

Abstract
Retroviral Gag targeting to the plasma membrane (PM) for assembly is mediated by the N-terminal matrix (MA) domain. For many retroviruses, Gag–PM interaction is dependent on phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2). However, it has been shown that for human T-cell leukemia virus type 1 (HTLV-1), Gag binding to membranes is less dependent on PI(4,5)P2 than HIV-1, suggesting that other factors may modulate Gag assembly. To elucidate the mechanism by which HTLV-1 Gag binds to the PM, we employed NMR techniques to determine the structure of unmyristoylated MA (myr(−)MA) and to characterize its interactions with lipids and liposomes. The MA structure consists of four α-helices and unstructured N- and C-termini. We show that myr(−)MA binds to PI(4,5)P2 via the polar head and that binding to inositol phosphates (IPs) is significantly enhanced by increasing the number of phosphate groups on the inositol ring, indicating that the MA–IP binding is governed by charge–charge interactions. The IP binding site was mapped to a well-defined basic patch formed by lysine and arginine residues. Using an NMR-based liposome binding assay, we show that PI(4,5)P2 and phosphatidylserine enhance myr(−)MA binding in a synergistic fashion. Confocal microscopy data revealed formation of puncta on the PM of Gag expressing cells. However, G2A-Gag mutant, lacking myristoylation, is diffuse and cytoplasmic. These results suggest that although myr(−)MA binds to membranes, myristoylation appears to be key for formation of HTLV-1 Gag puncta on the PM. Altogether, these findings advance our understanding of a key mechanism in retroviral assembly.
Keywords: Human T-cell leukemia virus type 1 (HTLV-1); human immunodeficiency virus type 1 (HIV-1); Gag myristoylation; phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2); nuclear magnetic resonance (NMR).
Graphical Abstract:
Introduction
During the late phase of retroviral replication, the virally encoded Gag polyproteins are targeted to the plasma membrane (PM) for assembly, virus budding and release [1–13]. During or subsequent to virus budding, the Gag and Gag-pol polyproteins are cleaved by the virally encoded protease into matrix (MA), capsid (CA), nucleocapsid (NC), and short peptides to form mature virions (reviewed in [9, 14, 15]). It is demonstrated that Gag binding to the PM is mediated by the MA domain, which for most retroviruses contains a bipartite signal consisting of an N-terminal myristoyl (myr) group and a highly basic region. Over the last three decades, studies have established that binding of retroviral Gag to membranes is regulated by many factors such as protein multimerization, cellular and viral RNA, and the type of lipids and degree of acyl chain saturation [6, 7, 16–34].
Targeting and localization of human immunodeficiency virus type 1 (HIV-1) Gag proteins to the PM was shown to be dependent on phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] [7], a signaling lipid that fulfills many cellular functions by acting as a substrate for numerous proteins [35, 36]. Over-expression of phosphoinositide 5-phosphatase IV (5ptaseIV), which reduces PI(4,5)P2 levels by hydrolyzing the phosphate at the D5 position of PI(4,5)P2, led to significant reduction in Gag–PM localization and attenuation of virus production [7]. PI(4,5)P2–dependent Gag assembly has also been shown for other retroviruses such as HIV-2 [12], Mason-Pfizer monkey virus (MPMV) [37, 38], murine leukemia virus [10], feline immunodeficiency virus [39], and avian sarcoma virus [40-42].
NMR structural studies of HIV-1 MA binding to PI(4,5)P2 containing truncated (tr) acyl chains have shown tr-PI(4,5)P2 binding to MA induced a conformational change that promoted myr exposure [43]. The structure of MA-tr-PI(4,5)P2 complex showed that both the polar head and the truncated 2’-acyl chain are involved in binding [43]. Subsequent studies have shown that PM lipids such as phosphatidylserine (PS), phosphatidylcholine (PC), and phosphatidylethanolamine (PE) with truncated acyl chains also bind to HIV-1 MA [44]. NMR studies confirmed MA binding to membrane mimetics such as bicelles, micelles and lipid nanodiscs [44, 45]. NMR studies of HIV-2 [12] and MPMV MA proteins [38] as well as surface plasmon resonance studies of HIV-1 Gag and/or MA proteins [46] have shown that MA proteins are capable of interacting with the acyl chains of phosphoinositides, and that increasing the length of the acyl chain resulted in stronger binding. Employing computational methods [47] and an NMR-based liposome assay [48], it was suggested that acyl chains of native PI(4,5)P2 are not involved in MA binding and that Gag–membrane interaction is mediated predominantly by dynamic, electrostatic interactions between conserved basic residues of MA and acidic phospholipids such as PI(4,5)P2 and PS [47, 48].
Most recent cryo-electron tomography data revealed that MA undergoes dramatic structural maturation to form very different lattices in immature and mature HIV-1 particles [49]. Mature MA forms a hexameric lattice in which the acyl chain of a phospholipid extends out of the membrane to bind a pocket in MA, consistent with the NMR studies [43]. Based on these studies, it was suggested that maturation of HIV-1 not only achieves assembly of the capsid surrounding the RNA genome, but it also extends to repurpose the MA lattice for an entry or post-entry function and causes partial removal of 2,500 acyl chains from the viral membrane [49]. Taken together, despite some differences in the proposed models, these studies have shed new insights on how various retroviral Gag proteins interact with the inner leaflet of the PM.
In this report, we focus on the molecular mechanism by which human T-cell leukemia virus type 1 (HTLV-1) Gag polyproteins are targeted to the PM for assembly. HTLV is a zoonotic virus with simian T-cell leukemia virus counterparts found in monkeys. HTLV-1 and HTLV-2 are the most studied subtypes of HTLV. Even though they share ~ 70% nucleotide identity, HTLV-1 is considered more pathogenic as it is associated with adult T-cell leukemia and HTLV-1 associated myelopathy/tropical spastic paraparesis [50-52]. HTLV-1 transmission occurs mainly through cell-to-cell contacts rather than cell-free virus particles [53, 54]. In addition, HTLV-1–infected T-cells can multiply by clonal expansion, consequently increasing the viral burden without the need for virus replication and reinfection [55, 56]. To develop effective antiretroviral treatments for HTLV-1, it is paramount to gain a more complete understanding of the molecular processes that govern HTLV-1 replication. However, many fundamental aspects of HTLV-1 replication, including particle assembly, are incompletely understood.
Previous studies have shown that HTLV-1 Gag is localized at the cell surface and intracellular compartments in HeLa cells [57-60]. More recently, studies have shown that, compared to HIV-1, HTLV-1 Gag binding to the PM of infected cells and to liposomes in vitro is less dependent on PI(4,5)P2 than that observed for HIV-1 [31]. Unlike HIV-1 Gag, subcellular localization of HTLV-1 Gag and subsequent virus-like particle (VLP) production in HeLa cells were minimally sensitive to 5ptaseIV overexpression, suggesting that the interaction of HTLV-1 MA with PI(4,5)P2 is not a key determinant for HTLV-1 particle assembly [31, 32]. It was also shown that although PI(4,5)P2 enhanced HTLV-1 Gag binding to liposomes, Gag proteins bound efficiently to liposomes lacking PI(4,5)P2 but containing PS if similar overall negative charge is maintained [31]. HTLV-1 Gag was found to bind to membranes with a higher affinity than that of HIV-1 Gag with no susceptibility to RNA-mediated inhibition of membrane binding [31]. Chimeric switching of MA domains between HIV-1 and HTLV-1 Gag proteins showed that these differences are mediated by the MA domain of Gag [31]. Subsequent studies have shown that single amino acid substitutions that confer a large basic patch rendered HTLV-1 MA susceptible to the RNA-mediated block, suggesting that RNA blocks MA containing a large basic patch [32]. These data supported a model in which HTLV-1 Gag localizes to the PM via the MA domain with higher efficiency but less specificity than for other retroviruses [31, 32].
Further comparison of the subcellular localization of HIV-1 with HTLV-1 Gag in HeLa cells using dual-color, z-scan fluorescence fluctuation spectroscopy and total internal reflection fluorescence microscopy revealed significant differences in the cytoplasmic threshold concentration of Gag required for PM binding [61, 62]. It was found that HIV-1 Gag membrane targeting required a concentration threshold of ~500 nM, a concentration also needed to form stable cytoplasmic Gag homo-complexes. At ~500 nM, Gag puncta on the PM become readily apparent [62]. On the other hand, HTLV-1 Gag puncta formation is observed at the PM regardless of cytoplasmic Gag concentration. Based on these findings, it was suggested that Gag associates with the PM as a multimer for HIV-1 and a monomer for HTLV-1 [62]. These differences in membrane association have also been recapitulated for the HIV-1 and HTLV-1 MA proteins [62]. Differential mechanisms for Gag–Gag and/or myr-mediated interaction with the PM between HIV-1 and HTLV-1 may explain the differences in their interaction with the PM. This dramatic difference in binding affinity highlights the need for a sharper understanding of retroviral Gag localization to the PM at the atomic and molecular levels [61, 62].
To provide structural insights into the mechanism by which HTLV-1 Gag is targeted to the PM for assembly, we characterized the interactions of the unmyristoylated MA protein (myr(−)MA) with lipids and liposomes by NMR, biophysical, and biochemical methods. We show that MA contains a PI(4,5)P2 binding site and that PI(4,5)P2 and PS enhance myr(−)MA binding in a synergistic fashion. Confocal microscopy data revealed formation of puncta on the PM of Gag expressing cells. However, the G2A-Gag mutant, lacking myristoylation, is diffuse and cytoplasmic, leading to severe attenuation of particle production. These findings advance our understanding of a key mechanism in retroviral assembly.
Results
Structure determination of HTLV-1 myr(−)MA99
The HTLV-1 MA domain consists of 130 residues and is naturally myristoylated. Due to technical challenges, we were unable to produce soluble, homogenous and monodisperse HTLV-1 MA for structural studies. Therefore, our studies were conducted with the HTLV-1 myr(−)MA protein. First, we generated a structural model of HTLV-1 myr(−)MA using I-TASSER, a web-based tool used for structure prediction of proteins [63, 64]. The resulting low-energy model indicated that the proline-rich C-terminal domain lacks an ordered structure. This model is consistent with the structural data of the closely related HTLV-2 myr(−)MA protein [65]. To avoid potential proteolytic cleavage of this tail during collection of multidimensional NMR data for lengthy periods, we truncated residues 100-130 to generate MA99. Solution properties of the full-length myr(−)MA and myr(−)MA99 proteins were analyzed by a gel filtration mobility assay (Fig. S1). A gel filtration mobility assay with known protein standards revealed that the estimated molecular weight of myr(−)MA and myr(−)MA99 proteins are ~24 and 10 kDa, respectively (Fig. S1). Whereas the estimated molecular weight of myr(−)MA appears to be higher than the calculated monomeric unit (~15 kDa), no evidence for protein self-association was observed at all tested protein concentrations. The migration behavior of myr(−)MA is likely attributed to its shape caused by the unstructured C-terminal 31 residues. A minor species (~10%) of disulfide cross-linked dimer via Cys61 was observed during purification and was eliminated by inclusion of reducing agents in buffers.
