

Abstract
Post‐transplant lymphoproliferative disorders (PTLDs) are life‐threatening neoplasms after organ transplantation. Because of their rarity and multiple grades of malignancy, the incidence, outcomes, and clinicopathological features affecting patient survival after liver transplantation (LT) remain unclear. We reviewed 1954 LTs in 1849 recipients (1990‐2020), including 886 pediatric (<18 years of age) and 963 adult recipients. The following clinicopathological factors were studied: age, sex, liver etiologies, malignancy grades, Epstein‐Barr virus status, performance status (PS), Ann Arbor stage, international prognostic index, and histopathological diagnosis. Of 1849 recipients, 79 PTLD lesions (4.3%) were identified in 70 patients (3.8%). After excluding 3 autopsy cases incidentally found, 67 (45 pediatric [5.1%] and 22 adult [2.3%]) patients were finally enrolled. Comorbid PTLDs significantly worsened recipient survival compared with non‐complicated cases (P < 0.001). The 3‐year, 5‐year, and 10‐year overall survival rates after PTLD diagnosis were 74%, 66%, and 58%, respectively. The incidence of PTLDs after LT (LT‐PTLDs) was significantly higher (P < 0.001) with earlier onset (P = 0.002) in children, whereas patient survival was significantly worse in adults (P = 0.002). Univariate and multivariate analyses identified the following 3 prognostic factors: age at PTLD diagnosis ≥18 years (hazard ratio [HR], 11.2; 95% confidence interval [CI], 2.63‐47.4; P = 0.001), PS ≥2 at diagnosis (HR, 6.77; 95% CI, 1.56‐29.3; P = 0.01), and monomorphic type (HR, 6.78; 95% CI, 1.40‐32.9; P = 0.02). A prognostic index, the “LT‐PTLD score,” that consists of these 3 factors effectively stratified patient survival and progression‐free survival (P = 0.003 and <0.001, respectively). In conclusion, comorbid PTLDs significantly worsened patient survival after LT. Age ≥18 years and PS ≥2 at PTLD diagnosis, and monomorphic type are independent prognostic factors, and the LT‐PTLD score that consists of these 3 factors may distinguish high‐risk cases and guide adequate interventions.
Abbreviations
- ALF
acute liver failure
- ANOVA
analysis of variance
- BA
biliary atresia
- BM
bone marrow
- CD
clusters of differentiation
- CI
confidence interval
- CIT
cold ischemic time
- CNS
central nervous system
- CT
computed tomography
- CMV
cytomegalovirus
- CNI
calcineurin inhibitor
- DLBCL
diffuse large B cell lymphoma
- EBER
Epstein‐Barr virus–encoded RNA
- EBV
Epstein‐Barr virus
- ECOG‐PS
Eastern Cooperative Oncology Group performance status
- FFH
florid follicular hyperplasia
- GRWR
graft‐to‐recipient weight ratio
- HCV
hepatitis C virus
- HR
hazard ratio
- IPI
International Prognostic Index
- IQR
interquartile range
- KT
kidney transplantation
- LDH
lactate dehydrogenase
- LDLT
living donor liver transplantation
- LT
liver transplantation
- LT‐PTLD
post‐transplant lymphoproliferative disorder after liver transplantation
- MELD
Model for End‐Stage Liver Disease
- NA
not applicable
- OS
overall survival
- PCR
polymerase chain reaction
- PELD
Pediatric End‐Stage Liver Disease
- PET
positron emission tomography
- PFS
progression‐free survival
- PH
plasmacytic hyperplasia
- PS
performance status
- PSC
primary sclerosing cholangitis
- PTLD
post‐transplant lymphoproliferative disorder
- sIL2R
soluble interleukin 2 receptor
- SOT
solid organ transplantation
- WIT
warm ischemic time
- WHO
World Health Organization
Post‐transplant lymphoproliferative disorders (PTLDs) are one of the most common malignancies after solid organ transplantation (SOT)( 1, 2, 3, 4, 5 ) and remain life‐threatening with 5‐year overall survival (OS) rates ranging 40% to 70%.( 6 ) These high mortalities and morbidities have highlighted the need to clarify the prognostic factors in PTLDs.
However, PTLDs have two difficult burdens to be investigated: rarity and heterogeneity. PTLDs develop only in transplant recipients, and their incidence rates have been reported to be 1.0% to 5.5% after liver transplantation (LT), 0.8% to 2.5% after kidney transplantation (KT), 0.5% to 5.0% for pancreas transplantation, 2.0% to 8.0% for heart transplantation, 3.0% to 10.0% for lung transplantation, and ≤ 20% for multiorgan/intestinal transplantation in adults.( 2, 7, 8 ) Because the numbers of PTLDs in each center are limited, previous studies were mostly conducted using relatively large post‐KT recipients or heterogeneous cohorts including various SOTs.( 6, 9, 10, 11, 12, 13, 14 ) PTLDs inherently have a wide range of clinicopathological characteristics, from indolent lymphoproliferation requiring only immunosuppressant modifications to malignant lymphomas that need chemotherapies.( 2 ) Moreover, their characteristics are reportedly different between adults and children.( 2, 15 ) Collectively, there is no widely accepted consensus on PTLDs so far, especially after LT (LT‐PTLDs).
This study thus aimed to clarify the clinicopathological features of LT‐PTLDs, identify the prognostic/risk factors therein, and compare their characteristics between pediatric and adult LT‐PTLDs by reviewing our relatively large cohort of almost 2,000 LTs.
Patients and Methods
Patients
We performed a total of 1954 LTs in 1849 recipients at our single center between June 1990 and March 2020, including 937 pediatric (<18 years of age) and 1017 adult (≥18 years of age) LTs in 886 children and 963 adult recipients, respectively. A total of 1874 of 1954 LTs were living donor LTs (LDLTs), whereas 80 were deceased donor LTs. Of 1849 recipients, 79 PTLD lesions (4.3%) were identified in 70 patients (3.8%). All lesions developed after LDLT. Of these, 9 metachronous lesions in the same patients and 3 autopsy cases incidentally found were excluded to identify prognostic factors in LT‐PTLDs. Thus, 67 (45 children [5.1%] and 22 adults [2.3%]) patients were finally enrolled (Supporting Fig. 1). Written informed consent was obtained from each patient or his/her parents. This study was approved by the Ethics Committee of Kyoto University (R1473) and was conducted in accordance with the institutional guidelines as well as the ethical guidelines mandated by the Declaration of Helsinki (2013).
Peritransplant Management
The selection criteria for donors and recipients, perioperative management, surgical procedures, and immunosuppression regimens are detailed elsewhere.( 16, 17, 18, 19, 20, 21 ) Briefly, the lower limit of the graft‐to‐recipient weight ratio (GRWR) in adult‐to‐adult LDLTs are as follows: ≥0.8% until November 2007, ≥0.7% from December 2007 until March 2009, and ≥0.6% from April 2009.( 20, 21 ) For biliary reconstruction, choledocho‐choledochostomy was our priority in adult LT. Modulations of portal‐venous pressure, such as splenectomy,( 20 ) was performed to keep 15 mm Hg or less at the end of surgery if needed.( 21 ) Recipients were postoperatively managed in intensive‐care/high‐care units during the first several days. Blood cell counts, biochemical and coagulation examinations, and Doppler ultrasonography were performed daily until stabilized. Tacrolimus or cyclosporine and steroids have been used since 1990. In the early period (1990‐2005), azathioprine or muromonab‐CD3 (OKT3) was given for acute rejection. Cyclophosphamide was added in recipients undergoing ABO‐incompatible LDLTs. In the late period (2006‐2020), however, these 3 drugs were all discontinued. The combination of tacrolimus, mycophenolate mofetil, and steroids became the standard regimen in the past 15 years. Recently, everolimus was added to reduce the trough level of tacrolimus, if necessary. In ABO blood‐type incompatible or donor‐specific antibody‐positive cases, recipients were preoperatively treated with anti‐CD20 monoclonal antibody (rituximab, 375 mg/m2) and plasma exchange to prevent antibody‐mediated rejection.( 22 ) Acute cellular and antibody‐mediated rejections were diagnosed according to the Banff criteria.( 23, 24 )
For pediatric LDLTs, briefly, the upper limit of GRWR was 4.0%. If the estimated GRWR exceeded 4.0%, a reduced, hyper‐reduced, or S2‐monosegment graft was selected.( 25 ) For biliary reconstruction, choledocho‐jejunostomy was mostly adopted because many patients underwent Kasai’s operation for biliary atresia (BA). A standard immunosuppression protocol consisting of tacrolimus and steroids was used. In ABO‐incompatible cases, recipients were pretreated with rituximab (≥2 years of age) or considered individually (1‐2 years of age). Exchange transfusion or plasma exchange was performed as needed.
