

Abstract
The purpose of this study was to assess whether short-term, mild exercise induces protection against myocardial infarction and, if so, what role the eNOS-PKCε-iNOS axis plays. Mice were subjected to 2 bouts/day of treadmill exercise (60 min at 15 m/min) for 2 consecutive days. At 24 h after the last bout of exercise, mice were subjected to a 30-min coronary artery occlusion and 24 h of reperfusion. In the exercise group (group III, wild-type mice), infarct size (25.5 ± 8.8% of risk region) was significantly (P < 0.05) reduced compared with the control groups (sham exercise, group II [63.4 ± 7.8%] and acute myocardial infarction, group I [58.6 ± 7.0%]). This effect was abolished by pretreatment with the NOS inhibitor L-NA (group VI, 56.1 ± 16.2%) and the PKC inhibitor chelerythrine (group VIII, 57.9 ± 12.5%). Moreover, the late PC effect of exercise was completely abrogated in eNOS−/− mice (group XIII, 61.0 ± 11.2%). The myocardial phosphorylated eNOS at Ser-1177 was significantly increased at 30 min after treadmill training (exercise group) compared with sham-exercised hearts. PKCε translocation was significantly increased at 30 min after exercise in WT mice but not in eNOS−/− mice. At 24 h after exercise, iNOS protein was upregulated compared with sham-exercised hearts. The protection of late PC was abrogated in iNOS−/− mice (group XVI, 56.4 ± 12.9%) and in wildtype mice given the selective iNOS inhibitor 1400 W prior to ischemia (group X 62.0 ± 8.8% of risk region). We conclude that 1) even short, mild exercise induces a delayed PC effect that affords powerful protection against infarction; 2) this cardioprotective effect is dependent on activation of eNOS, eNOS-derived NO generation, and subsequent PKCε activation during PC; 3) the translocation of PKCε is dependent on eNOS; 4) the protection 24 h later is dependent on iNOS activity. Thus, eNOS is the trigger and iNOS the mediator of PC induced by mild exercise.
Keywords: Exercise; Ischemia/reperfusion injury; Preconditioning; NOS, nitric oxide synthase; Protein kinase C
1. Introduction
Ischemic preconditioning (PC) is the phenomenon whereby the heart shifts to a cardioprotected phenotype in response to a mild ischemic stress. [1–4] This adaptation consists of an early phase, which begins within minutes but abates after 1–2 h, and a late phase, which begins 12–24 h after the stimulus and lasts for ~72 h. [1–4] Several studies have shown that a delayed cardioprotective effect similar to that induced by ischemia can be elicited by administration of various pharmacologic agents. [5–15] Although PC could be an attractive mechanism to protect myocardium in humans, [3,16–20] ischemic PC is not clinically feasible while pharmacologic PC is limited by side effects and practical considerations. [3,19,20] Recent evidence indicates that brief exposure to physical exercise also elicits a late PC effect. [21–30] Unlike ischemic or pharmacologic PC, exercise PC is triggered by a physiologic stimulus and thus would appear to be a natural means for achieving cardioprotection which would be clinically feasible. However, the mechanism of exercise-induced late PC is unclear.
Previous studies have demonstrated that the development of ischemia-induced late PC is triggered by enhanced release of NO from eNOS during the ischemia/reperfusion stress [31–33] and by the subsequent activation of protein kinase C epsilon (PKCε) [31,34,35] and p44 and p42 MAPKs. [31,36] These signaling events activate the transcription factors NF-kappa B, STAT1/3, and most likely other as yet unknown components, resulting in increased transcription of cardioprotective genes (including iNOS, COX-2, and HO-1), which confer protection during the second ischemic challenge. [3]
Physical exercise is known to be associated with enhanced generation of NO [37–43] and reactive oxygen species (ROS). [23,44,45] The reaction of NO with .O2− leads to the formation of ONOO−, a strong oxidant species [46] capable of activating cellular kinases. Previous studies in rats indicate that exercise induces delayed PC by activating PKC. [25] A recent study in mice concluded that exercise induces late PC by activating eNOS, which then leads to upregulation of iNOS, which mediates late PC. [30] A role of HSP 72 [21] and MnSOD [24] as mediators of late PC has been suggested by some [21,24] but disputed by others. [22,24,47] At present, the acute effects of short (1–2 days) periods of exercise on eNOS, NO, and PKC remain unclear.
The overall goal of the present study was to explore the molecular mechanisms responsible for late PC induced by a brief exposure (2 days) to moderate exercise. Specifically, we sought to determine (i) whether brief bouts of exercise induce a delayed infarct-sparing effect in mice; (ii) if so, whether the development of this adaptation occurs via a signaling pathway that involves the activation of eNOS and the translocation of PKCε; (iii) whether the NO that triggers late PC is generated as a result of increased eNOS activity, and (iv) whether the protective effects of exercise-induced late PC are mediated by upregulation of iNOS. The results of this investigation provide a new murine model of exercise-induced late PC and reveal an important role of a molecular pathway consisting of eNOS, PKC, and iNOS in this process.
2. Methods
2.1. Mice
eNOS−/− mice (genetic background, C57BL/6 J × 129/Sv [B6129], 28.5 ± 0.8 g, 21.0 ± 1.0 wk) were kindly provided by Dr. Thomas H. Hintze. iNOS−/− mice (genetic background, C57BL/6 J × 129/Sv [B6129], 29.5 ± 1.0 g, 17.6 ± 0.8 wk) and WT (wildtype) control mice (B6129F2/J mice, 29.7 ± 0.4 g, 13.2 ± 0.4 wk) were purchased from The Jackson Lab (Bar Harbor, ME). The experimental procedures and protocols used in this study were conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Louisville, School of Medicine (Louisville, KY; IACUC Number: IACUC 18336 approved on December 05, 2018) and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 86–23, Revised 1996). Mice were maintained in sterile microisolator cages under specific pathogen-free conditions in a low-stress environment (temperature of 22–23 °C, 55–65% relative humidity, low noise, and a 12 h light-dark cycle). eNOS−/− and iNOS−/− mice were genotyped by PCR at weaning as previously described. [48] The genotype of all mice was confirmed using DNA prepared from tissue samples taken at the end of the experiment.
2.2. Treadmill exercise
Mice were exercised using an Omni-Pacer LC4/M motor-driven treadmill (AccuScan Instruments, Inc., Columbus, OH) that had an adjustable belt speed (0–100 m/min), shock bars with adjustable amperage (0–2 mA), and an on-and-off shock switch for each lane. [49] The electric shock bars (0.1–0.2 mA) were kept on during the acclimation period but were turned off during the two days of exercise. In preliminary studies we found that mice that required a total of >250 shocks during the 4-day acclimation period (approximately 15% of all mice) failed to run adequately during the two days of exercise; consequently, these mice were prospectively excluded.
The exercise protocol consisted of four consecutive days of acclimation followed by two consecutive days of exercise. On each day of acclimation (days 1–4), mice ran at 15 m/min (0% incline) for 10 min and then were left on the treadmill for 2 h at 1 m/min with the electric shock bars on (0.1–0.2 mA). On the two days of exercise (days 5 and 6), mice ran at 15 m/min (0% incline) for 60 min twice a day (9 AM and 3 PM) (total of four sessions of exercise). Each session included a 10-min “warm-up” period at 7 m/min and a 5-min “cool off” period at 7 m/min before and after the 60-min run, respectively. This protocol represents a moderate (non-exhaustive) level of exercise for mice. [50–53] The goal of this study was to examine the delayed cardioprotective effects of a brief exposure to exercise. A two-day exercise protocol was used because preliminary studies showed that a one-day protocol conferred only partial protection. Sham-exercised mice received the same acclimation protocol but, instead of undergoing the four sessions of exercise, they were placed on the treadmill at 1 m/min (0% incline) for the same duration of time as the exercised mice (75 min). Untreated control mice were not placed on the treadmill.
2.3. Coronary occlusion and reperfusion
The protocol for producing coronary occlusion/reperfusion and measuring infarct size has been described in detail. [48,54,55] Briefly, mice were anesthetized with sodium pentobarbital (60 mg/kg i.p.) and ventilated by using carefully selected parameters. After administration of antibiotics, the chest was opened through a midline sternotomy, and a nontraumatic balloon occluder was implanted around the mid-left anterior descending coronary artery by using an 8–0 nylon suture. To prevent hypotension, blood from a donor mouse was given during surgery. Rectal temperature was carefully maintained between 36.8 and 37.2 °C throughout the experiment. In all groups, myocardial infarction was produced by a 30-min coronary occlusion followed by 24 h of reperfusion. At the conclusion of the study, the occluded/reperfused vascular bed and the infarct were identified by postmortem perfusion of the heart with phthalo blue dye and triphenyltetrazolium chloride, respectively. Infarct size was calculated by using computerized video-planimetry. [48,54,55]
2.4. Experimental protocols
The study consisted of two consecutive phases (Figs. 1 and B). The objective of phase I was to determine whether brief bouts of exercise activate eNOS and PKCε acutely and upregulate eNOS and iNOS protein 24 h later. The objective of phase II was to determine the functional significance of these events, i.e., whether activation of eNOS and PKCε immediately after exercise and upregulation of iNOS 24 h later play an important role in exercise-induced cardioprotection.
