

Abstract

Pyridyl tetrazines coordinated to metals like rhenium have been shown to be more reactive in [4 + 2] cycloadditions than their uncomplexed counterparts. Using density functional theory calculations, we found a more favorable interaction energy caused by stronger orbital interactions as the origin of this increased reactivity. Additionally, the high regioselectivity is due to a greater degree of charge stabilization in the transition state, leading to the major product.
The reaction between 1,2,4,5-tetrazines and alkenes was first described in 1959 by Carboni and Lindsey.1 These azines react with olefins in an inverse electron-demand Diels–Alder cycloaddition followed by a cycloreversion with loss of dinitrogen (Figure 1a). The formed dihydropyridazines can then further tautomerize or be oxidized to the corresponding pyridazine.2 In 1990, Sauer showed that tetrazines react very rapidly with strained alkenes, such as cyclopropanes and trans-cyclooctenes.3 This observation led to the introduction of tetrazine ligation as a biocompatible click reaction in 2008.4,5 This ligation has since been employed broadly in applications ranging from radiolabeling6−8 to material science.9−11 A targeted drug delivery system based on this cycloaddition is currently in phase 1 clinical studies.12,13
Figure 1.

[4 + 2] Cycloadditions of tetrazine 1 (Tz) and rhenium-coordinated tetrazine 5 (Re-Tz) with styrene (2, St) leads to the formation of intermediates 3 and 6, respectively. This is followed by a retro-[4 + 2] cycloaddition to form dihydropyridazines 4 and 7. The regioisomers in which the Ph group is ortho to the pyridyl substituent are shown. Subsequent oxidation of products 4 and 7 can form pyridazines.
Commonly used tetrazines include mono- and disubstituted derivatives with alkyl or aryl groups.14−17 Due to the inverse electron-demand nature of the initial rate-limiting [4 + 2] cycloaddition, electron-withdrawing substituents increase reactivity.18 2-Pyridyl- and 2-pyrimidyl-substituted tetrazine derivatives are among the most reactive dienes that can be used for these reactions.17,19
2-Pyridyl tetrazines can be used as ligands for metals, as analogues to 2,2′-bipyridine ligands.20,21 Recently, Ringenberg and co-workers investigated the click reactivity of a 2-pyridyl tetrazine ligand in a rhenium complex toward vinylferrocene, styrene, and trans-cyclooctene (Figure 1b).22 This reaction allows for efficient labeling with the metal complex, opening possible applications in the fields of chemical biology and medicine.23−26 Interestingly, Ringenberg et al. describe an increase in rate constants of 2 orders of magnitude compared to the reaction of tetrazine itself. The same behavior was observed by the same group for a ruthenium complex.27 This acceleration makes these systems particularly interesting as it would allow for labeling at ultralow concentrations. However, the origin of this increased reactivity could not be determined.
Here, we perform a computational analysis using distortion/interaction and energy decomposition analysis to unravel the underlying mechanisms that lead to the observed increase in reactivity. An initial version of this work was deposited in ChemRxiv on July 2, 2021.28
Computational Methods
Density functional theory (DFT) calculations were performed with Gaussian 16 RevA.03.29 For each structure, all possible conformers were considered. Geometry optimizations were performed with the B3LYP functional,30,31 which was shown to closely match experimental values, augmented with Grimme’s D3 empirical dispersion term,32−35 and the SDD basis set for Re and 6-311+G(d,p)36 for all other atoms. Dichloromethane solvation was modeled using the SMD solvation model.37 Frequency calculations confirmed the optimized structures as minima (zero imaginary frequencies) or transition state structures (one imaginary frequency) on the potential energy surface. A quasi-harmonic correction was applied using the GoodVibes program.38 Orbital energies were calculated at the same level of theory in the gas phase.
Distortion/interaction39 and energy decomposition analysis40 were performed in ADF (2019.304)41 with PyFrag 201942 using B3LYP-D3/TZ2P in the gas phase on structures that were optimized with the B3LYP-D3/6-311+G(d,p)/SDD-SMD(DCM) level of theory.
