

Abstract
Motivation
Biological systems function through dynamic interactions among genes and their products, regulatory circuits and metabolic networks. Our development of the Pathway Tools software was motivated by the need to construct biological knowledge resources that combine these many types of data, and that enable users to find and comprehend data of interest as quickly as possible through query and visualization tools. Further, we sought to support the development of metabolic flux models from pathway databases, and to use pathway information to leverage the interpretation of high-throughput data sets.
Results
In the past 4 years we have enhanced the already extensive Pathway Tools software in several respects. It can now support metabolic-model execution through the Web, it provides a more accurate gap filler for metabolic models; it supports development of models for organism communities distributed across a spatial grid; and model results may be visualized graphically. Pathway Tools supports several new omics-data analysis tools including the Omics Dashboard, multi-pathway diagrams called pathway collages, a pathway-covering algorithm for metabolomics data analysis and an algorithm for generating mechanistic explanations of multi-omics data. We have also improved the core pathway/genome databases management capabilities of the software, providing new multi-organism search tools for organism communities, improved graphics rendering, faster performance and re-designed gene and metabolite pages.
Availability
The software is free for academic use; a fee is required for commercial use. See http://pathwaytools.com.
Contact
Supplementary information
Supplementary data are available at Briefings in Bioinformatics online.
Keywords: Computational genomics, metabolic models, metabolic pathways, systems biology
1 Introduction
Pathway Tools is a software system that provides a wide range of tightly integrated capabilities from genome analysis to metabolic modeling to analysis of high-throughput data. Pathway Tools-derived databases exist for more than 20 000 organisms. Pathway Tools enables the creation and management of databases referred to as pathway/genome databases (PGDBs). These databases integrate a wide range of data types including genes, proteins, reactions, metabolites and pathways. Some PGDBs include additional data such as regulatory networks and growth phenotypes in experimental media. Pathway Tools can generate PGDBs from an input genome in GenBank format or GFF format. SRI makes available 14 500 PGDBs as part of our BioCyc database collection [1]; they can be downloaded with a few clicks within Pathway Tools via the PGDB registry [2]. Pathway Tools users have generated thousands of additional PGDBs. PGDBs are based on an expansive database schema that supports a mix of machine inferred and hand-curated information and evidence codes that distinguish the two. Pathway Tools includes components for viewing and analyzing PGDBs (Navigator), for creating new PGDBs (Pathologic), for editing existing PGDBs (Editors) and for building and running PGDB-based metabolic models (MetaFlux). Pathway Tools addresses a broad set of visualization and analysis needs in computational genomics and systems biology (see Figure 1):
Figure 1 .
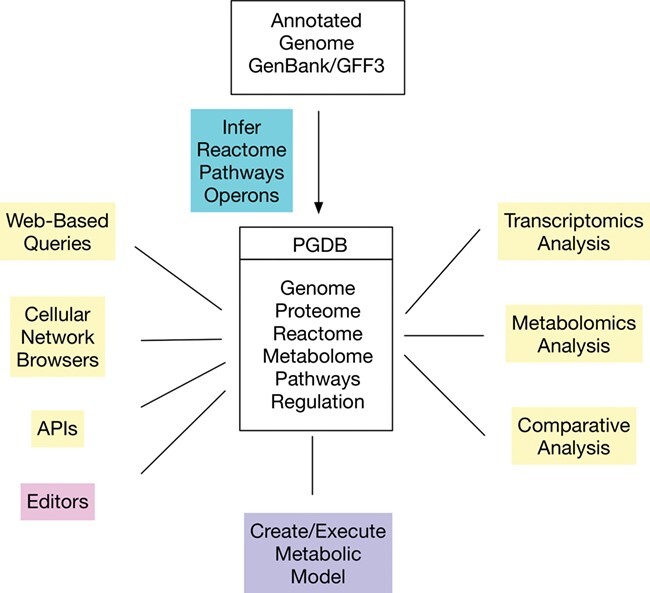
Major subsystems of Pathway Tools. Blue: PathoLogic. Yellow: Navigator. Purple: MetaFlux. Pink: Editors.
-

It performs computational inferences on annotated genomes, including metabolic reconstruction, filling of pathway holes (meaning reactions within a pathway that have no associated enzyme [3]) and prediction of operons.
-

It supports construction of steady-state and dynamic metabolic flux models. These models may include multiple organisms and spatial components.
-

It provides data analysis tools for gene-expression and metabolomics data. These include enrichment analysis of both genes for gene ontology (GO) terms, used by [4] and compounds enriched for pathways, used by [5].
-

It supports web-based dissemination of its databases with a range of search and display tools.
-

It supports comparative analysis across organisms for many data types including genes, reactions and pathways.
Metabolic reconstruction is the most commonly used feature of Pathway Tools in published studies, both as an end point (e.g. [6]) as well as a part of larger pipelines, such as [7] and [8]. There are more than 4000 externally developed databases listed at [9].
This article discusses new capabilities and refinements of the Pathway Tools environment developed during the past 4 years. A full description of Pathway Tools as of 2015 is available here [2]; an updated version of that article is under preparation. For a recent detailed comparison of BioCyc and Pathway Tools to software used in other microbial genome web portals, see [10].
The new capabilities presented here are listed in Table 1. There are many new and improved operations for managing PGDBs, such as new search, export and visualization operations (PGDB Management). A multi-organism metabolic route search and expanded metabolic pathway prediction algorithm are discussed in Pathway Informatics. MetaFlux presents improvements to the metabolic modeling capabilities of Pathway Tools. There are new capabilities in omics analysis including Pathway Covering, the Multi-Omics Explainer and the Omics Dashboard (Omics Analysis). Improvements to Pathway Tools metabolic map diagrams are discussed in Cellular Overview. In general we prefer new capabilities to be present in both Web mode and Desktop mode to reach the largest number of users, but this sometimes means implementing the user interface side of the tools twice, in Lisp and in Javascript (although for some user interface elements we can automatically convert Lisp-generated images to web graphics). The most common reasons for implementing a new feature for Desktop or Web but not both is that the feature pertains to a Pathway Tools component that is already specific to Web or Desktop only, such as to PathoLogic or to MetaFlux.
Table 1.