Fig. 1.

A. Experimental protocol for phase I. Six groups of WT or eNOS−/− mice underwent either the sham exercise or the exercise protocol. Myocardial samples were harvested either 30 min (groups I-IV, tissue assay for eNOS activity and PKCε) or 24 h (groups V and VI, tissue assay for eNOS and iNOS expression and NOx) after the last bout of sham exercise or exercise.
B. Experimental protocol for phase II. Seventeen groups of mice underwent five different protocols. Mice in groups I (wild-type [WT] naïve, n = 15), XI (eNOS−/−, naïve, n = 10) and XIV (iNOS−/−, naïve, n = 10) underwent a 30-min coronary occlusion followed by 24 h of reperfusion (Protocol I, Naïve). Mice in groups II (WT, sham exercise, n = 9), XII (eNOS−/−, sham exercise, n = 8) and XV (iNOS−/−, sham exercise, n = 12) underwent the same protocol as Protocol II (Sham Ex) except that on days 5 and 6 they ran at 1 m/min (instead of 15 m/min) for 75 min, twice each day. In groups III (WT, exercise PC, n = 17), XIII (eNOS−/−, exercise PC, n = 8), and XVI (iNOS−/−, exercise PC, n = 8) animals were subjected to Protocol III (Ex-PC), which consisted of three stages. In stage 1 (acclimation stage), all mice were acclimated to the treadmill for 4 consecutive days (days 1–4) by running at 15 m/min (0% grade) for 10 min and 1 m/min for 2 h/day (with electric shock on [0.1–0.2 mA]). An electronic counter counted the shock numbers. In stage 2 (exercise stage), on days 5 and 6, the mice ran for 10 min at 7 m/min (warm-up), then for 60 min at 15 m/min, and then for 5 min at 7 m/min (cooldown), twice a day (9:00 a.m. and 2:00 p.m.) without electrical shock (the distance covered by one bout of exercise was about 1000 m). In stage 3 (coronary occlusion stage), 24 h after exercise or sham exercise (day 7), mice underwent a 30-min occlusion followed by 24 h of reperfusion. Mice in groups V (WT, sham exercise + L-NA, n = 11) and VIII (WT, sham exercise + CHE, n = 8) received L-NA or chelerythrine, respectively, 30 min before each bout of sham exercise on days 5 and 6 (Protocol IV, Sham Ex + Drug). Mice in groups VI (WT, exercise + L-NA, n = 7), IX (WT, exercise + CHE, n = 9), IV (WT, exercise + NS, n = 9), and VII (WT, exercise + DMSO, n = 9) received L-NA, chelerythrine, or the respective vehicle 30 min before each bout of exercise on days 5 and 6 (Protocol V, Ex-PC + Drug). Moreover, mice in groups X (WT, exercise +1400 W) received 1400 W 30 min before coronary occlusion on day 7 and then subjected to a 30-min occlusion and followed by 24 h reperfusion.
2.4.1. Phase I: effect of exercise on eNOS and PKCε
To determine the effect of exercise on eNOS and PKCε, mice were assigned to six groups. Groups I (sham exercise, Ex-sham) and II (Ex-PC) were WT mice that underwent the protocols described above and then were euthanized at 30 min after the 4th session of exercise or sham exercise. Myocardial samples (anterior and posterior left ventricular [LV] wall) were rapidly removed and frozen in liquid nitrogen for assays of eNOS and PKCε. The phosphorylation of eNOS at Ser-1177 was assessed by immunoblotting with phosphorylation-specific antibodies. PKCε activity was detected by using immunoblotting with specific antibodies. [34,35,56] Gel transfer efficiency was recorded carefully by making photocopies of membranes dyed with reversible Ponceau staining; [35] gel retention was determined by Coomassie blue staining. [35] The Ser-1177 eNOS signals and the corresponding records of Ponceau stains of nitrocellulose membranes were quantitated by an image scanning densitometer, and each eNOS signal was normalized to the corresponding Ponceau stain signal. [35,57] In all samples, the content of Ser-1177 eNOS was expressed as a percentage of Ser-1177 eNOS in the anterior wall of group I (Ex-sham).
To determine the role of eNOS in exercise-induced activation and translocation of PKCε, two groups of eNOS−/− mice (groups III and IV) underwent the sham exercise and exercise protocols used in groups I and II, respectively. Both groups were euthanized 30 min after the 4th session of exercise or sham exercise, and myocardial samples were harvested as described above. Membranous and cytosolic fractions were prepared for immunoblotting with a specific anti-PKCε antibody (Santa Cruz Biotechnology, Inc., Dallas, Texas).
To determine the effect of exercise on eNOS expression, groups V and VI underwent the sham exercise or exercise protocols and were euthanized 24 h after the 4th session of exercise or sham exercise. Myocardial samples were harvested as described above. The expression of eNOS was assessed by Western immunoblotting. [31] Specific monoclonal anti-eNOS antibodies were purchased from Transduction Laboratories, Lexington, KY. NOS activity was determined by measuring the conversion of [14C] L-arginine to [14C] L-citrulline. [32,48]
2.4.2. Phase II: effect of exercise on infarct size
To determine the effect of exercise on infarct size, mice were assigned to three groups (Fig. 1B). Group I (WT control group, AMI [acute myocardial infarction]) was not placed on the treadmill and did not receive any intervention. After 4 days of acclimation stage, groups II (WT, sham) and III (WT, Ex-PC) underwent the sham exercise and exercise protocols described above, respectively, on days 5–6; on day 7 (18–24 h after the 4th session of exercise), they were subjected to a 30-min coronary occlusion followed by 24 h of reperfusion.
To determine the role of NO in triggering exercise-induced PC, on days 5–6, groups V (sham exercise + L-NA) and VI (exercise + L-NA) underwent the sham exercise and exercise protocols described above for groups II and III, respectively; both groups received Nω-nitro-L-arginine (L-NA; 1 mg/kg i.p) 30 min before each session of sham exercise or exercise and on day 7, underwent a 30-min coronary occlusion followed by 24 h of reperfusion. L-NA (Sigma Chemical Co., St. Louis, MO) was dissolved in normal saline. Group IV (exercise + NS) underwent the exercise protocol and received NS (same amount as group VI) 30 min before each session of exercise.
To determine the role of PKCε in exercise-induced late PC, groups VIII (sham exercise +CHE) and IX (exercise +CHE) underwent the protocols described above for groups II and III, respectively; 30 min before each session of sham exercise or exercise, group IX received chelerythrine chloride (chelerythrine [CHE]; 5 mg/kg i.v.) whereas group VII (exercise + DMSO) received the same amount of vehicle (DMSO). On day 7, mice were then subjected to the 30-min occlusion /24-h reperfusion protocol. CHE (Calbiochem, La Jolla, CA) was dissolved in 50% DMSO.
To determine the role of iNOS in mediating exercise-induced PC, on days 5–6, group X (exercise +1400 W) underwent the exercise protocols described above for group III, on day 7, this group received 1400 W, selective iNOS inhibitor (20 mg/kg i.p) 30 min before coronary occlusion and underwent a 30-min coronary occlusion followed by 24 h of reperfusion. 1400 W (Sigma Chemical Co., St. Louis, MO) was dissolved in normal saline.
To determine the role of eNOS in exercise-induced late PC, on days 5–6 three groups of eNOS−/− mice (groups XI-XIII) underwent the control, sham exercise, and exercise protocols described above for groups I, II, and III, respectively, and then, on day 7, were subjected to a 30-min occlusion followed by 4 h of reperfusion.
To determine the role of iNOS as a mediator of exercise-induced late PC, on days 5–6 three groups of iNOS−/− mice (groups XIV-XVI) underwent the control, sham exercise, and exercise protocols described above for groups I, II, and III, respectively, and then, on day 7, were subjected to a 30-min occlusion followed by 24 h of reperfusion.
2.5. Statistical analysis
Data are reported as means ± SD. Statistical analysis was performed as described previously. [58–60] Measurements were analyzed with a one-way or two-way repeated-measures ANOVA, as appropriate, followed by unpaired Student’s t-tests with the Bonferroni correction [58–60].
3. Results
A total of 228 mice were used. The reasons for exclusion are summarized in Supplemental Table 1.
3.1. Phase I
3.1.1. Effect of exercise PC on the eNOS phosphorylation status
eNOS activation was examined 30 min after the 4th session of exercise or sham exercise by using antibodies directed against pSer(1177)-eNOS. Fig. 2 shows that, in mice that underwent the four sessions of exercise, there was a robust (~70%) increase in the phosphorylation of eNOS at Ser-1177 in the membranous fraction of the cardiac homogenate compared with mice that underwent sham exercise, indicating activation of eNOS shortly (30 min) after exercise.
Fig. 2.
