

Abstract
Variable cellular composition of tissue samples represents a significant challenge for the interpretation of genomic profiling studies. Substantial effort has been devoted to modeling and adjusting for compositional differences when estimating differential expression between sample types. However, relatively little attention has been given to the effect of tissue composition on co-expression estimates. In this study, we illustrate the effect of variable cell-type composition on correlation-based network estimation and provide a mathematical decomposition of the tissue-level correlation. We show that a class of deconvolution methods developed to separate tumor and stromal signatures can be applied to two component cell-type mixtures. In simulated and real data, we identify conditions in which a deconvolution approach would be beneficial. Our results suggest that uncorrelated cell-type-specific markers are ideally suited to deconvolute both the expression and co-expression patterns of an individual cell type. We provide a Shiny application for users to interactively explore the effect of cell-type composition on correlation-based co-expression estimation for any cell types of interest.
Keywords: transcriptomics, deconvolution, cell-types, induced covariance, co-expression, tissue composition
Introduction
Cellular processes are governed by the interaction of genes and gene products. This produces shared patterns of expression between functionally related genes. Gene co-expression networks encode similarities in expression as edges in an undirected graph and have been used to identify genes that potentially share a regulatory relationship or common function [4, 6, 34, 36].
Co-expression network estimation
Statistical methods to estimate co-expression networks rely on gene expression measurements from biological replicates that share a common regulatory architecture. Relatively small variations in gene expression across these replicates are used to identify co-expressed genes. A major challenge is to design an experiment that is sufficiently large to distinguish true co-expression from noise while minimizing sources of technical variability, such as batch effects [15], that can easily overwhelm the biological signal [21]. As such, recent co-expression analyses have focused on data generated by large consortia such as the Gene Tissue Expression Project [23, 26].
The Pearson correlation coefficient is the most widelyused measure to capture linear dependencies between genes. For example, one of the most popular methods, WGCNA [13, 37], begins by constructing a gene co-expression network based on the absolute value of the Pearson correlation coefficient. A challenge in the application of network estimation methods to gene expression data is that the number of genes is usually much greater than the number of samples. A variety of approaches, from the ad hoc to the statistically rigorous, have been used to address this challenge. For example, Schäfer and Strimmer [28] proposed estimating the gene–gene covariance matrix by shrinking the empirical covariance matrix toward a diagonal matrix with unequal variance.
Tissue composition and statistical deconvolution
Tissues are comprised of multiple cell types with distinct molecular phenotypes, resulting from cell-type-specific gene expression. Many of the cellular processes encoded by gene regulatory networks are specific to a particular cell type, resulting in fundamental differences in these networks across cell types.
A substantial number of statistical methods have been developed to estimate cellular composition and/or cell-type-specific RNA expression from tissue-level expression data. The vast majority of these methods are based on a linear combination of cell-type-specific expression profiles, originally proposed by [32]. Specifically, these methods model the observed expression, 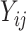 , of gene
, of gene 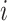 in tissue sample
in tissue sample 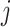 as a linear combination of cell-type-specific expression,
as a linear combination of cell-type-specific expression, 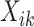 , of each of the
, of each of the 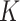 cell types that make up the tissue:
cell types that make up the tissue:
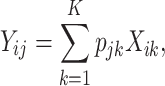 |
(1) |
where the compositional proportions, 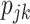 , have the constraint
, have the constraint 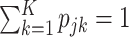 . A clear limitation to this model is that cell-type expression is assumed to be constant across tissue samples;
. A clear limitation to this model is that cell-type expression is assumed to be constant across tissue samples; 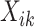 is the same for all
is the same for all 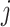 . This is biologically implausible and often in direct opposition to subsequent analyses that seek to identify genes whose expression differs between groups of samples.
. This is biologically implausible and often in direct opposition to subsequent analyses that seek to identify genes whose expression differs between groups of samples.
Two recent methods, focusing on tumor/normal mixtures, have improved upon these earlier approaches by allowing cell-type expression to vary across samples. One method, ISOpure, assumes that each normal expression profile is a convex combination of a set of reference normal profiles and that the cancer profiles are similar to one another [2]. The other method, DeMix, provides flexible modeling of mixed tissue samples across four different situations: with or without reference gene profiles and with or without matched tumor/normal samples. In each of these situations, DeMix assumes that the normal mixture component can be measured directly [1].
Approach
In this study, we begin by illustrating the challenges of estimating co-expression in the constituent cell types from heterogeneous tissue samples. We then assess the ability of deconvolution methods to facilitate the estimation of cell-type-specific co-expression. Finally, we conclude with a discussion of the conditions in which deconvolution improves estimation of cell-type-specific co-expression. For the purpose of this study, we focus on correlation-based co-expression networks, which are arguably the most commonly used [22, 33] and have been shown to perform comparably with more complex methods [31].
Methods
Co-expression network estimation
In the subsequent analyses, we use the absolute value of either Pearson’s correlation coefficient or a shrinkage correlation estimator [28] to assess the pairwise association between genes. The latter is a modification of Pearson’s correlation coefficient with a shrinkage intensity parameter, 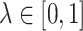 . Before constructing the shrinkage estimator, we define notation for the empirical variance–covariance matrix,
. Before constructing the shrinkage estimator, we define notation for the empirical variance–covariance matrix, 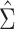 , with elements
, with elements 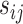 . Suppose
. Suppose 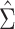 is an
is an 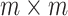 real, symmetric and positive definite matrix, then it can be expressed as:
real, symmetric and positive definite matrix, then it can be expressed as:
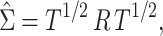 |
where
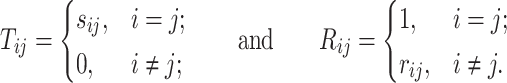 |
In other words, the off-diagonal elements (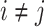 ) have the following expression:
) have the following expression:
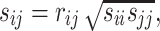 |
where 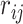 is Pearson’s correlation coefficient.
is Pearson’s correlation coefficient.
The modified correlation estimator is a linear shrinkage approach that combines the diagonal matrix with unequal variances, 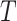 , and the empirical variance-covariance matrix,
, and the empirical variance-covariance matrix, 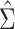 , such that
, such that
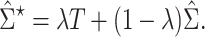 |
In the above equation, 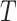 is the shrinkage target, and the optimal shrinkage parameter is
is the shrinkage target, and the optimal shrinkage parameter is
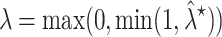 |
with
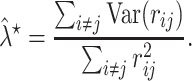 |
This optimal shrinkage parameter is determined analytically based on the minimal mean squared error (MSE) criterion, which was derived by Ledoit and Wolf [14]. Finally, the shrinkage estimator can be written explicitly as
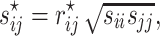 |
where 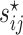 is the
is the 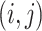 th element of
th element of 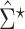 and
and 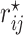 is the corresponding shrinkage correlation estimate. This approach is implemented in the R package GeneNet [27].
is the corresponding shrinkage correlation estimate. This approach is implemented in the R package GeneNet [27].
Sample-specific deconvolution
A sample-specific deconvolution model can be expressed as
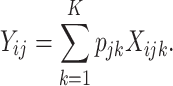 |
(2) |
Note that Equation (2) is a generalization of Equation (1) where cell-type-specific expression, 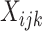 , is now allowed to vary across the sample index
, is now allowed to vary across the sample index 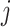 . Note that a co-expression analysis for a specific cell type is now possible because the expression within each cell type is allowed to vary across samples.
