

Abstract
Dabrafenib is an oral BRAF kinase inhibitor approved for the treatment of various BRAF V600 mutation–positive solid tumors. In vitro observations suggesting cytochrome P450 (CYP) 3A induction and organic anion transporting polypeptide (OATP) inhibition prompted us to evaluate the effect of dabrafenib 150 mg twice daily on the pharmacokinetics of midazolam 3 mg (CYP3A substrate) and rosuvastatin 10 mg (OATP1B1/1B3 substrate) in a clinical phase 1, open‐label, fixed‐sequence study in patients with BRAF V600 mutation–positive tumors. Repeat dabrafenib dosing resulted in a 2.56‐fold increase in rosuvastatin maximum observed concentration (Cmax), an earlier time to Cmax, but only a 7% increase in area under the concentration‐time curve from time 0 (predose) extrapolated to infinite time. Midazolam Cmax and AUC extrapolated to infinite time decreased by 47% and 65%, respectively, with little effect on time to Cmax. No new safety findings were reported. Exposure of drugs that are CYP3A4 substrates is likely to decrease when coadministered with dabrafenib. Concentrations of medicinal products that are sensitive OATP1B1/1B3 substrates may increase during the absorption phase.
Keywords: CYP3A4, dabrafenib, drug interaction, OATP, pharmacokinetics, transporters
Dabrafenib is a potent and selective inhibitor of BRAF kinase (a member of the RAF kinases), with a mechanism of action consistent with adenosine triphosphate–competitive inhibition. Dabrafenib has been approved as monotherapy for patients with BRAF V600 mutation–positive unresectable or metastatic melanoma in the United States, Australia, European Union, Canada, and several other countries. It is also approved in combination with the MEK inhibitor trametinib for patients with BRAF V600 mutation–positive unresectable or metastatic melanoma, as adjuvant treatment following melanoma resection, metastatic non–small cell lung cancer, and locally advanced or metastatic anaplastic thyroid cancer. The recommended starting dose for dabrafenib as a single agent and in combination with trametinib is 150 mg twice daily.
Following a single oral dose of dabrafenib, plasma concentrations peak approximately 2 hours after dosing followed by a biexponential decline, with oral bioavailability near complete (94.5%) relative to an intravenous microdose and a median terminal‐phase half‐life (t1/2) of ≈8 hours.1, 2 Dabrafenib is sequentially metabolized to hydroxy‐, carboxy‐, and desmethyl‐dabrafenib. Dabrafenib metabolism is mediated by cytochrome P450 (CYP) 2C8 and CYP3A4, whereas both its metabolites hydroxy‐dabrafenib and desmethyl‐dabrafenib are CYP3A4 substrates. Carboxy‐dabrafenib is decarboxylated via a nonenzymatic process to form desmethyl‐dabrafenib and is excreted in the bile and urine.3, 4 Following repeat‐dose administration of dabrafenib 150 mg twice daily, there is a decrease in exposure, likely due to induction of dabrafenib's own metabolism, and steady state is reached by day 15. Carboxy‐ and desmethyl‐dabrafenib accumulate with repeat dosing. Hydroxy‐dabrafenib terminal half‐life parallels that of parent with a half‐life of 9.7 hours, whereas the carboxy‐ and desmethyl‐metabolites exhibit longer half‐lives (21‐22 hours).5, 6
In vitro, dabrafenib is known to induce CYP3A4 and has been shown to be an inhibitor of the organic anion transporting polypeptide (OATP) 1B1/1B3; however, its effect after single or repeat dosing on an OATP1B1/1B3 substrate is unknown.4, 7, 8 In vivo, dabrafenib induces CYP3A4‐ and CYP2C9‐mediated metabolism. In an early clinical study involving 12 patients given single‐dose midazolam, a CYP3A substrate, maximum observed concentration (Cmax) and area under the concentration‐time curve (AUC) were decreased by 61% and 74%, respectively, with coadministration of repeat‐dose dabrafenib 150 mg twice daily using gelatin capsules. In a separate trial involving 14 patients, repeat‐dose dabrafenib (hydroxypropyl methylcellulose [HPMC] capsules) decreased the single‐dose AUC of S‐warfarin (a substrate for CYP2C9) and of R‐warfarin (a substrate for CYP3A4/CYP1A2) by 37% and 33%, respectively, with a small increase in Cmax (18%‐19%).6
Drugs in the statin class are the recommended substrates for evaluating potential inhibitors and inducers of OATP1B1/1B3. The involvement of CYP in statin metabolism varies, with a less significant role in rosuvastatin metabolism. Thus, rosuvastatin was selected as the OATP1B1/1B3 substrate for this study, while midazolam was selected because it is the preferred substrate to evaluate potential inhibitors and inducers of CYP3A4 catabolic activity.9, 10, 11
Previously, dabrafenib gelatin capsules were used to evaluate the effect of dabrafenib on midazolam pharmacokinetics (PK); however, in the current study, we used the marketed formulation of HPMC capsules.
The purpose of this study was to evaluate the effects of the approved dose of dabrafenib (150 mg twice daily) on the single‐dose PK of rosuvastatin and midazolam during the initiation of dabrafenib dosing and at steady state in patients with advanced BRAF V600 mutation–positive tumors.
Methods
Study Design
This study was conducted at 3 sites in Spain (Hospital Universitario Fundación Jiménez Díaz, Hospital General Universitario Vall d'Hebron, and Hospital Universitario Madrid Sanchinarro) between March 3, 2015, and August 1, 2016. The study protocol was reviewed and approved by the independent ethics committee or institutional review board at Fundación Jiménez Díaz (Madrid, Spain) for all sites. A signed, written informed consent form was obtained from each patient before any study‐specific procedures or assessments. This study was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice guidelines, patient privacy requirements, ethical principles outlined in the Declaration of Helsinki, and applicable local regulations.