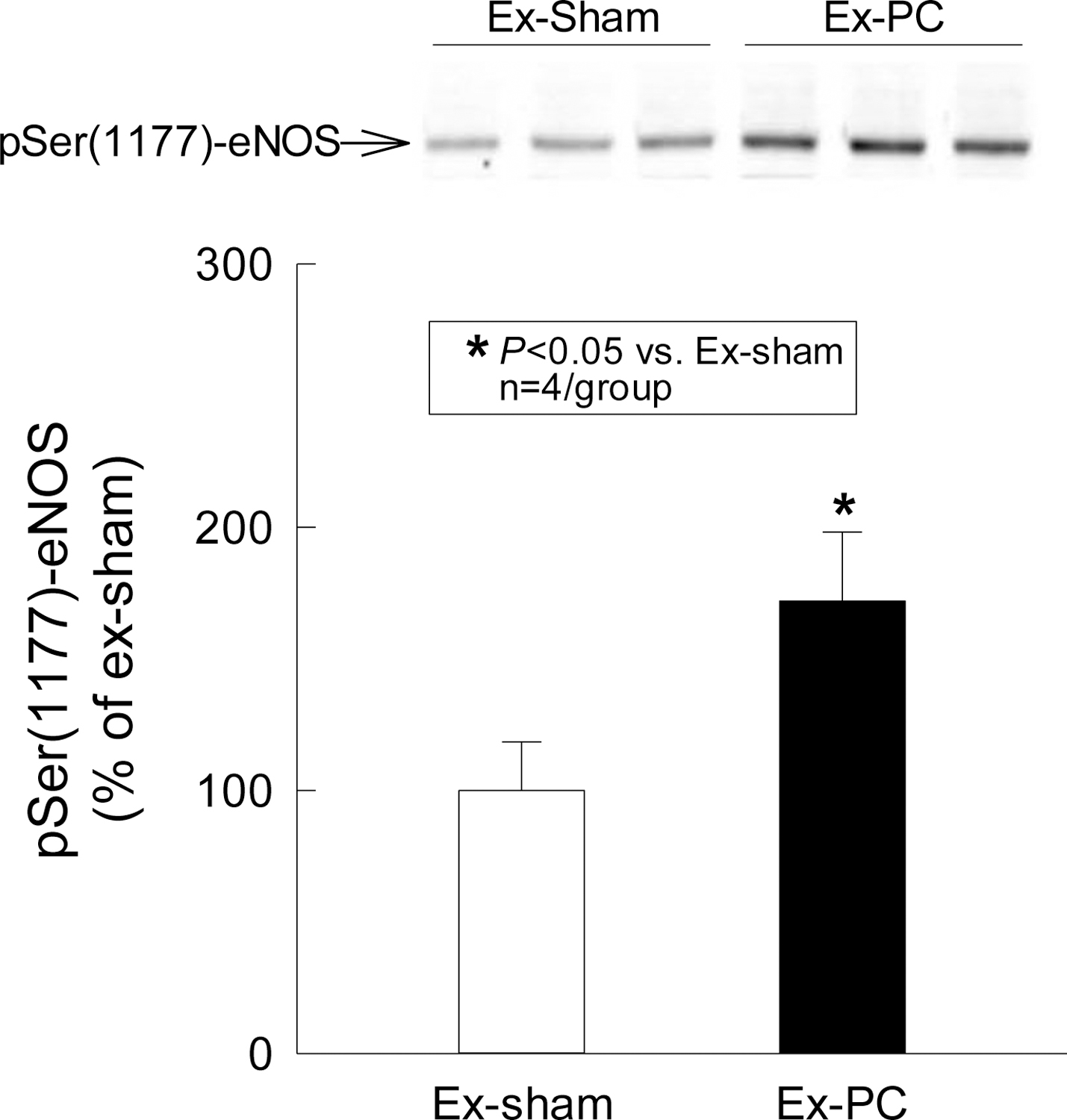
Serine phosphorylation of eNOS by exercise PC. Cardiac samples were obtained 30 min after the 4th session of sham exercise or exercise and the membranous fraction was prepared for immunoblotting with a specific anti-pSer(1177)-eNOS antibody. Exercise induced a 70% increase in pSer(1177)-eNOS. Data are means ± SD.
3.1.2. Effect of exercise PC on PKCε and role of eNOS in this process
As shown in Fig. 3, in WT mice the cytosolic content of PKCε decreased significantly at 30 min after the 4th session of exercise (−16% versus the sham exercise group [P = 0.043]) concomitant with an increase in the content of PKCε in the membranous fraction, indicating that PKCε is translocated from the cytosol to the membranous fraction shortly (30 min) after exercise. This phenomenon was completely abrogated in eNOS−/− mice, indicating that PKCε translocation is eNOS-dependent (Fig. 3).
Fig. 3.
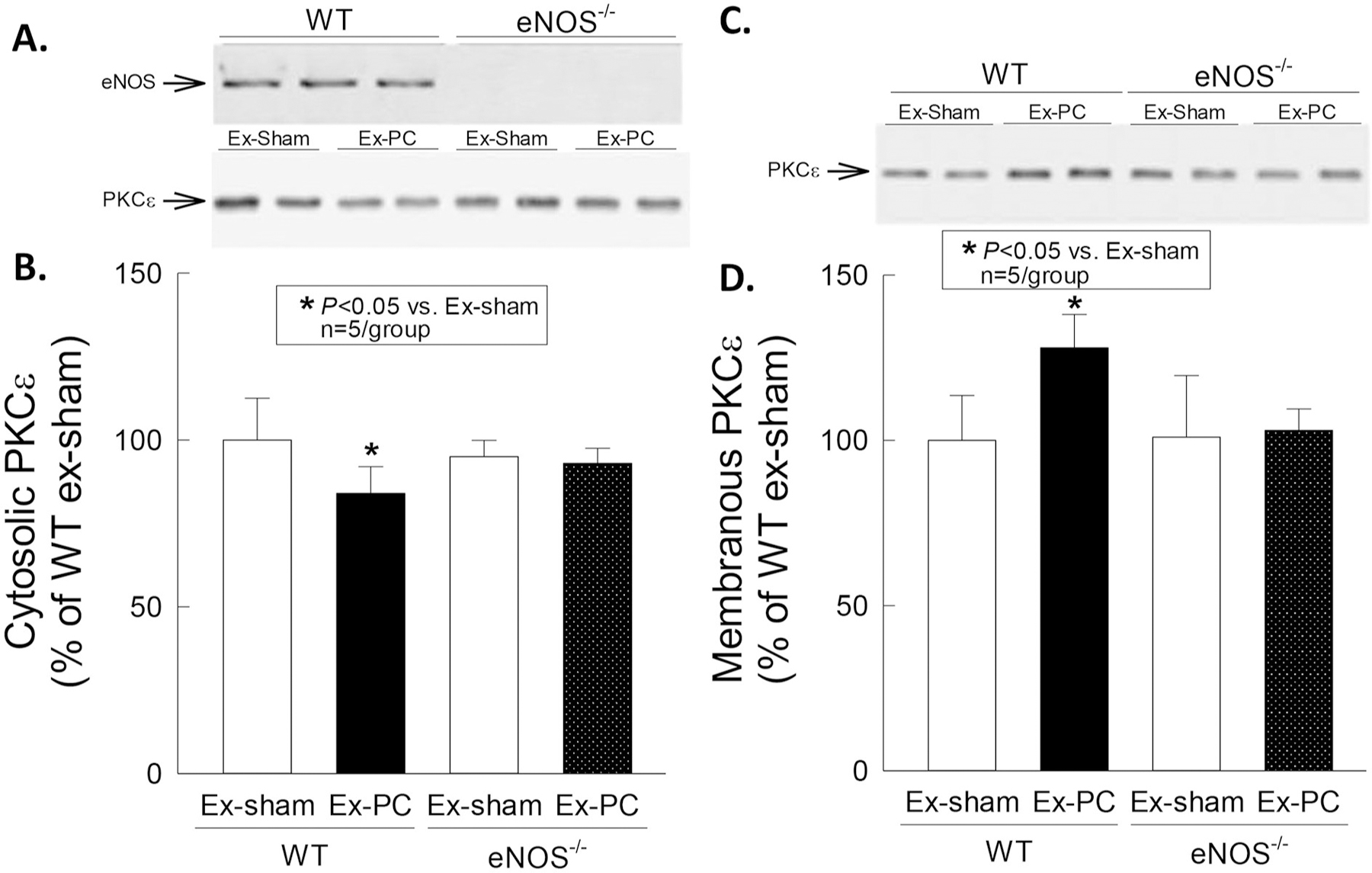
Membranous translocation of PKCε by exercise PC. Cardiac samples were obtained 30 min after the 4th session of sham exercise or exercise, and both membranous and cytosolic fractions were prepared for immunoblotting with a specific anti-PKCε antibody. Exercise induced a decrease in the cytosolic content of PKCε and a corresponding increase in the membranous content of this isoform in WT but not in eNOS−/− mice. Data are means ± SD.
Taken together, these data demonstrate that exercise PC activates PKCε as well as eNOS, and that eNOS is necessary for PKCε translocation.