. Note that a co-expression analysis for a specific cell type is now possible because the expression within each cell type is allowed to vary across samples.
However, in the most general case where only the 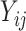 are observed, Equation (2) is an under-determined system of equations, i.e. the number of equations is less than the number of unknown parameters. With further assumptions, the deconvolution problem can be categorized into two types: (i) full deconvolution for unknown
are observed, Equation (2) is an under-determined system of equations, i.e. the number of equations is less than the number of unknown parameters. With further assumptions, the deconvolution problem can be categorized into two types: (i) full deconvolution for unknown 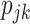 and
and 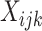 and (ii) partial deconvolution with known
and (ii) partial deconvolution with known 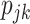 or
or 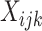 [19].
[19].
ISOpure deconvolution
ISOpure [2, 24] was originally developed for tumor profiles (pre-purification) that are considered as mixtures of cancer and normal profiles. By comparing the tumor profiles to a set of unmatched normal profiles, ISOpure estimates tumor purities (i.e. proportions in the mixtures) and individual cancer profiles (post-purification) for each tumor sample. The ISOpure statistical model is based on a Dirichlet-multinomial mixture model. The ISOpure algorithm estimates the proportions and the purified expression profiles in two steps. The 1st step is a Bayesian approach that iteratively updates the Dirichlet prior for the proportions and the compound multinomial distribution for the mixed profiles after appropriate data reorganization. Based on the outputs from the Bayesian model, the 2nd step is to estimate the individual profiles for the target cell type by maximum likelihood.
For a mixture of two cell types, we adopt ISOpure to decompose the compositional cell-type profiles using the R package ISOpureR [9]. For the two cell types, suppose cell type 1 is the target cell type, whose profile is in lieu of the cancer profile, and cell type 2 is the reference cell type, whose profile is in lieu of the normal profile. ISOpure assumes that there is a set of reference cell type profiles that can represent the cell type 2 expression profile. Thus, ISOpure inputs 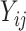 (i.e. the mixed profiles) and
(i.e. the mixed profiles) and 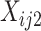 (i.e. the reference profiles), where the sample indices,
(i.e. the reference profiles), where the sample indices, 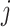 , from the two collections of profiles do not need to match; and, it returns estimated
, from the two collections of profiles do not need to match; and, it returns estimated 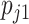 and
and 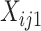 , where
, where 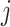 denotes the mixed sample index. Normalized but not log-transformed data are require for ISOpure. In our analyses, we use quantile normalization [5] to make expression profiles comparable from sample to sample. Technically, ISOpure depends on an initial random seed, and the algorithm may fail to converge occasionally. When this happened, we repeated the analysis with a new random seed.
denotes the mixed sample index. Normalized but not log-transformed data are require for ISOpure. In our analyses, we use quantile normalization [5] to make expression profiles comparable from sample to sample. Technically, ISOpure depends on an initial random seed, and the algorithm may fail to converge occasionally. When this happened, we repeated the analysis with a new random seed.
Limitations of regression-based composition adjustment
The full deconvolution problem is an under-determined system of linear equations, even in the case of a mixture of two cell types. Sometimes, investigators may have knowledge of 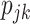 through complementary measurements (e.g. histological imaging). In this case, a regression-based deconvolution model [10] has been proposed such that
through complementary measurements (e.g. histological imaging). In this case, a regression-based deconvolution model [10] has been proposed such that
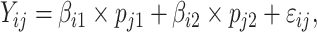 |
(3) |
where 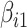 and
and 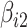 are the unknown average theoretical cell-type-specific gene expression and
are the unknown average theoretical cell-type-specific gene expression and 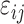 represents random error. This model suffers from two related limitations. First,
represents random error. This model suffers from two related limitations. First, 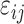 captures technical variation but not biological variation between samples. Second, as previously shown in the study by Jaffe and Irizarry [12], Equation (3) is only valid when the difference between gene expression in each sample and the cell-type average are the same across cell types, which is rarely a reasonable assumption.
captures technical variation but not biological variation between samples. Second, as previously shown in the study by Jaffe and Irizarry [12], Equation (3) is only valid when the difference between gene expression in each sample and the cell-type average are the same across cell types, which is rarely a reasonable assumption.
Simulations
We utilize simulations to access the effects of cell-type mixture and deconvolution on co-expression networks. Let 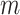 be the number of features, i.e.
be the number of features, i.e. 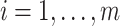 . Using vector forms, we denote
. Using vector forms, we denote 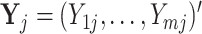 for the
for the 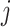 th mixed expression profile and
th mixed expression profile and 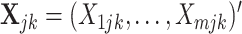 for the pure cell-type-
for the pure cell-type-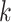 expression profile. Also, we denote the log2-transformed profiles as
expression profile. Also, we denote the log2-transformed profiles as 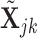 and
and 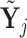 , such that
, such that
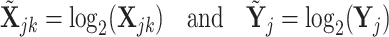 |
(4) |
and similarly for the mixed profiles. We emphasize this log-transformation because it has been shown that deconvolution models, such as Equations (1) or (2), should be built upon data that have not been log-transformed, since such a transformation introduces bias in the resulting profiles [38].
In our simulations, we generate the log-transformed data, 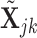 , from an
, from an 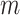 -dimensional multivariate normal distribution and use Equation (4) to obtain the appropriate data for deconvolution,
-dimensional multivariate normal distribution and use Equation (4) to obtain the appropriate data for deconvolution, 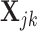 . Focusing on two cell types, we randomly draw
. Focusing on two cell types, we randomly draw 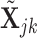 from the following multivariate normal distribution
from the following multivariate normal distribution
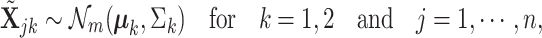 |
where 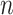 is the number of samples for each cell type. Suppose the cell-type-specific mean vectors,
is the number of samples for each cell type. Suppose the cell-type-specific mean vectors, 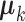 , are available. We design the cell-type-specific covariance matrix (i.e. co-expression structure),
, are available. We design the cell-type-specific covariance matrix (i.e. co-expression structure), 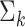 , according to Figure 1. For the two cell types, we set one as the reference cell type with an identity covariance matrix and the other cell type as the target with a block-diagonal covariance matrix. The block-diagonal covariance matrix is further characterized by the number of blocks, block size, correlation magnitude and co-varying features: the most differentially expressed, randomly selected or the least differentially expressed.
, according to Figure 1. For the two cell types, we set one as the reference cell type with an identity covariance matrix and the other cell type as the target with a block-diagonal covariance matrix. The block-diagonal covariance matrix is further characterized by the number of blocks, block size, correlation magnitude and co-varying features: the most differentially expressed, randomly selected or the least differentially expressed.
Figure 1 .