Pathway Tools capabilities introduced since Pathway Tools 19.0 (2015)
| Capability | Discussed in section | Web or Desktop |
|---|---|---|
| New search capabilities | PGDB Management | Both |
| GFF3 and Genbank export enhancements | PGDB Management | Desktop |
| Search for pseudogenes and cryptic prophages | PGDB Management | Web |
| Changed pseudogene representation | PGDB Management | Both |
| Show regulation details in pathway pages | PGDB Management | Both |
| Improved graphics and fonts | PGDB Management | Web |
| Revised protein feature coloring | PGDB Management | Both |
| Sequence coordinate mapping service | PGDB Management | Web |
| Redesigned gene pages | PGDB Management | Web |
| Redesigned metabolite pages | PGDB Management | Web |
| Notification of database updates | PGDB Management | Web |
| Scalability and performance | PGDB Management | Both |
| Multi-organism route search | Pathway Informatics | Web |
| Pathway prediction algorithm extended | Pathway Informatics | Desktop |
| SBML export | MetaFlux | Desktop |
| FVA | MetaFlux | Desktop |
| MetaFlux web solving mode | MetaFlux | Web |
| Send model fluxes to Dashboard | MetaFlux | Both |
| Send model outputs to GNUplot | MetaFlux | Desktop |
| Improvements to MetaFlux gap filler | MetaFlux | Desktop |
| Community models occupy spatial grid | MetaFlux | Desktop |
| Pathway covering | Omics Analysis | Web |
| Multi-omics explainer | Omics Analysis | Desktop |
| Omics Dashboard | Omics Analysis | Both |
| Import data from Metabolomics Workbench | Omics Analysis | Desktop |
| PPS | Omics Analysis | Web |
| Pathway collages | Omics Analysis | Both |
Note that Pathway Tools can operate in two distinct modes: desktop and web server modes; some tools are available only in one mode or the other. Features in web server mode are available through both the BioCyc.org web portal, and through Pathway Tools based web sites at other institutions (see [9]). Unless otherwise indicated, the capabilities described herein are present in both desktop and Web modes.
2 Comparison to related software
We recently published a comparison [10] between microbial genome web portals, including BioCyc (powered by Pathway Tools) [1], IMG [11], KBase [12], KEGG [13], PATRIC [14] and Ensembl Bacteria [15]. The comparison covered search capabilities, genomics capabilities, metabolic capabilities, table-analysis tools (e.g. SmartTables) and tools for analysis of transcriptomics and metabolomics data. BioCyc (Pathway Tools) was found to have the most extensive capabilities in all of these areas except for genomics tools, where it placed third.
The preceding comparison considers only the website functionality. Only the following tools support local downloading of the software as opposed to web-based operation: Pathway Tools, KEGG and KBase. Only the following tools enable users to process their own genomes to perform metabolic reconstructions: Pathway Tools, KEGG and KBase. Only the following tools provide an extensive toolkit of interactive editors allowing users to refine their PGDBs: Pathway Tools. Only the following tools enable users to operate their own websites using the software: Pathway Tools. Only the following tools enable construction of quantitative metabolic models: Pathway Tools, KBase and PATRIC. Only the following tools enable users to annotate genomes: PATRIC and KBase.
3 PGDB management
This section describes enhancements related to core operations of Pathway Tools such as PGDB search, export, visualization and performance.
3.1 Search
Pathway Tools has added several new search tools since version 19.0.
3.1.1 Multi-PGDB queries (Desktop only)
Many searches for entities such as pathways, compounds and genes can now be run against a collection of PGDBs, instead of against a single PGDB as previously. The user can define and save a named collection of PGDBs, which will become the subject of searches until changed. For example, this facility can be used to search multiple strains of the same species for the presence of a specified gene, or to search all organisms within a microbial community for the presence of a metabolite or a pathway. Searches return answer sets containing all objects that match the search across all the PGDBs in the collection.
3.1.2 Search for PGDBs by organism properties (Web only)
The web-based organism selection dialog now offers a search option for finding PGDBs based on properties of the organism. This tool can be used to select among the 14 500 PGDBs present within the BioCyc website. Example searches include searching for organisms with a given growth phenotype (e.g. presence of oxygen or temperature); the geographic region where the organism was collected; the human microbiome body site at which the organism was collected; or properties of the PGDB, such as the software used to annotate the genome, or the number of regulatory interactions present in the PGDB. This type of search will become more valuable as more metagenomics projects generate data tied to specific body sites (e.g. the Human Microbiome Project) or specific geographic regions or built environments (MetaSub [16]).
3.1.3 Search for DNA sites (Web only)
The web version of Pathway Tools now provides a Search for DNA sites option that enables users to search for 17 different types of DNA sites, including transcription units, prophages and pathogenicity islands. Likewise, the web version of Pathway Tools now supports searching for pseudogenes as an option of the Search/Filter by type/subunits of the Gene/Protein/RNA search.
3.1.4 Notification of database updates (Web only)
Users of the BioCyc website can now register interest in sets of genes, pathways, and/or GO terms. These interest areas are specified using Pathway Tools ontologies, such as using the pathway ontology to specify interest in cofactor biosynthesis, or using GO to specify interest in genes involved in the cellular process cell adhesion. When new information relevant to their interest areas is curated in BioCyc, the user is notified by email [17]. Email notifications are concise and targeted, with brief descriptions of what in the user’s interest area has changed, and links to the updated BioCyc web pages.
3.2 Export genome to GFF3 and GenBank formats (Desktop only)
Since 2005, we offered exporting a single replicon to the GenBank file format. Recently, we improved the export functionality to support inclusion of all the replicons in a PGDB’s genome into one GenBank or GFF3 file. GFF3 is a more stream-lined and cleaner format that is much easier to parse than is GenBank format, and is therefore recommended for use, going forward. GFF3 export is a new functionality. Within both exporters the user can select which replicons should be exported, and optionally, restricted start and end base-pair positions. Additionally, the desired types of features can be filtered, among choices such as genes, proteins, various RNAs, etc.
3.3 Show regulation details in pathway pages
Pathway diagrams can now be shown at a detail level that includes the transcriptional, translational and substrate-level regulators for each step, as computed from the regulatory data present within the PGDB. For each step, icons indicate the type of each regulator (protein, RNA, small molecule), and color indicates whether it activates or inhibits the reaction enzymes or genes. Mouseovers provide more information about the specific type and target of the regulation.
3.4 Improved graphics and fonts (Web only)
Pathway Tools has a large number of diagrams that are created automatically, some of them quite complex: the cellular overview (metabolic map), pathway diagrams at a variety of levels of detail, reaction diagrams including atom mappings, the genome browser, etc. All of these diagrams are laid out and drawn algorithmically, and rendered for the desktop or for the web. Previously, we rendered them for the Web as GIF images or using Scalable Vector Graphics (SVG).
We have implemented a modern web rendering system to generate what we call ‘web graphics’ files. A browser-based engine that we wrote renders these files to HTML5 canvases in the browser window. This means our diagrams are much higher resolution, the user can smoothly zoom the diagrams in real time, and we have decreased our dependence on less stable and widespread technologies such as SVG. At the same time, the core, robust algorithms for laying out and drawing our diagrams, which represent years of development, remain in place.