2D 1H-15N HSQC data obtained for myr(−)MA and myr(−)MA99 confirmed that truncation of the C-terminal 31 residues did not adversely affect the structure and/or fold of the globular domain (Figs. 1A and S2). Standard triple-resonance, NOESY-, and TOCSY-based experiments were collected for HTLV-1 myr(−)MA99, which were used to generate near-complete backbone and side-chain chemical shift assignments. Subsequently, an initial list of distance restraints was created using Unio’10 Atnos/Candid functionality of automated, iterative peak picking of raw NOESY spectra. The list of distance constrains was extended by manual analysis of NOE-based spectra, and structures were calculated using CYANA. Superposition of the 20 lowest-penalty myr(−)MA99 structures is shown in Figure S3 (see also Table S1). The globular domain of myr(−)MA99 extends from residue 21 to 93 and consists of four α-helices, similar to that observed for HTLV-2 myr(−)MA (Figs. 1B and S4) [65]. Not surprisingly, because HTLV-1 and HTLV-2 MA proteins share ~60% sequence identity, their structures exhibit an overall similar fold. However, differences were observed in helix packing and orientations (Fig. S4). Notably, we identified numerous unambiguous NOEs between residues Phe27 and Leu84/Leu87, and Tyr65 and Ile63/Leu87/Gln91, resulting in a tight packing of α-helix III against α-helix IV (Fig. 2). Additionally, NMR data did not support the existence of a stable 310 helix, previously observed between helices II and III in HTLV-2 myr(−)MA [65]. The myr(−)MA99 structure exhibits a well-defined right-handed turn in this area but lacks the critical amide (i, i+3) or amide (i, i+4) NOEs, indicating that this region is more flexible than in HTLV-2 myr(−)MA. Determination of the three-dimensional structure of HTLV-1 MA99 is a necessary step for characterizing MA–lipid and MA–liposome interactions.
Figure 1.
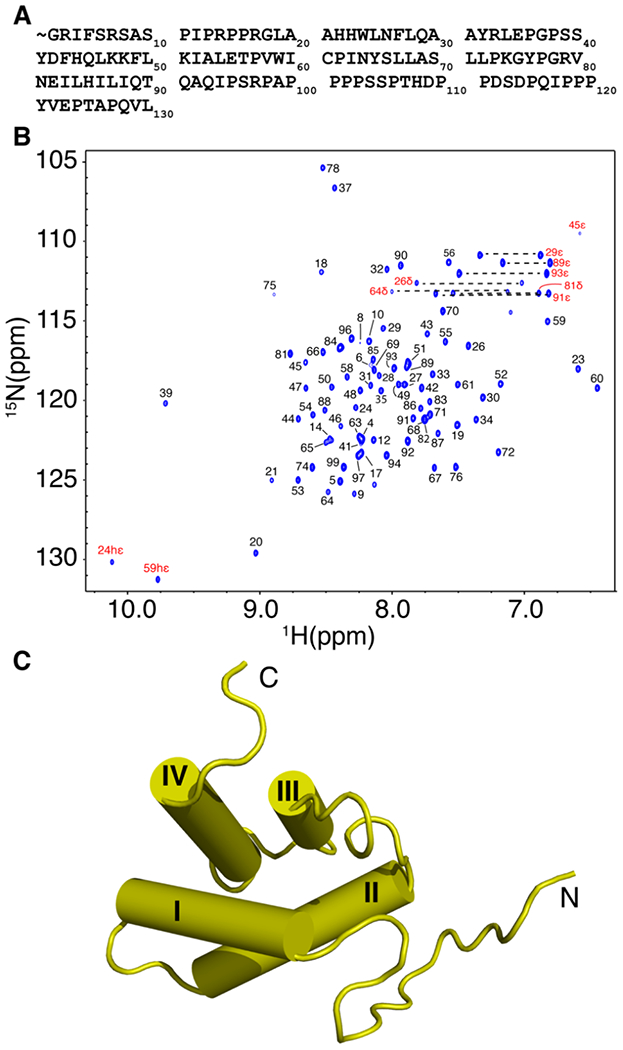
NMR data and structure of HTLV-1 myr(−)MA99. (a) HTLV-1 MA protein sequence. The myr group is denoted with the sign (~). (b) 2D 1H-15N HSQC NMR spectrum of the myr(−)MA99 protein at 35 °C in 50 mM sodium phosphates (pH 6), 100 mM NaCl and 2 mM TCEP. (c) Cartoon representation of the myr(−)MA99 structure.
Figure 2.
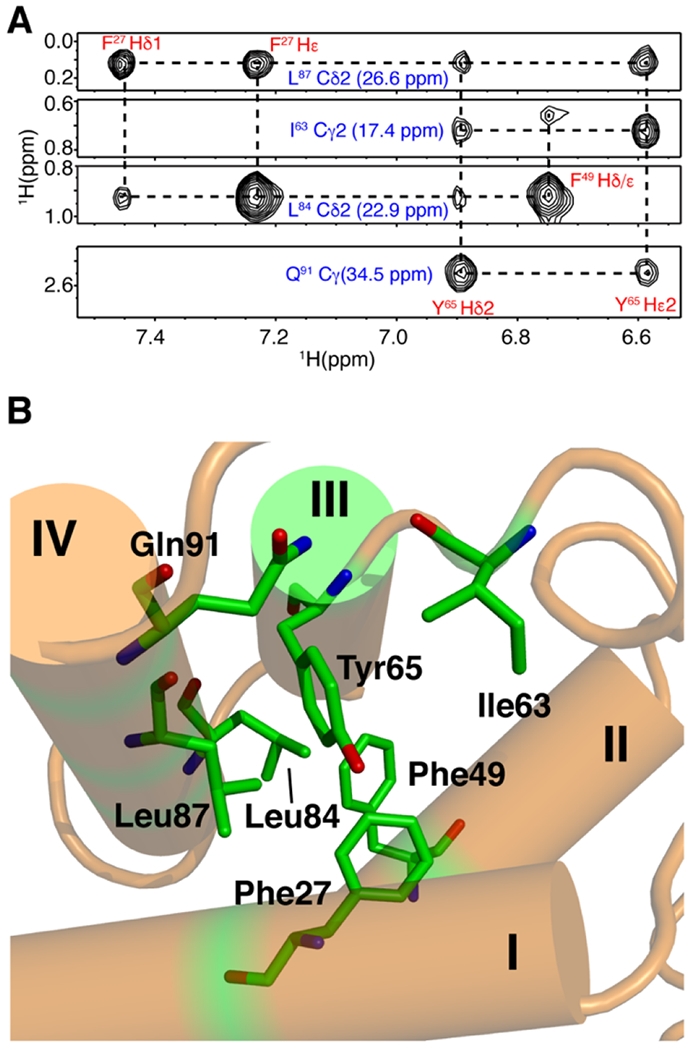
NMR data and structure of HTLV-1 myr(−)MA99. (a) Selected 1H–1H strips from the 13C-edited HMQC-NOESY spectrum showing unambiguous NOEs between residues on helices III and IV. (b) Cartoon and stick structural view showing relationship between residues represented by the NOEs above.
PI(4,5)P2 binding to HTLV-1 myr(−)MA
Native PI(4,5)P2 has a low critical micelle concentration threshold (~30 μM) in aqueous solution,[66, 67] which causes severe signal broadening in the NMR spectra as described [12, 43]. Therefore, we used dibutanoyl-PI(4,5)P2 (diC4-PI(4,5)P2), a soluble analog with truncated acyl chains (see Fig. S5 for chemical structures of lipids used in this study). This ligand was extensively used in previous studies of retroviral MA proteins [11, 12, 38, 41, 43, 46, 68]. Initial diC4-PI(4,5)P2 titration experiments with the myr(~)MA99 protein led to protein precipitation at high lipid stoichiometries, which precluded accurate analysis of data. Therefore, our lipid and liposome binding studies were all conducted with the full-length myr(~)MA protein.
Titration of HTLV-1 myr(−)MA with increasing amounts of diC4-PI(4,5)P2 led to significant chemical shift perturbations (CSPs) for a subset of1H and 15N resonances in the 2D 1H-15N HSQC spectra (Fig. 3). The incremental CSPs as function of increasing lipid concentration indicate a fast exchange regime, on the NMR timescale, between the free and bound states. CSPs data were used to determine the dissociation constant (Kd) by fitting the CSPs as a function of diC4-PI(4,5)P2 concentration, which yielded Kd values of 7 μM in the absence of salt (Fig. S6 and Table 1). At 100 mM NaCl, Kd was calculated as 306 μM (Table 1). Interestingly, in the absence of salt the affinity of diC4-PI(4,5)P2 to HTLV-1 myr(−)MA is ~20-fold tighter than those observed for the HIV-1 and HIV-2 MA proteins (Kd ~150 μM) [12, 43], and > 120-fold tighter than that observed for the ASV MA protein (Kd ~ 850 μM). CSPs were mapped on the structure of the myr(−)MA99 protein. Among the signals that exhibited significant CSPs are Lys47, Lys48, and Lys51 (Fig. 3), which reside n helix II and form a basic patch on the surface of the protein (Fig. 4). Additionally, apparent CSPs were observed for several residues within helix II (Ser39, Ser40, Phe43, His44, Gln45, Leu46, Phe49, Leu50, and Ile52; Fig. 3). Significant CSPs were also detected for signals corresponding to residues in the unstructured, N-terminal region of MA (Phe5, Ile12, Arg14, Arg17, Gly18, Leu19, Ala20, and Ala21; Figs. 3 and 4). Analysis of the surface electrostatic potential map of myr(−)MA99 shows that Arg14 and Arg17 (along with Arg3 and Arg7) form an extended second basic surface patch (Fig. 4).
Figure 3.
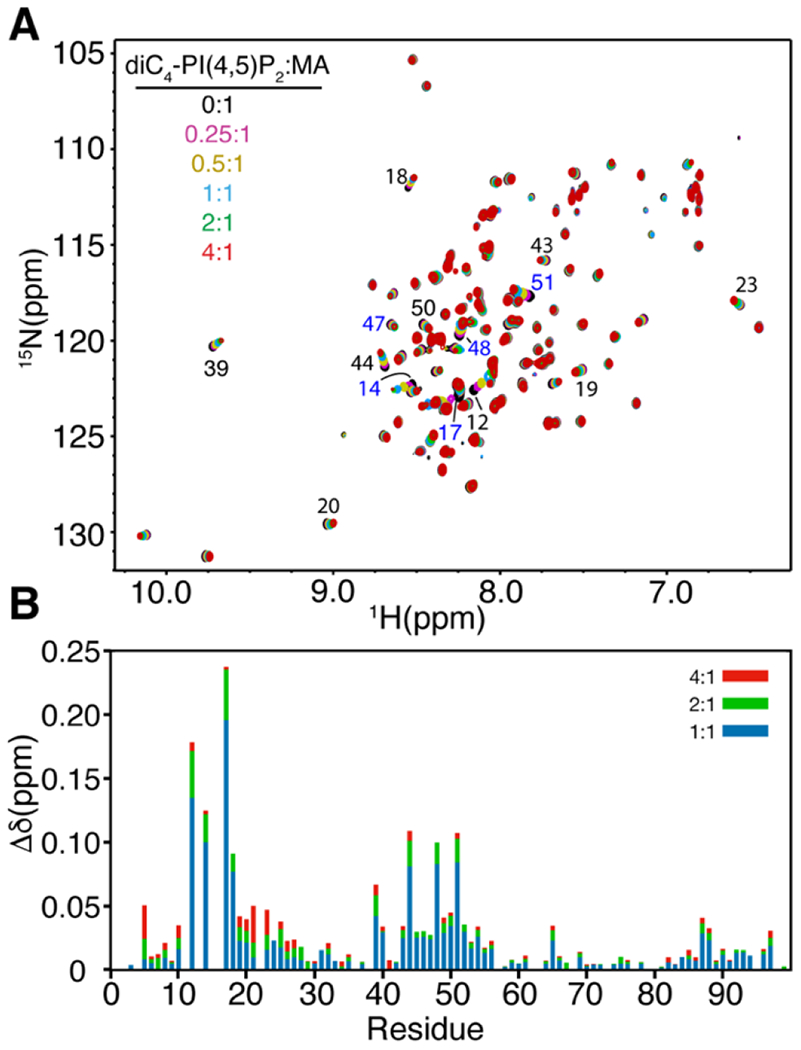
PI(4,5)P2 binding to myr(−)MA. (a) Overlay of 2D 1H-15N HSQC spectra upon titration of myr(−)MA with diC4-PI(4,5)P2 [100 μM, 35 °C; diC4-PI(4,5)P2:MA = 0:1 (black), 0.25:1 (magenta), 0.5:1 (olive), 1:1 (cyan), 2:1 (green), 4:1 (red)] in 50 mM phosphates (pH 6) and 2 mM TCEP. (b) Histogram of normalized 1H-15N chemical shift changes vs. residue number calculated from the HSQC spectra above.