Diagnosis of PTLDs
PTLDs were all histologically diagnosed by expert pathologists with excisional biopsies except for a case with needle biopsy. PTLDs were classified according to the World Health Organization (WHO) classification revised in 2017.( 26 ) After confirming histopathological diagnosis, patients underwent staging workup, including whole‐body computed tomography (CT), bone marrow (BM) biopsy/aspiration, [18F]‐fluorodeoxyglucose positron emission tomography (PET)/CT, and cerebrospinal fluid test to check central nervous system (CNS) invasion. Serological tests for Epstein‐Barr virus (EBV) and cytomegalovirus (CMV) infection were conducted preoperatively. Tumor EBV positivity was determined by in situ hybridization assays for EBV‐encoded RNA (EBER).( 27, 28 ) Serological status was confirmed by polymerase chain reaction (PCR) for the quantification of the EBV viral load.( 27, 28 )
PTLD‐Like Lesions
In this study, we defined “PTLD‐like lesions” as any post‐transplant lymphoproliferative lesion that showed clinical manifestations but did not fulfill the PTLD criteria according to the WHO classification.( 26 ) The following were included as PTLD‐like lesions: indolent small B cell lymphomas, including follicular lymphoma or marginal zone lymphoma( 29 ); EBV‐negative reactive lymphadenopathy; EBV‐positive mucocutaneous ulcer( 30 ); hairy cell leukemia( 31, 32 ); and EBV‐associated pleural effusion/ascites without malignant cells (Supporting Table 1). Although excluded from a strict definition of PTLDs,( 26 ) the clinical presentations of PTLD‐like lesions were similar to those of PTLDs, and patients with these lesions often required immunosuppressant modifications or chemotherapies that could affect patient prognosis.
EBV Monitoring
EBV status in both donors and recipients was determined serologically before LT. In pediatric recipients, EBV PCR was measured every month for the first 6 months regardless of EBV seropositivity. During the past decade, EBV PCR was performed every week before discharge after transplant, followed by biweekly to monthly after discharge for the first 6 months to detect not only EBV primary infection in naïve recipients but also EBV reactivation in seropositive patients as early as possible. If the result remained negative, intervals between EBV PCRs were prolonged. High‐risk recipients (recipients who are EBV naïve receive EBV‐infected donor livers) were more carefully followed by checking clinical symptoms (lymphadenopathy, fever, or hepatitis) and EBV PCR. In adults, EBV PCR was performed when recipients had some symptoms of EBV infection or unexplained fever.
Monitosring and Treatment of PTLDs
Recipients were usually followed up once a month for the first 6 months, every 2 months from 6 to 12 months, and every 3 months thereafter if the postoperative course was uneventful. Patients with high EBV viral load, unexplained fever, or lymphadenopathy underwent thorough examinations.
In patients with LT‐PTLDs, first of all, we considered to modify immunosuppressants, that is, cessation, reduction, or switching of calcineurin inhibitors (CNIs). In pediatric patients, we conducted first‐line chemotherapy according to the recommendation from the Japanese Pediatric Leukemia/Lymphoma Study Group( 33 ) since 2007 in combination with rituximab. For nonresponders to the first line, we used a more intensified second‐line chemotherapy, incorporating cisplatin (DECAL: dexamethasone, etoposide, cisplatin, cytarabine, l‐asparaginase) or high‐dose cytarabine.( 33 ) In adults, R‐CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone) have long been the mainstay, and dose‐adjusted (DA)‐EPOCH‐R (etoposide, vincristine, doxorubicin, cyclophosphamide, prednisolone, and rituximab) have recently been applied in high‐risk PTLDs. Surgical resections were performed for localized, perforated, and obstructed intestinal lesions. Radiation therapy was indicated in patients not eligible for chemotherapies, nonresponders to chemotherapies, or those with CNS involvement. Restaging CT scans were performed timely, and the therapeutic effects were assessed according to the Response Evaluation Criteria in Solid Tumors (RECIST) guidelines.( 34 )
Variables
The preoperative clinical variables included recipient/donor age at LT, sex, underlying liver etiologies, malignancy grades, ABO blood‐type compatibility, Pediatric End‐Stage Liver Disease (PELD) or Model for End‐Stage Liver Disease (MELD) scores, and pretransplant EBV/CMV serological status.
The operative variables included graft type (left or right lobe), GRWR, operation time, intraoperative blood loss, cold ischemic time (CIT), and warm ischemic time (WIT). As listed in Table 1, PTLD‐related variables are as follows: recipient age at PTLD diagnosis, serological and histopathological positivity of EBV, Eastern Cooperative Oncology Group performance status (ECOG‐PS),( 35 ) Ann Arbor stage,( 36 ) tumor size, presence/absence of extranodal lesions, soluble interleukin 2 receptor (sIL2R), International Prognostic Index (IPI),( 37 ) intervals between LT and PTLD diagnosis, histological classifications, and so on.
TABLE 1.
Patient Characteristics, Perioperative Variables, and PTLD‐Related Variables
| Characteristics | All (n = 67) | Pediatric (n = 43) | Adult (n = 24) | P Value |
|---|---|---|---|---|
| Male/female | 31/36 | 19/24 | 12/12 | 0.65 |
| Age at LT, years | 2.1 (0.9‐45.2) | 1.2 (0.6‐2.0) | 54.8 (31.5‐61.1) | <0.001 |
| Age at PTLDs, years | 6.1 (1.9‐53.4) | 2.8 (1.6‐4.2) | 60.0 (38.6‐66.3) | <0.001 |
| BA/metabolic/ALF/HCV/others | 38/5/6/8/10 | 35/3/2/0/3 | 3/2/4/8/7 | <0.001 |
| Malignant/benign liver etiology | 7/60 | 1/42 | 6/18 | 0.004 |
| ABO incompatible/not | 11/56 | 9/34 | 2/18 | 0.26 |
| EBV serology: +/− at LT | 39/10 | 19/10 | 20/0 | <0.001 |
| CMV serology: +/− at LT | 37/11 | 20/9 | 17/2 | 0.09 |
| EBV PCR: +/− | 31/9 | 25/1 | 6/8 | <0.001 |
| EBER: +/− | 35/24 | 30/9 | 5/15 | <0.001 |
| PS: 0/1/2/3/4 | 2/46/13/4/2 | 0/28/10/4/1 | 2/18/3/0/1 | 0.06 |
| Ann Arbor stage: I/II/III/IV | 35/9/10/12 | 22/4/8/8 | 13/5/2/4 | 0.44 |
| Bulky tumor (>5 cm)/not | 7/58 | 3/38 | 4/20 | 0.25 |
| Extranodal lesion ≥ 1/not | 28/39 | 13/30 | 15/9 | 0.01 |
| LDH at PTLD, U/L | 372 (258‐524) | 375 (262‐525) | 346 (244‐523) | 0.70 |
| sIL2R at PTLD, U/mL | 2480 (1605‐4225) | 2660 (1433‐4390) | 2420 (1710‐4150) | 0.96 |
| IPI: 0/1/2/3/4/5 | 7/21/16/8/5 | 5/13/9/5/2 | 2/8/7/3/3 | 0.85 |
| Tacrolimus/cyclosporine | 57/2 | 36/0 | 21/2 | 0.049 |
| Monomorphic (DLBCL/Burkitt)/polymorphic/nondestructive (PH/infectious mononucleosis/FFH)/PTLD‐like | 24 (13/7)/5/21 (5/14/2)/17 | 12 (6/4)/2/20 (5/13/2)/9 | 12 (7/3)/3/1 (0/1/0)/8 | 0.005 |
| Chemotherapy/not | 23/39 | 11/28 | 12/11 | 0.16 |
The data are presented as median (interquartile range) for continuous variables and number for categorical variables. P values < 0.05 are bold.
