

Abstract
Lipocalin 2 (LCN2) is a pleiotropic molecule that is induced in the central nervous system (CNS) in several acute and chronic pathologies. The acute induction of LCN2 evolved as a beneficial process, aimed at combating bacterial infection through the sequestration of iron from pathogens, while the role of LCN2 during chronic, non-infectious disease remains unclear, and recent studies suggest that LCN2 is neurotoxic. However, whether LCN2 is sufficient to induce behavioral and cognitive alterations remains unclear. In this paper, we sought to address the role of cerebral LCN2 on cognition in both acute and chronic settings. We demonstrate that LCN2 is robustly induced in the CNS during both acute and chronic inflammatory conditions, including LPS-based sepsis and cancer cachexia. In vivo, LPS challenge results in a global induction of LCN2 in the central nervous system, while cancer cachexia results in a distribution specific to the vasculature. Similar to these in vivo observations, in vitro modeling demonstrated that both glia and cerebral endothelium produce and secrete LCN2 when challenged with LPS, while only cerebral endothelium secrete LCN2 when challenged with cancer-conditioned medium. Chronic, but not short-term, cerebral LCN2 exposure resulted in reduced hippocampal neuron staining intensity, an increase in newborn neurons, microglial activation, and increased CNS immune cell infiltration, while gene set analyses suggested these effects were mediated through melanocortin-4 receptor independent mechanisms. RNA sequencing analyses of primary hippocampal neurons revealed a distinct transcriptome associated with prolonged LCN2 exposure, and ontology analysis was suggestive of altered neurite growth and abnormal spatial learning. Indeed, LCN2-treated hippocampal neurons display blunted neurite processes, and mice exposed to prolonged cerebral LCN2 levels experienced a reduction in spatial reference memory as indicated by Y-maze assessment. These findings implicate LCN2 as a pathologic mediator of cognitive decline in the setting of chronic disease.
Keywords: Lipocalin 2, hippocampus, cognitive decline, spatial reference memory, cachexia, sepsis, gliosis
Introduction
Neurocognitive decline is a common feature in patients suffering from chronic inflammatory diseases, including primary neurodegenerative diseases, cancers, and autoimmune manifestations1–3. Since cognitive decline reduces quality of life and ultimate survival, identification of novel mediators and pathways that incur cognitive deterioration is imperative in improving patient outcomes. As the population continues to age, the prevalence of disease-related cognitive decline will continue to rise, further necessitating the development of rational drug targets.
To this end, several studies describe elevated levels of Lipocalin 2 (LCN2) in the central nervous system (CNS) during both acute and chronic insults4–6, yet the literature is discordant concerning the biological role of LCN2 on behavioral and cognitive processes during disease5, 6. Since LCN2 is richly described as an anti-bacterial protein through its siderophilic properties, the acute induction of LCN2 during infectious disease is described as a beneficial process for clearing the pathogen7. However, in the context of chronic, non-infectious inflammatory conditions, the precise role of LCN2 is less clear, and recent evidence suggests LCN2 is neurotoxic8, 9. Given the clear utility of LCN2 in the context of acute disease, yet unclear role in a prolonged disease setting, we sought to address the temporal role of LCN2 on central nervous system (CNS) health and cognitive function.
Herein, utilizing both in vivo and in vitro models, we demonstrate that LCN2 is induced and secreted in the CNS in the context of LPS and cancer cachexia, with a unique neuroanatomical distribution dependent on the inflammatory insult. Mice given chronic intracerebroventricular (ICV) LCN2 displayed a reduction in hippocampal staining of mature neurons, while concurrently demonstrating an increase in newborn dentate gyrus neurons. In addition to hippocampal neuronal alterations, mice receiving ICV LCN2 displayed an increase in hippocampal microglia, as well as an increase in lymphoid and myeloid cells in the velum interpositum (VI) and nearby hippocampal structures. Recent studies demonstrate that LCN2 acts as an agonist at the type 4 melanocortin receptor in the CNS10. However, gene set analyses of hippocampi of LCN2-treated wild type and melanocortin-4 receptor knockout mice (Mc4r-KO) revealed similar terms across genotypes consistent with chemokine induction, activated microglia, and altered neuronal cytoskeletal dynamics, suggesting the observed in vivo effects of LCN2 are mediated through MC4R-independent pathways. We observe a distinct transcriptional profile in primary hippocampal neurons treated with LCN2 that demonstrates an unambiguous temporal effect of LCN2 exposure on the transcriptome, with an associated ontology analyses of altered neurite development and abnormal spatial learning. Indeed, LCN2-treated hippocampal neurons display a global reduction in neurite length, and mice receiving chronic ICV LCN2 demonstrated a reduction in spatial reference memory as indicated by Y-maze assessment. Our results implicate cerebral LCN2 as a pathologic mediator of cognitive decline in the setting of chronic, but not acute, exposure.
Results
LCN2 is robustly upregulated in the central nervous system during chronic and acute disease
While LCN2 is known to be induced in the CNS during inflammatory conditions11, 12, its neuroanatomical distribution and cellular source is widely variable depending on the underlying pathology. Using both chronic (cancer cachexia) and acute (LPS-based sepsis) murine models of neuroinflammation, we identified unique patterns of Lcn2 expression in the CNS. Specifically, Lcn2 is upregulated in the cerebral vasculature during cancer cachexia (Figure 1a), while LPS-sepsis resulted in an expression pattern that is consistent with global expression, including both vasculature and glial expression (Figure 1a). On closer examination, we noticed a particular region of high Lcn2 expression under cachectic conditions immediately ventral to the hippocampal formation, an area termed the velum interpositum (VI) (Figure 1b). By immunohistochemistry (IHC), we confirmed LCN2 staining in the cerebral vasculature during cachexia through its localization to CD31+ endothelial cells (Figure 1c–d; Supplementary figure 1), while we observe both vessel and diffuse parenchymal staining during LPS-based sepsis (Figure 1c–d). Utilizing a neuroinflammation transcript panel, we indeed identified Lcn2 as the most highly expressed gene in the hippocampus of cachectic mice (Figure 1e), while it was previously demonstrated that Lcn2 is the most elevated protein in the CNS after LPS challenge6. Finally, we also detect a large induction of LCN2 in the cerebrospinal fluid (CSF) during both cachexia and LPS (Figure 1f; Sham:tumor, p=0.0339, Sham:LPS, p<0.0001), suggesting that CNS cells not only produce, but also secrete LCN2 during acute and chronic inflammatory conditions.
Figure 1.

Lipocalin 2 is robustly upregulated in the central nervous system during LPS-based sepsis and cancer cachexia. (a) Representative coronal images of In situ hybridization of the brain of sham-operation controls, cancer cachexia, and LPS treated mice. (b) Lcn2 In-situ hybridization of the velum interpositum in sham operation controls, cancer cachexia, and LPS treated mice. (c) Representative immunohistochemistry images staining for CD31+ blood vessels (teal) and LCN2 (magenta), and (d) Iba1+ microglia (teal), GFAP+ astrocytes (yellow), and LCN2 (magenta) in sham operation controls, cancer cachexia, and LPS treated mice. (e) Volcano plot of inflammation-related transcript expression in the hippocampus of cancer cachexia mice compared to sham-operation controls using Nanostring™ neuroinflammation gene panel (n = 3 per group). (f) Terminal CSF levels in sham operation controls (n = 5), cachectic (n = 7) or LPS-treated (n = 5) mice. In situ images were taken using 20x magnification, while IHC images were taken using 10x or 40x (inset images). Data in (f) are expressed as mean ± SEM. Data represented in (e) were analyzed with one-way ANOVA with Bonferroni multiple comparisons. *p ≤ 0.05 and ****p ≤ 0.0001. LPS = lipopolysaccharide.
Soluble tumor factors induce LCN2 production and secretion in brain endothelial cells, while LPS results in both endothelial and glial expression patterns
Since we observed a robust induction of LCN2 in the brain in the context of both acute and chronic inflammatory disease, we sought to model LCN2 expression and secretion patterns in response to the respective in vitro inflammatory stimuli (pancreatic cancer or LPS). Specifically, we treated bEnd.3 brain endothelial cells and primary mixed glia with either cancer conditioned medium or LPS and performed an array of molecular assays to determine the expression, production, and secretion profiles of LCN2 in response to these inflammatory challenges (Figure 2a). As observed in vivo, we observe a specific amplification of Lcn2 in cultured brain endothelial cells, but not glia, in the context of cancer challenge by conditioned medium (CM; Figure 2b). Protein production and secretion patterns follow this RNA expression profile closely, as endothelium, but not glia, robustly produce and secrete LCN2 in response to CM (Figure 2c–e). Conversely, we observed a significant upregulation of Lcn2 in both endothelium and mixed glia in response to LPS challenge (Figure 2b), with a concurrent increase in LCN2 production and secretion (Figure 2c–e). These data confirm the unique expression and production pattern of LCN2 in the brain of mice under cachectic and LPS-treated conditions and suggests LCN2 may serve a distinct biological role amongst these conditions.
Figure 2.

Soluble tumor factors and LPS challenge results in unique LCN2 expression, production, and secretion profiles in brain endothelium and glia in vitro. (a) Experimental design for b-e. (b) Lcn2 gene expression in bEnd.3 endothelial cells and primary mixed glia after treatment with control media, cancer conditioned media or LPS (n = 3 biological replicates; bEnd.3 CM vs Control, p=0.0269). (c) LCN2 ELISA of bEnd.3 endothelial cells and primary mixed glia after treatment with control media, cancer conditioned media or LPS (n = 3 biological replicates). Representative immunohistochemistry images of (d) bEnd.3 cells and (e) mixed glia after cancer conditioned media or LPS challenge (20x objective). [LPS] = 10 ng/mL. Data in b-c are expressed as mean ± SEM. Data represented in b-c were analyzed with one-way ANOVA with Bonferroni multiple comparisons per cell type. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001, ****p ≤ 0.0001. b.End3 = Brain endothelium 3 cell line; CM = conditioned medium; LPS = lipopolysaccharide.
Genetic deletion of LCN2 improves sickness behaviors in chronic, but not acute, inflammatory disease
Since we observe a large induction of LCN2 in the CNS of both cancer cachexia and LPS-injected mice, and LCN2 is implicated as a neuropathologic agent, we sought to address whether genetic deletion of LCN2 broadly influences illness or cognitive behaviors during these acute and chronic inflammatory insults. To this end, we utilized home-cage nest building as a measure of overall well-being and cognitive function, as nest building is known to be impaired in various mouse models of illness, infection, and brain injury (including lesions to the hippocampus)13–16. We observed an improvement in nest building behaviors of Lcn2-KO mice under cancer cachectic conditions, but no such improvement in the context of LPS injection (Figure 3a–c; Supplementary figure 2a). While LCN2 may play a role in pancreatic cancer tumorigenesis, we did not observe a difference in tumor burden in this cohort nor previous studies17. We previously reported the improved nutritional status of Lcn2-KO mice during cancer cachexia17, but whether Lcn2 deletion improves nutrition after LPS challenge is not well described. We found that Lcn2 deletion did not improve illness-induced anorexia or loss of body mass and showed no effect on mortality (Supplementary Figure 2b–d). Consistent with these results, we previously reported that ICV injection of LCN2 did not influence food intake or body mass until after 4 days of exposure17. Since LCN2 was recently reported to be elevated after meal intake in fasted mice10, this presented a conundrum in the current context, as chronic and repeated elevations in LCN2, but not acute exposure, would be expected to induce neurotoxicity as demonstrated below. We ultimately observed results that are discordant with the prior report, as we found that refeeding after a fast did not result in an increase in LCN2 release (Supplementary figure 2e–f). With these data demonstrating Lcn2 deletion improved illness behaviors in the chronic inflammatory setting of cachexia but was dispensable during LPS challenge, we hypothesized LCN2 acts in a temporal fashion in the CNS on neurons regulating cognitive function.