3.5 Revised protein feature coloring
We have revised the coloring scheme within the protein-feature display, to assign a dozen colors according to our ontology of feature types. For example, metal binding sites are grey and sequence variations are ochre.
3.6 Re-designed gene and metabolite pages (Web only)
The gene and metabolite (compound) web pages have been redesigned with a more modern, tab-based interface (Figure 2)(note to reviewers: we are open to moving some of the figures to Supplemental Material). Each tab contains a subset of the information previously listed in one large page, reducing the amount of user scrolling required, and making more apparent the types of information available. Tabs for a gene page include reactions catalyzed by the gene product(s), and the GO terms associated with a gene. These tabbed sections display data in tables for easier reading and faster loading. The reactions tab is shown in Figure 3. The set of tabs available are sensitive to the information available for the gene and its product(s). A ‘Show All’ tab replicates the previous all in one display.
Figure 2 .
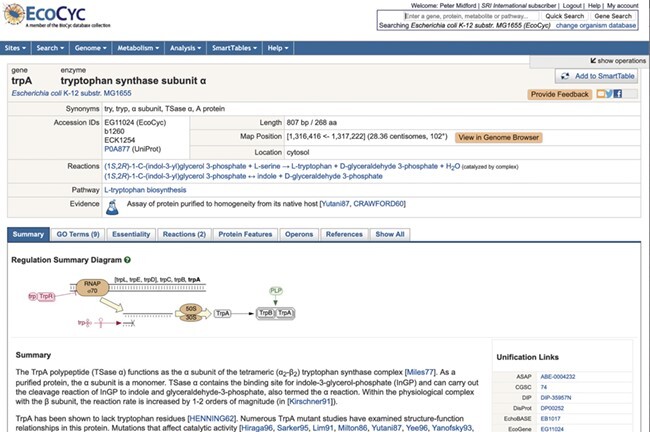
Top portion of trpA gene page showing tabbed sections and regulation summary diagram using new ‘web graphics’ rendering.
Figure 3 .
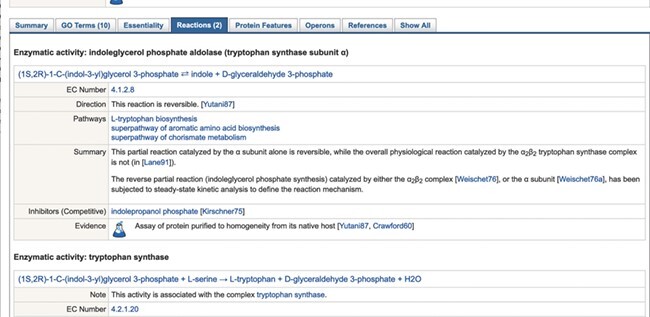
The top of the ‘Reactions’ tab of the trpA gene page.
Tabs for metabolite pages include ontology information, reactions catalyzed and regulated by the metabolite and the chemical structure.
Much of the information on these pages, and other object specific pages (e.g. pathways) are available via a REST API that returns XML[18], APIs for other languages (Lisp, Python, Java, etc.) or via flat files.
3.7 Sequence coordinate mapping service (Web only)
The authoritative or reference DNA sequence of a replicon is sometimes updated to fix sequencing errors. Because some of the errors can involve insertions or deletions, the base-pair coordinates further downstream will shift, compared to the uncorrected sequence. Such updates affect the positions of genes, promoter sites, and other regions of importance.
We implemented a web form that maps a user’s data files, which contain older coordinate information, to coordinates appropriate for the latest genome version. Note that this operation will not change the coordinates used in the PGDB itself, which always correspond to the latest version. However, it will enable updating older data files, so the revised file can be used for analyses against the latest sequence.
Pathway Tools provides a sequence editor that can be used to update the nucleotide sequence of a replicon. These individual edits are recorded in the replicon frame in the PGDB. The coordinate mapping works by chaining together these edits to produce the overall mapping between two specified versions of the replicon.
3.8 Scalability and performance
We improve the scalability and performance of Pathway Tools on an ongoing basis. Because gene and metabolite pages are the most highly referenced page types in the BioCyc website, we have made many changes to increase the speed with which these pages are generated, with the result that BioCyc gene pages are the second fastest (with KEGG being the fastest) among all microbial genome web portals [10]. We have also improved the speed of the comparative genome browser by a large factor. We have improved the speed of PathoLogic pathway inference substantially. We have improved the speed of many queries under the web search menu, including Search Genes, Proteins, or RNAs; Search Compounds; Search Reactions; Search Pathways; and Search DNA or mRNA Sites. After much study we determined that the large memory usage of BioCyc web servers was due in significant part to the use of Lisp symbols as the object identifiers by the Ocelot object-oriented database used within BioCyc. By changing Ocelot object identifiers to strings, which are garbage collected by Lisp, we decreased the memory usage of BioCyc web servers by ˜50%.
4 Pathway informatics
4.1 Multi-organism route search (Web only)
Multi-organism route search (MORS) [19] extends our previous single-organism metabolic route search to accept arbitrary sets of organisms, simultaneously, for searching across the union of the reactions in the selected organisms. A typical use case is searching HumanCyc plus the organisms in a microbiome body site, such as the gut, to investigate how a combination of organisms might synthesize a toxic compound, and to see which specific organisms are participating.
Originally, the route search operation would seek the best (least cost) route between user-specified start and goal compounds. A route is a linear series of reactions. The cost of a route involves a weighted combination of the length of the route (number of reactions) and the number of atoms lost from the start to the goal compound (computed using atom mapping information).
The MORS mode adds a multi-organism selector for selecting the set of BioCyc organisms to be searched; the set of reactions searched by MORS will be the union of reactions from that organism set. Additionally, a cost for ‘organism switching’ can be set. A switch occurs when the two organism sets of two consecutive reactions in a route have no overlap. In other words, if the first reaction is known to occur in one set of organisms and the second reaction is occurring in a different organism set, but there is no organism that contains both reactions simultaneously, then the route has to switch organisms by transferring the compound connecting both reactions, from one organism to another (by unspecified transport mechanisms). An organism switch is depicted in a route with a red vertical line. A SmartTable of the route can be generated, which shows the organism sets containing enzymes that catalyze each reaction along the route.
In addition to their direct utility as paths from one compound to another, routes identified by MORS could identify new alternative mult-organism pathways for use in analysis of meta-transcriptomics data, such as via enrichment analysis, or constraint based methods that use gene-expression data, such as implemented in Metabolizer [20].
