

Abstract
Background:
Skeletal muscle atrophy, whether caused by chronic disease, acute critical illness, disuse or aging, is characterized by tissue-specific decrease in oxidative capacity and broad alterations in metabolism that contribute to functional decline. However, the underlying mechanisms responsible for these metabolic changes are largely unknown. One of the most highly upregulated genes in atrophic muscle is AMP deaminase 3 (AMPD3: AMP→IMP+NH3), which controls the content of intracellular adenine nucleotides (AdN; ATP+ADP+AMP). Given the central role of AdN in signaling mitochondrial gene expression and directly regulating metabolism, we hypothesized that overexpressing AMPD3 in muscle cells would be sufficient to alter their metabolic phenotype similar to that of atrophic muscle.
Methods:
AMPD3 and GFP (control) were overexpressed in mouse tibialis anterior (TA) muscles via plasmid electroporation and in C2C12 myotubes using adenovirus vectors. TA muscles were excised one week later, and AdN were quantified by UPLC. In myotubes, targeted measures of AdN, AMPK/PGC-1α/mitochondrial protein synthesis rates, unbiased metabolomics, and transcriptomics by RNA sequencing were measured after 24 h of AMPD3 overexpression. Media metabolites were measured as an indicator of net metabolic flux. At 48 h, the AMPK/PGC-1α/mitochondrial protein synthesis rates, and myotube respiratory function/capacity were measured.
Results:
TA muscles overexpressing AMPD3 had significantly less ATP than contralateral controls (−25%). In myotubes, increasing AMPD3 expression for 24 h was sufficient to significantly decrease ATP concentrations (−16%), increase IMP, and increase efflux of IMP catabolites into the culture media, without decreasing the ATP/ADP or ATP/AMP ratios. When myotubes were treated with dinitrophenol (mitochondrial uncoupler), AMPD3 overexpression blunted decreases in ATP/ADP and ATP/AMP ratios but exacerbated AdN degradation. As such, pAMPK/AMPK, pACC/ACC, and phosphorylation of AMPK substrates, were unchanged by AMPD3 at this timepoint. AMPD3 significantly altered 191 out of 639 detected intracellular metabolites, but only 30 transcripts, none of which encoded metabolic enzymes. The most altered metabolites were those within purine nucleotide, BCAA, glycolysis, and ceramide metabolic pathways. After 48 h, AMPD3 overexpression significantly reduced pAMPK/AMPK (−24%), phosphorylation of AMPK substrates (-14%), and PGC-1α protein (−22%). Moreover, AMPD3 significantly reduced myotube mitochondrial protein synthesis rates (−55%), basal ATP synthase-dependent (−13%), and maximal uncoupled oxygen consumption (−15%).
Conclusions:
Increased expression of AMPD3 significantly decreased mitochondrial protein synthesis rates and broadly altered cellular metabolites in a manner similar to that of atrophic muscle. Importantly, the changes in metabolites occurred prior to reductions in AMPK signaling, gene expression, and mitochondrial protein synthesis, suggesting metabolism is not dependent on reductions in oxidative capacity, but may be consequence of increased AMP deamination. Therefore, AMP deamination in skeletal muscle may be a mechanism that alters the metabolic phenotype of skeletal muscle during atrophy and could be a target to improve muscle function during muscle wasting.
Keywords: muscle atrophy, AMP deaminase, metabolomics, mitochondrial biogenesis, AMP activated protein kinase, ATP
1. INTRODUCTION
Skeletal muscle atrophy is a detrimental consequence of chronic diseases, acute critical illnesses, disuse, and aging, that decreases patient quality of life and survival. In addition to the loss of contractile proteins and muscle strength, atrophic muscles have a profound difference in metabolism and increased fatigability. Common metabolic changes that occur in atrophying muscle include decreases in the content of adenine nucleotides (AdN: ATP, ADP, and AMP)[1] and mitochondrial oxidative enzymes[2, 3], increases in branched chain amino acid (BCAA) transamination to support increased de novo synthesis of glutamine and alanine[4–8], increases in polyol sugars (sorbitol and fructose)[9, 10], and increased de novo synthesis and accumulation of ceramides[11–13]. Collectively, these alterations partially characterize a common metabolic phenotype that distinguishes atrophic and healthy skeletal muscles. However, the precise molecular mechanisms and genes enabling these changes in skeletal muscle metabolism during atrophy are still unknown.
Skeletal muscle metabolism is regulated by absolute free concentrations of AdN, and their relative ratios to ATP, both acutely (i.e. post-translationally) and chronically (i.e. transcriptionally), through direct and indirect mechanisms. For example, ADP and the ATP/ADP ratio directly regulate mitochondrial ATP synthase activity, which subsequently has indirect effects on metabolism due to ATP synthase activity being the primary regulator of oxidative phosphorylation flux[14]. Additionally, ATP and AMP directly bind and allosterically modulate the activities of key enzymes that regulate metabolism, such as phosphofructokinase[15] and AMP-activated protein kinase (AMPK)[16, 17]. By regulating AMPK activity, AdN impact metabolism chronically due to AMPK having numerous downstream targets that selectively determine gene transcription. A well characterized example is AMPK’s ability to increase the transcription of mitochondrial oxidative enzymes by phosphorylating and activating the transcription factor coactivator PGC-1α[18], which results in chronic increases in mitochondrial oxidative enzyme levels and oxidative capacity of muscles[19].
A possible mechanism causing the decreases in intramuscular AdN and mitochondrial oxidative enzymes during atrophy is increased activity of the enzyme AMP Deaminase 3 (AMPD3). AMPD3 is a cytosolic enzyme that catalyzes the thermodynamically irreversible deamination of AMP to IMP and ammonia (AMP → IMP+NH3), and its expression is increased up to 100-fold in muscles undergoing atrophy[2, 3, 20, 21]. The irreversibility of this reaction, in conjunction with the near equilibrium of adenylate kinase (ADP+ADP↔ATP+AMP), allows cells to maintain favorable ATP/ADP and ATP/AMP ratios at the cost of depleting the total adenine nucleotide pool[22]. Therefore, increased AMP deamination due to upregulated AMPD3 expression may be a mechanism altering skeletal muscle metabolism during atrophy.
The main purpose of this study was to determine if increased levels of AMPD3 expression in skeletal muscle would be sufficient to alter their metabolite levels and mitochondria similar to that of atrophying muscles. To address this, we overexpressed AMPD3 in C2C12 myotubes and characterized their metabolic phenotype over time using targeted and unbiased approaches to determine how and when metabolism may be altered with changes in AMP deamination.
2. METHODS
2.1. Plasmid electroporation into mouse muscle
Eight (4 male and 4 female), 22-week old C57BL/6J mice from Jackson Laboratories were housed 2–3 per cage with free access to food and water. These studies were approved by the Indiana School of Medicine Animal Care and Use Committee.
Mice were anaesthetized via 2% isoflurane and subsequently provided a subcutaneous injection of sustained release buprenorphine as an analgesic (3.25 mg/kg body weight Ethiqa XR, Fidelis Pharmaceuticals). After the fur was shaved from the lower legs, the skin was scrubbed with a 10% povidone-iodine swab and wiped clean with 70% isopropyl alcohol. An ~1cm incision was made longitudinally in the skin over the belly of the tibialis anterior (TA), and a plate electrode was slid underneath the TA. pAMPD3 or pGFP lasmid DNA (25 μg in 40 μL 0.9% NaCl) was injected mid-belly with the needle inserted parallel to muscle fibers. (The pGFP consists of the pIRES-hrGFP II plasmid originally designed by Stratagene. The pAMPD3 plasmid consists of mouse AMPD3 cDNA clone (#4503933) from OpenBiosystems inserted into the pIRES-hrGFP II plasmid.) Another plate electrode was placed above the TA, and electrical pulses (five pulses, 15 V, each lasting 20 ms with 200 ms interval) were delivered using an ECM 830 Electroporator (BTX Harvard Apparatus). The incision was sutured using 5–0 Prolene (FS-2, Ethicon), and mice were monitored to confirm proper healing of the surgical site.
Muscles were collected 1 week after the electroporations. Mice were anesthetized by intraperitoneal injection of ketamine (20 mg/mL)/xylazine (2mg/mL) and placed on a 37°C heating pad. To maintain oxygen delivery to the muscles, the mice were administered 100% O2 at a flow rate of 1 L/min. Immediately following their removal, the TA muscles were blotted dry and immediately frozen using steel clamps cooled in liquid nitrogen.
2.2. Cell Culture
C2C12 mouse myoblasts (ATCC) were grown on gelatin-coated (Sigma G9391) 6-well plates in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose, L-glutamine, and sodium pyruvate, and supplemented with 10% heat inactivated fetal bovine serum (Gibco 16140-071), and penicillin (100 IU/mL)/streptomycin (100 μg/mL). When myoblasts were 75–90% confluent, media was switched to DMEM with 2% horse serum (Hyclone SH30074.03) and penicillin/streptomycin to induce differentiation into multi-nucleated myotubes. Cells were maintained at 37°C and 5% CO2, and media was changed every 48 h. Adenovirus constructs Ad-Ampd3-IRES-GFP and Ad-GFP (Vector Biolabs; Malvern, PA) were used to overexpress mouse AMPD3 + GFP or GFP alone as described previously[23]. Adenovirus constructs encoding mouse AMPD1 (ad-Ampd1-IRES-GFP; Vector Biolabs, Malvern, PA) was used to generate a loading control for western blots for positive identification of AMPD1 protein in C2C12 myotubes. DNP (2,4 Dinitrophenol; Sigma D198501) stock was prepared at 0.2 M concentration dissolved in 100% methanol. Stock was then diluted in warm DMEM before adding to culture wells, and equal volumes of methanol were used as vehicle control.
2.3. Western blots
Proteins were extracted from cells in radio-immunoprecipitation assay (RIPA) buffer including protease and phosphatase inhibitors (Roche) and quantified by BCA Assay (Pierce). Equal amounts of protein were separated by SDS-PAGE (6–12% BioRad TGX stain free gels) then transferred to polyvinylidene difluoride (PVDF) membranes. Equal loading and transfer of proteins was confirmed by imaging protein fluorescence after photoactivation of gels and membranes. Membranes were blocked for 1 hour at room temperature using a solution of 5% BSA (Sigma A7906) in TBS plus 0.1% tween-20 (TBS-T). Primary antibodies were purchased from ThermoFisher (AMPD3 PA5-76912), Santa Cruz (AMPD1 sc-393117), Abcam (PGC-1α ab54481, OXPHOS cocktail ab110413), and Cell Signaling (AMPKα #5831S, pAMPK Thr172 #2531S, ACC #8578S, pACC Ser79 #3661S, Phospho-AMPK Substrate Motif [LXRXX(pS/pT) #5759S). Antibodies were diluted in a solution of TBS-T with 2–5% BSA. Membranes were incubated in primary antibody overnight at 4°C. Secondary antibodies conjugated to horseradish peroxidase (Cell Signaling #7074, ThermoFisher #31444) were detected with Western Chemiluminescence HRP Substrate (EMD Millipore). Band intensities were captured using a Bio-Rad Chemi Doc XRS imager and analyzed using Image Lab Software 6.1 (Bio-Rad). Approximate molecular weights of protein were calculated relative to PageRuler Plus protein ladder (ThermoFisher).
2.4. Nucleotide Measurements
Skeletal muscles were weighed to the nearest 0.1 mg and then homogenized in ice cold 0.5 N perchloric acid + 5mM EDTA using a glass-on-glass tissue grinder. Homogenates were then centrifuged, and the supernatants neutralized with ice-cold 1N KOH. Perchlorate salts were removed by centrifugation. For C2C12 myotubes, metabolites were extracted using ice cold 0.5 N perchloric acid + 5mM EDTA, sonication, and centrifugation to remove precipitated proteins. Supernatants were neutralized with ice cold 1 N KOH and centrifuged at 4°C to remove the potassium-perchlorate salt. Protein pellets were re-suspended in 0.2 N NaOH for protein estimation by BCA Assay (Pierce). For media analysis, media aliquots were collected in ice-cold tubes and immediately treated with perchloric acid. Samples were processed exactly as above apart from protein assay. Adenine nucleotide (ATP, ADP, AMP) and degradation product (IMP, inosine, and hypoxanthine) concentrations were quantified by UV absorbance at 210 nm after separation by ultra-performance liquid chromatography (UPLC) using a Waters Acquity UPLC H-Class system and an Acquity UPLC HSS T3 1.8 μm, 2.1 mm × 150 mm column (Waters) as we have done previously [24].