Figure 3.

Genetic deletion of LCN2 improves nest building during cancer cachexia, but not LPS challenge. (a) Experimental design for pancreatic tumor and LPS studies. (b) Nest building scores in male Lcn2-KO and WT mice after sham operation or tumor implantation (Average of 2 consecutive scoring days; n = 6–12 per group; WT sham vs. tumor, p=0.0267). (c) Nest building scores in Lcn2-KO and WT mice 24 hours after LPS treatment (n = 5–7 per group; Lcn2-KO vehicle vs LPS, p=0.0259). Data in b-c are expressed as mean ± SEM. Data represented in b-c were analyzed with two-way ANOVA with Bonferroni multiple comparisons per cell type. LPS dose was 1 mg/kg. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001, ****p ≤ 0.0001. LPS = lipopolysaccharide; IP = intraperitoneal.
Chronic cerebral exposure to LCN2 alters hippocampal neuron composition
While we observed differences in nest building amongst Lcn2-KO and WT mice under cachectic conditions, it is difficult to disentangle the direct effects of LCN2 on cognitive outcomes during cancer cachexia since the robust inflammatory processes incurred during cachexia, including fatigue, are known to confound the results of advanced cognitive testing18. For this reason, we sought to explore the individual effects of cerebral LCN2 in the CNS by using an intracerebroventricular (ICV) treatment paradigm (Figure 4a). We also chose to focus on hippocampal alterations in these studies, since we observed a particularly large induction of Lcn2 immediately ventral to the dentate gyrus of the hippocampus in vivo (Figure 1a–b; Figure 4b). We observed that mice receiving high levels of ICV LCN2 for 10 days, but not 4 days, displayed a significant reduction in NeuN positive neuron staining intensity in the dentate gyrus (Figure 4c–e). Specifically, we administered 40 ng of LCN2 (2 ul injections) to achieve a similar degree of concentration-time exposure as cachectic mice (Figure 1f). Utilizing the Pomc-EGFP transgenic mouse, in which newborn neurons in the dentate gyrus express EGFP by the pro-opiomelanocortin (Pomc) promoter, we observed an increase in newborn neurons after 10 days, but not 4 days, of LCN2 treatment (Figure 4f–h)19. These data demonstrate that LCN2 acts in a temporal fashion to alter hippocampal neuron dynamics.
Figure 4.

Prolonged cerebral exposure to LCN2 results in reduced hippocampal density of mature neurons, but an increase in newborn neurons. (a) Experimental design of ICV LCN2 experiment in male mice. (b) Cartoon coronal section of the hippocampus and dentate gyrus, highlighting the quantified regions in d, e, g, h; the three red boxes on the dentate gyrus are the areas imaged on every slice at 40X for quantification analysis. (c) Representative images of NeuN+ neurons in the dentate gyrus of mice given ICV vehicle or LCN2. (d-e; 10-day Vehicle vs LCN2, p=0.0440) Quantification of NeuN+ neuronal staining intensity in selected regions of the dentate gyrus (n = 3–4 images per region of interest; n = 4–5 mice per group). (f) Representative images of Pomc-EGFP neurons in the dentate gyrus of mice given ICV vehicle or LCN2. (g-h; 10-day Vehicle vs LCN2, p=0.0409) Quantification of Pomc-EGFP neuron density in selected regions of the dentate gyrus (n = 3–4 images per region of interest; n = 3–4 mice per group). Data in d-e, g-h are expressed as mean ± SEM. Data represented in d-e, g-h were analyzed by two-tailed Students t-tests. Scale bar = 50 μm. *p ≤ 0.05. ICV = intracerebroventricular; ROI = region of interest.
Chronic LCN2 exposure induces hippocampal microgliosis
After observing changes in neuronal density and populations in the hippocampus, we sought to determine if other cell types in the hippocampus were influenced by LCN2 administration. Examining the same regions analyzed for neuronal alterations, we observed no change in microglial and astrocyte number or morphology after 4 days of cerebral LCN2 treatment (Figure 5a–e). After 10 days of cerebral LCN2 treatment, we observed an increase in microglial number and morphology score compared to vehicle only controls (Figure 5a–e)20. We next isolated primary microglia and treated them directly with LCN2 in vitro to determine if LCN2 is directly influencing microglial polarization (Figure 5f). Microglia treated with LCN2 did not display a significant change of expression in inflammatory-related genes, including Il6, Tnf-α, Nos2, and Arg1, but demonstrated a significant upregulation of several immune cell recruitment genes, including Ccl2 (p=0.0028), Ccl3 (p=0.0162), Cxcl1 (p=0.0105), Cxcl2 (p=0.0002), and Cxcl10 (p=0.0331) (Figure 5g–h). These data suggest that LCN2 mediates a distinct microglial polarization phenotype in vivo, characterized by increased microglial number and expression of immune cell recruitment genes, but does not impact expression of inflammation-related genes.
Figure 5.

Chronic cerebral LCN2 exposure results in an increase in hippocampal microglia and expression of chemokines. (a) Representative images of microglia (Iba1+) and astrocytes (GFAP+) around the dentate gyrus after 4 or 10 days of ICV vehicle or LCN2 treatment (40x magnification). (b) Quantification and (c) morphologic scoring of GFAP+ cells around the dentate gyrus after 4 and 10 days of ICV vehicle or LCN2 treatment. (d) Quantification and (e) morphologic scoring of Iba1+ cells around the dentate gyrus after 4 and 10 days of ICV vehicle or LCN2 treatment. (n = 4–5 per group; in d: 10 day vehicle vs LCN2, p=0.0027; in e: 10 day vehicle vs LCN2, p=0.0270). (f) Experimental design of primary microglia cultures treated with LCN2 for g-h. (g) Inflammatory transcriptional profile of primary microglia cultures treated with vehicle or LCN2 (100 ng/mL). (h) Immune cell recruitment transcriptional profile of primary microglia cultures treated with vehicle or LCN2 (100 ng/mL). n = 3 biological replicates per condition for g-h. Data in b-e, g-h are expressed as mean ± SEM. Data represented in b-e and g-h were analyzed by two-tailed Students t-tests amongst individual timepoints. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. ROI = region of interest. RQ = relative quantity.
LCN2 is a chemoattractant for immune cells in the CNS
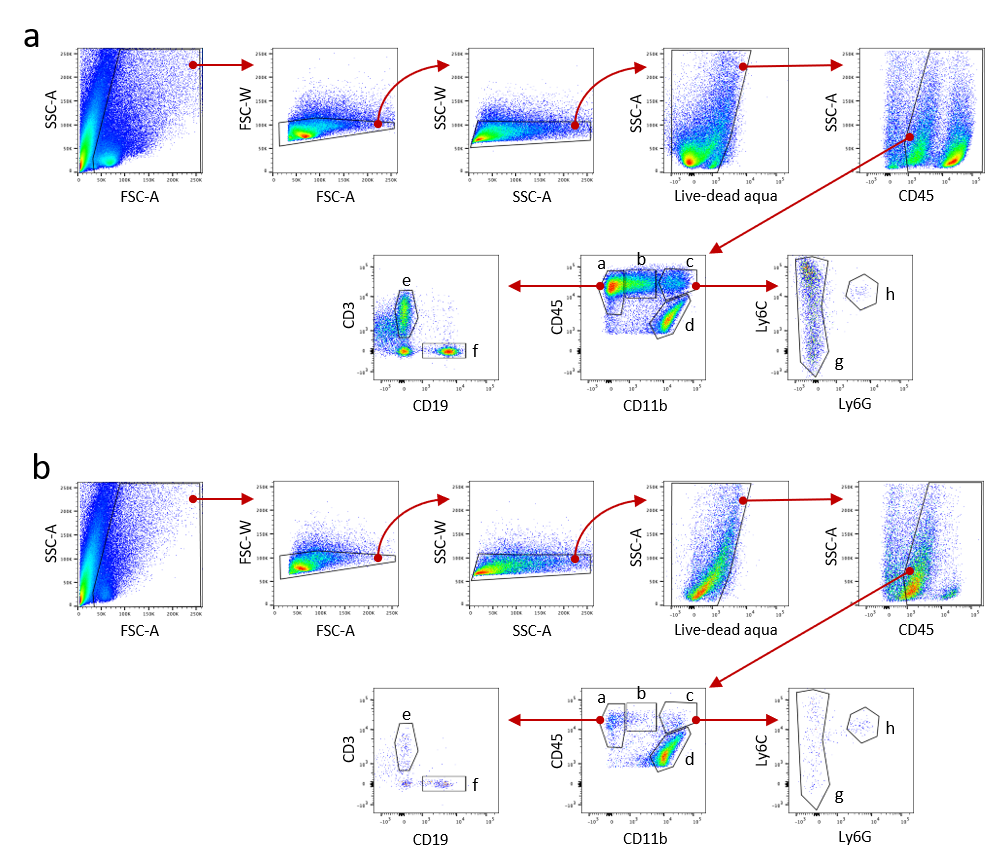
Given the consistent upregulation of immune cell recruitment genes in microglia treated with LCN2, we hypothesized that in addition to the observed neuronal and microglial changes, animals treated with cerebral LCN2 would demonstrate a concomitant increase in CNS immune cells. Indeed, we observed a modest increase in CD45+ cells in the hippocampus after 4 days of ICV LCN2 treatment, with a robust elevation after 10 days of treatment (Figure 6a,c,e,g,i; p=0.0038). Notably, we observed a non-significant increase in myeloperoxidase-positive neutrophils after 4 days of LCN2 treatment, but this increase disappeared after 10 days of treatment. (Figure 6b,d,f,h,j). To determine which immune cell subtypes of CD45+ leukocytes were infiltrating the CNS in the context of elevated LCN2, we performed flow cytometry on brain samples from animals treated with either LCN2 or vehicle for 10 days. Consistent with our histological analyses, we observed a robust increase of CD45+ cells in the brain of mice treated with LCN2 (Figure 6k [p=0.0209]; Supplementary figure 3). Furthermore, LCN2-treated mice displayed a significant increase in both lymphoid and myeloid immune cell populations (Figure 6l,o; p=0.0283 and p=0.0181, respectively). Amongst the lymphocyte population, we observed a significant increase in CD3+ T-cells (p=0.0074), and modest elevation of CD19+ B cells (Figure 6m–n), while CD11b-mid (p=0.0173) and non-neutrophil myeloid cells (p=0.0157) were both elevated amongst the myeloid cell subpopulations (Figure 6o–p). Consistent with our histological observations in the hippocampus, neutrophil count was unchanged between LCN2 and vehicle treated mice by flow cytometry (Figure 6r).
Figure 6.

Cerebral LCN2 results in the recruitment of immune cells to the CNS. (a-h) Representative confocal images of the dentate gyrus and velum interpositum (VI) in mice treated with ICV vehicle or LCN2 after 4 or 10 days. 10x magnification; scale bar = 300μm. (i-j) Imaging and quantification of CD45+ and MPO+ cells in mice treated with either ICV vehicle or LCN2 after 4 or 10 days (n = 3–4 per group). (k-r) Flow cytometry analysis of immune cell populations using hemi-brains of mice treated with ICV vehicle or LCN2 after 4 or 10 days (n = 4 per group). Data in i-r are expressed as mean ± SEM. Data represented in i-r were analyzed by two-tailed Students t-tests amongst treatment groups. *p ≤ 0.05, **p ≤ 0.01. MPO = myeloperoxidase.