Table 1.
Dissociation constants for lipids and IPs binding to HTLV-1 myr(−)MA obtained by plotting CSPs vs. lipid concentration.
| Lipid | Kd (μM) |
|---|---|
| diC4-PI(4,5)P2 (0 M NaCl) | 7.2 ± 2.1 |
| diC4-PI(4,5)P2 | 306 ± 30 |
| diC6-PI(4,5)P2 | 195 ± 19 |
| I(1,4,5) P3 | 338 ± 42 |
| I(1,4,5)P3 (0 M NaCl) | 4.0 ± 1.3 |
| I(1,3,4,5)P4 | 62 ± 14 |
| IP6 | 23 ± 11 |
| IP6 (0.2 M NaCl) | 36 ± 7 |
| IP6 (0.4 M NaCl) | 295 ± 56 |
Titrations were conducted in the presence of 0.1 M NaCl except when noted. Kds are average values obtained from plotting CSPs of 3-4 NMR signals.
Figure 4.
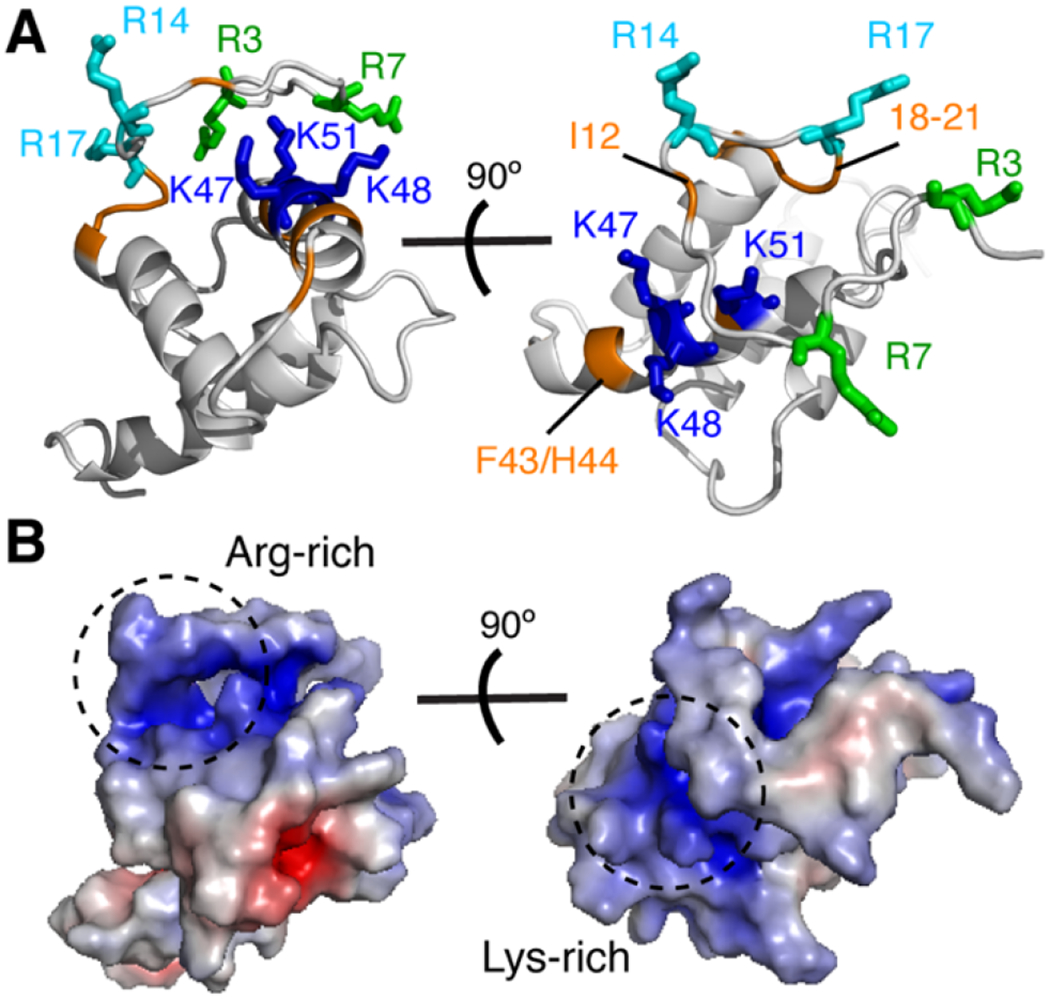
Chemical shift mapping of PI(4,5)P2 binding to myr(−)MA99. (a) Cartoon representation of the myr(−)MA99 structure highlighting basic residues (blue and cyan) that exhibited substantial CSPs upon binding of diC4-PI(4,5)P2. Signals of residues highlighted in orange are perturbed due to their proximity to the diC4-PI(4,5)P2 binding site. No CSPs have been detected for Arg3 and Arg7 (green sticks) (b) Electrostatic surface potential map of myr(−)MA99 showing the basic patches formed by the lysine-rich and arginine-rich regions.
Of note, several hydrophobic residues in the unstructured N-terminus (Phe5, Ile12, and Leu19) also exhibited CSPs upon titration of diC4-PI(4,5)P2 (Fig. 3). It is unclear whether these CSPs are a consequence of interactions with the polar head of diC4-PI(4,5)P2 via the N-terminal Arg14 and Arg17, or a result of direct contact with the acyl chains. It is possible that the flexibility of the N-terminus may allow for transient interactions with diC4-PI(4,5)P2 or other proximal regions. Thus, we assessed whether the acyl chains play a role in the interaction by conducting NMR titrations with dihexanoyl PI(4,5)P2 [diC6-PI(4,5)P2]. The CSPs for signals corresponding to hydrophobic residues in the N-terminus (Phe5, Ile12, and Leu19) and in helix I (Phe27, Leu28, Ala30, Ala31, Tyr32) were slightly larger than those observed for diC4-PI(4,5)P2, suggesting that the acyl chains may interact with the N-terminal hydrophobic residues (Fig. S7). Consequently, a slightly higher binding affinity was observed for diC6-PI(4,5)P2 binding to myr(−)MA (Table 1). We do not rule out that the interactions between the acyl chains and the myr(−)MA protein are nonspecific.
To examine whether the polar head of PI(4,5)P2 is sufficient for myr(−)MA binding, we conducted NMR titrations with inositol 1,4,5-trisphosphate (IP3). As shown in Figure S8, 1H-15N resonances that exhibited significant CSPs are similar to those observed upon binding of diC4-PI(4,5)P2 (Fig. S7). IP3 titration data afforded a Kd of 4 μM, which is similar to that observed for diC4-PI(4,5)P2 (Table 1). Altogether, our data demonstrate that PI(4,5)P2 binds directly to HTLV-1 myr(−)MA via interactions between the polar head and a basic patch formed by lysine and arginine residues and that the acyl chains of PI(4,5)P2 do not appear to be critical for the interaction.
Binding of myr(−)MA to IPs is governed by charge–charge interactions
To assess whether myr(−)MA binding to IPs is predominantly governed by electrostatic interactions, we conducted 2D 1H-15N HSQC NMR titration using IPs with varying number of phosphate groups on the inositol ring. Titration of myr(−)MA with inositol 1,3,4,5-tetrakisphosphate (IP4) yielded a Kd of 62 μM, which is ~5-fold tighter than that observed for IP3 at the same buffer conditions (Table 1). Similar titration experiments performed with inositol hexakisphosphate (IP6) yielded a Kd of 23 μM, which again indicates that binding affinity is enhanced by increasing the number of phosphate groups on the inositol ring. Expectedly, increasing salt concentrations led to decrease in the binding affinity of IP6 to myr(−)MA (Table 1). Mapping of the CSPs on the structure of myr(−)MA99 indicated that all tested IPs bind to the same site. These results demonstrate that HTLV-1 myr(−)MA binding to PI(4,5)P2 is governed by ionic forces.
Thermodynamics of IP binding to myr(−)MA
Having established the presence of a PI(4,5)P2 binding site on HTLV-1 MA, we sought to further investigate the nature of the interaction and the contribution of enthalpic and entropic factors. To determine stoichiometry (n), enthalpy change (∆H°) and entropic term (T∆S°), we conducted isothermal titration calorimetry (ITC) experiments upon titration of myr(−)MA with IP3. We used IP3 because it is the polar headgroup of PI(4,5)P2 and because the binding affinity is sufficiently strong to yield analyzable ITC data. Applying a single set of identical sites model to fit the data (Fig. 5) yielded the following parameters: Kd = 3.7 ± 0.7 μM, n = 1.04 ± 0.03, ∆H° = −8.3 ± 0.3 kcal/mol, and T∆S° = 1.4 ± 0.1 kcal/mol. The ITC indicated that the Kd obtained by ITC data is very consistent with that obtained from the NMR titration data, that myr(−)MA harbours a single IP binding site, and that the exothermic reaction is indicative of the electrostatic nature of the interaction.
Figure 5.
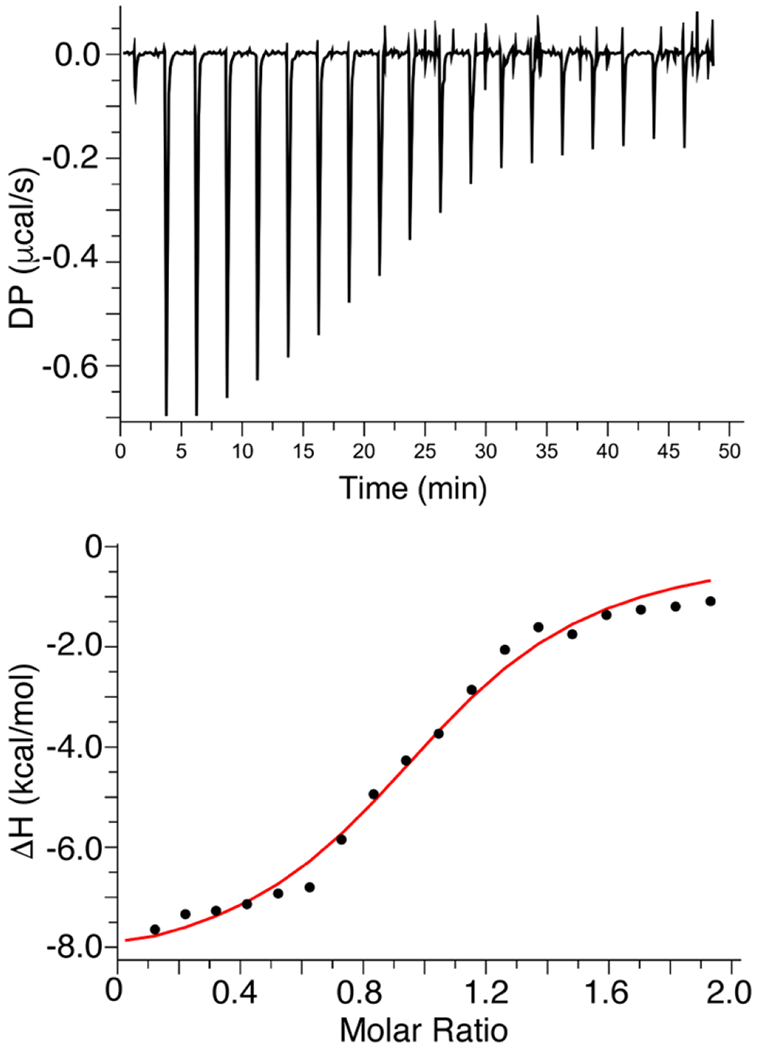
ITC data for binding of IP3 to myr(−)MA. ITC data obtained for titration of IP3 (400 μM) into myr(−)MA (40 μM) in 20 mM MES (pH 6.0) and 2 mM TCEP. Applying a single set of identical sites model to fit the data yielded a Kd of 3.7 ± 0.7 μM and stoichiometry value (n) of 1, indicating a single lipid binding site.