2.5. PGC-1α Promoter Luciferase Reporter assay
To measure PGC-1α promotor activity, myoblasts were transfected with plasmids encoding proximal 2kb PGC-1α promotor-firefly luciferase (Addgene, plasmid #8887). Twenty-four hours after transfection, myoblasts were switched to differentiation media. Luciferase activity was determined by following the Promega Dual-Luciferase Reporter Assay manufacturer’s protocol. Briefly, myotubes were washed twice with PBS, and then passive lysis buffer was added. After a 15–30-minute incubation at room temperature, cell lysates were collected. Firefly luciferase activity was measured and normalized to total protein content.
2.6. Transcriptomics
Myotubes were washed twice with PBS and 1 ml of TRIzol (Invitrogen) was added to each well of a 6-well plate. Myotubes were lysed by trituration, and the resulting solution was then passed through a syringe until clear and non-viscous. Tubes were snap frozen in liquid nitrogen and shipped to Novogene for transcriptomic analysis. Downstream analysis was performed using a combination of programs including STAR, HTseq, Cufflink, and our wrapped scripts. Alignments were parsed using Tophat program and differential expressions were determined through DESeq2/edgeR. GO and KEGG enrichment were implemented by the ClusterProfiler. Gene fusion and difference of alternative splicing event were detected by Star-fusion and rMATS software. FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. Differential expression analysis was performed using the DESeq2 R package (2_1.6.3). The resulting P-values were adjusted using the Benjamini and Hochberg’s approach for controlling the False Discovery Rate (FDR). Corrected P-value of 0.05 and absolute fold change of 1 were set as the threshold for significant differential expression.
2.7. Cellular Metabolomics
C2C12 myotubes were removed from plates with trypsin/EDTA, washed in DMEM, centrifuged to pellet the cells, and then flash frozen in liquid nitrogen. Metabolite extraction and metabolomics analysis were conducted by Metabolon Inc. Several recovery standards were added prior to the first step in the extraction process for QC purposes. Proteins were precipitated with methanol under vigorous shaking followed by centrifugation. The resulting extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/UPLC-MS/MS methods with positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC-MS/MS with negative ion mode ESI, one for analysis by HILIC/UPLC-MS/MS with negative ion mode ESI, and one sample was reserved for backup. Following normalization to Bradford protein concentration, log transformation and imputation of missing values, if any, with the minimum observed value for each compound, two-tail unpaired t-tests were used to identify significantly different biochemicals between experimental groups. Statistical analysis and graphs were performed using Viime (Viime.org).
2.8. Media Metabolomics
To determine the cellular uptake and release of endogenously produced metabolites, media samples were prepared by filtering 1 ml of media through a 10 kDa molecular weight cutoff filter to remove the macromolecules, e.g. proteins & lipoprotein particles. The filtrate was then lyophilized and the dried material resuspended in 600 ul of deuterated water containing 0.5 mM 2,2,-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) as a chemical shift and quantitation reference. NMR data were acquired on a Bruker Avance III 700MHz NMR spectrometer. This instrument has a TXI triple resonance probe operating at 25°C. Spectra were collected with a 1D NOESY pulse sequence covering 12 ppm. The spectra were digitized with 32768 points during a 3.9 second acquisition time. The mixing time was set to 100 ms and the relaxation delay between scans was set to 2.0 seconds. The data were processed using Advanced Chemistry Development Spectrus Processor (version 2016.1, Toronto, Canada). The spectra were zero filled to 65536 points and apodized using a 0.3Hz decaying exponential function and fast Fourier transformed. Automated phase correction and baseline correction were applied to all samples. Metabolite concentrations were quantified using the Chenomx NMR Suite (version 8.2, Edmonton, Canada). Quantitative fitting of each spectrum was carried out in batch mode, followed by manual adjustment for some spectra to correct for errors arising from spectral overlap.
2.9. Subfraction Protein Synthesis
Myotubes were pulsed for 2 h with L-[2,3,4,5,6-3H]-Phenylalanine (5 μCi/ml) in differentiation media (DMEM with 2% horse serum). After the pulse, myotubes were rinsed in cold PBS, trypsinized, and scraped off the dish. Samples were homogenized using Teflon pestles on glass tubes in ice cold STE buffer (250 mM sucrose, 5 mM Tris, 2 mM EGTA, pH 7.4). The samples were then centrifuged for 10 min at 4°C at low speed (1500 × g) to remove nuclei and cellular debris. The supernatant was collected and subsequently centrifuged for 15 min at 4°C at high speed (14,600 × g) to isolate the mitochondrial pellet. The supernatant was saved as the cytosolic fraction. Radioactivity was measured from the crude homogenate, the cytosolic fraction, and the mitochondrial fraction using a scintillation counter. An aliquot of each fraction was also saved to measure total protein concentration by Pierce BCA Protein Assay kit. The subfraction purities were confirmed by immunoblotting for nuclear (Histone 2b, Cell Signaling #cs2934), cytosolic (GAPDH, Abcam #ab8245), and mitochondrial (COXIV, Cell Signaling #cs4844) proteins.
2.10. Mitochondria Functional Analysis
Mitochondria functional analyses were performed 48 h after virus transduction using the XF Cell Mitostress test kit (Agilent) according to manufacturer’s protocol. Briefly, one hour prior to the assay, myotubes were washed and given warm Seahorse DMEM supplemented with 2 mM glutamine and 1 mM pyruvate and incubated at 37°C (no CO2) for 1 h. Oxygen consumption rates (OCR) were measured in an XF24 Analyzer at 37°C after sequential addition of 1 μM oligomycin, 1 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and 0.5 μM rotenone/antimycin A. OCR were then normalized to total protein per well, which was quantified using Pierce BCA Protein Assay Kit.
2.11. Statistics
Unless otherwise stated, all data are presented as mean ± standard deviation. Statistical comparisons were performed using two-tailed unpaired t-test between groups of two and two-way ANOVA with Tukey’s or Sidak’s multiple comparisons between groups of four. For two-way ANOVA analysis, data were tested for normality using Shapiro-Wilk test and Spearman’s test for heteroscedasticity. For data of unequal variance, data were log transformed and ANOVA was repeated. Data were analyzed using GraphPad Prism 9 software, and a p value < 0.05 was considered significantly different. All cell culture experiments were repeated completely independently (i.e. on different days using a different stock of frozen cells) at least twice in order to verify the reproducibility of our results.
3. RESULTS
3.1. Increased AMPD3 expression is sufficient to deplete ATP without decreasing ATP/ADP and ATP/AMP ratios
To study the effects of increased AMPD3 in muscle in vivo, we electroporated plasmids encoding GFP or AMPD3 into mouse tibialis anterior muscles. This resulted in increases in AMPD3 protein expression after one week (Fig 1A). TA muscles overexpressing AMPD3 had significantly less ATP and total adenine nucleotides compared to contralateral muscles expressing GFP (Fig 1B), but no significant differences were detected in muscle weights (Fig 1C).
Fig. 1. AMPD3 overexpression is sufficient to increase resting state adenine nucleotide degradation and accelerate degradation in response to mitochondrial uncoupling.
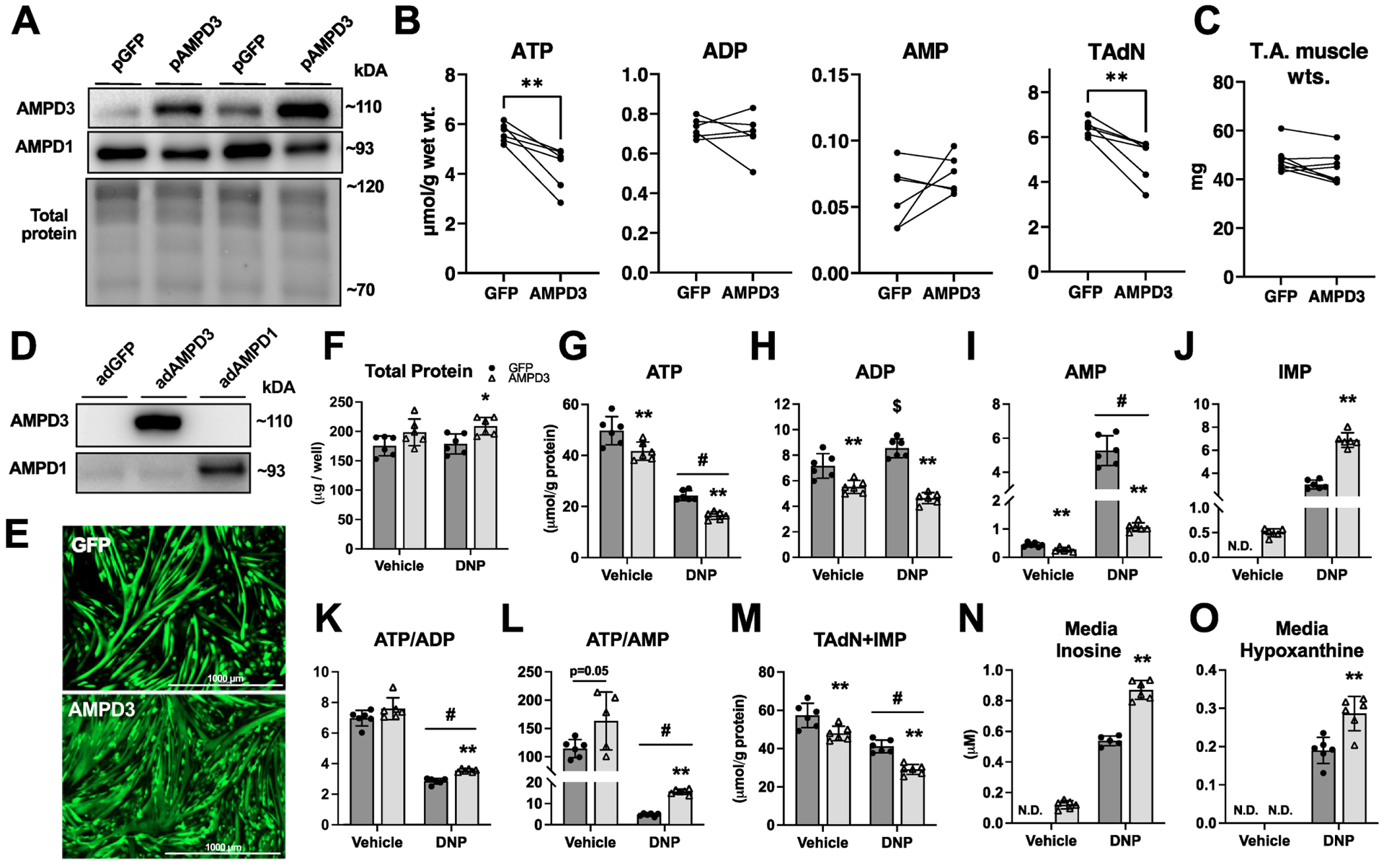
Plasmids encoding GFP or AMPD3 were electroporated into mouse TA muscles. One week later, muscles were excised for biochemical analysis. (A) Western blots for AMPD3 and AMPD1 in TA muscles. (B) Intramuscular concentrations of adenine nucleotides and the total adenine nucleotide pool (TAdN; ATP+ADP+AMP). (C) TA muscle wet weights. C2C12 myotubes were incubated with adenovirus encoding GFP or AMPD3+GFP for 24 h. During the final hour, myotubes were treated with either 0.6 mM 2,4 dinitrophenol (DNP) or vehicle (methanol). (D) AMPD3 and AMPD1 protein expression demonstrating specific upregulation of AMPD3. (E) Fluorescent images of C2C12 myotubes captured prior to DNP treatment. (F) Total protein content of experimental groups. (G-L) Intracellular concentrations of adenine nucleotides, IMP, and ratio of ATP to ADP and AMP. (M) Sum concentrations of TAdN plus IMP (ATP+ADP+AMP+IMP). (N-O) Media concentrations of inosine and hypoxanthine (purine nucleoside and base degradation products of IMP). All values are presented as mean±SD, n=5–6/condition, N.D.=none detected, two-way ANOVA with Tukey’s multiple comparisons or two-tailed unpaired t-test when comparing fewer than four groups, * p<0.05, ** p<0.01 vs GFP. $ p<0.05 vs vehicle. # p<0.01 main effect of DNP.
In order to more precisely identify potential metabolic alterations related specifically to AMPD3 upregulation in muscle cells and remove confounding variables such as exogenous hormones and other cell types, we utilized a cell culture model of differentiated skeletal myotubes (Fig 1D–O). C2C12 myotubes were incubated with adenoviruses encoding AMPD3+GFP or GFP alone (control) for 24 h. As expected, AMPD3 protein was upregulated, and we confirmed that we specifically increased the expression AMPD3 with no apparent changes in the amount of AMPD1, the predominant isoform typically expressed in adult skeletal muscle (Figure 1D). Next, we measured intracellular AdN and their breakdown products in the media after 24 h incubation with the adenoviruses. In addition, given that AMPD is best known in skeletal muscle for protecting the energetic state during periods of intense energy demands (contractions) or limited energy supply (ischemia)[22], we also treated myotubes with vehicle or the mitochondrial uncoupler 2,4 dinitrophenol (DNP) for 1 h to limit ATP production and increase AMP levels. This dose and duration of DNP treatment was selected based on findings from a dose-response experiment using myotubes grown under identical conditions but not incubated with adenoviruses (Sup Fig 1). A concentration of 0.6 mM was the lowest concentration able to significantly increase AMP without significantly decreasing the total adenine nucleotide pool plus IMP (TAdN+IMP) (Sup Fig 1).
