

Abstract
Napabucasin is an orally administered reactive oxygen species generator that is bioactivated by the intracellular antioxidant nicotinamide adenine dinucleotide phosphate:quinone oxidoreductase 1. Napabucasin induces cell death in cancer cells, including cancer stem cells. This phase 1 study (NCT03411122) evaluated napabucasin drug‐drug interaction potential for 7 cytochrome P450 (CYP) enzymes and the breast cancer resistance protein transporter/organic anion transporter 3. Healthy volunteers who tolerated napabucasin during period 1 received probe drugs during period 2, and in period 3 received napabucasin (240 mg twice daily; days 1‐11) plus a phenotyping cocktail containing omeprazole (CYP2C19), caffeine (CYP1A2), flurbiprofen (CYP2C9), bupropion (CYP2B6), dextromethorphan (CYP2D6), midazolam (CYP3A) (all oral; day 6), intravenous midazolam (day 7), repaglinide (CYP2C8; day 8), and rosuvastatin (breast cancer resistance protein/organic anion transporter 3; day 9). Drug‐drug interaction potential was evaluated in 17 of 30 enrolled volunteers. Napabucasin coadministration increased the area under the plasma concentration–time curve from time 0 extrapolated to infinity (geometric mean ratio [90% confidence interval]) of caffeine (124% [109.0%‐141.4%]), intravenous midazolam (118% [94.4%‐147.3%]), repaglinide (127% [104.7%‐153.3%]), and rosuvastatin (213% [42.5%‐1068.3%]) and decreased the area under the plasma concentration–time curve from time 0 extrapolated to infinity of dextromethorphan (71% [47.1%‐108.3%]), bupropion (79% [64.6%‐97.0%]), and hydroxybupropion (45% [15.7%‐129.6%]). No serious adverse events/deaths were reported. Generally, napabucasin is not expected to induce/inhibit drug clearance to a clinically meaningful degree.
Keywords: breast cancer resistance protein transporter, cytochrome P450, drug‐drug interactions, napabucasin, phase 1 trial
Napabucasin (Figure 1) is an orally administered reactive oxygen species (ROS) generator that is bioactivated by the intracellular antioxidant nicotinamide adenine dinucleotide phosphate:quinone oxidoreductase 1 (NQO1).1 Cancer cells, including cancer stem cells, often express high levels of NQO1 compared with healthy cells.2 Napabucasin exerts its antitumor activity by increasing levels of ROS beyond a cytotoxic threshold, causing cancer cell death.1, 3 In vitro assays have shown that NQO1‐expressing cancer cells are more sensitive to napabucasin.1, 3 Several clinical trials have investigated napabucasin as a single agent or in combination with several anticancer treatments (data reported in congress abstracts4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 or articles16, 17, 18, 19). A phase 3 study of napabucasin in combination with 5‐fluorouracil, leucovorin, and irinotecan vs 5‐fluorouracil, leucovorin, and irinotecan alone in patients with previously treated metastatic colorectal cancer (NCT02753127) is ongoing.20
Figure 1.
The structure of napabucasin and its major metabolite, dihydro‐napabucasin (M1).
Preclinical studies in bile duct cannulated rats have shown that napabucasin is highly membrane permeable and rapidly absorbed, with relatively high bioavailability (75.5%) following a single oral radiolabeled dose. Metabolism of napabucasin is qualitatively similar between species based on in vitro metabolic studies in cryopreserved hepatocytes from rats, dogs, rabbits, and humans (Sumitomo Dainippon Pharma Oncology, Inc., data on file). The major human metabolite of napabucasin is dihydro‐napabucasin (M1), which has reduced activity compared with napabucasin (Figure 1), reflected by reduced binding to NQO1 with a similar reduction in catalytic efficiency in a cell‐free system. Further, in a human lung cancer cell line (A549 cells), treatment with the M1 metabolite failed to produce ROS (a >10‐fold reduction compared with napabucasin) and diminished reduction of cell viability compared with treatment with napabucasin (Sumitomo Dainippon Pharma Oncology, Inc., data on file). Excretion of radioactivity following administration of a single radiolabeled napabucasin dose is rapid and primarily via the renal and fecal routes in rats, and the fecal route in dogs, with >90% of the dose eliminated within 48 and 24 hours after dosing, respectively (Sumitomo Dainippon Pharma Oncology, Inc., data on file). Absorption, metabolism, excretion, and pharmacokinetics (PK) of a single oral dose of napabucasin in humans was investigated in a phase 1 study (NCT03525405; manuscript in progress).
Patients with cancer often require multiple medications to manage disease symptoms, cancer drug side effects, and possible comorbid conditions, and are therefore susceptible to drug‐drug interactions (DDIs). DDIs increase the complexity of therapeutic management and can induce the development of adverse events (AEs) and/or impact clinical efficacy.21, 22, 23, 24, 25 In vitro data from preclinical studies in human liver microsomes indicate that napabucasin is an inhibitor of cytochrome P450 (CYP) 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4 isozymes, with half maximal inhibitory concentration (IC50) values of 0.207, 1.80, 1.02, 5.44, 1.70, 2.35, 1.56 (CYP3A4 probed with testosterone), and 1.37 μmol/L (CYP3A4 probed with midazolam), respectively (Sumitomo Dainippon Pharma Oncology, Inc., data on file). Additionally, in vitro data in porcine (LLC‐PK1), human (HEK293), or Drosophila melanogaster (S2) transporter‐expressing cell lines show that napabucasin is an inhibitor of the breast cancer resistance protein (BCRP) transporter (in LLC‐PK1 cells), organic cation transporter 2 (in HEK293 cells), organic anion transporters (OAT) 1 and 3 (in S2 cells), and multidrug and toxin extrusion protein 1 (in HEK293 cells) with IC50 values of 1.24, 9.56, 1.74, 0.724, and 11.7 μmol/L, respectively (Sumitomo Dainippon Pharma Oncology, Inc., data on file). In vivo maximum observed plasma concentration (Cmax) of napabucasin in healthy male volunteers after a single oral dose of napabucasin 240 mg is 1.78 μM (geometric mean; manuscript in progress), which is in the range of the IC50 values observed in vitro. The primary objective of this open‐label, phase 1 study was to quantitatively evaluate the DDI potential of napabucasin and its major metabolite (M1) (Figure 1) with respect to 7 major CYP enzymes and the BCRP transporter in healthy volunteers. DDIs were assessed using a phenotyping cocktail approach for the probe drugs, which enabled assessment of napabucasin DDI via several different pathways in a single study. Napabucasin was dosed orally to steady state, which is clinically relevant and which facilitated the examination of metabolite effects.
Methods
Study Design
The protocol was approved by the institutional review board at the study site (Avail Clinical Research, Deland, Florida) and all healthy volunteers provided written informed consent before participation.
This phase 1, single‐center, open‐label, single‐sequence, 3‐period, PK DDI study (NCT03411122) evaluated the effect of napabucasin on 8 probe drugs (7 CYP probe substrates and 1 BCRP transporter substrate) in healthy volunteers. The probe drugs were caffeine (CYP1A2 substrate), bupropion (CYP2B6 substrate), repaglinide (CYP2C8 substrate), flurbiprofen (CYP2C9 substrate), omeprazole (CYP2C19 substrate), dextromethorphan (CYP2D6 substrate), midazolam (CYP3A substrate), and rosuvastatin (BCRP/OAT3 substrate) (Table 1).
Table 1.
DDI Probe Drugs (Related CYP or Transporter)
| Drug | Dose (mg) | Administration Route |
|---|---|---|
| Caffeine (CYP1A2)a | 100 | Oral |
| Bupropion (CYP2B6)a | 150 | Oral |
| Repaglinide (CYP2C8) | 0.25 | Oral |
| Flurbiprofen (CYP2C9)a | 50 | Oral |
| Omeprazole (CYP2C19)a | 20 | Oral |
| Dextromethorphan (CYP2D6)a | 30 | Oral |
| Midazolam (CYP3A)a | 2 | Oral/IV |
| Rosuvastatin (BCRP/OAT3) | 10 | Oral |
BCRP, breast cancer resistance protein; CYP, cytochrome P450; DDI, drug‐drug interaction; IV, intravenous; OAT3, organic anion transporter 3.
Part of the phenotyping cocktail (oral doses only).
Healthy Volunteers
Healthy male or female adult volunteers aged 18 to 45 years with a body mass index of 18 to 34 kg/m2 were eligible for inclusion. Additional eligibility requirements included normal (or abnormal and clinically insignificant according to the investigator) laboratory values at screening and no significant abnormalities at the baseline physical examination. Healthy volunteers also agreed to abstain from taking any dietary supplements, herbal products, or nonprescription drugs (except as authorized by the investigator and medical monitor) and any foods with a known DDI impact (grapefruit, grapefruit juice, Seville oranges, and grapefruit‐ or Seville orange–containing products). Healthy volunteers also abstained from tobacco‐ and nicotine‐containing products, caffeine‐ or chocolate‐containing products, and alcohol‐containing beverages for up to 2 months before study admission through follow‐up. Healthy volunteers were excluded if they had a history of illicit drug abuse or positive findings on urine drug screen; were positive for human immunodeficiency virus, hepatitis B, and/or hepatitis C at screening; were poor metabolizers (ie, 2 inactive allele variants) for CYP2C19, CYP2C9, and/or CYP2D6; or had a hypersensitivity or allergy to napabucasin, any of the probe drugs or their ingredients, or other clinically significant allergy.