The reactivity of Tz (1) was first compared to that of Re-Tz (5) in the reaction with St (2). In these reactions, the formation of two regioisomers is possible: one in which the phenyl group ends up ortho to the pyridyl ring in the final dihydropyridazine (Figure 2a) and one in which these groups are meta to each other (Figure 2b). Experimentally, the ortho products are the major products observed. The energy barrier for the reaction leading to ortho intermediate 3 was calculated to be 23.5 kcal/mol (Figure 2a, TS-1a). Coordination of Re reduced this barrier to 21.4 kcal/mol (TS-2a) for the formation of ortho intermediate 6. This calculated activation barrier is in excellent agreement with experimental results by Ringenberg and co-workers, whose kinetic experiments correspond to a ΔG⧧ value of 22.0 kcal/mol. In these reactions, a total of four orientations of addition of St to Tz are possible; the lowest-energy TSs for the ortho and meta product are shown in Figure 2. Similarly, the two lowest-energy TSs for the reaction of St with Re-Tz for formation of the ortho and meta products out of a possible eight orientations are shown. The other higher-energy transition states and their energy barriers are included in the Supporting Information.
Figure 2.
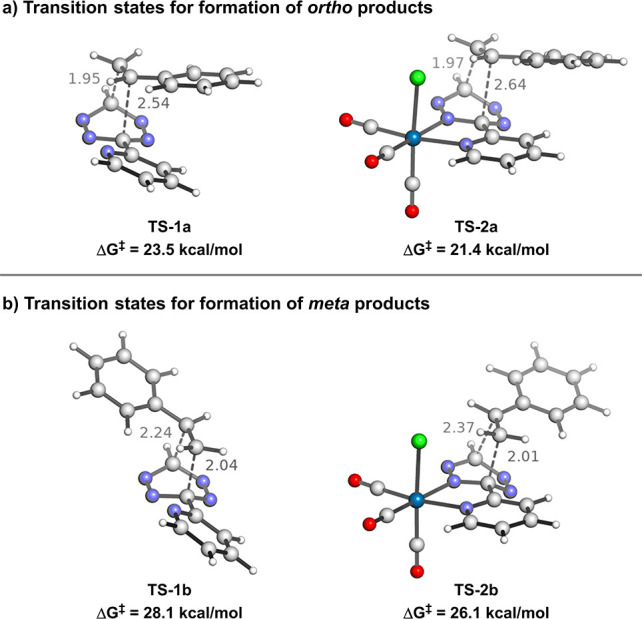
Transition states for the [4 + 2] cycloaddition to form the ortho products (a) and the meta products (b).
In order to determine what causes the lowering of the energy barrier upon coordination of Re, we performed distortion/interaction and energy decomposition analysis (Figure 3). For this analysis, we compared the geometries at the same point along the intrinsic reaction coordinate, in which the shorter bond-forming distance was 1.95 Å. In addition, we performed an analogous analysis of the two structures at the transition states, which can be found in the Supporting Information. At the so-called consistent geometries of 1.95 Å, the electronic energy was lower for the reaction of Re-Tz + St by 6.1 kcal/mol compared to that of Tz + St. This difference in electronic energy was decomposed into the interaction (ΔEint) and distortion (ΔEdist) components, both of which were decomposed further (Figure 3). Interaction energy was overall favorable for the Re-Tz reaction by 10.0 kcal/mol compared to that of the Tz reaction. The greatest contribution to this was orbital interactions (ΔEOI), which were stronger in the Re-Tz reaction by 7.2 kcal/mol. Electrostatic interactions (ΔVElstat) were also stronger in the reaction with Re-Tz, by 2.2 kcal/mol, as were dispersive interactions (ΔEDisp), by 1.9 kcal/mol. Conversely, Pauli repulsive interactions in the reaction of Tz were less unfavorable than that of Re-Tz by 1.3 kcal/mol.
Figure 3.
