

Abstract
Primase is an essential component of the DNA replication machinery, responsible for synthesizing RNA primers that initiate leading and lagging strand DNA synthesis. Bacterial primase activity can be regulated by the starvation-inducible nucleotide (p)ppGpp. This regulation contributes to a timely inhibition of DNA replication upon amino acid starvation in the Gram-positive bacterium Bacillus subtilis. Here, we characterize the effect of (p)ppGpp on B. subtilis DnaG primase activity in vitro. Using a single-nucleotide resolution primase assay, we dissected the effect of ppGpp on the initiation, extension, and fidelity of B. subtilis primase. We found that ppGpp has a mild effect on initiation, but strongly inhibits primer extension and reduces primase processivity, promoting termination of primer extension. High (p)ppGpp concentration, together with low GTP concentration, additively inhibit primase activity. This explains the strong inhibition of replication elongation during starvation which induces high levels of (p)ppGpp and depletion of GTP in B. subtilis. Finally, we found that lowering GTP concentration results in mismatches in primer base pairing that allow priming readthrough, and that ppGpp reduces readthrough to protect priming fidelity. These results highlight the importance of (p)ppGpp in protecting replisome integrity and genome stability in fluctuating nucleotide concentrations upon onset of environmental stress.
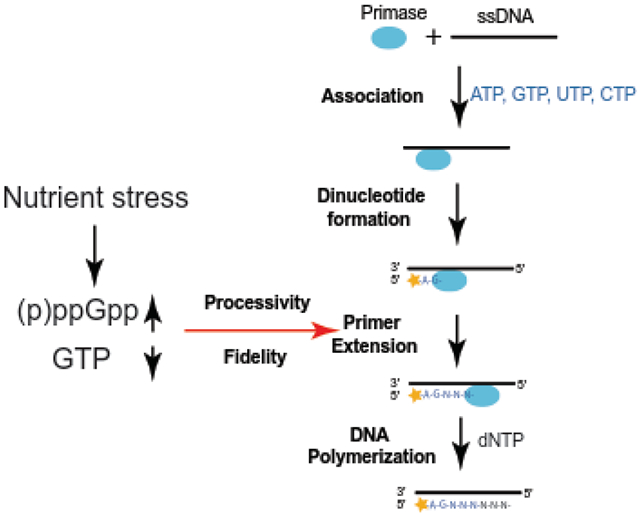
Graphical Abstract
INTRODUCTION
DNA replication is a highly regulated cellular process necessary to ensure faithful and complete duplication of genetic material (1). In most bacteria, DNA replication initiates at a single origin of replication on the circular chromosome. Bidirectional replication forks elongate from the origin until the terminus of replication is reached, producing two complete chromosomes. DNA replication is performed by a multicomponent cellular machine comprising the replicative helicase, the leading and lagging strand DNA polymerases, the DNA primase, and other accessory replication proteins. DNA primase is required to produce RNA primers for both leading and lagging strand DNA synthesis, functioning through a multi-step process (2-6). First, primase binds ssDNA and incorporates two ribonucleotides complementary to the second and third nucleotides of a trinucleotide primase initiation sequence. Next, primase extends the RNA primer in a 5′ to 3′ direction, before priming is terminated via hand-off of the RNA primer to the DNA polymerase.
Bacterial DNA primase (DnaG) is composed of three domains essential for its function and regulation: an N-terminal zinc-binding domain (ZBD) involved in recognizing and binding the DNA template, an RNA polymerase domain (RPD) containing the catalytic active site, and a C-terminal helicase interaction domain (CTD) that interacts with the replicative helicase and the single-stranded DNA binding protein (SSB) (7-13) (Figure 1A). The RPD domain is structurally unrelated to other DNA or RNA polymerases, instead being related to the active domain of type IA and type II topoisomerases. This domain is referred to as the TOPRIM (TOpoisomerase/PRIMase) domain (14-16). The interactions between primase and other replication enzymes, including the replicative helicase, SSB, or the clamp loader, regulates the activity of primase to allow its coordinated action with the rest of the replisome (7, 10, 17-22).
Figure 1. (p)ppGpp, coupled with depletion of GTP substrates, strongly inhibit B. subtilis DNA primase activity.

(A) The bacterial primase DnaG is composed of three domains: an N-terminal zinc binding domain (ZBD), an RNA polymerase domain (RPD), and a C-terminal helicase interacting domain (CTD).
(B) Fraction of [5’-α-32P]-pppGpp bound to 10 μM full-length or RPD-only B. subtilis primase DnaG, with or without single stranded DNA template oJW2384 (ssDNA, 10 μM), GTP (1 mM), or unlabeled pppGpp (1 mM). Representative DRaCALA images are shown above the graph. Error bars represent ± SEM for ≥ 3 replicates.
(C) Schematics of the fluorescence-based primase assay. NTPs are mixed with a ssDNA template and allowed to briefly incubate with or without (p)ppGpp before the addition of primase (DnaG). Aliquots of the reaction are taken at various time points after the addition of DnaG, and DNA-RNA heteroduplexes are measured with a dye that intercalates the double-stranded heteroduplex. The level of fluorescence is dependent upon the quantity of primers synthesized and is reported as relative fluorescent units (RFU).
(D-E) The initial velocity (V0) of full-length DnaG activity at indicated concentrations of ppGpp (D) or pppGpp (E). Reactions were run with 0, 12.5, 25, 50, 100, 200, or 400 μM GTP. Results are normalized to the enzyme concentration and are reported as relative fluorescent units per minute (RFU/min). Points represent averages of ≥ 3 replicates. Error bars represent ± SEM for ≥ 3 replicates. Curves were fit with an uncompetitive inhibition model for full-length DnaG with ppGpp and a competitive inhibition model for full-length DnaG with pppGpp, based on the highest goodness of fit values (R2). R2 values were obtained by fitting the data to different inhibition modes (Supplemental Figure 1-2).
(F) The initial velocity (V0) of DnaG(ZBD+RPD) activity at indicated concentrations of pppGpp. Reactions were run with 0, 12.5, 25, 50, 100, 200, or 400 μM GTP. Results are normalized to the enzyme concentration and are reported as relative fluorescent units per minute (RFU/min). Points represent averages of ≥ 3 replicates. Error bars represent ± SEM for ≥ 3 replicates. Curves were fit with an uncompetitive inhibition model, based on the goodness of fit values (R2). R2 values were obtained by fitting the data to different inhibition modes (Supplemental Figure 3).
(G) Primer synthesis rate by B. subtilis primase at indicated concentrations of GTP and (p)ppGpp (1 mM).
In addition to regulation by protein factors, bacterial primase activity can be regulated by the starvation-inducible nucleotide (p)ppGpp in response to environmental cues (23-26). (p)ppGpp, the collective name of the nucleotides guanosine tetraphosphate (ppGpp) and guanosine pentaphosphate (pppGpp), signals nutrient stress in bacteria by remodeling metabolism and regulating macromolecular machineries (27-32). In vitro analyses have shown that (p)ppGpp is capable of inhibiting the activity of DnaG primase from multiple bacterial species (23-26). Crystal structures of the active-site-containing RPD of Staphylococcus aureus DnaG bound to ppGpp or pppGpp reveal that (p)ppGpp binds to a site partially overlapping the entry point for NTP substrates, suggesting that (p)ppGpp obscures the DnaG active site to directly obstruct NTP binding (24). Because primase activity is strongly implicated in replicative helicase activity (20, 33), replication fork progression (34), and replisome stability (35), regulation of primase by (p)ppGpp may underlie a nutritional regulation of replication observed in the Gram-positive bacterium Bacillus subtilis, where replication elongation is strongly inhibited upon amino acid starvation in a (p)ppGpp-dependent, non-disruptive manner (23, 36, 37). This regulation is proposed to protect genome integrity from deleterious effects of nutrient stress (23).
Despite strong evidence for an interaction between (p)ppGpp and DnaG primase, key questions remain pertaining to the nature of this regulation. The inhibitory effect of (p)ppGpp has been established based on measurement of bulk RNA synthesis by DnaG (23, 24, 26); thus, it remains unclear whether (p)ppGpp regulates priming at initiation or affects the kinetics of extension. Furthermore, the similar in vitro effects of (p)ppGpp on DnaG from both E. coli and B. subtilis contrast with strong differences in vivo, where (p)ppGpp strongly inhibits replication elongation in B. subtilis but has only a modest effect in E. coli (23, 25, 38).
To provide a thorough understanding of how bacterial primase activity is regulated, we adapted an in vitro priming assay to examine the effect of (p)ppGpp on initiation, extension, processivity, and fidelity of B. subtilis DnaG primase. Interestingly, (p)ppGpp does not have a major impact on primer initiation (the formation of the first dinucleotide), but strongly reduces primer extension rate and processivity. Our results also suggest a surprising role of (p)ppGpp in preventing read-through of starved sites during primer extension. Because amino acid starvation in B. subtilis results in (p)ppGpp production and concomitant GTP depletion, the interplay between (p)ppGpp and GTP on priming was also examined, with implications for regulation of replication in nutrient-stressed bacterial cells.