3.1.3. Effect of exercise PC on eNOS and iNOS expression 24 h after exercise
Representative Western blot analysis of eNOS and iNOS is illustrated in Figs. 4 and 5, respectively. In WT mice subjected to exercise PC, 24 h after the 4th session of exercise the myocardial content of eNOS in the membranous fraction increased by 54% above the levels measured in sham exercised mice (P < 0.05) (Fig. 4), whereas the myocardial content of iNOS in the cytosolic fraction increased by 125% compared with the sham exercise group (P < 0.05) (Fig. 5). Supplemental Figs. 1&2 show the Ponceau S staining for the Western blots of eNOS and iNOS protein (Figs. 4& 5, respectively), and indicate that the amount of loaded protein in each lane on the Western blot membrane was equal. No immunoreactive nNOS was detected in either sham or exercise groups using commercially available nNOS monoclonal or polyclonal antibodies (Transduction Laboratories) (data not shown).
Fig. 4.

Upregulation of eNOS by exercise PC. Cardiac samples were obtained 24 h after the 4th session of sham exercise or exercise and the membranous fraction was subjected to immunoblotting with a specific anti-eNOS antibody. Exercise induced a 40% increase in eNOS protein expression. Data are means ± SD.
Fig. 5.

Upregulation of iNOS in exercise-induced PC. Cardiac samples were collected 24 h after the 4th session of sham exercise or exercise. The cytosolic fraction of protein was subjected to immunoblotting with a specific anti-iNOS antibody. Note the elevation of iNOS protein expression in exercise-induced PC. Data are means ± SD.
3.2. Phase II: effect of exercise PC on infarct size
3.2.1. Body temperature and heart rate
By experimental design, [48,54,55,61] rectal temperature remained within a narrow, physiologic range (36.9–37.2 °C) in all groups (supplemental Table 2). Five minutes before the 30-min coronary occlusion, the average heart rate in groups I-XVI ranged from 477 to 557 bpm (P NS). Heart rate did not differ significantly among the 16 groups at any time during the 30-min occlusion or the ensuing reperfusion (supplemental Table 3).
3.2.2. Infarct size
Examples of the infarcts observed in groups II and III are illustrated in Fig. 6A and B. There were no significant differences among the 17 groups with respect to body weight, LV weight, or weight of the region at risk (Table 1). In WT control mice (group I, AMI), infarct size averaged 58.6 ± 7.0% of the region at risk (Fig. 7 and Table 1). Infarct size was similar (63.4 ± 7.8% of the region at risk) in the sham group (group II) (Fig. 7 and Table 1), indicating that the four-day acclimation protocol and the exposure to the treadmill did not affect the extent of cell death induced by a subsequent 30-min coronary occlusion. However, when mice underwent four exercise sessions following the acclimation protocol (PC group [group III]), the size of the infarct produced by a 30-min coronary occlusion on the day after the 4th session was reduced by ~60% compared with the sham group (from 63.4 ± 7.8% to 25.5 ± 8.8% of the region at risk [P < 0.05]) (Fig. 7 and Table 1), indicating the development of a robust late PC effect. In contrast, when exercise-preconditioned mice were given L-NA 30 min prior to each session of exercise (group VI), infarct size was similar to that observed in sham exercised mice (56.1 ± 16.2% of the risk region), indicating that enhanced NOS activity is essential for the development of exercise PC. Pretreatment with L-NA had no effect on infarct size in the absence of exercise PC (sham exercise + L-NA group [group V]). Pretreatment with vehicle (NS) (exercise + NS group [group IV]) did not interfere with the infarct-sparing effects of exercise PC.
Fig. 6.

A, Representative example of a heart from group II (sham exercise group) subjected to a 30-min coronary occlusion and 24 h of reperfusion 24 h after a sham exercise protocol. The infarcted region was delineated by perfusing the aortic root with TTC; the region at risk was delineated by perfusing the aortic root with Phthalo blue after tying the previously occluded artery. As a result of this procedure, the nonischemic portion of the left ventricle was stained dark blue, the viable tissue within the region at risk was stained bright red, and the infarcted tissue was light yellow. Note the large, confluent areas of infarction spanning most of the thickness of the LV wall, with thin rims of viable subendocardial tissue. The scale at the bottom is in mm. B, Representative example of a heart from group III (exercise group), which was subjected to 2 sessions/day of treadmill exercise (60 min at 15 m/min) for 2 consecutive days and, 24 h later, to a 30-min occlusion and 24 h of reperfusion. Postmortem perfusion was performed as described in the legend to Fig. 1B. In contrast to the confluent infarction shown in Fig. 6A, in this heart the 30-min occlusion resulted in patchy areas of infarction, a pattern that was characteristic of both groups VI and VII, indicating that the sequence of four bouts of exercise-induced a powerful late PC effect against infarction. The scale at the bottom is in mm.
Table 1.
Size of Left Ventricle, Risk Region, and Infarct.
| Age | Body | Heart | H/B | LV | RR | Infarct | RR | Infarct | Infarct | Shock | |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
| (wk) | (g) | (mg) | (%) | (mg) | (mg) | (mg) | (% of LV) | (% of risk region) | (% of LV) | ||
| GroupI (WT, AMI) | 14 ± 5 | 29.2 ± 3.9 | 161 ± 30 | 0.55 ± 0.06 | 119 ± 25 | 43 ± 11 | 26 ± 9 | 36.8 ± 7.9 | 58.6 ± 7.0 | 21.7 ± 5.9 | N/A |
| Group II (WT, sham Ex) | 11 ± 1 | 28.5 ± 2.5 | 171 ± 12 | 0.60 ± 0.05 | 126 ± 12 | 43 ± 11 | 27 ± 8 | 34.1 ± 6.5 | 63.4 ± 7.8 | 21.5 ± 4.7 | 180 ± 51 |
| Group III (WT, Ex) | 13 ± 2 | 29.0 ± 2.4 | 157 ± 20 | 0.54 ± 0.06 | 112 ± 17 | 34 ± 11 | 9 ± 5* | 29.5 ± 7.4 | 25.5 ± 8.8* | 7.8 ± 3.7* | 139 ± 74 |
| Group IV (WT, Ex + NS) | 12 ± 3 | 27.2 ± 0.1.9 | 145 ± 13 | 0.54 ± 0.07 | 108 ± 15 | 30 ± 4 | 8 ± 4* | 28.2 ± 4.9 | 27.2 ± 14.0* | 7.3 ± 3.7* | 182 ± 62 |
| Group V (WT, sham Ex + L-NA) | 18 ± 4 | 32.6 ± 5.2 | 151 ± 18 | 0.47 ± 0.06 | 116 ± 13 | 38 ± 3 | 19 ± 4 | 33.5 ± 4.0 | 50.2 ± 10.4 | 16.7 ± 3.2 | N/A |
| Group VI (WT, Ex + L-NA) | 10 ± 1 | 29.1 ± 3.5 | 160 ± 24 | 0.55 ± 0.05 | 115 ± 17 | 42 ± 4 | 23 ± 6 | 36.8 ± 5.6 | 56.1 ± 16.2 | 20.3 ± 5.2 | 131 ± 75 |
| Group VII (WT, Ex + DMSO) | 11 ± 1 | 27.9 ± 1.4 | 146 ± 9 | 0.52 ± 0.03 | 111 ± 11 | 33 ± 5 | 9 ± 4* | 29.9 ± 5.5 | 27.9 ± 13.4* | 8.2 ± 4.0* | 44 ± 28 |
| Group VIII (WT, sham Ex + CHE) | 18 ± 4 | 34.5 ± 8.9 | 149 ± 29 | 0.44 ± 0.08 | 115 ± 22 | 42 ± 11 | 20 ± 7 | 36.3 ± 5.9 | 48.2 ± 8.5 | 17.3 ± 3.4 | N/A |
| Group IX (WT, Ex + CHE) | 14 ± 5 | 29.5 ± 2.8 | 147 ± 12 | 0.50 ± 0.03 | 109 ± 10 | 35 ± 9 | 21 ± 8 | 32.4 ± 10.1 | 57.9 ± 12.5 | 19.2 ± 8.1 | 81 ± 14 |
| Group X (WT, Ex +1400 W) | 11 ± 0 | 30.5 ± 2.1 | 175 ± 23 | 0.57 ± 0.05 | 134 ± 20 | 42 ± 11 | 26 ± 8 | 31.1 ± 5.2 | 62.0 ± 8.8 | 19.4 ± 4.6 | 127 ± 58 |
| Group XI (eNOS−/−, AMI) | 24 ± 6 | 28.4 ± 3.5 | 156 ± 22 | 0.55 ± 0.09 | 119 ± 17 | 46 ± 11 | 28 ± 8 | 38.8 ± 6.9 | 59.7 ± 4.6 | 23.3 ± 5.0 | N/A |
| Group XII (eNOS−/−, sham Ex) | 20 ± 2 | 26.7 ± 6.1 | 155 ± 24 | 0.59 ± 0.07 | 115 ± 15 | 46 ± 12 | 27 ± 6 | 40.1 ± 9.0 | 59.0 ± 9.6 | 23.2 ± 4.4 | 213 ± 24 |
| Group XIII (eNOS−/−, Ex) | 18 ± 4 | 30.5 ± 2.2 | 179 ± 13 | 0.59 ± 0.05 | 128 ± 7 | 41 ± 16 | 26 ± 14 | 32.0 ± 11.6 | 61.0 ± 11.2 | 20.3 ± 10.0 | 173 ± 68 |
| Group XIV (iNOS−/−, AMI) | 17 ± 4 | 30.8 ± 7.5 | 167 ± 46 | 0.54 ± 0.05 | 121 ± 30 | 44 ± 8 | 23 ± 6 | 38.0 ± 8.9 | 53.4 ± 11.2 | 20.3 ± 6.8 | N/A |
| Group XV (iNOS−/−, sham Ex) | 16 ± 3 | 29.1 ± 5.0 | 142 ± 32 | 0.48 ± 0.05 | 107 ± 24 | 40 ± 9 | 25 ± 8 | 37.7 ± 7.2 | 63.8 ± 8.8 | 24.0 ± 5.2 | 105 ± 69 |
| Group XVI (iNOS−/−, Ex) | 21 ± 4 | 28.4 ± 4.4 | 171 ± 18 | 0.61 ± 0.05 | 123 ± 15 | 44 ± 8 | 26 ± 10 | 36.0 ± 5.1 | 56.4 ± 12.9 | 20.8 ± 7.02.5 | 58 ± 39 |
Ex, exercise; Body, body weight; Heart, total heart weight (ventricles and atria); H/B, the ratio of heart weight with body weight; LV, left ventricle; RR, region at risk; Shock, number of shocks during acclimation. Data are means ± SD.