Primary hippocampal neurons differentially respond to LCN2 in a time-dependent manner
Previous literature suggests LCN2 acts in a time-dependent fashion in mediating neuronal apoptosis8, 21. However, an in-depth examination of neuronal gene expression in response to LCN2 is outstanding. Since we observe a progressive response of hippocampal neurons to LCN2 in vivo, we performed RNA sequencing of primary hippocampal neurons treated with LCN2 or vehicle for either 1 or 4 days. After 1 day of LCN2 treatment, neurons displayed an upregulation of several genes involved in transcriptional regulation and RNA splicing, including Pcsk1n, Basp1, and Dact3 (Figure 7a). Furthermore, ontology analysis did not reveal a significant association between 1 day of LCN2 treatment and cellular viability (Supplemental tables 1). These results are in contrast with the 4 day LCN2 treatment condition, as Hap1 (huntington-associated protein-1) and Hpcal4 (hippocalcin-like protein 4; also known as VILIP-2) are amongst the most up-regulated genes, and are known mediators of neurite development, calcium channel function, and synaptic release of neurotransmitter (Figure 7b)22–25. The third most up-regulated gene, Vgf, is highly upregulated after peripheral nerve injury and is suggested to be secreted by damaged neurons to activate nearby microglia (Figure 7b; Supplementary table 2)26, 27. Gene ontology analysis of biological and cellular processes demonstrated a significant association between multiple processes of neuron morphology, axogenesis, and cytoskeletal rearrangement in genes up-regulated in the 4-day treatment condition (Figure 7c; Supplementary table 3). Indeed, LCN2-treated primary hippocampal neurons demonstrated a decrease in total and relative neurite length after 4 days of treatment (Figure 7 d–g). Consistent with previous reports of LCN2’s apoptotic effects on neurons, several terms of programmed cell death were also significant, although both positive and negative regulation pathways were revealed (Supplementary table 3)8. Additionally, phenotypic ontology analysis of up-regulated genes in the 4-day treatment condition revealed a significant association with impaired synaptic plasticity, abnormal spatial learning, and hyperactivity (Figure 7c; Supplementary table 4). These results demonstrate a strong temporal effect of LCN2 on hippocampal neurons, with ontology analyses suggesting long-term exposure results in cytoskeletal remodeling (both axons and dendrites) and neuronal dysfunction.
Figure 7.

Primary hippocampal neurons respond to LCN2 in a temporal fashion, and prolonged exposure results in reduced neurite length. Differential gene expression assessed by RNA-seq in primary hippocampal neurons treated with LCN2 (100 ng/mL) or vehicle for either (a) 1 or (b) 4 days. (c) Gene ontology analysis performed on genes with increased expression after 4 days of LCN2 treatment. (d) Total neurite length, (e) number of neurons, and (f) average neurite length per neuron in a single field of view (FOV) of primary hippocampal neurons treated with 4 days of LCN2 (100 ng/mL) or vehicle (For f, p=0.0402). (g) Representative 10x field of view for neurons and neurites quantified in d-f (n = 26–27 fields per condition). Scale bar = 300μm.
LCN2-induced activation of hippocampal microglia and cell stress signatures is independent of Melanocortin 4 receptor signaling
Since LCN2 has three known receptors, including SLC22A17, megalin, and the type 4 melanocortin receptor (MC4R), we sought to address whether the observed neuronal, microglial, and immune cell alterations are mediated through the MC4R using gene pathway analysis. We chose to focus on MC4R signaling due to its known role in regulating hippocampal synaptic plasticity28, 29, but also because both the SLC22A17 and megalin knockout mice are embryonic lethal30. Utilizing the same in vivo treatment paradigm as in our previous experiments, we administered central LCN2 or vehicle control to both WT and Mc4r-KO mice and isolated hippocampi for transcript analyses. Utilizing the Nanostring Neuropathology gene panel, we observed similar gene induction and pathways between WT and Mc4r-KO after LCN2 treatment. Specifically, both WT and Mc4r-KO mice exhibited a significant upregulation of several chemokines in the hippocampi, including Cxcl10, Ccl5, and Ccl12, as well as the super-oxide generating enzyme Cybb (Figure 8a, c; Supplementary tables 5–6). We performed gene set analyses (GSA) and observed several overlapping pathways amongst WT and Mc4r-KO mice, including GSA terms of “cytokines” and “activated microglia” being the top two terms in both genotypes (Figure 8b, d; Supplementary tables 7–8). Consistent with our in vitro observations of altered hippocampal neurite outgrowth after LCN2 treatment, GSA terms of “neuronal cytoskeletal” was in the top 12 GSA terms in both genotypes, while Mc4r-KO mice also included “axon and dendrite structure” (Figure 8b, d). Finally, both genotypes showed “apoptosis” and “oxidative stress” terms. These results are broadly in line with our in vivo histologic analyses, including observations of reduced hippocampal neuron density, activated microglia, and immune cell infiltration, as well as our RNA-sequencing analyses of primary hippocampal neurons (ontology analyses including several terms related to neuronal cytoskeletal rearrangement), ultimately suggestive of these hippocampal molecular and cellular changes occur through Mc4r independent mechanisms.
Figure 8.

Chronic cerebral exposure to LCN2 results in similar, but not identical, gene set analyses in wild type and Mc4r-KO male mice. Volcano plot of neuropathology-related transcript expression in the hippocampus of LCN2-treated mice compared to vehicle-treated controls in (a) wild type or (c) Mc4r-KO mice using Nanostring™ Neuropathology gene panel. Concomitant gene set analyses of the top 12 regulated pathways in (b) wild type or (d) Mc4r-KO mice (n = 5–6 per group).
Prolonged cerebral LCN2 exposure results in impaired spatial recognition memory
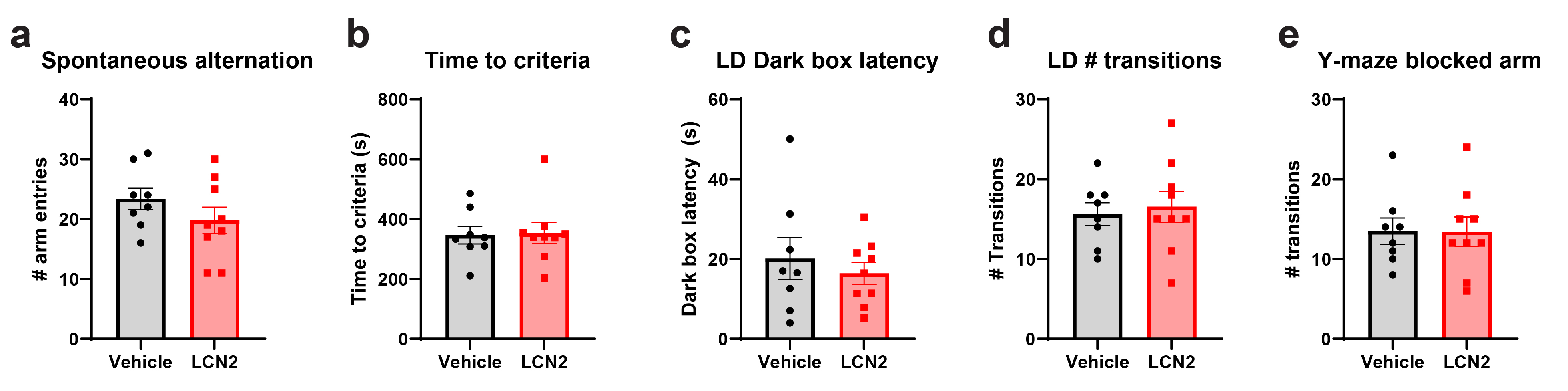
To determine whether cerebral LCN2 mediates alterations in cognitive function, we performed a battery of behavioral and cognitive assessments in mice treated with central LCN2. Specifically, we utilized an assessment paradigm that includes tests that present little to no intrinsic stress (except the final test of tail suspension), yet encompass several domains of cognitive and neuropsychiatric health31–33. To study working memory, we utilized spontaneous alternation in the Y-maze and observed no effect of cerebral LCN2 treatment on percentage of alternation (Figure 9a; Supplementary figure 4a). Similarly, LCN2 treated mice displayed no change in visual object recognition memory or anxiety-like behavior as indicated by the object recognition test and light-dark box test, respectively (Figure 9b–c; Supplementary figure 4b–d). We next utilized the two-trial Y-maze assessment to determine exploratory behavior of a novel arm after a short inter-trial interval (ITI). Specifically, mice were allowed to explore a Y-maze with a single arm blocked off, and after a short ITI, mice were re-introduced to the maze with the previously blocked arm open for exploration. Mice treated with central LCN2 showed impaired spatial memory upon this test, spending significantly less time in the previously-blocked arm as vehicle treated controls (Figure 9d; Supplementary figure 4e). Finally, we assessed depressive-like behavior by the tail suspension test and observed no difference between LCN2 and vehicle treated mice (Figure 9e). These data collectively suggest that increased cerebral LCN2 does not influence anxiety and depressive-like behaviors, working memory, or object recognition memory, but negatively impacts spatial recognition memory, a cognitive task largely attributable to hippocampal-dependent processes32.
Figure 9.

Chronic cerebral exposure to LCN2 results in impaired spatial recognition memory. (a) Y-maze spontaneous alternation test reporting percentage of consecutive arm entry alternations (b) Novel object recognition test for recognition memory showing ratio of time spent with novel object to familiar object. (c) Time spent in the light compartment on light-dark box anxiety test. (d) Y-maze blocked arm test reporting % of time spent in previously blocked arm after a short inter-trial intermission (p=0.0112). (e) Tail suspension test reporting time spent immobile over a 6-minute interval. n = 8–9 per group. *p ≤ 0.05.
Discussion
The purpose of this work was to evaluate the role of LCN2 in the CNS, specifically as it relates to cognitive outcomes after prolonged exposure. Herein, we describe a pathologic role of LCN2 in the CNS, in which LCN2 mediates distinct neuronal, glial, and immune cell alterations in a time-dependent and MC4R-independent fashion, ultimately resulting in cognitive impairment specific to spatial memory. Our findings implicate LCN2 as a pathologic molecule in the CNS during chronic inflammatory disease through its actions on the hippocampus, while the transient induction of LCN2 in the CNS during acute inflammation is generally tolerable (Figure 10).
Figure 10.

Graphical summary of the proposed role of LCN2 in disease-associated neurocognitive decline. During acute inflammatory disease, LCN2 is robustly induced by several CNS cells, including endothelium and glia, and protects the brain from invading pathogens. Acute exposure of LCN2 in the brain is not harmful to neurons. Conversely, during prolonged inflammatory disease, LCN2 is selectively produced and secreted by brain endothelium, particularly in pial vessels proximal to the hippocampus. This continued production and secretion of LCN2 during chronic disease results in a distinct microglial polarization phenotype, immune cell invasion, and prolonged neuronal stress, ultimately leading to neuronal dysfunction and impaired spatial memory.