MATERIALS AND METHODS
Protein Purification
A Bacillus subtilis DnaG full-length protein over-expression construct (pJW743) was generated via PCR amplification of genomic DNA using oligo templates oJW2646 and oJW702 (Integrated DNA Technologies, Supplemental Table 1) and inserted into the pLICtrPC-HA plasmid (39)(Supplemental Table 2). The ZBD+RPD (DnaGΔ440-603) construct pJW744 was generated similarly using oligos oJW2646 and oJW2649. Recombinant proteins were expressed in BL21 (DE3+) cells via induction with 0.1 mM IPTG prior to purification by Ni+ –affinity chromatography over a HisTrap column (GE Healthcare). Lysis buffer consisted of 300 mM NaCl, 20 mM imidazole, and 40 mM HEPES pH 7.8-8.0. Stepwise elution was carried out from 100% lysis buffer to 100% of an otherwise equivalent buffer containing 500 mM imidazole. Proteins were then dialyzed into a storage buffer that consisted of 40 mM HEPES pH 7.8, 300 mM NaCl, 10% glycerol, 1 mM EDTA, and 1 mM dithiothreitol (DTT). Purity was assessed using polyacrylamide gel electrophoresis and Coomassie staining, and protein preparations were confirmed to be ≥ 95% pure. Concentration was determined by absorption at 280 nm using the following extinction coefficients: 46,760 M−1cm−1 (full-length), 32,320 M−1cm−1 (DnaG(ZBD+RPD)). Extinction coefficients were determined using the ProtParam web tool (https://web.expasy.org/protparam/). Protein activity and binding to (p)ppGpp with and without the His-tag was assessed and no discernable difference was observed (data not shown).
Synthesis, Purification, and Quantification of (p)ppGpp
pppGpp was synthesized in vitro using RelSeq1–385 (40). 6 mM GTP was mixed with 8 mM ATP, 300 μg RelSeq1–385, 0.5 mM DTT, 25 mM bis-Tris propane pH 9.0, and 15 mM MgCl2 prior to incubation at 37°C for 2-5 hours to generate pppGpp. ppGpp was produced from pppGpp by GppA as previously described (40). (p)ppGpp was purified with an anion exchange column (HiTrap QFF 1 mL; GE Healthcare) using buffer (0.1 mM LiCl, 0.5 mM EDTA, 25 mM Tris pH 7.5). Stepwise elution was carried out by increasing LiCl from 0.1 mM to 500 mM. ppGpp and pppGpp were stored in desiccated form until immediately prior to use.
[5′ α-32P] (p)ppGpp was synthesized according to modified protocols of (p)ppGpp synthesis (41). The reaction contained 25 mM bis-Tris propane (pH 9.0), 15 mM MgCl2, 0.5 mM DTT, 2 mM ATP, 2 μM Relseq (1-385), and 37.5 μCi [α-32P]-GTP (Perkin Elmer). The reaction incubated at 37°C for 1 hour. ppGpp was produced via addition of the enzyme GppA for 1 hour. The reaction was diluted in 0.5 mL of Buffer A (0.1 mM LiCl, 0.5 mM EDTA, 25 mM Tris-HCl pH 7.5) prior to running through a 1 mL HiTrap QFF strong anion exchange column (GE Healthcare) equilibrated with 10 column volumes (CV) of 83% Buffer A + 17 % Buffer B (Buffer B: 1 M LiCL, 0.5 mM EDTA, 25 mM Tris-HCl pH 7.5). [5’-α-32P]-(p)ppGpp was eluted with a mixture of 50% Buffer A + 50% Buffer B. Fractions of 1 mL were collected from the elution.
Differential Radial Capillary Action of Ligand Assay (DRaCALA)
DRaCALA was performed following standard procedures (42). Briefly, reactions contained 40 mM HEPES pH 7.5-7.8, 1 mM MnCl2, 10 mM MgAc, 150 mM NaCl, 5% glycerol, 5 mM DTT, 10 μM purified DnaG or the isolated RPD of DnaG (diluted in 40 mM HEPES pH 7.5-7.8, 150 mM NaCl, 5% glycerol, and 5 mM DTT) and [5’-α-32P]-(p)ppGpp (1:100 final dilution of first elution fraction of [5’-α-32P]-(p)ppGpp purification). Reactions were incubated for 10 minutes at room temperature. Two microliters from each reaction were spotted in duplicate on Protran BA85 nitrocellulose (GE Healthcare) via pipette or a replicator pinning tool (VP 408FP6S2; V and P Scientific, Inc). Radioactivity was detected with phosphorimaging (Typhoon FLA9000) and fraction bound of [5’-α-32P]-(p)ppGpp was calculated as described (42).
Fluorescence-based Quantification of Primer Synthesis
Fluorometric quantifications of primer synthesis were based on a primase activity assay from ProFoldin (Hudson, MA) (43). All reactions were carried out in 50 mM Tris pH 7.5, 10 mM magnesium acetate, 25 mM potassium glutamate, 0.03% NP-40, 5 mM MnCl2, 5 mM CaCl2, and 10 mM DTT. Manganese is present in the reaction mixture as no activity was observed with magnesium alone. Reactions included 500 nM oJW2384 template (Supplemental Table 1), 0.4 mM ATP, CTP, UTP, the indicated concentrations of GTP, and the indicated concentrations of inhibitors. Reactions containing full-length DnaG included 100 nM enzyme, while reactions containing a truncated DnaG lacking the C-terminal domain (ZBD+RPD) included 200 nM enzyme. Following addition of the enzyme, reactions were incubated at 37°C for 30 minutes. For measurement of product, ProFoldin primase activity assay dye was added at 5-, 10-, 20-, and 30-minute time points after the start of the reaction and incubated without light for 5 minutes. Raw fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength of 528 nm in a BioTek Synergy2 multi-mode microplate reader. The data for DnaG and DnaG(ZBD+RPD) were normalized to the average maximum fluorescence intensity and fit by nonlinear regression through GraphPad Prism using competitive, uncompetitive, noncompetitive and mixed inhibition modes (Supplemental Figure 1-3). The best fits (uncompetitive inhibition) were presented (uncompetitive inhibition for full-length DnaG with ppGpp (Figure 1D) and DnaG(ZBD+RPD) with pppGpp (Figure 1F); competitive inhibition for full-length DnaG with pppGpp (Figure 1E)).
Competitive inhibition models were fit using the equations:
Uncompetitive inhibition models were fit using the equations:
Noncompetitive inhibition models were fit using the equations:
Mixed inhibition models were fit using the equations:
Vmax is the maximal enzyme rate (arbitrary fluorescence units (AFU)/min), K’M is the apparent KM of GTP in the absence of inhibitors, α is a value corresponding to the degree to which the binding of the inhibitor changes the substrate affinity of the enzyme, and Ki is the dissociation constant for inhibitor binding. Mean V0 and SEM were calculated from n ≥ 3 replicates.
Radiolabeled Primer Extension and Gel Separation
Primer extension assays were performed similarly to (3) with modifications. Reactions contained 40 mM HEPES, pH 7.5, 1% glycerol, 1 mM magnesium acetate, 1 mM MnCl2, 5 mM DTT, 6.4 μM DnaG, and 500 nM ssDNA templates listed in Supplemental Table 1. All primers were PAGE-purified standard oligos with the exception of oJW3668, which was blocked at the 3’ end as described in (43). Reaction mixtures had 373 nM [γ-32P]-ATP (5 μCi/nmol, Perkin Elmer). The reactions were briefly incubated at room temperature with ppGpp at the indicated concentrations before addition of the unlabeled NTPs. After addition of the unlabeled nucleotides (1 mM or 0.4 mM NTP, and 1 mM or 0.1 mM GTP as indicated in each figure), the reactions were incubated for 30 min at 30 °C. Reactions were stopped upon addition of 95% formamide, 5 mM EDTA, 0.09% xylene cyanol FF, heated for 5 min at 95 °C, and resolved in denaturing 20% polyacrylamide gels (acrylamide/bisacrylamide ratio, 19:1; 7 M urea) for 2 hours at 2000 V (an equivalent of 2.67 μL of reaction mix was loaded per lane). A Typhoon Phosphorimager was used to detect luminescence following exposure of the gel to a phosphor screen. Data were quantified using Image-J software. Lengths of RNA primers were compared against RNA standards with the predicted primer sequence derived from the template.
RNA standards were prepared using an RNA oligonucleotide with the complementary sequence to the primase template. The 5′-end was labeled with [γ-32P]-ATP (Perkin Elmer) and separated from ATP using a nucleotide clean up kit (Qiagen). 120 nM purified, labeled RNA was hydrolyzed for 5 minutes at 45°C in 300 nM NaOH. Hydrolysis reactions were neutralized with 2 volumes of 2 M Tris-HCl (pH 7-7.5). Stop buffer (95% formamide, 5 mM EDTA, 0.09% xylene cyanol FF) was added and the hydrolyzed fragments were heated for 2 minutes at 95°C and quickly cooled on ice. The RNA cleavage product can be hydrolyzed to either a 2'- or a 3'-nucleotide, which can be resolved at shorter RNA lengths, resulting in two bands per nucleotide length (Supplemental Figure 4).
RESULTS
(p)ppGpp, together with decreased GTP, strongly inhibits B. subtilis DnaG primase
We first applied the differential radial capillary action of ligand assay (DRaCALA) (42, 44) to confirm a direct interaction between B. subtilis primase (DnaG) and [5’-α-32P]-pppGpp. DRaCALA relies on the different migration properties of protein and ligand on nitrocellulose. Protein will diffuse slowly when a solution is spotted onto the membrane, while ligand will diffuse rapidly. However, when ligand interacts with protein, the ligand will co-migrate with the protein and can be quantified as the fraction of total ligand bound to the protein (Figure 1B). Here, the use of DRaCALA showed that pppGpp binds recombinantly expressed, purified DnaG. Among the three domains of DnaG, the RNA polymerase domain (RPD) was sufficient for strong binding (Figure 1B). Unlabeled pppGpp effectively competed for binding to DnaG, indicating that the observed DRaCALA result was not due to protein aggregation or precipitation of [5’-α-32P]-pppGpp. GTP was effective in competing with [5’-α-32P]-pppGpp for binding DnaG. These results are consistent with structural analyses with E. coli DnaG which indicate pppGpp binds the RPD at a site overlapping the substrate GTP (24).