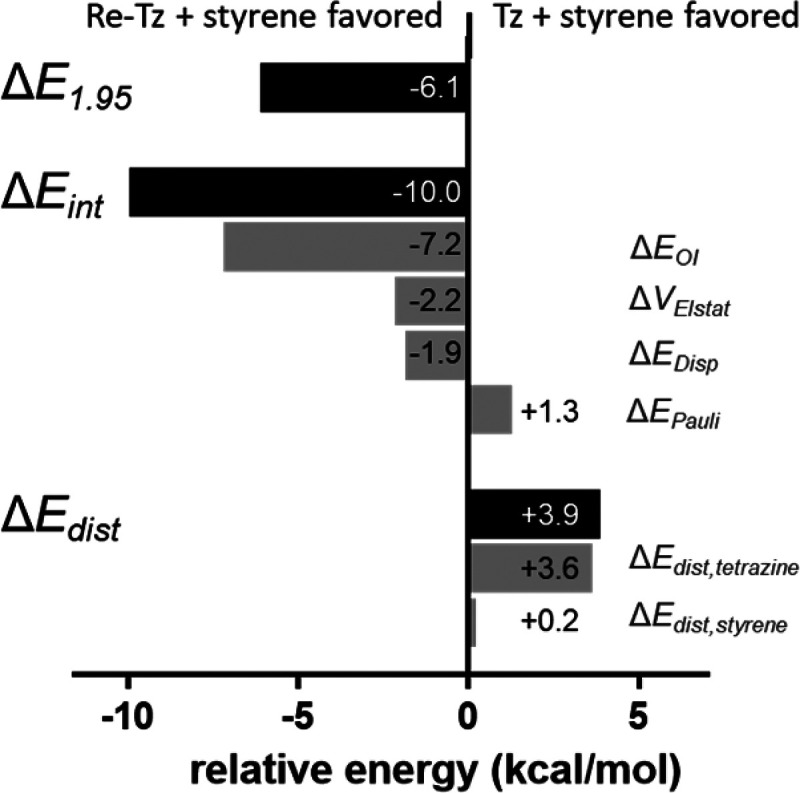
Distortion/interaction and energy decomposition analysis of TS-1a and TS-2a. Values are shown for TS-2a relative to those for TS-1a.
Distortion was more unfavorable in the case of the Re-Tz reaction by 3.9 kcal/mol. Distortion energies were calculated for the tetrazine/Re-tetrazine and styrene components, and the Re-Tz component was shown to be the main contributor to the difference in distortion energies between the two reactions by 3.6 kcal/mol more than the Tz component. Distortion in the St component was similar in both reactions, with a difference of only 0.2 kcal/mol.
As suggested by energy decomposition of the interaction energies, we performed further analysis of the orbital interactions at play in this reaction. The energies and visualizations of the relevant filled and empty molecular orbitals of the styrene and tetrazine components for the Tz and Re-Tz structures at the consistent geometry are depicted in Figure 4. The analogous energies for the starting materials and transition states are included in the Supporting Information. In the case of Re-Tz, the HOMO is located at the metal center; the Diels–Alder reactive filled orbital is the HOMO-7. As the reaction of tetrazines with dienophiles is an inverse electron-demand Diels–Alder reaction, the orbitals that contribute most to the reactivity are the LUMO of the diene and the HOMO of the dienophile. This can be seen by the HOMO/LUMO gap between the styrene and Tz/Re-Tz: 2.8 and 1.9 eV, respectively. In contrast, the normal electron-demand orbital gaps are 5.9 and 7.0 eV for Tz and Re-Tz, respectively (Figure 4). We thus conclude that the enhanced orbital interactions in the Re-Tz reaction, which is ultimately the major contributor to enhanced reactivity compared to the Tz reaction, originates from a lowered LUMO in the coordinated tetrazine.
Figure 4.
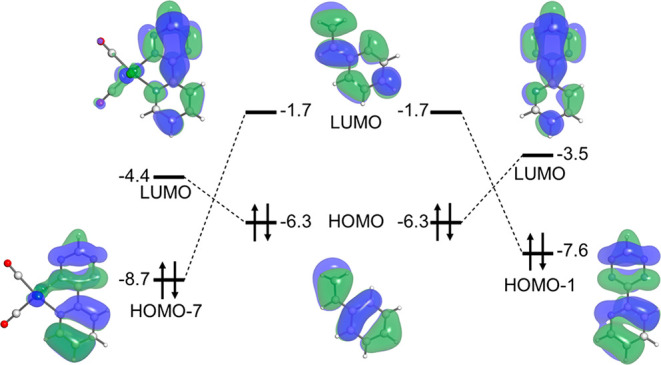
Reacting orbitals and orbital energies of fragments at the consistent geometry where the shorter forming bond is 1.95 Å. Reactions of Re-Tz (left) and Tz (right) with St are shown.
