

Abstract
In this study, we report the clinical features of Kelch-like protein 11 antibody-associated paraneoplastic neurological syndrome, design and validate a clinical score to facilitate the identification of patients that should be tested for Kelch-like protein 11 antibodies, and examine in detail the nature of the immune response in both the brain and the tumour samples for a better characterization of the immunopathogenesis of this condition. The presence of Kelch-like protein 11 antibodies was retrospectively assessed in patients referred to the French Reference Center for paraneoplastic neurological syndrome and autoimmune encephalitis with (i) antibody-negative paraneoplastic neurological syndrome [limbic encephalitis (n = 105), cerebellar degeneration (n = 33)] and (ii) antibody-positive paraneoplastic neurological syndrome [Ma2-Ab encephalitis (n = 34), antibodies targeting N-methyl-D-aspartate receptor encephalitis with teratoma (n = 49)]. Additionally, since 1 January 2020, patients were prospectively screened for Kelch-like protein 11 antibodies as new usual clinical practice. Overall, Kelch-like protein 11 antibodies were detected in 11 patients [11/11, 100% were male; their median (range) age was 44 (35–79) years], 9 of them from the antibody-negative paraneoplastic neurological syndrome cohort, 1 from the antibody-positive (Ma2-Ab) cohort and 1 additional prospectively detected patient. All patients manifested a cerebellar syndrome, either isolated (4/11, 36%) or part of a multi-system neurological disorder (7/11, 64%). Additional core syndromes were limbic encephalitis (5/11, 45%) and myelitis (2/11, 18%). Severe weight loss (7/11, 64%) and hearing loss/tinnitus (5/11, 45%) were common. Rarer neurologic manifestations included hypersomnia and seizures (2/11, 18%). Two patients presented phenotypes resembling primary neurodegenerative disorders (progressive supranuclear palsy and flail arm syndrome, respectively). An associated cancer was found in 9/11 (82%) patients; it was most commonly (7/9, 78%) a spontaneously regressed (‘burned-out’) testicular germ cell tumour. A newly designed clinical score (MATCH score: male, ataxia, testicular cancer, hearing alterations) with a cut-off ≥4 successfully identified patients with Kelch-like protein 11 antibodies (sensitivity 78%, specificity 99%). Pathological findings (three testicular tumours, three lymph node metastases of testicular tumours, one brain biopsy) showed the presence of a T-cell inflammation with resulting anti-tumour immunity in the testis and one chronic, exhausted immune response—demonstrated by immune checkpoint expression—in the metastases and the brain. In conclusion, these findings suggest that Kelch-like protein 11 antibody paraneoplastic neurological syndrome is a homogeneous clinical syndrome and its detection can be facilitated using the MATCH score. The pathogenesis is probably T-cell mediated, but the stages of inflammation are different in the testis, metastases and the brain.
Keywords: autoimmune encephalitis, paraneoplastic neurologic syndrome, immunopathology, antibodies, ataxia
Graphical Abstract
Graphical Abstract.

Vogrig et al. reported that the newly designed MATCH score (Male, Ataxia, Testicular, Cancer, Hearing alterations) successfully identified patients with KLHL11-Abs (sensitivity 78%, specificity 99%). The pathological substrate is an active T-cell inflammation at the primary tumour that leads to regression, in association with chronic, exhausted, inflammation in the brain.
Introduction
Antibodies targeting Kelch-like protein 11 (KLHL11-Abs) were initially identified in patients with a uniform clinical and oncological profile, namely young/middle-aged males with rhombencephalitis and testicular seminoma, potentially defining a new category of PNS.1,2 However, partially discordant findings suggesting that the clinical spectrum was more heterogeneous were recently reported, since half of the patients were female, some of them had co-occurring Abs targeting N-methyl-D-aspartate receptor and associated teratoma, and a few male patients had concomitant Ma2-Abs.3 Therefore, the exact clinical significance of KLHL11-Abs remains to be established clearly. Moreover, despite the detection of a T-cell-predominant infiltrate in brain samples and a KLHL11-specific T-cell response,2 the immunopathogenesis of this condition still remains elusive, in particular, the reason behind its frequent association with the otherwise rare spontaneously regressed (‘burned-out’) seminoma.
Herein, the aim was to describe the clinical spectrum of KLHL11-Abs among patients referred to the French Reference Center for Paraneoplastic Neurological Syndromes and Autoimmune Encephalitis (Lyon, France), in order to establish whether they define a distinct PNS group with a homogeneous phenotype. In addition, a clinical score was provided to facilitate the identification of patients that should be tested for KLHL11-Abs, and the nature of the immune response in both the brain and the tumour samples was examined in detail for a better characterization of the immunopathogenesis of this condition.
Materials and methods
Kelch-like protein 11 antibodies in reportedly seronegative paraneoplastic neurological syndrome
The presence of KLHL11-Abs was retrospectively assessed in a cohort of 138 patients with reportedly Ab-negative PNS [105 patients with limbic encephalitis and 33 with paraneoplastic cerebellar degeneration] referred to the French Reference Center for Paraneoplastic Neurological Syndromes and Autoimmune Encephalitis (exploratory cohort). All cases were previously examined using a comprehensive Ab-screening (anti-Hu, Ri, Yo, Ma2, CV2/CRMP5, amphiphysin, GAD65, AK5, NMDAR, GABAAR, GABABR, AMPAR, IgLON5, LGI1, CASPR2 and DPPX) with negative results, and their samples (CSF and/or serum) were stored at the NeuroBioTec biobank (Hospices Civils de Lyon, Lyon, France). KLHL11-Ab positivity was defined as a positive CBA result using HEK293 cells transfected with a human KLHL11 clone, as previously described.2,3 Subsequently, all positive samples were also examined by indirect IFA on rat brain sections in order to establish if a characteristic staining was present. Briefly, rat brain was cut in half, fixed during 1 h in paraformaldehyde 4% at 4°C, washed three times on phosphate buffer 0.1M, immersed in sucrose solution 30% for 36 h, then frozen in isopentane at −40°C for 2 min. The frozen brain was cut into 10-μm-thick sagittal sections. Brain sections were rehydrated with phosphate-buffered saline 1X during 10 min and blocked for 1 h in phosphate-buffered saline containing 3% bovine serum albumin and 3% normal goat serum. Patient CSF or serum was then incubated overnight at room temperature (dilution: CSF: 1/10; serum 1/100). Slides were washed three times in phosphate-buffered saline and incubated for 1 h with 488 Alexa Fluor-conjugated goat anti-human IgG (Thermo Fisher Scientific, Waltham, MA). After three washes, slides were mounted in Mowiol medium (Sigma-Aldrich, Saint-Louis, MO) and read using a Zeiss Axiophot microscope (Zeiss, Oberkochen, Germany). To assure that the immunostaining detected on IFA was related to the KLHL11-Abs, we compared it to the one obtained using a commercial KLHL11-Ab. In detail, brain slides were incubated with KLHL11 Polyclonal Ab (Thermo Fisher REF: PA5-62944, 1/1500) overnight, washed three times with phosphate-buffered saline 1X and highlighted with the antibody Alexa FluorTM 488 goat anti-rabbit IgG (H-L) (Invitrogen A11034, 1/1000).