Next, we used a fluorescence-based assay (43) to measure the effect of (p)ppGpp on the enzymatic activity of B. subtilis primase. Primase was mixed with NTPs and a single-stranded DNA template containing the preferred initiating sequence for B. subtilis primase, 5’-CTA (18, 45). The priming product, initiating from the formation of the 5’-AG dinucleotide, was measured via fluorescence of a dye that intercalates between the bases of RNA-DNA heteroduplexes (Figure 1C). We were initially unable to detect priming activity using the standard buffer with magnesium. However, in the presence of 1 mM or higher concentrations of manganese, we were able to detect significant priming activity. Using this assay, we assessed the ability of (p)ppGpp to inhibit primase activity with varying concentrations of GTP (Figure 1D-E). We then fit the data using uncompetitive, noncompetitive, and mixed inhibition models (Supplemental Figure 1, 2, 3). In order to evaluate which model fits best, we compared the goodness of fit values (R2), which showed a modest difference. The best fits agree with either a mixed inhibitory model previously established through characterization of (p)ppGpp’s effect on E. coli primase (24), or an uncompetitive inhibition model for ppGpp and a competitive inhibition model for pppGpp (Table 1).
Table 1. Kinetic parameters related to Figure 1.
Vmax is the maximal enzyme rate in arbitrary fluorescence units (AFU) in the absence of inhibitor. K’M is an apparent KM of GTP in the absence of (p)ppGpp. Ki is the inhibition constant for (p)ppGpp. Values were determined as described in Materials and Methods using an uncompetitive inhibition model for full-length DnaG with ppGpp and DnaG(ZBD+RPD) with pppGpp, and a competitive inhibition model for full-length DnaG with pppGpp.
| Full-Length DnaG, ppGpp (Uncompetitive inhibition) R2=0.869 | ||
|---|---|---|
| Vmax (AFU) ± SEM | K’M (μM) ± SEM | Ki (μM) ± SEM |
| 800.0 ± 49.1 | 78.7 ± 12.5 | 776.7 ± 110.3 |
| Full-Length DnaG, pppGpp (Competitive inhibition), R2= | ||
| Vmax (AFU) ± SEM | K’M (μM) ± SEM | Ki (μM) ± SEM |
| 804.4 ± 72.6 | 80.4 ± 20.4 | 162.6 ± 37.0 |
| DnaG(ZBD+RPD), pppGpp (Uncompetitive inhibition), R2= | ||
| Vmax (AFU) ± SEM | K’M (μM) ± SEM | Ki (μM) ± SEM |
| 295.2 ± 15.6 | 9.0 ± 2.8 | 115.0 ± 17.8 |
Next, we examined whether the interaction between the RPD domain and (p)ppGpp is sufficient to inhibit primase activity. Since in addition to the RPD, the N-terminal ZBD domain of DnaG is also required for primase activity, whereas the C-terminal domain of DnaG plays a regulatory role, we examined the specific effects of (p)ppGpp on a DnaG variant containing the ZBD and RPD domains, but lacking the C-terminal domain. This variant is also called P49 for E. coli primase, named after its molecular weight (46). For clarity, we refer to the B. subtilis variant as DnaG(ZBD+RPD). The ZBD+RPD variant was active in priming, with higher affinity to GTP, and its activity was inhibited by (p)ppGpp more strongly than full-length DnaG primase (Figure 1F, Table 1). This result agrees with the observed stronger binding of pppGpp to RPD than to full-length primase (Figure 1B). Taken together, our data suggest that (p)ppGpp binding of the RPD inhibits B. subtilis primase activity.
In B. subtilis, (p)ppGpp inhibits multiple purine nucleotide biosynthesis enzymes along the GTP biosynthesis pathway. Therefore, cellular nucleotide concentrations in B. subtilis during amino-acid starvation include both high (p)ppGpp (up to 1 - 2 mM) and very low GTP (down to ~0.1 mM) (32, 47). Because high levels of (p)ppGpp and low levels of GTP additively inhibit DnaG primase activity (Figure 1G), this could explain the nearly complete arrest of replication elongation during amino acid starvation in B. subtilis (23).
ppGpp inhibits extension of RNA primers by B. subtilis primase
The experiments above and prior published work evaluating the effects of (p)ppGpp on primase activity relied on measurement of total base pairs of all RNA-DNA heteroduplexes produced. However, there are various facets of DnaG primase action, including its binding to the DNA template, the initiation of primer synthesis (the successful production of the dinucleotide at the priming initiation sequence), the primer extension beyond the dinucleotide, and the processivity and fidelity of the enzyme.
To understand how (p)ppGpp regulates DnaG primase initiation and extension, we performed primase assays in the presence of [γ-32P]-ATP to specifically label the 5’ end of newly synthesized primer, then denatured and resolved the nascent primers via gel electrophoresis using urea-PAGE (Figure 2A). We were able to separate primers as small as dinucleotides (formed at the primer initiation step) to the maximum templated size (29 nucleotides). Detectable dinucleotide and longer primer formation depended strongly on the presence of DnaG primase and increased as primase concentration increased, until a stoichiometric ratio of primase to DNA template was reached (Figure 2, Supplemental Figure 5), in agreement with previous observations that following synthesis of dinucleotides the primase remains stably associated with the primer-template complex (22). The priming efficiency, which is the number of total primers synthesized relative to the number of templates, reaches ~100% as primase concentration surpasses DNA template. Thus, the initiation of priming is a rate-limiting step that strongly depends on primase association with the ssDNA template. Recently, B. subtilis primase was found to bind less frequently to the replication fork following nutrient downshift (which induces (p)ppGpp), suggesting that primase-template association is directly or indirectly affected by (p)ppGpp (48). Therefore, we next examined whether dinucleotide formation is directly controlled by (p)ppGpp.
Figure 2. Single nucleotide resolution primase assay with urea-PAGE denaturing gel.

(A) Schematics of radioactivity-based assay for detection of priming product of varying lengths, adapted from (3). Each detected primer is γ-32P-labelled only at the 5' end of the RNA. Products were denatured and resolved with 7M Urea 20% PAGE. The ssDNA template is 37 nucleotides long and can generate up to 29 nucleotide primers from the initiating site.
(B) Titration of DNA primase with 500 nM ssDNA, 0.4 mM NTPs (CTP, UTP, GTP) and ~0.4 μM [γ-32P]-ATP. Primers from 2 nucleotides (bottom) up to intermediate and full length (29 nucleotides) primers were observed. A small amount of primers longer than 29 nucleotides are due to impurity of the DNA template (Supplemental Figure 12). The sequence of the RNA standard is as follows: 5’-AGUGUGUGUGUGUGUGUGUGUGUGUGUCUG.
Using the single nucleotide resolution priming assay, we visualized production of primers over time using PAGE-purified DNA template oJW3319 (5’-AGAGAGAGAGAAGAGCCCCCTAGAGAGA, Supplemental Table 1) as a template. Dinucleotide (AG) formed rapidly within the first two minutes, followed by 9-13 nucleotide bands within 5-10 minutes, and finally the full-length primer of 21 nucleotides increasing in intensity throughout the 60 minutes duration of the reaction (Figure 3, Supplemental Figure 6). This range of B. subtilis primer lengths are similar to prior measurements of B. subtilis primase which produces RNA primers ranging from 5 nucleotides to full-length primers, with the majority being 10 nucleotide primers (45). Thus, we can monitor different rates of DnaG primase activity, including rapid primer initiation (the formation of the dinucleotide) (Figure 3C) and two phases of primer extension- an extension phase up to 9-13 nucleotides with intermediate rates (Figure 3D), and a slow extension rate up to full length (Figure 3E).
Figure 3. ppGpp inhibits primer extension when DNA template is in excess to primase.

(A) Representative gel of primer synthesis over the course of an hour (60 min) with time points taken at 2, 5, 10, 20, 30, and 60 minutes. Reactions contained 400 nM DnaG, 5 μM ssDNA, ~0.4 μM [γ-32P]-ATP, and 1 mM NTPs. Reactions on the left do not contain ppGpp, while reactions on the right contain 1 mM ppGpp.
(B) Quantification of the total number of primers synthesized over the course of an hour without ppGpp (black line) and with 1 mM ppGpp (blue line). Primer bands were quantified by comparing the signal of the primer(s) of interest to the signal of a known amount of RNA standard (Supplemental Figure 4). The same RNA standard was used to confirm the length of priming products. Points represent n = 3, and the SEM is indicated by error bars.
(C) Quantification of the initiating dinucleotide formed over the course of an hour without ppGpp (black line) and with 1 mM ppGpp (blue line). Points represent n = 3, and the SEM is indicated by error bars.
(D) Quantification of the intermediate primers 9-13 nucleotides long synthesized over the course of an hour without ppGpp (black line) and with 1 mM ppGpp (blue line). Points represent n = 3, and the SEM is indicated by error bars.