Increasing AMPD3 expression for 24 h in the vehicle group was sufficient to decrease intracellular ATP, ADP, and AMP (Fig 1G–I), due to increased AMPD activity, as evidenced by increased IMP (Fig 1J). Moreover, because ADP and ATP were decreased proportional to AMP deamination, AMPD3 groups had similar ATP/ADP (Fig 1K), and significantly higher ATP/AMP (Fig 1L), compared to GFP. AMPD3 groups also had significantly less TAdN+IMP (Fig 1M) and greater amounts of inosine in the culture media (Fig 1N), demonstrating that IMP produced by AMPD3 was further degraded.
As expected, GFP groups treated with 0.6 mM DNP had significant reductions in intracellular ATP, with reciprocal increases in ADP, AMP, and IMP (Fig 1G–J). However, AMPD3 groups treated with DNP had even greater reductions in intracellular ATP, but with significantly less ADP and AMP, and higher levels of IMP compared to GFP (Fig 1G–J). These differences in AMP deamination resulted in maintaining higher intracellular ATP/ADP and ATP/AMP (Fig 1K–L), at the cost of significantly decreasing TAdN+IMP (Fig 1M), and increasing media inosine and hypoxanthine (Fig 1N–O). Importantly, despite having less ATP and TAdN+IMP, myotubes overexpressing AMPD3 had no obvious qualitative differences in size or differentiation (Fig 1E), and protein content per well was actually higher in the DNP treated groups (Fig 1F).
3.2. AMPD3-mediated ATP depletion does not increase AMPK activity
Decreases in ATP are typically accompanied by reciprocal increases in ADP and AMP, and subsequent activation of AMPK[17]. Therefore, to determine if the decreases in ATP caused by AMPD3 (which do not increase ADP and AMP) would activate AMPK differentially than decreases in ATP caused by DNP (which do increase ADP and AMP), we repeated the experiment and performed western blots to determine the phosphorylation of AMPK and its downstream substrates (Fig 2A). DNP treatment caused a significant increase in pAMPKα(Thr172), pAMPK/AMPK ratio, pACC(Ser79), pACC/ACC ratio, and phosphorylated AMPK substrates [LXRXX(pS/pT)], versus vehicle treated groups (Fig 2 B–D). However, no differences were detected between AMPD3 and GFP groups. Thus, AMPK was not activated by 24 h of AMPD3 overexpression, despite a significant loss of ATP.
Fig. 2. AMPD3 overexpression does not activate AMPK and decreases PGC-1α promotor activity.
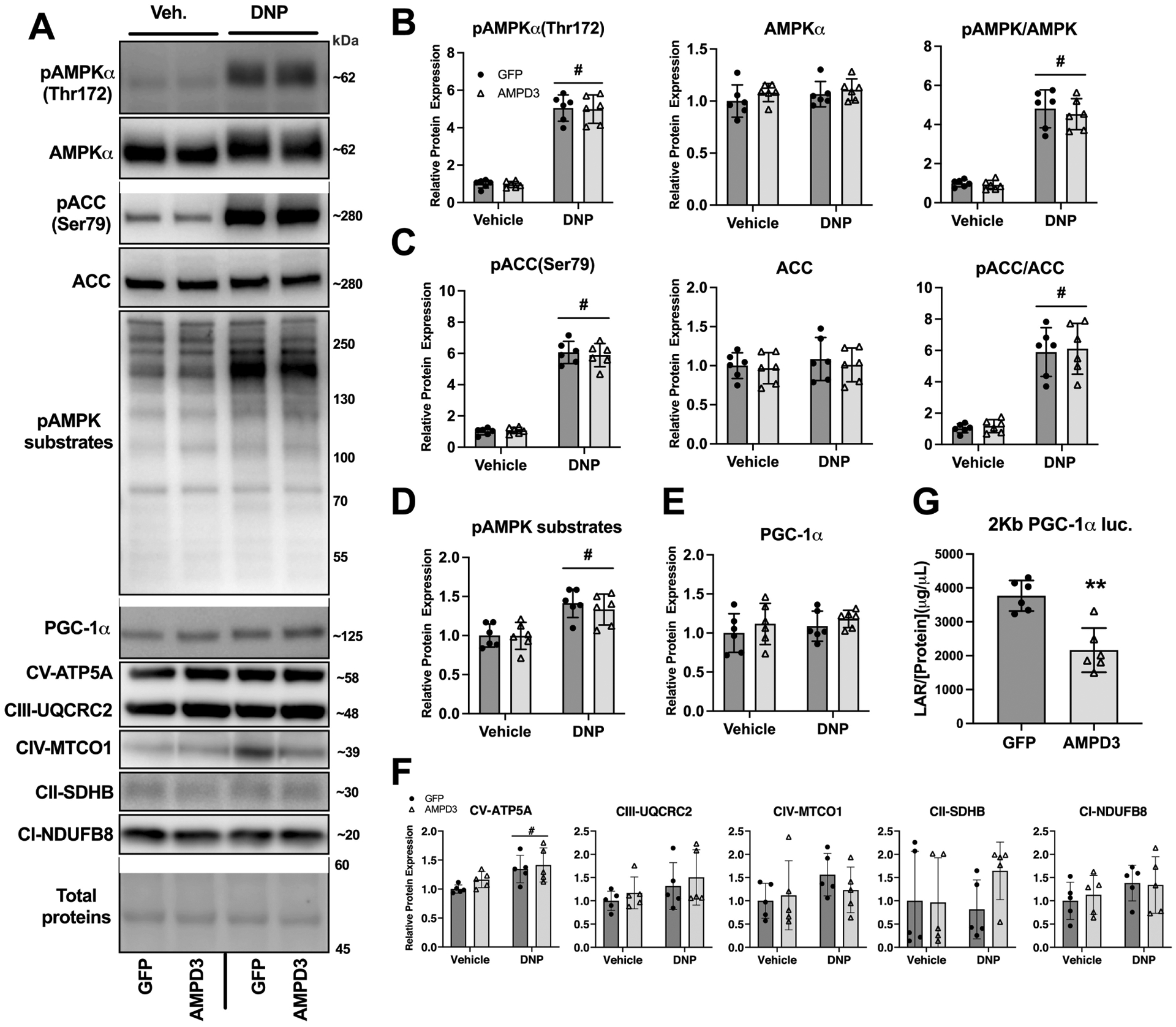
C2C12 myotubes were incubated with adenovirus encoding GFP or AMPD3+GFP for 24 h. During the final hour, myotubes were treated with either 0.6 mM 2,4 dinitrophenol (DNP) or vehicle (methanol). (A) Representative Western blot images for pAMPKα(Thr172), AMPKα, pACC(Ser79), ACC, phosphorylation of pAMPK targeted residues, PGC-1α, and OXPHOS proteins. (B-D) Densitometry analysis of Western blots demonstrating AMPD3 overexpression does not increase pAMPK(Thr172) expression, pACC(Ser79), or pAMPK targeted residues. (E-F) Densitometry analysis of PGC-1α and OXPHOS proteins expression. (G) Luciferase activity in C2C12 myotubes transfected with PGC-1α-promoter luciferase reporter plasmid. Mean±SD, n=6/condition, two-way ANOVA with Tukey’s multiple comparisons or two-tailed unpaired t-test when comparing fewer than four groups. # p<0.05 main effect of DNP, ** p<0.01 vs GFP.
Another downstream target of AMPK is PGC-1α, which upon phosphorylation by AMPK, translocates to the nucleus where it acts as a transcriptional coactivator for nuclear encoded mitochondrial genes, as well as its own gene in a positive feedback mechanism[18, 25]. No differences were detected in PGC-1α protein expression with DNP or AMPD3 groups (Fig 2E), and no differences were found in OXPHOS proteins between AMPD3 vs GFP groups (Fig 2F). However, AMPD3 (without DNP) overexpression led to approximately 50% less PGC-1α promotor activity quantified by luciferase-reporter activity (Fig 2G). Therefore, an observable decrease in AMPK phosphorylation/activation is not required for decreased PGC-1α promotor activity, which suggests that AMPD3 may elicit metabolic remodeling independent of AMPK.
3.3. AMPD3 alters the cellular metabolome independent of changes in gene expression
Given that AMPD3 was sufficient to decrease intracellular ATP and PGC-1α promotor activity, subsequent studies were focused at understanding the effects of AMPD3 alone, i.e. in the absence of DNP. Thus, we questioned whether global gene expression and cell metabolism would be altered by AMPD3 at this timepoint. After incubating myotubes with AMPD3 or GFP adenoviruses for 24 h, we isolated RNA for RNA-sequencing analysis, pelleted cells for intracellular metabolomics analysis, and collected media for extracellular metabolomics analysis.
RNA seq. revealed minimal differences between AMPD3 and GFP groups (Fig 3A). A total of only 30 out of 21,995 detected transcripts were significantly different between AMPD3 and GFP (Sup Table 1). Of these, 29 were significantly upregulated in AMPD3 groups while only 1 was downregulated. As would be predicted, AMPD3 was the most upregulated gene, but the modest upregulation of most other genes was associated with virus exposure and not AMPD3 per se. To our knowledge, none of the altered genes have known functions in cell metabolism.
Fig. 3. AMPD3 significantly alters the cellular metabolome independent of changes in gene expression.
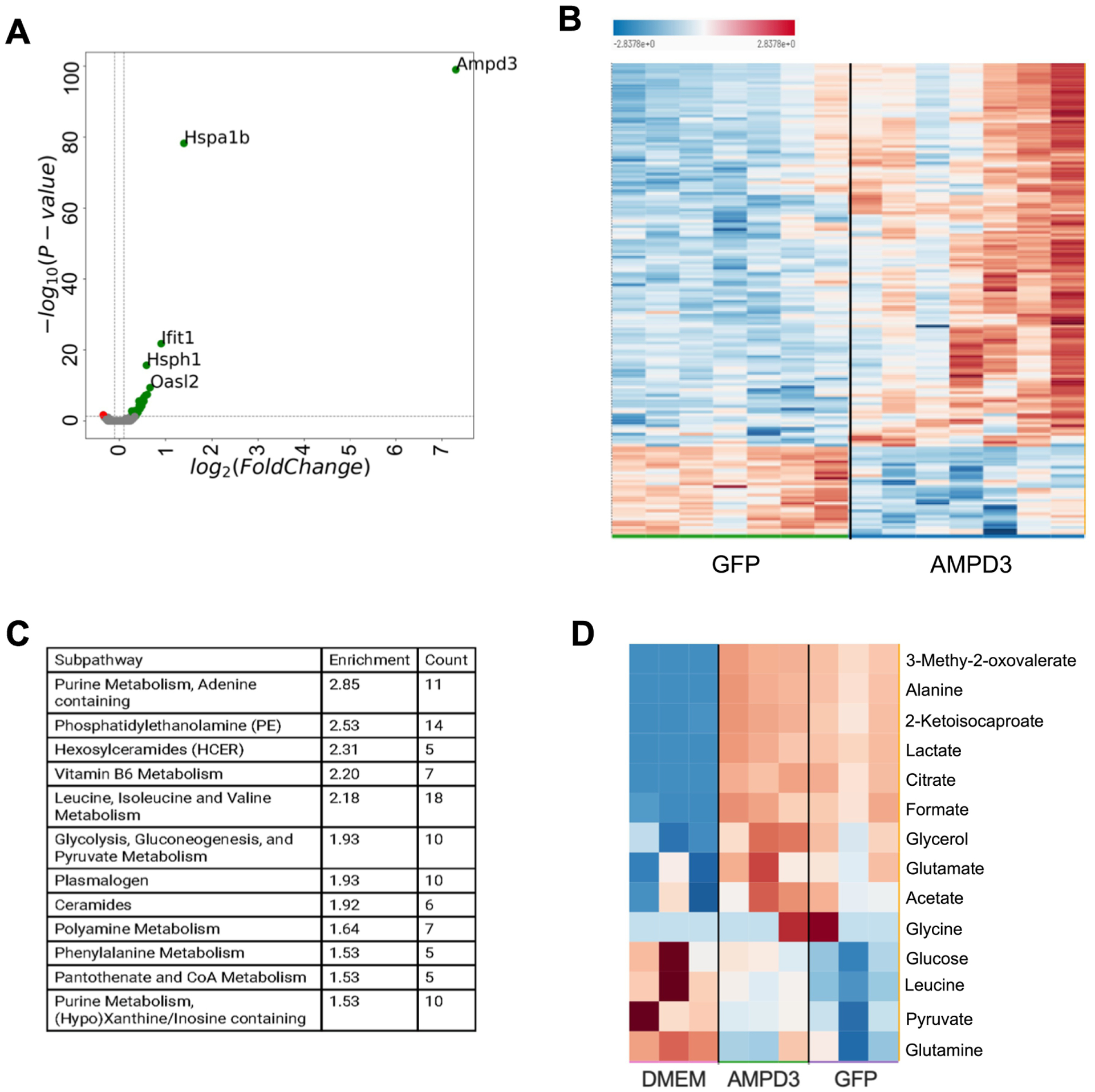
C2C12 myotubes were incubated with adenovirus encoding GFP or AMPD3+GFP for 24 h. (A) Volcano plot of significantly altered genes between AMPD3 and GFP controls as detected by RNAseq. n=3 (B) Heat map of significantly altered intracellular metabolites between AMPD3 and GFP as detected by metabolomics analysis. n=7 (C) Top 12 metabolic pathways altered by AMPD3 as determined by pathway enrichment analysis. (D) Heat map of significantly altered media metabolites between DMEM (untreated media), AMPD3 and GFP as detected by metabolomics analysis. n=3
Analysis of intracellular metabolites revealed substantial alterations in response to AMPD3 (Fig 3B). A total of 191 out of 639 identified metabolites were significantly different between AMPD3 and GFP (Sup Table 2). Pathway enrichment analysis was used to determine the most significantly altered metabolic pathways (Fig 3C). Not surprisingly, purine metabolism was the most altered by AMPD3. In addition, branched chain amino acid (leucine, isoleucine, valine), glycolysis, and ceramide metabolic pathways were also among the most highly enriched by AMPD3. Analysis of the media samples also revealed differences between AMPD3, GFP, and untreated media (Fig 3D). A total of 14 out of 26 measured metabolites were significantly different (Sup Table 2) suggesting that either metabolite consumption or production differed among the three conditions.
