forms of prion protein. Acta Neuropathol Commun 2014; 2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chesebro B, Race B, Meade-White K, et al. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010;6:e1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stohr J, Watts JC, Legname G, et al. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci USA 2011;108:21223–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Endres K, Fahrenholz F. Upregulation of the alpha-secretase ADAM10--risk or reason for hope? FEBS J. 2010;277:1585–96 [DOI] [PubMed] [Google Scholar]
- 147.Schulte M, Reiss K, Lettau M, et al. ADAM10 regulates FasL cell surface expression and modulates FasL‐induced cytotoxicity and activation‐induced cell death. Cell Death Differ 2007;14, 1040–9 [DOI] [PubMed] [Google Scholar]
- 148.Esselens CW, Malapeira J, Colome N, et al. Metastasis‐associated C4.4A, a GPI‐anchored protein cleaved by ADAM10 and ADAM17. Biol Chem 2008;389,1075–84 [DOI] [PubMed] [Google Scholar]
- 149.Waldhauer I, Goehlsdorf D, Gieseke F, et al. Tumor‐associated MICA is shed by ADAM proteases. Cancer Res. 2008;68, 6368–76 [DOI] [PubMed] [Google Scholar]
- 150.You B, Shan Y, Shi S, et al. Effects of ADAM10 upregulation on progression, migration, and prognosis of nasopharyngeal carcinoma. Cancer Sci. 2015;106(11):1506–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pasciuto E, Ahmed T, Wahle T, et al. Dysregulated ADAM10-mediated processing of APP during a critical time window leads to synaptic deficits in fragile X syndrome Neuron, 2015;87,382–39 [DOI] [PubMed] [Google Scholar]
- 152.Lo Sardo V, Zuccato C, Gaudenzi G et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci 2012;15, 713–21 [DOI] [PubMed] [Google Scholar]
- 153.Roberts TK, Eugenin EA, Morgello S, et al. PrPC, the cellular isoform of the human prion protein, is a novel biomarker of HIV-associated neurocognitive impairment and mediates neuroinflammation. Am J Pathol. 2010;177:1848–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Megra BW, Eugenin EA, Berman JW. The Role of Shed PrPc in the Neuropathogenesis of HIV Infection. J Immunol. 2017;199:224–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yuyama K, Sun H, Sakai S, et al. Decreased Amyloid-beta Pathologies by Intracerebral Loading of Glycosphingolipid-enriched Exosomes in Alzheimer Model Mice. Journal of Biochemistry. 2014a;289:24488–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yuyama K, Sun H, Usuki S, et al. A potential function for neuronal exosomes: Sequestering intracerebral amyloid-beta peptide. Febs Letters. 2014b;589:84–8 [DOI] [PubMed] [Google Scholar]
- 157.An K, Klyubin I, Kim Y, et al. Exosomes neutralize synaptic-plasticity-disrupting activity of Aβ assemblies in vivo. Mol. Brain 2013;6,47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Falker C, Hartmann A, Guett I, et al. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J Neurochem. 2016;137:88–100 [DOI] [PubMed] [Google Scholar]; **PrP on exosomes neutralize Aβ toxicity
- 159.Fevrier B, Vilette D, Archer F, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101:9683–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vella LJ, Sharples RA, Lawson VA, et al. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol. 2007;211:582–90 [DOI] [PubMed] [Google Scholar]
- 161.Cervenakova L, Saa P, Yakovleva O, et al. Are prions transported by plasma exosomes? Transfus Apher. Sci 2016;55,70–83 [DOI] [PubMed] [Google Scholar]
- 162.Emmanouilidou E, Melachroinou K, Roumeliotis T, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hu G, Yao H, Chaudhuri AD, et al. Exosome-mediated shuttling of microRNA-29 regulates HIV Tat and morphine-mediated neuronal dysfunction. Cell Death Dis. 2012;3:e381–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Saman S, Kim W, Raya M, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287: 3842–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li X, Corbett AL, Taatizadeh E, et al. Challenges and opportunities in exosome research-Perspectives from biology, engineering, and cancer therapy. APL bioengineering, 2019;3, 011503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kong Q RNAi: a novel strategy for the treatment of prion diseases. J Clin Invest. 2006; 116:3101–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pfeifer A, Eigenbrod S, Al-Khadra S, et al. Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J Clin Invest. 2006;116:3204–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pulford B, Reim N, Bell A, et al. Liposome-siRNA-peptide complexes cross the blood-brain barrier and significantly decrease PrPC on neuronal cells and PrPRES in infected cell cultures. PLoS One. 2010; 5:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bender H, Noyes N, Annis JL, et al. PrPC knockdown by liposome-siRNA-peptide complexes (LSPCs) prolongs survival and normal behavior of prion-infected mice immunotolerant to treatment. PLoS One. 2019;14:e0219995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Raymond GJ, Zhao HT, Race B, et al. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight. 2019;5:131175. [DOI] [PMC free article] [PubMed] [Google Scholar]