(E) Quantification of the full-length (21 nucleotide) primers synthesized over the course of an hour without ppGpp (black line) and with 1 mM ppGpp (blue line). Points represent n = 3, and the SEM is indicated by error bars.
We next examined the effect of 1 mM ppGpp on both the initiation and extension of primers. ppGpp has a significant but modest effect on total primer synthesis (Figure 3B). However, the step of dinucleotide synthesis (initiation) was not significantly affected by ppGpp (Figure 3C). Instead, ppGpp strongly reduced the subsequent extension steps compared to the reaction without ppGpp (Figure 3D-E). This suggests that the main effect of ppGpp is on primer extension.
In the above assay conditions, the ssDNA template was in 12.5-fold molar excess to the primase, and we observed 8-9% of priming efficiency (number of priming products per template), in good agreement with the hypothesis that primase performs one round of priming and remains associated with the primer-template complex. However, we cannot rule out a small fraction of primase performing multi-round priming, e.g. primase may finish priming a ssDNA template and then associate with a new one. To examine the effect of ppGpp on a single round of primer extension without the complication of DnaG primase re-initiation, we performed a reaction in which DnaG was in 12.8-fold molar excess to the template. Priming activity was measured 30 minutes following the start of the reaction (Figure 4, Supplemental Figure 7). Reactions were completed with either 0.1 mM or 1 mM of the substrate nucleotide GTP, as these are physiologically relevant concentrations with or without amino acid starvation (47). We verified that lower levels of substrate nucleotide GTP (0.1 mM) reduced primer extension compared to a higher concentration (1 mM GTP). Importantly, when increasing amounts of ppGpp were added, a clear and strong reduction in primer extension was observed (Figure 4B-C). In contrast, the total number of primers generated per template remains ~100% even in the presence of high (p)ppGpp concentrations. The dinucleotide formed via primer initiation remained mostly constant, and even arguably higher in intermediate ppGpp concentrations, likely due to the inhibition of extension without inhibition of initiation. Altogether, our data suggest that both lowering GTP and increasing ppGpp primarily inhibits primer extension and very modestly reduces primer initiation at high ppGpp concentrations.
Figure 4. ppGpp inhibits primer extension when primase is in excess to the DNA template.

(A) Representative gel showing 5' labeled RNA primers produced with 6.4 μM DnaG, 500 nM oJW3319, ~0.4 μM [γ-32P]-ATP, 1 mM each of UTP and CTP, and either 0.1 mM GTP (left) or 1 mM GTP (right) 30 minutes after the reaction started. RNA primer lengths were determined by comparison to a 21 nucleotide RNA standard.
(B, C) Quantification of primers synthesized over increasing amounts of ppGpp for the dinucleotide products, intermediate products, 21 nucleotide products (full-length primers), and total synthesized primers at 0.1 mM GTP (B) and 1 mM GTP (C). Points represent averages of 3 independent experiments, and the SEM is indicated by error bars.
ppGpp decreases the processivity of DnaG primase
We next sought to determine if the inhibition of primer extension by ppGpp can be attributed to decreased DnaG primase processivity, i.e. by reducing the length of primer before primase is released from the template. To ascertain the mechanism of inhibition, we applied an assay often used in characterizing RNA polymerase processivity - the heparin competition assay (49-52). Heparin is used to block protein rebinding to DNA or RNA through its helical structure and polyanionic nature (53). Therefore, we added heparin (~5 μg/mL) alongside the NTPs to a pre-incubated reaction mix of DnaG, ppGpp, and the ssDNA template (Figure 5). In these reactions, the ssDNA template was in 12.5-fold molar excess to DnaG. DnaG pre-bound to the template would initiate both the de novo synthesis of the primer and extension. Once DnaG fell off, it would be prohibited by heparin from rebinding to the partially extended RNA to continue extension. Compared to the same assay without heparin (Figure 3C-E), in which the primers gradually extend to full length, the reactions with heparin yielded primers which were predominantly 2 nucleotides long and only a very small amount of 7 - 21 nucleotide primers (Figure 5A-D, Supplemental Figure 8). When ppGpp was added to the reaction, the total number of primers was comparable to that without ppGpp (Figure 5B). However, primase extension was reduced in the presence of ppGpp, with a 40% decrease of 7 – 21 nucleotide primers (Figure 5D) and complete disappearance of the full-length, 21 nucleotide primers (Figure 5A). These data support the conclusion that ppGpp decreases primase processivity, allowing primase to be released from the template after generating a shorter RNA primer.
Figure 5. ppGpp reduces the processivity of DnaG primase.

(A) Representative gel of a primase activity time course in the presence of heparin (~5 μg/mL) with 400 nM DnaG and 5 μM ssDNA. Reactions on the left contain no ppGpp, while reactions on the right contain 1 mM ppGpp.
(B) Quantification of total primers synthesized without ppGpp (black line) and with 1 mM ppGpp (blue line) over the course of one hour. The total amount of primers synthesized remains largely unchanged with the addition of ppGpp. Points represent averages of n = 3, and the SEM is indicated by error bars.
(C) Quantification of primers 2-6 nucleotides in length without ppGpp (black line) and with 1 mM ppGpp (blue line) over the course of one hour. Synthesis of primers 2-6 nucleotides remains unchanged with the addition of ppGpp. Points represent averages of n = 3, and the SEM is indicated by error bars.
(D) Quantification of primers 7 - 21 nucleotides in length without ppGpp (black line) and with 1 mM ppGpp (blue line) over the course of one hour. Addition of ppGpp slightly reduced synthesis of primers 7 - 21 nucleotides long. Points represent averages of n = 3, and the SEM is indicated by error bars.
The precise mechanism of reducing the processivity of primase is unclear. Perhaps the most interesting possibility is direct (p)ppGpp incorporation at the 3’-end of the primer with subsequent chain-termination and dissociation of the enzyme caused by the high negative charge at the 3’-end. Indirect evidence for such an incorporation is the accumulation of a “shadow” band appearing one nucleotide shorter than the main intermediate length band in Figures 4A, 5A, and 6B. A short DNA oligo with an additional pyrophosphate at its 3’-end will migrate faster than a regular oligo of the same length. However, our preliminary assay with [5’-α-32P]-pppGpp did not detect significant incorporation of radioactivity into RNA primers by primase (data not shown). Alternatively, the “shadow” band may be due to mis-incorporated nucleotides. Further experiments will be required to differentiate the precise molecular mechanism of this regulation.
Figure 6. Low GTP promotes primase readthrough at starved CTP site, and high ppGpp reduces primase readthrough.

(A) Schematic of the readthrough assay using oligo template oJW3319, DnaG, [γ-32P]-ATP, UTP, and the indicated concentration of GTP as seen in (B). Priming starts from [γ-32P]-A at B. subtilis-preferred priming initiation sequence ATC, upstream of a G-site requiring CTP for base-pairing and incorporation. As CTP is withheld from these reactions, priming products should not be able to incorporate a nucleotide upon reaching the G starvation site, or readthrough via mismatches.
(B) A representative gel of primase reactions with 6.4 μM DnaG, 500 nM oJW3319, ~0.4 μM [γ-32P]-ATP, 1 mM UTP, and either 0.1 mM GTP (left) or 1 mM GTP (right). Primase reads through a CTP starvation site at low levels of GTP (expected product 7 nucleotides).
(C) Quantification of readthrough primers as a percentage of total primers synthesized at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Readthrough primer products constitute a higher percentage of the total primer products at 0.1 mM GTP, but decrease as ppGpp concentration increases. Points represent averages of n = 3, and the SEM is indicated by error bars.
(D) Quantification of total primers synthesized over increasing concentrations of ppGpp at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars.
ppGpp protects priming fidelity from lowering concentrations of GTP
Finally, we examined the effect of ppGpp on priming fidelity. To do so, we utilized a DNA template with a G-site requiring base-pairing with a CTP after a 7-nucleotide extension from the CTA initiating sequence (AGAGAGAGAGAAGAGCCCCCTAGAGAGA, Figure 6A). Therefore, when CTP was withheld from the reaction mix, DnaG primase should have halted at the starved site (G in the template), resulting in priming products 7 nucleotides or shorter. Yet, in addition to the expected prominent 7 nucleotide band, we also observed a significant fraction of primers longer than 7 nucleotides when GTP concentration was 0.1 mM (Figure 6B-D, Supplemental Figure 9). This result can be explained by incorporation of other NTPs by primase at the starved sites and extension further after the mismatch- the ~8 discrete bands longer than 7 nucleotides correspond to the 7 starvation sites and repeated readthrough up to full-length products. Alternatively, these longer primers can be primer repeat expansions due to a stretch of four C’s in the template or due to extension from the 3’ end of the DNA template. We ruled out these alternative possibilities by switching to an alternative template with a 3’-3’ linkage that prevents 3’ extension, lacking the stretch of Cs and containing a single starvation site instead of repeated starvation sites (Supplemental Table 1, oJW3668). With this alternative template, we still observed primers extended beyond the starved site up to the full length expected for a complete template-directed primer (Supplemental Figure 10). This outcome indicates that longer primers are indeed due to readthrough of the starved site via mismatch.
Interestingly, when ppGpp is added to the reactions, the readthrough products significantly decreased (Figure 6C), while the total number of primer products synthesized remains ~100% of the template and does not vary significantly with increasing ppGpp concentration (Figure 6D). These observations suggest that ppGpp has a stronger effect in preventing priming readthrough than inhibiting initiation of primer synthesis.