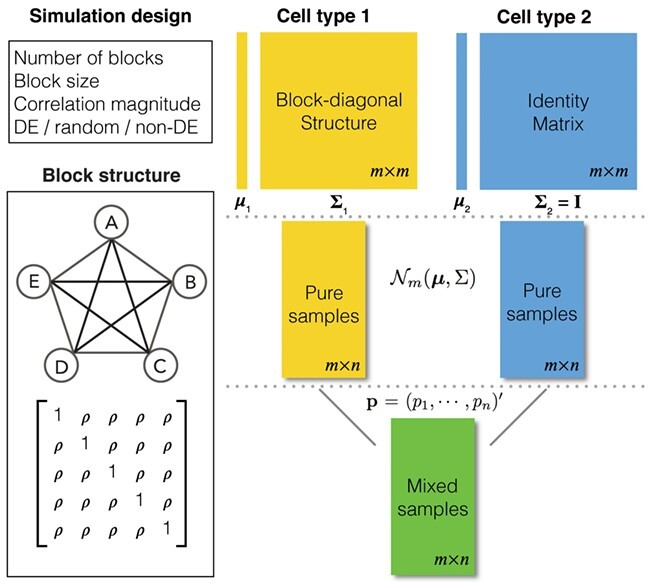
Overview of the simulation scheme. Consider two cell types: cell type 1 (the target cell type) in yellow with known mean vector and block-diagonal covariance matrix, 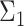 , and cell type 2 (the reference cell type) in blue with known mean vector and identity covariance matrix,
, and cell type 2 (the reference cell type) in blue with known mean vector and identity covariance matrix, 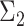 . The top-left panel characterizes the design of
. The top-left panel characterizes the design of 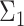 . The co-expression signal in the target cell type is concentrated on the most differentially expressed (DE) features randomly selected features, or the least differentially expressed (non-DE) features. The bottom-left panel is an example of the type of structure (i.e. co-expression network) applied to each signal-receiving block, where A–E are five genes, and the matrix beneath specifies the covariance structure. The right panel is a flow chart of the data generation procedure. Pure samples are generated from a multivariate normal distribution,
. The co-expression signal in the target cell type is concentrated on the most differentially expressed (DE) features randomly selected features, or the least differentially expressed (non-DE) features. The bottom-left panel is an example of the type of structure (i.e. co-expression network) applied to each signal-receiving block, where A–E are five genes, and the matrix beneath specifies the covariance structure. The right panel is a flow chart of the data generation procedure. Pure samples are generated from a multivariate normal distribution, 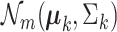 ; mixed samples are mixtures of the pure samples in given proportions,
; mixed samples are mixtures of the pure samples in given proportions, 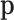 for the target cell type and
for the target cell type and 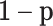 for the reference cell type.
for the reference cell type.
With 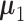 ,
, 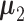 ,
, 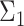 and
and 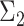 specified, we generate random samples for pure cell type 1,
specified, we generate random samples for pure cell type 1, 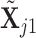 , and pure cell type 2,
, and pure cell type 2, 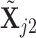 , using the corresponding multivariate normal distributions. Before mixing, we transform the data to obtain
, using the corresponding multivariate normal distributions. Before mixing, we transform the data to obtain 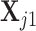 and
and 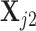 . Let
. Let 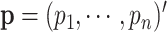 be the proportions of cell type 1 and
be the proportions of cell type 1 and 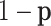 be the proportions of cell type 2 in the mixture. These proportions are generated in one of two ways: (1) equally spaced from zero to one or (2) randomly sampled from a beta distribution. Finally, expression in the mixed samples is generated as follows:
be the proportions of cell type 2 in the mixture. These proportions are generated in one of two ways: (1) equally spaced from zero to one or (2) randomly sampled from a beta distribution. Finally, expression in the mixed samples is generated as follows:
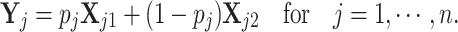 |
The microRNAome data
We obtained cell-type-specific microRNA expression data from the microRNAome Bioconductor package [18]. Simulated data were generated according to Figure 1 using 382 well-characterized microRNAs in pure aortic smooth muscle cells (ASM) and pure aortic endothelial cells (AEC). Without loss of generality, we set AEC as the reference cell type. For each cell type, we generated 20 samples and computationally mixed these to obtain 20 mixed samples. We designed the co-expression pattern for ASM as follows: 3 co-expression blocks, 10 features per block and a correlation magnitude of 0.7. The 20 mixture proportions were equally spaced from 0 to 1. In different simulations, the co-expressed features were selected as the most differentially expressed, as the least differentially expressed or randomly.
We applied the ISOpure deconvolution algorithm to the mixed samples and compared the simulation performance using receiver operating characteristic (ROC) curves based on the Pearson correlation and the shrinkage correlation for the pure ASM, mixed and deconvoluted samples.
To assess performance on a real data network, we selected two cell types from the microRNAome data: dendritic cells (18 samples) and fat cells (15 samples). We set the fat cells as the reference cell type. We identified 363 microRNAs, which were non-zero in at least half of the samples for each cell type. We mixed the pure cell-type samples with two different sequences of mixture proportions. In terms of the proportions of the dendritic cells, the 1st sequence is equally spaced from 0 to 1, and the 2nd sequence is equally spaced from 0.4 to 0.6. Based on Equation (2), we obtained computationally mixed samples by randomly selecting 15 samples of pure dendritic cells and combining them with the 15 samples of pure fat cells in the given proportions. This mixed sample generation was repeated 20 times to produce 20 complete mixed data sets.
A true edge in the real data network was defined based on the empirical correlation matrix from the data. We dichotomize the empirical correlation matrix and obtain the edge matrix as follows:
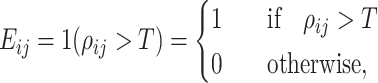 |
(5) |
where 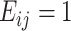 indicates an edge between the
indicates an edge between the 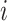 th and
th and 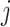 th features,
th features, 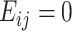 indicates no edge,
indicates no edge, 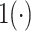 is an indicator function and
is an indicator function and 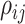 is the Pearson correlation between the pair of features. Unless otherwise stated, we set
is the Pearson correlation between the pair of features. Unless otherwise stated, we set 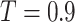 in these assessments.
in these assessments.
|
|

TCGA Data
We obtained 57 triple negative breast invasive carcinoma samples and 7 normal breast tissue samples from The Cancer Genome Atlas (TCGA) [20] via the TCGABiolinks Bioconductor package [7]. All samples were from white female individuals. The sample IDs are listed in Supplementary Table 1. These tumor samples have been previously shown to have variable purity [3, 35]. The ISOpure deconvolution algorithm was applied to these samples using the normal samples as a surrogate for the stromal component of the tumor samples. This represents the standard usage of the ISOpure algorithm.
Gene Set Analysis
Gene set analyses were performed using g:Profiler (database updated on 6 May 2019; version  ) [25]. g:Profiler queries the following databases: GO molecular function, GO cellular component, GO biological process, KEGG, Reactome, WikiPathways, TRANSFAC, miRTarBase, Human Protein Atlas, CORUM and HP.
) [25]. g:Profiler queries the following databases: GO molecular function, GO cellular component, GO biological process, KEGG, Reactome, WikiPathways, TRANSFAC, miRTarBase, Human Protein Atlas, CORUM and HP.
Assessments of performance
We used ROC curves [11] and area under curve (AUC) statistics to assess network estimation based on correlation measures. The ROC curve shows the relationship between the true positive rate (TPR) and the false positive rate (FPR) for a range of correlation thresholds, where
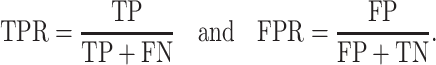 |
At each threshold, correlation values above the threshold are considered as positives (i.e. edges in a network), and correlation values below the threshold are negatives (i.e. no edge). In the simulations, true edges are defined as any non-zero off-diagonal element in 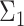 .
.