In addition to testing retrospectively collected cohorts, since 1 January 2020, samples sent to the reference centre were prospectively screened for KLHL11-Abs as new usual clinical practice.
Kelch-like protein 11 antibodies in well-characterized paraneoplastic neurological syndrome
In order to validate the findings regarding the clinical specificity of KLHL11-Abs, a retrospective cohort of 83 patients with well-characterized Ab-associated PNS diagnosed in the study centre was examined; this cohort consisted of Ma2-Ab encephalitis (n = 34) and Ab targeting N-methyl-D-aspartate receptor encephalitis with teratoma (n = 49) patients whose samples were stored at NeuroBioTec (validation cohort). The Ma2-Ab series included also patients (n = 4) with PNS triggered by immune checkpoint inhibitors.4
Clinical data
Clinical and oncological data of patients from both the exploratory and validation cohorts (total n = 221), stored in the database of the French Reference Center for Paraneoplastic Neurological Syndromes and Autoimmune Encephalitis, were collected by AV. Physicians in charge of the KLHL11-Ab positive patients were contacted in order to obtain information regarding the last follow-up, relevant neuroimaging data, pathological samples and—whenever available—videos of the last neurological examination.
Design and validation of a clinical score for predicting Kelch-like protein 11 antibodies’ positivity
Since there is no commercial test available at the moment, very few laboratories in the world are capable of identifying KLHL11-Abs. Therefore, a secondary aim of this study was to design a clinical score for detecting those cases that, despite being seronegative for other neuronal Abs, are most likely to harbour KLHL11-Abs, and hence need to be tested at reference centres for PNS. For this purpose, after identification of the relevant features of KLHL11-Ab syndrome, a clinical score was designed and its diagnostic performance was tested in reportedly seronegative cases (exploratory cohort, n = 138).
Pathological studies
Pathological samples (brain, testicular tumours and lymph node metastases) of patients with KLHL11-Ab syndrome were comprehensively examined by two experienced pathologists (S.P. and E.U.C.). The characteristics of tumour immune infiltrates were identified and compared to those of a control group consisting in testicular seminomas (n = 3) and ‘burned-out’ tumours (n = 2) of patients without PNS. In detail, 4 µm-thick sections of formalin-fixed-paraffin-embedded brain and testis samples were stained with haematoxylin and eosin and then immunohistochemically stained using Ventana Benchmark XT Immunostainer (Ventana, Tucson, AZ). Samples were stained with Abs against CD20, CD3, CD8, PD-1, PD-L1, T-cell immunoglobulin and mucin domain-3 and CD68. Haematoxylin and eosin slides were digitalized using Nanozoomer scanner and the distance between Purkinje cells was measured and considered as indirect measure of cerebellar atrophy using NDP.view2 (Hamamatsu, Shizuoka, Japan).
Statistical analysis
Categorical variables were expressed as frequencies and percentages and continuous variables as median and range. The sensitivity, specificity and accuracy of the newly designed clinical score were calculated using MedCalc, version 19.5.3 (MedCalc Software, Ostend, Belgium). A prevalence of 1.4 per 100 000 persons for KLHL11-Ab syndrome (previously described in the Olmsted County, MN, USA)1 was considered for the calculation of the diagnostic performance of the score.
Standard protocol approval and patient consent
Written consent for sample analysis and medical research was previously obtained from all patients, and the study was approved by the Ethical Committee of the Hospices Civils de Lyon.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article. Additional data not published are available and will be shared by request from any qualified investigator.
Results
Frequency of Kelch-like protein 11 antibodies in different clinical scenarios
KLHL11-Abs were detected in serum and/or CSF of 11 patients using CBA, 10 of them from retrospectively identified cases (exploratory and validation cohorts) and 1 additional prospectively detected patient.
In these 11 cases, IFA showed a distinct pattern (diffuse ‘leopard-like’ staining associated with ‘comb-like’ appearance in the CA3 region of the hippocampus, Fig. 1) not compatible with previously known Abs. In the retrospective exploratory cohort, the group with the highest rate of Ab-positivity was the paraneoplastic cerebellar degeneration group (7/33, 21% versus 2/105, 2% in the limbic encephalitis group). In the validation cohort, 1/34 (3%) patient with Ma2-Abs had concomitant KLHL11-Abs, and none (0/49, 0%) with Ab targeting N-methyl-D-aspartate receptor encephalitis and teratoma harboured KLHL11-Abs (Fig. 2).
Figure 1.

Immunostaining pattern in patients with KLHL11-Abs. Reactivity of rat brain tissue using a negative control (row 1) and with patient CSF positive for KLHL11-Abs (rows 2, 3 and 4). The CSF of patients showed extensive and diffuse immunostaining involving the cerebellar cortex and nuclei, putamen and hippocampus (in particular, the CA3 region, with a ‘comb-like’ staining). In the cerebellum and basal ganglia the immunostaining showed a ‘leopard-like’ appearance. For comparison, immunostaining using a commercial KLHL11-Abs is also shown (row 5), highlighting the involvement of the same regions.