The DnaG(ZBD+RPD) variant performed similarly to full-length DnaG (Supplemental Figure 11), with extensive priming readthrough at 0.1 mM GTP (Supplemental Figure 11A). Adding ppGpp decreased the readthrough primers observed, akin to the decrease observed with the full-length protein (Supplemental Figure 11C). Altogether, these results suggest that ppGpp, by binding to the RPD active site, protects priming fidelity.
DISCUSSION
Bacterial primase is a highly conserved essential replication enzyme required for both leading and lagging strand synthesis. In Gram positive bacteria, the activity of primase can be directly inhibited by the starvation-induced signaling nucleotide (p)ppGpp. Though there is strong evidence for the inhibitory effect of (p)ppGpp on primase activity (23, 24, 26), the specific stage(s) of primer synthesis inhibited by (p)ppGpp remained unclear. In this work, we found that (p)ppGpp strongly inhibits primer extension and reduces primase processivity. Furthermore, our work suggests (p)ppGpp promotes priming fidelity. In addition to (p)ppGpp, varying concentrations of the substrate nucleotide GTP also affect primase activity. (p)ppGpp and GTP occupy overlapping sites on DnaG (24) and increasing levels of (p)ppGpp are coupled with lowered levels of GTP in B. subtilis cells. We show that lowering GTP and increasing (p)ppGpp additively reduces DnaG primase activity, suggesting that the opposing changes in both nucleotide concentrations may underlie the stronger inhibition of replication elongation during starvation in Gram-positive bacteria. In contrast to their additive effect on primer extension, lowering GTP and (p)ppGpp have opposing impacts on priming fidelity. Lowering GTP promotes priming readthrough at a starved site likely via misincorporation of NTPs, while (p)ppGpp prevents priming readthrough to promote priming fidelity. Our results reveal an importance for (p)ppGpp in regulating DNA replication in the context of unbalanced nucleotide concentrations upon nutrient stress.
Diversity of (p)ppGpp regulation of DNA replication in different bacterial species
Our study offers an explanation for the difference in (p)ppGpp-dependent control of replication elongation between E. coli and B. subtilis. In vitro work has shown that (p)ppGpp inhibits DnaG similarly in both species (23, 24, 26). However, in vivo, ppGpp-dependent inhibition of replication elongation upon starvation is far stronger in B. subtilis than in E. coli. In E. coli, amino acid starvation regulates replication mainly through decreased initiation from the origin of replication (oriC) (54-56). (p)ppGpp is also involved in growth rate control of replication in E. coli, as its presence is necessary for maintaining reduced DNA replication initiation when growth rates are low (57). Although there is a dose-dependent inhibition of replication elongation by (p)ppGpp upon amino acid starvation in E. coli (up to ~10% in wild type cells, and ~30% in a mutant (ΔgppA) with elevated pppGpp) (25, 38), inhibition of B. subtilis replication elongation under similar starvation conditions is much stronger, completely pausing for extended periods of time under strong starvation (23, 25, 36, 37). This disparity between in vivo and in vitro results can be explained by the finding that in B. subtilis, inhibition of DnaG primase is strongest when (p)ppGpp levels are high and GTP levels are low (Figure 1). In B. subtilis, (p)ppGpp has a stronger inhibitory effect on GTP production than in E. coli, resulting in GTP depletion by more than 10-fold (32, 47, 58). This would result in far lower DnaG primase activity, and thus reduced replication elongation, in B. subtilis cells. In E. coli, amino acid starvation induces (p)ppGpp but does not reduce GTP levels to the same extent as in B. subtilis, explaining the mild, dose-dependent inhibition of replication elongation observed in starved E. coli cells (25).
Regulation of primase as a mechanism for preventing replication fork disruption
Throughout replication, forks may encounter deleterious events such as physical obstacles, DNA lesions, and starvation of NTPs, triggering replication fork breakage and requiring fork rescue and replication restart for cell survival (59). These deleterious events take place far more frequently during amino acid starvation, which has been shown to result in alteration of nucleotide pools and elevation of replication transcription conflicts (32, 60, 61). Failure to reactivate stalled or collapsed forks is a source of genome instability or lethality. Regulation of replication that results in non-disruptive fork arrest, however, can prevent these deleterious events from taking place. Since primase interacts with multiple components of the replisome, affecting helicase activity (20), recruitment of DNA polymerase and primer handoff (11, 22, 35), regulation of primase by (p)ppGpp may in turn regulate replication fork progression. Slowing down fork progression may prevent run-away replication in starved bacterial cells, preventing subsequent disruptive replication fork arrest due to lack of resources or barriers to replication such as transcription conflicts. Recent single molecule analysis in B. subtilis revealed that (p)ppGpp-inducing starvation results in a large increase of freely diffusive molecules for the primase (48), in agreement with a lack of helicase activity exposing new DNA template and primase initiation sites in a model of non-disruptive replication fork arrest. It also agrees with our in vitro result that high levels of (p)ppGpp reduce DNA primase processivity and leads to dissociation of primase from the DNA sooner, which may also halt progression of the replication fork.
In addition to confirming that regulation of DnaG by (p)ppGpp is mediated through its active site-containing RPD, our work also suggests an allosteric interaction between the RPD and CTD domains of primase. Removing the CTD domain strengthened the regulation of DnaG by (p)ppGpp (Figure 1F) and a higher fraction of (p)ppGpp was capable of binding DnaG when only the RPD was present (Figure 1B). Finally, higher affinity for the substrate GTP is observed in the ZBD+RPD variant compared to the full-length enzyme (Table 1). These results suggest that the allosteric interaction between the CTD and RPD lowers the affinity of RPD for both the GTP substrate and the (p)ppGpp inhibitor. This effect may be further regulated by CTD interaction with the replicative helicase or SSB. Conversely, (p)ppGpp, by interacting with the RPD and its potential allosteric effect on the CTD, may also regulate the helicase, SSB or the rest of the replisome. Further mechanistic work in the context of these components will be needed to elucidate the molecular details of this multifaceted interaction leading to controlled progression of the replication fork.
DnaG priming fidelity and genome stability in bacteria
We identified an unexpected effect of (p)ppGpp on priming readthrough at a starved NTP site, implicating (p)ppGpp in promoting DnaG priming fidelity. A common mechanism in promoting polymerization fidelity is via kinetic proofreading. The theoretical framework for kinetic proofreading was first established for ribosomes and DNA polymerase in which slower enzyme activity promotes higher enzyme fidelity (62). In transcription, (p)ppGpp has been shown to indirectly promote fidelity of RNA polymerase by affecting the transcription factor DksA (63). We propose that (p)ppGpp inhibits primer extension by DnaG primase, thus promoting priming fidelity via kinetic proofreading.
Low priming fidelity is not unexpected because bacterial primases contain the catalytic RPD domain related to the metal binding center of type IA and type II topoisomerases, some nucleases, and proteins involved in recombinational repair (15, 16), referred to as the TOPRIM domain. The open conformation of the TOPRIM active site of primase allows less discriminate selection of nucleotides, potentially contributing to error-prone synthesis ((14). In addition, the presence of manganese in the reaction buffer, which is required for primase activity in the absence of the helicase, may result in higher infidelity. Lack of priming fidelity has not been regarded as problematic, as RNA primers are eventually degraded and replaced by DNA during Okazaki fragment processing. However, the magnitude of the infidelity and its dependence on concentrations of GTP is unexpected. For example, at 1mM GTP levels, a concentration in nutrient replete B. subtilis cells, DnaG primase makes almost exclusively primers that stopped at a starved CTP site. However, at 0.1mM GTP levels, a concentration that B. subtilis encountered during starvation (47), priming readthrough occurs in 50% of the priming products, indicating that 50% of priming products contain mismatches (Figure 6). Such conditions can readily happen in vivo, when GTP levels are depleted during nutrient starvation, resulting in strongly elevated priming error. Although priming errors likely do not affect replication fidelity because RNA primers are eventually degraded and replaced by high fidelity DNA, our observation suggests that priming can be extremely inaccurate under certain conditions that may cause problems for replication. For example, we can speculate that complete loss of fidelity may affect how the primer-template duplex is handed off to the DNA polymerase during starvation. Since ppGpp accumulation reduces such priming infidelity, regulation of priming by (p)ppGpp may protect priming fidelity to ensure processive genome duplication and genomic integrity.
Supplementary Material
Supplemental Figure 1. Various inhibition models for reactions with ppGpp and full-length DnaG inFigure 1. The initial velocity (V0) of full-length DnaG activity at indicated concentrations of ppGpp. Reactions were run with 0, 12.5, 25, 50, 100, 200, or 400 μM GTP. Results are normalized to the enzyme concentration and are reported as relative fluorescent units per minute (RFU/min). Points represent averages of ≥ 3 replicates. Error bars represent ± SEM for ≥ 3 replicates. Curves were fit with a competitive inhibition (A), mixed model inhibition (B), noncompetitive inhibition (C), and an uncompetitive inhibition model (D). The best fit model is uncompetitive inhibition for full-length DnaG and ppGpp.
Supplemental Figure 2. Various inhibition models for reactions with pppGpp and full-length DnaG inFigure 1. The initial velocity (V0) of full-length DnaG activity at indicated concentrations of pppGpp. Reactions were run with 0, 12.5, 25, 50, 100, 200, or 400 μM GTP. Results are normalized to the enzyme concentration and are reported as relative fluorescent units per minute (RFU/min). Points represent averages of ≥ 3 replicates. Error bars represent ± SEM for ≥ 3 replicates. Curves were fit with a competitive inhibition (A), mixed model inhibition (B), noncompetitive inhibition (C), and an uncompetitive inhibition model (D). The best fit model is competitive inhibition for full-length DnaG and pppGpp.