This study was conducted in compliance with the Declaration of Helsinki; the ethical principles of Good Clinical Practice, according to International Conference on Harmonization, Harmonized Tripartite Guideline; and applicable national and local regulatory requirements.
Study Drug Administration
The selection of the probe drugs and doses used in the phenotyping cocktail was based on previous DDI studies conducted in healthy adult volunteers.26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37 Napabucasin 240 or 480 mg was administered orally twice daily (every 12 hours); healthy volunteers received the morning dose following an overnight fast, remained fasted for 4 hours after dosing, and fasted 1 to 2 hours before administration of the evening dose. Potential DDIs were assessed on the basis of the napabucasin 240‐mg dose only; DDIs were not assessed for the 480‐mg dose because healthy volunteers did not receive the 480‐mg twice‐daily regimen in conjunction with the probe drugs (due to a protocol amendment on October 12, 2017, that adjusted the dose to 240 mg).
Single doses of the phenotyping cocktail and then later repaglinide 0.25 mg and rosuvastatin 10 mg were administered orally with 240 mL of water following an overnight fast; healthy volunteers remained on an empty stomach for 4 hours after dosing. Intravenous (IV) midazolam 2 mg was administered over 1 minute and immediately followed by a 5‐mL normal saline IV bolus to flush the indwelling catheter. Water was permitted ad libitum for all doses except 1 hour before and after drug administration on fasting days; healthy volunteers remained at rest for 2 hours after dose administration (except in the event of AEs and for study procedures [eg, IV midazolam administration]).
Study Procedures
The study consisted of a screening period, 3 treatment periods, and a safety follow‐up visit occurring 7 to 10 days after the last dose of study drug (Figure 2). Healthy volunteers were screened within 28 days (day –28 to day –1) before admission to the clinical research unit. Baseline assessments were collected on day 0 or on day 1 before dosing.
Figure 2.
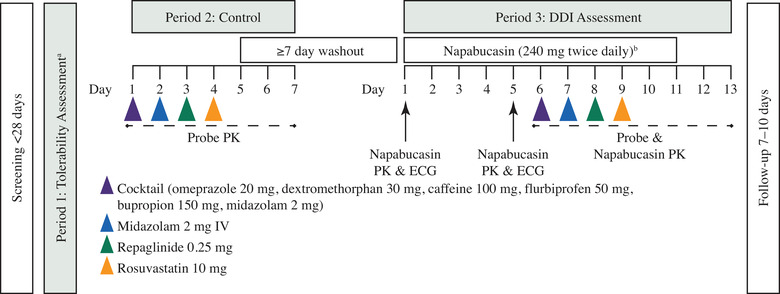
Study design. Dosing schedule of periods 1, 2, and 3. aHealthy volunteers dosed napabucasin in Protocol Amendment 1 received 480 mg twice daily for 2 days (4 doses) in period 1. Healthy volunteers dosed napabucasin in Protocol Amendment 2 received 240 mg twice daily for 2 days (4 doses) in period 1. bPotential DDIs were evaluated in the 17 healthy volunteers dosed with napabucasin 240 mg twice daily under Protocol Amendment 2. Healthy volunteers under Protocol Amendment 1 received a single dose of napabucasin 480 mg and were discontinued, so DDI could not be evaluated. Eleven healthy volunteers who received 480 mg in period 1 and one 480‐mg dose in period 3 before the Protocol Amendment continued receiving 240 mg in period 3 after the Protocol Amendment and were assessed for DDI. DDI, drug‐drug interaction; ECG, electrocardiogram; IV, intravenous; PK, pharmacokinetics.
Period 1: Tolerability Assessment
Napabucasin is associated with gastrointestinal (GI) AEs (eg, nausea, vomiting, diarrhea), and some patients who experience severe GI AEs require napabucasin dose hold, modification, or permanent discontinuation.17, 38 As the ability to assess drug tolerability in healthy volunteers is diminished by these typical napabucasin‐related GI events, only healthy volunteers who could tolerate napabucasin administered on days 1 and 2 of period 1 (no GI Common Terminology Criteria for Adverse Events [CTCAE] grade >1 AE) were permitted to continue on to period 2.
Period 2: Control
After a 7‐day washout, healthy volunteers received a single oral dose of a phenotyping cocktail (for DDI probe drugs, see Table 1) on day 1 of period 2. On day 2, IV midazolam 2 mg was administered, and on days 3 to 4, single oral doses of repaglinide 0.25 mg (day 3) and rosuvastatin 10 mg (day 4) were administered, which were followed by another 7‐day washout.
Period 3: DDI Assessment
On days 1 to 11 of period 3, napabucasin was administered. On days 6 to 9, healthy volunteers received single doses of the phenotyping cocktail (day 6), IV midazolam (day 7), repaglinide (day 8), and rosuvastatin (day 9). Following period 3, healthy volunteers participated in a safety follow‐up visit 7 to 10 days after administration of the last dose of study drug.
Safety Analysis
Safety analyses included the incidence of treatment‐emergent AEs (TEAEs), AEs by severity, and serious AEs (SAEs; periods 1‐3). Analyses also included changes from baseline in electrocardiogram parameters, clinical laboratory parameters, urinalysis, vital signs, and changes from predose physical exam findings to study discharge (periods 2 and 3).
Pharmacokinetic Sampling and Assays
In period 2, blood samples for the PK analysis of each of the probe drugs and their metabolites were obtained before dosing through 24 hours following administration of the phenotyping cocktail, IV midazolam, and repaglinide on days 1, 2, and 3, respectively, and through 72 hours following rosuvastatin administration on day 4.
Blood samples for the determination of napabucasin and M1 metabolite concentrations were collected before dosing, up to 10 hours following the morning doses on day 1 and day 5, and before the morning doses on days 2 to 4 in period 3. Blood samples for the PK analysis of each of the probe drugs were obtained before dosing through 24 hours following administration of the phenotyping cocktail, IV midazolam, and repaglinide on days 6, 7, and 8, respectively, and through 96 hours following rosuvastatin administration on day 9. PK samples collected on days 6 to 9 of period 3 were assayed for napabucasin, probe drugs, probe drug metabolites, and M1. M1 is the major circulating napabucasin metabolite; all other napabucasin metabolites are minor plasma components (≤7%) in healthy adult male human subjects.39 Thus, M1 was the metabolite measured in this study.
Napabucasin, M1, and probe drug plasma concentrations were quantified at Charles River Labs in Worcester, Massachusetts, using a validated liquid chromatography with tandem mass spectrometry bioanalytical method (ultra‐performance liquid chromatography with mass spectrometric detection). The lower limit of quantification was 5 ng/mL, and the upper limit of quantification was 500 ng/mL. Detailed descriptions of the PK assays are provided in the Supplemental Methods.
Pharmacokinetic Analysis
PK parameters for napabucasin, M1, and each of the probe drugs were calculated with Phoenix WinNonlin 6.3 (Certara USA, Inc., Princeton, New Jersey) using actual sampling time and noncompartmental analysis (data permitting). If actual times were missing, nominal times were used. PK parameters included Cmax, time to maximum concentration (tmax), terminal‐phase half‐life (t½), area under the plasma concentration‐time curve (AUC) from time 0 to last measurable plasma concentration (AUClast), AUC from time 0 extrapolated to infinity (AUCinf), percent AUC extrapolated beyond the last measurable concentration, systemic volume of distribution, and apparent systemic clearance.
Plasma drug concentrations for napabucasin, M1, and probe drugs were summarized using nominal PK sampling times. For the calculation of mean concentrations and generation of mean concentration vs time profiles, all values below the limit of quantification (BLQ) were set to 0.
For the PK analysis, a concentration that was BLQ was assigned a value of 0 if it occurred in a profile before the first measurable concentration. If a BLQ value occurred after a measurable concentration in a profile and was followed by a value above the lower limit of quantification, then the BLQ value was treated as missing data. If a BLQ value occurred at the end of the collection interval (after the last quantifiable concentration), it was treated as missing data. If 2 BLQ values occurred in succession after Cmax, the profile was deemed to have terminated at the first BLQ value, and any subsequent concentrations were omitted from the PK calculations by treating them as missing.
The PK results are reported as both the geometric mean with the associated coefficient of variation (CV%) and the arithmetic mean with the associated standard deviation (SD) or standard error of the mean (SEM) for each observed PK exposure measure with and without napabucasin. The DDI results are reported as geometric mean ratios with associated 90% confidence intervals (CIs). A linear mixed‐effects model was used to perform the DDI analysis comparing intravolunteer systemic exposures of each probe drug in the presence and absence of napabucasin. The analysis was performed on the natural log‐transformed AUClast, AUCinf, and Cmax of each probe drug. The model included treatment as a fixed effect and volunteer as a random effect.