Statistical Analysis
Data are expressed as median with interquartile range (IQR) for continuous variables and counts for categorical variables. Comparisons of continuous variables and categorical variables were performed using Mann‐Whitney U tests or chi‐square tests as appropriate. In cases with multiple LTs, the intervals from the first transplant to PTLD diagnosis were adopted to account for the duration of immunosuppressants exposure. Prognostic factors for LT‐PTLDs were analyzed using univariate and multivariate Cox regression analyses. Patient overall survival (OS) and progression‐free survival (PFS) were counted from the date of PTLD diagnosis to the patient’s death or the last follow‐up (OS) and to death or disease relapse/progression (PFS), respectively. These survivals were estimated by the Kaplan‐Meier curve method, followed by log‐rank tests. All analyses were 2‐sided, and P < 0.05 was considered statistically significant. Variables with P < 0.10 in the univariable analysis were included in the multivariable analysis. All statistical analyses were performed using JMP Pro14 (SAS Institute Inc., Cary, NC).
Results
Incidence, Timing, and Treatments of LT‐PTLDs
Overall, the incidence rates of pediatric PTLDs were significantly higher than in adults (n = 45 [5.1%] versus n = 22 [2.3%]; P < 0.001). The intervals between LT and PTLDs diagnosis varied widely, from 19 days to 24.5 years (median, 23 months; IQR, 5‐53); however, PTLD onsets were significantly earlier in children than in adults (14 [IQR, 4‐32] versus 57 [IQR, 25‐111] months; P = 0.002). Notably, almost half of the pediatric PTLDs (49%) developed within a year after LTs (Fig. 1A). Two pediatric recipients (<18 years of age at LT) developed PTLDs in adulthood (≥18 years of age at LT); therefore, a total of 43 pediatric and 24 adult PTLDs were identified. The most common treatment was immunosuppressant modifications (n = 27 [40%]) followed by chemotherapies (n = 23 [34%]).
FIG. 1.
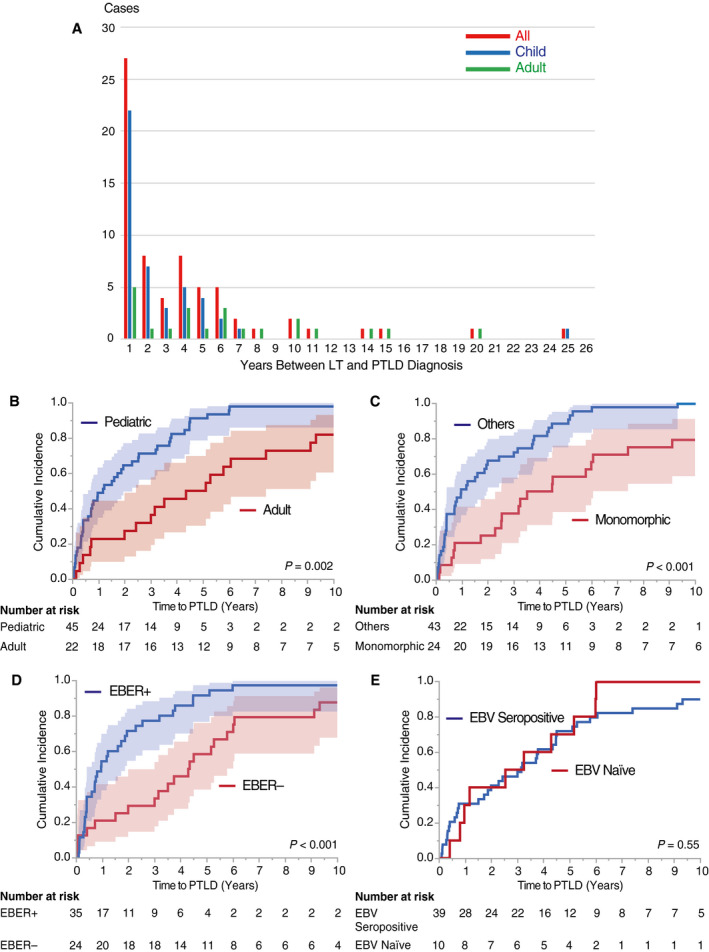
Time between LT and PTLD diagnosis. (A) Annual incidence of LT‐PTLDs. Annual incidence of LT‐PTLDs was counted in the overall, pediatric (<18 years of age at LT), and adult cohorts (≥18 years of age at LT) separately (n = 67, 45, 22, respectively). Although LT‐PTLDs occurred at any time from early to late time periods after LT, it is noteworthy that almost half (49%) of the pediatric LT‐PTLDs developed within the first year after LT. (B) Cumulative incidence of LT‐PTLDs: pediatric versus adult recipients. Pediatric LT‐PTLDs developed significantly earlier than the adult cases (P = 0.002 by a log‐rank test). The shaded areas show 95% CI hereafter unless otherwise indicated. (C) Cumulative incidence of LT‐PTLDs: monomorphic versus others. Monomorphic PTLDs developed significantly later than other types of LT‐PTLDs (P < 0.001). (D) Cumulative incidence of LT‐PTLDs: EBER+ versus EBER−. LT‐PTLDs with positive EBER developed significantly earlier than those without (P < 0.001). (E) Cumulative incidence of LT‐PTLDs: EBV seropositive versus EBV naïve. The curves for both cumulative incidence rates almost matched each other, indicating no significant difference regarding the timing of PTLD occurrence between recipients who were EBV seropositive and recipients who were EBV naïve (P = 0.55).
Patient Characteristics and Clinicopathological Variables
Patient characteristics, perioperative variables, and PTLD‐related variables are summarized in Table 1. The most common etiology was BA in 38 (57%), followed by hepatitis C virus (HCV) in 8 (12%) cases. Regarding blood‐type combinations, 11 cases (16%) were ABO‐incompatible, and the remaining 56 (84%) were identical or compatible. Pretransplant serological statuses for EBV and CMV were positive in 39 (58%) and 37 (55%) patients, respectively. The median age at PTLD diagnosis was 6.1 years (IQR, 1.9‐53.4). EBV PCR (blood) and histopathological EBV statuses were positive in 31 (46%) and 35 (52%), respectively. ECOG‐PS was 0‐1 in 48 (72%) and 2‐4 in 19 (28%) patients. Ann Arbor stage was 1‐2 in 44 (65%) and 3‐4 in 22 (33%) patients. Extranodal lesions were found in 28 (42%) patients. Lactate dehydrogenase (LDH) and sIL2R at PTLD diagnosis were 372 U/L (IQR, 258‐524) and 2480 U/mL (IQR, 1605‐4225), respectively. IPI was 0‐2 in 44 (65%) and 3‐4 in 13 (19%) patients. Histopathologically, monomorphic type was the most common (n = 24 [36%]), followed by PTLD‐like lesions (n = 17 [25%]) and infectious mononucleosis type (n = 14 [21%]). Monomorphic type included diffuse large B cell lymphoma (DLBCL; n = 13), Burkitt lymphoma (n = 7), and T cell neoplasms (n = 2).
Histopathological Types and Time to PTLDs
The median follow‐up period was 12.3 (range, 0.2‐29.2) years. Notably, non‐monomorphic PTLDs developed significantly earlier than monomorphic types (P < 0.001; Fig. 1C). Furthermore, EBV‐positive PTLD lesions developed significantly earlier than EBV‐negative PTLD lesions (P < 0.001; Fig. 1D), whereas there was no significant association between serological EBV positivity and the timing of PTLD onset (P = 0.55; Fig. 1E). These trends were observed in both pediatric and adult patients (Supporting Fig. 2).
Historical Transition of LT‐PTLD Incidence and Tacrolimus Trough Level
Then we compared the incidence of LT‐PTLDs between the early (1990‐2005) and the late (2006‐2020) periods. Although patient OS with LT‐PTLDs was not significantly different between the 2 periods in both children and adults (data not shown), the incidence of pediatric LT‐PTLDs decreased in the late period (P = 0.06; Fig. 2A,B). As a possible reason for this, the trough level of tacrolimus in pediatric patients with LT‐PTLDs was significantly higher in the early period compared with the late period (P < 0.001; Fig. 2C), whereas no significant differences were observed in adults between the 2 eras (Fig. 3).