This was an open‐label, multicenter, fixed‐sequence study conducted in patients with BRAF V600 mutation–positive tumors (NCT02082665). Rosuvastatin (10 mg) and midazolam (3 mg) were administered as a mini‐cocktail, allowing for 2 evaluations at the same time. The study consisted of 3 periods (Figure 1). During period 1 (day 1), patients received simultaneous doses of rosuvastatin 10 mg and midazolam 3 mg. During period 2 (day 8), patients received simultaneous doses of dabrafenib 150 mg twice daily, rosuvastatin 10 mg, and midazolam 3 mg (on the same day, patients were reminded to take the second dose of dabrafenib in the evening ≈12 hours after the morning dose). On days 8 to 23, patients continued to self‐administer dabrafenib 150 mg twice daily. During period 3 (day 22), patients received concurrent doses of dabrafenib 150 mg twice daily, rosuvastatin 10 mg, and midazolam 3 mg. On day 23, patients took the final morning dose of dabrafenib. Doses were administered after an overnight fast (at least 8 hours), and a meal was provided 4 hours after dosing. Patients received study treatment through day 23, unless disease progression, death, or unacceptable toxicity occurred. No efficacy assessments were performed.
Figure 1.
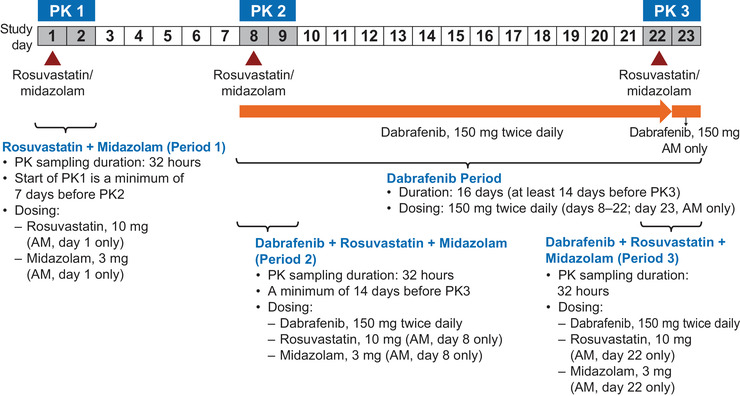
Study design. PK, pharmacokinetic.
Patients
Male or female patients aged between 18 and 65 years with a BRAF V600 mutation–positive tumor and an Eastern Cooperative Oncology Group performance status (ECOG PS) of ≤1 at screening were eligible to be enrolled. Patients were required to have a body weight of at least 45 kg, body mass index of ≥19 and <40 kg/m2, and adequate organ function.
Pharmacokinetic Assessments
Serial blood samples for PK analysis were collected over 32 and 24 hours for rosuvastatin and midazolam analysis, respectively, on days 1 to 2, 8 to 9, and 22 to 23. For rosuvastatin and midazolam analyses, the collection time points were before dosing and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, and 24 hours after dosing; additionally, samples for rosuvastatin analysis were collected at 26, 28, and 32 hours after dosing. Sparse PK samples were also collected before dosing and at 1, 2, 8, and 24 hours after dosing on days 8 to 9 and 22 to 23 for dabrafenib and metabolite analyses.
Analytical Methods
Dabrafenib (and metabolites) were measured using a validated liquid chromatography with tandem mass spectrometry (LC‐MS/MS) method, with an analytical range of 1 to 1000 ng/mL.3
Rosuvastatin was extracted from human plasma by liquid‐liquid extraction using ethyl‐acetate after the addition of methanol/water (50:50, v/v) containing [2H4 13C]‐rosuvastatin as internal standards, and of a solution of 1% (v/v) formic acid in water. After evaporation, the residue was reconstituted with acetonitrile/water (50:50, v/v). Extracts were then analyzed by LC‐MS/MS using a TurboIonSpray interface (Sciex, Framingham, Massachusetts) with positive ion multiple‐reaction monitoring. The m/z transitions were 482/258 for rosuvastatin and 487/263 for internal standard. Chromatographic separation was achieved using an Acquity ultra‐performance liquid chromatography HSS T3 1.8 μM (50 × 2.1 mm) column (Waters Corporation, Milford, Massachusetts), with an isocratic elution (water containing 0.1% v/v formic acid‐acetonitrile 57:43) and flow rate of 0.6 mL/min. This method was validated over the range of 0.2 to 30 ng/mL, and the lower limit of quantification was 0.2 ng/mL using 150 μL of human plasma. The precision (coefficient of variation) within and between runs was ≤11.8% and ≤2.4%, respectively.
Midazolam was extracted from human plasma by protein precipitation using acetonitrile containing [2H4]‐midazolam as an internal standard. Extracts were analyzed by LC‐MS/MS using a TurboIonSpray interface with positive ion multiple‐reaction monitoring. The m/z transitions were 326/291 for midazolam and 330/295 for internal standard. Chromatographic separation was achieved using a Waters Acquity ultra‐performance liquid chromatography HSS T3 1.8 μM (50 × 2.1 mm) column, with an isocratic elution (water containing 10 mM of ammonium acetate‐acetonitrile 52:48) and flow rate of 0.6 mL/min. This method was validated over the range of 0.1 to 100 ng/mL, and the lower limit of quantification was 0.1 ng/mL using 50 μL of human plasma. The precision (coefficient of variation) within and between runs was ≤4.8% and ≤4.3%, respectively.