Figure 2.

Frequency of KLHL11-Abs in different clinical scenarios. KLHL11-Abs positivity was retrospectively assessed in patients with Ab-negative PNS [limbic encephalitis (n = 105), cerebellar degeneration (n = 33)] and well-characterized PNS [Ma2-Ab syndrome (n = 34), Abs targeting N-methyl-D-aspartate receptor syndrome with teratoma (n = 49)].
Clinical features at symptom onset
All the patients with KLHL11-Abs were male (11/11, 100%), their median (range) age was 44 (35–79) years. At symptom onset, three patterns were observed: ataxia (8/11, 73%), neuropsychiatric symptoms (2/11, 18%) and isolated diplopia (1/11, 9%).
Among patients with ataxia, 3 of 8 (37%) experienced transient (few hours), paroxysmal episodes of vertigo, nausea/vomiting and unbalance, which lasted for ∼3 months (Patients 4 and 10), or up to 5 years (Patient 3), before they developed a permanent cerebellar syndrome. The remaining cases had a persistent ataxia with either a hyperacute (2/8, 25%) or subacute (3/8, 37%) onset.
A total of 7/11 (64%) patients also experienced severe weight loss at the beginning of the illness, and 2/11 (18%) patients reported a notable worsening of additional symptoms while standing (Patient 5: orthostatic headache, Patient 10: upper limb weakness).
Clinical features of the full-blown syndrome
The full-blown syndrome consisted of a cerebellar syndrome in all patients (11/11, 100%), either isolated (4/11, 36%) or, more commonly, part of a multi-system neurological disorder (7/11, 64%). Additional syndromes included limbic encephalitis (5/11, 45%), and longitudinally extensive myelitis (2/11, 18%). Among all cases, the symptoms were gait ataxia (11/11, 100%, Video 1), limb ataxia (9/11, 82%), dysarthria (9/11, 82%, Video 2), vertigo (8/11, 73%), cognitive disturbances (7/11, 64%), nystagmus (7/11, 64%), spasticity (6/11, 54%), oculomotor disturbances (5/11, 45%, Video 3), sensorineural hearing loss (5/11, 45%), tinnitus (5/11, 45%), intention tremor (4/11, 36%) and dysphagia (3/11, 27%). For patients with both gait and limb ataxia, axial involvement was always prevalent compared to the appendicular one (Videos 4 and 5). Personality change was reported in 7/11 (64%) patients, while 4/11 (36%) experienced a pseudobulbar syndrome, manifesting predominantly as pathological crying. Most of the patients with cognitive disorders were further studied with detailed neuropsychological examination (6/7, 86%), which disclosed deficits involving executive function (5/6, 83%), language (5/6, 83%) and spatial cognition (3/6, 50%). Neuropsychological reports are available in the Supplementary Table.
Rarer neurologic manifestations included hypersomnia and seizures (2/11, 18%). Patient 9, who initially presented apathy and depression, later developed a complex syndrome characterized by cognitive disturbance, vertical gaze palsy, tendency to fall backwards and tremor, which clinically resembled progressive supranuclear palsy (PSP). Patient 10, who manifested paroxysmal vertigo and tinnitus at onset, subsequently manifested prominent weakness in the upper limbs consistent with a clinical diagnosis of flail arm syndrome, comprising both upper and lower motor neuron signs in the upper limbs. Before a final diagnosis of PNS was made, 5/11 (45%) patients received alternative diagnoses that were genetic/degenerative ataxias (3/5, 60%), stroke (1/5, 20%) and depression (1/5, 20%).
Paraclinical studies
Brain MRI was abnormal in all but one patient (10/11, 91%) (Fig. 3). The most common neuroimaging abnormality was cerebellar atrophy (8/11, 73%), which was a late finding and was preceded by cerebellar T2-weighted hypersignal in 2/11 (18%) cases. A total of 5/11 (45%) patients had mesial temporal lobe hypersignal, either bilateral (3/11, 27%) or unilateral (2/11, 18%), and 4/5 (80%) patients later developed hippocampal atrophy. Less commonly, brain MRI showed hypersignal involving the brainstem (2/11, 18%) or both thalami (1/11, 9%). In addition, the presence of gadolinium enhancing lesions was detected in 4 of 11 (36%) cases: mesial temporal lobe in 2, brainstem in 1, leptomeningeal in 1. Two patients with clinical signs of myelopathy also underwent spine MRI that revealed longitudinally extensive myelitis in both. CSF was abnormal in all cases (11/11, 100%), the abnormalities found were: the presence of CSF-specific oligoclonal bands (8/9 tested, 89%), pleocytosis (8/11, 73%) and increased protein content (8/11, 73%). Pleocytosis was mild (median: 8 cells/µl, range: 0–83), as was the protein level (median: 0.6 mg/dl, range: 0.23–0.88). In two patients with progressive ataxia, a comprehensive panel of inflammatory and neurodegenerative biomarkers was performed, high levels of total tau and neopterin were detected in both of them.
Figure 3.

Neuroimaging findings in patients with KLHL11-Abs. Axial brain MRI (A), showing hypersignal asymmetrically involving both cerebellar hemispheres, colocalizing with an area of cerebellar hypometabolism on brain PET (B). Sagittal spine MRI showing spinal cord hypersignal in a patient with myelitis (C). Marked cerebellar atrophy is shown in both sagittal (D) and axial (E) brain MRI. The atrophy is more pronounced in the vermis and associates with an area of hypersignal involving the temporal lobe (E). Axial brain MRIs showing hypersignal involving the left mesial temporal lobe (F), the right thalamus (G) and coexistence of left mesial temporal lobe hypersignal and right hippocampal atrophy.