Supplemental Figure 3. Various inhibition models for reactions with pppGpp and DnaG(ZBD+RPD) inFigure 1. The initial velocity (V0) of DnaG(ZBD+RPD) activity at indicated concentrations of pppGpp. Reactions were run with 0, 12.5, 25, 50, 100, 200, or 400 μM GTP. Results are normalized to the enzyme concentration and are reported as relative fluorescent units per minute (RFU/min). Points represent averages of ≥ 3 replicates. Error bars represent ± SEM for ≥ 3 replicates. Curves were fit with a competitive inhibition (A), mixed model inhibition (B), noncompetitive inhibition (C), and an uncompetitive inhibition model (D). The best fit model is uncompetitive inhibition for DnaG(ZBD+RPD) and pppGpp.
Supplemental Figure 4. RNA standard ran alongside primase reactions. Standard was generated by labeling the 5' end of an RNA with a sequence identical to that of the primase reaction product. The labeled RNA was subjected to NaOH hydrolysis to generate the RNAs of different lengths. The hydrolysis was able to occur on either the 2' or 3'-OH which is why on the very short RNAs (2-4) there are two different species.
Supplemental Figure 5. Chromatograms representing band intensity from the gel inFigure 2. Reactions titrated DnaG with 500 nM ssDNA, 0.4 mM NTPs (CTP, UTP, GTP) and ~0.4 μM [γ-32P]-ATP. Primers from 2 nucleotides (bottom) up to intermediate and full-length (29 nucleotides) primers were observed. A small amount of primers longer than 29 nucleotides are due to impurity of the DNA template (Supplemental Figure 12) and result in disrupted peaks within the chromatograms near 29 nucleotides for concentrations of DnaG above 1600 nt.
Supplemental Figure 6. Chromatograms representing band intensity from the gel inFigure 3. Primer synthesis was measured over the course of one hour (60 min) without ppGpp (A) or with 1 mM ppGpp (B). Time points were taken at 2, 5, 10, 20, 30, and 60 minutes. Reactions contained 400 nM DnaG, 5 μM ssDNA template, ~0.4 μM [γ-32P]-ATP, and 1 mM NTPs (GTP, CTP, UTP). Diminished chromatogram peak size over time for primers longer than the dinucleotide show reduction in primer extension with 1 mM ppGpp (B) compared to no ppGpp (A).
Supplemental Figure 7. Chromatograms representing band intensity from the gel inFigure 4. Primer synthesis reactions contained 6.4 μM DnaG, 500 nM oJW3319, ~0.4 μM [γ-32P]-ATP, 1 mM each of CTP and UTP, and either 0.1 mM GTP (A) or 1 mM GTP (B). Reactions incubated 30 minutes after addition of NTPs. Higher GTP concentrations promote synthesis of priming products above 6 nucleotides, while higher ppGpp concentrations inhibit synthesis of full-length (21 nucleotide) products. Dinucleotide formation appears to be unaffected by GTP or ppGpp concentration. Together, this data suggests that primer extension is regulated by GTP and ppGpp concentrations.
Supplemental Figure 8. Chromatograms representing band intensity from the gel inFigure 5. Primer synthesis was measured over the course of one hour (60 minutes) in the presence of heparin (~5 μg/mL) either without ppGpp (A) or with 1 mM ppGpp (B). Time points were taken at 2, 5, 10, 20, 30, and 60 minutes. Reactions contained 400 nM DnaG, 5 μM ssDNA template, ~0.4 μM [γ-32P]-ATP, and 1 mM NTPs (GTP, CTP, UTP). Synthesis of 7 nucleotide and longer priming products is slightly reduced in the presence of ppGpp.
Supplemental Figure 9. Chromatograms representing band intensity from the gel inFigure 6. Primer synthesis reactions contained 6.4 μM DnaG, 500 nM oJW3319, ~0.4 μM [γ-32P]-ATP, 1 mM UTP, and either 0.1 mM GTP (A) or 1 mM GTP (B). Since CTP is withheld in these reactions, priming products should be 7 nucleotides long. Readthrough is observable at low GTP but diminishes as ppGpp concentrations increase.
Supplemental Figure 10. DNA primase reads through a starved site in an alternative oligonucleotide template. (A) Representative gel of primase reactions with 6.4 μM DnaG and 500 nM oJW3668 with CTP-starved site (expected product is 7 nucleotides). Oligo oJW3668, unlike oJW3319 in Figure 6, lacks a CCCC repeat in its sequence. (B) Chromatograms representing band intensity from (A) at 0.1 mM GTP (top) and 1 mM GTP (bottom) with increasing concentrations of ppGpp. (C) Quantification of readthrough primers as a percentage of total primers synthesized at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars. (D) Quantification of total primers synthesized over increasing concentrations of ppGpp at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars.
Supplemental Figure 11. ppGpp reduces readthrough of a starved site for the DnaG(ZBD+RPD) variant. (A) Representative gel of primase activity of the DnaG(ZBD+RPD) variant (6.4 μM, 500 nM oJW3319) shows that priming readthrough is still observed at 0.1 mM GTP and decreases upon addition of ppGpp. (B) Representative gel of primase activity of the DnaG(ZBD+RPD) variant (6.4 μM, 500 nM oJW3319) at 1 mM GTP shows that no priming readthrough is observed. (C) Quantification of readthrough primers as a percentage of total primers synthesized at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars. (D) Quantification of total primers synthesized over increasing concentrations of ppGpp at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars.
Supplemental Figure 12. Oligonucleotide used in the fluorescence-based primase assay. (Left) Run on 7M Urea 20% PAGE, the template used is not pure, but instead a heterogeneous population. (Right) PAGE-purified template shows the template is now a pure and purely homogenous population.
Research Highlights:
The starvation signal (p)ppGpp and GTP synergistically regulate the essential enzyme DNA primase in bacteria
(p)ppGpp inhibits primer extension and reduces primase processivity
(p)ppGpp protects priming fidelity by preventing mismatches during nucleotide fluctuation
ACKNOWLEDGEMENT
We thank Jim Keck, Wilma Ross, Rick Gourse, Albert Chen and Andrew Voter for technical help and useful advice. We thank the anonymous reviewers for insightful comments regarding the molecular mechanism of (p)ppGpp regulation.
FUNDING
This work was supported by NIH R35GM127088 and the Hatch Act Formula Fund WIS03010 to JDW, the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1256259 to CNG. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Kornberg AB and Baker T (1992) DNA Replication 2nd ed. W. H. Freeman and Company, New York. [Google Scholar]
- 2.Frick DN and Richardson CC (2001) DNA Primases. Annu. Rev. Biochem, 70, 39–80. [DOI] [PubMed] [Google Scholar]
- 3.Swart JR and Griep MA (1995) Primer Synthesis Kinetics by Escherichia coli Primase on Single-Stranded DNA Templates. Biochem, 34, 16097–16106. [DOI] [PubMed] [Google Scholar]
- 4.Kuchta RD and Stengel G (2010) Mechanism and evolution of DNA primases. Biochim. et Biophys. Acta (BBA) - Proteins and Proteomics, 1804, 1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu CA, Zechner EL and Marians KJ (1992) Coordinated leading- and lagging- strand synthesis at the Escherichia coli DNA replication fork. I. Multiple effectors act to modulate Okazaki fragment size. J. Biol. Chem, 267, 4030–4044. [PubMed] [Google Scholar]
- 6.Wu CA, Zechner EL, Reems JA, McHenry CS and Marians KJ (1992) Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. V. Primase action regulates the cycle of Okazaki fragment synthesis. J. Biol. Chem, 267, 4074–4083. [PubMed] [Google Scholar]
- 7.Naue N, Beerbaum M, Bogutzki A, Schmieder P and Curth U (2013) The helicase-binding domain of Escherichia coli DnaG primase interacts with the highly conserved C-terminal region of single-stranded DNA-binding protein. Nucleic Acids Res., 41, 4507–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey S, Eliason WK and Steitz TA (2007) Structure of Hexameric DnaB Helicase and Its Complex with a Domain of DnaG Primase. Science, 318, 459–463. [DOI] [PubMed] [Google Scholar]
- 9.Oakley AJ, Loscha K. v., Schaeffer PM, Liepinsh E, Pintacuda G, Wilce MCJ, Otting G and Dixon NE (2005) Crystal and Solution Structures of the Helicase-binding Domain of Escherichia coli Primase. J. Biol. Chem, 280, 11495–11504. [DOI] [PubMed] [Google Scholar]
- 10.Tougu K and Marians KJ (1996) The Interaction between Helicase and Primase Sets the Replication Fork Clock. J. Biol. Chem, 271, 21398–21405. [DOI] [PubMed] [Google Scholar]
- 11.Sun W and Godson GN (1996) Interaction of Escherichia coli primase with a phage G4ori(c)-E. coli SSB complex. J. Bacteriol, 178, 6701–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun W, Tormo J, Steitz TA and Godson GN (1994) Domains of Escherichia coli primase: functional activity of a 47-kDa N-terminal proteolytic fragment. Proc. Natl. Acad. Sci, 91, 11462–11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bird LE, Pan H, Soultanas P and Wigley DB (2000) Mapping Protein-Protein Interactions within a Stable Complex of DNA Primase and DnaB Helicase from Bacillus stearothermophilus †. Biochemistry, 39, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keck JL, Roche DD, Lynch AS and Berger JM (2000) Structure of the RNA Polymerase Domain of E. coli Primase. Science, 287, 2482–2486. [DOI] [PubMed] [Google Scholar]