As an example, let us examine how dietary L-tyrosine is transformed into toxic 4-methylphenyl sulfate, which is a protein fermentation product that has been modified in the liver and is implicated in kidney problems. As it is known that this toxin originates from L-tyrosine [21], the start compound was set to L-tyrosine and the goal compound to 4-methylphenyl sulfate. We selected all organisms in the human microbiome body site called ‘gastrointestinal-tract’ plus Homo sapiens. The total count of organisms was 675. The resulting top two routes are shown in Figure 4. Both routes retain eight atoms. The first route consists of two reactions, and the second of four reactions. The last reaction, after the organism switch, is only found in Homo sapiens. However, the reaction immediately before the switch occurs in 412 organisms in the first route and 80 organisms in the second.
Figure 4 .
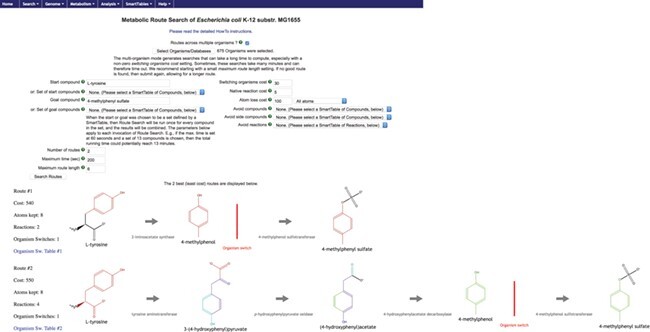
Result of MORS routes to 4-methylphenyl sulfate.
4.2 Pathway prediction algorithm revamped (Desktop only)
The PathoLogic algorithm for metabolic reconstruction takes as input an annotated genome and as output it predicts the metabolic reactions catalyzed by enzymes in the genome, and a set of metabolic pathways containing those metabolic reactions. In the past, the input genome could be provided in GenBank format or in PathoLogic format; the genome can now also be provided in GFF3 format.
PathoLogic was also updated to produce more accurate pathway predictions. Pathway prediction uses a rule-based expert system [22] that is more accurate than a machine-learning algorithm that we developed [23], and also produces explanations of its decisions. The expert system makes use of some of the more informative features used by the machine-learning system. We extended the expert system to now compute a score for each pathway. The pathway score depends on two factors: (1) the number of non-spontaneous pathway reactions for which enzymes are present in the genome, which is computed as a score for each reaction, and (2) whether the organism whose pathways are being predicted is in the expected taxonomic range of the pathway. The reaction score depends on whether an enzyme catalyzing the reaction is present in the genome, the uniqueness of the reaction (i.e. is the reaction found in one or a few pathways, or many pathways), and whether the reaction is considered a ‘key reaction’, that is, a reaction whose presence is a strong indicator for the presence of a given pathway.
The pathway-prediction expert system was tested after each update on gold-standard curated BioCyc PGDBs for Saccharomyces cerevisiae and for Synechococcus elongatus, both of which PGDBs are updated periodically to reflect new experimental information.
5 MetaFlux
MetaFlux is the metabolic modeling component of Pathway Tools. It supports creation, debugging and execution of metabolic models for individual organisms and for organism communities, using both flux balance analysis (FBA) and dynamic flux-balance analysis (dFBA). FBA is a modeling technique that computes steady-state fluxes for every reaction in an organism’s metabolic network. dFBA runs FBA simulations for a series of time steps to compute dynamic behavior of a metabolic network.
Advantages of MetaFlux compared to other constraint-based modeling software packages include (a) The desktop distribution of MetaFlux includes the SCIP solver [24], therefore the user need not install third-party LP or MILP solvers or determine appropriate parameters for these solvers; (b) MetaFlux can be run through a graphical user interface, therefore it can be used by users who are not programmers; (c) By coupling metabolic models with PGDBs, models become much more easily understandable and reusable since they can be explored using Pathway Tools’ extensive query and visualization tools, and they contain extensive information not present in other modeling tools that enriches the mode, such as chemical structures, reaction atom mappings, pathway definitions, and the genome sequence.
5.1 Analytic improvements
5.1.1 Improvements to MetaFlux gap filler (Desktop only)
Gap-filler software assists the developers of metabolic models by hypothesizing additional reactions to add to an organism’s metabolic network. Reactions are typically missing because of incompleteness in the genome annotation. We performed a study of gap-filler accuracy [25] in which we explored 13 variations of the 2 gap fillers initially present within MetaFlux. As a result of this study we replaced the MetaFlux GenDev gap filler with a different variation called ‘Technique C with Big M’ [25]. This variation produces fewer erroneous gap-filler solutions than the previous algorithm. A solution is erroneous if it does not in fact enable the metabolic model to produce all of the specified biomass metabolites. The new Technique C-based gap filler also has high accuracy (although not the highest of all variants tested), and it provides added information to the user beyond the information provided by other gap-filler variants, namely it identifies to the user which biomass metabolites can still not be produced even if all reactions from MetaCyc are added to the model.
5.1.2 Flux variability analysis (Desktop only)
Flux variability analysis (FVA) can be used to determine the robustness of metabolic models under different simulated growth conditions by computing the minimum and maximum possible flux values of each reaction. After FVA is run, a report is generated showing the reactions which are blocked (these reactions cannot carry flux), fixed (these reactions have identical, non-zero minimum and maximum flux values) or free (these reactions have flux values that span a range). Other uses of FVA may include classifying reactions as to whether they are fully-coupled, partially coupled or not coupled to biomass flux or investigating alternate solutions by fixing relevant reaction flux values.
5.1.3 MetaFlux web execution of metabolic models
MetaFlux can now execute metabolic models via its ‘solving mode’ through web mode in addition to desktop mode. This mode of operation is available for the EcoCyc metabolic model [26] via EcoCyc.org. The user can execute a metabolic model after optionally modifying its nutrients, secretions, biomass metabolites or enabled reactions.
5.2 New output visualizations
5.2.1 Visualize computed reaction fluxes on omics dashboard
MetaFlux has been extended so that the reaction fluxes computed by solving mode can be displayed on the Omics Dashboard [27] to speed user interpretation of fluxes. The Dashboard is an interactive tool for hierarchical visualization of large-scale data sets. The top level of the Dashboard graphs the aggregate behavior of every metabolic subsystem of the cell, from nucleotide biosynthesis to aromatic compound degradation to aerobic respiration. The user can drill down into any of these graphs to view the fluxes of individual pathways.
5.2.2 MetaFlux community models output to GNUplot (Desktop only)
When MetaFlux is used to model a community of organisms, its output results consist of organism biomass values and metabolite concentrations for each time point in the simulation for those metabolites supplied as nutrients or secreted into the extracellular space. If the simulation takes place across a spatial grid, then the output consists of organism biomass values and metabolite concentrations at each square within the grid, for each time point in the simulation. We developed several new visualization tools that aid the user in digesting this large amount of output data:
-

X-Y plot: aggregated biomasses/metabolites. This tool produces two separate X-Y plots. One graphs the organism biomasses (in gDW) accumulated in the grid as a function of time. The second graphs aggregate metabolite amounts (in mmol) across the grid as a function of time. The metabolites shown are for the extracellular compartment. The metabolites and organisms shown are selected by the user.