We subsequently analyzed the regioselectivity of this reaction. The reaction is highly regioselective, with formation of the ortho product favored over formation of the meta product. Transition states for formation of the meta products are shown in Figure 2b. For the reaction of Tz with styrene, the ortho TS is lower in energy than the meta TS by 4.6 kcal/mol. For the reaction of Re-Tz with styrene, this difference is 4.7 kcal/mol. Thus, both systems are highly regioselective. This is unusual for bioorthogonal tetrazine ligation, where poor regioselectivity is often observed.43 However, the high degree of regioselectivity in this reaction is not surprising, given the asymmetric electronic and steric nature of both the dienes and dienophile; furthermore, this selectivity has been reported before.44,45
The transition states leading to the favored ortho products are more asynchronous than the transition states leading to the meta products. In the ortho transition state, the more nucleophilic terminal carbon of styrene can interact strongly with the less sterically hindered tetrazine carbon, leading to a short carbon–carbon distance of about 1.96 Å. In addition, charges built up during this interaction can be well-stabilized by the aryl substituent at the ipso-carbon, leading to the highly asynchronous and energetically favored approach. In the transition state leading to the meta product, the terminal carbon of the styrene also participates in the initially forming bond. In this case, charge buildup on the unsubstituted tetrazine carbon could not be stabilized in a potential asynchronous transition state. This thus leads to a more synchronous transition state that is higher in energy. Comparing TS-2a and TS-2b using energy decomposition analysis shows that the interaction energy between these two pathways is essentially the same (Figure 5). The highly asynchronous nature of TS-2a leads to reduced Pauli repulsion, whereas orbital and electrostatic interactions are less favorable. The HOMOstyrene–LUMOtetrazine gap is essentially the same in both cases, but the more synchronous TS-2b shows an increased HOMOstyrene–LUMOtetrazine overlap of 0.137 compared to that of TS-2a with 0.131. However, distortion energies are considerably lower in TS-2a because of the asynchronous bond formation which results in an overall lower barrier.
Figure 5.

Distortion/interaction and energy decomposition analysis of TS-2a and TS-2b. Values are shown for TS-2a relative to TS-2b.
For the reaction of Re-Tz with St, we also studied the reaction profiles for the entire reaction sequence for formation of the ortho and meta products; these energy profiles are shown in Figure 6. It should be noted that, in both of these pathways, the preferred addition of styrene is from the face that contains the Cl ligand—this mode of addition is favored over addition to the CO face by 0.8 kcal/mol. Both pathways shown in Figure 5 correspond to addition to the Cl face. As discussed previously, the difference in energy barriers between TS-2a and TS-2b is 4.7 kcal/mol. These transition states lead to the formation of [4 + 2] cycloaddition products Int-1a and Int-1b, which are both at 6.2 kcal/mol. Subsequent retro-[4 + 2] cycloadditions have low energy barriers at 9.2 and 10.6 kcal/mol relative to starting materials SM and are highly exergonic, leading to low-energy products at −52.9 and −53.1 kcal/mol for Int-2a and Int-2b, respectively. Int-2a then preferentially undergoes isomerization to form a double bond between the two distal carbons of the dihydropyridazine (Int-3a). Int-3a, in which the Ph group is on the same face as the Cl ligand, is 0.2 kcal/mol lower in energy than the epimer in which the Ph group is on the same face as the CO ligand. It is likely that these two structures can reach a thermodynamic equilibrium at this stage through alkene isomerization.
Figure 6.

Free energy diagram for formation of ortho and meta dihydropyridazine products in the reaction of Re-Tz with St.
We also studied the possibility of a retro-[4 + 2] cycloaddition occurring with the N2 that is coordinated to the Re center. For the ortho pathway, the transition state for this reaction was at 22.3 kcal/mol, which is significantly higher than the 9.2 kcal/mol that is observed for retrocycloaddition to expel the distal N2. This is reflected in the greater stability and entropy of product Int-2a compared to that which would be formed in the case of the other retro-[4 + 2] pathway.