- 15.Aravind L, Leipe DD and Koonin E. v. (1998) Toprim--a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res., 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Podobnik M, McInerney P, O’Donnell M and Kuriyan J (2000) A TOPRIM domain in the crystal structure of the catalytic core of Escherichia coli primase confirms a structural link to DNA topoisomerases. J. Mol. Biol, 300. [DOI] [PubMed] [Google Scholar]
- 17.Corn JE and Berger JM (2006) Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acids Res., 34, 4082–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koepsell SA, Larson MA, Griep MA and Hinrichs SH (2006) Staphylococcus aureus Helicase but Not Escherichia coli Helicase Stimulates S. aureus Primase Activity and Maintains Initiation Specificity. J. Bacteriol, 188, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson SK, Bhattacharyya S and Griep MA (2000) DnaB Helicase Stimulates Primer Synthesis Activity on Short Oligonucleotide Templates †. Biochemistry, 39, 736–744. [DOI] [PubMed] [Google Scholar]
- 20.van Eijk E, Paschalis V, Green M, Friggen AH, Larson MA, Spriggs K, Briggs GS, Soultanas P and Smits WK (2016) Primase is required for helicase activity and helicase alters the specificity of primase in the enteropathogen Clostridium difficile. Open Biol., 6, 160272–160272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chintakayala K, Machón C, Haroniti A, Larson MA, Hinrichs SH, Griep MA and Soultanas P (2009) Allosteric regulation of the primase (DnaG) activity by the clamp-loader (τ) in vitro. Mol. Microbiol, 72, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuzhakov A, Kelman Z and O’Donnell M (1999) Trading places on DNA-A three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell, 96, 153–163. [DOI] [PubMed] [Google Scholar]
- 23.Wang JD, Sanders GM and Grossman AD (2007) Nutritional Control of Elongation of DNA Replication by (p)ppGpp. Cell, 128, 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rymer RU, Solorio FA, Tehranchi AK, Chu C, Corn JE, Keck JL, Wang JD and Berger JM (2012) Binding mechanism of metal·NTP substrates and stringent-response alarmones to bacterial DnaG-type primases. Structure (London, England : 1993), 20, 1478–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denapoli J, Tehranchi AK and Wang JD (2013) Dose-dependent reduction of replication elongation rate by (p)ppGpp in Escherichia coli and Bacillus subtilis. Mol Microbiol., 88, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maciąg M, Kochanowska M, Łyżeń R, Węgrzyn G, and Szalewska-Pałasz A (2010) ppGpp inhibits the activity of Escherichia coli DnaG primase. Plasmid, 63, 61–67. [DOI] [PubMed] [Google Scholar]
- 27.Cashel M, Gentry DR, Hernandez VJ and Vinella D (1996) The stringent response. In Neidhardt FC (ed), In Escherichia coli and Salmonella typhimurium Cellular and Molecular Biology. ASM Press, Washington D.C., Vol. 1, pp. 1458–1496. [Google Scholar]
- 28.Srivatsan A and Wang JD (2008) Control of bacterial transcription, translation and replication by (p)ppGpp. Curr. Microbiol, 11, 100–105. [DOI] [PubMed] [Google Scholar]
- 29.Irving SE, Choudhury NR and Corrigan RM (2021) The stringent response and physiological roles of (pp)pGpp in bacteria. Nature Reviews Microbiology, 19. [DOI] [PubMed] [Google Scholar]
- 30.Steinchen W, Zegarra V and Bange G (2020) (p)ppGpp: Magic Modulators of Bacterial Physiology and Metabolism. Frontiers In Microbiology, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gourse RL, Chen AY, Gopalkrishnan S, Sanchez-Vazquez P, Myers A and Ross W (2018) Transcriptional Responses to ppGpp and DksA. Annual Review of Microbiology, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kriel A, Bittner AN, Kim SH, Liu K, Tehranchi AK, Zou WY, Rendon S, Chen R, Tu BP and Wang JD (2012) Direct Regulation of GTP Homeostasis by (p)ppGpp: A Critical Component of Viability and Stress Resistance. Mol. Cell, 48, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanner NA, Hamdan SM, Jergic S, Loscha K.v, Schaeffer PM, Dixon NE and van Oijen AM (2008) Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat. Struct. Mol. Biol, 15, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J-B, Hite RK, Hamdan SM, Sunney Xie X, Richardson CC and van Oijen AM (2006) DNA primase acts as a molecular brake in DNA replication. Nature, 439, 621–624. [DOI] [PubMed] [Google Scholar]
- 35.Monachino E, Jergic S, Lewis JS, Xu Z-Q, Lo ATY, O’Shea VL, Berger JM, Dixon NE and van Oijen AM (2020) A Primase-Induced Conformational Switch Controls the Stability of the Bacterial Replisome. Mol. Cell, 79, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemon KP and Grossman AD (2000) Movement of Replicating DNA through a Stationary Replisome. Mol Cell, 6. [DOI] [PubMed] [Google Scholar]
- 37.Levine A, Vannier F, Dehbi M, Henckes G and Séror SJ (1991) The stringent response blocks DNA replication outside the ori region in Bacillus subtilis and at the origin in Escherichia coli. J. Mol. Biol, 219, 605–613. [DOI] [PubMed] [Google Scholar]
- 38.Maciąg-Dorszyńska M, Szalewska-Pałasz A and Węgrzyn G (2013) Different effects of ppGpp on Escherichia coli DNA replication in vivo and in vitro. FEBS Open Bio, 3, 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stols L, Gu M, Dieckman L, Raffen R, Collart FR and Donnelly MI (2002) A New Vector for High-Throughput, Ligation-Independent Cloning Encoding a Tobacco Etch Virus Protease Cleavage Site. Protein Expression and Purification, 25. [DOI] [PubMed] [Google Scholar]
- 40.Mechold U, Potrykus K, Murphy H, Murakami KS and Cashel M (2013) Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res., 41, 6175–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mechold U, Murphy H, Brown L and Cashel M (2002) Intramolecular Regulation of the Opposing (p)ppGpp Catalytic Activities of RelSeq, the Rel/Spo Enzyme from Streptococcus equisimilis. J. Bacteriol, 184, 2878–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roelofs KG, Wang J, Sintim HO and Lee VT (2011) Differential radial capillary action of ligand assay for high-throughput detection of protein-metabolite interactions. Proc. Natl. Acad. Sci, 108, 15528–15533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koepsell SA, Hanson S, Hinrichs SH and Griep MA (2005) Fluorometric assay for bacterial primases. Anal. Biochem, 339, 353–355. [DOI] [PubMed] [Google Scholar]
- 44.Orr MW and Lee VT (2017) Differential Radial Capillary Action of Ligand Assay (DRaCALA) for High-Throughput Detection of Protein–Metabolite Interactions in Bacteria. In Nordenfelt P, Collin M (eds), Bacterial Pathogenesis. Methods In Molecular Biology Humana Press, New York, Vol. 1535, pp. 25–41. [DOI] [PubMed] [Google Scholar]
- 45.Rannou O, le Chatelier E, Larson MA, Nouri H, Rengè Re Dalmais B, Laughton C, Janniè L and Soultanas P (2013) Functional interplay of DnaE polymerase, DnaG primase and DnaC helicase within a ternary complex, and primase to polymerase hand-off during lagging strand DNA replication in Bacillus subtilis. Nucleic Acids Res., 41, 5303–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tougu K, Peng H and Marians KJ (1994) Identification of a domain of Escherichia coli primase required for functional interaction with the DnaB helicase at the replication fork. Journal of Biological Chemistry, 269. [PubMed] [Google Scholar]
- 47.Liu K, Myers AR, Pisithkul T, Claas KR, Satyshur KA, Amador-Noguez D, Keck JL and Wang JD (2015) Molecular Mechanism and Evolution of Guanylate Kinase Regulation by (p)ppGpp. Mol. Cell, 57, 735–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hernández-Tamayo R, Schmitz H and Graumann PL (2021) Single-Molecule Dynamics at a Bacterial Replication Fork after Nutritional Downshift or Chemically Induced Block in Replication. mSphere, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liermo S and Gourse RL (1990) Factor-independent activation of Escherichia coli rRNA transcription: I. Kinetic analysis of the roles of the upstream activator region and supercoiling on transcription of the rrnB P1 promoter in vitro. J. Mol. Biol, 220, 555–568. [DOI] [PubMed] [Google Scholar]
- 50.Pfeffer SR, Stahl SJ and Chamberlin MJ (1977) Binding of Escherichia coli RNA Polymerase to T7 DNA. Displacement of holoenzyme from promoter complexes by heparin. J. Biol. Chem, 252, 5403–5407. [PubMed] [Google Scholar]
- 51.Reddy MK, Weitzel SE and von Hippel PH (1992) Processive Proofreading Is Intrinsic to T4 DNA Polymerase. J. Biol. Chem, 267, 14157–14166. [PubMed] [Google Scholar]
- 52.Reha-Krantz LJ and Nonay RL (1994) Motif A of Bacteriophage T4 DNA Polymerase: Role in Primer Extension and DNA Replication Fidelity. Isolation of new antimutator and mutator DNA polymerases. J. Biol. Chem, 269, 5635–5643. [PubMed] [Google Scholar]
- 53.Chammas O, Bonass WA and Thomson NH (2017) Effect of heparin and heparan sulphate on open promoter complex formation for a simple tandem gene model using ex situ atomic force microscopy. Methods, 120, 91–102. [DOI] [PubMed] [Google Scholar]