Pharmacokinetic and Statistical Analysis
PK parameters—Cmax, time to Cmax (tmax), AUC from time 0 to time t, AUC from time 0 (predose) extrapolated to infinite time (AUC0‐∞), and t1/2—for midazolam and rosuvastatin were calculated by standard noncompartmental analysis with Phoenix WinNonLin Pro version 6.4 (Certara, Princeton, New Jersey). All calculations were based on actual sampling times.
For the primary comparison of rosuvastatin and midazolam PK with dabrafenib (test; day 8 [initial dosing] or day 22 [steady state]) versus rosuvastatin and midazolam PK without dabrafenib (reference; day 1), log‐transformed Cmax and AUC0‐∞ of rosuvastatin or midazolam were analyzed separately using a mixed‐effects model, with treatment as a fixed effect and patient as a random effect. Point estimates and 90% confidence intervals (CIs) were constructed for the difference between the test and reference. The point estimates and associated 90%CIs were then back‐transformed to provide point estimates and 90%CIs for the geometric mean ratio (test/reference). For tmax, a nonparametric method was used to estimate the median difference and associated 90%CI between the test and reference.
Safety
Safety assessments included physical examinations, vital signs (blood pressure, temperature, and pulse rate), 12‐lead electrocardiograms, clinical laboratory tests (hematology and clinical chemistry), and monitoring of adverse events (AEs). AEs and relevant hematology and clinical chemistry data were graded by the investigator according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.
Results
Patient Characteristics
Sixteen patients were enrolled, and the same treatment sequence (Figure 1) of dabrafenib, rosuvastatin, and midazolam administration was followed for all patients. All patients had advanced disease, with colorectal cancer (19%), thyroid cancer (19%), non–small cell lung cancer (13%), and melanoma (13%) being the most common tumor types. Chemotherapy (63%) and biologic therapy (25%) were the most common prior anticancer therapies. Of the 16 patients, 12 (75%) had an ECOG PS of 0 at baseline, and 4 (25%) had an ECOG PS of 1 or 2 (Table 1).
Table 1.
Patient Demographics and Disease Characteristics
| Characteristic | (N = 16) |
|---|---|
| Age, y | 49.5 (29–64) |
| Sex, n (%) | |
| Female | 13 (81) |
| Male | 3 (19) |
| Height, cm | 164.5 (147–175) |
| Weight, kg | 66.2 (52–93) |
| BMI, kg/m2 | 24.7 (20–30) |
| Race, n (%) | |
| White/Caucasian/European | 14 (88) |
| Black/African | 1 (6) |
| Asian—Central/South Asian | 1 (6) |
| Primary tumor type, n (%) | |
| Colon/rectum cancer | 3 (19) |
| Thyroid cancer | 3 (19) |
| Melanoma | 2 (13) |
| NSCLC | 2 (13) |
| Breast cancer | 1 (6) |
| HCL | 1 (6) |
| LCH | 1 (6) |
| Liver cancer | 1 (6) |
| Lung adenocarcinoma | 1 (6) |
| SCLC | 1 (6) |
| Stage at screening, n (%) | |
| I | 2 (13) |
| IV | 13 (81) |
| Missing | 1 (6) |
| Prior anticancer therapy, n (%) | |
| Any therapy | 11 (69) |
| Chemotherapy | 10 (63) |
| Biologic therapy | 4 (25) |
| Hormonal therapy | 2 (13) |
| Immunotherapy | 2 (13) |
| Small‐molecule targeted therapy | 1 (6) |
| Time since diagnosis, days | 467.5 (21–7226) |
BMI, body mass index; HCL, hairy cell leukemia; LCH, Langerhans cell histiocytosis; NSCLC, non–small cell lung cancer; SCLC, small cell lung cancer.
Continuous data are presented as median (range).
All 16 patients received study treatments as scheduled, with the following exceptions: 1 patient experienced a serious adverse event (SAE) on day 22 and missed the last 2 scheduled 150‐mg dabrafenib doses (300 mg total); 1 patient skipped the last dose of midazolam on day 22 because of an SAE related to midazolam on day 8; and 2 patients took an extra unscheduled 150‐mg dabrafenib dose on days 23 and 24.
Pharmacokinetics
Effect of Dabrafenib on Rosuvastatin
PK parameters for rosuvastatin were calculated for days 1, 8, and 22 and are summarized descriptively (Table 2). The mean plasma rosuvastatin concentration‐time profiles after administration of rosuvastatin alone and with dabrafenib are displayed in Figure 2. An increase of 94% and 22% was observed in the single‐dose rosuvastatin Cmax and AUC0‐∞, respectively, with dabrafenib at the initiation of dosing. Repeat dabrafenib dosing resulted in a 2.56‐fold increase in rosuvastatin Cmax but only a 7% increase in AUC0‐∞. The administration of rosuvastatin with dabrafenib at the initiation of dabrafenib treatment resulted in an earlier tmax for rosuvastatin; this effect was more pronounced when rosuvastatin was administered with dabrafenib at steady state (Table 3).
Table 2.