Interaction of myr(−)MA with liposomes
It is established that the affinity of retroviral MA and/or Gag proteins to membranes is enhanced by incorporation of acidic phospholipids such as PS [6, 20, 23, 27, 69-73]. Previous liposome binding data revealed that HTLV-1 Gag can bind PS-containing membranes efficiently even in the absence of PI(4,5)P2 [31]. Herein, we employed a sensitive NMR–based assay to characterize binding of HTLV-1 myr(−)MA to large unilamellar vesicles (LUVs) containing native lipids. This NMR approach has been employed to characterize interaction of HIV-1 and ASV MA proteins to liposomes [41, 48]. The assay allows for measurement of the unbound protein population in solution under equilibrium conditions with the liposome–bound form, which can provide quantitative binding measurements such as Kd values and important information on the synergy of membrane components and cooperativity of binding [74, 75]. 1H NMR experiments were conducted on samples with fixed protein and total lipid concentrations, while varying lipid composition within LUVs. As predicted, myr(−)MA binding to LUVs made with 100% POPC was very weak, as judged by the negligible decrease in signal intensity (<10%) at saturating amounts of POPC LUVs (data not shown). However, NMR titrations of myr(−)MA with LUVs containing POPC and increasing amounts of PI(4,5)P2 led to increased signal attenuation (Fig. 6A), indicating that PI(4,5)P2 enhanced the affinity of myr(−)MA to liposomes. Data fitting using the Hill equation resulted in a Kd value of 36 μM (at 100 mM NaCl; Fig. 6B and Table 2), demonstrating that affinity of HTLV-1 myr(−)MA to membrane is enhanced by PI(4,5)P2 and in the absence of other acidic or charged lipids.
Figure 6.
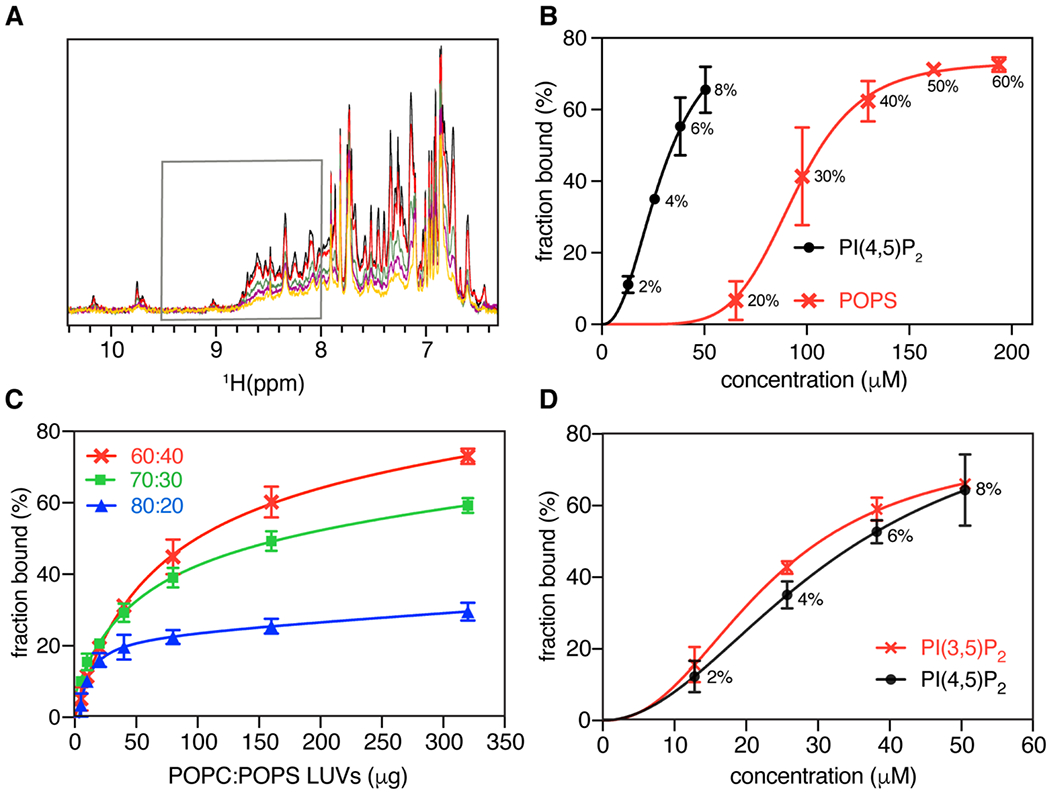
Interaction of myr(−)MA with LUVs. (a) Overlay of 1H NMR spectra of myr(−)MA (50 μM) in the presence of fixed amount of LUVs with (100 – x)% mol. POPC and x = 0 (black), 2 (red), 4 (green), 6 (purple), and 8 (yellow) molar % PI(4,5)P2. Gray box marks the area of spectra integration. (b) Isotherms of myr(−)MA binding to LUVs containing POPC and varying amounts of PI(4,5)P2 (black). Isotherms of myr(−)MA binding to LUVs containing POPC and varying %mol. of POPS is shown in red. Molar percentages of PI(4,5)P2 or POPS are indicated. Solid lines are Hill equation fits to the experimental data represented by points with error bars. (c) myr(−)MA binding to POPC:POPS LUVs with varying concentrations of POPS as a function of increasing amounts of LUVs. (d) Isotherms of myr(−)MA binding to PI(3,5)P2 and PI(4,5)P2 are shown in red and black respectively. Molar percentage of PI(4,5)P2 is indicated. Solid lines are fits of Hill equation to the experimental data represented by points with error bars.
Table 2.
Parameters for HTLV-1 myr(−)MA binding to LUVs determined using the Hill equation.
| LUV | Kd (μM) | n |
|---|---|---|
| POPC:POPS:PI(4,5)P2 | 10 ± 1 | 0.8 |
| POPC:PI(4,5)P2 | 36 ± 4 | 1.9 |
| POPC:diC16-PI(3,5)P2 | 32 ± 2 | 1.7 |
| POPC:POPS | 118 ± 16 | 2.9 |
Titrations were conducted in the presence of 0.1 M NaCl except when noted. Kds are average values of 2-3 replicates.
Next, we examined the effect of PS on myr(−)MA binding by conducting NMR titrations with LUVs containing POPC and increasing amounts of POPS. Interestingly, in contrast to what was observed for other retroviral myr(−)MA proteins [41, 48], binding was detectable upon incorporation of 20% mol. POPS (Figs. 6B and C). Fitting the binding isotherms with the Hill equation yielded a microscopic Kd of 118 μM for POPS (Table 2). Binding was highly cooperative as indicated by the n value (~3) (Table 2), suggesting that myr(−)MA engages multiple PS molecules. Next, we tested whether PI(4,5)P2 and PS compete for the same anionic binding site(s) or bind to distinct binding sites and synergistically enhance myr(−)MA binding. To do so, we conducted titrations with LUVs containing POPC, a fixed 20% mol. POPS and increasing amounts of PI(4,5)P2. Fitting of the titration data yielded a Kd value of 10 μM. Interestingly, this value is lower than those obtained for titration of LUVs containing PI(4,5)P2 or PS alone (36 and 118 μM, respectively; Table 2). These results suggest that PI(4,5)P2 and PS do not compete for the same binding site and, instead, bind to distinct sites or at least by different mechanisms, enhancing myr(−)MA binding in a synergistic fashion.
Specificity of myr(−)MA binding to phosphoinositides
To determine specificity of PI(4,5)P2 binding and whether the position of the phosphate groups on the inositol group affects myr(−)MA binding to membranes, titration experiments were conducted with POPC liposomes containing diC16-PI(3,5)P2, which differs from PI(4,5)P2 in the placement of a single phosphate. 1H NMR signals decreased in intensity upon addition of POPC/diC16-PI(3,5)P2 liposomes, similar to results obtained for myr(−)MA binding to POPC/PI(4,5)P2 liposomes. Fitting the binding data yielded a microscopic Kd of 32 μM, which is virtually identical to that of PI(4,5)P2 (Fig. 6D and Table 2). These results indicate that myr(−)MA binding to LUVs is not dependent on the positioning of the phosphate group (4th vs. 3rd carbon) and that the overall charge of the lipid appears to be the major determinant of binding.
Particle production and Gag subcellular localization
Previous studies observed a low level of HTLV-1 particle production with Gag containing the G2A mutation in 293T cells using a Gag only [62] and authentic particle system [59]. Furthermore, it has been shown that efficient membrane binding is requisite for robust particle production [76, 77]. In order to confirm that these results are observed in relevant cell lines, HTLV-1-like particles were produced in Jurkat (T cell line) and Raji/CD4 (B cell line) cells by nucleofection with the HTLV-1 Gag and Envelope (Env) expression plasmids. Particle production was driven by a 3:1 ratio of unlabeled and EYFP-labeled HTLV-1 Gag. WT and G2A-Gag and fluorescently-labeled particle production was monitored via confocal microscopy. Relative particle production efficiency was determined by calculating the number of fluorescently-labeled VLPs per total area imaged (μm2), normalized for transfection efficiency and expressed as a percentage of WT (Fig. 7A). Compared to WT Gag, the G2A-Gag mutant led to a 98% reduction of particle production from Jurkat cells and a 78% reduction in particle production from Raji/CD4 cells. Fewer than 0.095 particles/μm2 in Jurkat cells and 0.51 particles/μm2 from Raji/CD4 cells were detected in each sample (Table S2). The relatively low level of particle production observed was likely associated with low nucleofection efficiencies (Table S2).
Figure 7.
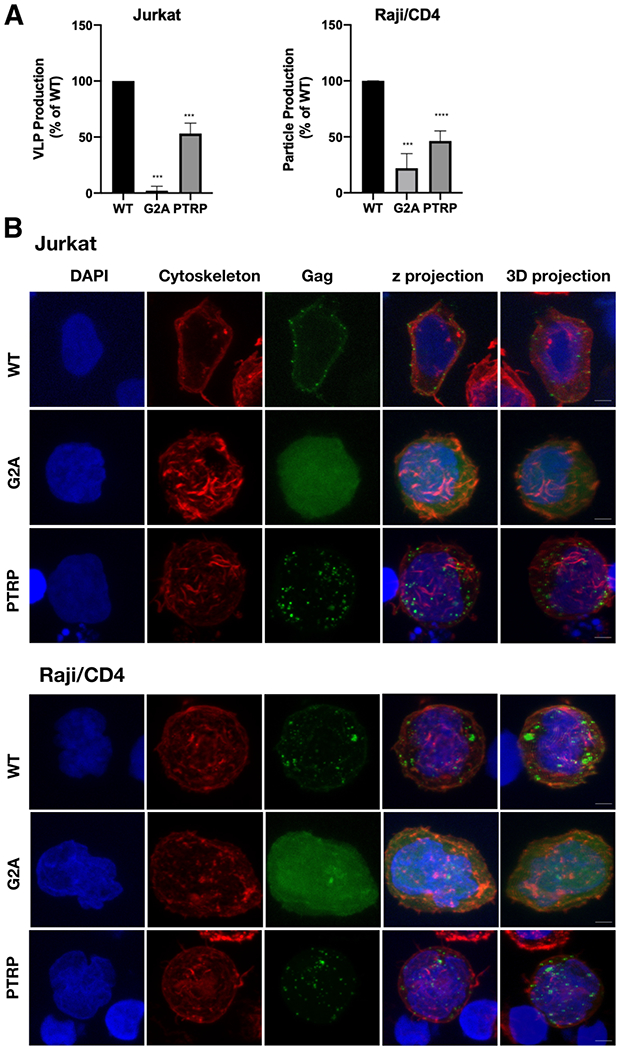
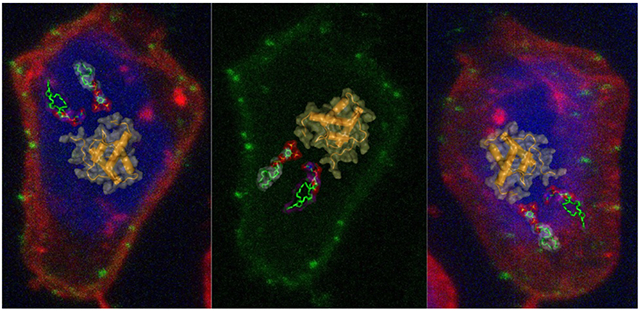
Analysis HTLV-1 Gag particle production and subcellular distribution. (a) Quantification of particle production from three independent biological replicates. Relative particle production of the G2A-Gag and PTRP-Gag mutant is indicated as a percentage of WT Gag. Relative particle production was determined by the number of fluorescently labeled particles per μm2 in concentrated cell culture supernatant detected by confocal microscopy. **** indicates a p value < 0.0001, *** indicates p value = 0.0001 (Student’s unpaired T-test). (b) Gag subcellular distribution. Representative z-projections from three independent biological replicates are shown in which Gag distribution in Jurkat and Raji/CD4 cells was determined for WT Gag, G2A-Gag, and PTRP-Gag. Gag localization was identified by green fluorescence; actin cytoskeleton was identified by red fluorescence; nuclei was identified by blue (DAPI) fluorescence. Scale bar represents 5 μm.