Each model included calculation of the least squares mean (LSM), the difference between treatment LSM, and the standard error associated with the difference. The geometric mean ratio of AUClast, AUCinf, and Cmax, along with the 2‐sided 90%CI, was derived from the LSM difference between each probe drug in the presence and absence of napabucasin in the model. This difference and the associated CI were back‐transformed to provide a ratio of geometric LSM and associated 90%CI. The end points of the bioequivalence interval of 80% to 125% were considered the no‐effect boundaries. If the 90%CI for the measured changes in systemic exposures fell completely within the no‐effect boundaries, it was interpreted that no DDI was present. If any of the 90%CI fell outside of the 80% to 125% range of the established no‐effect boundaries, the interpretation of a potential DDI relied on clinical understanding of the therapeutic range with regard to safety and efficacy.
Results
Baseline Demographics and Healthy Volunteer Disposition
A total of 30 healthy volunteers received ≥1 dose of napabucasin 240 or 480 mg between July 6, 2017, and January 23, 2018 (Figure S1). Twenty‐three healthy volunteers in period 1 received napabucasin 480 mg twice daily before the protocol amendment; 22 volunteers received 4 doses over 2 days, and 1 volunteer received 2 doses on day 1 and discontinued due to AEs. Eighteen of the 23 volunteers received a single dose of 480‐mg napabucasin on period 3, day 1, before the study was paused.
A protocol amendment that included formal stopping rules was added, which required termination of dosing in any healthy volunteer experiencing an event that was CTCAE grade ≥2 toxicity and termination of the entire study if ≥2 volunteers experienced any event that was CTCAE grade 3 toxicity or any single volunteer experienced an event that was grade 4 severity. Additionally, per the amendment, the dosing level of napabucasin was lowered from 480 mg twice daily to 240 mg twice daily, as this was the starting dose used in 2 pivotal registration trials (NCT02753127; NCT02993731).
When the study resumed, 11 of the 23 originally enrolled healthy volunteers reconsented and continued the study at the napabucasin 240‐mg dose level in period 3. As a result, these 11 volunteers received napabucasin 480 mg twice daily for 2 days in period 1, 1 dose of napabucasin 480 mg in period 3 before the protocol amendment, and napabucasin 240 mg twice daily for 22 doses over 11 days in period 3 following the protocol amendment. Seven additional healthy volunteers were recruited following the protocol amendment, and received napabucasin 240 mg twice daily in period 1. Of these 7 volunteers, 1 received 1 dose and discontinued prematurely due to AEs; thus, 6 volunteers received napabucasin 240 mg twice daily in period 3.
Of the 30 total healthy volunteers enrolled, 17 were evaluated for DDI potential and constitute the DDI population (Table 2). Of those volunteers in the DDI population, the mean (SD) age was 29 (6.3) years, the majority were men (64.7%) and black or African American (76.5%), and the mean (SD) body mass index was 27.0 (3.29) kg/m2. All patients in the DDI population had a normal CYP2C9 genotype, and the majority had normal CYP2D6 and CYP2C19 genotypes (82.4% and 52.9%, respectively).
Table 2.
Healthy Volunteer Demographics
| Characteristic | Safety Population (N = 30) | DDI Population (n = 17)a |
|---|---|---|
| Age, y, mean (SD) | 29 (5.9) | 29 (6.3) |
| Sex, male, n (%) | 20 (66.7) | 11 (64.7) |
| Race, n (%) | ||
| Black or African American | 16 (53.3) | 13 (76.5) |
| White | 14 (46.7) | 4 (23.5) |
| BMI, kg/m2, mean (SD) | 26.9 (3.9) | 27 (3.3) |
| CYP2D6 genotype, n (%) | ||
| Normal | 27 (90) | 14 (82.4) |
| Intermediate | 2 (6.7) | 2 (11.8) |
| Ultra‐rapid | 1 (3.3) | 1 (5.9) |
| CYP2C9 genotype, n (%) | ||
| Normal | 28 (93.3) | 17 (100) |
| Intermediate | 2 (6.7) | 0 |
| CYP2C19 genotype, n (%) | ||
| Normal | 16 (53.3) | 9 (52.9) |
| Intermediate | 7 (23.3) | 6 (35.3) |
| Extensive | 1 (3.3) | 1 (5.9) |
| Ultra‐rapid | 6 (20) | 1 (5.9) |
BMI, body mass index; CYP, cytochrome P450; DDI, drug‐drug interaction; PK, pharmacokinetics; SD, standard deviation.
All healthy volunteers with both phenotyping cocktail and napabucasin concentrations.
Drug‐Drug Interactions
The tested dose level of napabucasin 240 mg twice daily provided sufficient plasma exposure of napabucasin (Table 3) and its primary metabolite napabucasin M1 (Table 4) to assess potential DDI effects. Steady‐state exposure due to administration of napabucasin 240 mg twice daily had no impact on exposure to omeprazole, 5‐hydroxyomeprazole, paraxanthine, flurbiprofen, dextrorphan, oral midazolam, or 1‐hydroxymidazolam (both oral and IV) (Figure 3A‐C, Figure 4).
Table 3.
Noncompartmental Plasma Pharmacokinetic Parameters of Probes
| Period 2 (Probe Alone) | Period 3 (Probe With Napabucasin) | |||
|---|---|---|---|---|
| Arithmetic Mean (SD) | Geometric Mean (Geometric %CV) | Arithmetic Mean (SD) | Geometric Mean (Geometric %CV) | |
| Bupropion, oral, CYP2B6 | ||||
| AUClast, ng • h/mL | 639 (152) | 619 (28.0) | 524 (238) | 480 (44.0) |
| AUCext, % | 11.4 (6.38) | 9.81 (65.9) | 11.4 (4.86) | 10.5 (42.7) |