P < 0.05 vs. group II.
Fig. 7.

Myocardial infarct size in groups I (WT, control), II (WT, sham exercise), III (WT, exercise PC), VI (WT, exercise + NS), V (WT, sham exercise + L-NA), VI (WT, exercise + L-NA), VII (WT, exercise + DMSO), VIII (WT, sham exercise + CHE), IX (WT, exercise + CHE), X (WT, exercise + 1400 W), XI (eNOS−/−, control), XII (eNOS−/−, sham exercise), XIII (eNOS−/−, exercise PC), XIV (iNOS−/−, control), XV (iNOS−/−, sham exercise), XVI (iNOS−/−, exercise PC). The experimental protocols are specified in the legend to Fig. 1B. Open circles represent individual mice, whereas solid circles represent means ± SD.
When preconditioned mice were given CHE) 30 min prior to each bout of exercise (group IX), infarct size was not reduced compared with sham-exercised mice (57.9 ± 12.5% vs. 63.4 ± 7.8% of the risk region) (Fig. 7 and Table 1), indicating that the protective effect of exercise PC was completely abolished. CHE pretreatment had no effect in the absence of exercise PC (sham exercise + CHE group [group VIII]) and vehicle (DMSO) pretreatment did not limit the infarct-sparing effect of exercise PC (exercise + DMSO group [group VII]). These results indicate that the development of exercise PC requires activation of the PKCε signaling pathway.
When preconditioned mice were given 1400 W 24 h after exercise, 30 min prior to coronary occlusion on day 7 (groups X), infarct size was not reduced compared with sham-exercised mice (62.0 ± .8% vs. 63.4 ± 7.8% of the risk region in group II [WT, sham exercise]) (Fig. 7 and Table 1), indicating that the protective effect of exercise PC was completely abolished. These results indicate that iNOS mediates the protection of exercise PC.
The infarct-sparing effects of exercise-induced PC were abolished by genetic ablation of eNOS (group XIII, 61.0 ± 11.2% vs. 59.7 ± 4.6% of the risk region in group XI [eNOS−/−, AMI]) (Fig. 7 and Table 1), indicating that eNOS plays an essential role in this phenomenon. eNOS deletion had no effect on infarct size in the absence of exercise PC (eNOS−/− sham exercise group [group XII] or in AMI mice (group XI).
The infarct-sparing effects of exercise-induced PC were also abolished by genetic ablation of iNOS (group XVI, 56.4 ± 12.9% vs. 53.4 ± 11.2% of the risk region in group XIV [iNOS−/−, AMI]) (Fig. 7 and Table 1), indicating that iNOS is necessary in this phenomenon. iNOS deletion had no effect on infarct size in the absence of exercise PC (iNOS−/− sham exercise group [group XVI] and iNOS−/− AMI group [group XIV]).
4. Discussion
The salient findings of the present investigation can be summarized as follows: 1) a relatively brief exposure to exercise (four 30-min sessions of treadmill exercise over two days) is sufficient to elicit a robust infarct-sparing effect 18–24 h after the last bout of exercise in mice; 2) eNOS is activated very soon (30 min) after the fourth bout of exercise, as demonstrated by increased phosphorylation at Ser-1177; 3) at the same time, the ε isoform of PKC is activated, as demonstrated by its translocation from the cytosolic to the membranous fraction of the cardiac homogenate; 4) PKCε translocation is abrogated by genetic deletion of eNOS, indicating that it requires eNOS activity; 5) the activation of eNOS immediately after exercise is necessary for the development of the subsequent protective response, because administration of L-NA prior to exercise abrogates the infarct-sparing effects 24 h later; 6) administration of the PKC inhibitor CHE during each of the four bouts of exercise abrogates the infarct-sparing effects of exercise PC 24 h later, indicating that activation of PKCε immediately after exercise is obligatorily required for the development of the protective effect; 7) the two-day exercise protocol leads to upregulation of cardiac eNOS 24 h after the last bout of exercise, as demonstrated by increased eNOS protein expression and activity; 8) genetic ablation of eNOS completely abrogates the infarct-sparing effects of exercise-induced PC, indicating that eNOS plays an obligatory role in this phenomenon, as a trigger and possibly as a mediator; 9) genetic ablation of iNOS also completely abrogates the infarct-sparing effect of exercise-induced PC, indicating that iNOS plays an obligatory role in this phenomenon; 10) the role of iNOS is that of a mediator, because the infarct-sparing effects of exercise PC are abolished by inhibition of iNOS activity with 1400 W given on day 2, 24 h after exercise. Taken together, these results reveal an essential role of eNOS, iNOS, and PKCε in the delayed cardioprotective effects imparted by brief exposure to physical exercise. Previous studies have suggested a role of PKC in general (but not PKCε) in exercise-induced late PC in rats. [25,62] However, to our knowledge, no previous study has demonstrated a critical role of eNOS, iNOS, or PKCε in the delayed adaptive response of the heart to physical exercise.
Because the late phase of PC lasts for 72 h and protects against both myocardial stunning and infarction, [3] it may have considerable clinical relevance and potential therapeutic applications. However, translation of basic knowledge regarding late PC into therapeutic strategies has been hampered by the difficulty in identifying clinically-relevant, non-noxious stimuli that are capable of reproducing the sustained cardioprotective effects of late PC in patients. Exercise PC is conceptually attractive as a natural means for achieving cardioprotection that would be widely applicable in patients. At present, more than half of the US adult population is sedentary or inactive. [63–65] The identification of an endogenous adaptive mechanism that can provide sustained (several days) protection without the need for pharmacologic interventions or other manipulations may have major pathophysiological and therapeutic implications.
Our results demonstrate that physical exercise can elicit a delayed infarct-sparing effect equivalent to that observed during the late phase of ischemic PC. Interestingly, numerous epidemiologic and clinical investigations have shown that physical exercise reduces mortality and morbidity in patients with coronary artery disease. [63–71] The mechanism for these beneficial actions remains controversial, but it is clear that it is independent of risk factors for coronary artery disease. [63–71] Based on the present results and those of other studies, [23,30] we propose that the salutary effects of exercise observed in patients are mediated, at least in part, by direct cardioprotection via a PC mechanism.
This study provides a new murine model of exercise-induced late PC. Previous studies of this phenomenon have been conducted in rats [23,25] or mice subjected to a relatively long (7 days) exercise protocol. [30] Our murine model of 2 days of exercise is intended to address the effects of exercise per se, distinct from the long-term effects (LV hypertrophy and other cardiovascular adaptations) associated with sustained physical activity. Compared with other species, the use of a murine model is advantageous because it enables precise dissection of the components of exercise-induced PC by using genetically-engineered models with either deficiency or overexpression of specific proteins. Our murine model differs from that described by Akita et al [30] in several respects: (i) Akita et al used ketamine-xylazine for anesthesia (a combination that markedly depresses heart rate and arterial pressure), whereas we used pentobarbital, which does not significantly alter these variably in mice; (ii) in contrast to our protocol, Akita and coworkers did not include an acclimation phase in their exercise protocol and used electric shock driving exercise; (iii) the infarct model used by Akita et al included a 2-h reperfusion period vs. 24 h in our model; it is unknown whether an infarct achieves its final size after 2 h in the mouse; (iv) no indication is provided as to whether the investigators kept body temperature at physiologic values, whereas in this study body temperature was carefully kept within a narrow range of 36.9–37.2 (temperature is a major determinant of infarct size [72]).