-

X-Y Plot: metabolites used/produced per organism. The metabolites used and produced by each organism at each time step are shown in a separate X-Y plot for each organism. The metabolites are selected by the user. (The X-axis is time; the Y-axis shows aggregate metabolite concentrations across the grid for that organism.)
-

Static grids: biomasses/metabolites. A series of static 2D heatmaps depict the spatial grid in which the organism community grows, showing the organism biomasses (in gDW per grid box) and/or metabolites (in mmol per grid box) at several time steps. Each image of the grid depicts one organism and one metabolite among those selected by the user, at one time point.
-

Dynamic grids: biomasses/metabolites. This option is similar to the preceding static grids display, but an MPEG movie is created, in which each simulation time point corresponds to one step in the animation. An animation step is shown every three s. The FFmpeg program is used to create this MPEG movie, which will be shown using the default web browser installed on the user’s computer.
5.2.3 MetaFlux community models occupy spatial grid (Desktop only)
The organisms within a community model share the same physical space, which can now be specified by the user as a rectangular grid such as to simulate an animal digestive tract. At the start of the simulation, the user seeds the grid squares with different abundances of organisms and metabolite concentrations. Organisms and metabolites may also be introduced at specified grid squares at specified time points as the model runs. At each Dynamic FBA time step, metabolite concentrations are updated to reflect metabolites secreted by each organism, and by computed diffusion of metabolites across the grid (diffusion of organisms is also computed). This computation emulates the dispersion of metabolites and organisms due to Brownian collision. The diffusion coefficients of the metabolites and organisms are automatically selected by MetaFlux. An example showing the grid for two colonies of Escherichia coli K-12 is shown in Figure 5. The simulation shown here is anaerobic for the first five time steps and aerobic for the remaining time. The colony in the upper right is the wild type, but the colony in the lower left is missing a single reaction (NADH:ubiquinone reductase) which reduces its ability to use oxygen to speed growth, thus somewhat lower population and less CO2 production.
Figure 5 .

Example spatial grid output from a MetaFLux community model showing two colonies of E coli K-12 at the corners, and concentrations of four metabolites from the FBA model.
5.3 SBML export
The SBML (Systems Biology Markup Language) [28] export functionality in Pathway Tools has been updated. SBML is an XML-based, machine-readable format for representing models of reaction networks and their gene associations. SBML files can now be generated either for a selected set of reactions for a given PGDB, or for a flux-balance-analysis model by providing a MetaFlux FBA file.
6 Omics analysis
The current version of Pathway Tools significantly extends the set of tools available for analysis and display of metabolomics, gene expression, and proteomics data — gene expression and proteomics data are treated equivalently by these tools, it is simply a matter of the user providing gene names or identifiers versus protein names or identifiers.
6.1 Omics dashboard
The Omics Dashboard [27] is a novel tool for interactive exploration and analysis of omics data sets through a hierarchy of cellular systems. At its highest level the Dashboard contains panels for cellular systems such as biosynthesis, energy metabolism and cellular processes (see Figure 6). Each panel contains a series of X–Y plots depicting the amalgamated expression levels of genes (or quantities of metabolites, in the case of metabolomics data) within the subsystems of that panel. For example, the Response to Stimulus panel includes plots for its component subsystems starvation, DNA damage, osmotic stress and others. Clicking on any plot shows an expanded panel for that subsystem composed of either (a) plots for its component subsystems, if component subsystems are defined, or for ‘leaf’ subsystems (b) a graph of omics data values for all genes (or metabolites) in the subsystem (see Figure 7). For example, clicking on the Amino Acid Biosynthesis plot in the Biosynthesis panel brings up a panel consisting of plots for each individual amino acid. Clicking on the arginine biosynthesis plot then shows a graph of all the genes involved in arginine biosynthesis. This organization allows a user to quickly visually identify broad systems of interest, and then successively focus in on more specific areas of function. The set of systems and subsystems represented in the Omics Dashboard is derived from ontologies within Pathway Tools, primarily the pathway ontology and GO.
Figure 6 .
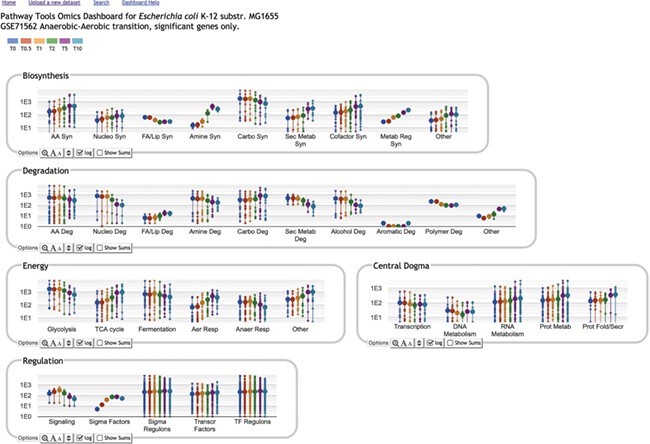
Truncated top-level dashboard view of a gene expression time series data set [29] for E. coli, showing changes in gene expression after transition to aerobic conditions at 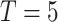 . Other panels include Regulation, Cellular Processes, Cell Exterior and Response to Stimulus
. Other panels include Regulation, Cellular Processes, Cell Exterior and Response to Stimulus
Figure 7 .
Dashboard drill down to expression of genes producing proteins involved in response to DNA damage, same data set as Figure 6.
In addition to panels and plots, the Omics Dashboard also provides access to pathway diagrams painted with omics data, diagrams showing the operon organization of all genes within a given biological system and plots of all known regulators for the genes within a given system. It is highly customizable, both in terms of appearance and content. Enrichment analysis (Hypergeometric approach) is also supported. Data for the Omics Dashboard and other omics analysis tools can come from a previously loaded data set, including data from GEO, a text file, or from a SmartTable.
6.2 Pathway covering (Web only)
Pathway Covering is a new approach to interpreting metabolomics data, available in the web version of Pathway Tools [30]. It addresses the same general question as enrichment scores of pathways for compounds, but takes an approach based on set theory rather than a statistical model such as the hypergeometric distribution. It takes a list of metabolites as input, and as output it reports a set of pathways that includes all metabolites in an input list as substrates (inputs, outputs or intermediates). The set returned by Pathway Covering will always be a minimum cost solution. Each pathway has a ‘cost’ that is calculated by a user selected cost function. The sum of the costs of each pathway is the cost of a pathway-covering solution set. The set of pathways returned will have the lowest total cost (through other possible sets may have the same cost).
