

Abstract
Metabolites regulate protein function via covalent and noncovalent interactions. However, manipulating these interactions in living cells remains a major challenge. Here, we report a chemical strategy for inducing cysteine S-succination, a nonenzymatic post-translational modification derived from the oncometabolite fumarate. Using a combination of antibody-based detection and kinetic assays, we benchmark the in vitro and cellular reactivity of two novel S-succination “agonists,” maleate and 2-bromosuccinate. Cellular assays reveal maleate to be a more potent and less toxic inducer of S-succination, which can activate KEAP1-NRF2 signaling in living cells. By enabling the cellular reconstitution of an oncometabolite–protein interaction with physiochemical accuracy and minimal toxicity, this study provides a methodological basis for better understanding the signaling role of metabolites in disease.
Graphical Abstract
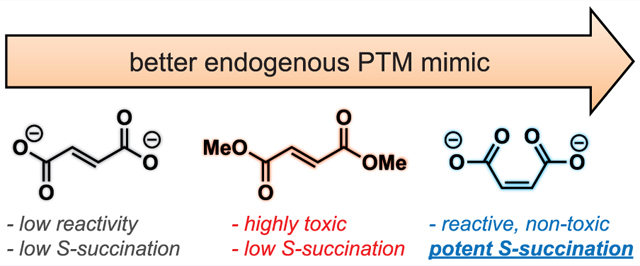
An emerging paradigm in cancer biology is that metabolism may function as an epigenetic signal in and of itself.1,2 A prototypical example of this phenomenon occurs in the genetic cancer syndrome hereditary leiomyomatosis and renal cell carcinoma (HLRCC). In this disorder, mutations in gene encoding the TCA cycle enzyme fumarate hydratase (FH) cause fumarate to accumulate to millimolar levels.3 These aberrant levels of fumarate are associated with chromatin hypermethylation and dysregulated gene expression;4,5 however, the molecular mechanisms by which this simple organic metabolite drives such profound changes in epigenetic signaling are not completely understood. Fumarate is unique among oncometabolites in that it has two physically distinct mechanisms by which it may alter post-translational modification (PTM) mediated signaling. First, fumarate can serve as a competitive inhibitor of 2-ketoglutarate (KG) utilizing enzymes, a mechanism it shares with other oncometabolites such as succinate and 2-hydroxyglutarate.6,7 Second, fumarate can react covalently with proteins to form the nonenzymatic modification cysteine S-succination (Figure 1a).8 This nonenzymatic PTM occurs exclusively in HLRCC and a few other pathophysiological contexts.9–11 While the targets and stoichiometries of protein S-succination are incompletely defined, at least one consequence of this PTM appears to be stabilization of the antioxidant transcription factor NRF2 due to covalent S-succination of its regulatory E3 ligase KEAP1.12,13
Figure 1.

(a) Nonenzymatic modification of cysteines by fumarate causes protein S-succination. (b) Structures of endogenous and synthetic inducers of cellular S-succination.
Recently, we reported a chemoproteomic approach for characterizing protein S-succination in patient-derived HLRCC cell lines.14 Investigation of the local sequence preferences of FH-sensitive cysteine residues and kinetic analyses led to the surprising discovery that fumarate itself appears to be chemically inert toward thiol addition, only becoming reactive upon protonation to hydrogen and dihydrogen fumarate.15 This leads to a paradoxical influence of pH on S-succination. However, fumarate’s low reactivity raises a technical challenge: it is very difficult to induce cysteine S-succination in cells. Most studies that have sought to explore fumarate’s reactivity using exogenous S-succination reagents have used dimethyl fumarate (DMF), an ester derivative that implements a physiochemically distinct PTM and is at least ∼100-fold more reactive than endogenous fumarate.16,17 Methods to temporally control cysteine S-succination have the potential to decouple fumarate’s covalent and noncovalent epigenetic mechanisms and provide new insights into the role of this oncometabolite in biology and disease.