FIG. 2.

Historical transition of the incidence rate of pediatric LT‐PTLDs. (A) Annual incidence rate of monomorphic and non‐monomorphic LT‐PTLDs in children (<18 years of age), given as the number of LT‐PTLDs occurrence/number of LTs per year, was investigated in the early (1990‐2005) and late periods (2006‐2020). (B) Cumulative incidence of pediatric LT‐PTLDs: early period versus late period. The incidence of pediatric LT‐PTLDs tended to decrease in the late period compared with those in the early period (P = 0.06 by a log‐rank test). (C) Historical transition of tacrolimus trough level: early period versus late period. The trough level of tacrolimus in pediatric patients with LT‐PTLDs was significantly higher in the early period compared with the late period (P < 0.001 by a 2‐way ANOVA).
FIG. 3.
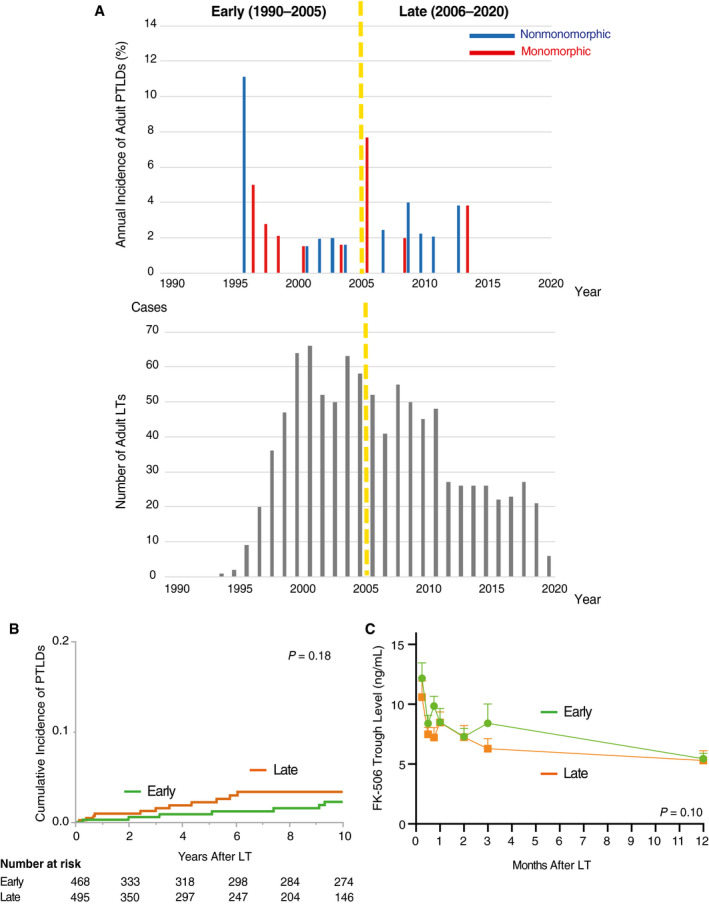
Historical transition of the incidence rate of adult LT‐PTLDs. (A) Annual incidence rate of monomorphic and non‐monomorphic LT‐PTLDs in adults (≥18 years of age), given as the number of LT‐PTLDs occurrence/number of LTs per year, was investigated in the early (1990‐2005) and late periods (2006‐2020). (B) Cumulative incidence of adult LT‐PTLDs: early period versus late period. In contrast to pediatric LT‐PTLDs, the incidence of adult LT‐PTLDs showed no significant differences in adults between the 2 eras (P = 0.18 by a log‐rank test). (C) Historical transition of tacrolimus trough level: early period versus late period. The trough level of tacrolimus in adult patients with LT‐PTLDs was not different between the 2 eras (P = 0.10 by a 2‐way ANOVA).
Patient Survival and Cause of Death
As shown in Fig. 4A‐C, comorbid PTLDs in the whole, pediatric, and adult cohorts significantly worsened OS after LT compared with non‐complicated cases (P < 0.001, P < 0.001, and P = 0.005, respectively). The 3‐year, 5‐year, and 10‐year OS rates in the whole, pediatric, and adult cohorts after PTLD diagnosis were 74%, 66%, and 58%; 81%, 79%, and 71%; and 61%, 38%, and 28%; respectively (Fig. 4D,E). Although the incidence of LT‐PTLDs was significantly lower in adults (P < 0.001), patient survival was significantly worse in adults than in pediatric patients (P = 0.002; Fig. 4E,F).
FIG. 4.
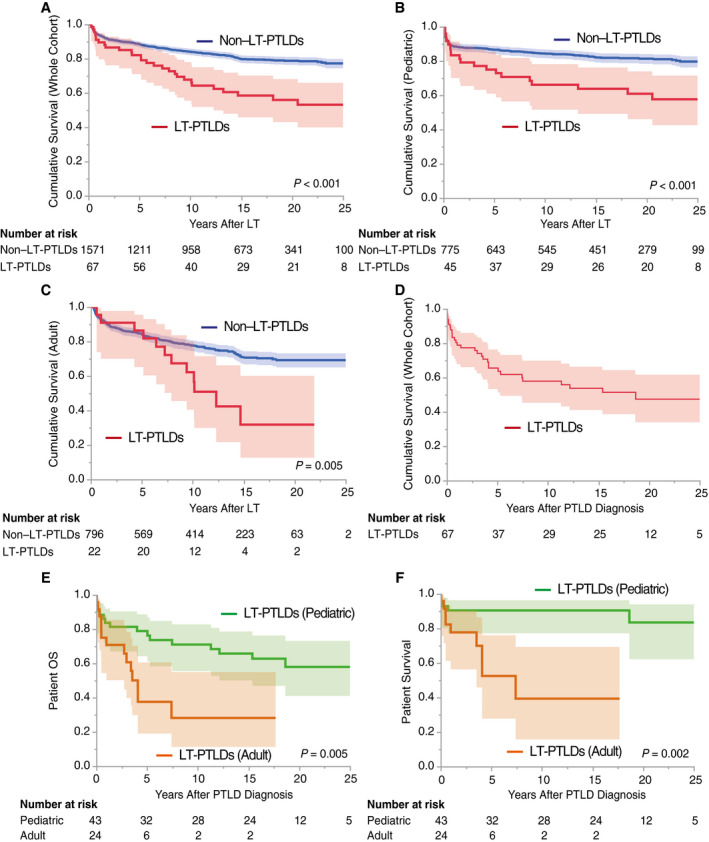
Patient survival after LT and PTLD diagnosis. The recipients who died within 3 months were excluded to eliminate the influence of early mortality from other causes, that is, the severe infections, refractory rejections, or intracranial bleeding in Fig. 2A‐C. The shaded areas show 95% CI. (A) Overall recipient survival: LT‐PTLDs versus non–LT‐PTLDs. Comorbid LT‐PTLDs significantly worsened recipient survival compared with those without (P < 0.001 by a log‐rank test). (B) Pediatric recipient survival: LT‐PTLDs versus non–LT‐PTLDs. Similarly, comorbid LT‐PTLDs in the pediatric cohort (<18 years of age) significantly worsened recipient survival compared with those without (P < 0.001). (C) Adult recipient survival: LT‐PTLDs versus non–LT‐PTLDs. In the adult cohort, comorbid PTLDs significantly worsened recipient survival compared with those without (P = 0.005). (D) Patient OS in LT‐PTLDs. The overall 3‐year, 5‐year, and 10‐year patient survival rates after LT‐PTLD diagnosis were 74%, 66%, and 58%, respectively. (E) Patient OS in LT‐PTLDs: pediatric versus adult cases. The 3‐year, 5‐year, and 10‐year pediatric patient survival rates after LT‐PTLD diagnosis were 81%, 79%, and 71%, respectively, whereas those of adults were 61%, 38%, and 28%, respectively. Pediatric LT‐PTLDs showed significantly better patient survival than adult PTLDs (P = 0.005). (F) Patient survival in LT‐PTLDs: pediatric versus adult cases. Patient deaths not related to PTLD were treated as "censored." LT‐PTLD–associated mortality was significantly lower in pediatric LT‐PTLDs than in adult LT‐PTLDs (P = 0.002).