Oncological associations
An associated testicular cancer was found in 9/11 (82%) patients, a spontaneously regressed (‘burned-out’) testicular germ cell tumour with lymph node (para-aortic) metastases in 7/9 (78%) of them (Table 1). No cancer was found in the remaining 2/11 (18%) patients after a follow-up of 21 and 33 months, respectively, but orchiectomy was not performed in these cases, and FDG-PET/CT imaging was done only in 1 of them. The first diagnostic test that raised the suspicion of cancer was testicular ultrasound (4/9, 44%), FDG-PET/CT (3/9, 33%) and whole-body CT scan (1/9, 11%), while this information was not available for one case. The onset of PNS preceded the discovery of cancer in 6/9 cases (67%), with a median (range) delay of 2 months (1–27). In the three cases in which cancer preceded PNS, median (range) delay was 74 months (2–156) but in the patient with the longest interval (13 years), PNS appearance coincided with cancer relapse. The oncological treatment consisted in surgery in all cases (9/9, 100%), chemotherapy in 6/9 (67%) and radiotherapy in 1/9 (11%).
Table 1.
Characteristics of patients with KLHL11-Ab paraneoplastic neurological syndrome
| Patient No. (Cohort) | Sex/age (years) | Presentation | Main clinical features | Cancer | Brain MRI | CSF [White cells per mm3/Protein (g/L)/oligoclonal bands] | Distinctive features | Immunotherapy | Modified Rankin Scale score before and after PNS treatment (length of follow-up) |
|---|---|---|---|---|---|---|---|---|---|
| 1 (Retrospective PCD) | M, 47 | Sudden onset of gait ataxia, with nausea and vomiting, weight loss (5 kg in 3 months) | Dysexecutive syndrome with behavioural disturbances, dysarthria, nystagmus, gait and limb ataxia | ‘Burned-out’ testicular germ cell tumour |
|
↑ (21)/↑ (0.87)/+ | Brain biopsy performed: microglial activation, perivascular epithelioid inflammatory infiltrate, rarefaction of Purkinje cells | Corticosteroids, IVIG, cyclophosphamide, rituximab | 5→4 (31 months) |
| 2 (Retrospective PCD) | M, 46 | Subacute onset of gait ataxia and dysarthria | Hypersomnia, ophthalmoplegia, bilateral deafness, anarthria, paraparesis and urinary disjunction, gait and limb ataxia | Non-seminomatous testicular germ cell tumours |
|
↑ (12)/↑ (0.74)/+ | Limbic encephalitis and myelitis developed after cerebellar syndrome | Corticosteroids, cyclophosphamide and rituximab | 3→5 (126 months) |
| 3 (Retrospective PCD) | M, 42 | Episodic ataxia, with vertigo, nausea and vomiting (transient episodes for 5 years) | Gait and limb ataxia, dysarthria, weight loss | ‘Burned-out’ testicular tumour | Cerebellar atrophy | ↑ (8)/n (0.46)/+ | Comprehensively studied for genetic causes of episodic ataxia | Corticosteroids, IVIG, rituximab | 3→3 (101 months) |
| 4 (Retrospective PCD) | M, 43 | Paroxysmal episodes of vertigo and gait imbalance |
|
Partial ‘burned-out’ testicular seminoma |
|
↑ (6)/↑ (0.58)/+ | Initial diagnosis of benign paroxysmal positional vertigo, later comprehensively studied for genetic ataxias | Corticosteroids, plasmapheresis, IVIG, cyclophosphamide | 4→6 (42 months) |
| 5 (Retrospective Ma2-Ab) | M, 35 | Sudden onset of oscillopsia, vertigo, headache, tinnitus, weight loss (10 kg in 1 month) | Dysexecutive syndrome, memory deficits, seizures, opsoclonus-myoclonus | Mixed testicular cancer (90% seminoma, 10% embryonic carcinoma) |
|
↑ (83)/n (0.29)/NA | Co-existence of Ma2-Abs | Corticosteroids | 4→3 (37 months) |
| 6 (Retrospective LE) | M, 44 | Memory deficits, psychomotor slowing, diplopia | Memory deficits, dysarthria, lower-limb spasticity, gait ataxia | ‘Burned-out’ testicular seminoma |
|
n (0)/↑ (0.58)/+ | Previous history of cryptorchidism | IVIG, cyclophosphamide, rituximab | 3→4 (62 months) |
| 7 (Retrospective PCD) | M, 64 | Gait ataxia, dysarthria, vomiting, weight loss (15 kg in 1 year) | Tetrapyramidal syndrome, transient episodes of diplopia, dysphagia, hyperacusis, tinnitus, gait ataxia | Not found (testicular ultrasound not performed) |
Initial: normal Evolution: cerebellar atrophy |
↑ (4)/↑ (0.46)/+ | Elevated CSF neopterin and total Tau | Corticosteroids, IVIG, rituximab, cyclophosphamide | 4→6 (33 months) |
| 8 (Retrospective PCD) | M, 41 | Gait and limb ataxia | Cognitive difficulties, hearing loss, dysarthria, vertical gaze palsy, nystagmus, spasticity | ‘Burned-out’ germ-cell tumour |
|
n (0)/↑ (0.8)/– | Significant hypometabolism on brain PET involving cerebellar vermis and L cerebellar hemisphere | IVIG | 4→4 (44 months) |
| 9 (Retrospective LE) | M, 79 | Apathy, weight loss (9 kg in 2 months) | Hypersomnia, memory disturbances, micrographia, vertical gaze palsy, tremor, gait instability, tendency to fall backwards | Not found (testicular ultrasound normal) | Hypersignal involving mesial temporal lobes and R hippocampal atrophy | ↑ (9)/↑ (0.7)/+ | PSP-like phenotype, elevated CSF neopterin and total Tau | Corticosteroids, IVIG | 4→6 (21 months) |
| 10 (Retrospective PCD) | M, 42 | Paroxysmal episodes of vertigo, nausea and tinnitus, weight loss (8 kg in 1 month) | Bilateral severe weakness of upper limbs, associated with amyotrophy, fasciculations, bilateral Hoffmann signs, gait ataxia | ‘Burned-out’ germ-cell tumour |
|
↑ (7)/↑ (0.6)/+ | Flail arm syndrome | Corticosteroids, IVIG, cyclophosphamide | 3→5 (51 months) |
| 11 (Prospective PCD) | M, 55 | Sudden onset of gait ataxia, vertigo, tinnitus and weight loss (2 kg in 4 months) | Gait and limb ataxia, dysarthria, nystagmus | ‘Burned-out’ germ-cell tumour | Initial: normal | ↑ (20)/↑ (0.88)/NA | Sudden onset | IVIG, corticosteroids | 3→4 (2 months) |
Treatment and outcome
The syndrome was severe in all patients, their median (range) modified Rankin Scale score was 4 (3–5) before immunotherapy treatment. All patients (11/11, 100%) were treated with at least a first-line immunotherapy, which consisted of corticosteroids (9/11, 82%), intravenous immunoglobulin (9/11, 82%), or plasma exchange (1/11, 9%).