| AUCinf, ng • h/mL | 727 (186) | 701 (30.2) | 566 (238) | 526 (39.7) |
| Cmax, ng/mL | 134 (38.1) | 130 (27.6) | 110 (67.7) | 94.3 (61.4) |
| Clast, ng/mL | 6.02 (2.55) | 5.45 (51.2) | 5.14 (2.62) | 4.69 (43.4) |
| CL/F, L/H | 224 (79.9) | 214 (30.2) | 303 (102) | 285 (39.7) |
| t½, h | 9.21 (3.24) | 8.74 (34.0) | 8.77 (1.90) | 8.59 (20.7) |
| Omeprazole, oral, CYP2C19 | ||||
| AUClast, ng • h/Ml | 1010 (468) | 894 (56.0) | 999 (568) | 828 (76.6) |
| AUCext, % | 3.16 (5.08) | 1.77 (122) | 1.85 (0.853) | 1.65 (58.4) |
| AUCinf, ng • h/mL | 1260 (409) | 1190 (37.8) | 1220 (495) | 1130 (44.1) |
| Cmax, ng/mL | 390 (194) | 338 (66.4) | 439 (236) | 379 (64.1) |
| Clast, ng/mL | 17.3 (17.1) | 13.2 (78.6) | 12.9 (7.17) | 11.3 (57.9) |
| CL/F, L/H | 17.9 (7.23) | 16.8 (37.8) | 19.3 (8.38) | 17.7 (44.1) |
| t½, h | 1.29 (0.448) | 1.24 (30.3) | 1.10 (0.226) | 1.08 (20.5) |
| Dextromethorphan, oral, CYP2D6 | ||||
| AUClast, ng • h/mL | 78.1 (56.6) | 60.4 (89.4) | 70.0 (64.5) | 45.4 (129) |
| AUCext, % | 20.6 (7.33) | 19.2 (43.2) | 17.3 (7.94) | 15.7 (47.7) |
| AUCinf, ng • h/mL | 77.1 (46.5) | 64.7 (76) | 89.9 (102) | 48.8 (170) |
| Cmax, ng/mL | 5.85 (3.93) | 4.66 (82.9) | 5.16 (4.41) | 3.57 (115) |
| Clast, ng/mL | 1.86 (1.84) | 1.25 (113) | 1.49 (1.70) | 0.772 (186) |
| CL/F, L/H | 558 (360) | 464 (70.6) | 1000 (840) | 615 (170) |
| t½, h | 9.16 (2.29) | 8.90 (26.1) | 8.29 (2.35) | 8.01 (27.2) |
| Midazolam, oral, CYP3A | ||||
| AUClast, ng • h/mL | 20.1 (7.62) | 19.0 (35.5) | 21.3 (7.06) | 20.2 (34.3) |
| AUCext, % | 14.3 (6.12) | 13.0 (47.8) | 13.3 (4.88) | 12.5 (37.3) |
| AUCinf, ng • h/mL | 23 (8.93) | 21.6 (36.9) | 24.8 (9.00) | 23.4 (36.7) |
| Cmax, ng/mL | 8.01 (4.09) | 7.28 (44.4) | 7.77 (2.61) | 7.37 (34.6) |
| Clast, ng/mL | 0.65 (0.25) | 0.612 (35.5) | 0.658 (0.317) | 0.607 (40.3) |
| CL/F, L/H | 98 (32.7) | 92.6 (36.9) | 90.8 (32.4) | 85.6 (36.7) |
| t½, h | 3.53 (1.46) | 3.25 (43.7) | 3.5 (1.09) | 3.33 (33.5) |
| Midazolam, IV, CYP3A | ||||
| AUClast, ng • h/mL | 74.7 (19.1) | 72.7 (23.3) | 94.0 (72.6) | 80.9 (53.6) |
| AUCext, % | 11.4 (5.43) | 10.2 (53.1) | 9.93 (6.41) | 7.86 (88.3) |
| AUCinf, ng • h/mL | 81.0 (16.1) | 79.6 (18.9) | 103 (72.2) | 90.0 (50.5) |
| Cmax, ng/mL | 42.4 (41.3) | 35.2 (54.2) | 75.9 (152) | 43.7 (91.5) |
| Clast, ng/mL | 1.49 (0.623) | 1.34 (53.7) | 1.30 (0.885) | 1.03 (82.8) |
| CL/F, L/H | 25.5 (4.61) | 25.1 (18.9) | 24.3 (9.98) | 22.2 (50.5) |
| t½, h | 4.57 (1.63) | 4.31 (36.3) | 5.30 (2.96) | 4.77 (46.7) |
| Vz, L | 162 (44.8) | 156 (28.9) | 189 (139) | 153 (80.0) |
| Caffeine (oral), CYP1A2 | ||||
| AUClast, ng • h/mL | 17 300 (10 700) | 15 000 (57.4) | 21 100 (11 400) | 18 400 (59.9) |
| AUCext, % | 8.65 (7.77) | 6.13 (105) | 9.70 (8.38) | 7.45 (83.4) |
| AUCinf, ng • h/mL | 20 100 (15 700) | 16 800 (62.3) | 24 800 (17 900) | 20 500 (69.9) |
| Cmax, ng/mL | 2 270 (905) | 2 110 (39.6) | 2 110 (756) | 2 000 (34.6) |
| Clast, ng/mL | 262 (296) | 164 (127) | 262 (306) | 172 (112) |
| CL/F, L/H | 6.82 (3.45) | 5.97 (62.3) | 5.91 (4.30) | 4.88 (69.9) |
| t½, h | 5.19 (2.59) | 4.81 (37.9) | 6.67 (3.06) | 6.15 (42.2) |
| Flurbiprofen, oral, CYP2C9 | ||||
| AUClast, ng • h/mL | 28 700 (8 310) | 27 500 (31.1) | 31 800 (6 890) | 31 100 (22.2) |
| AUCext, % | 3.95 (2.39) | 3.46 (53.6) | 4.15 (2.36) | 3.70 (49.0) |
| AUCinf, ng • h/mL | 30 900 (8 360) | 29 800 (28.0) | 33 300 (7 630) | 32 500 (23.5) |
| Cmax, ng/mL | 5 500 (1 840) | 5 210 (35.3) | 5 770 (1 300) | 5 630 (23.4) |
| Clast, ng/mL | 223 (261) | 160 (84.9) | 177 (102) | 155 (55.2) |
| CL/F, L/H | 1.74 (0.478) | 1.68 (28.0) | 1.58 (0.367) | 1.54 (23.5) |
| t½, h | 5.16 (0.822) | 5.11 (15.1) | 5.43 (0.761) | 5.38 (13.0) |
| Repaglinide, oral, CYP2C8 | ||||
| AUClast, ng • h/mL | 4.15 (2.01) | 3.72 (51.1) | 5.27 (2.54) | 4.75 (49.9) |
| AUCext, % | 8.14 (3.67) | 7.36 (50.0) | 7.51 (4.66) | 6.35 (65.1) |
| AUCinf, ng • h/mL | 4.45 (1.98) | 4.08 (45.1) | 6.01 (2.57) | 5.53 (45.4) |
| Cmax, ng/mL | 3.38 (1.58) | 3.09 (44.5) | 4.43 (2.58) | 3.85 (58.0) |
| Clast, ng/mL | 0.282 (0.0539) | 0.277 (18.3) | 0.332 (0.124) | 0.315 (33.5) |
| CL/F, L/H | 66.7 (28.4) | 61.3 (45.1) | 49.6 (24.1) | 45.2 (45.4) |
| t½, h | 0.770 (0.183) | 0.751 (23.5) | 0.795 (0.246) | 0.765 (28.2) |
| Rosuvastatin, oral, BCRP/OAT3‐mediated interactions | ||||
| AUClast, ng • h/mL | 38.4 (25.0) | 29.1 (107) | 49.3 (35.9) | 35.2 (136) |
| AUCext, % | 7.98 (2.71) | 7.61 (35.1) | 7.03 (5.63) | 5.41 (85.5) |
| AUCinf, ng • h/mL | 27.8 (19.4) | 21.1 (117) | 54.1 (36.1) | 40.3 (125) |
| Cmax, ng/mL | 3.91 (2.63) | 3.03 (93.8) | 4.49 (3.08) | 3.29 (122) |
| Clast, ng/mL | 0.202 (0.113) | 0.181 (49.3) | 0.230 (0.0782) | 0.216 (39.9) |
| CL/F, L/h | 693 (738) | 475 (117) | 518 (1120) | 248 (125) |
| t½, h | 9.29 (7.36) | 7.11 (103) | 7.66 (3.53) | 6.98 (46.8) |
%CV, percent coefficient of variation; AUC, area under the curve; AUCinf, AUC from time 0 extrapolated to infinity; AUCext, percent AUC extrapolated beyond the last measurable concentration; AUClast, AUC from time 0 to last measurable concentration; BCRP, breast cancer resistance protein; Clast, last observed quantifiable concentration; CL/F, apparent systemic clearance; Cmax, maximum concentration; CYP, cytochrome P450; DDI, drug‐drug interaction; IV, intravenous; LS, least squares; OAT3, organic anion transporter 3; SD, standard deviation; t½, half‐life; Vz, systemic volume of distribution following IV administration.