Overall, 30 patients (44.8%) died in the present series. Tumor progression was the leading cause of death (14 patients [47%]), followed by graft failure (7 patients [23.3%]), sepsis (2 patients [6.7%]), and cerebral hemorrhage (1 patients [3.3%]; Supporting Table 3).
Prognostic Factors in LT‐PTLDs
PTLD‐related mortality included deaths from tumor progression and treatment toxicities only. Univariate Cox regression analysis revealed that age at PTLD diagnosis ≥18 years (hazard ratio [HR], 5.69; 95% confidence interval [CI], 1.70‐19.0; P = 0.005), non‐BA (HR, 3.60; 95% CI, 1.17‐11.1; P = 0.03), PS ≥2 at PTLD diagnosis (HR, 3.21; 95% CI, 1.12‐9.17; P = 0.03), presence of extranodal lesions at diagnosis (HR, 5.00; 95% CI, 1.53‐16.3; P = 0.008), and monomorphic PTLDs (HR, 6.43; 95% CI, 1.98‐20.8; P = 0.002) were significant prognostic factors in LT‐PTLDs.
To eliminate confounding bias, “non‐BA” was excluded because it had a strong correlation with “age at PTLD diagnosis.” As summarized in Table 2, age at PTLD diagnosis ≥18 years (HR, 11.2; 95% CI, 2.63‐47.4; P = 0.001), PS ≥2 at PTLD diagnosis (HR, 6.77; 95% CI, 1.56‐29.3; P = 0.01), and monomorphic type (HR, 6.78; 95% CI, 1.40‐32.9; P = 0.02) were identified as independent prognostic factors in LT‐PTLDs.
TABLE 2.
Univariate and Multivariate Analyses of Clinical Factors Affecting Patient Survival With LT‐PTLDs
| Variables | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Value | HR | 95% CI | P Value | |
| Male sex | 0.84 | 0.29‐2.42 | 0.75 | |||
| Age at PTLDs ≥18 years | 5.69 | 1.70‐19.0 | 0.005 | 11.2 | 2.63‐47.4 | 0.001 |
| Primary disease: non‐BA | 3.60 | 1.17‐11.1 | 0.03 | |||
| ABO incompatible | 0.37 | 0.05‐2.82 | 0.38 | |||
| EBV naïve | NA* | NA* | NA* | |||
| Positive EBV PCR | 0.72 | 0.14‐3.76 | 0.70 | |||
| Positive EBER | 0.62 | 0.18‐2.17 | 0.46 | |||
| PS ≥2 | 3.21 | 1.12‐9.17 | 0.03 | 6.77 | 1.56‐29.3 | 0.01 |
| Ann Arbor stage ≥III | 0.88 | 0.27‐2.86 | 0.83 | |||
| Bulky tumor ≥5 cm | 2.11 | 0.46‐9.80 | 0.34 | |||
| Extranodal lesion ≥1 | 5.00 | 1.53‐16.3 | 0.008 | 0.38 | 0.07‐2.20 | 0.28 |
| LDH elevation | 3.69 | 0.48‐28.7 | 0.21 | |||
| IPI ≥3 | 1.36 | 0.36‐5.16 | 0.65 | |||
| Time to PTLDs ≥1 year | 1.23 | 0.41‐3.69 | 0.72 | |||
| Monomorphic PTLDs | 6.43 | 1.98‐20.8 | 0.002 | 6.78 | 1.40‐32.9 | 0.02 |
P values < 0.05 are bold.
NA because no patients died as a result of PTLDs in either or both groups of the analyzed variable.
Stratification of Patient Prognosis by LT‐PTLD score
We developed the LT‐PTLD score, which consists of the 3 prognostic factors identified by the multivariate analysis, that is, (A) age at PTLD diagnosis >18 years, (B) PS ≥2, and (C) monomorphic PTLDs. According to the HR of each factor (A = 11.2, B = 6.77, and C = 6.78), we constructed a prognostic scoring with a weighting of 2 points for age at PTLD diagnosis >18 years and 1 point for PS ≥2 and monomorphic PTLDs. Patients with LT‐PTLDs were classified into 0 to 4 points, by which the patient prognosis was effectively stratified (Fig. 5A). The LT‐PTLD patients with point 0 showed 100% survival. When 0, 1 to 2, and 3 to 4 points are regarded as low, intermediate, and high risk for PTLD‐related deaths, respectively (Fig. 5B), the prognostic score significantly stratified the patient survival with LT‐PTLD–related mortality (Fig. 5C), patient OS (Fig. 5D), and PFS (Fig. 5E). As shown in Fig. 5C, LT‐PTLD–related mortality significantly worsened as the prognostic score increased (P = 0.003 in 0 versus 1‐2, P = 0.04 in 1‐2 versus 3‐4, and P < 0.001 in 0 versus 3‐4). These results demonstrated that the LT‐PTLD score allows more precise estimation of patient prognosis with LT‐PTLDs.
FIG. 5.
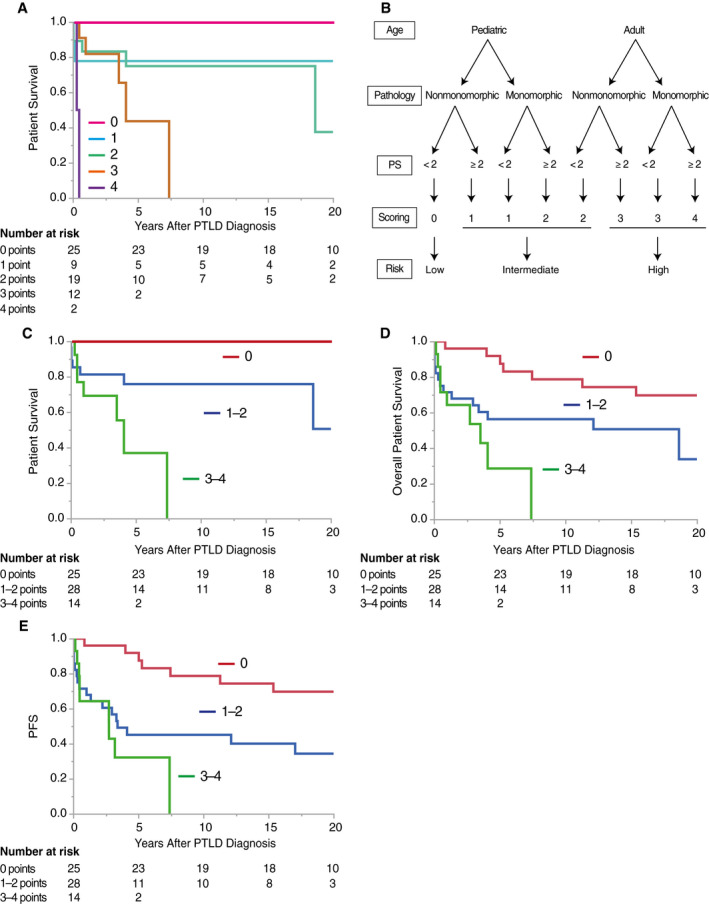
Significant stratification of patient prognosis by LT‐PTLD score. Patient survival and PFS proportions were analyzed according to LT‐PTLD score, which consists of the following 3 independent prognostic factors: age ≥18 years (2 points) and PS ≥2 (1 point) at LT‐PTLD diagnosis and monomorphic PTLDs (1 point). Patient deaths not related to PTLD were treated as “censored” in A and C. (A) Patients with LT‐PTLDs were classified into 0 to 4 points by the LT‐PTLD score, which effectively stratified the patient prognosis. As shown, LT‐PTLD patients with point 0 showed 100% survival. The higher the LT‐PTLD score, the worse the patient survival (P < 0.001 by a log‐rank test). (B) A flowchart illustrating the prognostic scoring system, by which 0, 1‐2, and 3‐4 points are regarded as low, intermediate, and high risk for PTLD‐related deaths, respectively. (C) LT‐PTLD score significantly stratified the patient prognosis. As shown, LT‐PTLD–related mortality significantly worsened as the score increased (P = 0.003 in 0 versus 1‐2, P = 0.04 in 1‐2 versus 3‐4, and P < 0.001 in 0 versus 3‐4). Similarly, (D) OS including other causes of death (P = 0.02 in 0 versus 1‐2, P = 0.17 in 1‐2 versus 3‐4, P < 0.001 in 0 versus 3‐4, P = 0.003 in 0 versus 1‐4) and (E) PFS (P = 0.003 in 0 versus 1‐2, P = 0.37 in 1‐2 versus 3‐4, P < 0.001 in 0 versus 3‐4, P < 0.001 in 0 versus 1‐4) were both significantly and effectively stratified by LT‐PTLD score. These results demonstrated that the LT‐PTLD score allows more precise estimation of patient prognosis with LT‐PTLDs.