Summary of Derived Plasma PK Parameters for Rosuvastatin and Midazolam
| Study Period/PK Day Treatmenta | AUC0‐∞, ng • h/mL | AUC0‐t, ng • h/mL | Cmax, (ng/mL) | tmax, h | t1/2, h | |
|---|---|---|---|---|---|---|
| Rosuvastatin | ||||||
| Period 1/PK day 1 Treatment A (N = 16) | n | 15 | 16 | 16 | 16 | 16 |
| Geo‐mean (CV%) | 58.5 (47) | 51.5 (48) | 5.1 (53) | 4.0 (1.5‐6)b | 9.6 (35) | |
| Mean | 64.1 | 56.6 | 5.7 | … | 10.2 | |
| (SD) | (28.1) | (25.7) | (2.9) | … | (3.7) | |
| Period 2/PK day 8 Treatment B (N = 16) | n | 15 | 16 | 16 | 16 | 16 |
| Geo‐mean (CV%) | 71.1 (51) | 62.4 (55) | 9.8 (63) | 3.0 (0.5‐4)b | 9 (26) | |
| Mean | 79.1 | 70.7 | 11.4 | … | 9.3 | |
| (SD) | (37.9) | (36.5) | (6.4) | … | (2.4) | |
| Period 3/PK day 22 Treatment C (N = 16) | n | 14 | 16 | 16 | 16 | 14 |
| Geo‐mean (CV%) | 59.6 (65) | 62.4 (74) | 13 (83) | 1.5 (1‐4)b | 8.2 (49) | |
| Mean | 68.3 | 74.9 | 16.2 | … | 9.2 | |
| (SD) | (32.6) | (46.0) | (10.3) | … | (4.9) | |
| Midazolam | ||||||
| Period 1/PK day 1 Treatment A (N = 16) | n | 16 | 16 | 16 | 16 | 16 |
| Geo‐mean (CV%) | 58.9 (64) | 56.2 (65) | 28.4 (45) | 0.5 (0.25‐0.58)b | 4.4 (45) | |
| Mean | 70.1 | 66.8 | 30.9 | … | 4.8 | |
| (SD) | (49.5) | (46.3) | (13.0) | … | (1.9) | |
| Period 2/PK day 8 Treatment B (N = 16) | n | 15 | 16 | 16 | 16 | 16 |
| Geo‐mean (CV%) | 61.7 (54) | 54.9 (62) | 28.1 (54) | 0.25 (0‐1)b | 4.9 (64) | |
| Mean | 69.5 | 63.6 | 31.5 | … | 6.3 | |
| (SD) | (37.2) | (37.1) | (15.0) | … | (6.9) | |
| Period 3/PK day 22 Treatment C (N = 16) | n | 15 | 15 | 15 | 15 | 15 |
| Geo‐mean (CV%) | 20.3 (64) | 19.4 (65) | 15.1 (72) | 0.25 (0.25‐0.5)b | 2.9 (63) | |
| Mean | 23.6 | 22.7 | 18.3 | … | 3.5 | |
| (SD) | (13.4) | (13.3) | (12.6) | … | (2.4) |
AUC, area under the concentration‐time curve; AUC0‐∞, AUC from time 0 (predose) extrapolated to infinite time; AUC0‐t, AUC from time 0 to time t (last measurable concentration); Cmax, maximum observed concentration; CV, coefficient of variation; geo‐mean, geometric mean; PK, pharmacokinetics; SD, standard deviation; t1/2, terminal phase half‐life; tmax, time to Cmax.
Treatments A, B, and C were as follows:
(A) Single doses of rosuvastatin 10 mg + midazolam 3 mg on PK day 1.
(B) Single doses of rosuvastatin 10 mg + midazolam 3 mg + dabrafenib 150 mg twice daily at initial dosing on PK day 8.
(C) Single doses of rosuvastatin 10 mg + midazolam 3 mg + dabrafenib 150 mg twice daily at steady state on PK day 22.
Median (minimum‐maximum) is presented.
Figure 2.
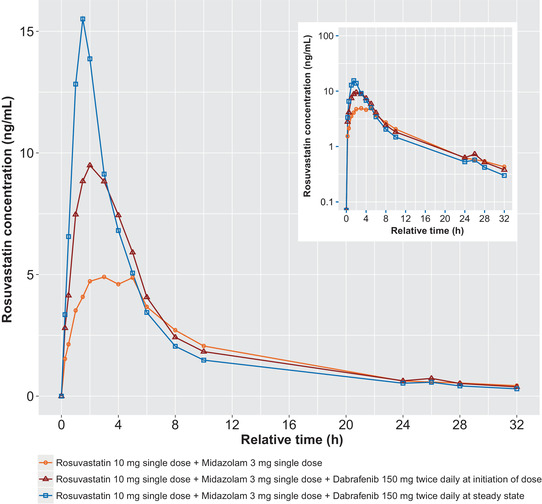
Mean rosuvastatin concentration‐time profiles after administration of rosuvastatin alone (PK day 1), coadministration with dabrafenib at initiation of dosing (PK day 8), and after repeat‐dose administration of dabrafenib at steady state (PK day 22). PK, pharmacokinetic.
Table 3.
Statistical Analysis Results of the Effect of Dabrafenib (Initial Dosing or Steady State) on Rosuvastatin PK Parameters
| PK Parametera | Treatment Comparisonb | Ratio or Difference (90%CI) |
|---|---|---|
| AUC0‐∞, ng • h/mL | B:A | 1.22 (1.07 to 1.38) |
| C:A | 1.07 (0.93 to 1.22) | |
| Cmax, ng/mL | B:A | 1.94 (1.65 to 2.27) |
| C:A | 2.56 (2.18 to 3.01) | |
| tmax, h | B−A | −1.04 (−1.94 to −0.49) |
| C−A | −2.25 (−2.79 to −1.5) |
AUC, area under the concentration‐time curve; AUC0‐∞, AUC from time 0 (predose) extrapolated to infinite time; CI, confidence interval; Cmax, maximum observed concentration; PK, pharmacokinetics; tmax, time to Cmax.