An ROC curve close to the top-left corner of the plotted area indicates high TPR and low FPR. On the contrary, if a curve is close to the diagonal line, it suggests that the performance is roughly the same as random chance. The AUC statistic is a quantitative summary of the ROC curve, which ranges from 0 to 1. As the ROC curve approaches the top-left corner, the corresponding AUC statistic approaches 1.
Additionally, we considered the MSE of the correlation matrix itself. Specifically, we used the integrated (cumulative) MSE, defined as the summation of all element-wise mean squared differences of two matrices, to summarize the discrepancy between the estimated correlation matrix from the mixed or deconvoluted samples and the correlation matrix from the pure samples. While the MSE does not quantify the quality of the co-expression network, it does provide the most direct assessment of the underlying correlation estimation.
Results
An illustrative model of tissue-level gene expression
Consider the following model of tissue-level gene expression arising from a mixture of two cell types:
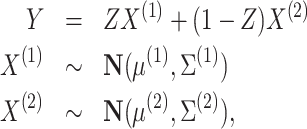 |
(6) |
where 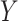 is a vector of tissue-level gene expression,
is a vector of tissue-level gene expression, 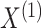 and
and 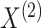 are random vectors of gene expression in cell types 1 and 2, respectively, and
are random vectors of gene expression in cell types 1 and 2, respectively, and 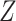 is a scalar random variable denoting the proportion of cell type 1 in the tissue. In contrast to a Gaussian mixture model, in which each observation comes from only one of the mixture components, i.e.
is a scalar random variable denoting the proportion of cell type 1 in the tissue. In contrast to a Gaussian mixture model, in which each observation comes from only one of the mixture components, i.e. 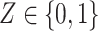 , here each observation is a convex combination of cell-type-specific expression vectors, i.e.
, here each observation is a convex combination of cell-type-specific expression vectors, i.e. 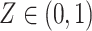 . Note that conditional on the mixing proportion,
. Note that conditional on the mixing proportion,
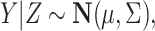 |
where
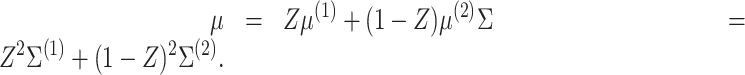 |
Thus, the covariance between any two genes 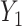 and
and 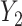 can be expressed as shown in Equation Box A. If tissue composition is variable, i.e.
can be expressed as shown in Equation Box A. If tissue composition is variable, i.e. 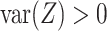 , the covariance between genes in the mixed tissue depends on the covariances in each cell type, as well as the difference in expression between the cell types. Finally, the correlation between any two genes
, the covariance between genes in the mixed tissue depends on the covariances in each cell type, as well as the difference in expression between the cell types. Finally, the correlation between any two genes 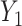 and
and 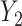 can be expressed as shown in Equation Box B.
can be expressed as shown in Equation Box B.
Note that even if the genes are positively correlated in both cell types, their tissue-level correlation can be negative if the differences in average expression between cell types for the two genes differ in sign and the variance in the mixing proportion is sufficiently large.
To further examine the effect of variable composition on tissue-level gene expression correlation, consider the following simplifications:
-

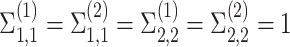 : the cell-type variances are all equal to 1.
: the cell-type variances are all equal to 1. -

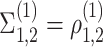 : the two genes are correlated in cell type 1 and have correlation magnitude equal to some non-zero
: the two genes are correlated in cell type 1 and have correlation magnitude equal to some non-zero 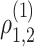 .
. -

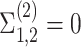 : the two genes are not correlated in cell type 2.
: the two genes are not correlated in cell type 2. -

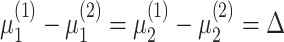 : the differences in expression between cell types are equal to
: the differences in expression between cell types are equal to 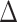 .
.
In this case, the tissue-level correlation can be expressed as shown Equation Box C. The 1st part of the summation represents the attenuated correlation due to the cell-type mixture. The 2nd part of the summation represents the correlation induced by variation in the mixing proportion. To be noted, the induced correlation also depends on how differently these genes are expressed in the different cell types.
Figure 2 provides an illustrative example of between-gene correlation induced by variable mixing of cell types in tissue samples. In this simulated experiment, we consider 10 genes (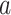 –
–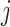 ) measured in two cell types (100 samples per cell type), where five of the genes are differentially expressed between the two cell types (Figure 2A). As in Equation (6), we assume that gene expression in each cell type follows a multivariate normal distribution. For illustrative purposes, we assume that
) measured in two cell types (100 samples per cell type), where five of the genes are differentially expressed between the two cell types (Figure 2A). As in Equation (6), we assume that gene expression in each cell type follows a multivariate normal distribution. For illustrative purposes, we assume that 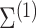 is block diagonal and
is block diagonal and 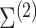 is the identity matrix (Figure 2B andC). We set
is the identity matrix (Figure 2B andC). We set 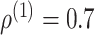 for the correlated gene pairs,
for the correlated gene pairs, 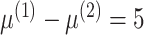 for the differentially expressed genes and
for the differentially expressed genes and 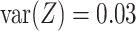 . Between the three mixtures (Figure 2D–F), we only vary the expected value of the mixing proportion,
. Between the three mixtures (Figure 2D–F), we only vary the expected value of the mixing proportion, 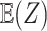 . Specifically, we consider generating the mixing proportions from three beta distributions (Figure 2G–I). As expected, in the mixed tissue samples, one can clearly see the effects of both the cell-type-specific correlation and the correlation induced by the mixing of cell types (comparison between Figure 2Band C and Figure 2Dand F). For genes
. Specifically, we consider generating the mixing proportions from three beta distributions (Figure 2G–I). As expected, in the mixed tissue samples, one can clearly see the effects of both the cell-type-specific correlation and the correlation induced by the mixing of cell types (comparison between Figure 2Band C and Figure 2Dand F). For genes 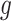 –
–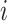 , which are not differentially expressed between the two cell types, we observe a clear attenuation of the correlation structure as the proportion of cell type 1 decreases (moving from Figure 2D to Figure 2F). For genes
, which are not differentially expressed between the two cell types, we observe a clear attenuation of the correlation structure as the proportion of cell type 1 decreases (moving from Figure 2D to Figure 2F). For genes 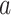 –
–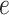 , which are differentially expressed between the two cell types, the correlation induced by the mixture masks the cell-type-specific correlation even in an equal mixture of the two cell types (Figure 2E). Finally, the amount of correlation induced by the mixture also depends on the statistical properties of the mixing distribution. Specifically, it is a non-monotone function of the 1st two moments of the distribution of the mixing proportion.
, which are differentially expressed between the two cell types, the correlation induced by the mixture masks the cell-type-specific correlation even in an equal mixture of the two cell types (Figure 2E). Finally, the amount of correlation induced by the mixture also depends on the statistical properties of the mixing distribution. Specifically, it is a non-monotone function of the 1st two moments of the distribution of the mixing proportion.
Figure 2 .
An example of correlation induced by cell type mixture. Consider 10 hypothetical genes (namely,  –
– ) that are expressed in two cell types. Expression profiles of 100 samples for each cell type are plotted in panel A. The 1st five genes (
) that are expressed in two cell types. Expression profiles of 100 samples for each cell type are plotted in panel A. The 1st five genes (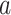 –
–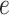 ) are differentially expressed between the two cell types, and the other five genes (
) are differentially expressed between the two cell types, and the other five genes (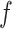 –
–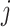 ) are not differentially expressed. Panels B–F are heatmaps of correlations. These genes form three co-expression blocks in cell type 1 (panel B) and are not co-expressed in cell type 2 (panel C). We generated three mixtures (panels D–F) with varying proportions of cell type 1 drawn from beta distributions (panels G–I).