Subgroup Analyses: Children Versus Adults
Pediatric and adult PTLDs were then separately analyzed. Patient characteristics and perioperative and PTLD‐related variables are summarized in Table 1. Similarly, PTLD‐associated mortality included deaths from tumor progression or treatment toxicities only. Univariate analysis demonstrated that PS ≥2 at PTLD diagnosis (HR, 9.64; 95% CI, 1.07‐86.8; P = 0.04) and monomorphic type (HR, 13.7; 95% CI, 1.51‐124.2; P = 0.02) were significant prognostic factors in pediatric PTLDs. Although statistically not significant, similar results were also obtained in adults (PS ≥2 at diagnosis: HR, 3.69 [95% CI, 0.89‐15.4], P = 0.07; monomorphic type: HR, 4.71 [95% CI, 0.87‐25.5], P = 0.07) (Table 3).
TABLE 3.
Univariate Analyses of Clinical Factors Affecting Pediatric and Adult Patient Survival With LT‐PTLDs
| Variables | Pediatric (n = 43) | Adult (n = 24) | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Value | HR | 95% CI | P Value | |
| Male sex | 1.67 | 0.28‐10.0 | 0.57 | 0.43 | 0.10‐1.75 | 0.24 |
| Primary disease: non‐BA | NA* | NA* | NA* | NA* | NA* | NA* |
| ABO incompatible | 0.95 | 0.11‐8.47 | 0.96 | NA* | NA* | NA* |
| PELD score | 1.04 | 0.90‐1.18 | 0.54 | NA* | NA* | NA* |
| MELD score | NA* | NA* | NA* | 1.02 | 0.94‐1.10 | 0.63 |
| EBV naïve | NA* | NA* | NA* | NA* | NA* | NA* |
| Positive EBV PCR | NA* | NA* | NA* | 2.71 | 0.36‐20.6 | 0.34 |
| Positive EBER | NA* | NA* | NA* | 0.93 | 0.16‐5.40 | 0.94 |
| PS ≥2 | 9.64 | 1.07‐86.8 | 0.04 | 3.69 | 0.89‐15.4 | 0.07 |
| Ann Arbor stage ≥III | 1.90 | 0.27‐13.5 | 0.52 | 0.64 | 0.13‐3.08 | 0.57 |
| Bulky tumor ≥5 cm | 5.94 | 0.54‐65.5 | 0.15 | 0.82 | 0.01‐6.66 | 0.85 |
| Extranodal lesion ≥1 | 4.46 | 0.73‐27.3 | 0.11 | 4.05 | 0.79‐20.8 | 0.09 |
| LDH elevation | NA* | NA* | NA* | 3.17 | 0.38‐26.1 | 0.28 |
| IPI ≥3 | NA* | NA* | NA* | 1.45 | 0.34‐6.14 | 0.61 |
| Time to PTLDs ≥1 year | 0.68 | 0.11‐4.14 | 0.68 | 1.14 | 0.23‐5.76 | 0.87 |
| Monomorphic PTLDs | 13.7 | 1.51‐124.2 | 0.02 | 4.71 | 0.87‐25.5 | 0.07 |
P values < 0.05 are bold.
NA because no patients died as a result of PTLDs in either or both groups of the analyzed variable.
Discussion
Because of high morbidity and mortality, PTLDs have been investigated in various SOTs; however, their rarity only in transplant recipients, as well as the heterogeneity, from nondestructive to destructive PTLDs( 26 ) have hampered detailed assessments of these critical complications. In the present study, using a relatively large cohort of 1954 LTs in 1849 patients, we found that the overall incidence of LT‐PTLDs was 3.8%. Of these, the incidence in pediatric recipients (<18 years of age at LT) was 5.1%, more than double the incidence of 2.3% in adults (≥18 years of age at LT). Moreover, almost half of the pediatric LT‐PTLDs developed within a year after transplant (Supporting Fig. 3), and the intervals between LT and PTLD occurrence were significantly shorter in pediatric than in adult recipients. These results may imply that LT‐PTLDs are more critical in pediatric rather than in adult recipients; however, the prognosis of adult LT‐PTLDs was significantly worse than that of pediatric cases. Notably, the 3‐year, 5‐year, and 10‐year OS rates after PTLD diagnosis were 81%, 79%, and 71% in children, respectively, whereas those in adults were as low as 61%, 38%, and 28%, respectively. These different oncological behaviors between children and adults may be attributable to the proportion of monomorphic PTLDs, which tended to occur more often in adults than in children (50% versus 28%; P = 0.07). In other words, pediatric recipients are more likely to develop non‐monomorphic PTLDs early after LT, which may have had a positive impact on patient survival.
As CNI is a well‐known risk factor for PTLD development,( 2 ) we investigated the relationship between the incidence of PTLDs and the tacrolimus concentration. Of note, the incidence of pediatric LT‐PTLDs decreased in the late period (2006‐2020). As a possible reason for this, the tacrolimus trough in pediatric patients with LT‐PTLDs was significantly higher in the early period than that in the late period. These results suggest that the high concentration of tacrolimus may, at least in part, be involved in the development of pediatric LT‐PTLDs.( 38 )
In this study, we identified the following 3 prognostic factors in LT‐PTLDs: age ≥18 years at PTLD diagnosis, PS ≥2 at PTLD diagnosis, and monomorphic PTLDs. The LT‐PTLD score, which consists of these 3 factors, significantly stratified patient survival and PFS in LT‐PTLDs. To date, the following prognostic factors have been reported for PTLDs after KT or various SOTs (Table 4): PS ≥2 or 3( 9, 10, 11, 12, 14 ); monomorphic PTLDs( 6, 10 ); age ≥16 years( 14 ), >55( 6 ), or >60( 11 ) years; LDH elevation( 6, 11, 12 ); hypoalbuminemia( 13 ); manifested B symptoms( 12 ); number of sites involved( 9 ); and involvement of transplanted organs,( 10 ) CNS,( 13 ) or BM.( 13 ) Taken together with the current results, age ≥18 years and PS ≥2 at PTLD diagnosis and monomorphic PTLDs may be universal prognostic factors for patient prognosis, regardless of transplanted organs.
TABLE 4.