Pathway Tools provides five cost functions for use with Pathway Covering:
(i) Constant - This returns a fixed value for all pathways.
(ii) Pathway size - This returns the number of reactions in the pathway.
(iii) Biosynthesis preferred - This starts with the number of reactions in the pathway, then reduces the cost by a factor of two if the pathway is classified as a biosynthesis pathway in the MetaCyc pathway ontology.
(iv) Compound sparseness - This considers all the substrate metabolites of a pathway and divides the number of substrate metabolites that were not in the input set by the number that were in input set. Because this is a cost function, returning large values for low proportions of compounds in the input set is appropriate.
(iv) Pathway Harmony - This loosely approximates the notion of pathway flux. It is sensitive to the direction of change in compound abundance. It takes all the compounds associated with the pathway and divides them into three groups: input compounds, output compounds, and intermediates. The function gives low scores if all or a majority of compounds in a group are changed in the same direction.
Pathway covering typically generates a different set of pathways than an enrichment, which ignores whether every metabolite is included in the result. Some weighting functions, e.g. Pathway Harmony, will also make use of the direction of change of a metabolite between a pair of experimental conditions.
Pathway Tools reports the set of covering pathways as a table. The table additionally includes, the subset of the user supplied compounds that are covered by each pathway, and a thumbnail showing the location of each user-supplied compound in the pathway that contains it. An example report page is shown in Figure 8.
Figure 8 .
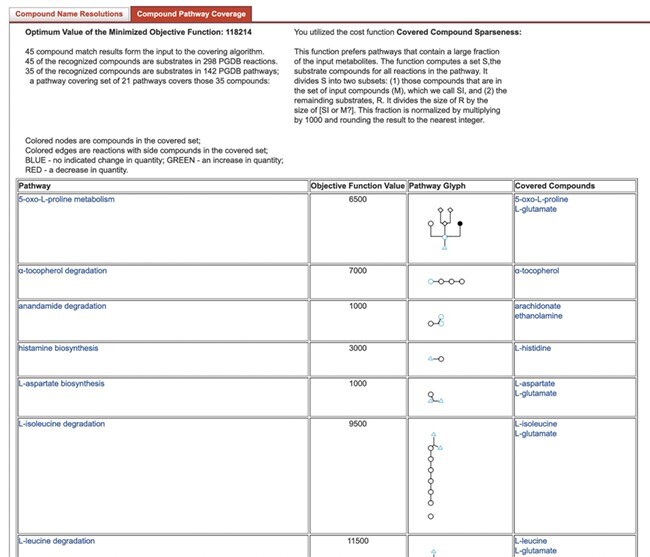
Results of Pathway Covering.
6.3 Import data from Metabolomics Workbench (Desktop only)
The desktop version of Pathway Tools can now import data for analysis from the Metabolomics Workbench [31]. The Metabolomics Workbench includes a repository of over 1000 metabolomics studies, many of which include identified compounds that can be imported into Pathway Tools. Once imported, the data can be overlaid on the cellular overview metabolic map diagram. The dialog for downloading data includes filters for species and keywords. It also supports grouping experimental results by factors common to multiple experiments such as subject sex and disease state, which simplifies working with clinical data sets with many replicates.
6.4 Pathway perturbation score (Web only)
In additional to visualization, and pathway covering for omics data, Pathway Tools provides another way to assess which pathways are affected by an omics experiment. Pathway Tools can compute a pathway perturbation score (PPS) for each pathway with omics data at a single timepoint. The PPS attempts to measure the overall extent to which a pathway is expressed by averaging the level of deviation from zero (after log transformation) over all the reactions in the pathway. For omics data representing a time series, the differential PPS (DPPS) measures the extent to which a pathway exhibits change between timepoints. Pathway Tools can use omics data from either gene expression or metabolite abundance to calculate these scores, we have not attempted to validate that the perturbations scores are consistent between gene and metabolite data in such mixed sets.
The PPS is based on the sum of the perturbation scores for each reaction in the pathway—the reaction perturbation score (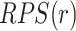 for each reaction
for each reaction 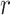 in the set of reactions
in the set of reactions 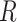 ). The reaction perturbation score in turn looks at each object associated with the reaction (e.g. genes for gene expression data, or compounds for metabolomics data). The perturbation score is a measure of deviation from zero across the participants or genes in the reaction. For each object, the data value is log transformed (if it was not previously) and then from the set of values for the reaction, the absolute values are computed and the largest absolute value is the reaction perturbation score.
). The reaction perturbation score in turn looks at each object associated with the reaction (e.g. genes for gene expression data, or compounds for metabolomics data). The perturbation score is a measure of deviation from zero across the participants or genes in the reaction. For each object, the data value is log transformed (if it was not previously) and then from the set of values for the reaction, the absolute values are computed and the largest absolute value is the reaction perturbation score.
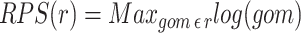 |
(1) |
where 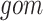 refers to the data representing the level of each “gene or metabolite” that is associated with (expressed as membership) of the reaction
refers to the data representing the level of each “gene or metabolite” that is associated with (expressed as membership) of the reaction 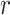 . The PPS is the root mean square of the reaction perturbation scores, or
. The PPS is the root mean square of the reaction perturbation scores, or
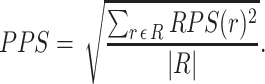 |
(2) |
where 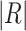 is the number of reactions in the pathway.
is the number of reactions in the pathway.
When the data is a time series, the DPPS is used to capture the overall magnitude of pathway activity change across time points; pathways with high DPPS scores exhibit stronger changes. The calculation is similar to the PPS except that the values for each component (gene, compound) associated with a reaction is the difference of the maximum and minimum value observed in the time series rather than a deviation from a baseline.
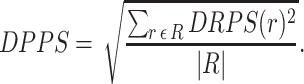 |
(3) |
where 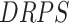 for each reaction is
for each reaction is
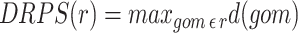 |
(4) |
and 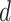 for each gene or metabolite associated with the reaction is
for each gene or metabolite associated with the reaction is
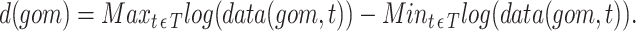 |
(5) |
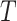 is the range of time points in the data set and
is the range of time points in the data set and 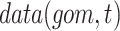 refers to the value of a gene or metabolite at a timepoint
refers to the value of a gene or metabolite at a timepoint 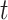 .
.
PPSs are displayed as a table with pathways sorted by decreasing PPS or DPPS depending on whether the data is a time series. The table for time series data includes a graph of the PPS at each time point as well as the DPPS across all time values.