To develop a reagent for manipulating oncometabolite-associated S-succination, we envisioned two straightforward modifications to fumarate’s structure. First, given our previous findings, we considered whether fumarate analogues with increased susceptibility to protonation may show coordinately increased cysteine reactivity. This led us to explore maleate, a cis isomer of fumarate (Figure 1b). Unlike fumarate, maleate has the unique ability to form an internal hydrogen bond upon protonation, leading to a substantial change in its pKa (maleic acid, pKa2 = 6.2; fumaric acid, pKa2 = 4.5).18 When present at equal concentrations in buffered solution, levels of electrophilic hydrogen maleate are over an order of magnitude higher than hydrogen fumarate, which would be expected to cause a concomitant increase in cysteine S-succination. Second, we hypothesized that the intrinsic electrophilicity of fumarate could be increased by replacing the α,β-unsaturated Michael acceptor with a bromoacetamide (Figure 1b). The cognate analogue, 2-bromosuccinate, has the potential to either label cysteines and form the desired PTM or undergo an elimination reaction to form electrophilically inert fumarate. Importantly, α-carboxyhaloacetamides have shown previous utility as cysteine labeling reagents in a number of settings.19–21
As an initial test of the ability of these oncometabolite analogues to induce S-succination, we incubated proteomes with fumarate, maleate, and 2-bromosuccinate and assessed covalent protein labeling using a recently developed anti-cysteine-S-succination antibody (Figure 2). Exposure of proteomes to maleate and 2-bromosuccinate led to protein S-succination that was detectable at concentrations as low as 0.5–1 mM (Figure 2b). In contrast, exposure of proteomes to fumarate at these low concentrations did not lead to detectable protein labeling, consistent with our previous studies. Comparison of the reagents revealed that maleate provided qualitatively greater protein labeling than 2-bromosuccinate when applied at an equivalent concentration (Figure S1). Both 2-bromosuccinate and maleate hyper-S-succinated proteomes in a time-dependent manner, with signal intensities increasing over 24 h (Figure 2c). This suggests that both of these reagents are stable in solution over prolonged time periods in cell lysates. These results establish 2-bromosuccinate and maleate as novel synthetic reagents capable of inducing oncometabolite-associated PTM cysteine-S-succination.
Figure 2.

(a) Structure of nonenzymatic S-succination reagents. (b) Dose-dependent effects of fumarate, maleate, and 2-bromosuccinate on protein S-succination in cell lysates (t = 15 h). (c) Time-dependent S-succination of proteomes upon the addition of maleate or 2-bromosuccinate.
Next, we aimed to quantitatively benchmark the reactivity of our chemical S-succination reagents. For these experiments, we incubated electrophiles (fumarate, maleate, or 2-bromosuccinate) with a model thiol (thiophenol, pKa 6.6) and assessed product formation via quantitative UV- RP-HPLC (Figure 3).14 Thiophenol was chosen due to its enhanced nucleophilicity compared to cysteine, which facilitates measurement of its reaction with fumarate prior to significant oxidation. Consistent with immunoblotting,, these studies revealed that maleate S-succinates thiols much faster (∼30×) than fumarate at neutral pH (Table 1). Examining the influence of pH on thiol modification we verified that fumarate’s reactivity is increased under more acidic conditions. In contrast, maleate and 2-bromosuccinate behave more like a conventional electrophile, with an increased reaction rate in neutral or mildly alkali solutions (Table 1). This suggests that unlike hydrogen fumarate (pKa 4.4), sufficient concentrations of hydrogen maleate (pKa 6.2) are present at neutral pH to enable thiol reactivity, the rate of which declines upon acidification due to reduced abundance of thiophenolate nucleophile.
Figure 3.

(a) Model reaction to assess thiol S-succination reaction kinetics. (b) Comparison of pH-dependent pseudo-first order rate constants for S-succination electrophiles.
Table 1.