- 54.Schreiber G, Ron EZ and Glaser G (1995) ppGpp-mediated regulation of DNA replication and cell division in Escherichia coli. Curr. Microbiol, 30, 27–32. [DOI] [PubMed] [Google Scholar]
- 55.Ferullo DJ and Lovett ST (2008) The Stringent Response and Cell Cycle Arrest in Escherichia coli. PLoS Genet., 4, e1000300–e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kraemer JA, Sanderlin AG and Laub MT (2019) The Stringent Response Inhibits DNA Replication Initiation in E. coli by Modulating Supercoiling of oriC. mBio, 10, e01330–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernández-Coll L, Maciag-Dorszynska M, Tailor K, Vadia S, Levin PA, Szalewska-Palasz A and Cashel M (2020) The Absence of (p)ppGpp Renders Initiation of Escherichia coli Chromosomal DNA Synthesis Independent of Growth Rates. mBio, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson BW, Liu K, Wolak C, Dubiel K, She F, Satyshur KA, Keck JL and Wang JD (2019) Evolution of (p)ppGpp-HPRT regulation through diversification of an allosteric oligomeric interaction. eLife, 8, e47534–undefined. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Michel B, Sinha AK and Leach DRF (2018) Replication Fork Breakage and Restart in Escherichia coli. Microbiol. Mol. Biol. Rev, 82, e00013–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang B, Grant RA and Laub MT (2020) ppGpp Coordinates Nucleotide and Amino-Acid Synthesis in E. coli During Starvation. Molecular Cell, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tehranchi AK, Blankschien MD, Zhang Y, Halliday JA, Srivatsan A, Peng J, Herman C and Wang JD (2010) The Transcription Factor DksA Prevents Conflicts between DNA Replication and Transcription Machinery. Cell, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hopfield JJ (1974) Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc. Natl. Acad. Sci, 71, 4135–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roghanian M, Zenkin N and Yuzenkova Y (2015) Bacterial global regulators DksA/ppGpp increase fidelity of transcription. Nucleic Acids Res., 43, 1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Various inhibition models for reactions with ppGpp and full-length DnaG inFigure 1. The initial velocity (V0) of full-length DnaG activity at indicated concentrations of ppGpp. Reactions were run with 0, 12.5, 25, 50, 100, 200, or 400 μM GTP. Results are normalized to the enzyme concentration and are reported as relative fluorescent units per minute (RFU/min). Points represent averages of ≥ 3 replicates. Error bars represent ± SEM for ≥ 3 replicates. Curves were fit with a competitive inhibition (A), mixed model inhibition (B), noncompetitive inhibition (C), and an uncompetitive inhibition model (D). The best fit model is uncompetitive inhibition for full-length DnaG and ppGpp.
Supplemental Figure 2. Various inhibition models for reactions with pppGpp and full-length DnaG inFigure 1. The initial velocity (V0) of full-length DnaG activity at indicated concentrations of pppGpp. Reactions were run with 0, 12.5, 25, 50, 100, 200, or 400 μM GTP. Results are normalized to the enzyme concentration and are reported as relative fluorescent units per minute (RFU/min). Points represent averages of ≥ 3 replicates. Error bars represent ± SEM for ≥ 3 replicates. Curves were fit with a competitive inhibition (A), mixed model inhibition (B), noncompetitive inhibition (C), and an uncompetitive inhibition model (D). The best fit model is competitive inhibition for full-length DnaG and pppGpp.
Supplemental Figure 3. Various inhibition models for reactions with pppGpp and DnaG(ZBD+RPD) inFigure 1. The initial velocity (V0) of DnaG(ZBD+RPD) activity at indicated concentrations of pppGpp. Reactions were run with 0, 12.5, 25, 50, 100, 200, or 400 μM GTP. Results are normalized to the enzyme concentration and are reported as relative fluorescent units per minute (RFU/min). Points represent averages of ≥ 3 replicates. Error bars represent ± SEM for ≥ 3 replicates. Curves were fit with a competitive inhibition (A), mixed model inhibition (B), noncompetitive inhibition (C), and an uncompetitive inhibition model (D). The best fit model is uncompetitive inhibition for DnaG(ZBD+RPD) and pppGpp.
Supplemental Figure 4. RNA standard ran alongside primase reactions. Standard was generated by labeling the 5' end of an RNA with a sequence identical to that of the primase reaction product. The labeled RNA was subjected to NaOH hydrolysis to generate the RNAs of different lengths. The hydrolysis was able to occur on either the 2' or 3'-OH which is why on the very short RNAs (2-4) there are two different species.
Supplemental Figure 5. Chromatograms representing band intensity from the gel inFigure 2. Reactions titrated DnaG with 500 nM ssDNA, 0.4 mM NTPs (CTP, UTP, GTP) and ~0.4 μM [γ-32P]-ATP. Primers from 2 nucleotides (bottom) up to intermediate and full-length (29 nucleotides) primers were observed. A small amount of primers longer than 29 nucleotides are due to impurity of the DNA template (Supplemental Figure 12) and result in disrupted peaks within the chromatograms near 29 nucleotides for concentrations of DnaG above 1600 nt.
Supplemental Figure 6. Chromatograms representing band intensity from the gel inFigure 3. Primer synthesis was measured over the course of one hour (60 min) without ppGpp (A) or with 1 mM ppGpp (B). Time points were taken at 2, 5, 10, 20, 30, and 60 minutes. Reactions contained 400 nM DnaG, 5 μM ssDNA template, ~0.4 μM [γ-32P]-ATP, and 1 mM NTPs (GTP, CTP, UTP). Diminished chromatogram peak size over time for primers longer than the dinucleotide show reduction in primer extension with 1 mM ppGpp (B) compared to no ppGpp (A).
Supplemental Figure 7. Chromatograms representing band intensity from the gel inFigure 4. Primer synthesis reactions contained 6.4 μM DnaG, 500 nM oJW3319, ~0.4 μM [γ-32P]-ATP, 1 mM each of CTP and UTP, and either 0.1 mM GTP (A) or 1 mM GTP (B). Reactions incubated 30 minutes after addition of NTPs. Higher GTP concentrations promote synthesis of priming products above 6 nucleotides, while higher ppGpp concentrations inhibit synthesis of full-length (21 nucleotide) products. Dinucleotide formation appears to be unaffected by GTP or ppGpp concentration. Together, this data suggests that primer extension is regulated by GTP and ppGpp concentrations.
Supplemental Figure 8. Chromatograms representing band intensity from the gel inFigure 5. Primer synthesis was measured over the course of one hour (60 minutes) in the presence of heparin (~5 μg/mL) either without ppGpp (A) or with 1 mM ppGpp (B). Time points were taken at 2, 5, 10, 20, 30, and 60 minutes. Reactions contained 400 nM DnaG, 5 μM ssDNA template, ~0.4 μM [γ-32P]-ATP, and 1 mM NTPs (GTP, CTP, UTP). Synthesis of 7 nucleotide and longer priming products is slightly reduced in the presence of ppGpp.
Supplemental Figure 9. Chromatograms representing band intensity from the gel inFigure 6. Primer synthesis reactions contained 6.4 μM DnaG, 500 nM oJW3319, ~0.4 μM [γ-32P]-ATP, 1 mM UTP, and either 0.1 mM GTP (A) or 1 mM GTP (B). Since CTP is withheld in these reactions, priming products should be 7 nucleotides long. Readthrough is observable at low GTP but diminishes as ppGpp concentrations increase.
Supplemental Figure 10. DNA primase reads through a starved site in an alternative oligonucleotide template. (A) Representative gel of primase reactions with 6.4 μM DnaG and 500 nM oJW3668 with CTP-starved site (expected product is 7 nucleotides). Oligo oJW3668, unlike oJW3319 in Figure 6, lacks a CCCC repeat in its sequence. (B) Chromatograms representing band intensity from (A) at 0.1 mM GTP (top) and 1 mM GTP (bottom) with increasing concentrations of ppGpp. (C) Quantification of readthrough primers as a percentage of total primers synthesized at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars. (D) Quantification of total primers synthesized over increasing concentrations of ppGpp at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars.
Supplemental Figure 11. ppGpp reduces readthrough of a starved site for the DnaG(ZBD+RPD) variant. (A) Representative gel of primase activity of the DnaG(ZBD+RPD) variant (6.4 μM, 500 nM oJW3319) shows that priming readthrough is still observed at 0.1 mM GTP and decreases upon addition of ppGpp. (B) Representative gel of primase activity of the DnaG(ZBD+RPD) variant (6.4 μM, 500 nM oJW3319) at 1 mM GTP shows that no priming readthrough is observed. (C) Quantification of readthrough primers as a percentage of total primers synthesized at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars. (D) Quantification of total primers synthesized over increasing concentrations of ppGpp at 0.1 mM GTP (green line) and 1 mM GTP (blue line). Points represent averages of n = 3, and the SEM is indicated by error bars.
Supplemental Figure 12. Oligonucleotide used in the fluorescence-based primase assay. (Left) Run on 7M Urea 20% PAGE, the template used is not pure, but instead a heterogeneous population. (Right) PAGE-purified template shows the template is now a pure and purely homogenous population.