Previously Reported Prognostic Factors in PTLDs
| Year | Journal | First Author and Citation | Transplanted Organs and Patient Numbers | Prognostic Factors |
|---|---|---|---|---|
| 2001 | Journal of Clinical Oncology | Leblond( 9 ) | Kidney: 35, heart: 19, lung: 5, liver: 3 | PS ≥2, number of involved sites |
| 2005 | Journal of Clinical Oncology | Ghobrial( 10 ) | Kidney: 36, kidney and pancreas: 7, pancreas: 5, liver: 35, lung: 4, heart: 15, multiorgan/other: 5 | PS ≥3, monomorphic PTLDs, graft organ involvement |
| 2007 | Annals of Hematology | Choquet( 11 ) | Heart: 16, kidney: 22, lung: 11, kidney and pancreas/heart and lung: 11 | Age >60 years, PS ≥2, elevated LDH |
| 2008 | British Journal of Haematology | Hourigan( 12 ) | Kidney: 42 | PS ≥2, elevated LDH, B symptoms |
| 2010 | Journal of Clinical Oncology | Evens( 13 ) | Kidney: 37, kidney and pancreas: 9, pancreas: 4, liver: 17, heart: 8, lung: 5 | CNS involvement, BM involvement, hypoalbuminemia |
| 2013 | Journal of Clinical Oncology | Caillard( 6 ) | Kidney: 500 | Age >55 years, serum creatinine >133 μmol/L, elevated LDH, disseminated PTLDs, CNS involvement, serous membrane invasion, T cell PTLDs, monomorphic PTLDs |
| 2015 | British Journal of Haematology | Montanari( 14 ) | Heart: 63, kidney: 32, liver: 22, other: 10 | Age ≥16 years, PS ≥2, CD20 negative |
Consistent with a previous report,( 2 ) we demonstrated that EBV‐positive PTLDs developed significantly earlier than EBV‐negative PTLDs (P < 0.001). In contrast, several reports have shown that recipients who are EBV naïve are at the highest risk to develop PTLDs( 39, 40, 41 ) and that primary EBV infection after SOT in recipients who are EBV naïve is a risk factor for early PTLDs.( 42 ) In this study, however, pretransplant EBV serologies were all positive in adults (100%), and even in the pediatric cohorts, 65.5% (19/29 cases) were EBV positive preoperatively. Notably, no significant difference was found between EBV‐seropositive and EBV‐negative patients in the timing of overall LT‐PTLD onset. Although EBV‐positive patients tended to develop LT‐PTLDs earlier than EBV‐naïve cases in children, this difference did not reach statistical significance (P = 0.06; Supporting Fig. 2E). It remains unclear whether these results are characteristic in LT‐PTLDs; however, given that more than 95% of adults worldwide are infected with EBV,( 43 ) further large‐scale studies are needed, especially focusing on pediatric LT‐PTLDs.
As for monitoring interventions for early detection of PTLDs, we focused on sIL2R. We examined sIL2R in 25 LT‐PTLD patients at diagnosis, in which 22 patients (88%) showed an increase in serum sIL2R. Notably, monomorphic PTLDs showed higher sIL2R than non‐monomorphic types (P = 0.09; Supporting Table 3). When the cutoff value of 1800 U/mL, calculated from the receiver operating characteristic curve (ROC), is indicated, serum sIL2R is significantly higher in monomorphic PTLDs than in the others (P = 0.02; Supporting Table 3). Although EBV PCR was used for monitoring the PTLD onset,( 44 ) early detection of monomorphic PTLDs seems difficult because EBV PCR viral load was significantly lower in monomorphic PTLDs than in the others (P = 0.03; Supporting Table 3). These results suggest that monomorphic PTLDs are less associated with EBV infection than non‐monomorphic types. Taken together, satisfying both high sIL2R and low viral load by EBV PCR may suggest the presence of monomorphic PTLDs. Although further large‐scale studies are needed, the combination of these 2 parameters may be useful for the early detection of monomorphic PTLDs that require intensive treatments including chemotherapies.
The current study has several limitations. First, this is a retrospective, single‐center study, which could not avoid potential selection bias. A multicenter study with a larger cohort is required to validate our findings. Second, we included PTLD‐like lesions in the current analysis, although they are excluded from PTLDs in the standard definition.( 26 ) However, their characteristics and required treatments are not different from those of PTLDs. Because PTLD‐like lesions accounted for as much as 25% of the current cohorts, we consider that they should be recognized more widely as important forms of PTLDs. Third, there were missing values in some variables, including pretransplant EBV/CMV serologies, EBV PCR, and sIL2R. Despite their significance in PTLD pathogenesis, they were not always measured, especially in the earlier era.
In conclusion, LT‐PTLDs occurred in 3.8% overall, 5.1% in pediatric, and 2.3% in adult LT recipients, which significantly worsened patient survival. Age ≥18 years and PS ≥2 at PTLD diagnosis and monomorphic PTLDs were identified as independent prognostic factors for patient survival with LT‐PTLDs. The LT‐PTLD score that consists of these 3 factors effectively stratified patient survival and PFS. Although required to be validated, the LT‐PTLD score may distinguish high‐risk cases of LT‐PTLDs and provide a potential guide for adequate interventions.
Supporting information
Fig S1
Fig S2
Fig S3
Table S1‐S3
This work was supported by the Medical Research and Development Programs Focused on Technology Transfer, Development of Advanced Measurement and Analysis Systems (SENTAN) from the Japan Agency for Medical Research and Development (AMED) (No. 20hm0102063h0003).
Momoko Nishikori has received grants from Eisai (no conflicts of interest) and Sumitomo Dainippon Pharma (no conflicts of interest).
Momoko Nishikori has grants with Eisai (no interest) and Sumitomo Dainippon Pharma (no interest).
[Correction added on July 29, 2021, after first online publication: commas in numerals listing thousands were removed from this article]
References
- 1.Penn I, Hammond W, Brettschneider L, Starzl TE. Malignant lymphomas in transplantation patients. Transplant Proc 1969;1:106‐112. [PMC free article] [PubMed] [Google Scholar]
- 2.Dierickx D, Habermann TM. Post‐transplantation lymphoproliferative disorders in adults. N Engl J Med 2018;378:549‐562. [DOI] [PubMed] [Google Scholar]
- 3.Bhat M, Mara K, Dierkhising R, Watt KD. Gender, race and disease etiology predict de novo malignancy risk after liver transplantation: insights for future individualized cancer screening guidance. Transplantation 2019;103:91‐100. [DOI] [PubMed] [Google Scholar]
- 4.Collett D, Mumford L, Banner NR, Neuberger J, Watson C. Comparison of the incidence of malignancy in recipients of different types of organ: a UK Registry audit. Am J Transplant 2010;10:1889‐1896. [DOI] [PubMed] [Google Scholar]
- 5.Miyazaki T, Sato S, Kondo T, Kusaka M, Gotoh M, Saiki Y, et al. National survey of de novo malignancy after solid organ transplantation in Japan. Surg Today 2018;48:618‐624. [DOI] [PubMed] [Google Scholar]
- 6.Caillard S, Porcher R, Provot F, Dantal J, Choquet S, Durrbach A, et al. Post‐transplantation lymphoproliferative disorder after kidney transplantation: report of a nationwide French registry and the development of a new prognostic score. J Clin Oncol 2013;31:1302‐1309. [DOI] [PubMed] [Google Scholar]
- 7.Opelz G, Döhler B. Lymphomas after solid organ transplantation: a collaborative transplant study report. Am J Transplant 2004;4:222‐230. [DOI] [PubMed] [Google Scholar]
- 8.Dierickx D, Tousseyn T, Sagaert X, Fieuws S, Wlodarska I, Morscio J, et al. Single‐center analysis of biopsy‐confirmed posttransplant lymphoproliferative disorder: incidence, clinicopathological characteristics and prognostic factors. Leuk Lymphoma 2013;54:2433‐2440. [DOI] [PubMed] [Google Scholar]
- 9.Leblond V, Dhedin N, Mamzer Bruneel MF, Choquet S, Hermine O, Porcher R, et al. Identification of prognostic factors in 61 patients with posttransplantation lymphoproliferative disorders. J Clin Oncol 2001;19:772‐778. [DOI] [PubMed] [Google Scholar]
- 10.Ghobrial IM, Habermann TM, Maurer MJ, Geyer SM, Ristow KM, Larson TS, et al. Prognostic analysis for survival in adult solid organ transplant recipients with post‐transplantation lymphoproliferative disorders. J Clin Oncol 2005;23:7574‐7582. [DOI] [PubMed] [Google Scholar]