S-Succination Reaction Kinetics As a Function of Electrophile and pH
| pH | fumarate | maleate | 2-Br-succinae | |
|---|---|---|---|---|
| rate (mM−1 min−1) | 5 | (1.1 ± 0.6) × 10−4 | (1.6 ± 0.78) × 10−4 | (0.08 ± 0.01) × 10−4 |
| 6 | (0.11 ± 0.03) × 10−4 | (2.9 ± 0.66) × 10−4 | (0.99 ± 0.23) × 10−4 | |
| 7 | (0.06 ± 0.01) × 10−4 | (3.9 ± 0.57) × 10−4 | (4.2 ± 0.59) × 10−4 |
The electrophilicity of maleate was less impacted by acidity (pH 5–6) than 2-bromosuccinate, consistent with the ability of protonation to increase its intrinsic reactivity (Table 1). To ensure that these trends were not an artifact of our kinetic analysis method, we performed equivalent pH-dependent labeling studies in whole proteomes and made identical observations for both fumarate and maleate (Figure S2). These studies define the chemical reactivity of the oncometabolite isomer maleate and quantitatively validate its utility as a rapid inducer of thiol S-succination.
Inactivation of the TCA cycle by FH mutation causes a multitude of cellular effects, only a subset of which may be related to protein S-succination. Previous studies have attempted to chemically induce this PTM to help isolate its influence on signaling but have been challenged by fumarate’s low reactivity and cell permeability.16,17 We wondered whether the hyper-reactivity of 2-bromosuccinate and maleate may overcome some of these challenges. To test this, we compared endogenous protein S-succination in FH-deficient cells (FH−/−) to that induced by treating an isogenic rescue (FH+/+) with 2-bromosuccinate, maleate, dimethyl fumarate, and ethyl fumarate (Figure 4, Figure S3). Analysis of treated cells revealed all four compounds were able to induce the PTM, as assessed by anti-S-succination immunoblotting (Figure 4c–f). However, notable differences were also observed. Dimethyl and ethyl fumarate led to only low levels of S-succination (Figure 4e,f). This is consistent with the chemical structures of these compounds, which would be expected to lead to the physiochemically distinct methyl- or ethyl-S-succination upon cysteine reaction.22,23 In contrast, 2-bromosuccinate and maleate modified a wide range of proteins, with a labeling pattern more akin to the endogenous modification found in FH−/− cells (Figure 4c,d). Interestingly, when dosed at equivalent concentrations, 2-bromosuccinate induced a stronger modification profile than maleate initially (3 h) but was superseded at extended time points. This may indicate that 2-bromosuccinate is metabolized or unstable in media over prolonged time periods.
Figure 4.

(a) Cellular evaluation of electrophilic inducers of S-succination. (b) Structure of electrophiles. (c) Cellular S-succination induced in UOK262 FH rescue (FH+/+) cells by maleate, (d) 2-bromosuccinate, (e) dimethyl fumarate, and (f) ethyl fumarate. Treatment time = 15 h.
During our cellular labeling studies, some treatments caused noticeable detachment of cells, suggesting toxicity. As cell death may introduce a variety of biological effects that are independent of protein S-succination, we compared the cytotoxicity of fumarate, dimethyl fumarate, ethyl fumarate, methyl fumarate, 2-bromosuccinate, and maleate. HEK-293T cells were used for these studies to avoid any confounding effects FH-deficiency may have on metabolism-dependent cell death assays. We confirmed the novel reagents investigated here, 2-bromosuccinate and maleate, both cause S-succination in this cell model (Figure S4). Analyzing dose-dependent cytotoxicity of these reagents, we found dimethyl fumarate to be the most potent inhibitor of cell growth, with a submicromolar IC50 at 48 h (Table 2, Figure S5). This is consistent with previous studies indicating the highly electrophilic nature of this compound15,24 and provides further evidence that caution should be used when interpreting biological assays using this reagent as an oncometabolite mimetic. The monoalkyl ester derivatives of fumarate, as well as 2-bromosuccinate, displayed an intermediate degree of toxicity, while fumarate and maleate were nontoxic up to millimolar concentrations. These studies specify maleate as an optimized nontoxic reagent for manipulation of cellular S-succination profiles.
Table 2.