Ratio of Cmax and AUC is the geometric least squares mean of test versus reference (90%CI); ratio of tmax is the median difference (90%CI).
Treatments A, B, and C were as follows:
(A) Single doses of rosuvastatin 10 mg + midazolam 3 mg on PK day 1.
(B) Single doses of rosuvastatin 10 mg + midazolam 3 mg + dabrafenib 150 mg twice daily at initial dosing on PK day 8.
(C) Single doses of rosuvastatin 10 mg + midazolam 3 mg + dabrafenib 150 mg twice daily at steady state on PK day 22.
Effect of Dabrafenib on Midazolam
PK parameters for midazolam were calculated for days 1, 8, and 22 and are summarized descriptively in Table 2. The mean plasma midazolam concentration‐time profiles after administration of midazolam alone and with dabrafenib are displayed in Figure 3. Initial administration of dabrafenib with a single dose of midazolam resulted in no change in midazolam AUC0‐∞ and 1% decrease in midazolam Cmax relative to midazolam alone. Repeat dabrafenib dosing decreased midazolam Cmax and AUC0‐∞ by 47% and 65%, respectively. No effect on tmax was observed (Table 4).
Figure 3.
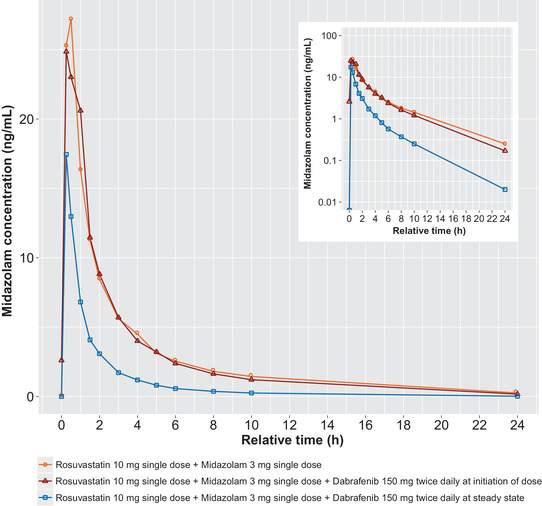
Mean midazolam concentration‐time profiles after administration of midazolam alone (PK day 1), coadministration with dabrafenib at initiation of dosing (PK day 8), and after repeat‐dose administration of dabrafenib at steady state (PK day 22). PK, pharmacokinetic.
Table 4.
Statistical Analysis Results of the Effect of Dabrafenib (Initial Dosing or Steady State) on Midazolam PK Parameters
| PK Parametera | Treatment Comparisonb | Ratio or Difference (90%CI) |
|---|---|---|
| AUC0‐∞, ng • h/mL | B:A | 1.00 (0.78 to 1.29) |
| C:A | 0.35 (0.27 to 0.45) | |
| Cmax, ng/mL | B:A | 0.99 (0.81 to 1.22) |
| C:A | 0.53 (0.43 to 0.66) | |
| tmax, h | B−A | −0.12 (−0.13 to 0) |
| C−A | −0.13 (−0.25 to 0) |
AUC, area under the concentration‐time curve; AUC0‐∞, AUC from time 0 (predose) extrapolated to infinite time; CI, confidence interval; Cmax, maximum observed concentration; PK, pharmacokinetics; tmax, time to Cmax.
Ratio of Cmax and AUC is the geometric least squares mean of test versus reference (90%CI); ratio of tmax is the median difference (90%CI).
Treatments A, B, and C were as follows:
(A) Single doses of rosuvastatin 10 mg + midazolam 3 mg on PK day 1.
(B) Single doses of rosuvastatin 10 mg + midazolam 3 mg + dabrafenib 150 mg twice daily at initial dosing on PK day 8.
(C) Single doses of rosuvastatin 10 mg + midazolam 3 mg + dabrafenib 150 mg twice daily at steady state on PK day 22.
Dabrafenib and Metabolite Concentrations
Dabrafenib and metabolite concentrations were consistent with historical data (data not shown).
Safety and Tolerability
Twelve of the 16 patients (75%) experienced at least 1 AE. The percentage of patients who reported any AE was lower during period 1 (31%) and increased with subsequent treatment in periods 2 (38%) and 3 (63%). The most common AEs reported in at least 2 patients during the study were somnolence, pyrexia, headache, fatigue, hyperkeratosis, nausea, and hypotension.
Most of the reported AEs were of grade ≤2. Four AEs were of grade ≥3, which included grade 4 hyponatremia, grade 4 presyncope, grade 3 decreased appetite, and grade 3 fatigue. Two patients discontinued dabrafenib treatment due to AEs; 1 patient had hypotension and pyrexia on day 22, while the other had presyncope following the second dose of midazolam on day 8. No deaths were reported during the study.
Hematology results reported as AEs included grade 2 anemia and grade 2 neutropenia, reported in 1 patient each. Anemia was considered to be related to study treatment, whereas neutropenia was considered unrelated to treatment. Clinical laboratory results reported as AEs included grade 2 hypocalcemia and an SAE of grade 4 hyponatremia, both reported in 1 patient; both events were considered to be not related to study treatment. No patients had liver signal events that suggested drug‐induced liver injury. No treatment‐related electrocardiographic changes were observed in these patients.
Discussion
This study evaluated the effects of the approved oral doses of dabrafenib (150 mg twice daily) on the single‐dose PK of rosuvastatin and midazolam during initiation of dabrafenib dosing and at steady state in patients with BRAF V600 mutation–positive tumors. At the time this study was designed, not enough safety data were available to be able to conduct the study in healthy volunteers. Since the study was to be conducted in patients with advanced cancer, a fixed‐sequence design was chosen, so that patients could receive the potentially active treatment after 1 week (and remain on treatment throughout the study and then continue to the rollover study).