We next performed confocal microscopy analysis to investigate the role of the G2A mutation on Gag-membrane association compared to that of WT. Z-projections of Jurkat and Raji/CD4 cells expressing HTLV-1 WT Gag were found to exhibit punctate Gag fluorescence (Fig. 7B). The punctate Gag fluorescence, particularly at earlier time points post transfection, is indicative of Gag multimerization which has been reported to occur at the membrane for HTLV-1 [62, 78]. Although it is possible that some Gag puncta are localized to internal membranes. In contrast, the diffuse pattern of fluorescence observed with the G2A-Gag mutant was markedly distinct from that of WT in both cell types. These observations indicate that WT Gag puncta are disrupted by the G2A mutation, leading to a diffuse and cytoplasmic localization. The relationship between membrane localization and punctate Gag fluorescence is further supported by the PTRP Gag mutant (PTRP-Gag), which contains a mutation in one of the known late (L) domains encoded in the HTLV-1 MA domain and found to negatively impacts virus particle budding and release [60]. In Jurkat and Raji/CD4 cells, an approximate 2-fold reduction in particle production was observed with the PTRP-Gag compared to WT, similar to that observed previously in other cell types [60] (Fig. 7A). The subcellular localization of the PTRP-Gag is comparable to WT Gag (i.e., punctate fluorescence) (Fig. 7B). These data implicate that the G2A-Gag mutant results in diffuse cytoplasmic expression that reduces Gag-membrane interaction and subsequent particle production. Taken together, these data help support the conclusion that HTLV-1 Gag myristoylation is needed for both Gag localization to the plasma membrane and particle production.
Discussion
Previous studies have shown that binding of HTLV-1 Gag to membranes is less dependent on PI(4,5)P2 than in HIV-1 [31]. Unlike HIV-1 Gag, subcellular localization of HTLV-1 Gag and VLP release were minimally sensitive to overexpression of 5ptaseIV, suggesting that the interaction of MA with PI(4,5)P2 is not essential for HTLV-1 particle assembly [31, 32]. On the other hand, it was found that HTLV-1 Gag binding to liposomes is enhanced upon incorporation of PI(4,5)P2 or PS when negative charge is equivalent to that of PI(4,5)P2 [31]. These results raised the questions whether the MA domain of HTLV-1 Gag contains a PI(4,5)P2 binding site, whether MA binds nonspecifically to PI(4,5)P2 and PS, and/or whether Gag binding to membranes is mediated exclusively by charge-charge interactions. This study was designed to address these questions and to draw a comparison to previously studied retroviral MA proteins. Several important points have emerged from this study: (i) The structure of HTLV-1 myr(−)MA99 revealed a single PI(4,5)P2 binding site formed by lysine and arginine residues. (ii) myr(−)MA binds to soluble analogs of PI(4,5)P2 with substantially higher affinity than previously studied retroviral MA proteins. (iii) The presence of PS in membranes enhances the binding affinity of myr(−)MA to PI(4,5)P2, suggesting that PI(4,5)P2 and PS do not compete for the same binding site and, instead, bind to distinct sites or at least by different mechanisms, enhancing myr(−) binding in a synergistic fashion. (iv) HTLV-1 myr(−)MA has no preference to differentially phosphorylated forms of PIP2 since it bound to PI(3,5)P2 with a similar affinity to that of PI(4,5)P2. (v) The acyl chains of PI(4,5)P2 have minimal role in the overall binding of PI(4,5)P2. (vi) Confocal microscopy data show that Gag is localized to the inner leaflet of the PM, while the G2A-Gag mutant lacking myristoylation is diffuse and cytoplasmic. These findings provided significant insights into the mechanism by which the Gag protein binds to the inner leaflet of the PM and the subsequent production and release of HTLV-1 particles.
HTLV-1 Gag has been shown to bind to membranes with a higher affinity than that of HIV-1 Gag [31]. Single amino acid substitutions that confer a large basic patch rendered HTLV-1 MA susceptible to the RNA-mediated block, suggesting that RNA blocks MA containing a large basic patch [32]. These data supported a model in which HTLV-1 Gag localizes to the PM via the MA domain with higher efficiency but less specificity than for other retroviruses [31, 32]. It was suggested that Gag targeting is mediated by electrostatic interactions with acidic lipids with no specificity to PI(4,5)P2. Here, we show that all tested analogs of PI(4,5)P2 bind to the same site on myr(−)MA, which consists of Arg14, Arg17, Lys47, Lys48, and Lys51. Interestingly, the positioning of the PI(4,5)P2 interacting residues in HTLV-1 myr(−)MA is similar of those observed for the HIV-1, HTLV-2 and ASV MA proteins (Fig. 8A) [41].
Figure 8.
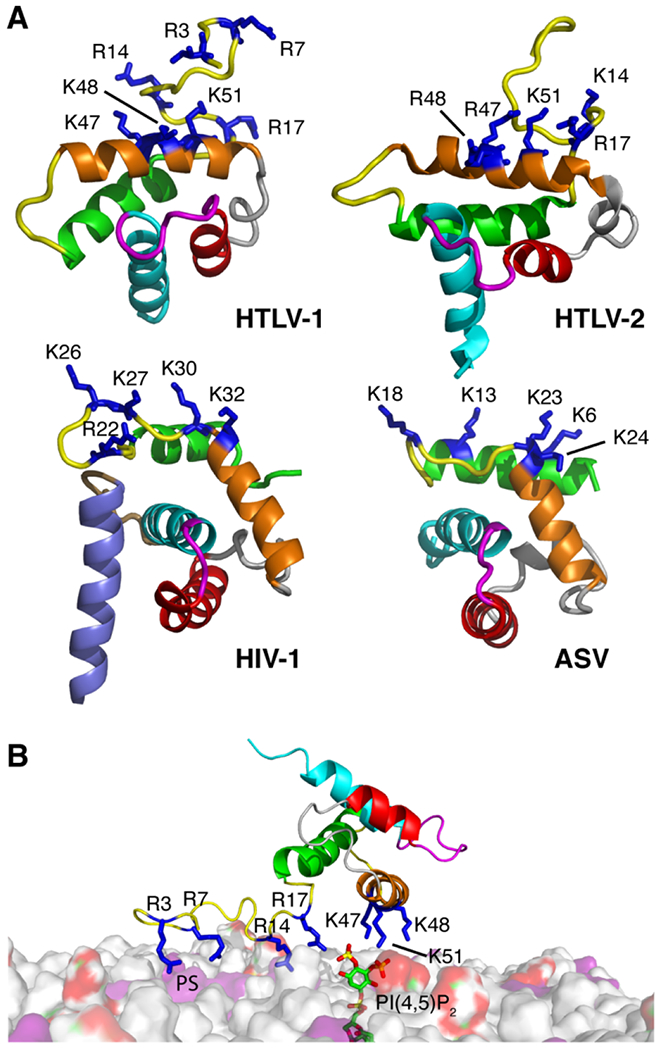
Comparison of membrane interacting residues in various retroviral MA structures and a model of HTLV-1 MA99 bound to membrane. (a) Comparison of the MA structures for HTLV-1, HTLV-2, HIV-1, and ASV (PDB codes 7M1W, 1JVR, 2H3I, and 6CCJ, respectively) highlighting the basic residues implicated in membrane binding (blue sticks). The following residues are not shown for clarity: HTLV-1 and HTLV-2 myr(−)MA99 (residues 1-2 and 94-99), and HIV-1 MA (myr group, residues 2-3 and 115-132). (b) A model of HTLV-1 myr(−)MA99 bound to membrane based on structural data. Favorable electrostatic interactions occur between the acidic polar head of PI(4,5)P2 (red and green surface) or PS (purple) and a basic patch formed by Arg3, Arg7, Arg14, Arg17, Lys47, Lys48, and Lys51 (blue). Membrane bilayer was constructed by CHARMM-GUI [96].
To our surprise, the affinity of HTLV-1 myr(−)MA to soluble analogs of PI(4,5)P2 was > 20-fold higher than that observed for HIV-1 and HIV-2 MA, and ~100-fold tighter than for ASV MA (Table 1). For the latter cases, Gag assembly has been shown to be dependent on PI(4,5)P2 [7, 12, 41]. Tighter binding is likely attributed to the extensive H-bond capabilities of the guanidinium group of arginine [79], supporting a model in which Arg14 and Arg17 may play a significant role in enhancing PI(4,5)P2 binding. The liposome binding assays, however, revealed that the affinity of HTLV-1 myr(−)MA to PI(4,5)P2 is similar to that observed for HIV-1 MA and ASV MA in the context of membrane [41, 48]. It is likely that when not incorporated in membranes, PI(4,5)P2 analogs are better accommodated in the binding site on HTLV-1 MA. Additionally, a higher fraction of myr(−)MA bound to membranes containing PI(4,5)P2 vs. PS membranes of equivalent negative charge (Fig. 6B), indicating a slight preference of myr(−)MA to PI(4,5)P2. However, increasing PS ratio in liposomes resulted in similar levels of protein bound. Altogether, our studies confirmed the presence of PI(4,5)P2 binding site on HTLV-1 MA and demonstrated that binding is governed by electrostatic interactions.