Using both in vitro and in vivo models, we observe a unique Lcn2 expression and production profile in the CNS dependent on the inflammatory insult. Specifically, the acute inflammatory model of LPS-based sepsis results in a large global induction of LCN2 in the CNS, while the chronic inflammatory model of cancer cachexia results in a specific induction in cerebral endothelium. We observe a particularly robust expression of Lcn2 in the velum interpositum (VI), a double-layered invagination of the pia matter immediately ventral to the hippocampus. Since both acute and chronic inflammatory conditions result in a large induction of LCN2 in the CNS, particularly in an area directly adjacent to the hippocampus, and LCN2 is implicated in the regulation of neuronal health and cognition6, 34, we sought to determine if genetic deletion of LCN2 influences illness behaviors during LPS or cancer challenge. We observed improved nest building in Lcn2-KO mice during cancer cachexia, but not LPS challenge, which led us to hypothesize that LCN2 acts in a temporal fashion in the CNS, with the acute exposure of LCN2 being less impactful on CNS processes than prolonged exposure. To formally test this hypothesis, we administered LCN2 to the CNS of mice for either 4 or 10 days, and observed a progressive decrease in hippocampal neuron density, increased number of hippocampal microglia, and a robust infiltration of leukocytes. Since LCN2 has three receptors (MC4R, SLC22A17, and megalin) all known to be expressed by hippocampal neurons, we performed gene set analyses of the hippocampi of WT and Mc4r-KO mice. These results revealed similar activated pathways across genotypes, suggesting the observed changed in hippocampal cellular dynamics are mediated through MC4R-independent mechanisms. RNA sequencing analysis of primary hippocampal neurons were consistent with the time-dependent action of LCN2 in the CNS observed in vivo, as neurons treated chronically demonstrated distinct biological, cellular, and phenotypic ontologies compared to short-term LCN2 treated neurons. Given these distinct alterations in hippocampal neuron, glia, and immune cell profiles in mice treated chronically with LCN2, along with ontology analyses suggesting prolonged exposure of LCN2 is associated with impaired synaptic plasticity and abnormal spatial learning, we performed behavioral and cognitive testing of mice after chronic central treatment with LCN2. Indeed, mice treated with LCN2 displayed an impairment in spatial memory, a cognitive process principally attributed to the hippocampus32, 35. Our data also demonstrate that LCN2 is not induced with meal intake which would be expected for a molecule with neurotoxic potential. These data are in contrast with previous reports, as we find a small but significant decrease in circulating LCN2 after refeeding10. It is currently unclear why these results are discordant, but our data suggest this molecule is a more robust mediator of appetite under pathological, rather than physiological, conditions.
LCN2 is classically described as a neutrophil-derived protein with an evolutionarily conserved role in sequestering iron containing siderophores during infection, thereby limiting the proliferative capacity of siderophilic pathogens7, 36. As such, it is generally accepted that the acute induction of LCN2 evolved as a beneficial process, aimed at combating infection and maintaining tissue homeostasis in the short term, as the protein is rapidly cleared after resolution of the infection37. Indeed, the acute induction of LCN2 in the CNS is proposed to be beneficial to the host6, although conflicting reports exist that suggest LCN2 is dispensable during LPS challenge5. In either case, it is clear that the central induction of LCN2 during acute inflammatory challenge is not pathological, and our results are consistent with these findings in that the cellular composition in the CNS and evolutionarily-conserved behaviors—specifically nest building—are broadly unaffected by short-term LCN2 exposure. In addition to infection, LCN2 is also upregulated in the brain during numerous non-infectious chronic diseases, during which the CNS would maintain continued exposure to high levels of LCN238–40. Recent work by Llorens et al demonstrate a robust association between CSF LCN2 levels and performance on clinical cognitive tests in patients with cerebrovascular disease, showing elevated CSF LCN2 levels were associated with poorer performance on the Mini Mental Status Examination4. Similar reports exist in examining peripheral LCN2 levels, such that increasing plasma LCN2 levels portends poorer neurocognitive testing performance41. Despite the described role of LCN2 in regulating cell viability in a temporal manner8, along with reports of elevated LCN2 in the CNS of patients with cognitive impairment4, whether or not LCN2 individually influences cognition has not been comprehensively examined. To our knowledge, this is the first report of the individual effects of LCN2 in the CNS, in which we observe a time-dependent effect of LCN2 on hippocampal cellular composition and cognition.
The cellular source of LCN2 in the brain is also disparately reported in the literature, ranging from astrocytes, microglia, endothelium, and neurons depending on underlying pathologic condition8, 42–46. Furthermore, the cell-specific expression profile of LCN2 may depend on cellular polarization, as microglial expression of Lcn2 was reported to be differentially expressed dependent on microglial inflammatory status44. Using LPS and cancer cachexia models of systemic inflammation, we observe a robust vascular expression of Lcn2 in the CNS under both conditions, suggesting systemic inflammatory mediators act at the point of contact between the periphery and brain to increase Lcn2 expression. Importantly, we confirm abluminal secretion or transmigration of LCN2 by detection of the molecule in the CSF of mice, demonstrating peripheral inflammatory insults are not only capable of increasing expression and production of LCN2 in the cerebral vasculature, but also result in release of the molecule into the CNS. Additionally, previous work also demonstrated the ability of LCN2 to cross the blood brain barrier10, 17. Therefore, our studies confirm that systemic inflammatory diseases can result in a robust induction of LCN2 into the brain, and under chronic conditions, may be partially responsible for progressive spatial memory cognitive decline through its actions in the hippocampus.
The effect of LCN2 on cognitive function using various murine models was examined in the past, demonstrating variable effects of Lcn2 genetic deletion on cognition depending on model of study. In experimental models of vascular dementia and diabetic encephalopathy, Lcn2 knockout mice demonstrated improved spontaneous alternation in the Y-maze test21, while our studies demonstrated that exogenous LCN2 did not affect spontaneous alternation performance, suggesting a combinatorial role of LCN2 and disease-specific factors in mediating a reduction in working memory. In a model of restraint stress, Lcn2 deficient mice displayed an enhancement of stress-induced anxiety as indicated by the elevated-plus maze and light-dark box tests45. Our study did not demonstrate an effect of exogenous LCN2 on anxiety using the light-dark box, suggesting that while a deficiency in CNS LCN2 induces anxiety-like behaviors, an excess of the molecule does not appear protective against anxiety. These findings may also be reconciled by the clear role LCN2 plays in hippocampal development, as Lcn2 null mice display significantly altered hippocampal neuron spine density and morphology47. Thus, while excessive CNS LCN2 appears neuropathological in several disease conditions, basal expression of the molecule is critical in normal hippocampal development, suggesting whole-body knockout conditions may not represent a biologically-relevant setting for the study of LCN2 on hippocampal function during disease. Using the J20 Alzheimer mouse model, Dekens et al observed no alterations in working and visual recognition memory by spontaneous alternation and novel-object recognition tests, respectively46. In contrast to our study, the authors observe no differences in spatial recognition memory using the Morris water maze, although our studies utilized the blocked arm version of the Y maze46. Collectively, these studies and more demonstrate that while LCN2 is induced in the CNS during several diseases associated with cognitive decline, its effects on cognitive function are strongly dependent on the specific disease state. Since our study demonstrates an individual effect of LCN2 on cognitive dysfunction in the absence of disease, it may be that the cognitive effects of LCN2 are masked by more robust mediators of neuronal dysfunction during primary neuropathologies with profound cognitive impairment, such as hyperphosphorylated tau in the context of tauopathies48.
Our study herein raises important questions, as well as limitations, concerning the precise mechanisms of LCN2-induced spatial memory decline. Since there are three putative receptors of LCN2 (SLC22A17, megalin, and the MC4R), it remains to be determined which ligand-receptor interaction mediates which molecular, cellular, and behavioral consequences observed in our study10, 49, 50. We chose to focus on MC4R-dependent and independent effects of LCN2 on hippocampal neurons for several reasons, including: 1) 1) both Slc22a17 and Megalin KO mice are embryonic lethal; 2) transfection and transduction of WT primary hippocampal neurons is especially difficult, as these cells are non-mitotic and particularly sensitive to these cytotoxic stressors; and 3) we examined gentamicin as a potential pharmacologic agent in disrupting LCN2’s binding to megalin, but realized that gentamicin’s low affinity for the receptor51—combined with the known neurotoxicity of gentamicin (this is especially observed in acoustic neurons)—made the confounding effects of gentamicin treatment on primary hippocampal neurons very difficult to disentangle from LCN2-megalin biology. Although our gene set analyses of LCN2 treated WT and Mc4r-KO mice revealed activation of similar hippocampal pathways, there were several disparately expressed genes between genotypes, suggesting that while MC4R signaling is not the predominant driver of our observed in vitro and in vivo effects, this receptor may provide a minor contribution to CA1 and CA3 hippocampal neuronal plasticity as described previously28, 29. Secondly, while our RNA sequencing analyses of LCN2-treated hippocampal neurons suggest LCN2 is individually detrimental to neurons, it is unclear whether the observed microgliosis and leukocyte infiltration into the CNS in vivo is beneficial or pathologic. In a recent report of HIV-1 induced neuropathology, it was demonstrated that LCN2’s neurotoxic effects were dependent on microglia34. Conversely, microglial depletion during cancer worsens cachexia illness behaviors, demonstrating a protective effect of microglia during chronic systemic inflammation52. Similar to microglia, brain-infiltrating immune cells are reported to be either pathologic or beneficial depending on the disease state53. Since we did not observe a change in expression of inflammation-related transcripts, including Il-6, Tnf-α, Nos2, and Arg1 in LCN2-treated microglia, it is plausible that the increased microglia, combined with their expression of chemokines, result in the recruitment of peripheral immune cells in attempt to repair the damaged hippocampus, although future investigation into these observations is warranted. It is also worth noting that it is unclear if the indwelling cannula, and its associated inflammation, amplifies the observe microglia and immune cell alterations in these studies. While the pancreatic cancer cachexia model utilized is associated with systemic inflammation, it is unclear at this time if this model is individually associated with alterations in hippocampal neurons in an LCN2-dependent manner. LCN2 is also known to modulate intracellular iron trafficking, as well as apoptosis depending on the iron-loading status of the molecule49. In our studies, is likely that LCN2 exists solely in the apo- form in our cachexia and LPS models, as we observe no evidence of bacterial infection (and therefore no bacterial siderophore production) during these conditions. Consistent with this idea, our ICV experimental paradigm utilized the apo- form of LCN2. Future studies should account for both apo- and holo- forms of LCN2 when considering its effects on CNS function. We utilize mixed sex studies in many places throughout this work, our battery of neurocognitive assessments includes only male mice—these data should be interpreted accordingly. Finally, our initial observations lead us to focus on the effects of LCN2 on hippocampal biology, but it is conceivable that central LCN2 mediates alterations elsewhere in the CNS. This remains an active area of investigation.
In summary, the data presented here demonstrate a temporal role of LCN2 in mediating neuronal dysfunction and subsequent cognitive impairment specific to spatial memory. Given the evolutionarily-conserved role of LCN2 in sequestering iron away from bacteria7, along with recent reports of LCN2’s beneficial effect in the CNS during acute inflammation6, it stands to reason that the rapid induction of LCN2 in the CNS is an adaptive host response. Furthermore, recent evidence demonstrates that LCN2 acts in a dose- and time-dependent fashion, as cells are able to tolerate high doses of LCN2 acutely, but activate apoptosis after prolonged exposure8, 21. These data demonstrate the dichotomous nature of LCN2, having protective effects acutely, yet detrimental effects with prolonged exposure (Figure 9). Consistent with the notion of LCN2 acting temporally, our study demonstrates LCN2 chronically disrupts hippocampal cellular composition, ultimately leading to a concomitant decline in cognitive function specific to spatial memory. Together, our data implicate LCN2 as a chronic mediator of CNS dysfunction and a potential therapeutic target for patients suffering from cognitive decline.
Methods
Mice
8–14 week-old male and female C57BL/6J wild type (WT, JAX catalog number # 000664), Lipocalin 2 knockout (Lcn2-KO, JAX catalog number # 024630), and Pomc-EGFP (JAX catalog number # 009593) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained in our animal facility. Sex of mice used in each study is specified in corresponding figure legends. Briefly, Lcn2-KO mice were generated on a C57BL/6J background. All mice were housed and bred in a dedicated mouse room with a temperature 26°C with a 12-hour light/dark cycle. Animals were provided ad libitum access to food and water (Rodent Diet 5001; Purina Mills). Lcn2-KO mice were genotyped according to the standard protocol from The Jackson Laboratory. In behavioral studies, animals were individually housed for acclimation at least 7 days prior to interventional studies. Mouse studies were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory animals, and approved by the Institutional Animal Care and Use Committee (IACUC) of Oregon Health & Science University.