Conclusion
With the considerable increase in Diels–Alder reactivity, the rhenium-mediated tetrazine ligation adds another interesting tool to the click chemistry toolbox. Using energy decomposition methods, we show that this higher reactivity is mainly due to a lowered LUMO of the tetrazine and therefore increased orbital interactions. These results also suggest that frontier molecular orbital interactions are a good indicator of cycloaddition reactivity of such tetrazine ligands in metal complexes. The high regioselectivity of the reaction with styrene is explained through a better stabilization of charges at the ipso-carbon of the monosubstituted tetrazine.
Acknowledgments
The authors are grateful to the National Science Foundation (Grant No. CHE-1764328). A.T. acknowledges the support of the National Institutes of Health under Ruth L. Kirschstein National Research Service Award F32GM134709. D.S. is grateful to the Austrian Science Funds (FWF, Grant No. J4216-N28) and the city of Vienna (H-331849/2018) for financial support. Calculations were performed on the Hoffman2 cluster at the University of California, Los Angeles, and the Vienna Scientific Cluster.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01564.
The authors declare no competing financial interest.
Supplementary Material
References
- Carboni R. A.; Lindsey R. V. Reactions of Tetrazines with Unsaturated Compounds. A New Synthesis of Pyridazines. J. Am. Chem. Soc. 1959, 81 (16), 4342–4346. 10.1021/ja01525a060. [DOI] [Google Scholar]
- Sauer J.; Mielert A.; Lang D.; Peter D. Eine Studie der Diels-Alder-Reaktion, III: Umsetzungen von 1.2.4.5-Tetrazinen mit Olefinen. Zur Struktur von Dihydropyridazinen. Chem. Ber. 1965, 98 (5), 1435–1445. 10.1002/cber.19650980514. [DOI] [Google Scholar]
- Thalhammer F.; Wallfahrer U.; Sauer J. Reaktivität einfacher offenkettiger und cyclischer dienophile bei Diels-Alder-reaktionen mit inversem elektronenbedarf. Tetrahedron Lett. 1990, 31 (47), 6851–6854. 10.1016/S0040-4039(00)97188-0. [DOI] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 2008, 130 (41), 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R.; Hilderbrand S. A. Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjugate Chem. 2008, 19 (12), 2297–2299. 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen E. J. L.; Jorgensen J. T.; Denk C.; Battisti U. M.; Norregaard K.; Edem P. E.; Bratteby K.; Shalgunov V.; Wilkovitsch M.; Svatunek D.; Poulie C. B. M.; Hvass L.; Simon M.; Wanek T.; Rossin R.; Robillard M.; Kristensen J. L.; Mikula H.; Kjaer A.; Herth M. M. Lipophilicity and Click Reactivity Determine the Performance of Bioorthogonal Tetrazine Tools in Pretargeted In Vivo Chemistry. ACS Pharmacol. Transl. Sci. 2021, 4 (2), 824–833. 10.1021/acsptsci.1c00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Svatunek D.; Rohlfing K.; Liu Y.; Wang H.; Giglio B.; Yuan H.; Wu Z.; Li Z.; Fox J. Conformationally Strained trans-Cyclooctene (sTCO) Enables the Rapid Construction of (18)F-PET Probes via Tetrazine Ligation. Theranostics 2016, 6 (6), 887–895. 10.7150/thno.14742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj R.; Giglio B.; Liu S.; Wang H.; Wang M.; Yuan H.; Chintala S. R.; Yap L. P.; Conti P. S.; Fox J. M.; Li Z. Improved metabolic stability for 18F PET probes rapidly constructed via tetrazine trans-cyclooctene ligation. Bioconjugate Chem. 2015, 26 (3), 435–442. 10.1021/acs.bioconjchem.5b00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Moore A. C.; Zerdoum A. B.; Zhang H.; Scinto S. L.; Zhang H.; Gong L.; Burris D. L.; Rajasekaran A. K.; Fox J. M.; Jia X. Cellular interactions with hydrogel microfibers synthesized via interfacial tetrazine ligation. Biomaterials 2018, 180, 24–35. 10.1016/j.biomaterials.2018.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo R.; Santos T.; Rivero D. S.; Perez-Perez Y.; Martin-Encinas E.; Pasan J.; Daranas A. H. Dynamic Nucleophilic Aromatic Substitution of Tetrazines. Angew. Chem., Int. Ed. 2021, 60 (34), 18783–18791. 10.1002/anie.202106230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q. H.; Zhou J.; Mao H.; Qiu Y.; Nguyen M. T.; Feng Y.; Liang J.; Shen D.; Li P.; Liu Z.; Wasielewski M. R.; Stoddart J. F. TetrazineBox: A Structurally Transformative Toolbox. J. Am. Chem. Soc. 2020, 142 (11), 5419–5428. 10.1021/jacs.0c01114. [DOI] [PubMed] [Google Scholar]
- Srinivasan S.; Yee N. A.; Wu K.; Zakharian M.; Mahmoodi A.; Royzen M.; Mejia Oneto J. M. SQ3370 Activates Cytotoxic Drug via Click Chemistry at Tumor and Elicits Sustained Responses in Injected & Non-injected Lesions. Adv. Ther. 2021, 4 (3), 2000243. 10.1002/adtp.202000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phase 1 Study of SQ3370 in Patients With Advanced Solid Tumors; https://clinicaltrials.gov/ct2/show/NCT04106492 (accessed 2021-07-06).