) are not differentially expressed. Panels B–F are heatmaps of correlations. These genes form three co-expression blocks in cell type 1 (panel B) and are not co-expressed in cell type 2 (panel C). We generated three mixtures (panels D–F) with varying proportions of cell type 1 drawn from beta distributions (panels G–I).
Co-expression estimation in pure, mixed and deconvoluted samples
Figure 3 shows the performance of two correlation-based network estimators on simulated data from pure cell-type samples, samples from a variable mixture of two cell types and the corresponding deconvoluted samples, with three types of co-expressed features. Both estimators performed best on the pure cell-type data (AUC 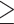 0.94), which suggests that both correlation measures are able to capture the co-expression signal well in pure samples. Compared to the pure samples, the mixed samples largely deviate from the true signals and have poor performance. Figure 3A shows the results when only the most differentially expressed features are co-expressed. Using the Pearson correlation, the mixed ROC curve is close to the pure curve. This is because the Pearson correlation is increased due to the correlation induced by the cell-type mixture, which does not represent cell-type-specific co-expression. In contrast, the shrinkage correlation estimator (Figure 3D) is a more conservative measure, which regularizes the empirical covariance matrix toward a diagonal matrix. This appears to greatly reduce the effect of the induced correlation. Finally, the deconvolution produces modest to substantial improvement over the mixed samples, except in the situation noted above. Across all scenarios, the deconvoluted samples result in stable performance (AUC > 0.8).
0.94), which suggests that both correlation measures are able to capture the co-expression signal well in pure samples. Compared to the pure samples, the mixed samples largely deviate from the true signals and have poor performance. Figure 3A shows the results when only the most differentially expressed features are co-expressed. Using the Pearson correlation, the mixed ROC curve is close to the pure curve. This is because the Pearson correlation is increased due to the correlation induced by the cell-type mixture, which does not represent cell-type-specific co-expression. In contrast, the shrinkage correlation estimator (Figure 3D) is a more conservative measure, which regularizes the empirical covariance matrix toward a diagonal matrix. This appears to greatly reduce the effect of the induced correlation. Finally, the deconvolution produces modest to substantial improvement over the mixed samples, except in the situation noted above. Across all scenarios, the deconvoluted samples result in stable performance (AUC > 0.8).
Figure 3 .
ROC curves from a simulation study with number of blocks = 3, block size = 10, number of samples = 20, and correlation magnitude = 0.7. The mixture proportions of cell type 1 were equally spaced from 0 to 1. Block correlation structure was imposed on the most differentially expressed features (left column), randomly selected features (middle column), and the least differentially expressed features (right column). Differential expression was quantified by the absolute difference between the cell type-specific mean vectors. Two measures of co-expression were used: the Pearson correlation (top row) and the shrinkage correlation implemented by GeneNet (bottom row). In each panel, the ROC curves for the pure samples (gray curve), the mixed samples (yellow curve), and the ISOpure deconvoluted samples (blue curve) are shown.
To further evaluate the deconvolution performance, we plotted the estimated proportions against the true proportions for the target cell type in Supplementary Figure S1. Recall that the true proportions in this simulation are equally spaced from 0 to 1. Based on the error measures, mean absolute difference and the root mean squared distance [19], the deconvolution estimation of mixing proportions appears stable and robust to the choice of co-expression features. However, the estimated proportions appear to consistently under-estimate the true proportions.
Additionally, we conducted simulation studies with mixing proportions randomly generated from a beta distribution. We show results similar to Figure 3 for these simulations in Supplementary Figures S2, S3 and S4. Overall, they reveal a similar pattern as that seen in Figure 3. The performance of the deconvoluted curves is stable across different scenarios and robust to the selection of co-expression features. They effectively recover the true co-expression signal from cell-type mixtures. Without deconvolution, the mixed curves vary dramatically from case to case. When only a small proportion of the target cell type is present in the mixture, the mixed curve is close to the diagonal line (Figure S2); when a large proportion of the target cell type is present in the mixture, the mixed curve performs similarly to the deconvoluted curve (Figure S4).
Appropriate cases for deconvolution
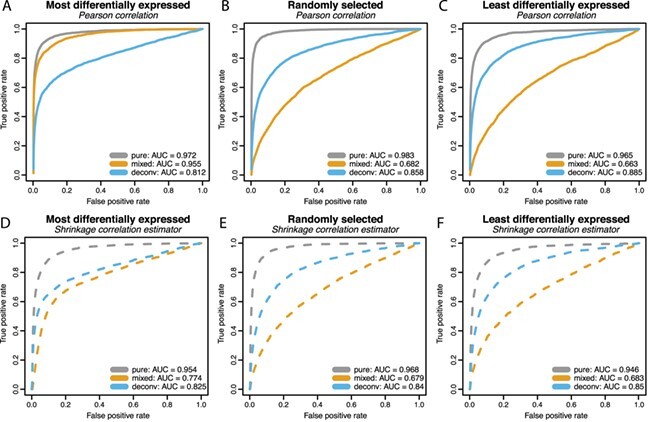
In order to further evaluate the situations in which deconvolution is able to recover the pure sample co-expression, we generated more simulation scenarios by varying block size, number of samples, and correlation magnitude in the same simulation design. While one parameter is varied, other parameters are kept fixed as before. The resulting AUC values are shown in Figure 4, and the corresponding MSE values for the correlation matrices are provided in Supplementary Table 2.
Figure 4 .
AUC values are reported with varying block size, number of samples and correlation magnitude. Co-expression association is measured by Pearson correlation. When one parameter is varied, the others are fixed at block size = 10, number of samples = 20 and correlation magnitude = 0.7. As before, three correlated blocks are generated with the mixture proportions of cell type 1 equally spaced from 0 to 1.
In the 1st column of Figure 4, in which the most differentially expressed genes are also those that are co-expressed, the AUC values for the mixed samples are higher than those for the deconvoluted samples and sometimes even higher than that of the pure samples. As shown in Equation Box C, if the correlated features are also differentially expressed between the two cell types, the differential expression ( ) induces correlation in the mixture if the mixture proportion varies across samples. The high AUC values are attributable to the induced correlation. When the true correlations are difficult to estimate due to small sample size (Figure 4D) or small magnitude (Figure 4G), the induced correlation dominates the attenuated co-expression signal in the mixture. This results in the mixed samples having higher AUC than the pure samples.
) induces correlation in the mixture if the mixture proportion varies across samples. The high AUC values are attributable to the induced correlation. When the true correlations are difficult to estimate due to small sample size (Figure 4D) or small magnitude (Figure 4G), the induced correlation dominates the attenuated co-expression signal in the mixture. This results in the mixed samples having higher AUC than the pure samples.
In the 2nd and 3rd columns of Figure 4, in which the co-expressed features are randomly selected or the least differentially expressed, the AUC values are always higher for the pure samples than the deconvoluted samples, which in turn are higher than the mixed samples. Overall, the AUC values increase as the number of samples or the correlation magnitude increases. The block size does not appear to impact the co-expression network estimation performance.