Toxicity of Electrophilic Fumarate Analogues and S-succination Agonist in HEK-293T Cells at 48 h.
| fumarate | 72 ± 11 |
| maleate | 11 ± 0.91 |
| 2-bromosuccinate | 1.2 ± 0.44 |
| ethyl fumarate | 0.95 ± 0.31 |
| methyl fumarate | 0.91 ± 0.48 |
| dimethyl fumarate | 0.059 ± 0.011 |
As indicated above, the structure and reactivity of maleate and 2-bromosuccinate differ in subtle—but important—ways from fumarate. Therefore, as a final metric, we set out to assess the effects of these two molecules on known covalent and noncovalent oncometabolite targets. As a prototypical covalent target, we chose the E3 ligase KEAP1, whose covalent modification by fumarate has previously been shown to cause stabilization of the transcription factor NRF2, a master regulator of the antioxidant response.12,13 Consistent with the hypothesis that maleate can recapitulate aspects of fumarate’s reactivity, incubation of HEK-293T cells with maleate induced higher levels of NRF2 (Figure 5a). To verify that this mechanism may be driven by maleate’s ability to covalently modify KEAP1, we overexpressed KEAP1 and subjected HEK-293T cells to different electrophile treatments. Transient transfection followed by lysis, KEAP1 immunoprecipitation, and anti-S-succination immunoblotting revealed detectable covalent modification of KEAP1 upon addition of maleate (Figure 5b). 2-Bromosuccinate also was able to trigger NRF2 stabilization and KEAP1 S-succination, although cytotoxicity was observed at the higher dosage (10 mM; Figure 5c). In contrast, DMF and fumarate itself did not induce detectable S-succination of KEAP1, presumably due to a lack of ester hydrolysis and cell permeability, respectively. Turning our attention to noncovalent targets of fumarate, it has previously been shown that inactivation of FH in HLRCC causes accumulation of the transcription factor HIF-1α, which is attributed to fumarate’s ability to competitively inhibit KG-dependent prolyl hydroxylases involved in HIF-1α degradation.6,7 In contrast to its effects on NRF2, incubation of HEK-293T cells with maleate (1–10 mM) for 24 h did not cause a substantial increase in HIF-1α levels. However, incubation with 2-bromosuccinate caused a detectible increase in HIF-1α levels (Figure 5a). This suggests that 2-bromosuccinate—or one of its metabolites—may function as a competitive inhibitor of prolyl hydroxylase activity, while maleate cannot. Previous crystallographic studies of prolyl hydroxylases complexed with KG-competitive ligands are more consistent with a trans-orientation near the Fe(II)-KG reaction center, which cannot be accessed by cis-fumarate.25,26 Together, these data suggest the unique structure of maleate may in certain instances allow selective induction of fumarate’s signature nonenzymatic PTM (S-succination) while limiting its noncovalent effects on KG-dependent enzymes such as prolyl hydroxylases.
Figure 5.

(a) Dose-dependent effects of maleate and 2-bromosuccinate on S-succination, NRF2, and HIF1-a in HEK-293T cells. Treatment time = 15 h. Loss of GAPDH in 2-bromosuccinate samples indicates increasing cell death. (b) Assay for assessing cellular S-succination of ectopically expressed FLAG-KEAP1. (c) Cellular S-succination of ectopically expressed KEAP1 by electrophiles (treatment time = 15 h). Br-succinate = 2-bromosuccinate. DMF = dimethyl fumarate.