Corticosteroid therapy typically consisted of IV steroid bolus (adopted in seven cases), with a dosage of 1 g daily of methylprednisolone for 3 or 5 days. Additionally, some patients were treated with second-line immunotherapies (6/11, 54% with rituximab and 5/11, 45% with cyclophosphamide). The beneficial effects of immunotherapy were poor: 2/11 (18%) patients improved, 2/11 (18%) stabilized and 7/11 (64%) worsened, including 3 who died (2 of them with no detected cancer). The median (range) follow-up was 42 (2–126) months and at last examination, the median (range) modified Rankin Scale score was 4 (3–6). The two patients that improved were treated aggressively: one underwent orchiectomy, treatment with intravenous immunoglobulin, corticosteroids, cyclophosphamide and rituximab in succession, while the other was treated with orchiectomy and chemotherapy, as well as corticosteroids (this latter case had coexisting Ma2-Abs). None of the patients with fatal outcome died due to cancer: one had severe neurological disability and requested medically assisted suicide outside France, one had severe bulbar involvement, and one had a subdural hematoma related to a fall.
Clinical score for identifying patients with Kelch-like protein 11 antibodies
A clinical score (MATCH score) was designed based on the relevant clinical findings identified from the present study and from reviewing published studies,1–3 and consisted of the following items: M [male sex (1 point)], A [ataxia or other cerebellar signs (1 point)], T and C [testicular tumour (2 points) or other cancer (1 point)], and H [sensorineural hearing loss or hearing alterations, including hyperacusis or tinnitus (1 point)].
It should be noted that, within the score, ataxia includes gait and/or limb ataxia, but also other cerebellar signs, comprising intention tremor, nystagmus, impaired smooth pursuits and slurred/scanning speech. Testicular tumours include also those detected in metastatic lymph nodes without primitive found (spontaneously regressed testicular cancer), or with indirect signs of a burned-out neoplasia (testicular fibrosis and microlithiasis).
A MATCH score cut-off ≥4 was retrospectively tested among patients from the exploratory cohort (n = 138): using this threshold, the score had a sensitivity of 78%, a specificity of 99% and an accuracy of 98% for identifying patients with KLHL11-Abs (Fig. 4).
Figure 4.

Diagnostic performance of the newly designed MATCH score. The MATCH score with a cut-off ≥4 identified patients with KLHL11-Abs in a cohort of 138 patients with reportedly seronegative PNS, with a sensitivity of 78%, a specificity of 99% and an accuracy of 98%.
In addition, we applied the score to the 13 patients of the initial publication of KLHL11-Abs,1 finding a median score (range) of 5 (4–5), which means that all the cases would have been detected by the MATCH score.
Immunopathology
Pathological samples were available for five patients: three ‘burned-out’ testicular tumours (Patients 1, 4 and 10), three lymph node metastases of testicular tumours (Patients 4, 6 and 8) and one brain biopsy (Patient 1).
Histological examination of the brain biopsy showed a cerebellar encephalitis. Numerous granulomas, consisting of macrophages, were located at the interface between the granular and molecular layers (Fig. 5A and B). A perivascular infiltrate was observed along with marked depletion of Purkinje cells (Fig. 5C). The distance between Purkinje cells ranged from 158 to 2 710 µm, while a normal gap of 174 ± 41 µm is observed in control subjects5 (Fig. 5C), indicating an extensive Purkinje cells loss. Immunohistochemical analyses revealed predominance of CD3+ T cells (Fig. 5D), with numerous CD8+ cytotoxic T cells (Fig. 5E). No Granzyme B expression was observed. Some T cells expressed the immune inhibitory checkpoint PD-1 (Fig. 5E), while a strong expression of two others immune inhibitory checkpoints (PD-L1 and T-cell immunoglobulin and mucin domain-3) was observed within granulomas (Fig. 5F and G). Taking together, these observations indicated that the immune reaction in the brain was a chronic inflammation at a phase of ‘exhaustion’—due to immune checkpoint expression—and most likely non-functional.
Figure 5.