When attempting to decipher how AMPD3 altered these metabolic pathways independent of changes in gene expression, we began by considering the metabolic fates of the AMPD products, IMP and ammonia. As presented in Fig. 1, the nucleoside (inosine) and purine base (hypoxanthine) catabolites of IMP were significantly higher in the culture media of AMPD3 groups due to their efflux from the cells. Metabolomics analysis also found a trend for higher intracellular levels of hypoxanthine (p=0.09), and a significant increase in its oxidation product uric acid, in AMPD3 versus GFP groups (Fig 4). Additionally, the levels of succinyl-AMP (sAMP), the intermediate metabolite produced in the process of aminating AMP from IMP, was significantly lower in AMPD3 groups (Fig 4) suggesting that less IMP was being converted to AMP. Hypoxanthine and xanthine are irreversibly oxidized to uric acid by the enzyme xanthine oxidoreductase (XOR), and this reaction can be a major source of reactive oxygen species production[26]. Levels of oxidized glutathione and methionine sulfoxide, which are markers of oxidative stress, were significantly higher in AMPD3 expressing cells (Sup Fig 2), which may be related to increased XOR activity.
Fig. 4. AMPD3 increases IMP flux into purine degradation pathway.
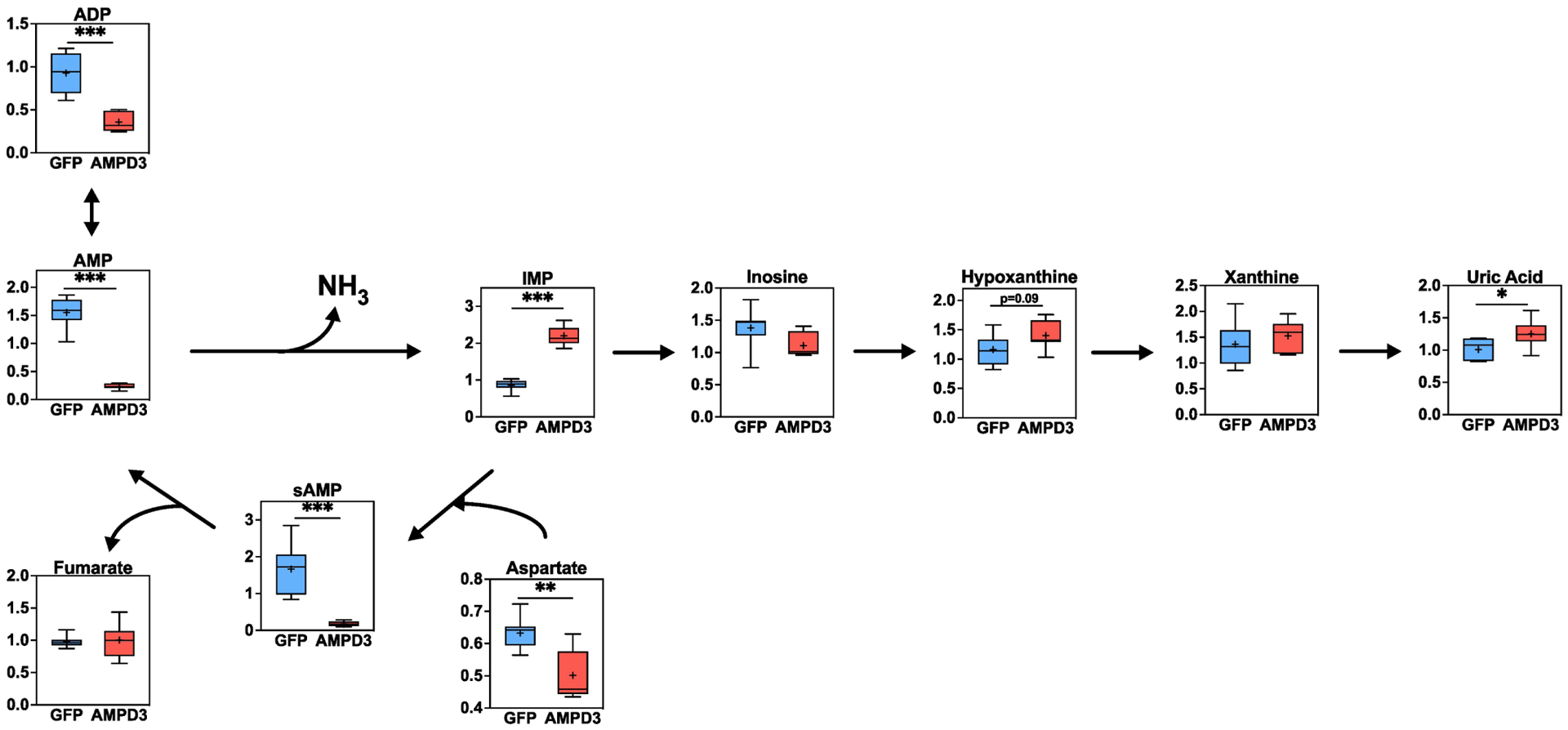
Box plots of intracellular adenine nucleotides, purine degradation metabolites, and the metabolites involved in IMP amination to AMP, determined by metabolomics analysis. Metabolites values were normalized to protein concentration. Statistical significance was determined by two-tailed unpaired t-test. * p<0.05, ** p<0.01, *** p<0.001. n=7/group
The other product of AMPD is free ammonia (NH3), which can have cytotoxic effects[27]. Detoxification of ammonia in muscle is accomplished exclusively by the enzyme glutamine synthetase (GS), which condenses free ammonia with glutamate forming glutamine[28, 29](Fig 5A). Glutamate consumed by GS originates from the transfer of BCAA’s amino group to α-ketoglutarate (α-KG), yielding branched chain keto-acids (BCKA’s) and glutamate in a reversible reaction catalyzed by branched chain aminotransferase 2 (BCAT2)[30]. Higher intracellular levels of BCAA’s and BCKA’s were found in AMPD3 vs. GFP groups (Fig 5B & C), yet no differences were detected in intracellular glutamate or glutamine (Fig 5E & D). Glutamate synthesized from BCAA catabolism is also the substrate for alanine aminotransferase (ALT), which transfers glutamate amino group to pyruvate, yielding alanine and α-KG[31](Fig 5A). Higher intracellular levels of pyruvate and α-KG, but no differences in intracellular alanine, were detected in AMPD3 vs GFP groups (Fig 5D). However, typically intramuscular alanine does not increase when ALT activity is elevated due to alanine being released into circulation[29]. Analysis of media metabolites revealed higher levels of BCKA’s and a trend for higher levels of alanine (p=0.06) in AMPD3 vs GFP groups (Fig 5F). Collectively these data indicate that AMPD3 increased the BCAT and ALT aminotransferase reactions and the flux of these pathways culminated in the release of BCKA’s and alanine into the culture media.
Fig. 5. AMPD3 increases BCAA transamination and BCKA efflux.
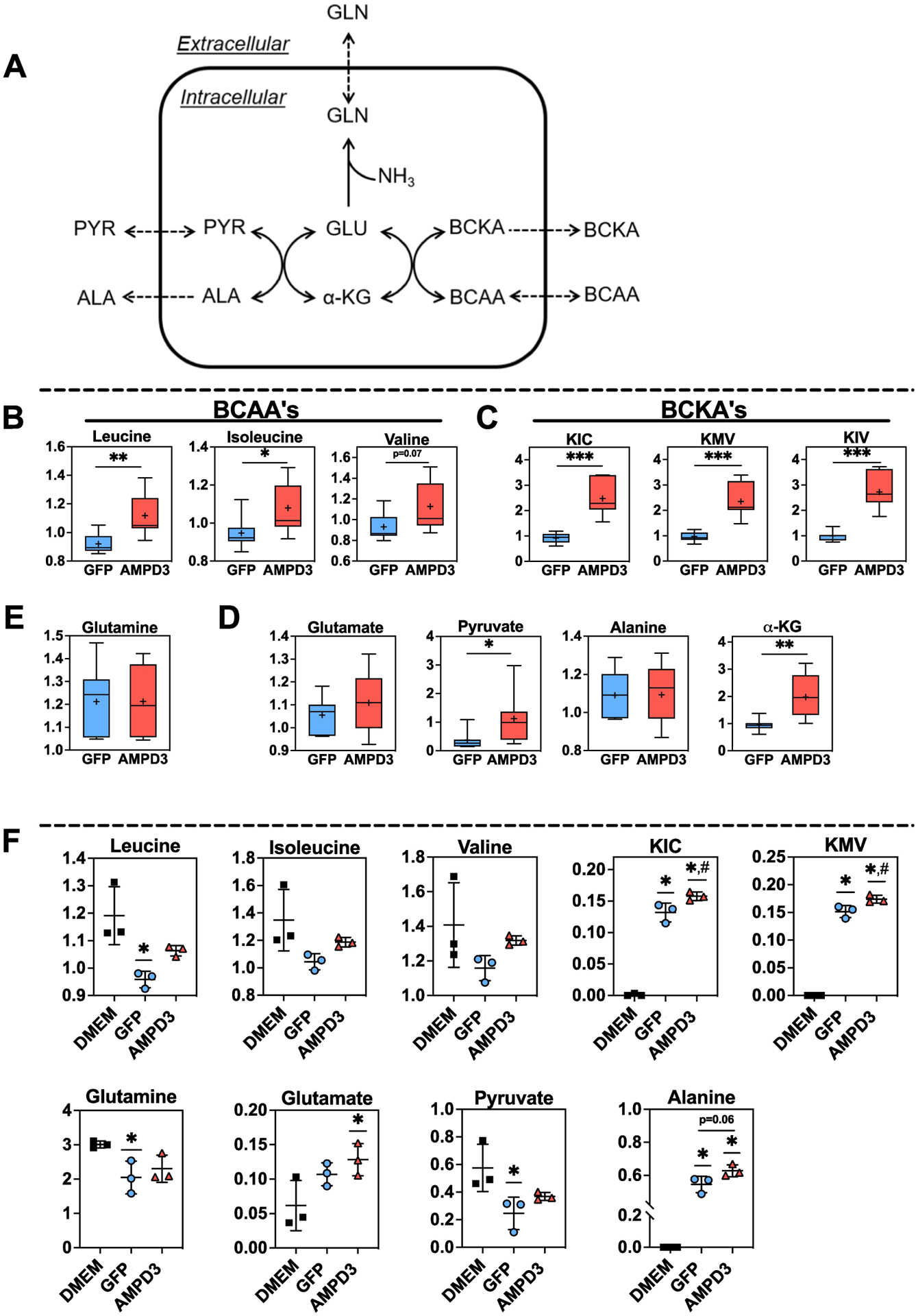
(A) Simplified schematic depicting the connection between de novo glutamine synthesis, de novo alanine synthesis, and BCAA transamination. (B-C) Intracellular branched chain amino acids (BCAA’s) and branched chain keto-acids (BCKA’s), KIC (α-ketoisocaproate), KMV (α-keto-β-methylvalerate), and KIV (α-ketoisovalerate) (E) Intracellular glutamine (D) Intracellular substrates (glutamate + pyruvate) and products (alanine + α-KG) of ALT. Metabolite values were normalized to protein concentration. Statistical significance was determined by two-tailed unpaired t-test between GFP vs AMPD3. * p<0.05, ** p<0.01, *** p<0.001. n=7/group (F) Extracellular (media) levels of BCAA’s, BCKA’s, glutamate, glutamine, alanine, and pyruvate. Statistical significance was determined by ordinary one-way ANOVA. * p<0.05 vs. DMEM, # p<0.05 vs. GFP. n=3/group
Another consequence of elevated resting state AMP deamination is depletion of AdN without decreasing ATP/ADP, ATP/AMP, and activating AMPK (Fig 1 & 2). Absolute concentrations of AdN, and their relative ratios to ATP, influence metabolism of glucose and fatty acids because they are required substrates/co-factors for hexokinase II, phosphoglycerate kinase, pyruvate kinase, and mitochondrial ATP synthase, as well as, allosteric regulators of rate limiting enzymes such as phosphofructokinase[15]. Additionally, they control AMPK activity, which regulates cellular uptake and mitochondrial oxidation of glucose and fatty acids by targeting key enzymes such as AS160 and ACC[32–35]. AMPD3 caused an approximately 57-fold increase in intracellular glucose, however, none of the glycolysis intermediates between glucose and 3-PG were changed by AMPD3 (Fig 6A). Moreover, the reduction in media glucose after 24 h exposure to myotubes was less in the AMPD3 vs. GFP groups suggesting a decrease in glycolytic flux (Fig 6B). If glucose is not catabolized through glycolysis, it can be metabolized by alternative pathways such as the polyol pathway, which involves the reduction glucose to sorbitol followed by sorbitol oxidation to fructose[36]. Sorbitol and fructose were significantly higher in AMPD3 groups (Fig 6C) suggesting increased glucose flux into the polyol pathway.