Deconvolution results in a decrease in the MSE of the correlation matrix for all situations (Supplementary Table 2). The MSE decreases substantially as the number of samples increases. However, the MSE appears to increase slightly as the block size increases and does not appear to be affected by changes in the correlation magnitude.
In summary, the ISOpure deconvolution method recovers the true co-expression signal from mixed samples. If the co-expressed features are differentially expressed between the two cell types, the induced correlation may lead to better estimation of the co-expression network; however, caution is required in interpreting the result as these features would appear correlated in the mixture regardless of whether they were truly co-expressed in the target cell type.
Co-expression estimation from computationally mixed and deconvoluted samples of purified cell types
Using the microRNAome data set, we computationally mixed pure dendritic cells with the same number of pure fat cells as described in the Methods. This mirrors a common disease process in which inflammatory cells, such as dendritic cells, are increased in adipose tissue. Treating the empirical network in the pure cell-type data as the true network, we plot the ROC curves for network reconstruction with the mixed and deconvoluted profiles (Figure 5Band D) with respect to two mixing proportion schemes. In both ROC plots, we focus on the region where the FPR is smaller than 0.5 and observe that the deconvoluted curve is uniformly better than the mixed curve. Over the whole range of FPR, the AUC statistics are also slightly higher for the deconvoluted curve in both cases: 0.711 versus 0.723 (Figure 5B) and 0.824 versus 0.845 (Figure 5D). The real data further demonstrate the improvements gained from deconvolution. For both sequences of mixing proportions, estimation of very small and very large proportions show less variation (Figure 5Aand C). Of note, when the reference cell type is rare, it is common for the estimated proportions to overestimate the purity of the mixed sample (Figure 5A). With regard to network estimation, the gain from deconvolution is larger when there is greater variation in the mixture proportions. However, the MSE of the correlation matrix is larger after deconvolution when the mixing proportions vary from zero to one (47 415 versus 57 494) but smaller when the mixing proportions vary from 0.4 to 0.6 (29 982 versus 24 582).
Figure 5 .
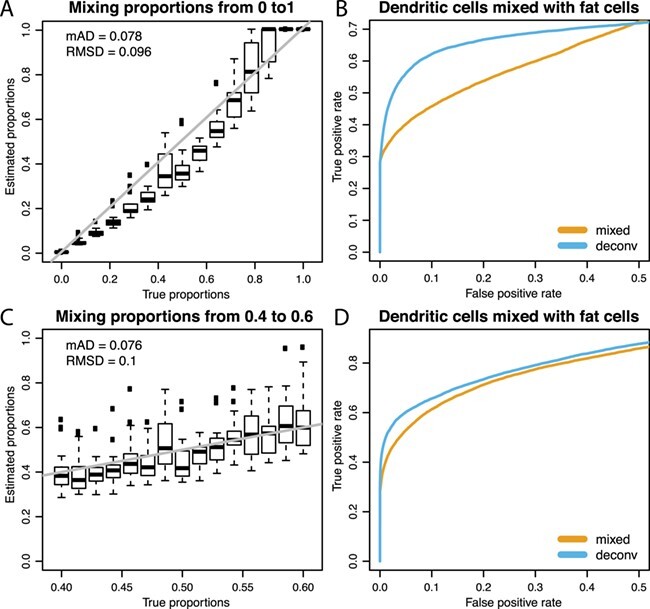
Deconvolution performance on real data. Top row: mixing proportions equally spaced from 0 to 1. Bottom row: mixing proportions equally spaced from 0.4 to 0.6. Panels A and C show the estimated proportions versus the true proportions; the gray line is the reference line for both proportions being equal. By defining a true edge if absolute correlation ¿ 0.9, panels B and D show dominating ROC curves for the deconvoluted samples over the mixed samples in the zoomed region where FPR ¡ 0.5.
Next, we varied the threshold used to define the true network based on the empirical correlation matrix. Supplementary Figures S5 and S6 show the performance for thresholds from 0.3 to 0.8. The co-expression estimates based on the deconvoluted samples are uniformly better than those based on the mixed samples across all thresholds. However, as the threshold used to define a true edge decreases, the AUC for both methods decreases and the difference in AUC decreases as well.
Lastly, we investigated the effect of deconvolution when the nominally mixed samples do not in fact contain any signal from the reference cell type. Specifically, we deconvoluted the pure dendritic cell samples using the fat samples as the reference. The estimated proportions of dendritic cells in these samples were all 0.9999997, indicating that ISOpure correctly identified these samples as essentially lacking any fat cell signal. Comparing the correlation between features pre- vs post-deconvolution, the minimum and maximum observed differences were −0.768 and 0.740; however, 90% of the differences in correlation fell between −0.082 and 0.039.
The effect of reference data size
In the previous assessments, we used 15 samples of adipose tissue as the pure reference data. We now consider the effect of reducing the number of reference samples (to 10, 5 or 2) on estimates of co-expression. If the mixing proportions range from 0 to 1, the deconvoluted AUC is always better than the mixed AUC across all reference data sizes (Supplementary Figure S7 top). On the other hand, if the mixing proportions are restricted to fall between 0.4 and 0.6, the deconvoluted AUC is less than the mixed AUC when the number of reference samples becomes sufficiently small (Supplementary Figure S7 bottom). These relationships do not appear to depend on the threshold used to define a ‘true’ edge; however, as seen previously the overall performance is affected by the edge threshold (Supplementary Figure S7). The performance following deconvolution for small reference data sizes appears to depend on the ability of ISOpure to accurately estimate the mixing proportions. When the mixing proportions range from 0 to 1, the estimated proportions are relatively accurate even down to a reference data size of 2 (Supplementary Figure S8). However, when the mixing proportions range from 0.4 to 0.6, the estimated proportions are become increasingly inaccurate as the reference data size decreases (Supplementary Figure S9).
An example of co-expressed microRNAs
Using real data, we illustrate what would happen to a pair of co-expressed microRNAs when mixing two cell types and deconvoluting the mixed samples. Figure 6 shows scatter plots of two microRNAs, hsa-let-7a-5p/7c-5p and hsa-miR-766-3p, in two different cell types. In dendritic cells (Figure 6A), these two features are strongly positively correlated (cor 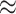 0.9); in fat cells (Figure 6B), these two features appear uncorrelated (cor
0.9); in fat cells (Figure 6B), these two features appear uncorrelated (cor 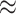 0). After computationally mixing the two cell types with mixing proportions ranging from zero to one, the mixed samples (Figure 6C) appear negatively correlated (cor
0). After computationally mixing the two cell types with mixing proportions ranging from zero to one, the mixed samples (Figure 6C) appear negatively correlated (cor 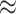 −0.3), which is entirely induced by the mixing. Finally, after ISOpure deconvolution targeting expression in the dendritic cells (Figure 6D), the positive correlation (cor
−0.3), which is entirely induced by the mixing. Finally, after ISOpure deconvolution targeting expression in the dendritic cells (Figure 6D), the positive correlation (cor 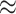 0.9) is re-established between the same pair of features.
0.9) is re-established between the same pair of features.
Figure 6 .
A real data example of two microRNAs (represented in the axes), which are highly correlated in dendritic cells (panel A) but uncorrelated in fat cells (panel B). Computationally mixed samples do not preserve the correlation (panel C). After deconvolution, strong correlation is recovered in the estimated expression of dendritic cells (panel D). Correlation values are shown in corresponding scatter plots. The color of each dot represents the proportion of dendritic cells in that sample, which were equally spaced between zero and one.