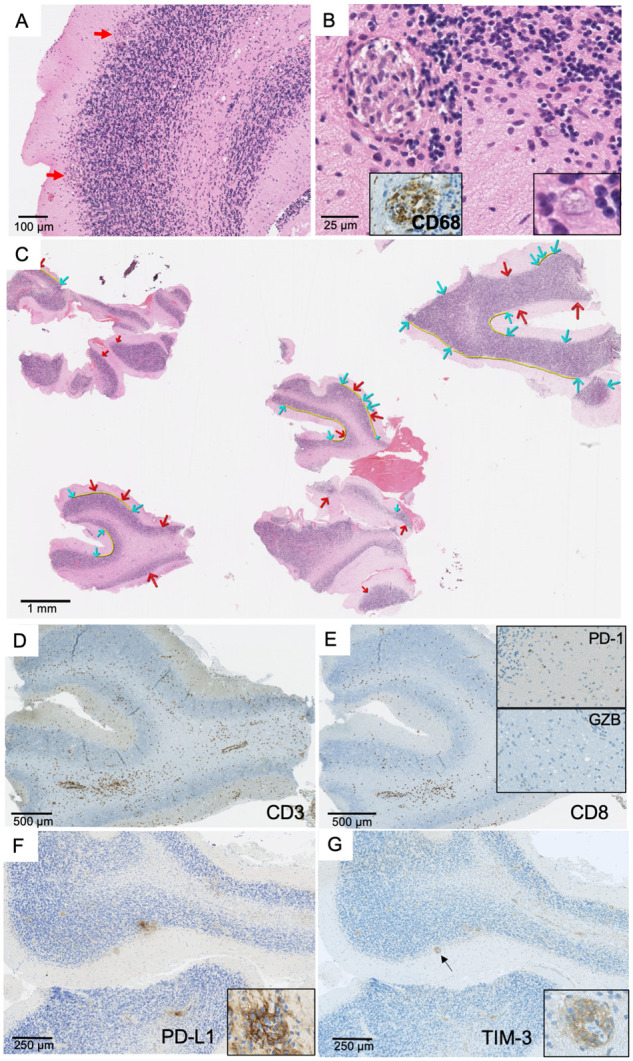
Brain biopsy in a patient with KLHL11-Ab encephalitis. Pathological brain findings demonstrating the presence of granulomas located at the interface between granular and molecular layers of the cerebellum (A, ×100). These granulomas were formed by epithelioid cells showing positive CD68 staining (B, left panel, ×200) in proximity to Purkinje cells with shrunk cytoplasm (B, right panel, ×300 and ×500). Panel C (×10): a huge neuronal loss was demonstrated by the increased distance (yellow line) between Purkinje cells (blue arrows); numerous granulomas were also observed (red arrows). CD3+ T cells infiltrate was observed predominantly in perivascular space (D, ×50) with numerous CD8+ T cells (E, ×50). Scattered lymphocytes expressing PD-1 (E, top insert, ×100) but not Granzyme B (E, bottom insert, ×150) were detected. A strong expression of PD-L1 was observed in granulomas (F, ×100), which was formed by macrophages (F, insert, ×400). A strong expression of TIM-3 was also detected in granulomas (arrow) (G, ×100; insert: ×400).
The haematoxylin and eosin staining of the testicular tumour in Patient 1 showed a hyaline scar with no tumour cell corresponding to a complete tumour regression (‘burned-out’ tumour) (Fig. 6A). This scar was surrounded by atrophic and inflammatory testicular parenchyma. The inflammatory infiltrate corresponded to CD3+CD8+ T cells often expressing Granzyme B, an activation marker (Fig. 6B–D). No or few cells expressed PD-1, PD-L1 and T-cell immunoglobulin and mucin domain-3. In this ‘burned-out’ tumour, the lymphocytic infiltrate could be classified as an efficient immune reaction. For Patients 4 and 10, the testicular tumours presented a lymphocytic infiltrate with predominance of CD3+ T cells, a strong expression of Granzyme B and a weak expression of PD-1, with no expression of PD-L1 (Fig. 7A–E).
Figure 6.

Pathological findings in ‘burned-out’ testicular tumours. Panel A (×100) shows a large area of fibrosis with no tumour cells (white star), surrounded by testicular parenchyma with abundant lymphocytic infiltrate. Numerous T cells expressed Granzyme B (B, ×200; insert: ×300). The infiltrate was represented by abundant T CD3+ cells (C, ×100, insert: ×200), most commonly by CD8+ (D, ×100; insert: ×200).
Figure 7.

Testicular tumour and lymph node metastasis. Testicular tumour (A–E). Partial fibrous involution of a testicular tumour resembling a teratoma (A, ×10) with staining of CD3+ T cells (B, ×150), Granzyme B+ cells (C, ×150), few scattered PD-1+ cells (D, ×100) and no expression of PD-L1 within the teratoma (E, ×150). Lymph node metastasis (F–J). Metastasis corresponding to the localization of a seminoma (red star: tumour) (F, ×100), with abundant CD3+ T cell infiltrate surrounding tumour cells (G, ×100), no Granzyme B expression (H, ×100), strong PD-1 expression by lymphocytes (I, ×100) and strong PD-L1 expression preferentially observed in macrophages (J, ×100).
Lymph node metastases (Fig. 7F–J) corresponded to lobules of seminomatous tumour cells surrounded by an abundant immune infiltrate, with a majority of CD3+ T cells with no expression of Granzyme B. PD-1 and PD-L1 were strongly expressed by lymphocytes and macrophages, in line with an escape phase of anti-tumour immunity.
In control patients (Supplementary Fig. 1), all tumours presented a CD3+ T cell infiltrate with numerous CD8+ T cells. The ‘burned-out’ tumours were also Granzyme B+ whereas seminomas were negative. ‘Burned-out’ tumours had few scattered PD-1+ lymphocytes and no expression of PD-L1, whereas lymphocytes and macrophages within seminoma tumours strongly expressed both PD-1 and PD-L1. These data indicated that ‘burned-out’ tumours exhibited a more efficient immune profile than classical forms of seminoma.