Table 4.
Plasma Noncompartmental PK Parameters of Probe Metabolites in Periods 2 and 3
| Period 2 (Probe Alone) | Period 3 (Probe With Napabucasin) | |||
|---|---|---|---|---|
| Arithmetic Mean (SD) | Geometric Mean (Geometric %CV) | Arithmetic Mean (SD) | Geometric Mean (Geometric %CV) | |
| 6‐hydroxybupropion, oral, CYP2B6 | ||||
| AUClast, ng • h/mL | 6 310 (3 110) | 5 630 (53.3) | 4 020 (2 310) | 3 430 (65.0) |
| AUCext, % | 33.2 (3.39) | 33.1 (10.5) | 36.9 (0.373) | 36.9 (1.01) |
| AUCinf, ng • h/mL | 6 210 (3 730) | 5 570 (60.1) | 3 340 (2 030) | 2 950 (66.8) |
| Cmax, ng/mL | 358 (165) | 323 (50.6) | 232 (125) | 200 (63.4) |
| Clast, ng/mL | 197 (112) | 169 (64.1) | 128 (76.4) | 108 (69.1) |
| t½, h | 13.9 (1.13) | 13.9 (8.32) | 16.4 (1.28) | 16.3 (7.66) |
| 5‐hydroxyomeprazole, oral, CYP2C19 | ||||
| AUClast, ng • h/mL | 388 (112) | 373 (29.6) | 444 (110) | 433 (23.5) |
| AUCext, % | 2.25 (0.974) | 2.07 (44.1) | 2.44 (1.33) | 2.20 (46.0) |
| AUCinf, ng • h/mL | 431 (110) | 420 (23.1) | 457 (114) | 445 (23.9) |
| Cmax, ng/mL | 124 (49.7) | 116 (41.3) | 153 (38.8) | 148 (26.5) |
| Clast, ng/mL | 6.88 (4.58) | 5.79 (63.0) | 5.81 (2.53) | 5.34 (43.3) |
| t½, h | 1.36 (0.242) | 1.34 (17.8) | 1.35 (0.190) | 1.33 (14.9) |
| Dextrorphan, oral, CYP2D6 | ||||
| AUClast, ng • h/mL | 1 400 (357) | 1 350 (29.7) | 1 490 (346) | 1 440 (26.6) |
| AUCext, % | 11.9 (7.23) | 10.3 (56.4) | 8.34 (6.42) | 6.36 (89.3) |
| AUCinf, ng • h/mL | 1 590 (382) | 1 540 (28.2) | 1 610 (331) | 1 580 (22.2) |
| Cmax, ng/mL | 137 (40.8) | 130 (36.5) | 162 (58.2) | 151 (43.0) |
| Clast, ng/mL | 17.6 (6.48) | 16.4 (42.2) | 13.5 (5.97) | 12.3 (47.1) |
| t½, h | 6.97 (2.06) | 6.73 (27.0) | 5.88 (1.89) | 5.63 (30.3) |
| 1‐hydroxymidazolam, oral, CYP3A | ||||
| AUClast, ng • h/mL | 10.9 (3.86) | 10.2 (44.8) | 10.7 (3.67) | 10.1 (34.8) |
| AUCext, % | 5.95 (3.31) | 5.12 (62.5) | 6.37 (2.77) | 5.78 (49.8) |
| AUCinf, ng • h/mL | 11.6 (3.97) | 10.8 (43.1) | 11.6 (3.95) | 11.0 (35.3) |
| Cmax, ng/mL | 4.55 (2.12) | 4.08 (52.9) | 4.45 (2.15) | 4.06 (44.5) |
| Clast, ng/mL | 0.164 (0.0544) | 0.157 (29.7) | 0.187 (0.0636) | 0.177 (34.4) |
| t½, h | 2.62 (1.09) | 2.44 (39.8) | 2.59 (0.812) | 2.47 (33.9) |
| 1‐hydroxymidazolam, IV, CYP3A | ||||
| AUClast, ng • h/mL | 11.4 (3.04) | 11.0 (31.2) | 10.7 (3.32) | 10.2 (33.6) |
| AUCext, % | 15.1 (6.30) | 14.0 (41.4) | 14.8 (7.21) | 13.0 (62.4) |
| AUCinf, ng • h/mL | 13.5 (3.41) | 13.0 (29.9) | 12.1 (3.18) | 11.7 (28.8) |
| Cmax, ng/mL | 2.94 (1.17) | 2.72 (43.8) | 3.05 (1.72) | 2.75 (46.0) |
| Clast, ng/mL | 0.286 (0.139) | 0.253 (57.3) | 0.287 (0.114) | 0.265 (44.9) |
| t½, h | 5.67 (2.88) | 5.06 (52.3) | 4.41 (2.17) | 4.02 (44.4) |
| Paraxanthine, oral, CYP1A2 | ||||
| AUClast, ng • h/mL | 8 930 (2 500) | 8 560 (31.9) | 9 550 (2 130) | 9 240 (29.8) |
| AUCext, % | 12.8 (8.79) | 10.3 (81.7) | 18.9 (8.15) | 17.6 (47.2) |
| AUCinf, ng • h/mL | 9 680 (3 190) | 9 230 (33.6) | 7 910 (2 430) | 7 610 (33.5) |
| Cmax, ng/mL | 610 (97.2) | 603 (15.7) | 572 (89.6) | 566 (14.9) |
| Clast, ng/mL | 204 (148) | 157 (90.7) | 271 (111) | 247 (49.7) |
| t½, h | 7.03 (2.47) | 6.68 (34.7) | 7.40 (2.13) | 7.19 (27.9) |
%CV, percent coefficient of variation; AUC, area under the curve; AUCinf, AUC from time 0 to infinity; AUCext, percent AUC extrapolated beyond the last measurable concentration; AUClast, AUC from time 0 to last measurable concentration; Clast, last observed quantifiable concentration; CL/F, apparent systemic clearance; Cmax, maximum concentration; CYP, cytochrome P450; DDI, drug‐drug interaction; IV, intravenous; LS, least squares; OAT3, organic anion transporter 3; SD, standard deviation; t½, half‐life.
Figure 3.
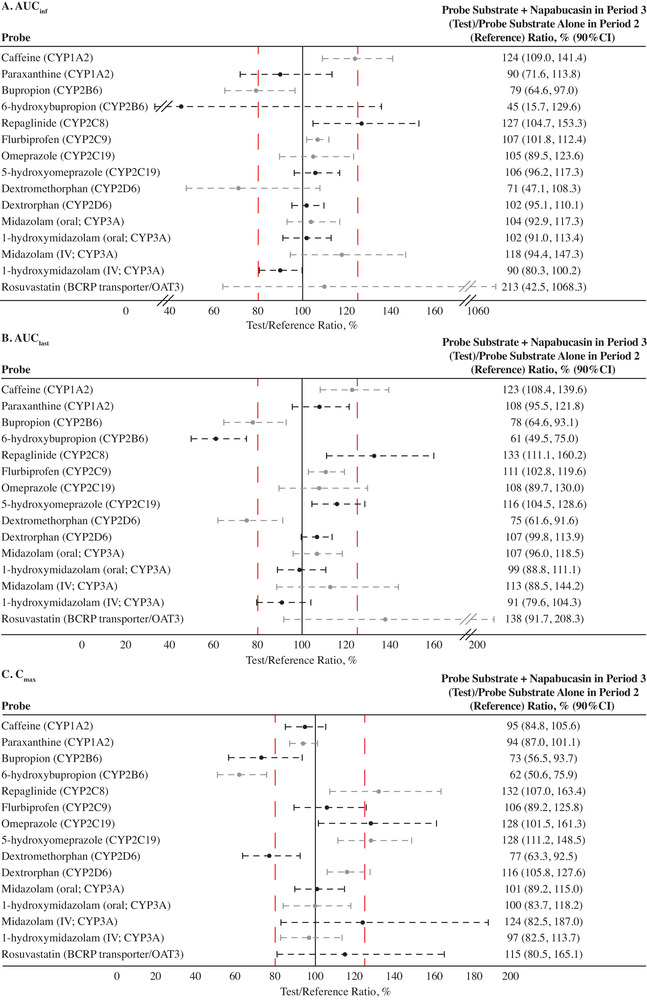
Drug‐drug interactions (DDIs) of napabucasin 240 mg and probe substrates and probe metabolites. Changes in (A) AUCinf, (B) AUClast, and (C) Cmax. The red dashed lines denote the no‐effect boundaries (80%‐125%). AUC, area under the concentration curve; AUCinf, AUC from time 0 extrapolated to infinity; AUClast, AUC from time 0 to time of last measurable plasma concentration; BCRP, breast cancer resistance protein; CI, confidence interval; Cmax, maximum observed plasma concentration; CYP, cytochrome P450; IV, intravenous; OAT3, organic anion transporter 3.
Figure 4.
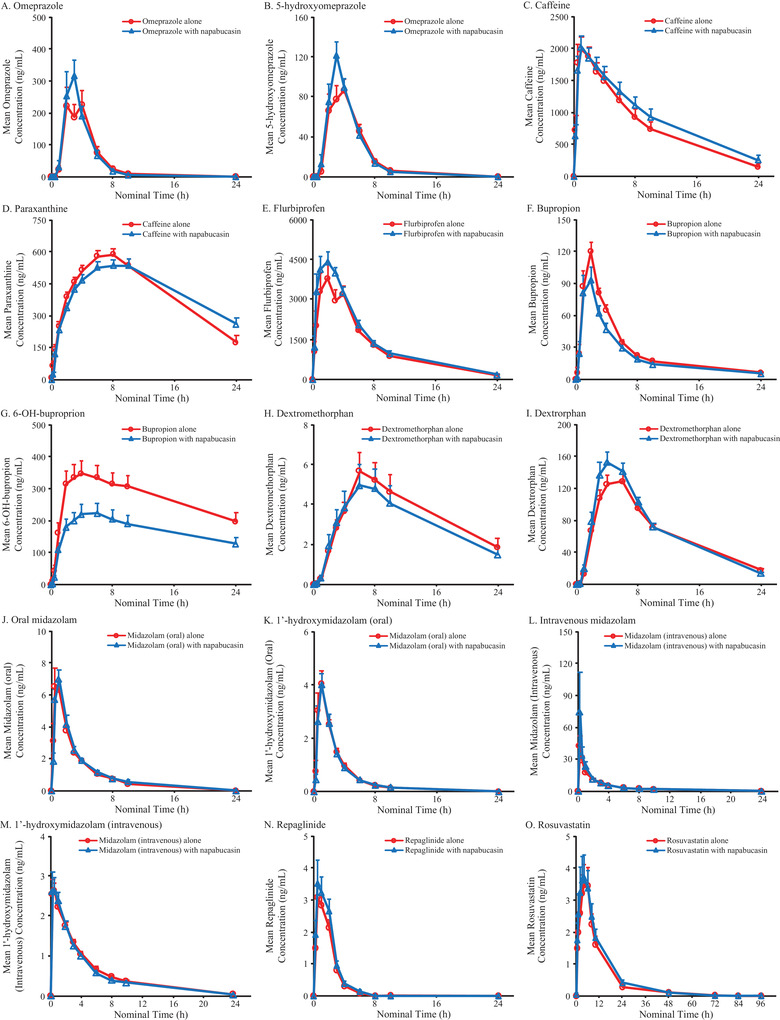
Mean plasma concentrations of probe substrates and probe metabolites over time when administered with (blue line, triangles) or without napabucasin (red line, circles). Mean concentrations of (A) omeprazole and (B) metabolite 5‐hydroxyomeprazole, (C) caffeine and (D) metabolite paraxanthine, (E) flurbiprofen, (F) bupropion and (G) metabolite 6‐OH‐buproprion, (H) dextromethorphan and (I) metabolite dextrorphan, (J) oral midazolam and (K) metabolite 1′‐hydroxymidazolam (oral), (L) intravenous midazolam and (M) metabolite 1′‐hydroxymidazolam (intravenous), (N) repaglinide, and (O) rosuvastatin are shown. Data are plotted as mean ± SEM.