Recent advances in chemoproteomics have enabled the detection and comprehensive profiling of metabolite–protein interactions.27–31 Translating these data sets into mechanistic knowledge requires methods to manipulate these interactions in living cells but is challenged by the limited permeability and tempered reactivity of many metabolites. Here, we report a new approach for the induction of cysteine S-succination, a nonenzymatic PTM produced by the covalent oncometabolite fumarate. In contrast to previous methods that utilize fumarate esters, the approach reported here recapitulates the endogenous PTM (rather than an ester-linked version) and can be applied in living cells with limited toxicity. Furthermore, our initial studies indicate that in HEK-293T cells one of these reagents, maleate, is able to differentially induce covalent (KEAP1 S-succination/NRF2 activation) versus noncovalent (PHD inhibition/HIF-1α stabilization) effects of fumarate. This property should be useful in studies seeking to define the relative influence of these two mechanisms on the plethora of biological phenomena in which fumarate has been implicated in HLRCC, which include altered gene expression, mitotic entry, and epithelial to mesenchymal transition.5,32,33 Finally, we note some limitations of our method as currently comprised. First, while maleate, 2-bromosuccinate, and fumarate induce the same PTM, they differ substantially in their reactivity and most notably show distinct pH-dependent labeling profiles. Our previous studies have provided evidence that hydrogen or dihydrogen fumarate is the reactive species underlying covalent S-succination in HLRCC,14 and if the S-succination agonists reported here are reactive in the absence of protonation, this may alter their proteomic reactivity profile. Future comparisons of these reagents and fumarate in competitive chemoproteomic experiments will help define the similarity (or distinctiveness) of their cysteine reactivity landscapes. Another key difference between FH-deficiency and this exogenous S-succination strategy is that in the former fumarate diffuses out of the mitochondria, while in the latter electrophiles diffuse in through the plasma membrane. Targeted delivery of maleate or 2-bromosuccinate to the mitochondria may be required to enable the study of S-succination in this organelle.34,35 In the immediate future, we anticipate the less toxic maleate reagent will be useful for determining whether S-succination elicits a distinct gene expression response relative to other NRF2-inducing electrophiles,5 as well as in isotopic labeling strategies designed to assess S-succination stoichiometry.36 In the longer term, we envision that maleate’s unique status as an anion electrophile that is relatively insensitive to bulk pH may prove useful in the construction of covalent fragment libraries designed to interrogate the ligandability of distinct subsets of the proteome.37 By expanding our inventory of methods for the study of metabolite-derived PTMs, these studies provide a foundation for defining and manipulating the signaling role of metabolism in cancer.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, The Center for Cancer Research (ZIA BC011488-06).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.0c00044
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00044.
Figures S1–S5 and supporting materials and methods (PDF)
The authors declare no competing financial interest.
Contributor Information
Sarah E. Bergholtz, Chemical Biology Laboratory, National Cancer Institute, Frederick, Maryland 21702, United States.
Chloe A. Briney, Chemical Biology Laboratory, National Cancer Institute, Frederick, Maryland 21702, United States.
Susana S. Najera, Chemical Biology Laboratory and Urologic Oncology Branch, National Cancer Institute, Frederick, Maryland 21702, United States
Minervo Perez, Chemical Biology Laboratory, National Cancer Institute, Frederick, Maryland 21702, United States.