Because previous studies demonstrated that PI(4,5)P2 binding to retroviral MA proteins is independent of the myr group [12, 41, 43, 68], we examined the role of the myr group in HTLV-1 Gag colocalization on the PM and particle production in mammalian cells. The finding that G2A-Gag mutant led to severe reduction of particle production compared to WT Gag (Fig. 7A) indicates that the myr group is required for proper Gag localization even though the myr(−)MA domain is still capable of binding to PI(4,5)P2/PS-containing liposomes. This observation is consistent with previous data that a low level of HTLV-1 particle production is detected with Gag containing the G2A mutation [62], and that efficient membrane binding is requisite for robust particle production [76, 77]. Confocal microscopy analysis of Jurkat and Raji/CD4 cells expressing HTLV-1 WT Gag revealed formation of Gag puncta, particularly at earlier time points post transfection, indicative of Gag multimerization. In contrast, the G2A-Gag mutant had a diffuse pattern of fluorescence (Fig. 7B). This result is also consistent with previous studies which have shown cytosolic localization of G2A-Gag [59, 80]. The phenotype of puncta disassembly and cytosolic localization of HTLV-1 G2A-Gag was observed for other retroviral G2A-Gag proteins including HIV-1 [62, 81, 82]. The relationship between puncta formation and affinity to membranes is not well understood. For example, previous studies indicated that HIV-1 myr(−)MA is able to bind (albeit weaker than the myristoylated protein) to liposomes and membrane bilayer surfaces containing PS and PI(4,5)P2 [28, 48]. On the other hand, we and others have shown that ASV Gag which is naturally unmyristoylated is able to assemble as puncta on the inner leaflet of the PM [32, 40, 42]. ASV MA is able to bind liposomes containing PS and PI(4,5)P2 [41]. The relationship between puncta formation and localization to the inner leaflet of the PM is also supported by decreased PTRP-Gag particle production and punctate Gag fluorescence similar to what is observed with WT Gag. This implies that the observed G2A-Gag diffuse, cytoplasmic localization is related to limited membrane interaction. The 2-fold reduction in particle production for PTRP-Gag was predicted since the second L domain in HTLV-1 MA (i.e., the PPPY motif) was intact [60]. Altogether, these observations suggest that proper Gag assembly on the inner leaflet of the PM may require an affinity threshold mediated by the basic residues, acidic phospholipids and myr group.
The effect of individual basic amino acid substitutions in the HTLV-1 MA protein on cell-to-cell transmission of the virus was previously examined. WT phenotype was only obtained for mutant viruses with mutations of Arg7 and Arg97 [59]. However, point mutations of residues Arg3, Arg14, Arg17, Arg33, Lys47, Lys48, Lys51, Lys74, and Arg79 completely abolished viral infectivity and impacted various steps of the replication cycle, including events following membrane targeting of Gag. It was found that most of the mutations allowed normal synthesis, transport, and cleavage of the Gag precursor, but particle release was greatly affected for seven mutants (R3L, R14L, R17L, K48I, K74I, and R79L) [59]. Interestingly, in situ immunofluorescence analysis of the distribution of the HTLV-1 Gag proteins in transfected cells revealed that the intracellular distribution of Gag with these point mutations was similar to that of the WT protein. Limited membrane binding studies using cell fractionation have shown that Gag-R17L or K48I mutants bound to membranes with a similar affinity to the WT protein, suggesting that these two residues are important for infectivity at various stages of the viral replication cycle but do not play a major role, at least individually, in targeting the Gag precursor to the PM [59]. Our structural data show that the majority of the basic residues are located in the basic patch implicated in PI(4,5)P2 binding (Fig. S9) and that 5-7 basic residues can potentially contribute to membrane binding. Therefore, it is unlikely that substitution of a single basic amino acid may not be detrimental to membrane binding.
Another observation in our study is the finding that the binding affinity of PI(4,5)P2 to HTLV-1 myr(−)MA is not dependent on the presence/lack of or length of acyl chain (Table 1). A very modest increase in affinity (~2-fold) is observed upon increasing the length of the acyl chain by two methylene groups. As revealed by the CSPs (Fig. S7), this slight increase in the affinity is probably caused by interactions between the acyl chains and hydrophobic residues in the unstructured N-terminal region and α-helix I of myr(−)MA. A recent study demonstrated the ability of HIV-1 MA to partially displace the acyl chains from the bilayer membrane via interaction with hydrophobic regions of MA and stabilization of the MA-lattice ([49]). This result is consistent with our previous structural studies that the 2’-acyl chain of PI(4,5)P2 is sequestered in a pre-exisitng cleft in HIV-1 MA [43]. Further studies are needed to investigate whether the interaction of MA with the acyl chain can occur in infected cells and can overcome the energy penalty resulting from displacement of part of the acyl chain from the membrane bilayer, as has been shown for HIV-1 [49].
Another interesting observation in this study is the finding that PI(4,5)P2 does not preferentially bind to myr(−)MA as PI(3,5)P2 is capable of binding with a similar affinity. This result indicates that the position of the phosphate groups (3 vs. 4) is not a key determinant for binding of HTLV-1 myr(−)MA. This result is similar to those obtained for HIV-1 and ASV MA in which the affinity to PI(3,5)P2 was found to be relatively similar to PI(4,5)P2 [41, 48]. It has been suggested that since PI(3,5)P2 abundance in cells is significantly lower than that of PI(4,5)P2 (~100-fold lower [83]), targeting of Gag to the PM results from the high relative concentration of PI(4,5)P2 rather than differences in affinity of MA for these phosphoinositides [48]. This hypothesis is perhaps applicable to HTLV-1 Gag since it appears that the total negative charge on lipids is more important than the positioning of the phosphate groups.
In summary, our data support a model in which HTLV-1 MA binding to membranes is governed by charge-charge interactions and that the affinity of MA binding to membranes is enhanced by acidic lipids such as PI(4,5)P2 and PS (Fig. 8B). Based on the findings that PI(4,5)P2 and PS do not compete for the same binding site, and that Arg14, Arg17, Lys47, Lys48, and Lys51 form a PI(4,5)P2 binding site, it is conceivable that Arg3 and Arg7 interact with PS in a synergistic fashion (Fig. 8B). Because retroviral MA proteins have similar functions and can share similar structural features, we analyzed the membrane-interacting motifs of HTLV-1 to those of HIV-1 and the naturally unmyristoylated ASV MA protein. As shown in Figure 8B, HIV-1 and ASV MA share a similar fold with membrane-interacting regions located on similar regions of the protein (α-helices I and II and the connecting loop). Interestingly, the membrane interacting motif in HTLV-1 MA is formed by residues located on different regions (unstructured N-terminus and helix II). Altogether, our findings provide a structural framework for HTLV-1 Gag binding to the inner leaflet of the PM and advance our understanding of the basic mechanisms of retroviral assembly.
Materials and Methods
Sample Preparation
Plasmid construction.
The MA genes encoding for amino acids 1-130 or 1-99 were generated via PCR using a plasmid containing the full-length HTLV-1 Gag gene as a template [84]. MA genes were inserted into a pET11a vector using standard cloning techniques, yielding a construct that is fused to a His6-tag coding gene on the 3’-end. Plasmid sequencing was performed at the Heflin Genomics Core at the University of Alabama at Birmingham.
Protein expression and purification.
The myr(−)MA and myr(−)MA99 proteins were overexpressed in Escherichia coli BL21 Codon Plus-RIL cells (Agilent Technologies). Cells were grown in LB broth supplemented with 100 mg/L ampicillin at 37 °C. Cells were induced with isopropyl β-d-1-thiogalactoside when the OD600 was ~0.7 and grown at 22 °C overnight. Cells were harvested via centrifugation and stored at −80 °C. Cell pellet was resuspended in lysis buffer (50 mM sodium phosphates, pH 8, 500 mM NaCl, 40 mM imidazole, 1% Triton, and 1 mM phenylmethylsulfonyl fluoride). Cells were lysed via sonication and lysate was spun down at 35,000xg for 30 min. The supernatant was subjected to cobalt affinity chromatography, washed with a buffer containing 50 mM sodium phosphates (pH 8), 500 mM NaCl, and 40 mM imidazole, and eluted with imidazole gradient using a buffer containing 50 mM sodium phosphates (pH 8), 500 mM NaCl, and 300 mM imidazole. Proteins were further purified by cation-exchange [Binding buffer: 50 mM sodium phosphates (pH 7) and 5 mM dithiothreitol (DTT); elution buffer: 50 mM sodium phosphates (pH 7), 700 mM NaCl, and 5 mM DTT], and size-exclusion chromatography [buffer: 50 mM sodium phosphates, pH 7, 100 mM NaCl, and 5 mM DTT]. Protein samples were dialyzed in NMR buffer [20 mM MES (pH 6), 100 mM NaCl, and 2 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP)] or LUV buffer [50 mM sodium phosphates (pH 7.4), and 100 mM NaCl) and concentrated using 3 kDa cut-off centrifugal filter units. Uniformly 15N and 15N-,13C-labeled MA samples were prepared by growing cells in M9 minimal medium containing 15NH4Cl and glucose-13C6. Protein purification was performed as described above.
Gel filtration assay.
The mobility and approximate molecular weight of myr(−)MA and myr(−)MA99 proteins were analyzed by a gel filtration assay. Briefly, 0.5 mL of 100 μM protein samples was loaded on ENrich SEC 70 column (BioRad) in a buffer containing 50 mM phosphates (pH 7.4), 300 mM NaCl and 2mM TCEP. Protein fractions were analyzed by SDS-PAGE and stained by Coomassie brilliant blue. The approximate molecular weights of the loaded proteins were determined by molecular weight calibration kits (GE Healthcare).
Preparation of Large Unilamellar Vesicles (LUVs)
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), porcine brain PI(4,5)P2 (Avanti Polar Lipids), and dipalmitoyl-phosphatidylinositol 3,5-bisphosphate (diC16-PI(3,5)P2) (Echelon Biosciences) were used as received. Lipids were mixed in appropriate ratios and solvent was evaporated under a stream of air, followed by lyophilization. Dried lipids were then resuspended in a buffer containing 50 mM sodium phosphates (pH 7.4) and 100 mM NaCl by repeated brief vortexing and allowed to rehydrate for 45 min at room temperature. Lipid suspension was then passed 30 times through a 100 nm pore filter in an extruder (Avanti Polar Lipids). LUV’s were stored at 4 °C and used within 24h. Final total lipid concentration in LUV stocks was 10 mg/ml.
NMR Spectroscopy
NMR data were collected at 35 °C on a Bruker Avance II (700 MHz 1H) equipped with a cryogenic triple-resonance probe, processed with NMRPIPE [85] and analyzed with NMRVIEW [86] or CCPN Analysis [87]. 13C-, 15N-, or 13C-/15N-labeled protein samples were prepared at ~ 300–500 μM in 50 mM sodium phosphates (pH 6.0), 100 mM NaCl, and 1 mM TCEP. 1H, 13C and 15N resonances were assigned using 1H–15N HSQC, HNCA, HN(CO)CA, HNCACB, HN(CO)CACB, HNCO, 15N-edited HSQC-TOCSY, 15N-edited HSQC-NOESY and (H)CCH-TOCSY experiments. 15N-edited NOESY-HSQC and 13C-edited HMQC-NOESY data were collected with a mixing time of 120 ms.
Lipid NMR titrations
1H-15N HSQC NMR titrations were conducted with 50-100 μM samples of 15N-labeled myr(−)MA99 or myr(−)MA in 20 mM MES (pH 6.0), 2 mM TCEP, and varying NaCl concentrations. Stock solutions of lipids were prepared in water at 10–50 mM. The pH of IP6 stock solution was adjusted to 6.0 by using NaOH prior to titrations. CSPs were calculated as where ∆δH and ∆δN are 1H and 15N chemical shift changes, respectively. Dissociation constants were calculated by non-linear least-square fitting algorithm in gnuplot software (http://www.gnuplot.info) using the equation:
where is chemical shift difference between complex and free protein, [L]0 total concentration of lipid, and [P]0 total concentration of protein.
LUV NMR titration
Individually prepared samples for NMR titration contained 25 or 50 μM myr(−)MA in 50 mM sodium phosphates (pH 7.4), 100 mM NaCl, 2 mM TCEP, 250 or 500 μg LUVs with varying POPS and/or PI(4,5)P2 concentrations, and 5% D2O (vol/vol) in a total volume of 500 μL. 1H NMR spectra with excitation sculpting water suppression were recorded for each sample and integral intensity measured in the region 9.5–8.0 ppm. The amount of protein bound to LUVs was determined as the difference between integrals of samples with and without LUVs. The binding data (averages of 2-3 experiments) were fitted in Matlab 2015b (MathWorks) using Hill equation where θ is fraction of bound protein, [L] concentration of available lipid in LUV, Kd microscopic dissociation constant, and n cooperativity constant.