A limitation of the PPS and DPPS is that they do not take into account the consistency of expression changes, e.g. if a regulator 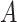 increased in expression, and the expression of a protein
increased in expression, and the expression of a protein 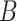 that was negatively regulated by
that was negatively regulated by 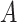 also increased, this could be viewed as an inconsistency. This concern applies only to signaling pathways since metabolic pathways do not contain regulators.
also increased, this could be viewed as an inconsistency. This concern applies only to signaling pathways since metabolic pathways do not contain regulators.
6.5 MultiOmics explainer (Desktop only)
High-throughput experiments can reveal associations between genes, proteins and/or metabolites whose source is not immediately obvious to researchers, but which can be explained by existing knowledge. The MultiOmics Explainer is a new tool that leverages what is known about an organism’s metabolic and regulatory network to suggest explanations for some of the results of omics experiments (Figure 9). Its goal is to speed understanding of experimental results, find explanations that scientists might otherwise overlook and aid researchers in differentiating that effects can and cannot be explained by existing knowledge.
Figure 9 .
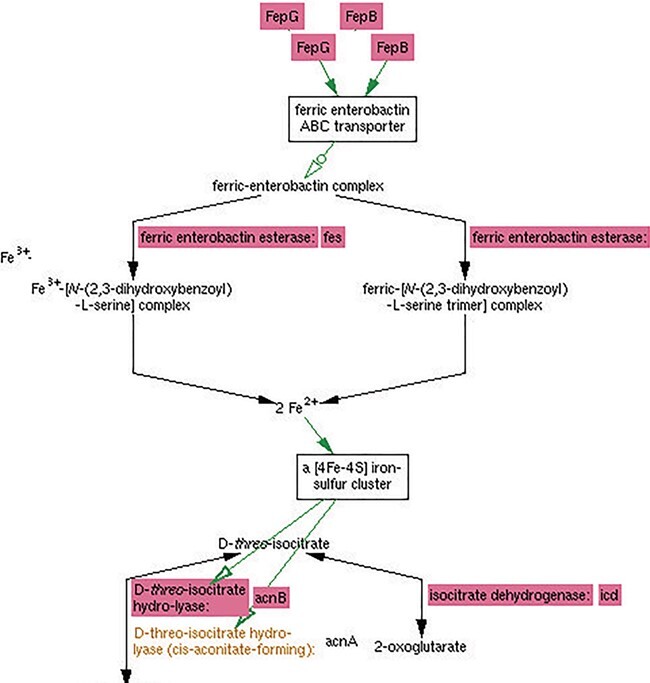
MultiOmics Explainer graph showing genes from [32] that, when knocked out, cause levels of cis-aconitate to increase. In particular, these genes indicate a connection between enterobactin-related genes and cis-aconitate metabolism that might not otherwise have been obvious.
Given a small number of effect entities (genes, proteins or metabolites), the tool constructs a network of known influences on those entities. If one or more condition entities (for example, a knocked-out gene or a changed metabolite in the medium) are supplied, the tool attempts to find causal paths in the network that connect the conditions to the effects. If no condition entities are supplied, then the tool attempts to identify common influencers (for example, a regulatory gene) that can be linked to the effect entities. The output of the MultiOmics Explainer is an interactive diagram that illustrates one or a small number of possible routes by which the condition entities, or common influencers, influence the effect entities.
The combined metabolic and regulatory network used by the MultiOmics Explainer comprises a set of interactions assembled from an organism’s (a) metabolic, transport and protein modification reactions; (b) enzymes and enzyme activators, inhibitors and cofactors; (c) transcriptional regulators such as sigma factors, transcription factors and the small molecules that bind them; and (d) translational regulators such as attenuators, regulatory proteins and RNAs and small molecule riboswitches. The MultiOmics Explainer is unique in the wide range of potential causal relationships it considers, reflecting the depth and richness of the Pathway Tools ontology.
6.6 Pathway Collages
The Pathway Collages facility enables the user to create multi-pathway diagrams that can be annotated, printed and shared. A set of pathways can be specified in several ways in Pathway Tools, and the pathways are then exported to a separate web application. Although the web application requires a browser, sets of pathways can be created and exported from the desktop version of Pathway Tools, which will launch a browser after the export is complete.
In the web version of Pathway Tools, pathway sets for a collage can be specified by including a column of pathways in a SmartTable, from data uploaded into the Cellular Overview, by creating a collage with a single pathway from the pathway’s page, or by selecting pathways from a checklist on the page describing pathway collages. In the desktop version of Pathway Tools pathways can be selected from a SmartTable or from a select pathways tool available in the Cellular Overview.
Within the Pathway Collage tool, the user initially sees a high-level drawing of the pathways they selected. The user can zoom in (and show more detail), move a pathway by dragging it, and perform editing operations (changing drawing properties or labels, deleting) on compounds or reactions within the pathway. Compound editing also includes the option to edit all occurrences of the compound within the diagram or import other pathways that involve the compound. Compounds can be connected with links, either linking all occurrences of a compound, or linking pairs of the users choosing. Omics data can also be added to a collage while in the collage editor.
Finally, the collage can be saved for further editing or sharing, or it can be exported as a PNG file.
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Peter D. Karp is the Director of the Bioinformatics Research Group at SRI International. He received the PhD degree in Computer Science from Stanford University. SRI International is a multi-disciplinary non-profit research institute headquartered in the San Francisco area.
Peter E. Midford is a Computational Biologist at SRI International. He received an MS in Computer Science from Yale University and a PhD in Zoology from University of Wisconsin-Madison.
Richard Billington received a MS from the University of Pennsylvania in Computer and Information Science. He is a Senior Software Developer at SRI.
Anamika Kothari is a Software Developer at SRI International. She holds a graduate Degree in Chemistry and Biochemistry from Mumbai University, India.
Markus Krummenacker a Scientific Programmer, has worked on Pathway Tools/BioCyc for 15 years. His interests reside in the intersection and synergy between (bio-)chemistry and computer science, ultimately culminating in atomically precise manufacturing.
Mario Latendresse received a PhD in Computer Science and has published in bioinformatics, programming language compilation, malware detection, functional languages and high-performance computing.
Wai Kit Ong is a Computational Biologist in the Bioinformatics Research Group at SRI. He received his PhD in Chemical Engineering from University of Wisconsin-Madison with a focus in Systems Biology.
Pallavi Subhraveti is a Scientific Programmer/Release Manager at SRI International. She received a BS in Biology from UC Riverside and a MS in Computer Science from Cal State Northridge.
Ron Caspi curator of the MetaCyc database, received his PhD in biology from the Scripps Institution of Oceanography, La Jolla, CA. He is secretary of the IUPAC-IUBMB Joint Commission on Biochemical Nomenclature (JCBN) and a member of the Enzyme Commission (EC).
Carol Fulcher is a Scientific Database Curator at SRI International. She received a PhD in Molecular Genetics from the University of Virginia.