Cell lines
The pancreatic cancer cell line used herein, generously provided by Dr. Elizabeth Jaffee, is derived from a C57BL/6J mouse with pancreatic-specific conditional alleles KRASG12D and TP53R172H expression driven by the PDX-1-Cre promoter (KPC). The KPC model is a highly characterized and published model of PDAC cachexia due to its close biological semblance of human disease54–57. KPC cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum, 1% minimum essential medium non-essential amino acids, 1 mM sodium pyruvate, and 50 U/mL penicillin/streptomycin (Gibco). bEnd.3 cells were purchased from ATCC (ATCC CRL-2299) and maintained in DMEM supplemented with 10% fetal bovine serum, 1% minimum essential medium non-essential amino acids, 1 mM sodium pyruvate, and 50 U/mL penicillin/streptomycin (Gibco). All cells were grown in cell incubators maintained at 37° Celsius and 5% CO2 and were routinely tested and confirmed negative for mycoplasma contamination prior to experimentation.
Primary glia culture and conditioned media studies
Primary mixed-glial cultures containing microglia and astrocytes were prepared from 1–3 day old WT C57BL/6J mouse pups. Briefly, cortices were dissected, freed of the meninges, and then digested with papain (Worthington Biochemical Corporation). Mixed cortical cells were passed through a 70-μm cell strainer and seeded in 75-cm2 flasks for generating mixed-glia. Cultures were maintained 6 in DMEM media with low glucose supplemented with l-glutamine, 10% FBS and 1% penicillin/streptomycin. Media was refreshed every 3–4 days for 14–16 days. Primary microglia were isolated from mixed-glia by shaking flasks at 200 rpm for 2 hours in an incubator-shaker. More than 90% of the isolated cells were confirmed as microglia by Iba1 staining and flow cytometry (CD45+ CD11b+ cells, data not shown).
bEnd.3 cells, mixed-glia and isolated primary microglia were re-plated into 6-well plates (for mRNA analysis) and poly-D-lysine/laminin coated glass coverslips (Corning Biocoat,) in 24-well plates (for immunocytochemistry) for 1–2 days prior to treatment. KPC cells were cultured in a 75-cm2 flask until 80–90% confluent. 24 hours prior to treatment, 13 ml fresh media (RPMI supplemented with 10% FBS and 1% penicillin–streptomycin) was added for generating KPC-conditioned media. On the treatment day, 4 parts KPC-conditioned media were mixed with 1 part fresh RPMI media prior to adding to mixed glia, primary microglia, and b3nd.3 cells plated in 6-well plates. Cells were treated with either KPC conditioned media, LPS (10 ng/mL), or control media (fresh RPMI media supplemented with 10% FBS and 1% penicillin–streptomycin). Each treatment condition was performed in triplicate. Cells were treated for 16 hours prior to RNA extraction using a Qiagen RNAEasy kit or fixation with 4% paraformaldehyde for immunofluorescent staining with primary antibodies LCN2 (R&D Systems, AF1857, goat, 1:500); GFAP (Calbiochem, IF03L-100UG, Mouse, 1:1000); Iba1 (Wako, 019–19741, Rabbit, 1:1000); CD31/PECAM (BD Pharmingen, 550274, Rat, 1:250); NeuN (Millipore, MAB377, Mouse, 1:1000). Secondary antibodies: donkey anti-goat AF630 (1:500); donkey anti-goat AF555 (1:500); donkey anti-mouse AF488 (1:1000); donkey anti-rabbit AF555 (1:1000); donkey anti-rat AF488 (1:500); goat anti-mouse AF594 (1:500). Cell nuclei were labeled with DAPI from the mount media (ProLong Gold Antifade media, Invitrogen).
Primary hippocampal neuronal culture
Wells of a 12-well plate were treated with Poly-D-Lysine (PDL, Sigma) overnight in a cell incubator on the day prior to hippocampal culturing. The morning of hippocampal dissection, PDL was aspirated from plates and wells were washed for 3 times with sterile water prior to adding hippocampal plating medium (NeuroQ basal medium [Global Stem] supplemented with 10% FBS, 1% GlutaMAX [Gibco], 1% Sodium pyruvate [Gibco], and 1% penicillin–streptomycin).
Pregnant (18 days post-fertilization) wild type C57BL/6J were rapidly killed by CO2 euthanasia and cervical dislocation. After sterilizing, an incision along the middle abdomen was made to expose uterine horns. Using sterile instruments, the uterus was opened and embryos were rapidly decapitated and heads placed in Hanks’ balanced salt solution (HBSS) buffer (Gibco 14175). Whole brains were carefully removed from the skull base and immediately placed in fresh HBSS buffer. Ventrally, hippocampi were freed from midbrain through gentle blunt dissection along meningeal foldings between the diencephalon and striatum (rostrally) to the caudal hippocampi (approaching base of the brain). Hippocampi were then blunt-dissected from the cortex and corpus callosum, and transferred to a 15 ml conical tube filled with 9 ml of HBSS. After hippocampi were allowed to settle to the bottom of the tube, 4.5 ml of HBSS was removed from the tube and replaced with 0.5 ml 2.5% trypsin (final concentration of 0.25%) and 50 μl of DNAse1 (final concentration of 1 mg/ml). Hippocampi were digested for 15 minutes in a 37C water bath swirling every 5 minutes. Supernatant was removed and enzymatic digestion was neutralized with plating medium (NeuroQ basal medium supplemented with 10% FBS, 1% GlutaMAX, 1% sodium pyruvate, and 1% penicillin–streptomycin). Hippocampi were dissociated and then passed through a 40 μm cell strainer. The resulting cell suspension was passed through another 40 μm cell strainer, and plated with plating medium in a 12-well plate at a density of 1–1.5 ×106 cell/well (for mRNA analysis). 4 hours later after plating, plating medium was replaced with culture medium (NeuroQ basal medium supplemented with 1% GlutaMAX, 1% penicillin–streptomycin, and B-27 Plus supplement, Gibco). Day 3 after plating, half of the medium was replaced with culture medium containing 10 μM 5-fluoro-2’-deoxyuridine (FdU, Gibco). Day 6 after plating, one-third of the medium was replaced with fresh culture medium containing 10 μM FdU. All assays were started on day 7 after plating. Neuron and neurite staining was performed using a MAP2 antibody (Sigma; M9942; 1:800), and entire somas and neurite processes were image and compressed to a max-intensity Z stack. The resulting max-intensity image was subjected to the ImageJ plugin NeuriteTracer58. Parameters were as follows: 12 on neuronal images, 120 on nuclear images, and nuclear size of 5–850. Total neurite length, total neuron count, and neurite length per neuron (as indicated by DAPI staining) were calculated per field of view.
Nanostring gene expression methods
Gene expression analyses were performed on dissected hippocampi utilizing 50 ng of RNA on either Nanostring Neuroinflammation or Neuropathology gene panels per the manufacturer’s recommendations. The resulting dataset was analyzed by the proprietary ROSALIND® software, in which the producers of this software include the following bioinformatics analyses: “The resulting data was analyzed by ROSALIND® (https://rosalind.onramp.bio/), with an architecture developed by ROSALIND, Inc. (San Diego, CA). Read Distribution percentages, violin plots, identity heatmaps, and sample MDS plots were generated as part of the QC step. Normalization, fold changes and p-values were calculated using criteria provided by Nanostring. ROSALIND® follows the nCounter® Advanced Analysis protocol of dividing counts within a lane by the geometric mean of the normalizer probes from the same lane. Housekeeping probes to be used for normalization are selected based on the geNorm algorithm as implemented in the NormqPCR R library59. Abundance of various cell populations is calculated on ROSALIND using the Nanostring Cell Type Profiling Module. ROSALIND performs a filtering of Cell Type Profiling results to include results that have scores with a p-Value greater than or equal to 0.05. Fold changes and pValues are calculated using the fast method as described in the nCounter® Advanced Analysis 2.0 User Manual. P-value adjustment is performed using the Benjamini-Hochberg method of estimating false discovery rates (FDR). Clustering of genes for the final heatmap of differentially expressed genes was done using the PAM (Partitioning Around Medoids) method using the fpc R library60 that takes into consideration the direction and type of all signals on a pathway, the position, role and type of every gene, etc. Hypergeometric distribution was used to analyze the enrichment of pathways, gene ontology, domain structure, and other ontologies. The topGO R library61, was used to determine local similarities and dependencies between GO terms in order to perform Elim pruning correction. Several database sources were referenced for enrichment analysis, including Interpro62, NCBI63, MSigDB64, 65, REACTOME66, WikiPathways67. Enrichment was calculated relative to a set of background genes relevant for the experiment.”
RNA Sequencing and bioinformatics analyses
Primary hippocampal neurons were treated with 100 ng/mL LCN2 or vehicle control for either 1 or 4 days. Total RNA was purified with the RNeasy Mini Kit (Qiagen). Library preparation and sequencing was performed by BGI Genomics at 100 base pair paired-end sequencing. Sequencing libraries were analyzed using the methods, parameters, and software versions, described this pipeline written in SnakeMake format. Briefly, reads were trimmed with bbduk and high-quality reads were screened for contamination with fastq_screen and aligned to the mm10 genome with STAR. Alignment features were filtered for protein-coding genes, and individual sample counts were aggregated into a counts table for input to DESeq2. Specific contrasts were constructed to facilitate differential expression analysis, such as LCN2_1day-vs-Veh_1day and LCN2_4day-vs-Veh_4day. Lowly-expressed genes with counts equal to or less than 1 across all samples were filtered away, and differential analysis was run with default settings. Results were extracted from the DESeq2 object by contrast and further analyzed with topGO with a BH p-value correction. Results were visualized using ggplot2. Pathway and ontology analyses were performed using Enrichr68–70. Below is a comprehensive list of bioinformatics tools utilized in the aforementioned pipeline, as well as hyperlinks to URL locations:
FastQ Screen: https://pubmed.ncbi.nlm.nih.gov/30254741/
Bbduk: BBtools may be cited using the primary website: BBMap – Bushnell B. – sourceforge.net/projects/bbmap/
STAR: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3530905/
DESeq2: https://genomebiology.biomedcentral.com/articles/10.1186/s13059–014-0550–8
topGO: https://bit.ly/3cHsbAz
snake make: https://snakemake.readthedocs.io/en/stable/project_info/citations.html
ggplot2: https://cran.r-project.org/web/packages/ggplot2/citation.html
BH FDR adjustment: https://www.jstor.org/stable/2346101?seq=1
Intracerebroventricular cannulation and injections
Mice were anesthetized using isoflurane and gently placed on a stereotactic alignment instrument (Kopf Instruments). Using sterile technique, bregma was exposed with a 3 mm incision and a 26-gauge lateral ventricle cannula was placed at 1.0 mm X, −0.5 mm Y, and −2.25 mm Z relative to bregma. Cannulas were secured to the skull with embedded screws and crosslinked flash acrylic. Mice were allowed 8 days for recovery after cannulation surgery. Cannula placement was verified through two means: 1) after recovery from cannulation procedure, cannula caps were removed to allow for ventricular pressure equilibration—in many cases, a small amount of CSF was observed flowing through the cannula; 2) smooth and non-disrupted microinjections through the cannula were observed for every mouse in these studies, consistent with cannula placement in the ventricle (compared to a blocked cannula or cannula lodged in brain parenchyma). Recombinant mouse LCN2 (R&D Systems, 40 ng) or vehicle were injected in a total volume of 2 uL. For repeated or single injection experiments of LCN2 or vehicle, mice received injections under restriction, and received restriction training every day for 5 days prior to initial injections.
Cerebrospinal fluid extraction
Mice were anesthetized using isoflurane and placed on a stereotactic alignment instrument (Kopf Instruments). A 2 cm incision was made over the cisterna magna and the trapezius and paraspinal muscles were reflected. Blood and extracellular fluid lying over the cisterna magna were carefully removed to avoid CSF contamination. A glass micropipette (tip diameter of approximately 400–800 μm) was stereotactically inserted into the cisterna magna for capillary action-based CSF collection, transferred to a LoBind (Eppendorf) microcentrifuge tube, and immediately flash frozen.