Co-expression estimation in breast cancer samples
Using triple negative breast invasive carcinoma data from TCGA, we compared co-expression estimates before and after deconvolution with ISOpure. Our analysis focused on co-expression among the 1414 genes with variance greater than one in the mixed tumor samples. We identified a subset of these genes that showed large and frequent changes in their spearman correlation with other genes after deconvolution. Specifically, we identified 116 genes whose spearman correlation decreased by more than 0.4 with at least 10 other genes following deconvolution (Supplementary Table 3). We also identified 334 genes whose spearman correlation increased by more than 0.4 with at least 10 other genes following deconvolution (Supplementary Table 4).
The 116 genes that were highly correlated in the mixed tumor samples but mostly uncorrelated following deconvolution were enriched for collagen, integrin and fibronectin binding as well as structural and organizational components of the extracellular matrix (Supplementary Table 5). This suggests that the observed correlation in the tumor samples is likely due to variable tumor purity, for which deconvolution is able to mostly adjust. Closer examination of co-expression patterns of these genes revealed wide-spread positive correlation before deconvolution, as well as subsets of these genes that remained highly correlated following deconvolution (Figure 7). Of particular note, the small cluster of highly correlated genes present before and after deconvolution consists of immune response genes (Supplementary Table 6), likely capturing variable immune infiltration, which was not adjusted for by ISOpure. The 334 genes that were mostly uncorrelated in the mixed tumor samples but highly correlated following deconvolution were enriched for nuclear receptor signaling, triglyceride catabolism and metabolism, as well as NF- B and AP-2
B and AP-2 binding motifs, in addition to components of the extracellular matrix (Supplementary Table 7).
binding motifs, in addition to components of the extracellular matrix (Supplementary Table 7).
Figure 7 .
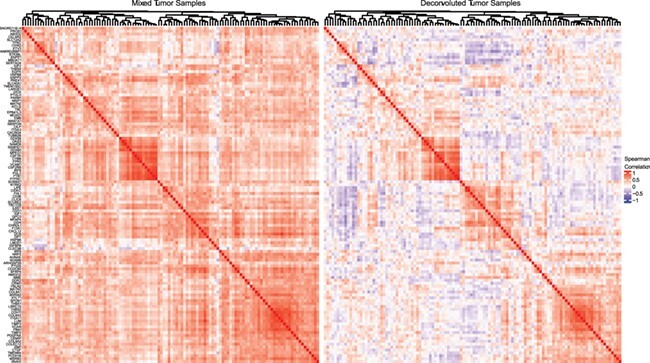
Heatmaps showing the spearman correlation in the mixed tumor samples (left) and the deconvoluted tumor samples (right) for 116 genes whose spearman correlation decreased by more than 0.4 with at least 10 other genes following deconvolution. Hierachical clustering was performed using average linkage and one minus the spearman correlation to define the distance between genes. The clustering of both heatmaps is identical and represents the clustering of the mixed tumor samples. Horizontal white lines separate the six major subclusters.
Interactive exploration of correlation induced by tissue composition
To further explore tissue composition induced co-expression, we developed a Shiny application that allows the user to easily evaluate the degree of induced or attenuated correlation due to cell-type mixtures. The app allows users to select values for the parameters indicated in Figure 1. The user must first select a data set to work with, which will automatically update the list of cell types to choose from in the two dropdown menus for selecting cell types. The user can then proceed to select other parameters for the simulation. We have also added the option to generate beta-distributed proportions, for which the user can specify the mean and variance. Once all parameters have been selected, the user can generate an ROC curve to compare performance based on mixed and pure samples.
An R package containing the web application is hosted on GitHub at yunzhang813/simDeNet-R-Package-Shiny. A vignette is available after installation of the package, which includes a short tutorial describing how to launch and use the Shiny web application.
Discussion
In the absence of targeted perturbation experiments, measurement of co-expression between genes is the primary method of assessing gene–gene interactions. The Pearson correlation between genes is usually the 1st step in co-expression network reconstruction, such as the widelyused WGCNA algorithm. By using this fundamental measure, our results are applicable to a wide range of network algorithms based on the Pearson correlation.
As we have shown, gene co-expression in tissue samples is often dominated by varying cellular composition. When applied to tissue gene expression data, methods that rely on co-expression to define gene modules, primarily identify groups of genes specific to a given component cell type. Variation in these gene modules therefore is capturing changing tissue composition. Some of these compositional differences may arise from biological variation or disease processes that affect the entirety of the tissue from which the sample was obtained. However, others may be due to spatial heterogeneity within a tissue, such that the proportion of cell types within the tissue sample is not representative of the proportion of cell types within the entire tissue. For example, variable sampling of highly localized cellular structures within in a tissue can result in substantial variation in tissue sample composition [17]. These latter sources of co-expression reflect technical rather than biological variation.
In a mixture of two cell types, current deconvolution methods can be used to estimate cell-type-specific expression within each sample. Their ability to provide accurate estimates of gene–gene correlation depends upon the covariance between marker genes within each component cell type. When the genes used to identify the abundance of a specific cell type are all highly correlated with each other (e.g. members of a cell-type-specific pathway), current methods are unable to distinguish between changes in composition and changes in expression. Therefore, ideal marker genes are those that are cell type specific but uncorrelated within each cell type. The identification of such ideal marker genes remains an open question; however, it may be possible to use our current understanding of the biological pathways and processes to restrict selection to a single gene from each pathway or process. Alternatively, one could attempt to identify approximately uncorrelated marker genes via thresholding of the empirical gene–gene correlation matrix for each component cell type.
The accuracy of correlation-based network estimates following computational deconvolution is dependent on the performance of the deconvolution algorithm as well as factors that affect network estimation even in pure samples, such as sample size and the strength of the true co-expression. When the true network structure was defined to include weaker relationships (down to a correlation of 0.3), performance decreased substantially. This may be due to the deconvolution algorithm performing poorly or a limitation of correlation-based network estimation with limited sample size. The complex interplay between deconvolution methodology and correlation-based network estimation warrants further investigation.
In this manuscript, we have focused on correlation-based network estimation; however, two other co-expression network estimation categories should be noted: information theoretic and Bayesian [33]. Mutual information (MI) is an information-theoretic measure that captures nonlinear dependences between genes. Another popular network estimation method, ARACNE [4, 16], begins by calculating the MI between each pair of genes. Finally, Bayesian networks encode causal relationships or hierarchical structure between genes in a directed acyclic graph. A rich collection of Bayesian network learning algorithms are implemented in the R package bnlearn [29]. We suspect that these methods of estimating co-expression networks are also susceptible to tissue-level co-expression induced by variable cellular composition. It would be relatively straightforward to apply the assessments described in this manuscript to other types of co-expression networks.
An alternative to analyzing complex tissue samples is to measure cellular expression in a more homogeneous population obtained from cell culture, laser capture microdissection, centrifugation or fluorescence-activated cell sorting. These methods simplify the assessment of cell-type-specific co-expression; however, they often fail to determine the true biology of an organ where cell–cell interactions are critical to transcriptomic expression. Moreover, these methods often result in residual compositional heterogeneity, the introduction of technical artifacts, expression changes due to cell culture and/or RNA degradation [8, 30]. Therefore, it is often necessary to estimate cell-type-specific co-expression from tissue gene expression data.