Fig. 6. AMPD3 does not increase glucose uptake and increases polyol pathway metabolites.
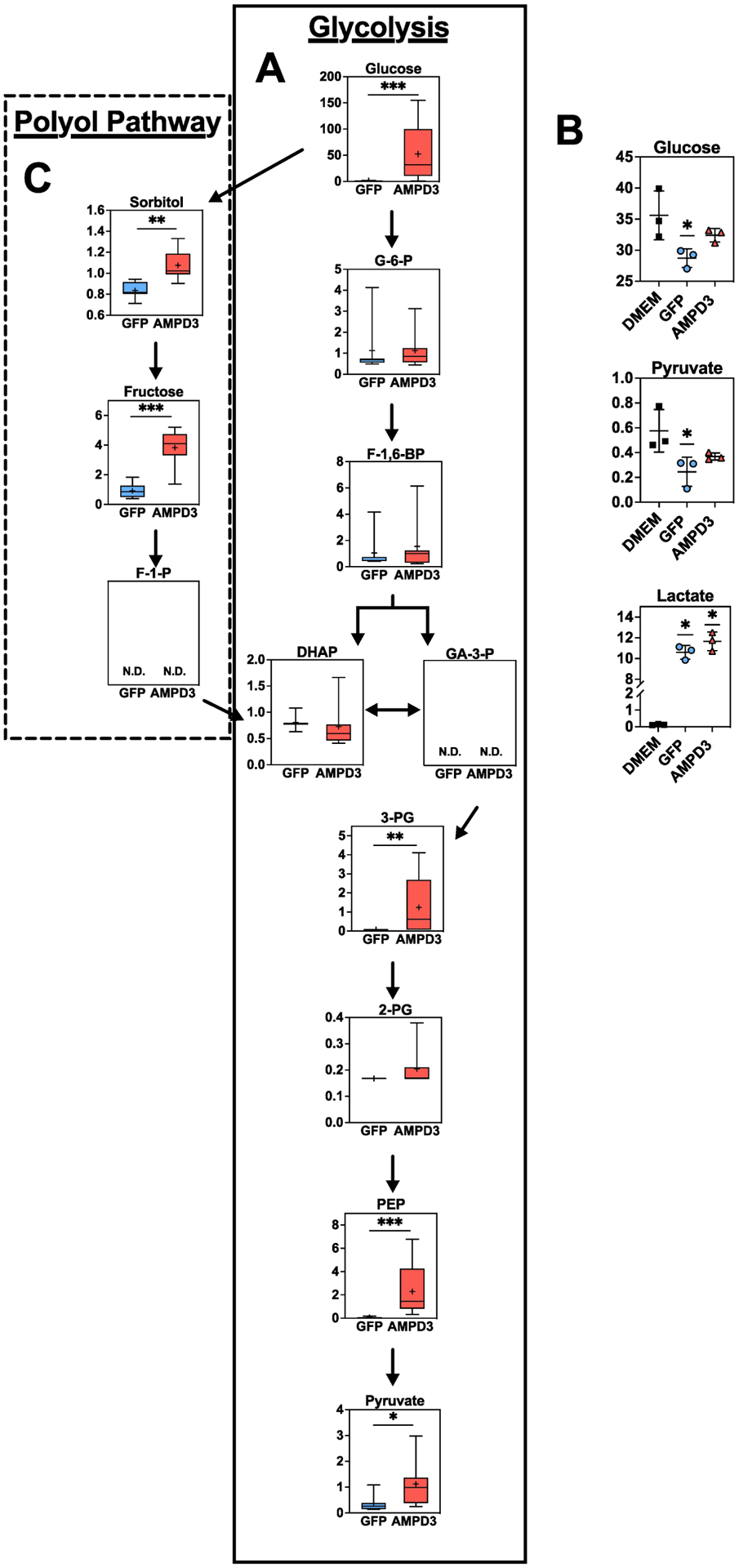
(A) Intracellular metabolites of the glycolysis pathway. Metabolite values were normalized to protein concentration. Statistical significance was determined by two-tailed unpaired t-test. * p<0.05, ** p<0.01, *** p<0.001. n=7/group. (B) Media concentrations of substrates and products of glycolysis (glucose, pyruvate, and lactate). Statistical significance was determined by one-way ANOVA with Tukey post-hoc test, if required. * p<0.05 vs. DMEM. n=3 (C) Intracellular metabolites of the polyol pathway. Metabolite values were normalized to protein concentration. Statistical significance was determined by two-tailed unpaired t-test. * p<0.05, ** p<0.01, *** p<0.001. n=7/group.
A heat map of detected lipids shows an overall increase in lipids in AMPD3 groups (Fig 7A), with particularly higher levels of ceramides and metabolites within the de novo ceramide synthesis pathway (Fig 7B–C). Additionally, palmitoylcarnitine was significantly higher in AMPD3 (Sup. Table 2), but this was the only difference detected in acylcarnitine’s between AMPD3 and GFP, indicating that fatty acid oxidation was unaffected by AMPD3 at this timepoint.
Fig. 7. AMPD3 increases intracellular ceramides and metabolites of de novo ceramide synthesis pathway.
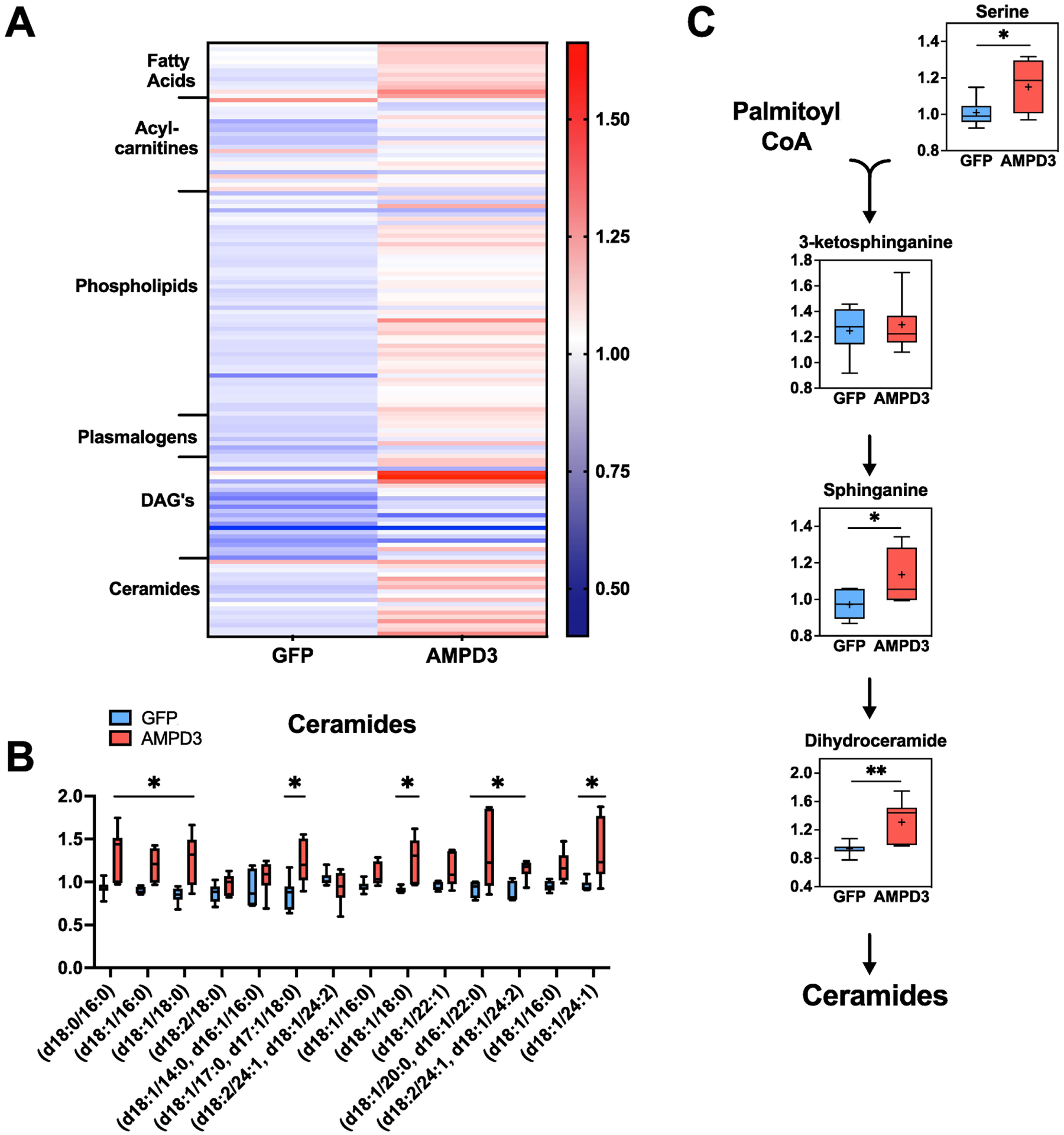
(A) Heat map containing all detected intracellular lipids. (B) Ceramide levels (C) Metabolites of the de novo ceramide synthesis pathway. Metabolites were normalized to protein concentration. Statistical significance was determined by two-tailed unpaired t-test. * p<0.05, ** p<0.01, *** p<0.001. n=7
3.4. Prolonged AMPD3 expression decreases PGC-1a protein, mitochondrial protein synthesis, and basal and maximal mitochondrial respiration
The previous data demonstrated that 24 hours of AMPD3 overexpression led to a major shift in cell metabolism, without measurable differences in markers of AMPK activation, PGC-1α protein expression, or mRNA of mitochondrial genes. However, PGC-1α promotor activity was significantly decreased by 24 h which suggested that longer durations of AMPD3 expression may be required to detect changes in the AMPK/PGC-1α/mitochondrial protein synthesis signaling pathway. Therefore, we performed similar measures of AMPK, PGC-1α and OXPHOS protein expression (Fig 8A). Protein levels of PGC-1α, pAMPKα(thr172), pAMPK/AMPK ratio, and phosphorylation of AMPK substrates were all significantly lower in AMPD3 vs GFP (Fig 8B). Yet, pACC(Ser79) and pACC(Ser79)/ACC ratio was not different between groups. Further, no significant differences were observed with AMPD3 overexpression among the five mitochondrial proteins tested, each from a different electron transport chain subunit. Protein synthesis rates of the total cell homogenate, cytosolic, and mitochondrial subfractions were similar between AMPD3 and GFP at 24 h (Fig 8C). However, at 48 h AMPD3 expressing cells had significantly lower mitochondrial protein synthesis rates compared to GFP, but no difference in total homogenate and cytosolic subfraction protein synthesis rates (Fig 8C). Additionally, basal oxygen consumption trended lower (p=0.07), while ATP synthase-dependent and maximal uncoupled oxygen consumption rates were significantly lower in myotubes expressing AMPD3 vs. GFP (Fig 8 E–H). AMPD3 overexpression did not alter total protein per well (Fig 8I).