In situ hybridization
Mice from control, LPS (acute inflammation, 24 hours after IP injection), or and pancreatic cancer (chronic inflammation, day 12 after tumor implantation) were sacrificed and underwent transcardial perfusion with 20 mL PBS to ensure robust vasculature flushing. In situ hybridization was then performed on fresh frozen brains as previously described71. Antisense 33P-labeled mouse LCN2 (Lcn2) riboprobe (corresponding to bases 276–676 of murine Lcn2; GenBank accession no. NM_008491.1 (.25 pmol/ml) was denatured, dissolved in hybridization buffer along with tRNA (1.7 mg/ml), and applied to slides. Slides were glass coverslipped, placed in a humidified chamber, and incubated overnight at 55 °C. The following day, slides were treated with RNase A and washed under conditions of increasing stringency. Slides were dipped in 100% ethanol, air dried, and then dipped in NTB liquid emulsion (Carestream Health Inc., Rochester, NY). Slides were developed 4d later and coverslipped.
Histology and Immunohistochemistry
Mice were deeply anesthetized using a ketamine/xylazine/acepromazine cocktail and sacrificed by transcardial perfusion with 20 mL PBS followed by ice cold 4% paraformaldehyde (PFA). Tissues were post-fixed in 4% PFA overnight at 4°C prior to sectioning protocols. After post-fixation, brains were cryoprotected in 20% sucrose for 24 hours at 4°C prior to 30 μM microtome sectioning. Free-floating sections were incubated in blocking solution for 1 hour at room temperature, followed by primary antibody incubation overnight at 4°C. Sections were washed with PBS between steps. Sections were then mounted on gelatin-coated slides and coverslipped with Prolong Gold anti-fade media (Thermofisher). Fluorescent-based images were acquired on a Nikon confocal microscope, while chromogen-based images were acquired using a Leica microscope (model DM 4000B).
Primary antibodies utilized above are listed with company, clone, host, species, and concentration defined in parentheses, respectively: LCN2 (R&D Systems, AF1857, goat, 1:500); GFAP (Calbiochem, IF03L-100UG, Mouse, 1:1000); Iba1 (Wako, 019–19741, Rabbit, 1:1000); CD31/PECAM (BD Pharmingen, 550274, Rat, 1:250); NeuN (Millipore, MAB377, Mouse, 1:1000). Secondary antibodies: donkey anti-goat AF630 (1:500); donkey anti-goat AF555 (1:500); donkey anti-mouse AF488 (1:1000); donkey anti-rabbit AF555 (1:1000); donkey anti-rat AF488 (1:500); goat anti-mouse AF594 (1:500)
Image acquisition and analysis
Fluorescent images were acquired using a Nikon confocal microscope. 5-layer flattened Z-stack images of the hippocampus were procured using a 10× objective for wide view in the Lcn2 costains and CD45/MPO stain, 20x for the chronic Lcn2 gliosis staining, and 40x for the chronic Lcn2 NeuN quantification and Lcn2 costain insets. Images were 2048 × 2048 pixels, with a pixel size of 0.63 μm for 10x images, 0.32 μm for 20x images, and 0.16 μm for 40x images. Images of POMC-GFP mice dentate gyri were taken using using a 40x objective. The hippocampus was identified by the granule cell layer of the dentate gyrus, which was positioned at the left end of each image.
Microglia, astrocytes, immune cells (CD45+ and MPO+), mature neurons, and GFP-positive newly born granule neurons dentate gyrus of POMC-GFP mice were quantified using the Fiji (ImageJ, NIH) plugin “Cell counter”. Microglia and astrocytic morphology were scored using a validated activation scale described by Harrison et al20. All images were quantified or scored by a blinded observer.
Quantitative real time PCR
Snap-frozen tissues or cell pellet were rapidly homogenized and RNA was purified with the RNeasy Mini Kit (Qiagen). Samples were then reverse-transcribed with the High Capacity cDNA Reverse Transcription Kit (Life Technologies). qRT-PCR was performed using reagents and TaqMan primer probes listed in Supplementary table 1. Tissues were normalized to 18S using the ddCT method.
Enzyme linked immunosorbent assay
LCN2 concentrations in the CSF were assayed by ELISA according to the manufacturer’s protocol (R&D Systems, Catalog # DY1857).
Flow cytometry
Contralateral brain halves from ICV treated, PBS-perfused mice were dispersed into a single cell suspension by straining through a steel mesh screen in RPMI. CNS mononuclear cells were isolated at the 40%−80% interface of a Percoll (Cytiva 17089101) step gradient after centrifugation at 500 x g for 45 minutes. Cells were washed, then resuspended in 100μl RPMI + 5%FBS and blocked for Fc receptor binding (TruStain FcX Plus, BioLegend 156604). Cells were stained with antibodies to the following surface markers for 1hr at 4oC (cat. no., dilution): CD11b-BB515 from BD Biosciences (564454, 1:125). All remaining antibodies were obtained from BioLegend: CD3-PE (100206, 1:100); CD45-PerCP-Cy5.5(103132, 1:125); CD19-APC (115512, 1:100); Ly6C-APC-Cy7 (128026, 1:200); Ly6G-BV421 (127628, 1:200). Live/Dead Fixable Aqua (ThermoFisher L34957) was used for the gating of viable cells. Samples were fixed (BD Cytofix, BD Biosciences 554655) for 15mins at 4oC and run on the LSR II flow cytometer (BD Biosciences, San Jose, CA, USA). Gating and analysis were performed using FlowJo software version 10.1r7 (FlowJo LLC, Ashland, OR, USA).
Behavioral analysis
Y-maze spontaneous alternation assessment: 12-week old male C57BL/6J mice were tested in a Y-maze apparatus consisting of three enclosed arms set at a 120° angle to one another, measuring 50 cm long, 11 cm wide, and 11 cm high. Visual cues were placed in the testing room around the Y-maze apparatus and kept constant throughout testing sessions. Mice were placed in the center of the maze and allowed to explore for a total of 5 minutes. Percentage alternation was calculated at the end of the study by the total number of successive alternations, defined as sequential entry into three different arms, divided by the total number of arm entries minus 2. Y-maze blocked arm assessment: utilizing the same Y-maze described above, mice were placed at the end of a randomly chosen arm and allowed to explore the maze for 5 minutes with one arm closed. After a 2-hour intertrial interval, mice were re-introduced to the maze (in the different arm than the acquisition trial, but not the novel arm) with all arms open and allowed to explore again for 5 minutes. Time spent in each arm was recorded, and the percentage of time spent in the novel arm was calculated (time spent in novel arm/total time spent in all three arms). Novel object recognition: Mice were subjected to a novel object recognition test utilizing the well-established protocol by Leger et al with some minor modifications72. Briefly, we tested mice with two identical objects placed in an open-field apparatus composed of a black wooden box measuring 33 × 33 × 20 cm. We utilized two different objects (Falcon tissue culture flask filled with sand and an equally-sized LEGO tower) in triplicate to minimize olfactory and sensory cues between the familiarization and test sessions. During the familiarization trial, mice were allowed to explore two identical objects in the open field until a total object exploration time of 20 seconds was achieved. After a 6-hour intertrial interval, mice were re-introduced to the open field with the familiar and novel object and allowed to explore for a total of 10 minutes. Preference index was calculated as time spent with novel object minus time spent with familiar object divided by total exploration time. Light-dark box: Mice were placed in the light compartment of a light-dark box and allowed to traverse the apparatus for 5 minutes. The test apparatus consisted of two equally-sized plexiglass compartments (one opaque/dark compartment, and one clear/light-penetrating compartment) with a small opening through which mice could enter and leave. Total time in each compartment, latency to enter the dark compartment after placement in the light compartment, as well as total number of transitions, were recorded. Tail suspension test: Four mice at a time were suspended by the tail using 17 cm tape strands (allowing for the last 2 cm to loop around the animal’s tail) from a wooden dowel, separated by a wooden contraption that does not allow for mice to visualize one another. Prior to taping the tail, a small rigid plastic tube was placed proximal to the end of the tail where tape was then fastened to prevent mice from climbing. Mice were suspended for a total of 6 minutes with total time immobile being recorded. Nest building: Nest building and blinded scoring was performed as previously reported73. Briefly, mice were supplied a new 3.0 g nestlet along with the removal of used enrichment items one hour before the dark phase. The next morning (approximately 15 hours later unless otherwise indicated) nests were scored on a rating scale of 1–5 as described by Deacon73. Where appropriate, all behavioral testing equipment was thoroughly cleaned with 70% ethanol between trials and mice to minimize olfactory cues.
Statistics
All statistical analyses for murine data were performed in GraphPad Prism 8.0 software. Quantitative data are reported as mean +/− standard error. Two-tailed Students t-tests were performed when comparing two groups. When comparing more than two groups of a single genotype, One-way ANOVA was utilized. Two-way ANOVA with Bonferroni multiple comparisons test was utilized when comparing multiple genotypes and treatment groups (sham and tumor; sham and LPS) unless otherwise specified in figure legends. Where appropriate, normality (Shapiro-Wilk; alpha=0.05), lognormality, and outlier tests were performed to ensure normal distributions of datasets herein. For all analyses, a p value of < 0.05 was considered to be statistically significant.
Supplementary Material
Supplementary Figure 2. Related to Figure 3. (a) Nest building score in male Lcn2-KO and WT mice 48 hours after LPS challenge (n = 5-6 per group). (b) Food intake as a percentage of initial body mass in Lcn2-KO and WT mice 48 hours after vehicle injection or LPS challenge (n = 5-7 per group). (c) Body mass change body mass in Lcn2-KO and WT mice 24 and 48 hours after vehicle injection or LPS challenge (n = 5-7 per group). (d) Survival analysis of Lcn2-KO and WT mice after a lethal dose of LPS (n = 10 per group). (e) Hourly food intake after re-feeding male WT mice after 16 hour fast (n = 4-5 per group). (f) Plasma LCN2 levels in mice after 16 hour of fasting (16 F), 16 hour fasting + 1 hour after re-feeding (1 HRF), and 16 hour fasting + 4 hours after re-feeding (4 HRF) (n = 11-12 per group; 5-6 males, 5-6 females; 16 F vs 1 HRF, p=0.0136).
{kind=link}
Supplementary figure 3. Related to Figure 6. Flow cytometry gating strategy for quantifying immune cell subpopulations in the CNS of mice receiving (a) LCN2 or (b) vehicle. Cells were initially gated to remove debris, doublets and dead cells before gating on CD45+ cells. Immune cell markers were used to define the following subpopulations: (a) lymphoid cells = CD45+ CD11b−; (b) CD45+ CD11bmid; (c) myeloid cells = CD45+ CD11b+; (d) microglia = CD45lo CD11b+; (e) T cells = CD3+; (f) B cells = CD19+; (g) myeloid cells (non-neutrophils) = Ly6C+ Ly6G−; (h) neutrophils = Ly6C+ Ly6G+.
{kind=link}
Supplementary figure 4. Related to Figure 9. (a) Total number of arm entries in the spontaneous alternation test. (b) Time to achieve novel object recognition criteria as defined by a total of 20 seconds of exploration with novel or familiar objects. (c) Latency to enter dark compartment of the light-dark box. (d) Number of transitions between light and dark compartments in the light-dark box text. (e) Total number of transitions between Y-maze arms in the Y-maze blocked arm test.