Key Points
-

The observed correlation between genes in tissue samples can be decomposed into the attenuated cell-type-specific correlation and the correlation induced by variance into cellular composition.
-

Co-expression in tissue samples is often dominated by varying cellular composition.
-

The ability of current deconvolution methods to provide accurate estimates of gene–gene correlation depends upon the covariance between marker genes within each component cell type.
-

Uncorrelated cell-type-specific markers appear to be ideally suited to deconvolute both the expression and co-expression patterns of an individual cell type.
Supplementary Material
Yun Zhang is a staff scientist-biostatistician in the Informatics Department at the J. Craig Venter Institute. She received a PhD in Statistics from the University of Rochester Medical Center. Dr. Zhang’s research interests include statistical modeling and methodology development for big data produced by advanced biotechnologies.
Jonavelle Cuerdo is a data analyst at Origent Data Sciences, Inc. She received a BA in Data Science from the University of Rochester. Her research interests are focused on the application of machine learning algorithms and the use of predictive modeling techniques.
Marc K. Halushka is a professor in the Department of Pathology at Johns Hopkins University. As a genomicallytrained anatomic pathologist, his research focuses on the cellular location of expression signal. His laboratory has created the miRge2.0 and HPASubC software tools and co-developed xMD-miRNA-seq to isolate and sequence small RNAs from specific cells.
Matthew N. McCall is an associate professor in the Departments of Biostatistics and Computational Biology and Biomedical Genetics. Research in his laboratory focuses on developing methods to estimate gene regulatory networks from gene perturbation experiments, to address within-subject heterogeneity in genomic tumor biomarkers, to preprocess and analyze genomic data and to examine the effect of cellular composition on tissue-level gene expression.
Funding
National Institutes of Health (R00HG006853, R01HL137811, T32ES007271 and HHSN272201200005C); University of Rochester CTSA (UL1TR002001) from the National Center for Advancing Translational Sciences of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Ahn J, Yuan Y, Parmigiani G, et al. DeMix: deconvolution for mixed cancer transcriptomes using raw measured data. Bioinformatics 2013; 29(15): 1865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anghel CV, Quon G, Haider S, et al. Isopurer: an r implementation of a computational purification algorithm of mixed tumour profiles. BMC Bioinformatics 2015; 16(1): 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun 2015; 6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basso K, Margolin AA, Stolovitzky G, et al. Reverse engineering of regulatory networks in human B cells. Nat Genet 2005; 37(4): 382–90. [DOI] [PubMed] [Google Scholar]
- 5.Bolstad BM, Irizarry RA, Astrand M, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003; 19(2): 185–93. [DOI] [PubMed] [Google Scholar]
- 6.Butte AJ, Tamayo P, Slonim D, et al. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proc Natl Acad Sci U S A 2000; 97(22): 12182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colaprico A, Silva TC, Olsen C, et al. Tcgabiolinks: an r/bioconductor package for integrative analysis of tcga data. Nucleic Acids Res 2015; 44(8): e71–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Debey S, Schoenbeck U, Hellmich M, et al. Comparison of different isolation techniques prior gene expression profiling of blood derived cells: impact on physiological responses, on overall expression and the role of different cell types. Pharmacogenomics J 2004; 4(3): 193–207. [DOI] [PubMed] [Google Scholar]
- 9.Quon G, Anghel CV, Haider S, et al. ISOpureR: deconvolution of tumour profiles. R package version 1.1.2 2018. [Google Scholar]
- 10.Glass ER, Dozmorov MG. Improving sensitivity of linear regression-based cell type-specific differential expression deconvolution with per-gene vs. global significance threshold. BMC Bioinformatics 2016; 17(13): 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (roc) curve. Radiology 1982; 143(1): 29–36. [DOI] [PubMed] [Google Scholar]
- 12.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 2014; 15(2): R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langfelder P, Horvath S. Wgcna: an r package for weighted correlation network analysis. BMC Bioinformatics 2008; 9(1): 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ledoit O, Wolf M. Improved estimation of the covariance matrix of stock returns with an application to portfolio selection. J Empir Financ 2003; 10(5): 603–21. [Google Scholar]
- 15.Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 2010; 11(10): 733–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margolin AA, Nemenman I, Basso K, et al. Aracne: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 2006; 7(1): S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCall MN, Illei PB, Halushka MK. Complex sources of variation in tissue expression data: analysis of the gtex lung transcriptome. Am J Hum Genet 2016; 99(3): 624–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCall MN, Kim M-S, Adil M, et al. Toward the human cellular microRNAome. Genome Res 2017; 27(10): 1769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohammadi S, Zuckerman N, Goldsmith A, et al. A critical survey of deconvolution methods for separating cell types in complex tissues. Proc IEEE 2017; 105(2): 340–66. [Google Scholar]
- 20.The Cancer Genome Atlas Network . Comprehensive molecular portraits of human breast tumours. Nature 2012; 490(7418): 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parsana P, Ruberman C, Jaffe AE, et al. Addressing confounding artifacts in reconstruction of gene co-expression networks. Genome Biol 2012; 20(1): 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petereit J, Smith S, Harris FC, et al. Petal: co-expression network modelling in r. BMC Syst Biol2016; 10(2): 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierson E, GTEx Consortium, Koller D, et al. Sharing and specificity of co-expression networks across 35 human tissues. PLoS Comput Biol 2015; 11(5): e1004220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quon G, Haider S, Deshwar AG, et al. Computational purification of individual tumor gene expression profiles leads to significant improvements in prognostic prediction. Genome Med 2013; 5(3): 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raudvere U, Kolberg L, Kuzmin I, et al. G: profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019; 47:W191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saha A, Kim Y, Gewirtz ADH, et al. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res 2017; 27(11): 1843–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaefer J, Opgen-Rhein R, Strimmer K. GeneNet: modeling and inferring gene networks. R package version 1.2.13 2015. [Google Scholar]
- 28.Schäfer J, Strimmer K. A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat Appl Genet Mol Biol 2005; 4(1). Retrieved 4 Nov. 2019, from doi:10.2202/1544-6115.1175. [DOI] [PubMed] [Google Scholar]
- 29.Scutari M. Learning bayesian networks with the bnlearn R package. 2009; preprint arXiv:0908.3817.
- 30.Shen-Orr SS, Gaujoux R. Computational deconvolution: extracting cell type-specific information from heterogeneous samples. Curr Opin Immunol 2013; 25(5): 571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song L, Langfelder P, Horvath S. Comparison of co-expression measures: mutual information, correlation, and model based indices. BMC Bioinformatics 2012; 13(1): 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venet D, Pecasse F, Maenhaut C, et al. Separation of samples into their constituents using gene expression data. Bioinformatics 2001; 17(Suppl 1): S279–87. [DOI] [PubMed] [Google Scholar]
- 33.Villaverde AF, Banga JR. Reverse engineering and identification in systems biology: strategies, perspectives and challenges. J R Soc Interface 2014; 11(91): 20130505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474(7351): 380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whiteside T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008; 27(45): 5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Han L, Yuan Y, et al. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat Commun 2014; 5:3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 2005; 4(1): 1128. [DOI] [PubMed] [Google Scholar]
- 38.Zhong Y, Liu Z. Gene expression deconvolution in linear space. Nat Methods 2012; 9(1): 8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.