Fig. 8. Prolonging AMPD3 expression reduces AMPK/PGC-1α/mitochondrial protein synthesis pathway activity and decreases cellular respiration.
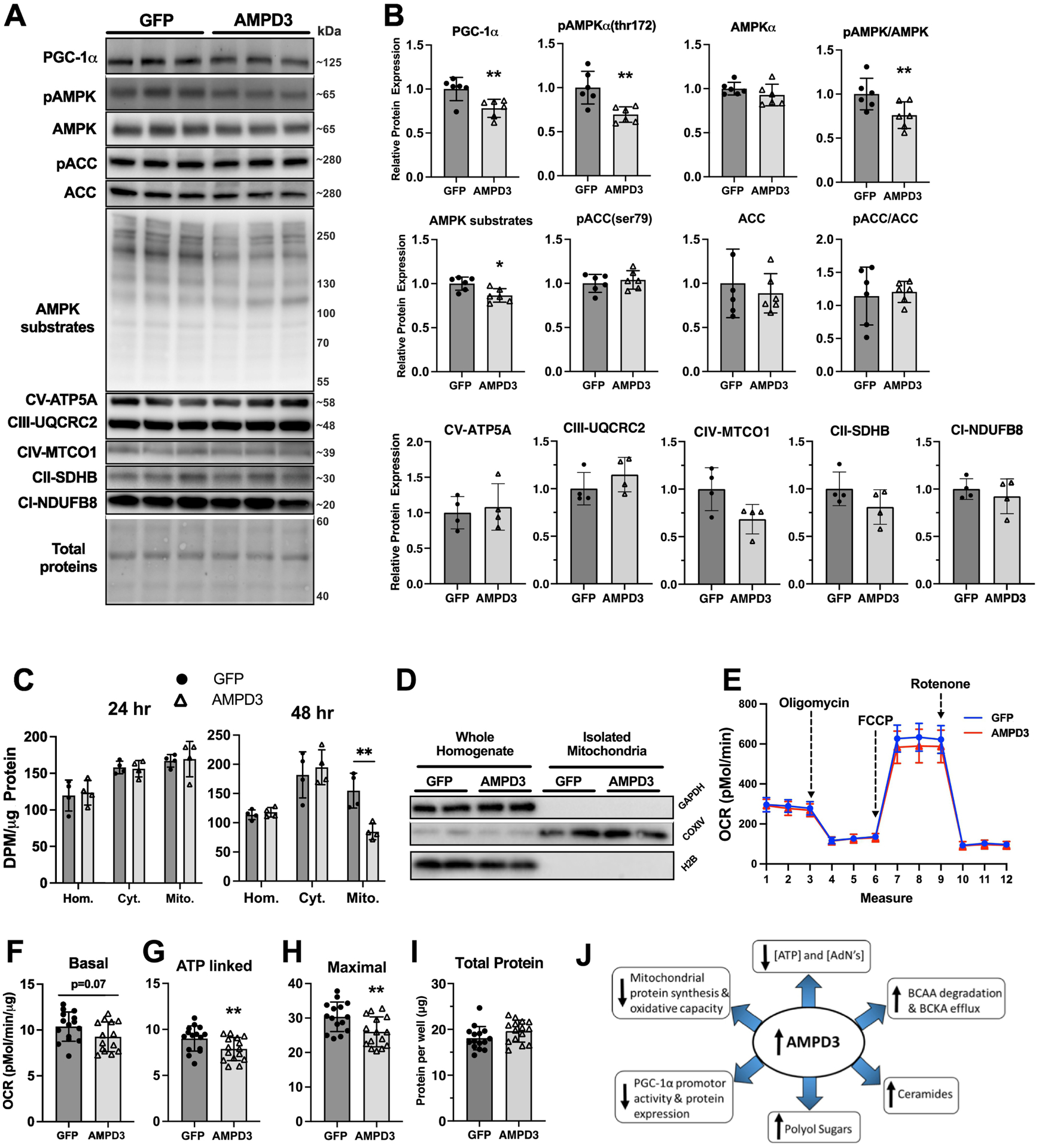
C2C12 myotubes were incubated with adenovirus encoding GFP or AMPD3+GFP for 48 h. (A) Representative Western blot images for PGC-1α, pAMPKα(thr172), AMPKα, pACC(ser79), ACC, phosphorylation of pAMPK targeted residues, and OXPHOS proteins. (B) Densitometry analysis of western blots demonstrating significant reductions in PGC-1α, pAMPK(thr172), pAMPK/AMPK, and pAMPK targeted residues. n=6/group. Two-tailed unpaired t-test. ** p<0.01. (C) Quantifications of total homogenate, cytosolic, and mitochondrial protein synthesis rates after 24 or 48 h of AMPD3 or GFP overexpression. n=4/group. Two-way ANOVA with Sidak’s multiple comparison ** p<0.01 vs GFP. (D) Representative Western blots of proteins specific to cytosolic (GAPDH), mitochondrial (COXIV), and nuclear (H2B) cellular subfractions, demonstrating the efficacy of our mitochondrial isolation method. (E) Representative tracing of oxygen consumption rates by C2C12 myotubes overexpressing AMPD3 or GFP for 48 h and sequentially treated with an electron transport chain complex V inhibitor (oligomycin), inner-membrane uncoupler (FCCP), or complex I inhibitor (Rotenone). (F) Quantification of basal oxygen consumption (measure 3 minus the minimal rate between measures 10–12) normalized to protein concentration. (G) Quantification of complex V (ATP synthase) dependent oxygen consumption (measure 3 minus the minimal rate between measures 4–6) normalized to protein concentration. (H) Quantification of maximal uncoupled oxygen consumption (maximal rate between measures 7–9 minus the minimal rate between measures 10–12) normalized to protein concentration. (I) Total protein determined by BCA assay. n=15/group. Two-tailed unpaired t-test. ** p<0.01. (J) Schematic summarizing the main findings of the study.
4. DISCUSSION
AMPD3 is highly upregulated in atrophying muscle, however, the effects of increased AMPD3 expression on muscle metabolism are unknown. The main purpose of this study was to determine if high levels of AMPD3 expression in skeletal muscle cells would be sufficient to alter their metabolic phenotype similar to that of atrophying muscles. Increasing AMPD3 expression acutely (24 h), was sufficient to cause profound alterations in the intra- and extracellular metabolomes, and some of the notable alterations were: 1) Decreased intracellular ATP, TAdN, and increased intracellular and extracellular AdN degradation products; 2) Increased intracellular and extracellular BCKA concentrations; 3) Increased intracellular glucose, sorbitol, and fructose levels without differences in media glucose; 4) Increased intracellular ceramides and de novo ceramide synthesis pathway metabolites (Fig 8J). These metabolome changes occurred despite no differences in mRNA levels of metabolic enzymes, markers of AMPK activity, PGC-1α protein expression, or mitochondrial protein synthesis. However, increasing AMPD3 expression chronically (48 h), was sufficient to cause significant reductions in markers of AMPK activity, PGC-1α protein expression, mitochondrial protein synthesis, and basal ATP synthase-dependent and maximal uncoupled oxygen consumption. Therefore, high levels of AMPD3 expression in skeletal muscle cells was sufficient to alter their metabolic phenotype similarly to that of atrophying muscles.
Reductions in intramuscular ATP and TAdN, and increases in intramuscular and/or blood concentrations of AdN degradation products (IMP, inosine, hypoxanthine, xanthine, uric acid) have been found across a broad variety of atrophy conditions[1]. We have previously shown that AMPD3 overexpression is sufficient to decrease intracellular ATP and the TAdN+IMP in C2C12 myotubes[23], and in this study we expand upon these findings by showing that IMP produced by AMPD3 is further degraded to its purine nucleoside (inosine) and purine base (hypoxanthine, xanthine, uric acid) degradation products. This was evidenced by our UPLC measures of increased media concentrations of inosine and hypoxanthine which typically do not accumulate in muscle fibers due to their efflux through membrane transporters and/or further oxidation[37, 38]. Moreover, intracellular metabolomics analysis found higher levels of uric acid which is produced exclusively from oxidation of purine bases by xanthine oxidoreductase (XOR) whose activity is increased in muscles during atrophy[39–42]. Therefore, our data suggests that increased IMP degradation caused by increased AMPD3 mediated AMP deamination may be a mechanism linking decreased adenine nucleotides and elevated XOR activity during muscle atrophy.
Increased de novo synthesis and release of glutamine and alanine by skeletal muscles occurs during diabetes[43–45], cancer cachexia[46], acute critical illnesses[47], denervation[6, 48], hindlimb suspension[49], chronic uremia[4], aging[50], glucocorticoid treatment[5, 51–53], TNF-α treatment[54], sepsis[55], and fasting/starvation[29, 56, 57]. Increased de novo synthesis of these amino acids creates a heightened demand for glutamate that is met by transferring a BCAA amino group to the carbon backbone of α-ketoglutarate, yielding a BCKA and glutamate, in a reversible reaction catalyzed by branched chain aminotransferase 2 (BCAT2)[30, 31]. To form glutamine, glutamate is condensed with free ammonia in an irreversible reaction catalyzed by glutamine synthetase (GS). To form alanine, the glutamate amino group is transferred to pyruvate, yielding α-KG and alanine, in a reaction catalyzed by alanine aminotransferase (ALT). It is not fully understood what causes the increase in GS and ALT reactions during atrophy conditions. However, glutamine synthetase is the only enzyme capable of de novo glutamine synthesis and it exclusively uses free ammonia, thus, in order for its activity to be increased an increased source of free ammonia must also be available. Indeed, by comparing the arterial-venous differences across the hindquarters of wild-type and skeletal muscle specific GS knockout mice during prolonged fasting, it was found that only GS knockout muscles released ammonia, and they also failed to release glutamine and alanine[29]. At least two insights can be made from this finding: 1) GS is required for muscles to handle increases in free ammonia produced endogenously by an unknown source; and 2) De novo synthesis and release of alanine is dependent on, and likely a secondary effect of, increased glutamine synthetase activity. Considering that AMPD3 is highly upregulated in muscle during fasting[2], and AMP deamination is a major producer of free ammonia in muscle[58], increased deamination of AMP by AMPD3 may be a mechanism stimulating/supporting de novo glutamine and alanine synthesis during atrophy. Moreover, this process may play a causative role in increasing the transamination of BCAA’s and α-KG to support the heightened demand for glutamate. Based on our findings that AMPD3 increased intracellular BCAA’s, BCKA’s, pyruvate, and α-KG, with concomitant increased release of BCKA’s and a trend for alanine (p=0.06), it is likely that increased AMPD3 activity drove the BCAT2 and ALT reactions. The increase in BCAT2 an ALT activity may have been caused by a heightened demand for glutamate due to elevated ammonia production and GS activity. However, we cannot definitively attribute the effects to increases in GS activity because we did not detect intra- or extracellular differences in glutamate or glutamine between AMPD3 and GFP. This may have been due to supplementing our culture media with exogenous glutamine which has been found to oppose increases in GS activity[59, 60] and thus may have weakened our sensitivity to detect changes in glutamine.
Increased de novo ceramide synthesis and accumulation of ceramides has been found in myotubes after TNF-α treatment[11], and in rodent skeletal muscles after C26 carcinoma implantation[11], hindlimb unloading[12], glucocorticoid treatment[61], and obesity induced insulin resistance[61]. Palmitoyl-CoA availability is the rate limiting factor in de novo ceramide synthesis. This is based on multiple studies showing elevated palmitoyl-CoA levels, due to either excess fatty acid consumption (e.g. high-fat diet[13], leptin receptor deficiency[13], exogenous palmitate supplementation[61]), enhanced de novo fatty acid synthesis[34], or insufficient/impaired fatty acid oxidation (e.g. CPT-1 KO[62]), result in increased de novo ceramide synthesis and accumulation. Conversely, ceramide accumulation is prevented by increasing mitochondrial respiration and fatty acid oxidation with AICAR or resveratrol treatments[63–65], or in transgenic mice with elevated fatty acid oxidation rates[66]. Our finding that AMPD3 caused an increase in many different ceramide species and metabolites of the de novo ceramide synthesis pathway, suggests that it elevated free palmitoyl-CoA levels which may have arisen from increased de novo fatty acid synthesis from TCA metabolites, triacylglycerol lipolysis, and/or reduced fatty acid oxidation.