Ingrid M. Keseler is a Senior Scientific Database Curator in the Bioinformatics Research Group at SRI International. She has an MS in Microbiology from the University of Georgia and a PhD in Biochemistry from Stanford University.
Suzanne M. Paley is a Senior Computer Scientist in the Bioinformatics Research Group at SRI International, and a developer of Pathway Tools since the project’s inception. She holds an MS degree in Computer Science from UC Berkeley, and an MS degree in Chemistry from Stanford University.
KEY POINTS
In the past 4 years Pathway Tools has seen major expansions in PGDB management, pathway informatics, metabolic modeling and omics data analysis.
In the area of PGDB management, a number of new queries and visualizations were added, modern graphics and fonts were added to Web visualizations, and a new database update-notification service was added. New multi-organism query capabilities were added for querying genomes from organism communities.
Pathway informatics enhancements include a MORS tool, and enhancements to the pathway prediction accuracy.
Many improvements were made to the MetaFlux metabolic modeling tool, including improvements to SBML export, addition of FVA, improvements to the reaction gap filler, ability to execute models through the web and the ability to send model outputs to the Omics Dashboard and to GNUplot.
Multiple new omics-data analysis tools were introduced, including the Omics Dashboard, the pathway covering algorithm, the MultiOmics Explainer, the pathway-perturbation score and pathway collages.
Funding
Development of Pathway Tools was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers GM075742, GM080746, and GM077678.
References
- 1.Karp PD, Billington R, Caspi R, et al. The BioCyc collection of microbial genomes and metabolic pathways. Briefings in Bioinformatics 2017; 20:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karp PD, Latendresse M, Paley SM, et al. Pathway Tools version 19.0: Integrated software for pathway/genome informatics and systems biology. arXiv, 2015, 1–79.
- 3.Green ML, Karp PD. A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics 2004; 5(1): 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duru IC, Laine P, Andreevskaya M, et al. Metagenomic and metatranscriptomic analysis of the microbial community in swiss-type maasdam cheese during ripening. Int J Food Microbiol 2018; 281:10–23. [DOI] [PubMed] [Google Scholar]
- 5.Andreevskaya M, Jaaskelainen E, Johansson P, et al. Food spoilage-associated leuconostoc, lactococcus, and lactobacillus species display different survival strategies in response to competition. Appl Environ Microbiol 2018; 84(13): e00554-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schatschneider S, Schneider J, Blom J, et al. Systems and synthetic biology perspective of the versatile plant-pathogenic and polysaccharide-producing bacterium xanthomonas campestris. Microbiology-SGM 2017; 163(8): 1117–44. [DOI] [PubMed] [Google Scholar]
- 7.Hahn AS, Altmann T, Konwar KM, et al. A geographically-diverse collection of 418 human gut microbiome pathway genome databases. Scientific Data 2017; 4. 10.1038/sdata.2017.35. 170035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Francis TB, Kruger K, Fuchs BM, et al. Candidatus prosiliicoccus vernus, a spring phytoplankton bloom associated member of the flavobacteriaceae. Syst Appl Microbiol 2019; 42(1): 41–53. [DOI] [PubMed] [Google Scholar]
- 9.Pathway/Genome Database Websites . https://BioCyc.org/otherpgdbs.shtml.
- 10.Karp PD, Ivanova N, Krummenacker M, et al. A comparison of microbial genome web portals. Front Microbiol 2019; 10:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen IA, Markowitz VM, Chu K, et al. IMG/M: integrated genome and metagenome comparative data analysis system. Nuc Acids Res 2017; 45(D1): D507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arkin AP, Cottingham RW, Henry CS, et al. KBase: the United States Department of Energy Systems Biology Knowledgebase. Nat Biotechnol 2018; 36(7): 566–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nuc Acids Res 2017; 45(D1): D353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wattam AR, Abraham D, Dalay O, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nuc Acids Res 2014; 42(Database issue): D581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kersey PJ, Allen JE, Allot A, et al. Ensembl genomes 2018: an integrated omics infrastructure for non-vertebrate species. Nuc Acids Res 2018; 46(1): D802–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The MetaSUB International Consortium . The metagenomics and metadesign of the subways and urban biomes (metasub) international consortium inaugural meeting report. Microbiome 2016; 4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paley S, Karp PD. Update notifications for the BioCyc collection of databases. Database 2017;2017:bax086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.BioCyc Web Services . https://biocyc.org/web-services.shtml.
- 19.Krummenacker M, Latendresse M, Karp PD. Metabolic route computation in organism communities. Microbiome 2019; 7:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Çubuk C, Hidalgo MR, Amadoz A, et al. Differential metabolic activity and discovery of therapeutic targets using summarized metabolic pathway models. npj Systems Biology and Applications 2019; 5:7. 10.1038/s41540-01900087-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selmer T, Andrei PI. P-Hydroxyphenylacetate decarboxylase from Clostridium difficile. A novel glycyl radical enzyme catalysing the formation of p-cresol Eur J Biochem 2001; 268(5): 1363–72. [DOI] [PubMed] [Google Scholar]
- 22.Karp PD, Latendresse M, Caspi R. The pathway tools pathway prediction algorithm. Stand Genomic Sci Dec 2011; 5(3): 424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dale JM, Popescu L, Karp PD. Machine learning methods for metabolic pathway prediction. BMC Bioinformatics 2010; 11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.SCIP Software Home Page . http://scip.zib.de/.
- 25.Latendresse M, Karp P. Evaluation of reaction gap-filling accuracy by randomization. BMC Bioinformatics 2018; 19:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weaver DS, Keseler IM, Mackie A, et al. A genome-scale metabolic flux model of E. coli K–12 derived from the EcoCyc database. BMC Syst Biol 2014; 8:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paley SM, Parker K, Spaulding A, et al. The Omics dashboard for interactive exploration of gene-expression data. Nuc Acids Res 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hucka M, Bergmann FT, Drager A, et al. The systems biology markup language (sbml): language specification for level 3 version 2 core. J Integr Bioinform 2018; 15(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Wulffen J, Ulmer A, Jager G, et al. Rapid sampling of Escherichia coli after changing oxygen conditions reveals transcriptional dynamics. Genes (Basel) 2017; 8(3): 90–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Midford PE, Latendresse M, O’Maille P, et al. Using pathway covering to explore connections among metabolites. Metabolites 2019; 9(5): 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sud M, Fahy E, Cotter D, et al. Metabolomics workbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nuc Acids Res 2015; 44:D463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuhrer T, Zampieri M, Sevin DC, et al. Genome-wide landscape of gene-metabolome associations in Escherichia coli. Mol Syst Biol 2017; 13(1): 907. [DOI] [PMC free article] [PubMed] [Google Scholar]