All 16 patients who were enrolled in the study received the investigational product, completed the study, and were included in the safety and PK analyses. Dabrafenib and its metabolites hydroxy‐dabrafenib, carboxy‐dabrafenib, and desmethyl‐dabrafenib are inhibitors of OATP1B1 and OATP1B3 in vitro. In vitro data also demonstrated that dabrafenib induces CYP3A4 and CYP2B6 via activation of the pregnane X receptor and constitutive androstane receptor, which also regulates OATP1B1/1B3 expression.12, 13
Rosuvastatin is a known OATP1B1 substrate, 90% of which is excreted unchanged in the feces and the remaining through the CYP2C9 pathway. It is possible that repeat dosing of dabrafenib could induce OATP1B1/1B3 expression, leading to reduced plasma concentrations of rosuvastatin. In addition to being an OATP1B1 substrate, rosuvastatin has been identified as a substrate of the breast cancer resistance protein (BCRP).14, 15 BCRP is an efflux transporter that facilitates hepatobiliary excretion and decreases intestinal absorption of BCRP substrates.16 Dabrafenib and desmethyl‐dabrafenib are inhibitors of BCRP in vitro.7 Concomitant administration of a single dose of rosuvastatin 10 mg with dabrafenib after the initiation of dosing resulted in a 22% increase in rosuvastatin AUC0‐∞ and a 94% increase in rosuvastatin Cmax compared with the administration of rosuvastatin alone. The t1/2 was similar between the 2 treatment periods; however, the median tmax was earlier (3 vs 4 hours) when administered with dabrafenib. The increased exposure is in line with inhibition of hepatic uptake of rosuvastatin through OATP inhibition.17
Gemfibrozil is an OATP1B1 inhibitor, and the interaction between gemfibrozil and rosuvastatin in a clinical study was similar to the interaction between rosuvastatin and dabrafenib with regard to Cmax (2.21‐fold) but not AUC (1.9‐fold).18 The interaction between the combination of tipranavir/ritonavir 500 mg/200 mg twice daily for 11 days and rosuvastatin was more similar to the interaction between dabrafenib and rosuvastatin; rosuvastatin Cmax was increased by 2‐fold and AUC only by 26%.16 Similar to dabrafenib, tipranavir and ritonavir are BCRP inhibitors in vitro,19, 20 and ritonavir is an OATP1B1/1B3 inhibitor.21 The potential mechanism of interaction observed in the current study may be similar to that proposed with tipranavir/ritonavir, which is dual inhibition of hepatic OATP1B1/1B3 uptake and inhibition of BCRP, leading to decreased hepatobiliary excretion and increased absorption.16
Clinical drug interaction studies with gemfibrozil and other 3‐hydroxy 3‐methylglutaryl‐coenzyme A (HMG‐CoA) reductase inhibitors have shown increases in the plasma concentration of other HMG‐CoA reductase inhibitors.22, 23, 24, 25, 26 However, this effect was observed only to a very minor extent with atorvastatin, where coadministration with gemfibrozil increased plasma atorvastatin AUC0‐∞ by 24%,27 and with fluvastatin, where coadministration with gemfibrozil did not alter plasma fluvastatin PK.28 Dabrafenib is also an inducer of CYP3A4 and may reduce the exposure of many HMG‐CoA reductase inhibitors that are metabolized by CYP450 enzymes. Therefore, the net effect of dabrafenib on OATP1B1/1B3 inhibition and enzyme induction is unknown.
The same trend was observed for rosuvastatin PK parameters after dabrafenib administration for 15 days; however, Cmax was further increased (2.6‐fold vs 1.9‐fold) and the median tmax was earlier (1.5 versus 3 hours). For AUC, the 90%CI included 1 and was within the bioequivalence range. It appears that after repeat dabrafenib dosing, there is further inhibition instead of induction. The lack of observed transporter induction is consistent with data reported from a healthy volunteer study that showed no significant alteration in the PK parameters of rosuvastatin after 6 days of rifampin (inducer) dosing.29 With repeat dabrafenib dosing, both carboxy‐ and desmethyl‐dabrafenib accumulate, and both these metabolites have been shown to also inhibit OATP1B1/1B3 in vitro. Desmethyl‐dabrafenib has also been shown to inhibit BCRP. The further increase observed in rosuvastatin Cmax after repeat‐dose dabrafenib may be due to the increased metabolite concentrations at steady state. The hypothesis on the mechanism could be explored further by applying an in silico approach such as physiologically based PK modeling. A physiologically based PK model for dabrafenib has been published and verified30; in addition, a rosuvastatin compound library file is available in the Simcyp Simulator software (Certara, Princeton, New Jersey) that could be used for these follow‐up studies.
Midazolam is the preferred substrate for evaluating potential inhibitors and inducers of CYP3A catabolic activity. Concomitant administration of a single dose of midazolam 3 mg with dabrafenib after the initiation of dosing did not result in any significant changes in exposure or t1/2. Dabrafenib did not appear to inhibit CYP3A4‐mediated metabolism after initiation of treatment (1‐2 days). Coadministration of dabrafenib 150 mg twice daily for 15 days and a single dose of midazolam 3 mg decreased midazolam AUC by 65%. These results were consistent with previously reported results from the phase 1 dose‐escalating study,31 wherein midazolam exposure was decreased by 74%. The previous study used dabrafenib gelatin capsules, whereas the current study used the marketed HPMC capsules, which were shown to have a higher exposure (1.8‐fold AUC HPMC/gelatin).1 The reduction in midazolam exposure and decreased t1/2 are consistent with dabrafenib‐inducing CYP3A4‐mediated metabolism after 15 days of dabrafenib dosing (steady state).