Discussion
In this study, the clinical and immunopathological features of a French cohort of patients with KLHL11-Abs were examined in detail, which provided important information with practical and mechanistic implications. Firstly, the results herein suggest that the patients displayed a homogeneous phenotype, which was characterized by specific core features that were included in the newly designed MATCH score (male sex, ataxia, testicular cancer and hearing alterations). Since few laboratories in the world are capable of testing KLHL11-Abs, this clinical score can be helpful in deciding which samples need to be sent for testing in reference centres, as it had a good sensitivity (78%) and a very high specificity (99%) in identifying KLHL11-Abs patients. Secondly, the reported number of cases with this syndrome is very low, but probably largely underestimated, as approximately one-fifth of patients with reportedly seronegative paraneoplastic cerebellar degeneration harboured Abs against KLHL11 herein. While it is known that most female patients with paraneoplastic cerebellar degeneration carry Yo-Abs,6,7 male patients display a more heterogeneous Ab-association, including some seronegative cases.7 An important message is therefore to test with testicular ultrasound all adult male patients with new-onset, progressive, gait ataxia with no alternative aetiology found, as some patients will display signs of an active or spontaneously regressed tumour (in the context of a KLHL11 or Ma2-Abs syndromes). Thirdly, all patients herein presented a distinctive immunostaining, and probably the differences observed across previous studies are related to the role of IFA in the inclusion of patients. Since Ab-testing performed in non-compatible clinical contexts, or without confirmation using other techniques, can lead to a large proportion of false positive results,8,9 we advise to perform IFA always in conjunction with CBA, as suggested by others.2,10
We did not find any patients positive for KLHL11-Abs on CBA, but negative on IFA. Again, this is probably linked to the inclusion of patients with a high pre-test probability of PNS, as only samples from patients with a compatible clinical presentation were stored in our dedicated biobank. It should also be noted that the immunostaining pattern detected here (‘leopard-like’ with CA3 ‘comb-like’ appearance) is different from the one shown in the initial description of KLHL11-Abs (‘sparkles’ pattern),2 which could reflect the differences in the tissue fixation methodology adopted. Nevertheless, the pattern observed here was validated using a commercial Ab, while the antigenic specificity was confirmed using a CBA. In addition, the expression pattern of the KLHL11 protein in the mouse brain supports the prominent hippocampal and cerebellar staining (Supplementary Fig. 2).
From a clinical standpoint, several clinical specificities beyond the initial described features of this disorder were noticed, namely the severe weight loss in a substantial proportion of patients, as well as the rare occurrence of central hypersomnia and phenotypes resembling primary neurodegenerative disorders, including atypical parkinsonism (progressive supranuclear palsy-like) and motor neuron syndrome (flail arm syndrome-like). The occurrence of these features has been previously reported in other types of PNS, including motor neuron syndrome in 10% of patients with Ma2-Abs,11 parkinsonism in 17% of Ri-Ab12 and 23% of IgLON5-Ab syndromes.13 Similarly to IgLON5 encephalitis, for which the pathological findings are suggestive of a convergence of autoimmune and neurodegenerative mechanisms, high levels of total tau in the CSF of some patients and a clear cerebellar atrophy (from both neuroimaging and pathological evidence) were observed, pointing towards a two-step course of the disease, with inflammation at onset (when Purkinje cells dysfunction is probably not yet irreversible) and subsequent development of neuronal loss. In agreement with this hypothesis, three patients exhibited transient, paroxysmal episodes of vertigo, nausea/vomiting and unbalance that lasted for several months or years prior to the development of a permanent cerebellar syndrome. This atypical course has also been previously documented in other autoimmune ataxias, including those linked to Abs targeting CASPR2 and GAD65.14,15
On the opposite side of the spectrum, two patients developed abruptly severe and persistent cerebellar syndrome (‘stroke-like’ onset).16 In all patients, a cerebellar syndrome was present in the full-blown disease, and consisted of a severe gait ataxia, commonly associated with a less prominent limb ataxia, and variable combinations of dysarthria, oculomotor disturbance and nystagmus. A minority of the patients developed limbic encephalitis and others myelitis, confirming previous observations.2 Importantly, evidence of cognitive disturbance was observed in most cases, manifested by prominent impairment of executive functions, language deficits, as well as difficulties in spatial cognition and behavioural abnormalities. These features are the core elements of ‘cerebellar cognitive affective syndrome’, a distinctive neuropsychological disorder observed in patients with lesion restricted to the posterior cerebellar lobes or the vermis.17 In addition, in some cases, a hippocampal memory dysfunction is also suggested. Further prospective studies are needed to delineate the precise neuropsychological spectrum of KLHL11-Ab patients, and to validate our observations on cerebellar cognitive affective syndrome in larger cohorts.
The immunopathological studies performed in tumours, lymph nodes and brain of KLHL11-Ab patients permitted to shed light on a very rare and intriguing phenomenon observed in oncology—that is, the spontaneous regression of a primary testicular tumour and the concomitant development of a PNS. Despite the extreme rarity of burned-out testicular tumours,18 most of the patients with KLHL11-Abs had this type of tumour. An involvement of anti-tumour immunity as the cause of tumour regression is probable,19 as it has been observed in other cancers, such as melanoma.20 Interestingly, seminoma cells from primary testicular tumours have been shown to express the KLHL11 antigen,2 but it is not clear whether the immune breakdown leading to the PNS is initiated in the primary tumour or in the lymph node metastases. We speculate that the arrival of tumour clones in the lymph node could stimulate an anti-tumour immunity via the selection and cascade activation of CD8+ T lymphocytes directed against several tumour antigens, including KLHL11. This stimulation of a large lymphocyte population would allow the efficient destruction of the primary tumour, leading to the observed total regression. However, metastatic tumour clones may resist to this destruction, potentially via the expression of molecules that inhibit the immune attack such as PD-L1, as found herein. In parallel, the systemic circulation of anti-KLHL11 CD8+ T lymphocytes is thought to be responsible for the destruction of Purkinje neurons. As long as the tumour clones located in the lymph node are not destroyed, they could be responsible for the chronic stimulation of self-reactive anti-KLHL11 CD8+ lymphocytes. The chronicity of this auto-reactive immune response is well underlined by the ‘exhausted/chronic’ lymphocyte phenotype observed in metastases and in the cerebellum of patients.
Since the final consequence of this immune response is the irreversible destruction of Purkinje cells, it is of major importance to identify readily and treat these patients. In this context, the MATCH score proposed herein can be a practical tool for physicians. In addition, tumour removal and second-line immunosuppression should be considered early, especially given the probable T-cell-mediated pathogenesis, although larger series are needed to support this recommendation. In this study, ∼40% of patients did not receive aggressive immunosuppression, probably because in most of the cases, at time of initial diagnosis, no associated Abs were found and many cases received an initial misdiagnosis.
Along with the small sample size and availability of only one brain sample for pathological analysis, the other limitations of the present study are its retrospective design and the potential referral bias towards more severe cases. Therefore, it is important to propose a prospective validation of the MATCH score with more patients to assess its clinical value.