- Johann K.; Svatunek D.; Seidl C.; Rizzelli S.; Bauer T. A.; Braun L.; Koynov K.; Mikula H.; Barz M. Tetrazine- and trans-cyclooctene-functionalised polypept(o)ides for fast bioorthogonal tetrazine ligation. Polym. Chem. 2020, 11 (27), 4396–4407. 10.1039/D0PY00375A. [DOI] [Google Scholar]
- Denk C.; Wilkovitsch M.; Aneheim E.; Herth M. M.; Jensen H.; Lindegren S.; Mikula H. Multifunctional Clickable Reagents for Rapid Bioorthogonal Astatination and Radio-Crosslinking. ChemPlusChem 2019, 84 (7), 775–778. 10.1002/cplu.201900114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Devaraj N. K. Advances in Tetrazine Bioorthogonal Chemistry Driven by the Synthesis of Novel Tetrazines and Dienophiles. Acc. Chem. Res. 2018, 51 (5), 1249–1259. 10.1021/acs.accounts.8b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eising S.; Engwerda A. H. J.; Riedijk X.; Bickelhaupt F. M.; Bonger K. M. Highly Stable and Selective Tetrazines for the Coordination-Assisted Bioorthogonal Ligation with Vinylboronic Acids. Bioconjugate Chem. 2018, 29 (9), 3054–3059. 10.1021/acs.bioconjchem.8b00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svatunek D.; Denk C.; Mikula H. A computational model to predict the Diels-Alder reactivity of aryl/alkyl-substituted tetrazines. Monatsh. Chem. 2018, 149 (4), 833–837. 10.1007/s00706-017-2110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darko A.; Wallace S.; Dmitrenko O.; Machovina M. M.; Mehl R. A.; Chin J. W.; Fox J. M. Conformationally Strained trans-Cyclooctene with Improved Stability and Excellent Reactivity in Tetrazine Ligation. Chem. Sci. 2014, 5 (10), 3770–3776. 10.1039/C4SC01348D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulton K. G.; Beagan D. M.; Maciulis N. A.; Pink M.; Carta V.; Huerfano I. J.; Chen C. H. A Redox-active Tetrazine-based Pincer Ligand for the Reduction of N-oxyanions Using a Redox-inert Metal. Chem. - Eur. J. 2021, 27, 11676. 10.1002/chem.202101302. [DOI] [PubMed] [Google Scholar]
- Stetsiuk O.; Abherve A.; Avarvari N. 1,2,4,5-Tetrazine based ligands and complexes. Dalton Trans. 2020, 49 (18), 5759–5777. 10.1039/D0DT00827C. [DOI] [PubMed] [Google Scholar]
- Schnierle M.; Blickle S.; Filippou V.; Ringenberg M. R. Tetrazine metallation boosts rate and regioselectivity of inverse electron demand Diels-Alder (iEDDA) addition of dienophiles. Chem. Commun. 2020, 56 (80), 12033–12036. 10.1039/D0CC03805A. [DOI] [PubMed] [Google Scholar]
- Tang T. S.; Liu H. W.; Lo K. K. Monochromophoric iridium(iii) pyridyl-tetrazine complexes as a unique design strategy for bioorthogonal probes with luminogenic behavior. Chem. Commun. 2017, 53 (23), 3299–3302. 10.1039/C7CC00427C. [DOI] [PubMed] [Google Scholar]
- Capper M. S.; Packman H.; Rehkamper M. Rhenium-Based Complexes and in Vivo Testing: A Brief History. ChemBioChem 2020, 21 (15), 2111–2115. 10.1002/cbic.202000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer E. B.; Haase A. A.; Reich R. M.; Crans D. C.; Kühn F. E. Organometallic and coordination rhenium compounds and their potential in cancer therapy. Coord. Chem. Rev. 2019, 393, 79–117. 10.1016/j.ccr.2019.04.014. [DOI] [Google Scholar]