An increase in polyol pathway metabolites sorbitol and fructose have been found in skeletal muscle during diabetes[9, 67, 68], after nerve damage[10], and hindlimb ischemia[69]. The polyol pathway involves the reduction of glucose to sorbitol by aldose reductase, followed by sorbitol oxidation to fructose by sorbitol dehydrogenase[36]. That the polyol pathway is induced by ischemia is interesting because hypoxia is a known stimulus of rapid AMP deamination and purine nucleotide degradation that culminates in elevated uric acid production[70]. In hepatocytes, uric acid has been shown to activate aldose reductase which caused glucose flux into the polyol pathway and correlated with lipid accumulation[71]. Therefore, our finding that AMPD3 increased the polyol metabolites along with various lipids may be related to the effects of increased uric acid on aldose reductase.
Although RNAseq analysis did not reveal any differences in metabolic genes, AMPD3 overexpression did cause an increase in HSPa1a and HSPa1b mRNA. These transcripts encode for heat shock protein 70 (Hsp70), which is a critical chaperone protein that functions to maintain protein homeostasis during periods of cell stress[72]. Hsp70 mRNA typically has a half-life of fifty minutes, but upon loss of ATP its half-life increases dramatically[73]. Therefore, AMPD3 mediated increases in Hsp70 mRNA and the expected increase in chaperone capacity may partly explain our previous findings that AMPD3 is capable of slowing protein degradation[23].
In addition to acutely regulating metabolism, AdN chronically influence the metabolic phenotype of muscles due to their regulation of enzymes involved in signaling pathways that selectively determine gene expression. The AMPK/PGC-1α/mitochondrial protein synthesis pathway is a well characterized example that is critical for muscles to chronically match the transcription of nuclear encoded mitochondrial genes with the energy demands of the muscle fiber[34, 74–76]. While the evidence for AMPK activity during atrophy are inconsistent, reductions in mRNA and protein expression of PGC-1α and mitochondrial oxidative enzymes are consistently found during atrophy[2, 3, 77, 78]. Moreover, treating animals during atrophy with chemicals that activate AMPK indirectly by decreasing the ATP/AMP, such as resveratrol or AICAR[79], can preserve PGC-1α expression, mitochondrial enzymes, and/or oxidative capacity of muscles[65, 78, 80–82]. The effects of these chemicals are dependent on the activation of AMPK[35, 83]. These studies suggest that the AMPK/PGC-1α/mitochondrial protein synthesis pathway is reduced during atrophy but still capable of being activated. Here we show that increased AMPD3 expression for 48 hours leads to reductions in pAMPKα(Thr172)/AMPKα and phosphorylation of AMPK targeted residues, PGC-1α promotor activity and protein expression, mitochondrial protein synthesis, and both ATP synthase dependent and maximal uncoupled oxygen consumption rates of myotubes. However, we were unable to detect significant decreases in mitochondrial proteins. This apparent discrepancy is likely due to the kinetics of mitochondrial loss. Mitochondrial protein synthesis rates were decreased at 48h but not at 24h, which suggests that changes in synthesis rates may have only been present for a short duration. Only sustained changes in mitochondrial protein synthesis rates would be expected to result in measurable changes in mitochondrial protein content. Thus, increased AMPD3 and AMP deamination may contribute to decreases in oxidative metabolism and oxidative capacity of skeletal muscles during atrophy by chronically suppressing mitochondrial ATP synthase activity and the AMPK/PGC-1α/mitochondrial protein synthesis signaling pathway.
This current study using overexpression of AMPD3 in cultured C2C12 myotubes has at least two limitations. First, common C2C12 differentiation media (DMEM with 2% horse serum) has high levels of glucose (25 mM), pyruvate (1 mM), and glutamine (4_mM) and low levels of fatty acids as compared to mouse plasma. Since media composition can impact mitochondrial content and perhaps other aspects of metabolism[84], our studies of AMPD3 overexpression in myotubes should be interpreted in the context of a different metabolic baseline as compared to adult muscle. A second potential limitation is the use of adenoviruses to induce high levels of AMPD3 overexpression (AMPD3 mRNA was 157-fold greater than GFP controls). However, in skeletal muscles undergoing atrophy AMPD3 has repeatedly been identified as one of the most highly upregulated genes. For example, AMPD3 mRNA is increased 100-fold in streptotozcin-induced diabetes[2], 28-fold in chronic renal failure [2], and 46-fold in denervation [21]. Therefore, our adenoviral-induced overexpression is greater than that found in vivo but is comparable in magnitude. Future studies, conducted in vivo utilizing preclinical animal models of muscle atrophy will help to further elucidate the function of AMPD3 upregulation in skeletal muscles during atrophy conditions.
Because of the broad metabolic effects induced by AMPD3, targeting this gene may have therapeutic potential. Increased accumulation of intramuscular ceramides[13, 61, 85], increased oxidative stress[41, 86], and increased glucose metabolism via the polyol pathway[9, 36, 67] have been shown to reduce muscle insulin signaling pathways and glucose uptake, and promote systemic hyperglycemia Since AMPD3 can drive the accumulation of ceramides and the polyol pathway, inhibition of AMPD3 may allow for the attenuation of these unfavorable metabolic alterations and improve muscle and systemic metabolic health. This has been partly tested using AMPD1 (the predominant AMPD isoform in skeletal muscle) knockout mice fed a high fat diet. In this study, loss of AMPD1 did not attenuate rises in blood glucose in response to oral glucose tolerance tests compared to wild-type controls [87]. However, AMPD3 is upregulated in muscles during hypoinsulinemia induced diabetes [2]. Therefore, a dual knockout of both AMPD isoforms may be required to test whether AMPD inhibition would be a molecular target for increasing muscle glucose uptake. Atrophic muscles have severe reductions in oxidative capacity[88–90], which results in enhanced fatigability during daily physical activity and reduced quality of life of affected individuals[91–93]. The decrease in oxidative capacity is greatly mediated by reductions in total mitochondria content of the muscle fibers. Therefore, therapeutic approaches aimed at inhibiting AMPD3 in skeletal muscle, possibly in combination with tolerable contractile activity if feasible, to prevent the loss of mitochondria, may represent a therapeutic strategy to preserve oxidative metabolism and oxidative capacity of muscle fibers during atrophy conditions.
In conclusion, our study shows that increasing AMPD3 expression in C2C12 myotubes is sufficient to replicate many of the metabolic changes that occur in skeletal muscles during atrophy. These changes occur temporally, such that the changes in metabolism occur prior to substantial changes in gene expression, AMPK signaling, and mitochondrial protein synthesis rates, suggesting that changes in metabolism are not dependent on reductions in oxidative capacity. Therefore, increased AMP deamination due to upregulated AMPD3 expression may be a cellular mechanism altering the metabolic phenotype of skeletal muscles during atrophy.
Supplementary Material
Highlights.
AMP deaminase (AMPD) decreases ATP without activating AMPK or its substrates
AMPD alters the intracellular metabolome similar to atrophic muscle
AMPD slows mitochondria synthesis and oxygen consumption similar to atrophic muscle
Metabolome shift is independent of metabolic genes and precede mitochondria changes
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (R01 AR070200 to JJB; R01 DK103562 to CAW; and P01 AG039355, P30 AR072581, SBIR HHSN26120600064C to TMOC), the American Physiological Society (Postdoctoral Fellowship to PSH), and the Indiana Clinical and Translational Sciences Institute (to DLL).
We gratefully thank Dr. Espen Spangenburg and Dr. Michael Tarpey for their expert assistance with the myotube respiration studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTEREST: none
References
- [1].Miller SG, Hafen PS, Brault JJ. Increased Adenine Nucleotide Degradation in Skeletal Muscle Atrophy. Int J Mol Sci. 2019;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- [3].Ibebunjo C, Chick JM, Kendall T, Eash JK, Li C, Zhang Y, et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol Cell Biol. 2013;33:194–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Garber A Skeletal muscle protein and amino acid metabolism in experimental chronic uremia in the rat. Accelerated alanine and glutamine formation and release. J Clin Invest. 1978:623–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Darmaun D, Matthews DE, Bier DM. Physiological hypercortisolemia increases proteolysis, glutamine, and alanine production. AJP Endocrinol Metab. 1988;18:E366–E73. [DOI] [PubMed] [Google Scholar]
- [6].Hundal HS, Babij P, Watt PW, Ward MR, Rennie MJ. Glutamine transport and metabolism in denervated rat skeletal muscle. AJP Endocrinol Metab. 1990;22:E148–E54. [DOI] [PubMed] [Google Scholar]
- [7].Hutson SS A; LaNoue K Branched-Chain amino acid metabolism: implications for establishing safe intakes. The Journal of Nutrition. 2005;135:1557S–64S. [DOI] [PubMed] [Google Scholar]
- [8].Ruderman NB, Berger M. The formation of glutamine and alanine in skeletal muscle. The Journal of Biological Chemistry. 1974;249:5500–6. [PubMed] [Google Scholar]
- [9].Cotter MA, Cameron NE, Robertson S, Erwing I. Polyol pathway-related skeletal muscle contractile and morphological abnormalities in diabetic rats. Experimental Physiology. 1993;78:139–55. [DOI] [PubMed] [Google Scholar]
- [10].Langer HT, Afzal S, Kempa S, Spuler S. Nerve damage induced skeletal muscle atrophy is associated with increased accumulation of intramuscular glucose and polyol pathway intermediates. Sci Rep. 2020;10:1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].De Larichaudy J, Zufferli A, Serra F, Isidori AM, Naro F, Dessalle K, et al. TNF-alpha- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet Muscle. 2012;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Salaun E, Lefeuvre-Orfila L, Cavey T, Martin B, Turlin B, Ropert M, et al. Myriocin prevents muscle ceramide accumulation but not muscle fiber atrophy during short-term mechanical unloading. J Appl Physiol (1985). 2016;120:178–87. [DOI] [PubMed] [Google Scholar]
- [13].Ussher JR, Koves TR, Cadete VJ, Zhang L, Jaswal JS, Swyrd SJ, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes. 2010;59:2453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab. 2012;23:142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bruser A, Kirchberger J, Kloos M, Strater N, Schoneberg T. Functional linkage of adenine nucleotide binding sites in mammalian muscle 6-phosphofructokinase. J Biol Chem. 2012;287:17546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci. 2008;65:3737–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gowans GJ, Hardie DG. AMPK: a cellular energy sensor primarily regulated by AMP. Biochem Soc Trans. 2014;42:71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jager S, Handschin C, St.-Pierre J, Spiegelman B. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGG-1α PNAS. 2007;104:12017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–26. [DOI] [PubMed] [Google Scholar]
- [20].Brocca L, Toniolo L, Reggiani C, Bottinelli R, Sandri M, Pellegrino MA. FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J Physiol. 2017;595:1143–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hancock CR, Brault JJ, Terjung R. Protecting the cellular energy state during contractions: role of AMP deaminase. Journal of Physiology and Pharmacology. 2006;57. [PubMed] [Google Scholar]
- [23].Davis PR, Miller SG, Verhoeven NA, Morgan JS, Tulis DA, Witczak CA, et al. Increased AMP deaminase activity decreases ATP content and slows protein degradation in cultured skeletal muscle. Metabolism. 2020;108:154257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Brault JJ, Pizzimenti NM, Dentel JN, Wiseman RW. Selective inhibition of ATPase activity during contraction alters the activation of p38 MAP kinase isoforms in skeletal muscle. J Cell Biochem. 2013;114:1445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Handschin C, Rhee J, Lin J, Tarr P, Spiegelman B. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. PNAS. 2003;100:7111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hakvoort TB, He Y, Kulik W, Vermeulen JL, Duijst S, Ruijter JM, et al. Pivotal role of glutamine synthetase in ammonia detoxification. Hepatology. 2017;65:281–93. [DOI] [PubMed] [Google Scholar]
- [28].Hod G, Chaouat M, Haskel Y, Lernau OZ, Mayer M, Nissan S. Ammonia uptake by skeletal muscle in the hyperammonaemic rat. European Journal of Clinical Investigation. 1982;12:445–50. [DOI] [PubMed] [Google Scholar]