W. Marston Linehan, Urologic Oncology Branch, National Cancer Institute, Bethesda, Maryland, United States.
Jordan L. Meier, Chemical Biology Laboratory, National Cancer Institute, Frederick, Maryland 21702, United States
REFERENCES
- (1).Meier JL (2013) Metabolic mechanisms of epigenetic regulation. ACS Chem. Biol 8, 2607–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kinnaird A, Zhao S, Wellen KE, and Michelakis ED (2016) Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 16, 694–707. [DOI] [PubMed] [Google Scholar]
- (3).Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, and Tomlinson IP (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet 14, 2231–2239. [DOI] [PubMed] [Google Scholar]
- (4).Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, and Guan KL (2012) Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MG, Rajeeve V, Cardaci S, Foster S, Yun H, Cutillas P, Warren A, Gnanapragasam V, Gottlieb E, Franze K, Huntly B, Maher ER, Maxwell PH, Saez-Rodriguez J, and Frezza C. (2016) Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, and Gottlieb E. (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77–85. [DOI] [PubMed] [Google Scholar]
- (7).Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, and Neckers L. (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–153. [DOI] [PubMed] [Google Scholar]
- (8).Alderson NL, Wang Y, Blatnik M, Frizzell N, Walla MD, Lyons TJ, Alt N, Carson JA, Nagai R, Thorpe SR, and Baynes JW (2006) S-(2-Succinyl)cysteine: a novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch. Biochem. Biophys 450, 1–8. [DOI] [PubMed] [Google Scholar]
- (9).Frizzell N, Thomas SA, Carson JA, and Baynes JW (2012) Mitochondrial stress causes increased succination of proteins in adipocytes in response to glucotoxicity. Biochem. J 445, 247–254. [DOI] [PubMed] [Google Scholar]
- (10).Piroli GG, Manuel AM, Clapper AC, Walla MD, Baatz JE, Palmiter RD, Quintana A, and Frizzell N. (2016) Succination is Increased on Select Proteins in the Brainstem of the NADH dehydrogenase (ubiquinone) Fe-S protein 4 (Ndufs4) Knockout Mouse, a Model of Leigh Syndrome. Mol. Cell. Proteomics 15, 445–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ternette N, Yang M, Laroyia M, Kitagawa M, O’Flaherty L, Wolhulter K, Igarashi K, Saito K, Kato K, Fischer R, Berquand A, Kessler BM, Lappin T, Frizzell N, Soga T, Adam J, and Pollard PJ (2013) Inhibition of Mitochondrial Aconitase by Succination in Fumarate Hydratase Deficiency. Cell Rep. 3, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, and Pollard PJ (2011) Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kinch L, Grishin NV, and Brugarolas J. (2011) Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell 20, 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kulkarni RA, Bak DW, Wei D, Bergholtz SE, Briney CA, Shrimp JH, Alpsoy A, Thorpe AL, Bavari AE, Crooks DR, Levy M, Florens L, Washburn MP, Frizzell N, Dykhuizen EC, Weerapana E, Linehan WM, and Meier JL (2019) A chemoproteomic portrait of the oncometabolite fumarate. Nat. Chem. Biol 15, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schmidt TJ, Ak M, and Mrowietz U. (2007) Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine–preparation of S-substituted thiosuccinic acid esters. Bioorg. Med. Chem 15, 333–342. [DOI] [PubMed] [Google Scholar]
- (16).Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, and Chandel NS (2013) The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zheng L, Cardaci S, Jerby L, MacKenzie ED, Sciacovelli M, Johnson TI, Gaude E, King A, Leach JD, Edrada-Ebel R, Hedley A, Morrice NA, Kalna G, Blyth K, Ruppin E, Frezza C, and Gottlieb E. (2015) Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun 6, 6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bruice TC, and Bradbury WC (1965) The gem Effect. III. The Influence of 3-Mono- and 3-gem Substitution on the Acid Dissociation Constants of Glutaric Acid. A Comparison of the Sensitivity of [UNK]pKa of Dicarboxylic Acids and the Rate of Ring Closure of Their Monoesters to the Intramolecular Distance Separating Carboxyl Functions. J. Am. Chem. Soc 87, 4851–4855. [DOI] [PubMed] [Google Scholar]
- (19).Barnard EA, and Stein WD (1959) Histidine Residue in the Active Centre of Ribonuclease. 1. Specific Reaction with Bromoacetic Acid. J. Mol. Biol 1, 339–349. [Google Scholar]
- (20).Wang H, Vath GM, Gleason KJ, Hanna PE, and Wagner CR (2004) Probing the mechanism of hamster arylamine N-acetyltransferase 2 acetylation by active site modification, site-directed mutagenesis, and pre-steady state and steady state kinetic studies. Biochemistry 43, 8234–8246. [DOI] [PubMed] [Google Scholar]