Isothermal Titration Calorimetry
Thermodynamic parameters of IP3 binding to myr(−)MA were determined using a MicroCal PEAQ-ITC (Malvern Instruments). ITC experiments were conducted in a buffer containing 20 mM MES (pH 6.0) and 2 mM TCEP. IP3 prepared at 400 μM in the same buffer was titrated into 40 μM myr(−)MA. Heat of reaction was measured at 25 °C for 19 injections. Heat of dilution was measured by titrating IP3 into buffer and was subtracted from the heat of binding. Data analysis was performed using PEAQ analysis software. The thermodynamic parameters were determined by fitting baseline-corrected data by a binding model for a single set of identical sites.
Structure Calculations
Structure calculations were performed using Unio’10 software [88] that utilizes Atnos/Candid functionality for automated iterative peak picking of raw NOESY spectra, peak assignments and calibration, in conjunction with CYANA structure calculation engine [89, 90]. Backbone φ and ψ dihedral angle constraints were generated by Unio based on the chemical shifts. Tolerance windows for direct and indirect dimensions were set to 0.04. NOESY spectra were converted to XEASY format using CARA [91]. Structures were visualized in Pymol (Schrodinger, LLC.). Electrostatic potential maps were generated using PDB2PQR and APBS software complied within Pymol [92, 93].
Analysis of HTLV-1 particle production and Gag subcellular distribution
Jurkat (E6-1) cells (NIH HIV Reagent Program) and Raji/CD4 cells, graciously provided by Vineet N. Kewal-Ramani (NCI-Frederick), were grown in RPMI1640 medium (Corning) supplemented with 10% fetal clone III (Cytiva). For cell culture assays, a 3:1 molar ratio of unlabeled HTLV-1 Gag to EYFP-labeled HTLV-1 Gag expression plasmids were co-transfected into cells in order to analyze the release of morphologically-correct fluorescently-labeled VLPs [94]. Three sets of HTLV-1 Gag expression plasmids were used: 1) a codon-optimized pN3 HTLV-1 Gag and pN3 HTLV-1 Gag-EYFP [84]; 2) a derivative containing the G2A mutation (pN3 HTLV-1 G2A-Gag and pN3 HTLV-1 G2A-Gag-EYFP) [62]; or 3) a derivative containing the PTRP mutation (pN3 HTLV-1 PTRP-Gag and pN3 HTLV-1 PTRP-Gag-EYFP). The unlabeled/labeled HTLV-1 Gag plasmids were co-transfected at a molar ratio 10:1 with a HTLV-1 Env expression construct, which was graciously provided by Kathryn Jones and Marie-Christine Dokhelar [95]. Cells were co-transfected with 10 μg total plasmid DNA by nucleofection using the Neon Transfection System (Thermo Fisher Scientific) using protocols provided by the manufacturer. Specifically, 2 million cells were electroporated in 100 μl using 3 pulses at 1,350 V for 10 ms or 1 pulse at 1,350 V for 30 ms for Jurkat and Raji/CD4 cells respectively.
For particle production assays, 16 hours post-transfection, cell culture supernatants were collected, passed through a 0.22 μm filter, and concentrated by centrifugation at 20,000xg for 90 minutes using a F-45–30-11 rotor (Eppendorf). Pellets were resuspended in PBS at a 50-fold reduction of the initial volume of the collected cell culture supernatant. The analysis of fluorescently-labeled VLPs was done by using a Zeiss LSM700 confocal laser scanning microscope with a Plan-Apochromat 100x/1.4 aperture (NA) oil objective.
For Gag subcellular distribution assays, 16 hours post-transfection, cells were fixed with 4% paraformaldehyde and permeabilized. The cytoskeleton was visualized by using the ActinRed 555 ReadyProbes reagent (Life Technologies) and cell nuclei were visualized by using the NucBlue Fixed Cell ReadyProbes reagent (Life Technologies) following the protocols provided by the manufacturer. Images of cells were collected by using a Zeiss LSM700 confocal laser scanning microscope with a Plan-Apochromat 63x/1.4 aperture (NA) oil objective.
Supplementary Material
Highlights.
Structure of HTLV-1 myr(−)MA share a similar globular fold to HTLV-2 myr(−)MA.
HTLV-1 myr(−)MA structure revealed a membrane-interacting basic patch.
HTLV-1 myr(−)MA contains a well-defined PI(4,5)P2 binding site.
Phosphatidylserine and PI(4,5)P2 enhance myr(−) binding in a synergistic fashion.
HTLV-1 Gag’s myristoyl group promotes Gag puncta formation in Gag expressing cells.
Acknowledgments
This work was supported by grants 9 R01 AI150901-10 from the National Institutes of Health (NIH) to JSS, R01 GM098550 (to LMM), T32 AI083196 and F31 AI147805 (to HMH), and T90 DE022732 (to NAW). The High-Field NMR facility at the University of Alabama at Birmingham was established through NIH grant 1S10RR026478 and is currently supported through the comprehensive cancer center (NCI grant P30 CA013148). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Iwen F. Grigsby for creating the plasmids pN3 HTLV-1 PTRP-Gag and pN3 HTLV-1 PTRP-Gag-EYFP.
Abbreviations:
- HTLV-1
human T-cell leukemia virus type 1
- MA
myristoylated matrix
- myr(−)MA
unmyristoylated matrix
- PI(4,5)P2
phosphatidylinositol 4,5-bisphosphate
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single quantum coherence
- CSP
chemical shift perturbation
- ITC
isothermal titration calorimetry
- PI(4,5)P2
phosphatidylinositol 4,5-bisphosphate
- PI(3,5)P2
phosphatidylinositol 3,5-bisphosphate
- IP3
inositol 1,4,5-trisphosphate
- IP4
inositol 1,3,4,5-tetrakisphosphate
- IP6
inositol hexakisphosphate
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine
- LUV
large unilamellar vesicle.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
The atomic coordinates of HTLV-1 myr(−)MA99 (code 7M1W) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The NMR chemical shift data for HTLV-1 myr(−)MA are available from the Biological Magnetic Resonance Data Bank under BMRB accession number 30880.
References
- [1].Finzi A, Orthwein A, Mercier J, Cohen EA. Productive Human Immunodeficiency Virus Type 1 Assembly Takes Place at the Plasma Membrane. J Virol. 2007;81:7476–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gousset K, Ablan SD, Coren LV, Ono A, Soheilian F, Nagashima K, et al. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 2008;4:e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Joshi A, Ablan SD, Soheilian F, Nagashima K, Freed EO. Evidence that productive human immunodeficiency virus type 1 assembly can occur in an intracellular compartment J Virol. 2009;83:5375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jouvenet N, Neil SJD, Bess C, Johnson MC, Virgen CA, Simon SM, et al. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, Kräusslich H-G. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 2007;3:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chukkapalli V, Hogue IB, Boyko V, Hu W-S, Ono A. Interaction between HIV-1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag-membrane binding. J Virol. 2008;82:2405–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A. 2004;101:14889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ghanam RH, Samal AB, Fernandez TF, Saad JS. Role of the HIV-1 matrix protein in Gag intracellular trafficking and targeting to the plasma membrane for virus assembly. Front Microbiol. 2012;3:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vlach J, Saad JS. Structural and molecular determinants of HIV-1 Gag binding to the plasma membrane. Front Microbiol. 2015;6:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hamard-Peron E, Juillard F, Saad JS, Roy C, Roingeard P, Summers MF, et al. Targeting of murine leukemia virus gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J Virol. 2010;84:503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Prchal J, Kroupa T, Ruml T, Hrabal R. Interaction of Mason-Pfizer monkey virus matrix protein with plasma membrane. Front Microbiol. 2014;4:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Saad JS, Ablan SD, Ghanam RH, Kim A, Andrews K, Nagashima K, et al. Structure of the myristylated HIV-2 MA protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J Mol Biol. 2008;382:434–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eichorst JP, Chen Y, Mueller JD, Mansky LM. Distinct Pathway of Human T-Cell Leukemia Virus Type 1 Gag Punctum Biogenesis Provides New Insights into Enveloped Virus Assembly. mBio. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Freed EO. HIV-1 assembly, release and maturation. Nat Rev Microbiol. 2015;13:484–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ganser-Pornillos BK, Yeager M, Sundquist WI. The structural biology of HIV assembly. Curr Opin Struct Biol. 2008;18:203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chukkapalli V, Inlora J, Todd GC, Ono A. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J Virol. 2013;87:7155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chukkapalli V, Ono A. Molecular Determinants that Regulate Plasma Membrane Association of HIV-1 Gag. J Mol Biol. 2011;410:512–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Purohit P, Dupont S, Stevenson M, Green MR. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA. 2001;7:576– 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li H, Dou J, Ding L, Spearman P. Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J Virol. 2007;81:12899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dalton AK, Ako-Adjei D, Murray PS, Murray D, Vogt VM. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J Virol. 2007;81:6434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dick RA, Goh SL, Feigenson GW, Vogt VM. HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc Natl Acad Sci USA. 2012;109:18761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Waheed AA, Freed EO. Lipids and membrane microdomains in HIV-1 replication. Virus Res. 2009;143:162–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chukkapalli V, Oh SJ, Ono A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A. 2010;107:1600–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Waheed AA, Freed EO. The role of lipids in retrovirus replication. Viruses. 2010;2:1146–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ono A HIV-1 assembly at the plasma membrane. Vaccine. 2010;28Suppl 2:B55–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chan J, Dick RA, Vogt VM. Rous Sarcoma Virus Gag Has No Specific Requirement for Phosphatidylinositol-(4,5)-Bisphosphate for Plasma Membrane Association In Vivo or for Liposome Interaction In Vitro. J Virol. 2011;85:10851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Alfadhli A, Still A, Barklis E. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol. 2009;83:12196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Barros M, Heinrich F, Datta SA, Rein A, Karageorgos I, Nanda H, et al. Membrane Binding of HIV-1 Matrix Protein: Dependence on Bilayer Composition and Protein Lipidation. J Virol. 2016;90:4544–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Olety B, Veatch SL, Ono A. Phosphatidylinositol-(4,5)-Bisphosphate Acyl Chains Differentiate Membrane Binding of HIV-1 Gag from That of the Phospholipase Cdelta1 Pleckstrin Homology Domain. J Virol. 2015;89:7861–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gaines CR, Tkacik E, Rivera-Oven A, Somani P, Achimovich A, Alabi T, et al. HIV-1 Matrix Protein Interactions with tRNA: Implications for Membrane Targeting. J Mol Biol. 2018;430:2113– 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Inlora J, Chukkapalli V, Derse D, Ono A. Gag localization and virus-like particle release mediated by the matrix domain of human T-lymphotropic virus type 1 Gag are less dependent on phosphatidylinositol-(4,5)-bisphosphate than those mediated by the matrix domain of HIV-1 Gag. J Virol. 2011;85:3802–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Inlora J, Collins DR, Trubin ME, Chung JY, Ono A. Membrane binding and subcellular localization of retroviral Gag proteins are differentially regulated by MA interactions with phosphatidylinositol-(4,5)-bisphosphate and RNA. mBio. 2014;5:e02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Thornhill D, Olety B, Ono A. Relationships between MA-RNA binding in cells and suppression of HIV-1 Gag mislocalization to intracellular membranes. J Virol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thornhill D, Murakami T, Ono A. Rendezvous at Plasma Membrane: Cellular Lipids and tRNA Set up Sites of HIV-1 Particle Assembly and Incorporation of Host Transmembrane Proteins. Viruses. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hammond GR, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, et al. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science. 2012;337:727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kolay S, Basu U, Raghu P. Control of diverse subcellular processes by a single multi-functional lipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]. Biochem J. 2016;473:1681–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stansell E, Apkarian R, Haubova S, Diehl WE, Tytler EM, Hunter E. Basic residues in the Mason-Pfizer monkey virus gag matrix domain regulate intracellular trafficking and capsid-membrane interactions. J Virol. 2007;81:8977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Prchal J, Srb P, Hunter E, Ruml T, Hrabal R. The Structure of Myristoylated Mason-Pfizer Monkey Virus Matrix Protein and the Role of Phosphatidylinositol-(4,5)-Bisphosphate in Its Membrane Binding. J Mol Biol. 2012;423:427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Brown LA, Cox C, Baptiste J, Summers H, Button R, Bahlow K, et al. NMR structure of the myristylated feline immunodeficiency virus matrix protein. Viruses. 2015;7:2210–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nadaraia-Hoke S, Bann DV, Lochmann TL, Gudleski-O’Regan N, Parent LJ. Alterations in the MA and NC domains modulate phosphoinositide-dependent plasma membrane localization of the Rous sarcoma virus Gag protein. J Virol. 2013;87:3609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vlach J, Eastep GN, Ghanam RH, Watanabe SM, Carter CA, Saad JS. Structural basis for targeting avian sarcoma virus Gag polyprotein to the plasma membrane for virus assembly. J Biol Chem. 2018;293:18828–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Watanabe SM, Medina GN, Eastep GN, Ghanam RH, Vlach J, Saad JS, et al. The matrix domain of the Gag protein from avian sarcoma virus contains a PI(4,5)P2-binding site that targets Gag to the cell periphery. J Biol Chem. 2018;293:18841–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A. 2006;103:11364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vlach J, Saad JS. Trio engagement via plasma membrane phospholipids and the myristoyl moiety governs HIV-1 matrix binding to bilayers. Proc Natl Acad Sci U S A. 2013;110:3525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Murphy RE, Samal AB, Vlach J, Mas V, Prevelige PE, Saad JS. Structural and biophysical characterizations of HIV-1 matrix trimer binding to lipid nanodiscs shed light on virus assembly. J Biol Chem. 2019;294:18600–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Anraku K, Fukuda R, Takamune N, Misumi S, Okamoto Y, Otsuka M, et al. Highly sensitive analysis of the interaction between HIV-1 Gag and phosphoinositide derivatives based on surface plasmon resonance. Biochemistry. 2010;49:5109–16. [DOI] [PubMed] [Google Scholar]
- [47].Charlier L, Louet M, Chaloin L, Fuchs P, Martinez J, Muriaux D, et al. Coarse-Grained Simulations of the HIV-1 Matrix Protein Anchoring: Revisiting Its Assembly on Membrane Domains. Biophys J. 2014;106:577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mercredi PY, Bucca N, Loeliger B, Gaines CR, Mehta M, Bhargava P, et al. Structural and Molecular Determinants of Membrane Binding by the HIV-1 Matrix Protein. J Mol Biol. 2016;428:1637–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Qu K, Ke Z, Zila V, Anders-Össwein M, Glass B, Müller B, et al. Maturation of the matrix and viral membrane of HIV-1. bioRxiv. 2021: 10.1101/2020.09.23.309542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Martinez MP, Al-Saleem J, Green PL. Comparative virology of HTLV-1 and HTLV-2. Retrovirology. 2019;16:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Poiesz BJ, Ruscetti FW, Reitz MS, Kalyanaraman VS, Gallo RC. Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cell leukaemia. Nature. 1981;294:268–71. [DOI] [PubMed] [Google Scholar]
- [52].Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci U S A. 1982;79:2031–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ceccaldi PE, Delebecque F, Prevost MC, Moris A, Abastado JP, Gessain A, et al. DC-SIGN facilitates fusion of dendritic cells with human T-cell leukemia virus type 1-infected cells. J Virol. 2006;80:4771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gross C, Wiesmann V, Millen S, Kalmer M, Wittenberg T, Gettemans J, et al. The Tax-Inducible Actin-Bundling Protein Fascin Is Crucial for Release and Cell-to-Cell Transmission of Human T-Cell Leukemia Virus Type 1 (HTLV-1). PLoS Pathog. 2016;12:e1005916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gillet NA, Melamed A, Bangham CR. High-Throughput Mapping and Clonal Quantification of Retroviral Integration Sites. Methods Mol Biol. 2017;1582:127–41. [DOI] [PubMed] [Google Scholar]
- [56].Watanabe T Adult T-cell leukemia: molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood. 2017;129:1071–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Heidecker G, Lloyd PA, Soheilian F, Nagashima K, Derse D. The role of WWP1-Gag interaction and Gag ubiquitination in assembly and release of human T-cell leukemia virus type 1. J Virol. 2007;81:9769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Le Blanc I, Blot V, Bouchaert I, Salamero J, Goud B, Rosenberg AR, et al. Intracellular distribution of human T-cell leukemia virus type 1 Gag proteins is independent of interaction with intracellular membranes. J Virol. 2002;76:905–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Le Blanc I, Rosenberg AR, Dokhelar MC. Multiple functions for the basic amino acids of the human T-cell leukemia virus type 1 matrix protein in viral transmission. J Virol. 1999;73:1860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang H, Machesky NJ, Mansky LM. Both the PPPY and PTAP motifs are involved in human T-cell leukemia virus type 1 particle release. J Virol. 2004;78:1503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Fogarty KH, Zhang W, Grigsby IF, Johnson JL, Chen Y, Mueller JD, et al. New Insights into HTLV-1 Particle Structure, Assembly, and Gag-Gag Interactions in Living Cells. Viruses. 2011;3:770–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fogarty KH, Berk S, Grigsby IF, Chen Y, Mansky LM, Mueller JD. Interrelationship between cytoplasmic retroviral Gag concentration and Gag-membrane association. J Mol Biol. 2014;426:1611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5:725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Christensen AM, Massiah MA, Turner BG, Sundquist WI, Summers MF. Three-dimensional structure of the HTLV-II matrix protein and comparative analysis of matrix proteins from the different classes of pathogenic human retroviruses. J Mol Biol. 1996;264:1117–31. [DOI] [PubMed] [Google Scholar]
- [66].Janmey PA, Iida K, Yin HL, Stossel TP. Polyphosphoinositide micelles and polyphosphoinositide-containing vesicles dissociate endogenous gelsolin-actin complexes and promote actin assembly from the fast-growing end of actin filaments blocked by gelsolin. J Biol Chem. 1987;262:12228–36. [PubMed] [Google Scholar]
- [67].Moens PD, Bagatolli LA. Profilin binding to sub-micellar concentrations of phosphatidylinositol (4,5) bisphosphate and phosphatidylinositol (3,4,5) trisphosphate. Biochim Biophys Acta. 2007;1768:439–49. [DOI] [PubMed] [Google Scholar]
- [68].Saad JS, Loeliger E, Luncsford P, Liriano M, Tai J, Kim A, et al. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J Mol Biol. 2007;366:574–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ehrlich LS, Fong S, Scarlata S, Zybarth G, Carter C. Partitioning of HIV-1 Gag and Gag-related proteins to membranes. Biochemistry. 1996;35:3933–43. [DOI] [PubMed] [Google Scholar]
- [70].Scarlata S, Ehrlich LS, Carter CA. Membrane-Induced Alterations in HIV-1 Gag and Matrix Protein-Protein Interactions. J Mol Biol. 1998;277:161–9. [DOI] [PubMed] [Google Scholar]
- [71].Wen Y, Dick RA, Feigenson GW, Vogt VM. Effects of Membrane Charge and Order on Membrane Binding of the Retroviral Structural Protein Gag. J Virol. 2016;90:9518–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Doktorova M, Heberle FA, Kingston RL, Khelashvili G, Cuendet MA, Wen Y, et al. Cholesterol Promotes Protein Binding by Affecting Membrane Electrostatics and Solvation Properties. Biophys J. 2017;113:2004–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wen Y, Feigenson GW, Vogt VM, Dick RA. Mechanisms of PI(4,5)P2 Enrichment in HIV-1 Viral Membranes. J Mol Biol. 2020;432:5343–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bodner CR, Dobson CM, Bax A. Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J Mol Biol. 2009;390:775–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ceccon A, D’Onofrio M, Zanzoni S, Longo DL, Aime S, Molinari H, et al. NMR investigation of the equilibrium partitioning of a water-soluble bile salt protein carrier to phospholipid vesicles. Proteins. 2013;81:1776–91. [DOI] [PubMed] [Google Scholar]
- [76].Hayakawa T, Miyazaki T, Misumi Y, Kobayashi M, Fujisawa Y. Myristoylation-dependent membrane targeting and release of the HTLV-I gag precursor, Pr53gag, in yeast. Genes & Development. 1992;119:273–7. [DOI] [PubMed] [Google Scholar]
- [77].Rayne F, Bouamr F, Lalanne J, Mamoun RZ. The NH2-terminal domain of the human T-cell leukemia virus type 1 capsid protein is involved in particle formation. J Virol. 2001;75:5277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Fogarty KH, Chen Y, Grigsby IF, Macdonald PJ, Smith EM, Johnson JL, et al. Characterization of cytoplasmic Gag-gag interactions by dual-color z-scan fluorescence fluctuation spectroscopy. Biophys J. 2011;100:1587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li L, Vorobyov I, Allen TW. The different interactions of lysine and arginine side chains with lipid membranes. J Phys Chem B. 2013;117:11906–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bryant M, Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A. 1990;87:523–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Spearman P, Horton R, Ratner L, Kuli-Zade I. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J Virol. 1997;71:6582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hermida-Matsumoto L, Resh MD. Localization of Human Immunodeficiency virus Type 1 Gag and env at the Plasma Membrane by Confocal Imagine. J Virol. 2000;74:8670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Behnia R, Munro S. Organelle identity and the signposts for membrane traffic. Nature. 2005;438:597–604. [DOI] [PubMed] [Google Scholar]
- [84].Grigsby IF, Zhang W, Johnson JL, Fogarty KH, Chen Y, Rawson JM, et al. Biophysical analysis of HTLV-1 particles reveals novel insights into particle morphology and Gag stochiometry. Retrovirology. 2010;7:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. [DOI] [PubMed] [Google Scholar]
- [86].Johnson BA, Blevins RA. NMRview: a Computer Program for the Visualization and Analysis of NMR Data. J Biomol NMR. 1994;4:603–14. [DOI] [PubMed] [Google Scholar]
- [87].Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–96. [DOI] [PubMed] [Google Scholar]
- [88].Guerry P, Herrmann T. Comprehensive automation for NMR structure determination of proteins. Methods Mol Biol. 2012;831:429–51. [DOI] [PubMed] [Google Scholar]
- [89].Güntert P Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–78. [DOI] [PubMed] [Google Scholar]
- [90].Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for protein structure calculations with a new program, DYANA. J Mol Biol. 1997;273:283–98. [DOI] [PubMed] [Google Scholar]
- [91].Keller RLJ. The Computer Aided Resonance Assignment Tutorial. Goldau, Switzerland: CANTINA Verlag; 2004. [Google Scholar]
- [92].Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Maldonado JO, Angert I, Cao S, Berk S, Zhang W, Mueller JD, et al. Perturbation of Human T-Cell Leukemia Virus Type 1 Particle Morphology by Differential Gag Co-Packaging. Viruses. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Delamarre L, Rosenberg AR, Pique C, Pham D, Dokhelar MC. A novel human T-leukemia virus type 1 cell-to-cell transmission assay permits definition of SU glycoprotein amino acids important for infectivity. J Virol. 1997;71:259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem. 2014;35:1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates of HTLV-1 myr(−)MA99 (code 7M1W) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The NMR chemical shift data for HTLV-1 myr(−)MA are available from the Biological Magnetic Resonance Data Bank under BMRB accession number 30880.