Coadministration of napabucasin increased the AUCinf (geometric mean ratio as percentage [90%CI]) of caffeine (124% [109.0%‐141.4%]), IV midazolam (118% [94.4%‐147.3%]), repaglinide (127% [104.7%‐153.3%]), and rosuvastatin (213% [42.5%‐1 068.3%]) and decreased the AUCinf of dextromethorphan (71% [47.1%‐108.3%]), bupropion (79% [64.6%‐97.0%]), and 6‐hydroxybupropion (45% [15.7%‐129.6%]) (Figure 3A). Similar results were found for AUClast (Figure 3B; and presented as arithmetic means [SEM] in Figure 4).
Coadministration of napabucasin increased the Cmax (geometric mean ratio as percentage [90%CI]) of caffeine (95% [84.8%‐105.6%]), IV midazolam (124% [82.5%‐187.0%]), repaglinide (132% [107.0%‐163.4%]), and rosuvastatin (115% [80.5%‐165.1%]) and decreased the Cmax of dextromethorphan (77% [63.3%‐92.5%]), bupropion (73% [56.5%‐93.7%]), and hydroxybupropion (62% [50.6%‐75.9%]) (Figure 3C; and presented as arithmetic means [SEM] in Figure 4).
Generally, napabucasin did not induce drug clearance to a clinically meaningful degree for all measured components, with the possible exception of hydroxybupropion, for which post‐napabucasin AUC exposures were 45% of control (representing a 55% reduction). Per the US Food and Drug Administration, reductions between 50% and 80% are considered moderate reduction effects.40 This may be a result of decreased formation of hydroxybupropion from bupropion, rather than an induction of elimination.
Pharmacokinetics—Period 3
Among healthy volunteers who received a single dose of napabucasin 240 mg, the geometric mean Cmax (CV%) was 375 (26.6) ng/mL, and the geometric mean AUClast (CV%) was 1660 (28.8) ng • h/mL. PK parameters following repeated napabucasin 240 mg twice‐daily administration were comparable across all days sampled (days 5‐9) (Table S1), with geometric mean Cmax ranging from 287 to 652 ng/mL, geometric mean AUClast ranging from 1570 to 4160 ng • h/mL, and median tmax ranging from 2.98 to 5.00 hours. Geometric mean t½ ranged from 1.64 to 2.66 hours (Table S1).
Similar results were observed for key napabucasin M1 PK parameters (Table S1), with geometric mean Cmax ranging from 226 to 544 ng/mL, geometric mean AUClast ranging from 1 290 to 3 690 ng • h/mL, and the median tmax value ranging from 2.46 to 6.00 hours. Geometric mean t½ ranged from 1.54 to 2.68 hours (Table S1).
Safety
TEAEs were experienced by most healthy volunteers while receiving repeated napabucasin doses in period 1 (240‐mg or 480‐mg doses: 70.0% [21/30 volunteers]) and period 3 (240‐mg dose: 70.6% [12/17 volunteers]), and most were grade 1 (experienced by 63.3% [19/30] of volunteers in period 1 and 37.5% [9/24] in period 3) or grade 2 in severity (experienced by 6.7% [2/30] of volunteers in period 1 and 29.2% [7/24] in period 3) (Table 5). Two volunteers experienced grade 2 AEs after the protocol amendment; however, treatment was not discontinued because the events were originally reported as grade 1. Study monitoring queried the grade of these 2 AEs because the source data described the AEs as “moderate.” Based on these queries, the site changed the grade to grade 2. This occurred after the volunteers had already completed the study. All other grade 2 events (n = 7 patients) occurred before the amendment and therefore before the requirement to stop treatment. Only 3 of 26 healthy volunteers had a TEAE during period 2, when napabucasin was not administered. Overall, 19 volunteers in period 1 and 9 in period 3 experienced mild (grade 1) TEAEs. One grade 3 TEAE (severe; low neutrophil count) was observed during period 3 in a healthy volunteer who was reenrolled after the study was paused. The volunteer's neutrophil count was toward the low range of normal (1.4 × 109 cells/L) at rescreening, when the study resumed, and decreased to 0.9 × 109 cells/L at follow‐up after completion of period 3.
Table 5.
Summary of TEAEs Regardless of Relationship to Study Drug in Periods 1 and 3
| Period 1 | Period 3 | |||||
|---|---|---|---|---|---|---|
| Summary of TEAEs, Healthy Volunteers, n (%) | All (N = 30) | Napabucasin 240 mg Twice Daily (n = 7) | Napabucasin 480 mg Twice Daily (n = 23) | All (N = 24)a | Napabucasin 240 mg Twice Daily (n = 17) | Napabucasin 480 mg (n = 18)b |
| ≥1 TEAE | 21 (70.0) | 5 (71.4) | 16 (69.6) | 17 (70.8) | 12 (70.6) | 11 (61.1) |
| Severity | ||||||
| Grade 1 | 19 (63.3) | 4 (57.1) | 15 (65.2) | 9 (37.5) | 9 (52.9) | 4 (22.2) |
| Grade 2 | 2 (6.7) | 1 (14.3) | 1 (4.3) | 7 (29.2) | 2 (11.8) | 7 (38.9) |
| Grade 3 | 0 | 0 | 0 | 1 (4.2)c | 1 (5.9)c | 0 |
| Healthy volunteers with TEAEs, n (%) | ||||||
| Diarrhea | 14 (46.7)d | 4 (57.1)e | 10 (43.5)f | 15 (62.5) | 9 (52.9)g | 10 (55.6)h |
| Chromaturia | 9 (30.0) | 1 (14.3) | 8 (34.8) | 1 (4.2) | 1 (5.9) | 0 |
| Abdominal pain | 8 (26.7) | 0 | 8 (34.8) | 6 (25.0) | 0 | 6 (33.3) |
| Vomiting | 4 (13.3) | 1 (14.3) | 3 (13) | 2 (8.3) | 2 (11.8) | 1 (5.6) |
| Nausea | 2 (6.7) | 0 | 2 (8.7) | 3 (12.5) | 3 (17.6) | 0 |
| Abdominal pain, upper | 1 (3.3) | 1 (14.3) | 0 | 0 | 0 | 0 |
| Diarrhea, hemorrhagic | 0 | 0 | 0 | 1 (4.2) | 1 (5.9) | 0 |
| Headache | 0 | 0 | 0 | 3 (12.5) | 2 (11.8) | 1 (5.6) |
| Pollakiuria | 0 | 0 | 0 | 1 (4.2) | 1 (5.9) | 0 |
TEAE, treatment emergent adverse event.
All healthy volunteers enrolled in period 3 include the 7 enrolled after the protocol amendment who always received the 240‐mg dose and the 18 enrolled before the protocol amendment who received one 480‐mg dose in period 3.
Eighteen healthy volunteers who were administered napabucasin in the protocol amendment received a single 480‐mg dose on day 1, period 3, and resumed dosing in period 3 at napabucasin 240 mg twice daily. Exposure of healthy volunteers to napabucasin 480 mg in period 3 was for one dose only.
Low neutrophil count (severe) was observed in a healthy volunteer who was reenrolled after the study was paused.
Thirteen grade 1 and 1 grade 2.
All grade 1.
Nine grade 1 and 1 grade 2.
Eight grade 1 and 1 grade 2.
Three grade 1 and 7 grade 2.
The most frequent TEAEs reported in >1 healthy volunteer who received napabucasin 240 mg twice daily were diarrhea (57.1% [4/7]) in period 1 and diarrhea (52.9% [9/17]), nausea (17.6% [3/17]), headache (11.8% [2/17]), and vomiting (11.8% [2/17]) in period 3 (Table 5). All events resolved, except for the event of low neutrophil count (severe) in 1 volunteer for which the outcome was unknown (ie, no additional hematologic laboratory values were obtained for this volunteer). Two healthy volunteers experienced TEAEs that led to study withdrawal: 1 occurred during the tolerability assessment period (period 1) in a volunteer who had received 2 doses of napabucasin 480 mg and experienced nausea and diarrhea (both grade 2; both resolved 13 days after study drug discontinuation); the other occurred in a volunteer who received 1 dose of napabucasin 240 mg in period 1 and experienced vomiting (grade 2; resolved 15 minutes after it began) and stomach pain (grade 2; resolved 2 hours after it began). No deaths or SAEs were reported, and no clinically meaningful changes from baseline were observed in vital signs or electrocardiogram results.