- 11.Choquet S, Oertel S, LeBlond V, Riess H, Varoqueaux N, Dörken B, et al. Rituximab in the management of post‐transplantation lymphoproliferative disorder after solid organ transplantation: proceed with caution. Ann Hematol 2007;86:599‐607. [DOI] [PubMed] [Google Scholar]
- 12.Hourigan MJ, Doecke J, Mollee PN, Gill DS, Norris D, Johnson DW, et al. A new prognosticator for post‐transplant lymphoproliferative disorders after renal transplantation. Br J Haematol 2008;141:904‐907. [DOI] [PubMed] [Google Scholar]
- 13.Evens AM, David KA, Helenowski I, Nelson B, Kaufman D, Kircher SM, et al. Multicenter analysis of 80 solid organ transplantation recipients with post‐transplantation lymphoproliferative disease: outcomes and prognostic factors in the modern era. J Clin Oncol 2010;28:1038‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montanari F, Radeski D, Seshan V, Alobeid B, Bhagat G, O'Connor OA. Recursive partitioning analysis of prognostic factors in post‐transplant lymphoproliferative disorders (PTLD): a 120 case single institution series. Br J Haematol 2015;171:491‐500. [DOI] [PubMed] [Google Scholar]
- 15.Jain A, Nalesnik M, Reyes J, Pokharna R, Mazariegos G, Green M, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20‐year experience. Ann Surg 2002;236:429‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka K, Kiuchi T, Kaihara S. Living related liver donor transplantation: techniques and caution. Surg Clin North Am 2004;84:481‐493. [DOI] [PubMed] [Google Scholar]
- 17.Morioka D, Egawa H, Kasahara M, Ito T, Haga H, Takada Y, et al. Outcomes of adult‐to‐adult living donor liver transplantation: a single institution's experience with 335 consecutive cases. Ann Surg 2007;245:315‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubota T, Hata K, Sozu T, Ueda Y, Hirao H, Okamura Y, et al. Impact of donor age on recipient survival in adult‐to‐adult living‐donor liver transplantation. Ann Surg 2018;267:1126‐1133. [DOI] [PubMed] [Google Scholar]
- 19.Kusakabe J, Hata K, Tanaka S, Omae K, Okamura Y, Tajima T, et al. Prognostic index consisting of early post‐transplant variables <2 weeks in adult living‐donor liver transplantation. Hepatol Res 2020;50:741‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uemura T, Wada S, Kaido T, Mori A, Ogura Y, Yagi S, et al. How far can we lower graft‐to‐recipient weight ratio for living donor liver transplantation under modulation of portal venous pressure? Surgery 2016;159:1623‐1630. [DOI] [PubMed] [Google Scholar]
- 21.Ogura Y, Hori T, El Moghazy WM, Yoshizawa A, Oike F, Mori A, et al. Portal pressure <15 mm Hg is a key for successful adult living donor liver transplantation utilizing smaller grafts than before. Liver Transpl 2010;16:718‐728. [DOI] [PubMed] [Google Scholar]
- 22.Egawa H, Ohmori K, Haga H, Tsuji H, Yurugi K, Miyagawa‐Hayashino A, et al. B‐cell surface marker analysis for improvement of rituximab prophylaxis in ABO‐incompatible adult living donor liver transplantation. Liver Transpl 2007;13:579‐588. [DOI] [PubMed] [Google Scholar]
- 23.Banff schema for grading liver allograft rejection: an international consensus document. Hepatology 1997;25:658‐663. [DOI] [PubMed] [Google Scholar]
- 24.Demetris AJ, Bellamy C, Hübscher SG, O'Leary J, Randhawa PS, Feng S, et al. 2016 Comprehensive update of the Banff Working Group on liver allograft pathology: introduction of antibody‐mediated rejection. Am J Transplant 2016;16:2816‐2835. [DOI] [PubMed] [Google Scholar]
- 25.Shehata MR, Yagi S, Okamura Y, Iida T, Hori T, Yoshizawa A, et al. Pediatric liver transplantation using reduced and hyper‐reduced left lateral segment grafts: a 10‐year single‐center experience. Am J Transplant 2012;12:3406‐3413. [DOI] [PubMed] [Google Scholar]
- 26.Swerdlow SH, Webber SA, Chadburn A, Ferry JA. Post‐transplant lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th edn. Lyon: IARC; 2017:453‐462. [Google Scholar]
- 27.Gulley ML, Tang W. Laboratory assays for Epstein‐Barr virus‐related disease. J Mol Diagn 2008;10:279‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gulley ML, Tang W. Using Epstein‐Barr viral load assays to diagnose, monitor, and prevent posttransplant lymphoproliferative disorder. Clin Microbiol Rev 2010;23:350‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galera P, Flavin R, Savage NM, Saksena A, Gong S, Wang HY, et al. Epstein‐Barr virus‐negative marginal zone lymphoma as an uncommon form of monomorphic posttransplant lymphoproliferative disorder. Am J Surg Pathol 2020;44:1340‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hart M, Thakral B, Yohe S, Balfour HH Jr, Singh C, Spears M, et al. EBV‐positive mucocutaneous ulcer in organ transplant recipients: a localized indolent posttransplant lymphoproliferative disorder. Am J Surg Pathol 2014;38:1522‐1529. [DOI] [PubMed] [Google Scholar]
- 31.Parker A, Bowles K, Bradley JA, Emery V, Featherstone C, Gupte G, et al. Diagnosis of post‐transplant lymphoproliferative disorder in solid organ transplant recipients—BCSH and BTS guidelines. Br J Haematol 2010;149:675‐692. [DOI] [PubMed] [Google Scholar]
- 32.Tsao L, Chu KE, Bhagat G, Alobeid B. Development of hairy cell leukemia in a patient after cardiac transplantation. Leuk Lymphoma 2006;47:361‐363. [DOI] [PubMed] [Google Scholar]
- 33.Tsurusawa M, Mori T, Kikuchi A, Mitsui T, Sunami S, Kobayashi R, et al. Improved treatment results of children with B‐cell non‐Hodgkin lymphoma: a report from the Japanese Pediatric Leukemia/Lymphoma Study Group B‐NHL03 study. Pediatr Blood Cancer 2014;61:1215‐1221. [DOI] [PubMed] [Google Scholar]
- 34.Tsuchida Y, Therasse P. Response evaluation criteria in solid tumors (RECIST): new guidelines. Med Pediatr Oncol 2001;37:1‐3. [DOI] [PubMed] [Google Scholar]
- 35.Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982;5:649‐655. [PubMed] [Google Scholar]
- 36.Carbone PP, Kaplan HS, Musshoff K, Smithers DW, Tubiana M. Report of the committee on Hodgkin's disease staging classification. Cancer Res 1971;31:1860‐1861. [PubMed] [Google Scholar]
- 37.International Non‐Hodgkin's Lymphoma Prognostic Factors Project . A predictive model for aggressive non‐Hodgkin's lymphoma. N Engl J Med 1993;329:987‐994. [DOI] [PubMed] [Google Scholar]
- 38.Narkewicz MR, Green M, Dunn S, Millis M, McDiarmid S, Mazariegos G, et al. Decreasing incidence of symptomatic Epstein‐Barr virus disease and posttransplant lymphoproliferative disorder in pediatric liver transplant recipients: report of the studies of pediatric liver transplantation experience. Liver Transpl 2013;19:730‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knight JS, Tsodikov A, Cibrik DM, Ross CW, Kaminski MS, Blayney DW. Lymphoma after solid organ transplantation: risk, response to therapy, and survival at a transplantation center. J Clin Oncol 2009;27:3354‐3362. [DOI] [PubMed] [Google Scholar]
- 40.Ho M, Miller G, Atchison RW, Breinig MK, Dummer JS, Andiman W, et al. Epstein‐Barr virus infections and DNA hybridization studies in posttransplantation lymphoma and lymphoproliferative lesions: the role of primary infection. J Infect Dis 1985;152:876‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas JA, Hotchin NA, Allday MJ, Amlot P, Rose M, Yacoub M, et al. Immunohistology of Epstein‐Barr virus‐associated antigens in B cell disorders from immunocompromised individuals. Transplantation 1990;49:944‐953. [DOI] [PubMed] [Google Scholar]
- 42.Cockfield SM. Identifying the patient at risk for post‐transplant lymphoproliferative disorder. Transpl Infect Dis 2001;3:70‐78. [DOI] [PubMed] [Google Scholar]
- 43.Luzuriaga K, Sullivan JL. Infectious mononucleosis [published correction appears in N Engl J Med 2010 Oct 7;363(15):1486]. N Engl J Med 2010;362:1993‐2000. [DOI] [PubMed] [Google Scholar]
- 44.McDiarmid SV, Jordan S, Kim GS, Toyoda M, Goss JA, Vargas JH, et al. Prevention and preemptive therapy of postransplant lymphoproliferative disease in pediatric liver recipients [published correction appears in Transplantation 1999 Sep 27;68(6):909. Lee GS [corrected to Kim GS]]. Transplantation 1998;66:1604‐1611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Table S1‐S3