{kind=link}
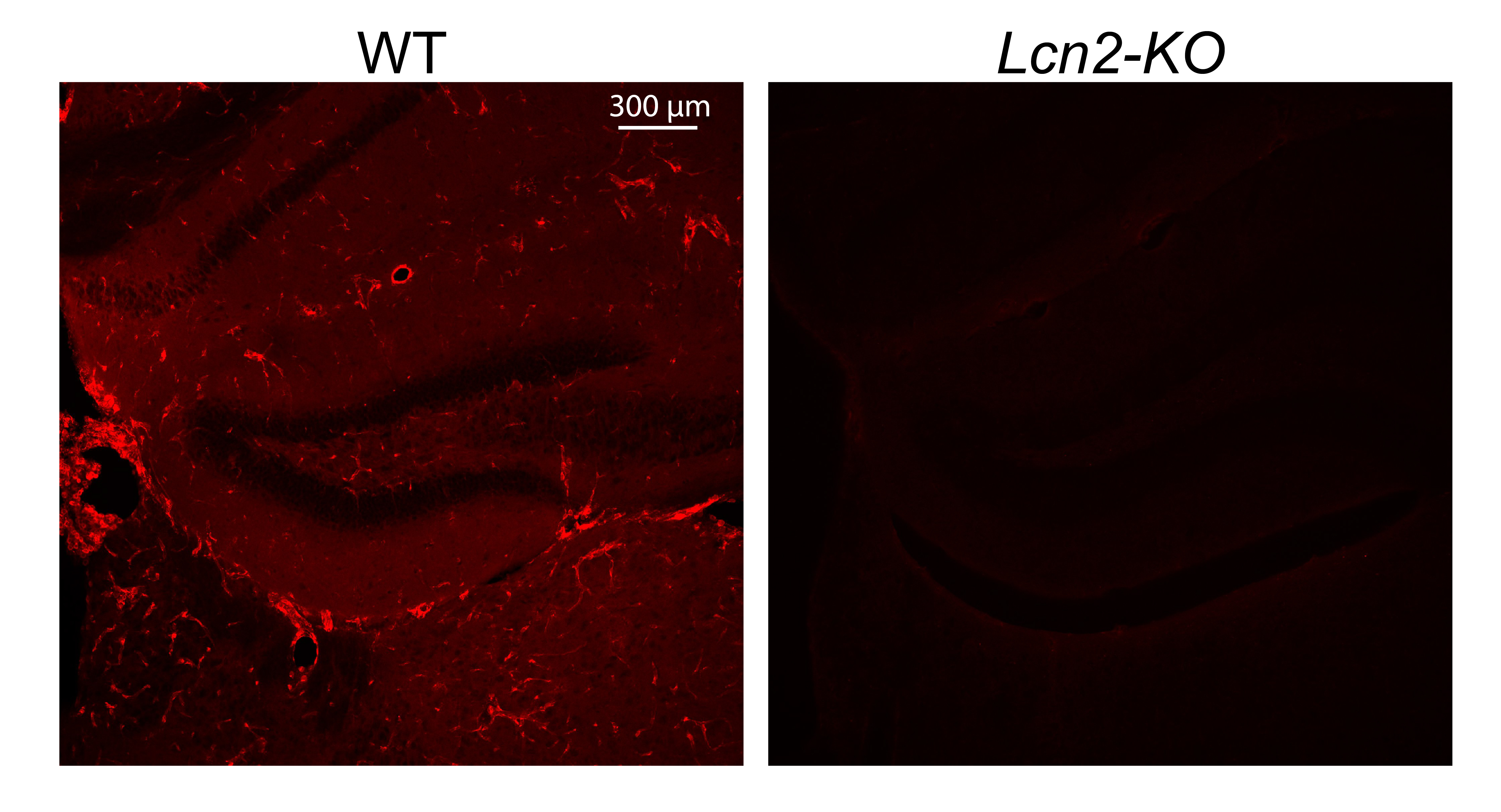
Supplementary Figure 1. Related to figure 1. Confirmation of LCN2 antibody specificity by IHC in WT and Lcn2-KO mice were injected with 200 μg/kg LPS. Images representative of 3 independently injected mice. 10x magnification.
{kind=link}
LCN2 is robustly upregulated in the central nervous system during acute and chronic inflammation
-
Chronic, but not acute, central LCN2 treatment alters hippocampal cell composition
Decreased mature neurons, but increased newborn neurons
Microgliosis
Immune cell invasion
Cellular composition alterations in the hippocampus occur through MC4R-independent pathways
LCN2 treated neurons display blunted neurite outgrowth
Mice treated with chronic LCN2 display impaired spatial recognition memory
Acknowledgements
We are grateful for the help of several investigators and core laboratories, including Ashley J. Olson, Stephanie Krasnow, the OHSU flow cytometry core, and the OHSU advanced light microscopy core. This work was supported by NCI R01CA184324 (Marks), the Brenden-Colson Center for Pancreatic Care (Marks), and NIH F30CA254033 (Olson).
DM is a consultant for Pfizer, Inc. and Alkermes, Inc. DM is a consultant, has received grant funding, and has equity in Tensive Controls, Inc.
Footnotes
All other authors declare no conflicts of interest.
References
- 1.Olson B, Marks DL. Pretreatment Cancer-Related Cognitive Impairment-Mechanisms and Outlook. Cancers (Basel). May162019;11(5)doi: 10.3390/cancers11050687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kayser MS, Dalmau J. The emerging link between autoimmune disorders and neuropsychiatric disease. J Neuropsychiatry Clin Neurosci. Winter2011;23(1):90–97. doi: 10.1176/jnp.23.1.jnp90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gan L, Cookson MR, Petrucelli L, La Spada AR. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat Neurosci. October2018;21(10):1300–1309. doi: 10.1038/s41593-018-0237-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llorens F, Hermann P, Villar-Piqué A, et al. Cerebrospinal fluid lipocalin 2 as a novel biomarker for the differential diagnosis of vascular dementia. Nat Commun. January302020;11(1):619. doi: 10.1038/s41467-020-14373-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vichaya EG, Gross PS, Estrada DJ, et al. Lipocalin-2 is dispensable in inflammation-induced sickness and depression-like behavior. Psychopharmacology (Berl). October2019;236(10):2975–2982. doi: 10.1007/s00213-019-05190-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang SS, Ren Y, Liu CC, et al. Lipocalin-2 protects the brain during inflammatory conditions. Molecular psychiatry. February2018;23(2):344–350. doi: 10.1038/mp.2016.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flo TH, Smith KD, Sato S, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. December162004;432(7019):917–21. doi: 10.1038/nature03104 [DOI] [PubMed] [Google Scholar]
- 8.Bi F, Huang C, Tong J, et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci U S A. March52013;110(10):4069–74. doi: 10.1073/pnas.1218497110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao N, Xu X, Jiang Y, et al. Lipocalin-2 may produce damaging effect after cerebral ischemia by inducing astrocytes classical activation. J Neuroinflammation. August192019;16(1):168. doi: 10.1186/s12974-019-1556-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosialou I, Shikhel S, Liu JM, et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature. March162017;543(7645):385–390. doi: 10.1038/nature21697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ip JP, Noçon AL, Hofer MJ, Lim SL, Müller M, Campbell IL. Lipocalin 2 in the central nervous system host response to systemic lipopolysaccharide administration. Journal of neuroinflammation. September262011;8:124. doi: 10.1186/1742-2094-8-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nam Y, Kim JH, Seo M, et al. Lipocalin-2 protein deficiency ameliorates experimental autoimmune encephalomyelitis: the pathogenic role of lipocalin-2 in the central nervous system and peripheral lymphoid tissues. J Biol Chem. June132014;289(24):16773–89. doi: 10.1074/jbc.M113.542282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deacon RM, Croucher A, Rawlins JN. Hippocampal cytotoxic lesion effects on species-typical behaviours in mice. Behav Brain Res. May142002;132(2):203–13. doi: 10.1016/s0166-4328(01)00401-6 [DOI] [PubMed] [Google Scholar]
- 14.Deacon RM, Penny C, Rawlins JN. Effects of medial prefrontal cortex cytotoxic lesions in mice. Behav Brain Res. February172003;139(1–2):139–55. doi: 10.1016/s0166-4328(02)00225-5 [DOI] [PubMed] [Google Scholar]
- 15.Gaskill BN, Karas AZ, Garner JP, Pritchett-Corning KR. Nest building as an indicator of health and welfare in laboratory mice. Journal of visualized experiments : JoVE. December242013;(82):51012. doi: 10.3791/51012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neely CLC, Pedemonte KA, Boggs KN, Flinn JM. Nest Building Behavior as an Early Indicator of Behavioral Deficits in Mice. Journal of visualized experiments : JoVE. October192019;(152)doi: 10.3791/60139 [DOI] [PubMed] [Google Scholar]
- 17.Olson B, Zhu X, Norgard MA, et al. Lipocalin 2 mediates appetite suppression during pancreatic cancer cachexia. Nat Commun. April62021;12(1):2057. doi: 10.1038/s41467-021-22361-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olson B, Marks DL, Grossberg AJ. Diverging metabolic programmes and behaviours during states of starvation, protein malnutrition, and cachexia. J Cachexia Sarcopenia Muscle. December2020;11(6):1429–1446. doi: 10.1002/jcsm.12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overstreet LS, Hentges ST, Bumaschny VF, et al. A transgenic marker for newly born granule cells in dentate gyrus. The Journal of neuroscience : the official journal of the Society for Neuroscience. March312004;24(13):3251–9. doi: 10.1523/jneurosci.5173-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison L, Pfuhlmann K, Schriever SC, Pfluger PT. Profound weight loss induces reactive astrogliosis in the arcuate nucleus of obese mice. Mol Metab. June2019;24:149–155. doi: 10.1016/j.molmet.2019.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JH, Ko PW, Lee HW, et al. Astrocyte-derived lipocalin-2 mediates hippocampal damage and cognitive deficits in experimental models of vascular dementia. Glia. September2017;65(9):1471–1490. doi: 10.1002/glia.23174 [DOI] [PubMed] [Google Scholar]
- 22.Li SH, Li H, Torre ER, Li XJ. Expression of huntingtin-associated protein-1 in neuronal cells implicates a role in neuritic growth. Mol Cell Neurosci. August2000;16(2):168–83. doi: 10.1006/mcne.2000.0858 [DOI] [PubMed] [Google Scholar]
- 23.Few AP, Lautermilch NJ, Westenbroek RE, Scheuer T, Catterall WA. Differential regulation of CaV2.1 channels by calcium-binding protein 1 and visinin-like protein-2 requires N-terminal myristoylation. The Journal of neuroscience : the official journal of the Society for Neuroscience. July272005;25(30):7071–80. doi: 10.1523/jneurosci.0452-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lautermilch NJ, Few AP, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by the neuronal calcium-binding protein visinin-like protein-2. The Journal of neuroscience : the official journal of the Society for Neuroscience. July272005;25(30):7062–70. doi: 10.1523/jneurosci.0447-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leal K, Mochida S, Scheuer T, Catterall WA. Fine-tuning synaptic plasticity by modulation of Ca(V)2.1 channels with Ca2+ sensor proteins. Proc Natl Acad Sci U S A. October162012;109(42):17069–74. doi: 10.1073/pnas.1215172109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss A, Ingram R, Koch S, et al. Origins, actions and dynamic expression patterns of the neuropeptide VGF in rat peripheral and central sensory neurones following peripheral nerve injury. Mol Pain. December102008;4:62. doi: 10.1186/1744-8069-4-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riedl MS, Braun PD, Kitto KF, et al. Proteomic analysis uncovers novel actions of the neurosecretory protein VGF in nociceptive processing. The Journal of neuroscience : the official journal of the Society for Neuroscience. October212009;29(42):13377–88. doi: 10.1523/jneurosci.1127-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen Y, Fu WY, Cheng EY, Fu AK, Ip NY. Melanocortin-4 receptor regulates hippocampal synaptic plasticity through a protein kinase A-dependent mechanism. The Journal of neuroscience : the official journal of the Society for Neuroscience. January92013;33(2):464–72. doi: 10.1523/jneurosci.3282-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen Y, Tian M, Zheng Y, Gong F, Fu AKY, Ip NY. Stimulation of the Hippocampal POMC/MC4R Circuit Alleviates Synaptic Plasticity Impairment in an Alzheimer’s Disease Model. Cell Rep. November82016;17(7):1819–1831. doi: 10.1016/j.celrep.2016.10.043 [DOI] [PubMed] [Google Scholar]
- 30.Willnow TE, Hilpert J, Armstrong SA, et al. Defective forebrain development in mice lacking gp330/megalin. Proc Natl Acad Sci U S A. August61996;93(16):8460–4. doi: 10.1073/pnas.93.16.8460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conrad CD, Galea LA, Kuroda Y, McEwen BS. Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behavioral neuroscience. December1996;110(6):1321–34. doi: 10.1037//0735-7044.110.6.1321 [DOI] [PubMed] [Google Scholar]
- 32.Sarnyai Z, Sibille EL, Pavlides C, Fenster RJ, McEwen BS, Toth M. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc Natl Acad Sci U S A. December192000;97(26):14731–6. doi: 10.1073/pnas.97.26.14731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Messier C Object recognition in mice: improvement of memory by glucose. Neurobiology of learning and memory. March1997;67(2):172–5. doi: 10.1006/nlme.1996.3755 [DOI] [PubMed] [Google Scholar]