Overall, dabrafenib and metabolite exposures observed on day 1 and at steady state were similar to those reported previously after administration of dabrafenib 150 mg twice daily after a single dose and at steady state.6
Overall, the AEs, SAEs, and laboratory abnormalities reported during this study are consistent with the safety profile of dabrafenib and the patient population under study. No unusual or unexpected events were reported. Grade 4 presyncope was reported in 1 patient; this, however, was attributed to midazolam dosing.
Additionally, the mini‐cocktail approach to administer midazolam and rosuvastatin significantly decreased the length of time the patient was required to participate in the drug interaction trial. Historical internal data had shown that the PK of each drug was not affected when administered as part of a 7‐probe cocktail compared with administering each drug alone.
Conclusions
This study, which followed a mini‐cocktail approach, showed that the exposure of drugs that are CYP3A4 substrates is likely to decrease when coadministered with dabrafenib, and that the concentrations of medicinal products that are sensitive OATP1B1/1B3 substrates (eg, atorvastatin) may increase during the absorption phase.
Conflicts of Interest
N.N., C.S.W., and D.‐Y.L. are former employees of Novartis and have nothing to disclose beyond their current affiliation. V.M. reports personal fees from Bristol Myers Squibb, Janssen, and Bayer, outside the submitted work. E.G. is an employee of Novartis and reports stocks in Novartis. E.B. is an employee of Novartis. E.M.‐C. declares no conflict of interest.
Funding
This study was sponsored by GlaxoSmithKline; dabrafenib and trametinib are assets of Novartis AG as of March 2, 2015.
Author Contributions
N.N., C.S.W., V.M., E.M.‐C., D.‐Y.L., E.G., and E.B. wrote the manuscript. N.N., V.M., and E.M.‐C. performed the research. N.N., C.S.W., V.M., D.‐Y.L., and E.B. analyzed the data.
Data Accessibility
This trial is registered at https://www.clinicaltrials.gov/. The identifier is NCT02082665.
Acknowledgments
The authors thank the patients and their caregivers for participating in the study. The authors thank Sharol Janice Rodrigues (Novartis Healthcare Pvt Ltd) for providing medical writing support/editorial support, which was funded by Novartis Pharmaceuticals Corporation in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Noelia Nebot: Former employee. Current affiliation: GlaxoSmithKline, Ermington, New South Wales, Australia
Christina S. Won: Former employee. Current affiliation: Bristol Myers Squibb, Summit, New Jersey, USA
Dung‐Yang Lee: Former employee. Current affiliation: University of Texas School Health Science Center, Houston, Texas, USA
Note: This work was presented as a poster at the Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics (ASCPT), March 21 to 24, 2018; Orlando, Florida, USA.
References
- 1.Ouellet D, Grossmann KF, Limentani G, et al. Effects of particle size, food, and capsule shell composition on the oral bioavailability of dabrafenib, a BRAF inhibitor, in patients with BRAF mutation‐positive tumors. J Pharm Sci. 2013;102(9):3100‐3109. [DOI] [PubMed] [Google Scholar]
- 2.Denton CL, Minthorn E, Carson SW, et al. Concomitant oral and intravenous pharmacokinetics of dabrafenib, a BRAF inhibitor, in patients with BRAF V600 mutation–positive solid tumors. J Clin Pharmacol. 2013;53(9):955‐961. [DOI] [PubMed] [Google Scholar]
- 3.Bershas DA, Ouellet D, Mamaril‐Fishman DB, et al. Metabolism and disposition of oral dabrafenib in cancer patients: proposed participation of aryl nitrogen in carbon‐carbon bond cleavage via decarboxylation following enzymatic oxidation. Drug Metab Dispos. 2013;41(12):2215‐2224. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence SK, Nguyen D, Bowen C, Richards‐Peterson L, Skordos KW. The metabolic drug‐drug interaction profile of dabrafenib: in vitro investigations and quantitative extrapolation of the P450‐mediated DDI risk. Drug Metab Dispos. 2014;42(7):1180‐1190. [DOI] [PubMed] [Google Scholar]
- 5.Ouellet D, Gibiansky E, Leonowens C, et al. Population pharmacokinetics of dabrafenib, a BRAF inhibitor: effect of dose, time, covariates, and relationship with its metabolites. J Clin Pharmacol. 2014;54(6):696‐706. [DOI] [PubMed] [Google Scholar]
- 6.Suttle AB, Grossmann KF, Ouellet D, et al. Assessment of the drug interaction potential and single‐ and repeat‐dose pharmacokinetics of the BRAF inhibitor dabrafenib. J Clin Pharmacol. 2015;55(4):392‐400. [DOI] [PubMed] [Google Scholar]
- 7.Ellens H, Johnson M, Lawrence SK, Watson C, Chen L, Richards‐Peterson LE. Prediction of the transporter‐mediated drug‐drug interaction potential of dabrafenib and its major circulating metabolites. Drug Metab Dispos. 2017;45(6):646‐656. [DOI] [PubMed] [Google Scholar]
- 8.Creusot N, Gassiot M, Alaterre E, et al. The anti‐cancer drug dabrafenib is a potent activator of the human pregnane X receptor. Cells. 2020;9(7):1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.US Food and Drug Administration . Clinical drug interaction studies — cytochrome P450 enzyme‐ and transporter‐mediated drug interactions. 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed January 22, 2020.