- Lo K. K.-W. Luminescent Rhenium(I) and Iridium(III) Polypyridine Complexes as Biological Probes, Imaging Reagents, and Photocytotoxic Agents. Acc. Chem. Res. 2015, 48 (12), 2985–2995. 10.1021/acs.accounts.5b00211. [DOI] [PubMed] [Google Scholar]
- Schnierle M.; Leimkuhler M.; Ringenberg M. R. [(eta(6)-p-Cymene)[3-(pyrid-2-yl)-1,2,4,5-tetrazine]chlororuthenium(II)](+), Redox Noninnocence and Dienophile Addition to Coordinated Tetrazine. Inorg. Chem. 2021, 60 (9), 6367–6374. 10.1021/acs.inorgchem.1c00094. [DOI] [PubMed] [Google Scholar]
- Turlik A.; Houk K. N.; Svatunek D.. Origin of Increased Reactivity in Rhenium-Mediated Cycloadditions of Tetrazines. ChemRxiv 2021, 10.33774/chemrxiv-2021-9s0r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, revision A.03; Gaussian Inc.: Wallingford, CT, 2016. [Google Scholar]
- Vosko S. H.; Wilk L.; Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58 (8), 1200–1211. 10.1139/p80-159. [DOI] [Google Scholar]
- Head-Gordon M.; Pople J. A.; Frisch M. J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153 (6), 503–506. 10.1016/0009-2614(88)85250-3. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37 (2), 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98 (45), 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Krishnan R.; Binkley J. S.; Seeger R.; Pople J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72 (1), 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113 (18), 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Luchini G.; Alegre-Requena J. V.; Funes-Ardoiz I.; Paton R. S. GoodVibes: automated thermochemistry for heterogeneous computational chemistry data. F1000Research 2020, 9, 291. 10.12688/f1000research.22758.1. [DOI] [Google Scholar]
- Bickelhaupt F. M.; Houk K. N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed. 2017, 56 (34), 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickelhaupt F. M.; Baerends E. J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. In Rev. Comput. Chem. 2007, 1–86. 10.1002/9780470125922.ch1. [DOI] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22 (9), 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Sun X.; Soini T. M.; Poater J.; Hamlin T. A.; Bickelhaupt F. M. PyFrag 2019-Automating the exploration and analysis of reaction mechanisms. J. Comput. Chem. 2019, 40 (25), 2227–2233. 10.1002/jcc.25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J. C. T.; Mikula H.; Weissleder R. Unraveling Tetrazine-Triggered Bioorthogonal Elimination Enables Chemical Tools for Ultrafast Release and Universal Cleavage. J. Am. Chem. Soc. 2018, 140 (10), 3603–3612. 10.1021/jacs.7b11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meresz O.; Foster-Verner P. A. Synthesis of 3-monosubstituted s-tetrazines and their reactions with monosubstituted acetylenes. J. Chem. Soc., Chem. Commun. 1972, (16), 950–951. 10.1039/c39720000950. [DOI] [Google Scholar]
- Boger D. L.; Schaum R. P.; Garbaccio R. M. Regioselective Inverse Electron Demand Diels-Alder Reactions of N-Acyl 6-Amino-3-(methylthio)-1,2,4,5-tetrazines. J. Org. Chem. 1998, 63 (18), 6329–6337. 10.1021/jo980795g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.