- 34.Ojeda-Juárez D, Shah R, Fields JA, et al. Lipocalin-2 mediates HIV-1 induced neuronal injury and behavioral deficits by overriding CCR5-dependent protection. Brain Behav Immun. October2020;89:184–199. doi: 10.1016/j.bbi.2020.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winters BD, Forwood SE, Cowell RA, Saksida LM, Bussey TJ. Double dissociation between the effects of peri-postrhinal cortex and hippocampal lesions on tests of object recognition and spatial memory: heterogeneity of function within the temporal lobe. J Neurosci. June302004;24(26):5901–8. doi: 10.1523/jneurosci.1346-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kjeldsen L, Johnsen AH, Sengeløv H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. May151993;268(14):10425–32. [PubMed] [Google Scholar]
- 37.Zhao H, Konishi A, Fujita Y, et al. Lipocalin 2 bolsters innate and adaptive immune responses to blood-stage malaria infection by reinforcing host iron metabolism. Cell host & microbe. November152012;12(5):705–16. doi: 10.1016/j.chom.2012.10.010 [DOI] [PubMed] [Google Scholar]
- 38.Viau A, El Karoui K, Laouari D, et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. The Journal of clinical investigation. November2010;120(11):4065–76. doi: 10.1172/jci42004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu G, Li H, Fang Q, et al. Elevated circulating lipocalin-2 levels independently predict incident cardiovascular events in men in a population-based cohort. Arterioscler Thromb Vasc Biol. November2014;34(11):2457–64. doi: 10.1161/atvbaha.114.303718 [DOI] [PubMed] [Google Scholar]
- 40.Wieser V, Tymoszuk P, Adolph TE, et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J Hepatol. April2016;64(4):872–80. doi: 10.1016/j.jhep.2015.11.037 [DOI] [PubMed] [Google Scholar]
- 41.Eruysal E, Ravdin L, Kamel H, Iadecola C, Ishii M. Plasma lipocalin-2 levels in the preclinical stage of Alzheimer’s disease. Alzheimer’s & dementia (Amsterdam, Netherlands). December2019;11:646–653. doi: 10.1016/j.dadm.2019.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamzic N, Blomqvist A, Nilsberth C. Immune-induced expression of lipocalin-2 in brain endothelial cells: relationship with interleukin-6, cyclooxygenase-2 and the febrile response. J Neuroendocrinol. March2013;25(3):271–80. doi: 10.1111/jne.12000 [DOI] [PubMed] [Google Scholar]
- 43.Weng YC, Huang YT, Chiang IC, Tsai PJ, Su YW, Chou WH. Lipocalin-2 mediates the rejection of neural transplants. Faseb j. February2021;35(2):e21317. doi: 10.1096/fj.202001018R [DOI] [PubMed] [Google Scholar]
- 44.Jang E, Lee S, Kim JH, et al. Secreted protein lipocalin-2 promotes microglial M1 polarization. Faseb j. March2013;27(3):1176–90. doi: 10.1096/fj.12-222257 [DOI] [PubMed] [Google Scholar]
- 45.Mucha M, Skrzypiec AE, Schiavon E, Attwood BK, Kucerova E, Pawlak R. Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc Natl Acad Sci U S A. November82011;108(45):18436–41. doi: 10.1073/pnas.1107936108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dekens DW, Naudé PJW, Keijser JN, Boerema AS, De Deyn PP, Eisel ULM. Lipocalin 2 contributes to brain iron dysregulation but does not affect cognition, plaque load, and glial activation in the J20 Alzheimer mouse model. Journal of neuroinflammation. November302018;15(1):330. doi: 10.1186/s12974-018-1372-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferreira AC, Pinto V, S DM, et al. Lipocalin-2 is involved in emotional behaviors and cognitive function. Front Cell Neurosci. 2013;7:122. doi: 10.3389/fncel.2013.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orr ME, Sullivan AC, Frost B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol Sci. July2017;38(7):637–648. doi: 10.1016/j.tips.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. December292005;123(7):1293–305. doi: 10.1016/j.cell.2005.10.027 [DOI] [PubMed] [Google Scholar]
- 50.Hvidberg V, Jacobsen C, Strong RK, Cowland JB, Moestrup SK, Borregaard N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS letters. January312005;579(3):773–7. doi: 10.1016/j.febslet.2004.12.031 [DOI] [PubMed] [Google Scholar]
- 51.Dagil R, O’Shea C, Nykjær A, Bonvin AMJJ, Kragelund BB. Gentamicin binds to the megalin receptor as a competitive inhibitor using the common ligand binding motif of complement type repeats: insight from the nmr structure of the 10th complement type repeat domain alone and in complex with gentamicin. The Journal of biological chemistry. 2013;288(6):4424–4435. doi: 10.1074/jbc.M112.434159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burfeind KG, Zhu X, Norgard MA, et al. Microglia in the hypothalamus respond to tumor-derived factors and are protective against cachexia during pancreatic cancer. Glia. July2020;68(7):1479–1494. doi: 10.1002/glia.23796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. February2017;20(2):136–144. doi: 10.1038/nn.4475 [DOI] [PubMed] [Google Scholar]
- 54.Michaelis KA, Zhu X, Burfeind KG, et al. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle. October2017;8(5):824–838. doi: 10.1002/jcsm.12225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burfeind KG, Zhu X, Levasseur PR, Michaelis KA, Norgard MA, Marks DL. TRIF is a key inflammatory mediator of acute sickness behavior and cancer cachexia. Brain Behav Immun. October2018;73:364–374. doi: 10.1016/j.bbi.2018.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu X, Burfeind KG, Michaelis KA, et al. MyD88 signalling is critical in the development of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle. April2019;10(2):378–390. doi: 10.1002/jcsm.12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michaelis KA, Norgard MA, Zhu X, et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat Commun. October152019;10(1):4682. doi: 10.1038/s41467-019-12657-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pool M, Thiemann J, Bar-Or A, Fournier AE. NeuriteTracer: a novel ImageJ plugin for automated quantification of neurite outgrowth. J Neurosci Methods. February152008;168(1):134–9. doi: 10.1016/j.jneumeth.2007.08.029 [DOI] [PubMed] [Google Scholar]
- 59.Perkins JR, Dawes JM, McMahon SB, Bennett DL, Orengo C, Kohl M. ReadqPCR and NormqPCR: R packages for the reading, quality checking and normalisation of RT-qPCR quantification cycle (Cq) data. BMC Genomics. July22012;13:296. doi: 10.1186/1471-2164-13-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hennig C Cran-package fpc. https://cran.r-project.org/web/packages/fpc/index.html.
- 61.A Alexia RJ. topGO: Enrichment Analysis for Gene Ontology. R package version 1.38.1 (2019). [Google Scholar]
- 62.Mitchell AL, Attwood TK, Babbitt PC, et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. January82019;47(D1):D351–d360. doi: 10.1093/nar/gky1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sayers EW, Barrett T, Benson DA, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. January2010;38(Database issue):D5–16. doi: 10.1093/nar/gkp967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. October252005;102(43):15545–50. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. June152011;27(12):1739–40. doi: 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fabregat A, Jupe S, Matthews L, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. January42018;46(D1):D649–d655. doi: 10.1093/nar/gkx1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slenter DN, Kutmon M, Hanspers K, et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. January42018;46(D1):D661–d667. doi: 10.1093/nar/gkx1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie Z, Bailey A, Kuleshov MV, et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc. March2021;1(3):e90. doi: 10.1002/cpz1.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. April152013;14:128. doi: 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. July82016;44(W1):W90–7. doi: 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grossberg AJ, Zhu X, Leinninger GM, et al. Inflammation-induced lethargy is mediated by suppression of orexin neuron activity. The Journal of neuroscience : the official journal of the Society for Neuroscience. August32011;31(31):11376–86. doi: 10.1523/jneurosci.2311-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leger M, Quiedeville A, Bouet V, et al. Object recognition test in mice. Nature protocols. December2013;8(12):2531–7. doi: 10.1038/nprot.2013.155 [DOI] [PubMed] [Google Scholar]
- 73.Deacon RM. Assessing nest building in mice. Nature protocols. 2006;1(3):1117–9. doi: 10.1038/nprot.2006.170 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 2. Related to Figure 3. (a) Nest building score in male Lcn2-KO and WT mice 48 hours after LPS challenge (n = 5-6 per group). (b) Food intake as a percentage of initial body mass in Lcn2-KO and WT mice 48 hours after vehicle injection or LPS challenge (n = 5-7 per group). (c) Body mass change body mass in Lcn2-KO and WT mice 24 and 48 hours after vehicle injection or LPS challenge (n = 5-7 per group). (d) Survival analysis of Lcn2-KO and WT mice after a lethal dose of LPS (n = 10 per group). (e) Hourly food intake after re-feeding male WT mice after 16 hour fast (n = 4-5 per group). (f) Plasma LCN2 levels in mice after 16 hour of fasting (16 F), 16 hour fasting + 1 hour after re-feeding (1 HRF), and 16 hour fasting + 4 hours after re-feeding (4 HRF) (n = 11-12 per group; 5-6 males, 5-6 females; 16 F vs 1 HRF, p=0.0136).
Supplementary figure 3. Related to Figure 6. Flow cytometry gating strategy for quantifying immune cell subpopulations in the CNS of mice receiving (a) LCN2 or (b) vehicle. Cells were initially gated to remove debris, doublets and dead cells before gating on CD45+ cells. Immune cell markers were used to define the following subpopulations: (a) lymphoid cells = CD45+ CD11b−; (b) CD45+ CD11bmid; (c) myeloid cells = CD45+ CD11b+; (d) microglia = CD45lo CD11b+; (e) T cells = CD3+; (f) B cells = CD19+; (g) myeloid cells (non-neutrophils) = Ly6C+ Ly6G−; (h) neutrophils = Ly6C+ Ly6G+.
Supplementary figure 4. Related to Figure 9. (a) Total number of arm entries in the spontaneous alternation test. (b) Time to achieve novel object recognition criteria as defined by a total of 20 seconds of exploration with novel or familiar objects. (c) Latency to enter dark compartment of the light-dark box. (d) Number of transitions between light and dark compartments in the light-dark box text. (e) Total number of transitions between Y-maze arms in the Y-maze blocked arm test.
Supplementary Figure 1. Related to figure 1. Confirmation of LCN2 antibody specificity by IHC in WT and Lcn2-KO mice were injected with 200 μg/kg LPS. Images representative of 3 independently injected mice. 10x magnification.