- (21).Murray RI, Gunsalus IC, and Dus KM (1982) Active-Site Studies of Cytochrome-P-450. 1. Specific Cysteine Labeling with the Affinity Reagent Isobornyl Bromoacetate As a Model for Substrate Binding. J. Biol. Chem 257, 2517–2525. [PubMed] [Google Scholar]
- (22).Manuel AM, and Frizzell N. (2013) Adipocyte protein modification by Krebs cycle intermediates and fumarate ester-derived succination. Amino Acids 45, 1243–1247. [DOI] [PubMed] [Google Scholar]
- (23).Piroli GG, Manuel AM, Patel T, Walla MD, Shi L, Lanci SA, Wang J, Galloway A, Ortinski PI, Smith DS, and Frizzell N. (2019) Identification of Novel Protein Targets of Dimethyl Fumarate Modification in Neurons and Astrocytes Reveals Actions Independent of Nrf2 Stabilization. Mol. Cell. Proteomics 18, 504–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Blewett MM, Xie JJ, Zaro BW, Backus KM, Altman A, Teijaro JR, and Cravatt BF (2016) Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci. Signaling 9, rs10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).McDonough MA, Li V, Flashman E, Chowdhury R, Mohr C, Lienard BM, Zondlo J, Oldham NJ, Clifton IJ, Lewis J, McNeill LA, Kurzeja RJ, Hewitson KS, Yang E, Jordan S, Syed RS, and Schofield CJ (2006) Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. U. S. A 103, 9814–9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chowdhury R, McDonough MA, Mecinovic J, Loenarz C, Flashman E, Hewitson KS, Domene C, and Schofield CJ (2009) Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure 17, 981–989. [DOI] [PubMed] [Google Scholar]
- (27).Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, Chin EN, Ahmad I, Chatterjee AK, Lairson LL, Schultz PG, and Moellering RE (2018) A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature 562, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Qin W, Qin K, Zhang Y, Jia W, Chen Y, Cheng B, Peng L, Chen N, Liu Y, Zhou W, Wang YL, Chen X, and Wang C. (2019) S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat. Chem. Biol 15, 983–991. [DOI] [PubMed] [Google Scholar]
- (29).Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, and Cravatt BF (2013) Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 10, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Levy MJ, Montgomery DC, Sardiu ME, Montano JL, Bergholtz SE, Nance KD, Thorpe AL, Fox SD, Lin Q, Andresson T, Florens L, Washburn MP, and Meier JL (2020) A Systems Chemoproteomic Analysis of Acyl-CoA/Protein Interaction Networks. Cell Chem. Biol 27, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kulkarni RA, Worth AJ, Zengeya TT, Shrimp JH, Garlick JM, Roberts AM, Montgomery DC, Sourbier C, Gibbs BK, Mesaros C, Tsai YC, Das S, Chan KC, Zhou M, Andresson T, Weissman AM, Linehan WM, Blair IA, Snyder NW, and Meier JL (2017) Discovering Targets of Non-enzymatic Acylation by Thioester Reactivity Profiling. Cell Chem. Biol 24, 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Johnson TI, Costa ASH, Ferguson AN, and Frezza C. (2018) Fumarate hydratase loss promotes mitotic entry in the presence of DNA damage after ionising radiation. Cell Death Dis. 9, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tyrakis PA, Yurkovich ME, Sciacovelli M, Papachristou EK, Bridges HR, Gaude E, Schreiner A, D’Santos C, Hirst J, Hernandez-Fernaud J, Springett R, Griffiths JR, and Frezza C. (2017) Fumarate Hydratase Loss Causes Combined Respiratory Chain Defects. Cell Rep. 21, 1036–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chalmers S, Caldwell ST, Quin C, Prime TA, James AM, Cairns AG, Murphy MP, McCarron JG, and Hartley RC (2012) Selective uncoupling of individual mitochondria within a cell using a mitochondria-targeted photoactivated protonophore. J. Am. Chem. Soc 134, 758–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kulkarni RA, Briney CA, Crooks DR, Bergholtz SE, Mushti C, Lockett SJ, Lane AN, Fan TW, Swenson RE, Marston Linehan W, and Meier JL (2019) Photoinducible Oncometabolite Detection. ChemBioChem 20, 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Prus G, Hoegl A, Weinert BT, and Choudhary C. (2019) Analysis and Interpretation of Protein Post-Translational Modification Site Stoichiometry. Trends Biochem. Sci 44, 943–960. [DOI] [PubMed] [Google Scholar]
- (37).Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, and Cravatt BF (2016) Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.