As mentioned above, our murine model was developed to dissect the effects of exercise in itself from the cardiovascular adaptations associated with it. Accordingly, we sought to use the smallest number of treadmill sessions that could provide reproducible protection. A two-day exercise protocol was selected because, in preliminary studies, we found that one day of exercise (two 30-min sessions of treadmill exercise) did not provide consistent cardioprotection 24 h later. The level of exercise selected for this model is a non-exhaustive, moderate level of physical activity for mice. [73] Particular care was taken to ensure that mice would be allowed to be acclimated to the treadmill and to the environment, so as to separate the effects of the new environment from those of exercise in itself. A four-day acclimation protocol was therefore implemented before the exercise started. Furthermore, each bout of exercise was preceded by a 5 min warm-up period (at 7 m/min) and followed by a 60-min running period (at 15 m/min) and a 5 min cooldown period (at 7 m/min). The four-day acclimation protocol was also useful to screen out mice that were not likely to exercise adequately. As detailed in Methods, electric shocks were delivered during the acclimation protocol but not during the four sessions of exercise. Two facts demonstrate that the electric shocks used during the acclimation protocol cannot account for the reduction in infarct size observed after exercise: i) the total number of shocks delivered during the acclimation protocol was similar in sham exercised (group II) and exercised (group III) mice (180 ± 51 vs. 139 ± 74, respectively), but only the exercised mice were preconditioned; ii) there was no correlation between the number of shocks during the acclimation protocol and infarct size. We found that mice requiring a high number (>250) of shocks during acclimation were generally not able to exercise adequately; these mice (15% of those initially enrolled into the study) were excluded. In summary, the murine model of exercise-induced late PC presented herein was carefully designed and should be useful in future studies to investigate the molecular mechanisms of this phenomenon in genetically-engineered mice.
When addressing the phenomenon of late PC, it is important to distinguish the triggers of this adaptation (i.e., the signals that initiate the cascade of events that culminates in subsequent cardioprotection) from the mediators of protection (i.e., the proteins that are responsible for the relative resistance to ischemia 24–72 h after the PC stimulus). [3] Although pharmacologic and genetic studies have implicated eNOS and PKCε as triggers of the late phase of ischemia-induced PC, [16,31,32,35] the role of these enzymes in exercise-induced late PC remains unclear. To address this issue, mice were subjected to our exercise protocol and myocardial samples were harvested very soon (30 min) after the last session of exercise. Our results reveal a crucial role of eNOS in triggering exercise-induced late PC, based upon the fact that (i) the myocardial content of pSer(1177)-eNOS was markedly increased 30 min after exercise PC (Fig. 2), indicating activation of this enzyme, [74] and (ii) non-isoform-selective inhibition of NOS with L-NA during the exercise protocol completely abrogated the infarct-sparing effects of exercise (Fig. 7). A critical role of PKCε in signaling downstream of eNOS is documented by our observation that PKCε was translocated to the membranous fraction at 30 min after the last session of exercise (Fig. 3A and B), and that administration of the PKCε inhibitor CHE during exercise completely abrogated the development of protection 24 h later (Fig. 7). Because the translocation of PKCε to the membranous fraction was abolished in eNOS null mice (Fig. 3C and D), this signaling event appears to be downstream of eNOS activation. Therefore, we propose that exercise triggers delayed cardioprotection by activating constitutively-expressed eNOS, resulting in increased generation of NO that subsequently activates PKCε. The mechanism whereby exercise activates eNOS remains to be elucidated, but it could include shear-stress induced activation of endothelial eNOS and/or activation of myocyte or endothelial eNOS by reactive oxygen species generated during physical exercise. [23,44,45,75] The signaling pathway downstream of PKCε remains to be elucidated.
In searching for the mediator of exercise-induced late PC, we found that eNOS was upregulated (by ~54%) 24 h after exercise (Fig. 4). To our knowledge, this is the first indication that a relatively brief exposure to physical activity results in increased eNOS expression in the myocardium. Since nNOS was not detected but iNOS was upregulated (Fig. 5), the increased NO generation 24 h after exercise PC could reflect increased activity of either eNOS or iNOS. To determine the functional significance of eNOS and iNOS upregulation, we studied the effect of genetic ablation of eNOS or iNOS on exercise PC-induced cardioprotection. Our finding that the cardioprotective effects were completely abrogated in eNOS null mice (Fig. 7) demonstrates an obligatory role of eNOS in this phenomenon but does not distinguish between its role as a trigger or as a mediator of exercise-induced late PC. Our data with L-NA, however, clearly demonstrate that eNOS serves as a necessary trigger for this phenomenon (Fig. 7). Whether eNOS is also an obligatory mediator of late PC (i.e., whether eNOS activity is obligatorily required to confer protection 24 h after the exercise protocol) remains to be determined. Nevertheless, our results in eNOS null mice point to a fundamental role of eNOS in the phenomenon of exercise-induced late PC. Moreover, our finding that the cardioprotective effects were also completely abrogated in iNOS null mice and with selective iNOS inhibitor (1400 W) demonstrates an important role of iNOS in this phenomenon (Fig. 7). Our data clearly demonstrate that iNOS serves as a necessary mediator for this phenomenon.
In conclusion, we describe a new murine model of exercise-induced late PC that should be useful to interrogate the molecular mechanism of this phenomenon in genetically-engineered animals. Our observations demonstrate a critical role of eNOS as a trigger and of iNOS as a mediator of exercise-induced late PC. The possible role of as eNOS, a mediator of this phenomenon, remains to be verified. Our findings further demonstrate that eNOS triggers late PC by activating the ε isoform of PKC. The identification of eNOS and iNOS as specific proteins responsible for exercise-induced late PC provides a target for therapeutic manipulations aimed at mimicking the infarct-sparing effects of exercise PC with eNOS and/or iNOS gene therapy and/or other strategies for increasing cardiac NOS activity.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant P01 HL078825 USA.
Abbreviations:
- AMI
acute myocardial infarction
- CHE
chelerythrine
- eNOS
endothelial NOS
- eNOS−/−
eNOS gene knockout
- Ex
exercise
- iNOS
inducible NOS
- iNOS−/−
iNOS gene knockout
- LV
left ventricle or left ventricular
- NOS
nitric oxide synthase
- PC
preconditioning
- PKC
protein kinase C
- WT
wild-type
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijcard.2021.08.021.
References
- [1].Bolli R, The early and late phases of preconditioning against myocardial stunning and the essential role of oxyradicals in the late phase: an overview, Basic Res. Cardiol 91 (1996) 57–63, 10.1007/BF00788866. [DOI] [PubMed] [Google Scholar]
- [2].Yellon DM, Downey JM, Preconditioning the myocardium: from cellular physiology to clinical cardiology, Physiol. Rev 83 (2003) 1113–1151, 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- [3].Bolli R, The late phase of preconditioning, Circ. Res 87 (2000) 972–983, 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- [4].Dawn B, Bolli R, Role of nitric oxide in myocardial preconditioning, Ann. N. Y. Acad. Sci 962 (2002) 18–41, 10.1111/j.1749-6632.2002.tb04053.x. [DOI] [PubMed] [Google Scholar]
- [5].Guo Y, Stein AB, Wu WJ, Zhu X, Tan W, Li Q, et al. , Late preconditioning induced by NO donors, adenosine A1 receptor agonists, and delta1-opioid receptor agonists is mediated by iNOS, Am. J. Physiol. Heart Circ. Physiol 289 (2005) H2251–H2257, 10.1152/ajpheart.00341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shinmura K, Nagai M, Tamaki K, Tani M, Bolli R, COX-2-derived prostacyclin mediates opioid-induced late phase of preconditioning in isolated rat hearts, Am. J. Physiol. Heart Circ. Physiol 283 (2002) H2534–H2543, 10.1152/ajpheart.00209.2002. [DOI] [PubMed] [Google Scholar]