Discussion
DDIs are common in patients undergoing cancer treatment.21 Risk factors for potential DDIs include the number and type of medications used and age‐related changes in metabolism.41, 42, 43, 44 Most drugs, including anticancer drugs, are primarily metabolized by CYPs, and the majority of DDIs are due to inhibition or induction of different CYPs.45
In vitro, napabucasin is an inhibitor of the CYP2C9, CYP2C19, CYP3A, CYP1A2, CYP2D6, CYP2C8, and CYP2B6 isozymes, as well as the BCRP/OAT3 (Sumitomo Dainippon Pharma Oncology, Inc., data on file). This was the first study to quantitatively assess the in vivo DDI potential of napabucasin with respect to these major human drug CYP enzymes and the BCRP/OAT3. This study employed a phenotyping cocktail, which permitted the simultaneous but independent evaluation of multiple probe drugs in the same healthy volunteer; this design reduced the study duration, number of volunteers needed, and intervolunteer variability.46, 47, 48
The results from this study suggest minimal in vivo DDI potential of napabucasin or its metabolite with respect to the 7 major human CYP enzymes and the BCRP/OAT3. DDI results were interpreted as clinically meaningful if the magnitude of the exposure change was >2‐fold, a common cutoff used to define a clinically significant effect.40 The exposure of bupropion (substrate of CYP2B6) and its metabolite hydroxybupriopion decreased when coadministered with napabucasin; the magnitude of decrease was <2‐fold for bupropion and slightly >2‐fold for hydroxybupriopion. The exposure of rosuvastatin (BCRP/OAT3) increased following coadministration of napabucasin, but these changes are not expected to be clinically meaningful. Collectively, except for the sensitive substrate of CYP2B6 (bupropion), whereby the metabolite hydroxybupriopion exposure changed by 55%, the changes observed with respect to the other 6 major human CYP enzymes and the BCRP/OAT3 are not expected to be clinically meaningful because their DDI did not result in a change that exceeded a factor of 2.
As patients with cancer often receive combination treatment, it is important to diminish DDI effects and maintain the optimal exposure of each cancer agent. In this study, the PK of napabucasin and M1 were comparable across all days sampled in period 3, when both napabucasin 240 mg twice daily and the probe drugs were administered. A phase 1 study of 6 Japanese patients with advanced/recurrent gastric cancer (JapicCTI‐142420) found that the PK and safety profiles of napabucasin were not affected by paclitaxel, a CYP2C8 and CYP3A4 substrate.18, 49, 50 A phase 1b study of napabucasin combined with gemcitabine and the CYP2C8 and CYP3A4 substrate nab‐paclitaxel in patients with metastatic pancreatic adenocarcinoma (NCT02231723; N = 59) also showed no notable PK interactions or dose‐limiting toxicities.8, 49, 50
There were no SAEs, deaths, or fatal AEs reported in this study. The majority of reported TEAEs involved the GI system and were mild to moderate in severity, which is similar to the safety profile reported in previous studies of napabucasin.8, 18, 51 These results suggest that napabucasin is generally tolerable in healthy volunteers when administered at a dose of 240 mg twice daily.
Limitations
Bupropion (CYP2B6 probe) inhibits CYP2D6 activity (as measured by dextromethorphan in this study) and therefore could confound the DDI potential of napabucasin. However, previous phenotyping cocktail studies showed a low risk for clinically relevant DDI following single‐dose administration.30 Furthermore, we observed a decrease rather than an increase in exposure to dextromethorphan and bupropion.
Conclusions
Generally, per the data from this study in healthy volunteers, napabucasin shows possible moderate inhibition of CYP2B6 vis‐à‐vis metabolite exposure, but it is not expected to induce or inhibit CYP1A2, 2C8, 2C9, 2C19, 2D6, 3A, or the BCRP/OAT3 to a clinically meaningful degree. Coadministration of napabucasin with CYP and transporter substrates was tolerable and associated with a low incidence of grade 3 TEAEs.
Conflicts of Interest
X.D., M.D.K., and M.H. are employees of Sumitomo Dainippon Pharma Oncology, Inc. C.F.M. and S.J.B. were paid consultants of Boston Biomedical, Inc (now Sumitomo Dainippon Pharma Oncology, Inc.). M.L.H. and M.T.G. previously served as paid consultants to Boston Biomedical, Inc (currently Sumitomo Dainippon Pharma Oncology, Inc.).
Funding
This study was funded by Sumitomo Dainippon Pharma Oncology, Inc.
Supporting information
Supplementary information
Acknowledgments
The authors acknowledge the contribution of Keri L. Hamilton, PhD, and Zachary F. Walls, PhD, of Nuventra, Inc., for assistance in writing the clinical study report. Editorial and medical writing support for this manuscript were provided by Rob Steger, PhD, and Christina Khodr, PhD, of Ashfield MedComms, an Ashfield Health Company, and were funded by Sumitomo Dainippon Pharma Oncology, Inc.
References
- 1.Chang A‐Y, Hsu E, Patel J, et al. Evaluation of tumor cell‐tumor microenvironment component interactions as potential predictors of patient response to napabucasin. Mol Cancer Res. 2019;17(7):1429‐1434. [DOI] [PubMed] [Google Scholar]
- 2.Madajewski B, Boatman MA, Chakrabarti G, Boothman DA, Bey EA. Depleting tumor‐NQO1 potentiates anoikis and inhibits growth of NSCLC. Mol Cancer Res. 2016;14(1):14‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Froeling FEM, Swamynathan MM, Deschênes A, et al. Bioactivation of napabucasin triggers reactive oxygen species‐mediated cancer cell death. Clin Cancer Res. 2019;25(23):7162‐7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becerra C, Braiteh FS, Spira AI, et al. A phase Ib/II study of cancer stemness inhibitor napabucasin (BB608) combined with weekly paclitaxel in advanced triple‐negative breast cancer. J Clin Oncol. 2016;34(suppl 15):1094. [Google Scholar]
- 5.Becerra C, Garcia AA, Hays JL, et al . A phase 1b/2 study of napabucasin with weekly paclitaxel in advanced, previously treated platinum resistant ovarian cancer. J Clin Oncol. 2017;35(suppl 15):5548. [Google Scholar]
- 6.Becerra C, Hanna WT, Richey SL, et al. A phase 1b/2 study of napabucasin with weekly paclitaxel in advanced, previously treated non‐squamous non‐small cell lung cancer. J Clin Oncol. 2017;35(suppl 15):9052. [Google Scholar]
- 7.Bekaii‐Saab TS, Li C‐P, Okusaka T, et al. CanStem111P trial: a phase III study of napabucasin (BBI‐608) plus nab‐paclitaxel (nab‐PTX) with gemcitabine (gem) in adult patients with metastatic pancreatic adenocarcinoma (mPDAC). J Clin Oncol. 2017;35(suppl 15):TPS4148. [Google Scholar]
- 8.Bekaii‐Saab TS, Starodub A, El‐Rayes BF, et al. Phase 1b/2 trial of cancer stemness inhibitor napabucasin (NAPA) + nab‐paclitaxel (nPTX) and gemcitabine (Gem) in metastatic pancreatic adenocarcinoma (mPDAC). J Clin Oncol. 2018;36(suppl 15):4110. [Google Scholar]
- 9.Edenfield WJ, Becerra C, Langleben A, et al. A phase 2 study of napabucasin with weekly paclitaxel in previously treated metastatic breast cancer. J Clin Oncol. 2017;35(suppl 15):1084. [Google Scholar]
- 10.Edenfield WJ, Becerra C, Braiteh FS, et al. A phase Ib study of napabucasin plus weekly paclitaxel in patients with advanced melanoma. J Clin Oncol. 2017;35(suppl 15):9553. [Google Scholar]
- 11.El‐Rayes BF, Richards DA, Cohn AL, et al. BBI608‐503‐103HCC: a phase Ib/II clinical study of napabucasin (BBI608) in combination with sorafenib or amcasertib (BBI503) in combination with sorafenib (Sor) in adult patients with hepatocellular carcinoma (HCC). J Clin Oncol. 2017;35(suppl 15):4077. [Google Scholar]
- 12.Kalra M, Cote GM, Heist RS, et al. A phase 1b study of napabucasin (NAPA) + weekly paclitaxel (PTX) in patients (pts) with advanced thymoma and thymic carcinoma. J Clin Oncol. 2018;36(suppl 15):e20578. [Google Scholar]
- 13.Larson T, Ortuzar WF, Bekaii‐Saab TS, et al. BBI608‐224: a phase Ib/II study of cancer stemness inhibitor napabucasin (BBI‐608) administered with panitumumab in KRAS wild‐type patients with metastatic colorectal cancer. J Clin Oncol. 2017;35(suppl 4):677. [Google Scholar]
- 14.Mason WP, de Robles P, Borodyansky L, et al. BBI608‐201GBM: a phase Ib/II clinical study of napabucasin (BBI608) in combination with temozolomide (TMZ) for adult patients with recurrent glioblastoma (GBM). J Clin Oncol. 2017;35(suppl 15):e13525. [Google Scholar]
- 15.Shah MA, Shitara K, Lordick F, et al. The BRIGHTER trial: a phase 3 randomized double‐blind study of napabucasin (NAPA) plus paclitaxel (PTX) versus placebo (PBO) plus PTX in patients (pts) with pretreated advanced gastric and gastroesophageal junction (GEJ) adenocarcinoma. J Clin Oncol. 2018;36(suppl 15):4010. [Google Scholar]