- [29].He Y, Hakvoort TB, Kohler SE, Vermeulen JL, de Waart DR, de Theije C, et al. Glutamine synthetase in muscle is required for glutamine production during fasting and extrahepatic ammonia detoxification. J Biol Chem. 2010;285:9516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aoki TT, Brennan MF, Fitzpatrick GF, Knight DC. Leucine meal increases glutamine and total nitrogen release from forearm muscle. J Clin Invest. 1981;68:1522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Simmons PS, Miles JM, Gerich JE, Haymond MW. Increased proteolysis. An effect of increases in plasma cortisol within the physiologic range. J Clin Invest. 1984;73:412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. AJP Endocrinol Metab. 1996;33:E299–E304. [DOI] [PubMed] [Google Scholar]
- [33].Smith AC, Bruce CR, Dyck DJ. AMP kinase activation with AICAR simultaneously increases fatty acid and glucose oxidation in resting rat soleus muscle. J Physiol. 2005;565:537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].O’Neill HM, Lally JS, Galic S, Thomas M, Azizi PD, Fullerton MD, et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia. 2014;57:1693–702. [DOI] [PubMed] [Google Scholar]
- [35].Treebak JT, Glund S, Deshmukh A, Klein DK, Long YC, Jensen TE, et al. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes. 2006;55:2051–8. [DOI] [PubMed] [Google Scholar]
- [36].Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol. 2012;3:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hellsten Y, Richter EA, Kiens B, Bangsbo J. AMP deamination and purine exchange in human skeletal muscle during and after intense exercise. Journal of Physiology. 1999;529:909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bone DB, Choi DS, Coe IR, Hammond JR. Nucleoside/nucleobase transport and metabolism by microvascular endothelial cells isolated from ENT1−/− mice. Am J Physiol Heart Circ Physiol. 2010;299:H847–56. [DOI] [PubMed] [Google Scholar]
- [39].Derbre F, Ferrando B, Gomez-Cabrera MC, Sanchis-Gomar F, Martinez-Bello VE, Olaso-Gonzalez G, et al. Inhibition of xanthine oxidase by allopurinol prevents skeletal muscle atrophy: role of p38 MAPKinase and E3 ubiquitin ligases. PLoS One. 2012;7:e46668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Konishi M, Pelgrim L, Tschirner A, Baumgarten A, von Haehling S, Palus S, et al. Febuxostat improves outcome in a rat model of cancer cachexia. J Cachexia Sarcopenia Muscle. 2015;6:174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Whidden MA, McClung JM, Falk DJ, Hudson MB, Smuder AJ, Nelson WB, et al. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J Appl Physiol (1985). 2009;106:385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Aranda R, Domenech E, Rus AD, Real JT, Sastre J, Vina J, et al. Age-related increase in xanthine oxidase activity in human plasma and rat tissues. Free Radic Res. 2007;41:1195–200. [DOI] [PubMed] [Google Scholar]
- [43].Feng B, Banner C, Max S. Effect of diabetes on glutamine synthetase expression in rat skeletal muscles. AJP Endocrinol Metab. 1990;258:E762–E6. [DOI] [PubMed] [Google Scholar]
- [44].Karl I, Garber A, Kipnis D. Alanine and glutamine synthesis and release from skeletal muscle. The Journal of Biological Chemistry. 1976;251:844–50. [PubMed] [Google Scholar]
- [45].Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pedersen KS, Gatto F, Zerahn B, Nielsen J, Pedersen BK, Hojman P, et al. Exercise-Mediated Lowering of Glutamine Availability Suppresses Tumor Growth and Attenuates Muscle Wasting. iScience. 2020;23:100978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Biolo G, Zorat F, Antonione R, Ciocchi B. Muscle glutamine depletion in the intensive care unit. Int J Biochem Cell Biol. 2005;37:2169–79. [DOI] [PubMed] [Google Scholar]
- [48].Feng B, Konagaya M, Konagaya Y, Thomas J, Banner C, Mill J, et al. Neural control of glutamine synthetase activity in rat skeletal muscles. Am J Physiol Endocrinol Metab 1990;221:E757–E61. [DOI] [PubMed] [Google Scholar]
- [49].Jaspers S, Jacob S, Tischler M. Metabolism of amino acids by the atrophied soleus of tail-casted, suspended rats. Metabolism. 1986;35:216–23. [DOI] [PubMed] [Google Scholar]
- [50].Pinel C, Coxam V, Mignon M, Taillandier D, Cubizolles C, Lebecque P, et al. Alterations in glutamine synthetase activity in rat skeletal muscle are associated with advanced age. Nutrition. 2006;22:778–85. [DOI] [PubMed] [Google Scholar]
- [51].Max S Glucocorticoid-mediated induction of glutamine synthetase in skeletal muscle. Medicine and Science in Sports and Exercise. 1990;22:325–30. [PubMed] [Google Scholar]
- [52].Hickson R, Wegrzyn L, Osborne D, Karl I. Alanyl-glutamine prevents muscle atrophy and glutamine synthetase induction by glucocorticoids. AJP Regulatory Integrative Comp Physiol. 1996;40:R1165–R72. [DOI] [PubMed] [Google Scholar]
- [53].Salleh M, Ardawi M, Jamal Y. Glutamine metabolism in skeletal muscle of glucocorticoid-treated rats. Clinical Science. 1990;79. [DOI] [PubMed] [Google Scholar]
- [54].Chakrabarti R Transcriptional regulation of the rat glutamine synthetase gene by tumor necrosis factor-α. Eur J Biochem 1998;254:70–4. [DOI] [PubMed] [Google Scholar]
- [55].Karinch A, Pan M, Lin C, Strange R, Souba W. Glutamine Metabolism in Sepsis and Infection. American Society of Nutritional Sciences. 2001. [DOI] [PubMed] [Google Scholar]
- [56].Marliss EB, Aoki TT, Pozefsky T, Most AS, Cahill GF Jr.. Muscle and splanchnic glutamine and glutamate metabolism in postabsorptive and starved man. J Clin Invest. 1971;50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Odessey R, Khairallah E, Goldberg A. Origin and possible significance of alanine production by skeletal muscle. Journal of Biologial Chemistry. 1974;249:7623–9. [PubMed] [Google Scholar]
- [58].Sahlin K, Broberg S. Adenine nucleotide depletion in human muscle during exercise: causality and significance of AMP Deamination. Int J Sports Med. 1990;11. [DOI] [PubMed] [Google Scholar]
- [59].Wang Y, Watford M. Glutamine, insulin and glucocorticoids regulate glutamine synthetase expression in C2C12 myotubes, Hep G2 hepatoma cells and 3T3 L1 adipocytes. Biochim Biophys Acta. 2007;1770:594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hickson R, Wegrzyn L, Osborne D, Karl I. Glutamine interferes with glucocorticoid-induced expression of glutamine synthetase in skeletal muscle. AJP Endocrinol Metab. 1996;33:E912–E7. [DOI] [PubMed] [Google Scholar]
- [61].Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–79. [DOI] [PubMed] [Google Scholar]
- [62].Wicks SE, Vandanmagsar B, Haynie KR, Fuller SE, Warfel JD, Stephens JM, et al. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc Natl Acad Sci U S A. 2015;112:E3300–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Erickson KA, Smith ME, Anthonymuthu TS, Evanson MJ, Brassfield ES, Hodson AE, et al. AICAR inhibits ceramide biosynthesis in skeletal muscle. Diabetol Metab Syndr. 2012;4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, et al. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180–91. [DOI] [PubMed] [Google Scholar]
- [65].Momken I, Stevens L, Bergouignan A, Desplanches D, Rudwill F, Chery I, et al. Resveratrol prevents the wasting disorders of mechanical unloading by acting as a physical exercise mimetic in the rat. FASEB J. 2011;25:3646–60. [DOI] [PubMed] [Google Scholar]
- [66].Dobrzyn A, Dobrzyn P, Lee SH, Miyazaki M, Cohen P, Asilmaz E, et al. Stearoyl-CoA desaturase-1 deficiency reduces ceramide synthesis by downregulating serine palmitoyltransferase and increasing beta-oxidation in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E599–607. [DOI] [PubMed] [Google Scholar]
- [67].Cameron NE, Cotter MA, Robertson S. Changes in skeletal muscle contractile properties in streptozocin-induced diabetic rats and role of polyol pathway and hypoinsulinemia. Diabetes. 1990;39:460–5. [DOI] [PubMed] [Google Scholar]
- [68].Sanchez OA, Walseth TF, Snow LM, Serfass RC, Thompson LV. Skeletal muscle sorbitol levels in diabetic rats with and without insulin therapy and endurance exercise training. Exp Diabetes Res. 2009;2009:737686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yagihashi S, Mizukami H, Ogasawara S, Yamagishi S, Nukada H, Kato N, et al. The role of the polyol pathway in acute kidney injury caused by hindlimb ischaemia in mice. J Pathol. 2010;220:530–41. [DOI] [PubMed] [Google Scholar]
- [70].Lindsay TF, Liauw S, Romaschin AD, Walker PM. The effect of ischemia/reperfusion on adenine nucleotide metabolism and xanthine oxidase production in skeletal muscle. J Vasc Surg. 1990;12:8–15. [DOI] [PubMed] [Google Scholar]
- [71].Sanchez-Lozada LG, Andres-Hernando A, Garcia-Arroyo FE, Cicerchi C, Li N, Kuwabara M, et al. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J Biol Chem. 2019;294:4272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Silver JT, Noble EG. Regulation of survival gene hsp70. Cell Stress Chaperones. 2012;17:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Benjamin IJ, Horie S, Greenberg ML, Alpern RJ, Williams RS. Induction of stress proteins in cultured myogenic cells. Molecular signals for the activation of heat shock transcription factor during ischemia. J Clin Invest. 1992;89:1685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Fentz J, Kjobsted R, Birk JB, Jordy AB, Jeppesen J, Thorsen K, et al. AMPKalpha is critical for enhancing skeletal muscle fatty acid utilization during in vivo exercise in mice. FASEB J. 2015;29:1725–38. [DOI] [PubMed] [Google Scholar]
- [75].Lee-Young RS, Griffee SR, Lynes SE, Bracy DP, Ayala JE, McGuinness OP, et al. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem. 2009;284:23925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Okamoto S, Asgar NF, Yokota S, Saito K, Minokoshi Y. Role of the alpha2 subunit of AMP-activated protein kinase and its nuclear localization in mitochondria and energy metabolism-related gene expressions in C2C12 cells. Metabolism. 2019;90:52–68. [DOI] [PubMed] [Google Scholar]
- [77].Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. [DOI] [PubMed] [Google Scholar]
- [78].Brault JJ, Jespersen JG, Goldberg AL. Peroxisome proliferator-activated receptor gamma coactivator 1alpha or 1beta overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem. 2010;285:19460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bennett BT, Mohamed JS, Alway SE. Effects of resveratrol on the recovery of muscle mass following disuse in the plantaris muscle of aged rats. PLoS One. 2013;8:e83518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Huang Y, Zhu X, Chen K, Lang H, Zhang Y, Hou P, et al. Resveratrol prevents sarcopenic obesity by reversing mitochondrial dysfunction and oxidative stress via the PKA/LKB1/AMPK pathway. Aging 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–22. [DOI] [PubMed] [Google Scholar]
- [83].Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59:554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Elkalaf M, Andel M, Trnka J. Low glucose but not galactose enhances oxidative mitochondrial metabolism in C2C12 myoblasts and myotubes. PLoS One. 2013;8:e70772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, et al. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem. 2003;278:10297–303. [DOI] [PubMed] [Google Scholar]
- [86].Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Admyre T, Amrot-Fors L, Andersson M, Bauer M, Bjursell M, Drmota T, et al. Inhibition of AMP deaminase activity does not improve glucose control in rodent models of insulin resistance or diabetes. Chem Biol. 2014;21:1486–96. [DOI] [PubMed] [Google Scholar]
- [88].Wicks K, Hood D. Mitochondrial adaptations in denervated muscle: relationship to muscle performance. American Journal of Physiology Cell Physiology. 1990. [DOI] [PubMed] [Google Scholar]
- [89].Kwon OS, Nelson DS, Barrows KM, O’Connell RM, Drummond MJ. Intramyocellular ceramides and skeletal muscle mitochondrial respiration are partially regulated by Toll-like receptor 4 during hindlimb unloading. Am J Physiol Regul Integr Comp Physiol. 2016;311:R879–R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Roberts BM, Frye GS, Ahn B, Ferreira LF, Judge AR. Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochem Biophys Res Commun. 2013;435:488–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Curt G, Breitbart W, Cella D, Groopman J, Horning S, Itri L, et al. Impact of Cancer-Related Fatigue on the Lives of Patients: New Findings From the Fatigue Coalition. The Oncologist. 2000;5:353–60. [DOI] [PubMed] [Google Scholar]
- [92].Johansen KL, Shubert T, Doyle J, Soher B, Sakkas GK, Kent-Braun JA. Muscle atrophy in patients receiving hemodialysis: effects on muscle strength, muscle quality, and physical function. Kidney Int. 2003;63:291–7. [DOI] [PubMed] [Google Scholar]
- [93].Iwashyna T, Wesley Ely E, Smith D, Langa K. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.