- 10.McFeely SJ, Ritchie TK, Yu J, Nordmark A, Levy RH, Ragueneau‐Majlessi I. Identification and evaluation of clinical substrates of organic anion transporting polypeptides 1B1 and 1B3. Clin Transl Sci. 2019;12(4):379‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halama B, Hohmann N, Burhenne J, Weiss J, Mikus G, Haefeli WE. A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin Pharmacol Ther. 2013;93(6):564‐571. [DOI] [PubMed] [Google Scholar]
- 12.Ihunnah CA, Jiang M, Xie W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim Biophys Acta. 2011;1812(8):956‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer zu Schwabedissen HE, Kim RB. Hepatic OATP1B transporters and nuclear receptors PXR and CAR: interplay, regulation of drug disposition genes, and single nucleotide polymorphisms. Mol Pharm. 2009;6(6):1644‐1661. [DOI] [PubMed] [Google Scholar]
- 14.Hirano M, Maeda K, Matsushima S, Nozaki Y, Kusuhara H, Sugiyama Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol Pharmacol. 2005;68(3):800‐807. [DOI] [PubMed] [Google Scholar]
- 15.Huang L, Wang Y, Grimm S. ATP‐dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab Dispos. 2006;34(5):738‐742. [DOI] [PubMed] [Google Scholar]
- 16.Pham PA, la Porte CJ, Lee LS, et al. Differential effects of tipranavir plus ritonavir on atorvastatin or rosuvastatin pharmacokinetics in healthy volunteers. Antimicrob Agents Chemother. 2009;53(10):4385‐4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higgins JW, Bao JQ, Ke AB, et al. Utility of Oatp1a/1b‐knockout and OATP1B1/3‐humanized mice in the study of OATP‐mediated pharmacokinetics and tissue distribution: case studies with pravastatin, atorvastatin, simvastatin, and carboxydichlorofluorescein. Drug Metab Dispos. 2014;42(1):182‐192. [DOI] [PubMed] [Google Scholar]
- 18.Schneck DW, Birmingham BK, Zalikowski JA, et al. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2004;75(5):455‐463. [DOI] [PubMed] [Google Scholar]
- 19.Matsson P, Englund G, Ahlin G, Bergstrom CA, Norinder U, Artursson P. A global drug inhibition pattern for the human ATP‐binding cassette transporter breast cancer resistance protein (ABCG2). J Pharmacol Exp Ther. 2007;323(1):19‐30. [DOI] [PubMed] [Google Scholar]
- 20.Weiss J, Rose J, Storch CH, et al. Modulation of human BCRP (ABCG2) activity by anti‐HIV drugs. J Antimicrob Chemother. 2007;59(2):238‐245. [DOI] [PubMed] [Google Scholar]
- 21.Alam K, Crowe A, Wang X, et al. Regulation of organic anion transporting polypeptides (OATP) 1B1‐ and OATP1B3‐mediated transport: an updated review in the context of OATP‐mediated drug‐drug interactions. Int J Mol Sci. 2018;19(3):855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Backman JT, Kyrklund C, Kivistö KT, Wang JS, Neuvonen PJ. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin Pharmacol Ther. 2000;68(2):122‐129. [DOI] [PubMed] [Google Scholar]
- 23.Kyrklund C, Backman JT, Kivistö KT, Neuvonen M, Laitila J, Neuvonen PJ. Plasma concentrations of active lovastatin acid are markedly increased by gemfibrozil but not by bezafibrate. Clin Pharmacol Ther. 2001;69(5):340‐345. [DOI] [PubMed] [Google Scholar]
- 24.Kyrklund C, Backman JT, Neuvonen M, Neuvonen PJ. Gemfibrozil increases plasma pravastatin concentrations and reduces pravastatin renal clearance. Clin Pharmacol Ther. 2003;73(6):538‐544. [DOI] [PubMed] [Google Scholar]
- 25.Mathew P, Cuddy T, Tracewell WG, Salazar D. An open‐label study on the pharmacokinetics (PK) of pitavastatin (NK‐104) when administered concomitantly with fenofibrate or gemfibrozil in healthy volunteers. Clin Pharmacol Ther. 2004;75(2):33. [Google Scholar]
- 26.Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80(6):565‐581. [DOI] [PubMed] [Google Scholar]
- 27.Backman JT, Luurila H, Neuvonen M, Neuvonen PJ. Rifampin markedly decreases and gemfibrozil increases the plasma concentrations of atorvastatin and its metabolites. Clin Pharmacol Ther. 2005;78(2):154‐167. [DOI] [PubMed] [Google Scholar]
- 28.Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol. 1995;76(2):80A‐83A. [DOI] [PubMed] [Google Scholar]
- 29.Zhang W, Deng S, Chen XP, et al. Pharmacokinetics of rosuvastatin when coadministered with rifampicin in healthy males: a randomized, single‐blind, placebo‐controlled, crossover study. Clin Ther. 2008;30(7):1283‐1289. [DOI] [PubMed] [Google Scholar]
- 30.Rowland A, van Dyk M, Hopkins AM, et al. Physiologically based pharmacokinetic modeling to identify physiological and molecular characteristics driving variability in drug exposure. Clin Pharmacol Ther. 2018;104(6):1219‐1228. [DOI] [PubMed] [Google Scholar]
- 31.Falchook GS, Long GV, Kurzrock R, et al. Dose selection, pharmacokinetics, and pharmacodynamics of BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res. 2014;20(17):4449‐4458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This trial is registered at https://www.clinicaltrials.gov/. The identifier is NCT02082665.