- 16.Kawazoe A, Kuboki Y, Shinozaki E, et al. Multicenter phase I/II trial of napabucasin and pembrolizumab in patients with metastatic colorectal cancer (EPOC1503/SCOOP Trial). Clin Cancer Res. 2020;26(22):5887‐5894. [DOI] [PubMed] [Google Scholar]
- 17.Jonker DJ, Nott L, Yoshino T, et al. Napabucasin versus placebo in refractory advanced colorectal cancer: a randomised phase 3 trial. Lancet Gastroenterol Hepatol. 2018;3(4):263‐270. [DOI] [PubMed] [Google Scholar]
- 18.Shitara K, Yodo Y, Iino S. A phase I study of napabucasin plus paclitaxel for Japanese patients with advanced/recurrent gastric cancer. In Vivo. 2019;33(3):933‐937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawazoe A, Kuboki Y, Bando H, et al. Phase 1 study of napabucasin, a cancer stemness inhibitor, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2020;85(5):855‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shah M, Grothey A, Tebbutt N, et al. CanStem303C trial: a phase 3 study of napabucasin (NAPA) in combination with 5‐fluorouracil (5‐FU), leucovorin, and irinotecan (FOLFIRI) in adult patients (pts) with previously treated metastatic colorectal cancer (mCRC)—trial in progress. Ann Oncol. 2018;29(suppl 5):v84. [Google Scholar]
- 21.Riechelmann RP, Tannock IF, Wang L, Saad ED, Taback NA, Krzyzanowska MK. Potential drug interactions and duplicate prescriptions among cancer patients. J Natl Cancer Inst. 2007;99(8):592‐600. [DOI] [PubMed] [Google Scholar]
- 22.Van Leeuwen RWF, Brundel DHS, Neef C, et al. Prevalence of potential drug‐drug interactions in cancer patients treated with oral anticancer drugs. Br J Cancer. 2013;108(5):1071‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Given BA, Given CW, Sikorskii A, Vachon E, Banik A. Medication burden of treatment using oral cancer medications. Asia Pac J Oncol Nurs. 2017;4(4):275‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riechelmann RP, Moreira F, Smaletz O, Saad ED. Potential for drug interactions in hospitalized cancer patients. Cancer Chemother Pharmacol. 2005;56(3):286‐290. [DOI] [PubMed] [Google Scholar]
- 25.Riechelmann RP, Krzyzanowska MK, O'Carroll A, Zimmermann C. Symptom and medication profiles among cancer patients attending a palliative care clinic. Support Care Cancer. 2007;15(12):1407‐1412. [DOI] [PubMed] [Google Scholar]
- 26.Backman JT, Honkalammi J, Neuvonen M, et al. CYP2C8 activity recovers within 96 hours after gemfibrozil dosing: estimation of CYP2C8 half‐life using repaglinide as an in vivo probe. Drug Metab Dispos. 2009;37(12):2359‐2366. [DOI] [PubMed] [Google Scholar]
- 27.Ermer J, Corcoran M, Martin P. Lisdexamfetamine dimesylate effects on the pharmacokinetics of cytochrome P450 substrates in healthy adults in an open‐label, randomized, crossover study. Drugs R D. 2015;15(2):175‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Dose‐dependent interaction between gemfibrozil and repaglinide in humans: strong inhibition of CYP2C8 with subtherapeutic gemfibrozil doses. Drug Metab Dispos. 2011;39(10):1977‐1986. [DOI] [PubMed] [Google Scholar]
- 29.Martin P, Gillen M, Ritter J, et al. Effects of fostamatinib on the pharmacokinetics of oral contraceptive, warfarin, and the statins rosuvastatin and simvastatin: results from phase I clinical studies. Drugs R D. 2016;16(1):93‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nordmark A, Andersson A, Baranczewski P, Wanag E, Ståhle L. Assessment of interaction potential of AZD2066 using in vitro metabolism tools, physiologically based pharmacokinetic modelling and in vivo cocktail data. Eur J Clin Pharmacol. 2014;70(2):167‐178. [DOI] [PubMed] [Google Scholar]
- 31.Polli JW, Hussey E, Bush M, et al. Evaluation of drug interactions of GSK1292263 (a GPR119 agonist) with statins: from in vitro data to clinical study design. Xenobiotica. 2013;43(6):498‐508. [DOI] [PubMed] [Google Scholar]
- 32.Turpault S, Brian W, Van Horn R, et al. Pharmacokinetic assessment of a five‐probe cocktail for CYPs 1A2, 2C9, 2C19, 2D6 and 3A. Br J Clin Pharmacol. 2009;68(6):928‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zadoyan G, Rokitta D, Klement S, et al. Effect of Ginkgo biloba special extract Egb 761(R) on human cytochrome P450 activity: a cocktail interaction study in healthy volunteers. Eur J Clin Pharmacol. 2012;68(5):553‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zgheib NK, Frye RF, Tracy TS, Romkes M, Branch RA. Validation of incorporating flurbiprofen into the Pittsburgh cocktail. Clin Pharmacol Ther. 2006;80(3):257‐263. [DOI] [PubMed] [Google Scholar]
- 35.Ancrenaz V, Déglon J, Samer C, et al. Pharmacokinetic interaction between prasugrel and ritonavir in healthy volunteers. Basic Clin Pharmacol Toxicol. 2013;112(2):132‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snyder BD, Rowland A, Polasek TM, Miners JO, Doogue MP. Evaluation of felodipine as a potential perpetrator of pharmacokinetic drug‐drug interactions. Eur J Clin Pharmacol. 2014;70(9):1115‐1122. [DOI] [PubMed] [Google Scholar]
- 37.Bosilkovska M, Samer C, Déglon J, et al. Evaluation of mutual drug‐drug interaction within Geneva cocktail for cytochrome P450 phenotyping using innovative dried blood sampling method. Basic Clin Pharmacol Toxicol. 2016;119(3):284‐290. [DOI] [PubMed] [Google Scholar]
- 38.Laurie SA, Jonker DJ, Edenfield WJ, et al. A phase 1 dose‐escalation study of BBI503, a first‐in‐class cancer stemness kinase inhibitor in adult patients with advanced solid tumors. J Clin Oncol. 2014;32(15_suppl):2527. [Google Scholar]
- 39.Dai X, Karol MD, Hitron M, et al. Mass balance and pharmacokinetics of an oral dose of 14C‐napabucasin in healthy adult male subjects. Pharmacol Res Perspect. 2021:9(1):e00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.US Food and Drug Administration . Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Silver Spring, MD: US Food and Drug Administration; 2020. [Google Scholar]
- 41.Gujjarlamudi HB. Polytherapy and drug interactions in elderly. J Midlife Health. 2016;7(3):105‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borad MJ, Curtis KK, Babiker HM, et al. The impact of concomitant medication use on patient eligibility for phase I cancer clinical trials. J Cancer. 2012;3:345‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salwe KJ, Kalyansundaram D, Bahurupi Y. A study on polypharmacy and potential drug‐drug interactions among elderly patients admitted in department of medicine of a tertiary care hospital in Puducherry. J Clin Diagn Res. 2016;10(2):FC06‐FC10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viktil KK, Blix HS, Moger TA, Reikvam A. Polypharmacy as commonly defined is an indicator of limited value in the assessment of drug‐related problems. Br J Clin Pharmacol. 2007;63(2):187‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beijnen JH, Schellens JHM. Drug interactions in oncology. Lancet Oncol. 2004;5(8):489‐496. [DOI] [PubMed] [Google Scholar]
- 46.Streetman DS, Bleakley JF, Kim JS, et al. Combined phenotypic assessment of CYP1A2, CYP2C19, CYP2D6, CYP3A, N‐acetyltransferase‐2, and xanthine oxidase with the “Cooperstown cocktail.” Clin Pharmacol Ther. 2000;68(4):375‐383. [DOI] [PubMed] [Google Scholar]
- 47.Christensen M, Andersson K, Dalén P, et al. The Karolinska cocktail for phenotyping of five human cytochrome P450 enzymes. Clin Pharmacol Ther. 2003;73(6):517‐528. [DOI] [PubMed] [Google Scholar]
- 48.Ryu JY, Song IS, Sunwoo YE, et al. Development of the “Inje cocktail” for high‐throughput evaluation of five human cytochrome P450 isoforms in vivo. Clin Pharmacol Ther. 2007;82(5):531‐540. [DOI] [PubMed] [Google Scholar]
- 49.Harris JW, Rahman A, Kim BR, Guengerich FP, Collins JM. Metabolism of taxol by human hepatic microsomes and liver slices: participation of cytochrome P450 3A4 and an unknown P450 enzyme. Cancer Res. 1994;54(15):4026‐4035. [PubMed] [Google Scholar]
- 50.Rahman A, Korzekwa KR, Grogan J, Gonzalez FJ, Harris JW. Selective biotransformation of taxol to 6 alpha‐hydroxytaxol by human cytochrome P450 2C8. Cancer Res. 1994;54(21):5543‐5546. [PubMed] [Google Scholar]
- 51.Bendell JC, Hubbard JM, O'Neil BH, et al. Phase 1b/II study of cancer stemness inhibitor napabucasin (BBI‐608) in combination with FOLFIRI +/− bevacizumab (bev) in metastatic colorectal cancer (mCRC) patients (pts). J Clin Oncol. 2017;35(15_suppl):3529.28796588 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information