- [7].Zhao TC, Hines DS, Kukreja RC, Adenosine-induced late preconditioning in mouse hearts: role of p38 MAP kinase and mitochondrial K(ATP) channels, Am. J. Physiol. Heart Circ. Physiol 280 (2001) H1278–H1285, 10.1152/ajpheart.2001.280.3.H1278. [DOI] [PubMed] [Google Scholar]
- [8].Maldonado C, Qiu Y, Tang XL, Cohen MV, Auchampach J, Bolli R, Role of adenosine receptors in late preconditioning against myocardial stunning inconscious rabbits, Am. J. Phys 273 (1997) H1324–H1332, 10.1152/ajpheart.1997.273.3.H1324. [DOI] [PubMed] [Google Scholar]
- [9].Baxter GF, Yellon DM, Time course of delayed myocardial protection after transient adenosine A1-receptor activation in the rabbit, J. Cardiovasc. Pharmacol 29 (1997) 631–638, 10.1097/00005344-199705000-00011. [DOI] [PubMed] [Google Scholar]
- [10].Takano H, Tang XL, Qiu Y, Guo Y, French BA, Bolli R, Nitric oxide donors induce late preconditioning against myocardial stunning and infarction in conscious rabbits via an antioxidant-sensitive mechanism, Circ. Res 83 (1998) 73–84, 10.1161/01.res.83.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Banerjee S, Tang XL, Qiu Y, Takano H, Manchikalapudi S, Dawn B, et al. , Nitroglycerin induces late preconditioning against myocardial stunning via a PKC-dependent pathway, Am. J. Phys 277 (1999) H2488–H2494, 10.1152/ajpheart.1999.277.6.H2488. [DOI] [PubMed] [Google Scholar]
- [12].Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, et al. , Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning, Circ. Res 84 (1999) 587–604, 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- [13].Hill M, Takano H, Tang XL, Kodani E, Shirk G, Bolli R, Nitroglycerin induces late preconditioning against myocardial infarction in conscious rabbits despite development of nitrate tolerance, Circulation 104 (2001) 694–699, 10.1161/hc3201.092218. [DOI] [PubMed] [Google Scholar]
- [14].Patel HH, Hsu AK, Gross GJ, COX-2 and iNOS in opioid-induced delayed cardioprotection in the intact rat, Life Sci 75 (2004) 129–140, 10.1016/j.lfs.2003.10.036. [DOI] [PubMed] [Google Scholar]
- [15].Iliodromitis EK, Gaitanaki C, Lazou A, Aggeli IK, Gizas V, Bofilis E, et al. , Differential activation of mitogen-activated protein kinases in ischemic and nitroglycerin-induced preconditioning, Basic Res. Cardiol 101 (2006) 327–335, 10.1007/s00395-006-0594-3. [DOI] [PubMed] [Google Scholar]
- [16].Bolli R, Dawn B, Tang XL, Qiu Y, Ping P, Xuan YT, et al. , The nitric oxide hypothesis of late preconditioning [editorial; see comments], Basic Res. Cardiol 93 (1998) 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tomai F, Perino M, Ghini AS, Crea F, Gaspardone A, Versaci F, et al. , Exercise-induced myocardial ischemia triggers the early phase of preconditioning but not the late phase, Am. J. Cardiol 83 (1999) 586–588 (A7–8). [DOI] [PubMed] [Google Scholar]
- [18].Leesar MA, Stoddard MF, Dawn B, Jasti VG, Masden R, Bolli R, Delayed preconditioning-mimetic action of nitroglycerin in patients undergoing coronary angioplasty, Circulation 103 (2001) 2935–2941, 10.1161/01.cir.103.24.2935. [DOI] [PubMed] [Google Scholar]
- [19].Bolli R, Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research, J. Mol. Cell. Cardiol 33 (2001) 1897–1918, 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- [20].Kloner RA, Jennings RB, Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2, Circulation 104 (2001) 3158–3167, 10.1161/hc5001.100039. [DOI] [PubMed] [Google Scholar]
- [21].Locke M, Tanguay RM, Klabunde RE, Ianuzzo CD, Enhanced postischemic myocardial recovery following exercise induction of HSP 72, Am. J. Phys 269 (1995) H320–H325, 10.1152/ajpheart.1995.269.1.H320. [DOI] [PubMed] [Google Scholar]
- [22].Taylor RP, Harris MB, Starnes JW, Acute exercise can improve cardioprotection without increasing heat shock protein content, Am. J. Phys 276 (1999) H1098–H1102. [DOI] [PubMed] [Google Scholar]
- [23].Yamashita N, Hoshida S, Otsu K, Asahi M, Kuzuya T, Hori M, Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation, J. Exp. Med 189 (1999) 1699–1706, 10.1084/jem.189.11.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hamilton KL, Powers SK, Sugiura T, Kim S, Lennon S, Tumer N, et al. , Short-term exercise training can improve myocardial tolerance to I/R without elevation in heat shock proteins, Am. J. Physiol. Heart Circ. Physiol 281 (2001) H1346–H1352, 10.1152/ajpheart.2001.281.3.H1346. [DOI] [PubMed] [Google Scholar]
- [25].Yamashita N, Baxter GF, Yellon DM, Exercise directly enhances myocardial tolerance to ischaemia-reperfusion injury in the rat through a protein kinase C mediated mechanism, Heart 85 (2001) 331–336, 10.1136/heart.85.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guo Y, Wu WJ, Zhu XP, Li QH, Tang XL, Bolli R, Exercise-induced late preconditioning is triggered by generation of nitric oxide, J. Mol. Cell. Cardiol 33 (2001) A41. [Google Scholar]
- [27].Guo Y, Xuan YT, Wu WJ, Li QH, Tang XL, Bolli R, Nitric oxide plays a dual role in exercise-induced late preconditioning, Circulation 104 (2001). II–60(288). [Google Scholar]
- [28].Demirel HA, Powers SK, Zergeroglu MA, Shanely RA, Hamilton K, Coombes J, et al. , Short-term exercise improves myocardial tolerance to in vivo ischemia-reperfusion in the rat, J. Appl. Physiol 91 (1985) 2205–2212, 10.1152/jappl.2001.91.5.2205, 2001. [DOI] [PubMed] [Google Scholar]
- [29].Domenech R, Macho P, Schwarze H, Sanchez G, Exercise induces early and late myocardial preconditioning in dogs, Cardiovasc. Res 55 (2002) 561–566, 10.1016/s0008-6363(02)00334-6. [DOI] [PubMed] [Google Scholar]
- [30].Akita Y, Otani H, Matsuhisa S, Kyoi S, Enoki C, Hattori R, et al. , Exercise-induced activation of cardiac sympathetic nerve triggers cardioprotection via redox-sensitive activation of eNOS and upregulation of iNOS, Am. J. Physiol. Heart Circ. Physiol 292 (2007) H2051–H2059, 10.1152/ajpheart.01102.2006. [DOI] [PubMed] [Google Scholar]
- [31].Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Bolli R, Endothelial nitric oxide synthase plays an obligatory role in the late phase of ischemic preconditioning by activating the protein kinase C epsilon p44/42 mitogen-activated protein kinase pSer-signal transducers and activators of transcription1/3 pathway, Circulation 116 (2007) 535–544, 10.1161/CIRCULATIONAHA.107.689471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xuan YT, Tang XL, Qiu Y, Banerjee S, Takano H, Han H, et al. , Biphasic response of cardiac NO synthase isoforms to ischemic preconditioning in conscious rabbits, Am. J. Physiol. Heart Circ. Physiol 279 (2000) H2360–H2371. [DOI] [PubMed] [Google Scholar]
- [33].Bolli R, Dawn B, Xuan YT, Role of the JAK-STAT pathway in protection against myocardial ischemia/reperfusion injury, Trends Cardiovasc Med 13 (2003) 72–79, 10.1016/s1050-1738(02)00230-x. [DOI] [PubMed] [Google Scholar]
- [34].Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, et al. , Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning, Circ. Res 84 (1999) 587–604. [DOI] [PubMed] [Google Scholar]
- [35].Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, et al. , Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity, Circ. Res 81 (1997) 404–414. [DOI] [PubMed] [Google Scholar]
- [36].Ping P, Zhang J, Cao X, Li RC, Kong D, Tang XL, et al. , PKC-dependent activation of p44/p42 MAPKs during myocardial ischemia-reperfusion in conscious rabbits, Am. J. Phys 276 (1999) H1468–H1481, 10.1152/ajpheart.1999.276.5.H1468. [DOI] [PubMed] [Google Scholar]
- [37].Bode-Boger SM, Boger RH, Schroder EP, Frolich JC, Exercise increases systemic nitric oxide production in men, J. Cardiovasc. Risk 1 (1994) 173–178. [PubMed] [Google Scholar]
- [38].Jungersten L, Ambring A, Wall B, Wennmalm A, Both physical fitness and acute exercise regulate nitric oxide formation in healthy humans, J. Appl. Physiol 82 (1985) 760–764, 10.1152/jappl.1997.82.3.760, 1997. [DOI] [PubMed] [Google Scholar]
- [39].Maxwell AJ, Schauble E, Bernstein D, Cooke JP, Limb blood flow during exercise is dependent on nitric oxide, Circulation 98 (1998) 369–374, 10.1161/01.cir.98.4.369. [DOI] [PubMed] [Google Scholar]
- [40].Hambrecht R, Fiehn E, Weigl C, Gielen S, Hamann C, Kaiser R, et al. , Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure, Circulation 98 (1998) 2709–2715, 10.1161/01.cir.98.24.2709. [DOI] [PubMed] [Google Scholar]
- [41].Cuzzolin L, Lussignoli S, Crivellente F, Adami A, Schena F, Bellavite P, et al. , Influence of an acute exercise on neutrophil and platelet adhesion, nitric oxide plasma metabolites in inactive and active subjects, Int. J. Sports Med 21 (2000) 289–293, 10.1055/s-2000-13308. [DOI] [PubMed] [Google Scholar]