Moreover, incomplete or atypical presentations not captured by this study and the previous clinical series that investigated this disorder may exist.
In conclusion, the present study demonstrated that patients with KLHL11-Abs share a homogeneous clinical phenotype, whose main features are summarized in the newly designed MATCH score, which can be used by clinicians to select the patients that need to be tested for KLHL11-Abs. The pathological substrate is represented by an active T-cell inflammation at the primary tumour that leads to regression, in association with chronic, exhausted, inflammation in the brain with massive Purkinje cell loss, which probably explains the treatment-refractory nature of this syndrome.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
The authors thank NeuroBioTec, Hospices Civils de Lyon BRC (France, AC-2013–1867, NFS96-900) for banking sera and CSF samples. They also thank Dr Elsa Poullot for providing a pathological sample. We gratefully acknowledge Dr Hélène Boyer for English language editing (Direction de la Recherche Clinique et de l’Innovation, Hospices Civils de Lyon).
Funding
This study is supported by research grants from FRM (Fondation pour la recherche médicale, DQ20170336751) and has been developed within the frameworks of the LABEX CORTEX (reference ANR-11-LABX-0042) and the BETPSY project (reference ANR-18-RHUS-0012), both operated by the French National Research Agency (ANR).
Competing interests
A.V. received a fellowship grant from the European Academy of Neurology (EAN). M.M. received funding from the Luxembourg National Research Fund (FNR PEARL P16/BM/11192868). No other disclosure was reported.
Glossary
- CBA
cell-based assay
- IFA
immunofluorescence assay
- IVIG
intravenous immunoglobulin
- KLHL11
Kelch-like protein 11
- LE
limbic encephalitis; PCPurkinje cell
- PCD
paraneoplastic cerebellar degeneration
- PD-1
Programmed cell death-1
- PD-L1
Programmed cell death-ligand 1
- TIM-3
T-cell immunoglobulin and mucin domain-3
References
- 1.Mandel-Brehm C, Dubey D, Kryzer TJ, et al. Kelch-like protein 11 antibodies in seminoma-associated paraneoplastic encephalitis. N Engl J Med. 2019;381(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubey D, Wilson MR, Clarkson B, et al. Expanded clinical phenotype, oncological associations, and immunopathologic insights of paraneoplastic Kelch-like protein-11 encephalitis. JAMA Neurol. 2020;77(11):1420–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maudes E, Landa J, Muñoz-Lopetegi A, et al. Clinical significance of Kelch-like protein 11 antibodies. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e666. doi: 10.1212/NXI.0000000000000666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vogrig A, Fouret M, Joubert B, et al. Increased frequency of anti-Ma2 encephalitis associated with immune checkpoint inhibitors. Neurol Neuroimmunol Neuroinflamm. 2019;6(6):e604. doi: 10.1212/NXI.0000000000000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louis ED, Rabinowitz D, Choe M, et al. Mapping Purkinje cell placement along the Purkinje cell layer: An analysis of postmortem tissue from essential tremor patients vs. controls. Cerebellum. 2016;15(6):726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hébert J, Riche B, Vogrig A, et al. Epidemiology of paraneoplastic neurologic syndromes and autoimmune encephalitides in France. Neurol Neuroimmunol Neuroinflamm. 2020;7(6):e883. doi: 10.1212/NXI.0000000000000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vogrig A, Gigli GL, Segatti S, et al. Epidemiology of paraneoplastic neurological syndromes: A population-based study. J Neurol. 2019;267(1):26–35. [DOI] [PubMed] [Google Scholar]
- 8.Déchelotte B, Muñiz-Castrillo S, Joubert B, et al. Diagnostic yield of commercial immunodots to diagnose paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e701. doi: 10.1212/NXI.0000000000000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graus F, Vogrig A, Muñiz-Castrillo S, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. 2021;8(4):e1014. doi: 10.1212/NXI.0000000000001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75(8):1135–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogrig A, Joubert B, Maureille A, et al. Motor neuron involvement in anti-Ma2-associated paraneoplastic neurological syndrome. J Neurol. 2019;266(2):398–410. [DOI] [PubMed] [Google Scholar]
- 12.Simard C, Vogrig A, Joubert B, et al. Clinical spectrum and diagnostic pitfalls of neurologic syndromes with Ri antibodies. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e699. doi: 10.1212/NXI.0000000000000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaig C, Graus F, Compta Y, et al. Clinical manifestations of the anti-IgLON5 disease. Neurology. 2017;88(18):1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joubert B, Gobert F, Thomas L, et al. Autoimmune episodic ataxia in patients with anti-CASPR2 antibody-associated encephalitis. Neurol Neuroimmunol Neuroinflamm. 2017;4(4):e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muñiz-Castrillo S, Vogrig A, Joubert B, et al. Transient neurological symptoms preceding cerebellar ataxia with glutamic acid decarboxylase antibodies. Cerebellum. 2020;19(5):715–721. [DOI] [PubMed] [Google Scholar]
- 16.Vogrig A, Bernardini A, Gigli GL, et al. Stroke-like presentation of paraneoplastic cerebellar degeneration: A single-center experience and review of the literature. Cerebellum. 2019;18(5):976–982. [DOI] [PubMed] [Google Scholar]
- 17.Schmahmann JD, Sherman JC.. The cerebellar cognitive affective syndrome. Brain. 1998;121 (Pt 4):561–579. [DOI] [PubMed] [Google Scholar]
- 18.Dorantes-Heredia R, Motola-Kuba D, Murphy-Sanchez C, et al. Spontaneous regression as a “burned-out” non-seminomatous testicular germ cell tumor: A case report and literature review. J Surg Case Rep. 2019;1:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogrig A, Muñiz-Castrillo S, Desestret V, et al. Pathophysiology of paraneoplastic and autoimmune encephalitis: Genes, infections, and checkpoint inhibitors. Ther Adv Neurol Disord. 2020;13:1756286420932797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukherji B. Immunology of melanoma. Clin Dermatol. 2013;31(2):156–165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article. Additional data not published are available and will be shared by request from any qualified investigator.