- [42].Niebauer J, Maxwell AJ, Lin PS, Tsao PS, Kosek J, Bernstein D, et al. , Impaired aerobic capacity in hypercholesterolemic mice: partial reversal by exercise training, Am. J. Phys 276 (1999) H1346–H1354, 10.1152/ajpheart.1999.276.4.H1346. [DOI] [PubMed] [Google Scholar]
- [43].Varin R, Mulder P, Richard V, Tamion F, Devaux C, Henry JP, et al. , Exercise improves flow-mediated vasodilatation of skeletal muscle arteries in rats with chronic heart failure. Role of nitric oxide, prostanoids, and oxidant stress, Circulation 99 (1999) 2951–2957, 10.1161/01.cir.99.22.2951. [DOI] [PubMed] [Google Scholar]
- [44].Woods JA, Davis JM, Mayer EP, Ghaffar A, Pate RR, Effects of exercise on macrophage activation for antitumor cytotoxicity, J. Appl. Physiol 76 (1985) 2177–2185, 10.1152/jappl.1994.76.5.2177, 1994. [DOI] [PubMed] [Google Scholar]
- [45].Bejma J, Ji LL, Aging and acute exercise enhance free radical generation in rat skeletal muscle, J. Appl. Physiol 87 (1985) 465–470, 10.1152/jappl.1999.87.1.465, 1999. [DOI] [PubMed] [Google Scholar]
- [46].Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA, Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide, Proc. Natl. Acad. Sci. U. S. A 87 (1990) 1620–1624, 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lennon SL, Quindry JC, Hamilton KL, French JP, Hughes J, Mehta JL, et al. , Elevated MnSOD is not required for exercise-induced cardioprotection against myocardial stunning, Am. J. Physiol. Heart Circ. Physiol 287 (2004) H975–H980, 10.1152/ajpheart.01208.2003. [DOI] [PubMed] [Google Scholar]
- [48].Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, et al. , The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene, Proc. Natl. Acad. Sci. U. S. A 96 (1999) 11507–11512, 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fewell JG, Osinska H, Klevitsky R, Ng W, Sfyris G, Bahrehmand F, et al. , A treadmill exercise regimen for identifying cardiovascular phenotypes in transgenic mice, Am. J. Phys 273 (1997) H1595–H1605, 10.1152/ajpheart.1997.273.3.H1595. [DOI] [PubMed] [Google Scholar]
- [50].Woods JA, Ceddia MA, Wolters BW, Evans JK, Lu Q, McAuley E, Effects of 6 months of moderate aerobic exercise training on immune function in the elderly, Mech. Ageing Dev 109 (1999) 1–19, 10.1016/s0047-6374(99)00014-7. [DOI] [PubMed] [Google Scholar]
- [51].Murphy EA, Davis JM, Brown AS, Carmichael MD, Van Rooijen N, Ghaffar A, et al. , Role of lung macrophages on susceptibility to respiratory infection following short-term moderate exercise training, Am. J. Phys. Regul. Integr. Comp. Phys 287 (2004) R1354–R1358, 10.1152/ajpregu.00274.2004. [DOI] [PubMed] [Google Scholar]
- [52].Camacho RC, Donahue EP, James FD, Berglund ED, Wasserman DH, Energy state of the liver during short-term and exhaustive exercise in C57BL/6J mice, Am. J. Physiol. Endocrinol. Metab 290 (2006) E405–E408, 10.1152/ajpendo.00385.2005. [DOI] [PubMed] [Google Scholar]
- [53].Lu Q, Ceddia MA, Price EA, Ye SM, Woods JA, Chronic exercise increases macrophage-mediated tumor cytolysis in young and old mice, Am. J. Phys 276 (1999) R482–R489, 10.1152/ajpregu.1999.276.2.R482. [DOI] [PubMed] [Google Scholar]
- [54].Guo Y, Stein AB, Wu WJ, Tan W, Zhu X, Li QH, et al. , Administration of a CO-releasing molecule at the time of reperfusion reduces infarct size in vivo, Am. J. Physiol. Heart Circ. Physiol 286 (2004) H1649–H1653, 10.1152/ajpheart.00971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R, Demonstration of an early and a late phase of ischemic preconditioning in mice, Am. J. Phys 275 (1998) H1375–H1387, 10.1152/ajpheart.1998.275.4.H1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, et al. , Formation of protein kinase C(epsilon)-Lck signaling modules confers cardioprotection, J. Clin. Invest 109 (2002) 499–507, 10.1172/JCI13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Xuan YT, Guo Y, Han H, Zhu Y, Bolli R, An essential role of the JAK-STAT pathway in ischemic preconditioning, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 9050–9055, 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dawn B, Tiwari S, Kucia MJ, Zuba-Surma EK, Guo Y, Sanganalmath SK, et al. , Transplantation of bone marrow-derived very small embryonic-like stem cells attenuates left ventricular dysfunction and remodeling after myocardial infarction, Stem Cells 26 (2008) 1646–1655, 10.1634/stemcells.2007-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Takano H, Bolli R, Black RG Jr., Kodani E, Tang XL, Yang Z, et al. , A(1) or A (3) adenosine receptors induce late preconditioning against infarction in conscious rabbits by different mechanisms, Circ. Res 88 (2001) 520–528, 10.1161/01.res.88.5.520. [DOI] [PubMed] [Google Scholar]
- [60].Tang XL, Qiu Y, Park SW, Sun JZ, Kalya A, Bolli R, Time course of late preconditioning against myocardial stunning in conscious pigs, Circ. Res 79 (1996) 424–434, 10.1161/01.res.79.3.424. [DOI] [PubMed] [Google Scholar]
- [61].Guo Y, Bao W, Wu WJ, Shinmura K, Tang XL, Bolli R, Evidence for an essential role of cyclooxygenase-2 as a mediator of the late phase of ischemic preconditioning in mice, Basic Res. Cardiol 95 (2000) 479–484, 10.1007/s003950070024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Carson LD, Korzick DH, Dose-dependent effects of acute exercise on PKC levels in rat heart: is PKC the heart’s prophylactic? Acta Physiol. Scand 178 (2003) 97–106, 10.1046/j.1365-201X.2003.01131.x. [DOI] [PubMed] [Google Scholar]
- [63].Bijnen FC, Caspersen CJ, Mosterd WL, Physical inactivity as a risk factor for coronary heart disease: a WHO and international society and Federation of Cardiology position statement, Bull. World Health Organ 72 (1994) 1–4. [PMC free article] [PubMed] [Google Scholar]
- [64].Miller TD, Balady GJ, Fletcher GF, Exercise and its role in the prevention and rehabilitation of cardiovascular disease, Ann. Behav. Med 19 (1997) 220–229, 10.1007/BF02892287. [DOI] [PubMed] [Google Scholar]
- [65].Considine RV. Invited editorial on “Acute and chronic effects of exercise on leptin levels in humans”. J Appl Physiol (1985). 1997;83:3–4.doi: 10.1152/jappl.1997.83.1.3. [DOI] [PubMed] [Google Scholar]
- [66].Fletcher GF, Blair SN, Blumenthal J, Caspersen C, Chaitman B, Epstein S, et al. , Statement on exercise. Benefits and recommendations for physical activity programs for all Americans. A statement for health professionals by the Committee on Exercise and Cardiac Rehabilitation of the Council on Clinical Cardiology, American Heart association, Circulation 86 (1992) 340–344. [DOI] [PubMed] [Google Scholar]
- [67].Jennings GL, Mechanisms for reduction of cardiovascular risk by regular exercise, Clin. Exp. Pharmacol. Physiol 22 (1995) 209–211, 10.1111/j.1440-1681.1995.tb01982.x. [DOI] [PubMed] [Google Scholar]
- [68].Sandvik L, Erikssen J, Thaulow E, Erikssen G, Mundal R, Rodahl K, Physical fitness as a predictor of mortality among healthy, middle-aged Norwegian men [see comments], N. Engl. J. Med 328 (1993) 533–537. [DOI] [PubMed] [Google Scholar]
- [69].Powell KE, Thompson PD, Caspersen CJ, Kendrick JS, Physical activity and the incidence of coronary heart disease, Annu. Rev. Public Health 8 (1987) 253–287, 10.1146/annurev.pu.08.050187.001345. [DOI] [PubMed] [Google Scholar]
- [70].O’Connor GT, Buring JE, Yusuf S, Goldhaber SZ, Olmstead EM, Paffenbarger RS Jr., et al. , An overview of randomized trials of rehabilitation with exercise after myocardial infarction, Circulation 80 (1989) 234–244, 10.1161/01.cir.80.2.234. [DOI] [PubMed] [Google Scholar]
- [71].Desai KH, Sato R, Schauble E, Barsh GS, Kobilka BK, Bernstein D, Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease, Am. J. Phys 272 (1997) H1053–H1061. [DOI] [PubMed] [Google Scholar]
- [72].Chien GL, Wolff RA, Davis RF, van Winkle DM, “Normothermic range” temperature affects myocardial infarct size, Cardiovasc. Res 28 (1994) 1014–1017, 10.1093/cvr/28.7.1014. [DOI] [PubMed] [Google Scholar]
- [73].Davis JM, Murphy EA, Brown AS, Carmichael MD, Ghaffar A, Mayer EP, Effects of moderate exercise and oat beta-glucan on innate immune function and susceptibility to respiratory infection, Am. J. Phys. Regul. Integr. Comp. Phys 286 (2004) R366–R372, 10.1152/ajpregu.00304.2003. [DOI] [PubMed] [Google Scholar]
- [74].Wu KK, Regulation of endothelial nitric oxide synthase activity and gene expression, Ann. N. Y. Acad. Sci 962 (2002) 122–130, 10.1111/j.1749-6632.2002.tb04062.x. [DOI] [PubMed] [Google Scholar]
- [75].Venditti P, Bari A, Di Stefano L, Di Meo S, Role of mitochondria in exercise-induced oxidative stress in skeletal muscle from hyperthyroid rats, Arch. Biochem. Biophys 463 (2007) 12–18, 10.1016/j.abb.2007.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.